 Alireza Saidi-Mehrabad
Alireza Saidi-Mehrabad Patrick Neuberger
Patrick Neuberger Morteza Hajihosseini
Morteza Hajihosseini Duane Froese
Duane Froese Brian D. Lanoil
Brian D. Lanoil- 1Department of Biological Sciences, University of Alberta, Edmonton, AB, Canada
- 2School of Public Health, University of Alberta, Edmonton, AB, Canada
- 3Department of Earth and Atmospheric Sciences, University of Alberta, Edmonton, AB, Canada
Despite the presence of well-documented changes in vegetation and faunal communities at the Pleistocene-Holocene transition, it is unclear whether similar shifts occurred in soil microbes. Recent studies do not show a clear connection between soil parameters and community structure, suggesting permafrost microbiome-climate studies may be unreliable. However, the majority of the permafrost microbial ecological studies have been performed only in either Holocene- or Pleistocene-aged sediments and not on permafrost that formed across the dramatic ecosystem reorganization at the Pleistocene-Holocene transition. In our study, we used permafrost recovered in proximity to the Pleistocene-Holocene transition subsampled under strict sterile conditions developed for ancient DNA studies. Our ordination analyses of microbial community composition based on 16S RNA genes and chemical composition of the soil samples resulted into two distinct clusters based on whether they were of late Pleistocene or Holocene age, while samples within an epoch were more similar than those across the boundary and did not result in age based separation. Between epochs, there was a statistically significant correlation between changes in OTU composition and soil chemical properties, but only Ca and Mn were correlated to OTU composition within Holocene aged samples; furthermore, no chemical parameters were correlated to OTU composition within Pleistocene aged samples. Thus, the results indicate that both soil chemical and microbial parameters are fairly stable until a threshold, driven by climate change in our study, is crossed, after which there is a shift to a new steady state. Modern anthropogenic climate change may lead to similar transitions in state for soil biogeochemical systems and microbial communities in Arctic regions.
Introduction
The Pleistocene – Holocene transition was accompanied by rapid, extensive global climate and ecosystem changes (Barnosky et al., 2004; Guthrie, 2006). These rapid changes forced the Earth system to cross a climate threshold causing a major transition in terrestrial biosphere and pedogenesis from one stable state to another stable state and coincided with extinction of megafauna, reorganization of plant communities, and accompanying changes in soil chemistry and sedimentation (Alley et al., 2003; Steffen et al., 2018; Lacelle et al., 2019). At high latitudes, these changes may be preserved in relict permafrost (Shapiro and Cooper, 2003; Froese et al., 2009; Gaglioti et al., 2016). These natural biological and chemical archives have been used to reconstruct late Quaternary paleoclimate (Porter et al., 2019), vegetation (Willerslev et al., 2014), and faunal communities (Haile et al., 2009; Lorenzen et al., 2011). Despite the presence of well-documented changes in vegetation and faunal community structure across the Pleistocene-Holocene transition (Guthrie, 2006; Mann et al., 2016), it is still unclear whether these changes were associated with concomitant restructuring of soil microbial communities.
Recent studies have shown that the main factors governing microbial community structure in permafrost appear to be the age of the samples (Mackelprang et al., 2017; Burkert et al., 2019; Liang et al., 2019), ice content (Burkert et al., 2019), dispersal limitation and physical/thermodynamic constraints (Bottos et al., 2018), with little to no correlation with soil chemical parameters. For example, one recent study indicated that microbes within three Pleistocene-aged permafrost chronosequences changed in composition in response to increasing age, with a corresponding increase in survival strategies (Mackelprang et al., 2017). These correlations suggest that these environmental pressures, arising from harsh permafrost conditions, select for a subset of species and that the community structure deviates from the community present at the time the permafrost was formed (Willerslev et al., 2004; Kraft et al., 2015; Mackelprang et al., 2017; Liang et al., 2019). However, another study performed on two Pleistocene-aged permafrost samples of a similar age but differing origins (lake-alluvial sediments and Yedoma/ice complex sediments) indicated the presence of two distinct microbial communities, which were formed in response to differing environmental settings and not due to the age of the samples alone (Rivkina et al., 2016). As a result, it is not clear whether relict permafrost microbial communities provide a window into past soil microbial communities, or are the strict remnants of selective pressures of the permafrost environment, or some combination of the two.
In this study, we selected samples on either side of the Pleistocene-Holocene transition from a permafrost core extracted from central Yukon, a part of Eastern Beringia. The permafrost of Eastern Beringia contains exceptional proxy records of climate change (Froese et al., 2009). Furthermore, the Pleistocene-Holocene transition zone preserved in Eastern Beringian permafrost is prominent in the core stratigraphy, allowing for detailed microbiological and soil physicochemical analyses (Porter et al., 2019). The core in this study was collected about 1.5 m adjacent to, and shares stratigraphy with a well-dated and chemically and physically characterized core spanning the last ca. 16,000 years (Davies et al., 2018; Porter et al., 2019).
Materials and Methods
Permafrost Coring Location and Analysis of the Soil Texture
The topography of our sampling site (DHP174-13L) along with the paleoenvironmental setting, stratigraphy and coring strategies were described previously (Davies et al., 2018; Porter et al., 2019). We obtained a 3.97 m long permafrost core (called DHL_16, collected in May 2016) about 1.5 m lateral to the core reported by Porter et al. (2019). Three intervals from depths of 174–210, 296–319, and 327–365 cm (termed DHL_16_1, DHL_16_2, and DHL_16_3 in this manuscript, respectively) were selected from the DHL_16 core for microbiological and chemical analyses. We were careful to sample near the Pleistocene-Holocene transition zone while avoiding the transition zone itself to minimize any effects of water migration in the paleoactive layer [detailed in Porter et al. (2019) for the stable isotopes of the pore waters in that study]. Active layer (or seasonally thawed soils) can allow water leaching from the paleo-surface to deeper depths (<∼50 cm at this site today) and result in mixing of the surface water, potentially impacting microbial and edaphic properties. We used the depleted water isotopes to provide further support that the waters (and thus the microbes) have not been mobilized since they froze (detailed in Porter et al., 2019).
The core stratigraphy, reported by Davies et al. (2018) and Porter et al. (2019), documents three prominent units that are readily recognized in both cores: units 1, 2, and 3 from the bottom of the core to the surface. The youngest unit sampled, corresponding to unit 3 of Porter et al. (2019) consists of peat (sampled interval 174–210 cm; DHL_16_1) and dates to the early Holocene (∼8–10.5 k cal yr BP), whereas DHL_16_2 and DHL_16_ 3 are loessal silts (eolian silt) (sampled at 296–319 cm and 327–365 cm respectively) both associated with the late Pleistocene (∼14.3–15 k cal yr BP, and ∼15–16 k cal yr BP, respectively), based on the age model of Porter et al. (2019). At the site, the peat/silt boundary was determined to be at 244 cm (which was dated to ∼10.5 k cal yr BP, Supplementary Figure S1). These three selected intervals were vertically cut into 1/3 and 2/3 subsections with the aid of a masonry saw. The 2/3 sections were used for DNA extraction, while the 1/3 sections were used for chemical analyses (Saidi-Mehrabad et al., 2020).
Soil textures of Holocene-aged samples from 210 cm depth and Pleistocene-aged samples from 296 cm depth were examined and photographed under 4.5× and 67× magnifications of a zoom stereomicroscope (Olympus-Life Sciences SZ61, Japan). These two depths were near the 244 cm peat/silt boundary. Samples were allowed to thaw and dry at 70°C in a table top oven for 48 h prior to microscopy.
Permafrost Edaphic Parameter Analysis
Selected intervals of DHL_16_1, DHL_16_2, and DHL_16_3 were sub-sectioned for soil chemical analysis based on a previously described method (Saidi-Mehrabad et al., 2020). Briefly, the 1/3 core sections were cut into 1 cm3 cubes with a hand saw in a 4°C cold room for measuring the basic chemical parameters of these samples, including organic carbon (OC), pH and gravimetric water content (GW) (Saidi-Mehrabad et al., 2020). Total carbon (TC) and total nitrogen (TN) were measured from 0.5 g of subsections based on a dry combustion method with the aid of an Elemental Analyzer (EA 4010; Costech International Strumatzione, Italy) (Sparks et al., 1996). Nitrate/nitrite (NO–3/NO2–) was analyzed using ∼ 5 g of soil material with a colorimetric diazo coupling method by a SmartChem Discrete Wet Chemistry analyzer (model 200, Westco Scientific, United States) (Maynard et al., 1993). Ammonium (NH4+) was measured from 0.5 g of subsections based on a colorimetric Berthelot protocol (Maynard et al., 1993). Electrical conductivity (EC) was measured at 1:5 soil:water (w/v) ratio, calibrated at 20°C with 0.01M KCl solution (Hardie and Doyle, 2012) using an EC500 ExStik® II conductivity measuring device (EXTECH instruments, United States). Dissolved metals were extracted from 0.5 g of sample by HNO3/HCl digestion and measured via inductively coupled plasma-optical emission spectroscopy (iCAP6300, Thermofisher Scientific, Canada) (Skoog et al., 2007).
DNA Extraction and Sequencing
Prior to DNA extraction the 2/3 sections associated with the DHL_16_1, DHL_16_2, and DHL_16_3 intervals were cut into ∼6 × 3.5 × 2.5 cm rectangular pieces (n = 16) and the exterior of these rectangles were painted with a total of 3 × 108 cells/ml of Escherichia coli (E. coli) (strain DH10B) suspended in 50 ml of 1 × PBS using a 25 mm paintbrush (Saidi-Mehrabad et al., 2020). The E. coli strain used in our contamination monitoring procedure harbored a mNeonGreen-expressing and ampicillin-resistant pBAD/His B vector, which allowed us to trace the contamination at a much smaller levels via direct PCR targeting the vector or macro-photography of the glowing mNeonGreen protein under 470 nm wavelength (Saidi-Mehrabad et al., 2020). All rectangular pieces were decontaminated by the combination of bleach washing and scraping under sterilize conditions designed for ancient DNA analysis in a class 1000 clean lab with no history of sample processing (Saidi-Mehrabad et al., 2020). Genomic DNA was extracted from a total of ∼10 g of homogenized thawed soil at room temperature from each 6 cm subsection using a modified ZymoBIOMICS DNA Microprep kit (Zymo Research, United States) protocol (Saidi-Mehrabad et al., 2020). DNA concentration was measured with a Qubit dsDNA HS Assay Kit according to manufacturer’s protocol (Thermofisher Scientific, Canada). The extracted DNA was tested for the presence of the biological tracer used in our contamination monitoring procedure via PCR (performed in triplicate and in a variety of dilutions) as described previously (Saidi-Mehrabad et al., 2020). We were unable to obtain sufficient DNA devoid of the contamination tracer for sequencing from DHL_16_3 sub-sections, as a result, these samples were excluded from further molecular analysis (Supplementary Figure S2). Thus, a total of six segments, three from DHL_16_1 (174–179, 179–184, and 189–194 cm) and three from DHL_16_2 (296–301, 305–310, and 314–319 cm), passed our quality control steps (Saidi-Mehrabad et al., 2020) for microbial community analysis.
Clean genomic DNA extracted from the permafrost samples, a lab-constructed mock community (Supplementary Figure S3), and DNA extraction blanks (Supplementary Table S1) were sequenced based on the V4 region of the 16S rRNA genes by using standard Earth Microbiome Project (EMP) primers 515F and 806R (Caporaso et al., 2011). The platform used by the commercial sequencing provider (Microbiome Insights, Canada) was an Illumina MiSeq machine using a 250-bp paired-end kit (V2 500-cycle PE Chemistry; Illumina, United States). No additional library preparation kits were used, as the libraries were constructed in a single PCR step. Generated sequences were demultiplexed via bcl2fastq2 2.20 according to Illumina guidelines (Illumina, United States).
Quality Control of Raw Sequences and OTU Construction
A combination of USEARCH 10.0.240 (Edgar, 2010) and Mothur 1.39.5 (Schloss et al., 2009) pipelines were utilized for analyzing the sequences. USEARCH was used to merge the reads and to form contigs based on the criteria recommended in USEARCH 10 manual for analyzing sequences generated from the V4 region of the 16S rRNA genes. Mothur was used to merge the raw reads into a single merge file and to normalize the reads to the lowest read counts via random subsampling. OTU picking, chimera, and universal singleton checks were performed with the UPARSE pipeline via de novo/greedy heuristic algorithm (Edgar, 2013). The threshold used for OTU clustering was 97% similarity. Constructed OTUs were mapped to taxonomy via SINTAX algorithm by using a trained V4 16S rRNA RDP (v16; 13k sequences) database (Cole et al., 2014; Edgar, 2016). The confidence level cut-off at each taxonomic level was set to 0 to prevent data loss. In addition to the bootstrap values obtained from SINTAX algorithm, the taxonomic identity of the sequences were manually investigated via Basic Local Alignment Search Tool (BLAST) in the NCBI database (Altschul et al., 1990). All strains detected in the blank controls, Eukarya, Archaea species and Cyanobacteria/Chloroplast (termed anomalous sequences) were removed from downstream community analyses (Supplementary Table S2).
In summary, 98.1% of the reads passed the filtering Q score parameters (with expected error < 1.0) and resulted in 87843 filtered reads. Of these, 11417 reads were unique (de-replicated) and 8287 were singletons (72.6%). OTU counts were rarified to 1768 sequences per sample by random subsampling. UPARSE picked 778 OTUs, with 612 OTUs assigned to domain Bacteria via SINTAX after removal of anomalous sequences. Despite a relatively small number of OTUs and sample size, the constructed rarefaction curve demonstrated enough sampling depth for downstream diversity analyses (Supplementary Figure S4). Raw sequences obtained in this study have been archived in NCBI Sequence Read Archive under the ascension number of PRJNA607368, and BioSample accession of SAMN14130768.
Community Analysis
All the statistical computations that utilized the R language were performed in R Studio environment 1.1.442. The soil parameters were log10 (x + 1) transformed and were later used to create a variable matrix for PcOrd version 6.22 (Wild Blueberry Media, Inc., United States) and a Phyloseq 1.16.2 object for alpha and beta diversity analyses (McMurdie and Holmes, 2013). The possible differences in OTU structure between groups were calculated with Bray-Curtis dissimilarity matrix and clustering was calculated based on average linkage in PcOrd version 6.22. Results were visualized in a principal coordinate analysis biplot (PCoA) and statistical support for differences between groups was calculated with the aid of Multi-Response Permutation Procedure (MRPP) with 999 randomized permutation tests in PcOrd version 6.22 (Mielke et al., 1976). The coefficient of determination of chemical parameters to sample groups (Holocene or Pleistocene) were assessed based on the “envfit” function of vegan ver 2.4-2 package with max p < 0.05 and 999 permutation tests (Oksanen et al., 2007). Differences in chemical composition between samples were visualized in a principal component biplot (PCA). Samples were clustered based on average linkage and statistical significance between clusters were assessed based on MRPP in PcOrd version 6.22 (Mielke et al., 1976). The shared and unique OTUs between and within clusters detected in PCoA analysis were investigated with a co-occurrence network test based on a maximum ecological distance of 0.8 and distance matrix of Jaccard by the aid of Phyloseq 1.16.2 package (McMurdie and Holmes, 2013). To assess which taxa were significantly differentially abundant in Pleistocene and Holocene groups, the DESeq2 differential abundance method (Love et al., 2014) with a threshold of 0.01 and p < 0.05 was used on the top 10 dominant phyla within all samples combined (microbiomeSeq package 0.1). In order to assess the sequence similarity of the Pleistocene- and Holocene-aged bacteria to published bacteria, each representative sequence of an OTU was compared to publicly available sequences via BLAST in NCBI (standard nucleotide (nr/nt) database). Uncultured/environmental sample sequences were excluded from our search criteria. The top sequence hit similarity values for each individual group were averaged and compared with each other. The possible effect of epoch-based separation on the relationship between soil chemistry and changes in OTU structure were investigated using a negative binomial many-generalized linear model (ManyGLM) (Warton et al., 2012). The model performed multiple random re-samplings via Monte Carlo algorithm to investigate many correlated variables while considering the strong mean-variance relationship due to the presence of rare species and high zero inflation (Warton, 2011; Warton et al., 2012). The statistical significance of this model was based on Wald’s test, which employed a generalized estimating equations approach (Warton et al., 2012). The model was executed with the mvabund 4.0.1 package (Wang et al., 2012). This model was also used to assess the differential abundance of OTUs shared between Pleistocene- and Holocene-aged samples with the same criteria described above.
Live/Dead Microscopy Assay
The viability assay was performed with a Live/Dead BacLight Bacterial Viability Kit (Invitrogen Thermo Fisher, Canada) and a DM RXA fluorescence microscope (Leica Microsystems, Germany). The viable cells were enumerated via green fluorescence at 470 nm excitation and the dead cells were enumerated via red fluorescence at 530 nm excitation. A 36 cm section from DHL_16_1 and a 23 cm section from DHL_16_2 were cut, homogenized and subsampled in quadruplicate as representative Holocene and Pleistocene samples, respectively. For each replicate subsample, 15 fields of view were counted under both wavelengths and the resulting n = 60 mean observation was used to calculate the abundance and live/dead proportion of the cells. The detailed protocol is available in the supporting information.
Results
The Holocene-aged permafrost sediments had properties distinct from Pleistocene-aged permafrost sediments, matching the descriptions of Fraser and Burn (1997) and Kotler and Burn (2000). The most noticeable difference was Holocene sediments were dominated by peat, while Pleistocene sediments were comprised of silty loess with frozen rare lenses of ice and largely lacking visible plant remains (Supplementary Figures S2A–D). No clear differences in texture were observed for samples within each epoch (Supplementary Figure S2). The presence of well-preserved plant macroremains from the peat suggest a relatively uninterrupted aggradation of permafrost with the plant material since the time of deposition, and that the permafrost of our sampling region formed syngenetically, or as the surface aggraded, without signs of previous thaw; this is also reflected by the water isotopes from these cores reported by Porter et al. (2019).
All measured chemical parameters differed significantly between Pleistocene and Holocene sediments (Tables 1, 2 and Supplementary Figures S1, S5). Similar to texture, none of the measured chemical parameters showed statistically significant changes within epochs. Holocene samples had significantly higher organic carbon (p < 0.001), nitrate/nitrite (p < 0.01), water (p < 0.01), and total nitrogen (p < 0.01) as well as significantly lower metals (p < 0.05), electrical conductivity (p < 0.05), and pH (p < 0.001) than Pleistocene samples (Tables 1, 2).
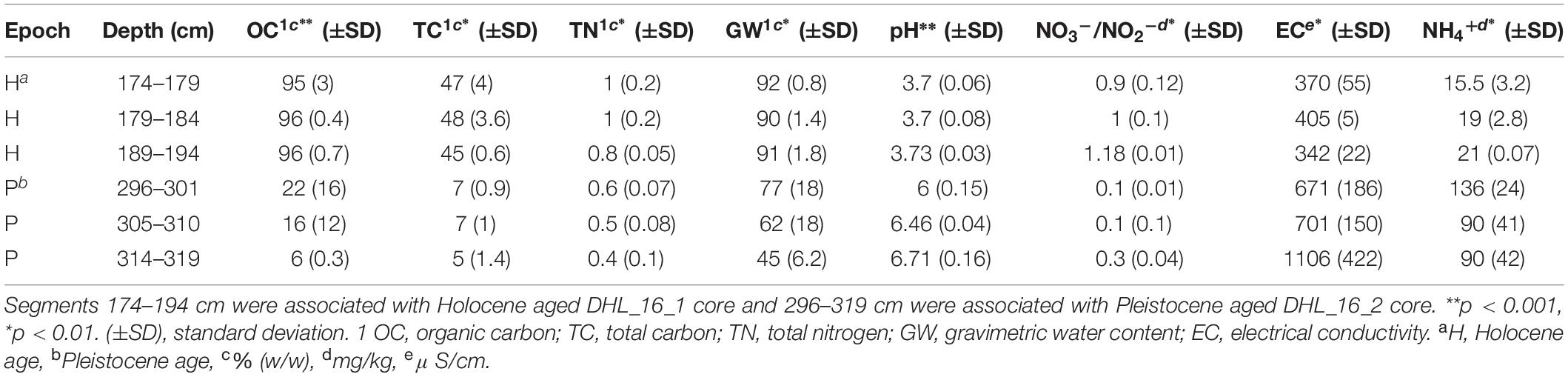
Table 1. Basic soil edaphic parameters of the selected segments for microbial analysis.
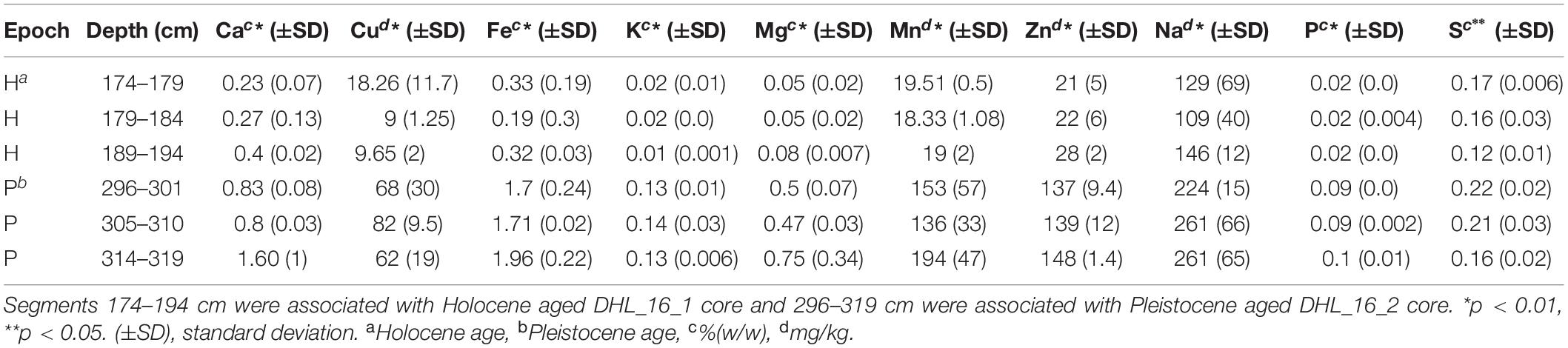
Table 2. Measured dissolved metals of the selected segments for microbial analysis.
In addition to dramatic changes in soil composition and chemistry, there was significantly higher cell viability and cell abundance in Holocene samples than in Pleistocene samples, with ∼8 fold higher viable cells (p < 0.001), ∼2 fold fewer dead cells (p < 0.001) and ∼2 fold higher total cells (p < 0.001) (Figure 1). Direct microscopic cell counts demonstrated Holocene samples had a total of 1.12 × 107 (±4.44 × 106) cells g–1 of wet soil, with 84% viable cells and 16% non-viable cells. Pleistocene samples had a total of 6.47 × 106 (±9.57 × 105) cells g–1 of wet soil, with 17% viable cells and 83% non-viable cells (Figure 1).
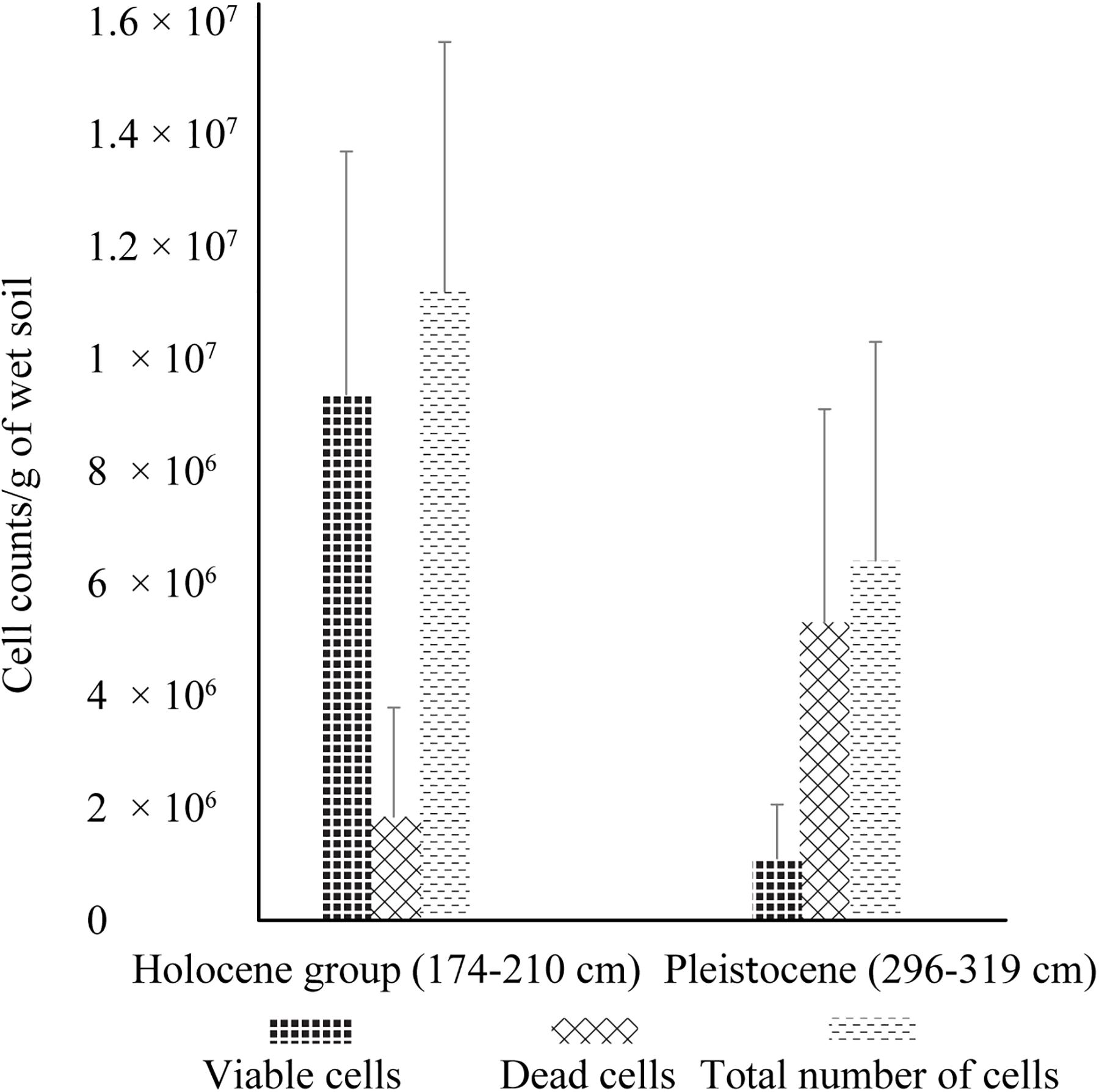
Figure 1. Live/dead cell counts of Holocene and Pleistocene aged samples. Error bars represent one standard deviation from the mean (n = 60 mean observation). For this experiment, samples from 174–210 cm (n = 4) and 296–319 cm (n = 4) were selected to investigate cell viability in Holocene aged and Pleistocene aged soils. These depths were selected based on their close proximity to the Pleistocene-Holocene transition zone of 244 cm. The statistical significance (Student’s t-test) of fold change in cell counts from Holocene to Pleistocene is as follows: fold change in viable cells (p < 0.001), fold change in dead cells (p < 0.001), and fold change in total cells (p < 0.001).
Overall microbial diversity based on 16S rRNA genes was either not significantly different between Holocene and Pleistocene samples (as measured by observed OTUs, Shannon diversity index, Chao1 richness estimator, Simpson evenness, and Fisher’s alpha diversity index), or the statistical support for differences was weak (as for the Mann–Whitney U test) (Supplementary Table S3). In a PCoA biplot, composite Holocene and Pleistocene samples demonstrated a dramatic dissimilarity in OTU composition. The microbial communities formed two clusters based on epoch; there was no clear differentiation within the epoch-based groups (Figure 2). Similar to soil texture, chemistry and cell abundance, each epoch-based cluster had a unique set of OTUs not present in the other epoch, which are probably selected by the environmental condition of each permafrost soil. The OTUs within and between epochs demonstrated that of 336 OTUs within Holocene samples, 84.5% were unique to this group (which we termed core Holocene OTUs, 284 OTUs), and only 15.5% were shared with the Pleistocene group (which we termed Pleistocene-Holocene shared, 52 OTUs) (Supplementary Figure S6). Similarly, of a total of 328 OTUs within the Pleistocene cluster, 84.1% of the OTUs were unique to this group (which we termed core Pleistocene OTUs, 276 OTUs), and only 15.8% of the OTUs were in Pleistocene-Holocene shared group (Supplementary Figure S6). The three dominant OTUs that formed the top 30% of the composite Pleistocene and Holocene samples were associated with the Pleistocene-Holocene shared group. In general, OTUs found within this cluster were slightly more dominant in Pleistocene samples (52.1%) than Holocene samples (47.8%).
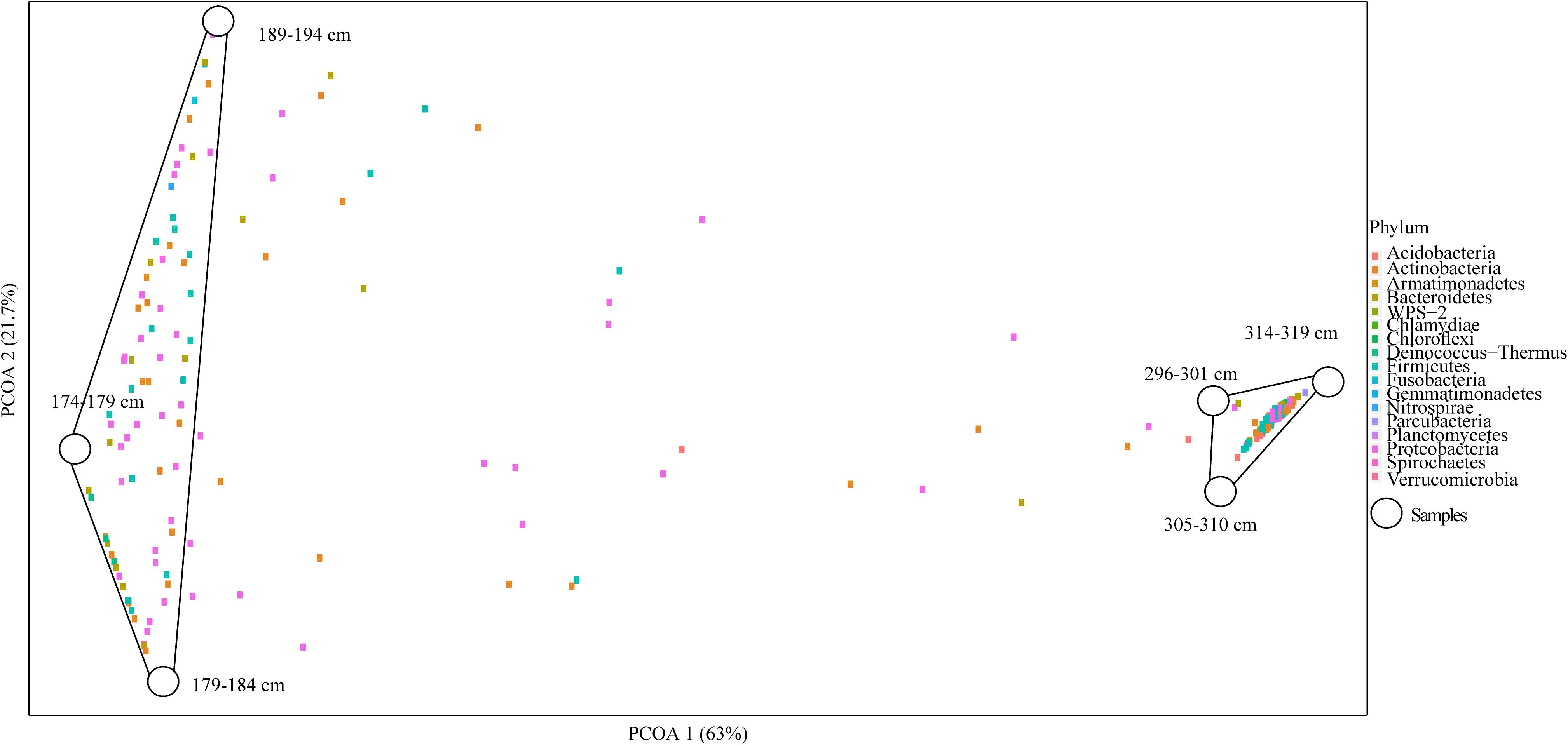
Figure 2. PCoA biplot of the OTU composition in Holocene (n = 3) and Pleistocene (n = 3) aged samples. Combined Axis 1 (PCOA1) and Axis 2 (PCOA2) components explained a total of 84.7% of the dissimilarity between the clusters in the system (p = 0.022). Samples 174–179, 179–184, 189–194 cm formed the Holocene cluster (8–10 k cal yr BP) and 296–301, 305–310, 314–319 cm formed the Pleistocene aged cluster (14–15 k cal yr BP). In the ordination space, samples (5 cm segments of DHL_16-1 and 2) have been shown with white circles and OTUs with colored squares. Colors of OTUs represent their taxonomic identity at the phylum level.
Core Pleistocene and Holocene sequences were individually compared to the standard reference DNA sequence database in NCBI via BLAST. The average percent similarity of core Pleistocene OTU 16S rRNA gene sequences to published bacterial sequences was 92.1% (±0.05%; n = 276 OTUs) in comparison to Holocene OTUs, which was 97.5% (±0.03%; n = 284 OTUs) (p < 0.001). Since the average of the 16S rRNA gene sequence similarity value of Pleistocene OTUs were less than the 97% identity boundary between unknown and known species (Stackebrandt and Goebel, 1994), they are uncharacterized and are more different from extant bacterial communities in databases than are Holocene OTUs. Pleistocene-Holocene shared OTUs (n = 52) demonstrated similarity to characterized strains similar to that for the Holocene OTUs (97.3% ± 0.05%) (p > 0.05) and not Pleistocene OTUs (p < 0.001).
Differential abundance analysis of the top 10 dominant phyla in composite Holocene and Pleistocene samples identified five phyla (Actinobacteria, Acidobacteria, Proteobacteria, Deinococcus-Thermus and Fusobacteria) as the main drivers of differences in taxonomy (p < 0.001) between Pleistocene and Holocene samples (Supplementary Figures S7, S8). In composite Holocene samples, 94% of the sequences fell within the Proteobacteria (56%), Actinobacteria (17%), Firmicutes (15%), and Bacteroidetes (5%) (Figure 3). Deinococcus-Thermus and Fusobacteria were part of the core Holocene samples and together constituted 3.58% of the community. On the other hand, 98% of the composite Pleistocene sequences were Actinobacteria (43%), Firmicutes (24%), Proteobacteria (16%), Acidobacteria (8%), and Bacteroidetes (6%). A similar distinction in taxonomic composition between composite Holocene and Pleistocene samples was also evident at the class level (Supplementary Figure S9). In the Pleistocene-Holocene shared group, 98% of the OTUs were associated with Proteobacteria (88%), Actinobacteria (8%), Firmicutes (2%) and Bacteroidetes (1%) respectively (Supplementary Figure S10a). Although these OTUs were shared between Pleistocene and Holocene samples the abundance of these OTUs were significantly different between epochs (p < 0.001) (Supplementary Figure S10b).
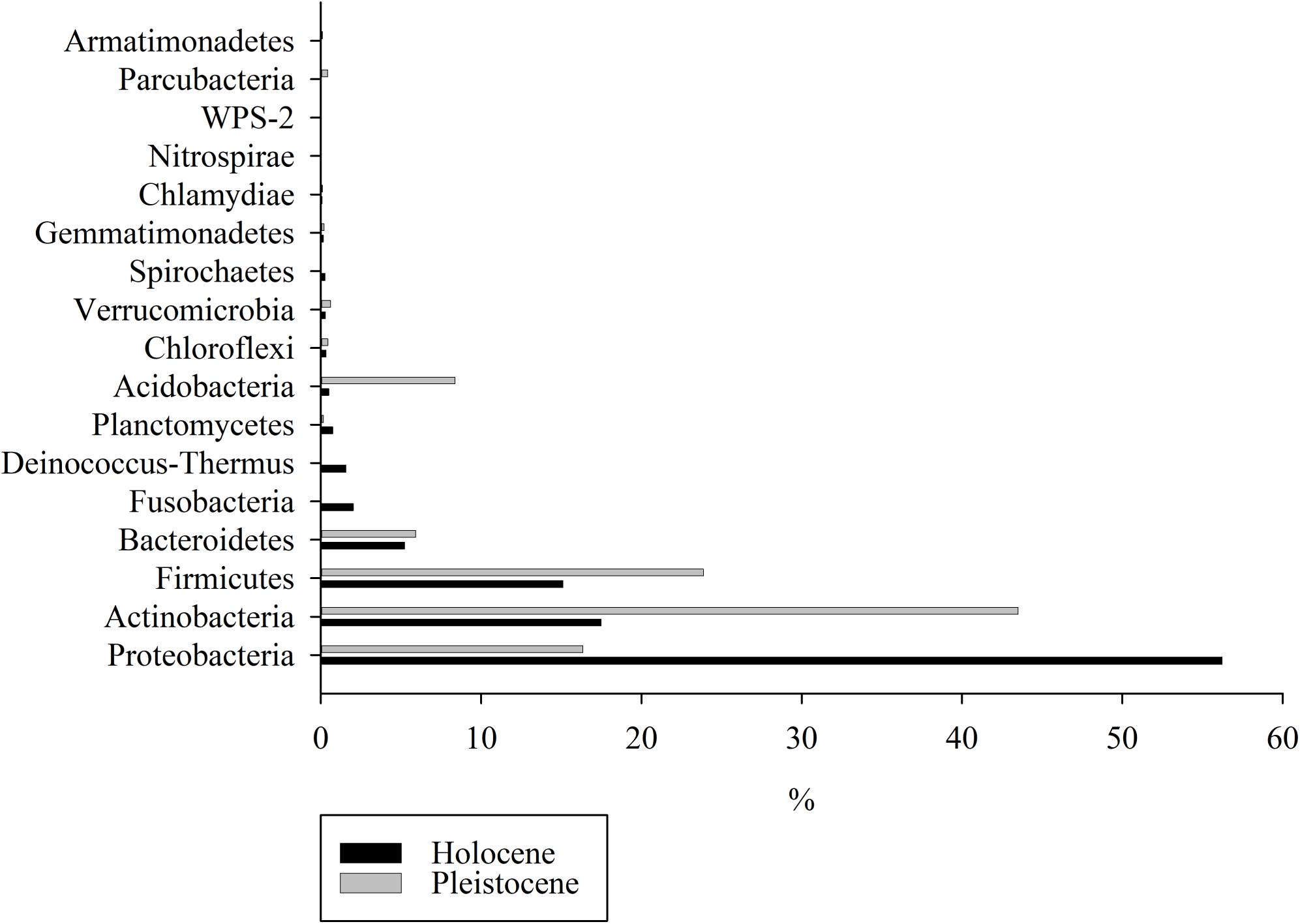
Figure 3. Relative abundance analyses of the OTUs detected in Holocene-aged and Pleistocene-aged permafrost samples at the phylum level.
To test for possible correlation between epoch-based separation of the microbial community structure and changes in soil chemical parameters, a ManyGLM model was utilized. The ManyGLM indicated that within the composited Holocene samples, only Ca and Mn had a statistically significant correlation (p < 0.05) to the core OTUs. The other 20 measured variables, including depth of the samples, did not show any significant relationship to the core OTUs (Table 3). Within Pleistocene samples, no significant correlation was found between core OTUs and any measured variable, including depth of the samples. However, between the Pleistocene and Holocene groups, there was a clear correlation between shift in epoch, changes in soil chemical parameters and reorganization in core OTU compositions (Table 3).
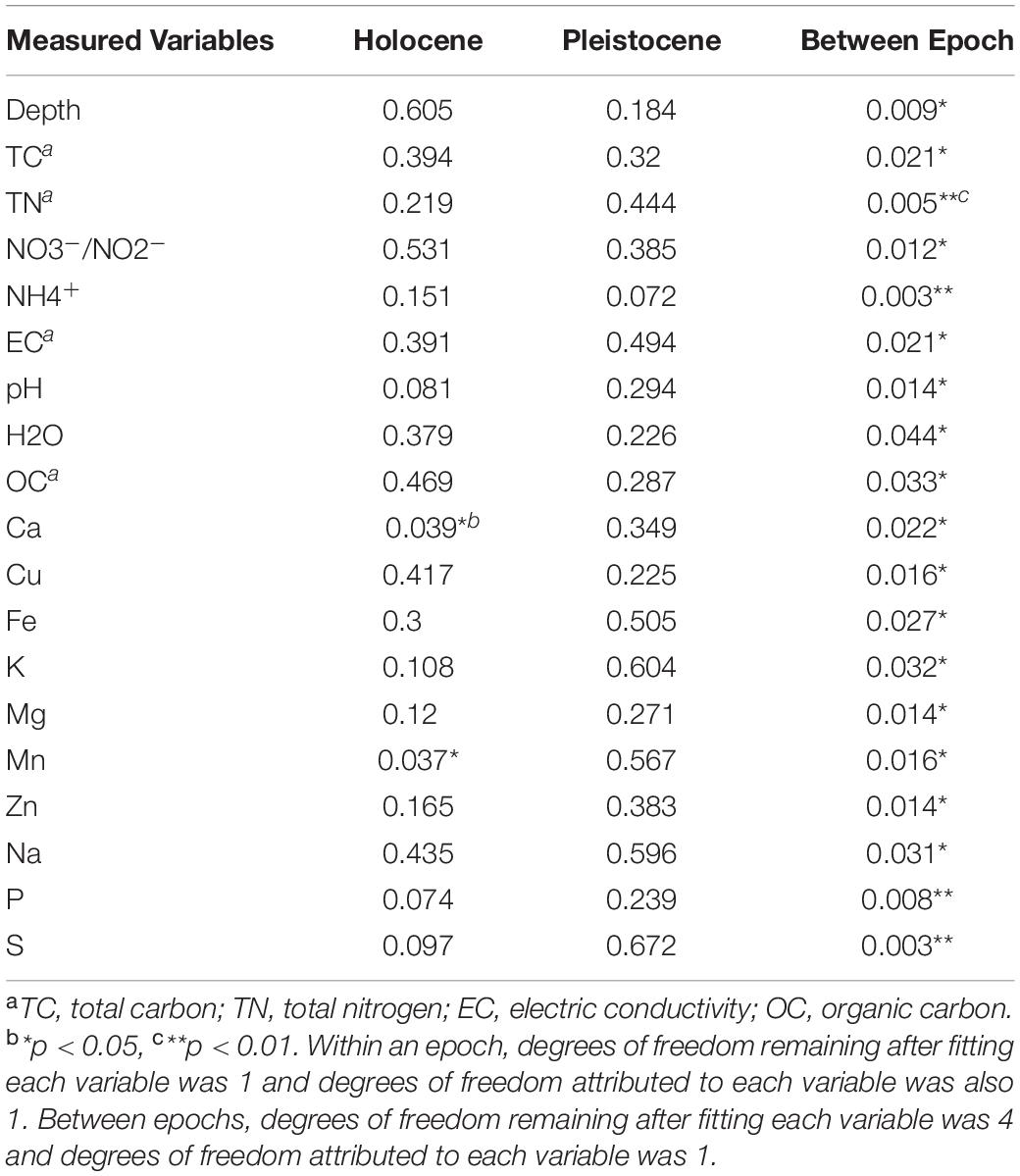
Table 3. The probability values calculated based on Wald’s generalized estimating equation in the context of a negative binomial many generalized linear model (ManyGLM).
Discussion
The perennially frozen nature of permafrost prevents movement of entrapped bacteria, creating a dispersal limitation. Furthermore, it is a challenging environment in terms of energy and nutrient availability (thermodynamic constraints) and damage to cells (physical constraints) (Mackelprang et al., 2017; Bottos et al., 2018; Burkert et al., 2019; Liang et al., 2019). Therefore, permafrost creates an environmental filter that only allows species resistant to these effects to survive, dramatically changing the composition of the community (Mackelprang et al., 2017; Bottos et al., 2018). Furthermore, these effects are compounded with time, further shifting the communities as the permafrost ages (Mackelprang et al., 2017). Hence, it is not clear if the surviving community members can be used as a paleoenvironmental tool.
Most previous studies of permafrost microbial community structure or community-level function have been conducted on permafrost chronosequences from a single epoch (e.g., Mackelprang et al., 2011, 2017; Hultman et al., 2015). Therefore, it is not known whether there is a shift in the permafrost microbial community structure close to the Pleistocene-Holocene transition. We observed dramatic differences in soil chemistry and OTU composition in samples from either side of the Pleistocene-Holocene transition (Figure 2, Supplementary Figures S1, S5, S6, and Tables 1, 2). A small number of OTUs (n = 52) that were able to persist despite the dramatic change during the Pleistocene-Holocene transition and the permafrost freezing-filtering effect were shared among the Pleistocene and Holocene samples (Pleistocene-Holocene shared group; Supplementary Figure S6). However, their abundance differed dramatically between epochs (Supplementary Figure S10b). No significant differences were observed between samples within an epoch (despite a difference in age between the samples). The lack of clear observable correlations between chemistry and microbiology within epochs is consistent with prior observations (e.g., Mackelprang et al., 2017; Bottos et al., 2018).
Based on our data, microbial community structure within an epoch is relatively stable over time, likely due to physiological flexibility and community stability of the microbial community (Shade et al., 2013). Thus, the environmental filter of the frozen and isolated conditions in the permafrost creates a strong selection pressure on microbial communities that are initially similar. Any relatively small signal of microbial community differences at the time of formation are lost. However, once the system surpasses a threshold, both chemical and microbial parameters rapidly shift to a new stable state. Some OTUs are able to persist, despite the dramatic change during the Pleistocene-Holocene transition and the permafrost freezing filtering effect, and were shared among the Pleistocene and Holocene samples (Pleistocene-Holocene shared group; Supplementary Figure S6) (Mandakovic et al., 2018). This scenario resembles the threshold hypothesis (also known as the tipping point hypothesis) (Scheffer et al., 2001), wherein a critical threshold exists in a system beyond which perturbations cause extensive changes (called “catastrophic” by Scheffer et al., 2001) and the system shifts from one stable state to another (Allison and Martiny, 2008; Shade et al., 2012; Mikkelson et al., 2016). Many paleoclimatic records indicate that the Pleistocene-Holocene transition was rapid and caused the climate system to cross a threshold, triggering a rapid transition to a warmer Holocene climate (Alley et al., 2003; Guthrie, 2006; Boers et al., 2018). Such an abrupt change is consistent with our ManyGLM model (Table 2), which did not detect any statistically significant correlation between OTU composition and soil chemical parameters within an epoch, with the exception of weak statistical support for correlations with Mn and Ca in Holocene samples (Table 2). However, between epochs, the ManyGLM model indicated a statistically significant change in microbial community structure and chemical profile (Table 2). Hence, we hypothesize that both soil chemical and microbial parameters were relatively stable until a threshold was reached at the Pleistocene-Holocene transition, after which there was a drastic shift to a new set of chemical and microbial parameters.
One implication of the threshold hypothesis described above is that it may explain why some decades-long in situ soil warming experiments have shown little or no change in microbial community structure (e.g., Kuffner et al., 2012), while lab-based incubations have shown dramatic shifts in community structure in a matter of days (e.g., Mackelprang et al., 2011). This difference between field and lab outcomes is likely because field-based studies generally involve moderate heating of the surface active layer of soil and thus do not strongly affect the underlying permafrost. In these experiments, microbes are able to gradually acclimate to moderate warming and the community resists shifts in composition (Allison and Martiny, 2008; Bradford et al., 2008; Rousk et al., 2012; Metcalfe, 2017). In contrast, in laboratory-based experiments, small amounts of permafrost thaw rapidly in response to increased temperature, leading to significant changes in the physicochemical parameters of the soil (Vonk et al., 2015) accompanied by dramatic changes in microbial community structure (e.g., Mackelprang et al., 2011; Schostag et al., 2019). Even changes at subzero temperatures in laboratory microcosms have been reported to alter microbial community structure (Tuorto et al., 2014).
Changes in OTU and chemical composition between epochs were accompanied by major changes in taxonomy (Supplementary Figures S7, S8). Proteobacteria-related species were dominant in Holocene samples, whereas Actinobacteria-related species dominated Pleistocene samples (Figure 3 and Supplementary Figures S7, S8). Species associated with phyla Proteobacteria and Actinobacteria are the most dominant species in soil worldwide (Janssen, 2006), including cold environments such as permafrost (e.g., Gittel et al., 2014; Zhang et al., 2016). Although phylogeny cannot be used to directly infer physiology and broad taxonomic groups (such as phyla) can include organisms with many different physiological features, strains affiliated with Actinobacteria have been characterized as oligotrophs adapted to nutrient-poor environments, while strains associated with Proteobacteria have been characterized as copiotrophs adapted to carbon-rich systems (De Vries and Shade, 2013; Itcus et al., 2018). Competition for nutrients among species associated with these two phyla have been reported (Goldfarb et al., 2011). According to a recent revision in environmental filtering theory, abiotic factors initially select certain strains, and then the second phase of selection pressure occurs based on species competition for resources (Freedman and Zak, 2015; Kraft et al., 2015). Hence, competition for resources could be a major consequence of threshold-crossing events, which might drastically alter ecosystem services, in particular when Pleistocene-aged permafrost thaws and releases the entrapped microorganisms to the surrounding environment due to climate warming (Castro et al., 2010; De Vries and Shade, 2013; Strauss et al., 2017; Schostag et al., 2019). Based on metatranscriptome analysis of frozen permafrost, Coolen and Orsi (2015) demonstrated a shift in microbial community structure from Firmicutes, Acidobacteria, Actinobacteria and Proteobacteria dominant community to a Firmicutes, Bacteroidetes, and Euryarchaeota dominant community upon thaw. Permafrost thaw in their study was in parallel in increase in complex biopolymer degradation, translation and biogenesis transcripts, indicating a general increase in microbial activity (Coolen and Orsi, 2015).
Another, less likely, scenario to explain the microbial community shift between epochs is the effect of the relict extracellular DNA on our diversity analyses. In this scenario, relict preserved DNA released from dead cells differs between Holocene and Pleistocene samples and bias increases with relict DNA pool size (similar to Carini et al., 2016). Within the Pleistocene samples, 83% of the enumerated cells were non-viable and Holocene samples had ∼8 fold higher viable cells (Figure 1). Hence, it is possible that relict DNA might be an important factor in epoch based separation of Holocene and Pleistocene microbial community structure. However, this scenario seems unlikely for two reasons. First, Burkert et al. (2019) did not find a significant change in microbial community structure after removal of relict DNA preserved within Beringian permafrost. Second, Lennon et al. (2018) reported a minimal effect of relict DNA on estimates of microbial alpha diversity as long as the species abundance distribution of the relict and intact DNA pools are equivalent. Hence, since our alpha diversity indices did not indicate any statistically significant variation between epochs (Supplementary Table S3), the species abundance distribution of the relict and intact DNA are likely equivalent. However, it is not possible to fully exclude this scenario.
Conclusion
Anthropocene warming in the western Arctic exceeds the dramatic warming associated with the Early Holocene thermal maximum, and arguably represents the warmest temperatures of the last ca. 14,000 years (Porter et al., 2019). As a result fragile cold-adapted systems have been predicted to be near a threshold- similar to the Pleistocene-Holocene transition (Miller et al., 2010). Similar changes observed in this study might occur in modern cold soils, which could alter ecosystem services with unknown consequences. The single core and six samples from this study only represent a snap shot of the effects of dramatic climate change on microbial population dynamics across the Pleistocene-Holocene transition at one location. Future investigations should focus on different types of permafrost-affected regions with different terrain types or different permafrost stratigraphy, both at microbial community and functional levels. It is yet to be seen if our findings are more generally representative of the microbial community dynamics across the Pleistocene-Holocene transition at a pan-Arctic scale. If our threshold hypothesis is supported by more regional studies in diverse settings, it may provide a framework for a better understanding of potential changes in important archaeal and bacterial functional guilds, which could in turn improve climate models by predicting microbial responses in a warmer world.
Also, we noticed that the bacteria entrapped in Pleistocene aged permafrost are distantly related to the known reference sequences, this suggest that the microbial members of the Eastern Beringian permafrost are mainly unknown. Future research demands to isolate these strains and study them at a phylogenetic and genome levels for possible biotechnological applications or for expanding our knowledge regarding bacterial evolution/succession over geologically meaningfully timescales.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA607368; https://www.ncbi.nlm.nih.gov/, SAMN14130768.
Author Contributions
AS-M collected the samples, performed the experiments, analyzed the data, and wrote the manuscript. PN performed the experiments. MH performed and analyzed the statistical tests. DF and BL designed the project, obtained the funding, and aided with the interpretation of the data. All the authors reviewed and edited the manuscript.
Funding
This research was funded by the NSERC Discovery Grants (BL and DF) and NSERC Northern Research Supplements (DF) and ArcticNet project 2019-13 (BL). A Yukon Scientists and Explorers License was acquired from the Yukon Government for coring the permafrost samples.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge Dr. Lauren Davies for assistance with radiocarbon dating and chronology of the cores, and Sasiri Bandara, Casey Buchanan, and Joseph Young for assisting us in sampling. We would like to extend our thanks to the University of Alberta (UANRA grant), the Polar Knowledge Canada (NSTP grant), and the Alberta Innovates Technology (AITF) for their student financial support. In addition, we would like to thank Dr. Martin Sharp, Dr. Alberto Reyes, Dr. Derek MacKenzie, and Ashley Gilliland for their insightful comments, support, and allowing us to use their labs.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2020.00133/full#supplementary-material
References
Alley, R. B., Marotzke, J., Nordhaus, W. D., Overpeck, J. T., Peteet, D. M., Pielke, R. A., et al. (2003). Abrupt climate change. Science 299, 2005–2010.
Allison, S. D., and Martiny, J. B. (2008). Resistance, resilience, and redundancy in microbial communities. PNAS 105, 11512–11519. doi: 10.1073/pnas.0801925105
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Barnosky, A. D., Koch, P. L., Feranec, R. S., Wing, S. L., and Shabel, A. B. (2004). Assessing the causes of late Pleistocene extinctions on the continents. Science 306, 70–75. doi: 10.1126/science.1101476
Boers, N., Ghil, M., and Rousseau, D. D. (2018). Ocean circulation, ice shelf, and sea ice interactions explain Dansgaard–Oeschger cycles. PNAS 115, E11005–E11014.
Bottos, E. M., Kennedy, D. W., Romero, E. B., Fansler, S. J., Brown, J. M., Bramer, L. M., et al. (2018). Dispersal limitation and thermodynamic constraints govern spatial structure of permafrost microbial communities. FEMS Microbiol. Ecol. 94:fiy110.
Bradford, M. A., Davies, C. A., Frey, S. D., Maddox, T. R., Melillo, J. M., Mohan, J. E., et al. (2008). Thermal adaptation of soil microbial respiration to elevated temperature. Ecol. Lett. 11, 1316–1327. doi: 10.1111/j.1461-0248.2008.01251.x
Burkert, A., Douglas, T. A., Waldrop, M. P., and Mackelprang, R. (2019). Changes in the active, dead, and dormant microbial community structure across a Pleistocene permafrost chronosequence. Appl. Environ. Microbiol. 85: e02646–18.
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS 108, 4516–4522. doi: 10.1073/pnas.1000080107
Carini, P., Marsden, P. J., Leff, J. W., Morgan, E. E., Strickland, M. S., and Fierer, N. (2016). Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat. Microbiol. 2, 1–6.
Castro, H. F., Classen, A. T., Austin, E. E., Norby, R. J., and Schadt, C. W. (2010). Soil microbial community responses to multiple experimental climate change drivers. Appl. Environ. Microbiol. 76, 999–1007. doi: 10.1128/aem.02874-09
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., McGarrell, D. M., Sun, Y., et al. (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642.
Coolen, M. J., and Orsi, W. D. (2015). The transcriptional response of microbial communities in thawing Alaskan permafrost soils. Front. Microbiol. 6:197. doi: 10.3389/fmicb.2015.00197
Davies, L. J., Appleby, P., Jensen, B. J., Magnan, G., Mullan-Boudreau, G., Noernberg, T., et al. (2018). High-resolution age modelling of peat bogs from northern Alberta, Canada, using pre-and post-bomb 14C, 210Pb and historical cryptotephra. Quat. Geochronol. 47, 138–162. doi: 10.1016/j.quageo.2018.04.008
De Vries, F. T., and Shade, A. (2013). Controls on soil microbial community stability under climate change. Front. Microbiol. 4:265. doi: 10.3389/fmicb.2013.00265
Edgar, R. (2016). SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. BioRxiv [Preprint]. doi: 10.1101/074161
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10:996. doi: 10.1038/nmeth.2604
Fraser, T. A., and Burn, C. R. (1997). On the nature and origin of “muck” deposits in the Klondike area, Yukon Territory. Can. J. Earth Sci. 34, 1333–1344. doi: 10.1139/e17-106
Freedman, Z., and Zak, D. R. (2015). Soil bacterial communities are shaped by temporal and environmental filtering: evidence from a long-term chronosequence. Environ. Microbiol. 17, 3208–3218. doi: 10.1111/1462-2920.12762
Froese, D. G., Zazula, G. D., Westgate, J. A., Preece, S. J., Sanborn, P. T., Reyes, A. V., et al. (2009). The Klondike goldfields and Pleistocene environments of Beringia. GSA Today 19, 4–10. doi: 10.1130/gsatg54a.1
Gaglioti, B. V., Mann, D. H., Jones, B. M., Wooller, M. J., and Finney, B. P. (2016). High-resolution records detect human-caused changes to the boreal forest wildfire regime in interior Alaska. Holocene 26, 1064–1074. doi: 10.1177/0959683616632893
Gittel, A., Bárta, J., Kohoutová, I., Mikutta, R., Owens, S., Gilbert, J., et al. (2014). Distinct microbial communities associated with buried soils in the Siberian tundra. ISME 8, 841–853. doi: 10.1038/ismej.2013.219
Goldfarb, K. C., Karaoz, U., Hanson, C. A., Santee, C. A., Bradford, M. A., Treseder, K. K., et al. (2011). Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front. Microbiol. 2:94. doi: 10.3389/fmicb.2011.00094
Guthrie, R. D. (2006). New carbon dates link climatic change with human colonization and Pleistocene extinctions. Nature 441, 207–209. doi: 10.1038/nature04604
Haile, J., Froese, D. G., MacPhee, R. D., Roberts, R. G., Arnold, L. J., Reyes, A. V., et al. (2009). Ancient DNA reveals late survival of mammoth and horse in interior Alaska. PNAS 106, 22352–22357. doi: 10.1073/pnas.0912510106
Hardie, M., and Doyle, R. (2012). Measuring Soil Salinity In Plant Salt Tolerance 2012. Totowa, NJ: Humana Press.
Hultman, J., Waldrop, M. P., Mackelprang, R., David, M. M., McFarland, J., Blazewicz, S. J., et al. (2015). Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 521, 208–212. doi: 10.1038/nature14238
Itcus, C., Pascu, M. D., Lavin, P., Perşoiu, A., Iancu, L., and Purcarea, C. (2018). Bacterial and archaeal community structures in perennial cave ice. Sci. Rep. 8, 1–4.
Janssen, P. H. (2006). Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72, 1719–1728. doi: 10.1128/aem.72.3.1719-1728.2006
Kotler, E., and Burn, C. R. (2000). Cryostratigraphy of the Klondike “muck” deposits, west-central Yukon Territory. Can. J. Earth Sci. 37, 849–861. doi: 10.1139/e00-013
Kraft, N. J., Adler, P. B., Godoy, O., James, E. C., Fuller, S., and Levine, J. M. (2015). Community assembly, coexistence and the environmental filtering metaphor. Funct. Ecol. 29, 592–599. doi: 10.1111/1365-2435.12345
Kuffner, M., Hai, B., Rattei, T., Melodelima, C., Schloter, M., Zechmeister-Boltenstern, S., et al. (2012). Effects of season and experimental warming on the bacterial community in a temperate mountain forest soil assessed by 16S rRNA gene pyrosequencing. FEMS. Microbiol. Ecol. 82, 551–562. doi: 10.1111/j.1574-6941.2012.01420.x
Lacelle, D., Fontaine, M., Pellerin, A., Kokelj, S. V., and Clark, I. D. (2019). Legacy of holocene landscape changes on soil biogeochemistry: a perspective from paleo-active layers in Northwestern Canada. J. Geophys. Res. Biogeol. 124, 2662–2679.
Lennon, J. T., Muscarella, M. E., Placella, S. A., and Lehmkuhl, B. K. (2018). How, when, and where relic DNA affects microbial diversity. Am. Soc. Microbiol. 9:e00637-18.
Liang, R., Lau, M., Vishnivetskaya, T., Lloyd, K. G., Wang, W., Wiggins, J., et al. (2019). Predominance of anaerobic, spore-forming bacteria in metabolically active microbial communities from ancient Siberian permafrost. Appl. Environ. Microbiol. 85:e00560-19.
Lorenzen, E. D., Nogués-Bravo, D., Orlando, L., Weinstock, J., Binladen, J., Marske, K. A., et al. (2011). Species-specific responses of Late Quaternary megafauna to climate and humans. Nature 479, 359–364.
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 15:550.
Mackelprang, R., Burkert, A., Haw, M., Mahendrarajah, T., Conaway, C. H., Douglas, T. A., et al. (2017). Microbial survival strategies in ancient permafrost: insights from metagenomics. ISME 11, 2305–2318. doi: 10.1038/ismej.2017.93
Mackelprang, R., Waldrop, M. P., DeAngelis, K. M., David, M. M., Chavarria, K. L., Blazewicz, S. J., et al. (2011). Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371. doi: 10.1038/nature10576
Mandakovic, D., Rojas, C., Maldonado, J., Latorre, M., Travisany, D., Delage, E., et al. (2018). Structure and co-occurrence patterns in microbial communities under acute environmental stress reveal ecological factors fostering resilience. Sci. Rep. 8:5875.
Mann, P. J., Spencer, R. G., Hernes, P. J., Six, J., Aiken, G. R., Tank, S. E., et al. (2016). Pan-Arctic trends in terrestrial dissolved organic matter from optical measurements. Front. Earth Sci. 4:25. doi: 10.3389/feart.2016.00025
Maynard, D. G., Kalra, Y. P., and Crumbaugh, J. A. (1993). Nitrate and Exchangeable Ammonium Nitrogen. Soil Sampling and Methods of Analysis. Washington, DC: Lewis Publishers.
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Metcalfe, D. B. (2017). Microbial change in warming soils. Science 358, 41–42. doi: 10.1126/science.aap7325
Mielke, P. W. Jr., Berry, K. J., and Johnson, E. S. (1976). Multi-response permutation procedures for a priori classifications. Commun. Stat Theor. 5, 1409–1424. doi: 10.1080/03610927608827451
Mikkelson, K. M., Lozupone, C. A., and Sharp, J. O. (2016). Altered edaphic parameters couple to shifts in terrestrial bacterial community structure associated with insect-induced tree mortality. Soil Biol. Biochem. 95, 19–29. doi: 10.1016/j.soilbio.2015.12.001
Miller, G. H., Alley, R. B., Brigham-Grette, J., Fitzpatrick, J. J., Polyak, L., Serreze, M. C., et al. (2010). Arctic amplification: can the past constrain the future? Quat. Sci. Rev. 29, 1779–1790. doi: 10.1016/j.quascirev.2010.02.008
Oksanen, J., Kindt, R., Legendre, P., O’Hara, B., Stevens, M. H., Oksanen, M. J., et al. (2007). The vegan package. Commun. Ecol. Package 10, 631–637.
Porter, T. J., Schoenemann, S. W., Davies, L. J., Steig, E. J., Bandara, S., and Froese, D. G. (2019). Recent summer warming in northwestern Canada exceeds the Holocene thermal maximum. Nat. Commun. 10, 1–10.
Rivkina, E., Petrovskaya, L., Vishnivetskaya, T., Krivushin, K., Shmakova, L., Tutukina, M., et al. (2016). Metagenomic analyses of the late Pleistocene permafrost–additional tools for reconstruction of environmental conditions. Biogeosciences 13, 2207–2219. doi: 10.5194/bg-13-2207-2016
Rousk, J., Frey, S. D., and Bååth, E. (2012). Temperature adaptation of bacterial communities in experimentally warmed forest soils. Glob. Change Biol. 18, 3252–3258. doi: 10.1111/j.1365-2486.2012.02764.x
Saidi-Mehrabad, A., Neuberger, P., Cavaco, M., Froese, D., and Lanoil, B. D. (2020). Optimization of subsampling, decontamination, and DNA extraction of difficult peat and silt permafrost samples. bioRxiv [Preprint]. doi: 10.1101/2020.01.02.893438
Scheffer, M., Carpenter, S., Foley, J. A., Folke, C., and Walker, B. (2001). Catastrophic shifts in ecosystems. Nature 413, 591–596. doi: 10.1038/35098000
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Schostag, M., Priemé, A., Jacquiod, S., Russel, J., Ekelund, F., and Jacobsen, C. S. (2019). Bacterial and protozoan dynamics upon thawing and freezing of an active layer permafrost soil. ISME. 13, 1345–1359. doi: 10.1038/s41396-019-0351-x
Shade, A., Caporaso, J. G., Handelsman, J., Knight, R., and Fierer, N. (2013). A meta-analysis of changes in bacterial and archaeal communities with time. ISME 7:1493. doi: 10.1038/ismej.2013.54
Shade, A., Peter, H., Allison, S. D., Baho, D., Berga, M., Bürgmann, H., et al. (2012). Fundamentals of microbial community resistance and resilience. Front. Microbiol. 3:417. doi: 10.3389/fmicb.2012.00417
Shapiro, B., and Cooper, A. (2003). Beringia as an ice age genetic museum. Quat. Res. 60, 94–100. doi: 10.1016/s0033-5894(03)00009-7
Skoog, D. A., Holler, F. J., and Crouch, S. R. (2007). Atomic Emission Spectrometry: Chapter 10 in Principles of Instrumental Analysis, 6th Edn. Belmont, CA: Saunders College Publication.
Sparks, D. L., Page, A. L., Helmke, P. A., Loeppert, R. H., Soltanpour, P. N., Tabatabai, M. A., et al. (1996). Methods of Soil Analysis, part 3, Chemical Methods. Madison: SSSA.
Stackebrandt, E., and Goebel, B. M. (1994). A place for DNA DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Steffen, W., Rockström, J., Richardson, K., Lenton, T. M., Folke, C., Liverman, D., et al. (2018). Trajectories of the earth system in the anthropocene. PNAS 115, 8252–8259.
Strauss, J., Schirrmeister, L., Grosse, G., Fortier, D., Hugelius, G., Knoblauch, C., et al. (2017). Deep Yedoma permafrost: a synthesis of depositional characteristics and carbon vulnerability. Earth Sci. Rev. 172, 75–86. doi: 10.1016/j.earscirev.2017.07.007
Tuorto, S. J., Darias, P., McGuinness, L. R., Panikov, N., Zhang, T., Häggblom, M. M., et al. (2014). Bacterial genome replication at subzero temperatures in permafrost. ISME 8, 139–149. doi: 10.1038/ismej.2013.140
Vonk, J. E., Tank, S. E., Bowden, W. B., Laurion, I., Vincent, W. F., Alekseychik, P., et al. (2015). Reviews and syntheses: effects of permafrost thaw on Arctic aquatic ecosystems. Biogeosciences 12, 7129–7167. doi: 10.5194/bg-12-7129-2015
Wang, Y. I., Naumann, U., Wright, S. T., and Warton, D. I. (2012). mvabund–an R package for model-based analysis of multivariate abundance data. Methods. Ecol. Evol. 3, 471–474. doi: 10.1111/j.2041-210x.2012.00190.x
Warton, D. I. (2011). Regularized sandwich estimators for analysis of high-dimensional data using generalized estimating equations. Biometrics 67, 116–123. doi: 10.1111/j.1541-0420.2010.01438.x
Warton, D. I., Wright, S. T., and Wang, Y. (2012). Distance-based multivariate analyses confound location and dispersion effects. Methods. Ecol. Evol. 3, 89–101. doi: 10.1111/j.2041-210x.2011.00127.x
Willerslev, E., Davison, J., Moora, M., Zobel, M., Coissac, E., Edwards, M. E., et al. (2014). Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 506, 47–51.
Willerslev, E., Hansen, A. J., Rønn, R., Brand, T. B., Barnes, I., Wiuf, C., et al. (2004). Long-term persistence of bacterial DNA. Curr. Biol. 14, R9–R10.
Keywords: permafrost, microbial community, Pleistocene-Holocene, climate change, soil physicochemical parameters
Citation: Saidi-Mehrabad A, Neuberger P, Hajihosseini M, Froese D and Lanoil BD (2020) Permafrost Microbial Community Structure Changes Across the Pleistocene-Holocene Boundary. Front. Environ. Sci. 8:133. doi: 10.3389/fenvs.2020.00133
Received: 04 April 2020; Accepted: 16 July 2020;
Published: 19 August 2020.
Edited by:
Adam Jerold Reed, University of Southampton, United KingdomReviewed by:
Pernille Bronken Eidesen, The University Centre in Svalbard, NorwayJianfang Hu, Guangzhou Institute of Geochemistry, CAS, China
Lyle Whyte, McGill University, Canada
Copyright © 2020 Saidi-Mehrabad, Neuberger, Hajihosseini, Froese and Lanoil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian D. Lanoil, bGFub2lsQHVhbGJlcnRhLmNh