 Andreas Makiola1
Andreas Makiola1 Zacchaeus G. Compson2,3
Zacchaeus G. Compson2,3 Donald J. Baird2
Donald J. Baird2 Matthew A. Barnes4
Matthew A. Barnes4 Sam P. Boerlijst5,6
Sam P. Boerlijst5,6 Agnès Bouchez7
Agnès Bouchez7 Georgina Brennan8Alex Bush2,9
Georgina Brennan8Alex Bush2,9 Elsa Canard10
Elsa Canard10 Tristan Cordier11
Tristan Cordier11 Simon Creer8
Simon Creer8 R. Allen Curry12
R. Allen Curry12 Patrice David13Alex J. Dumbrell14
Patrice David13Alex J. Dumbrell14 Dominique Gravel15
Dominique Gravel15 Mehrdad Hajibabaei16
Mehrdad Hajibabaei16 Brian Hayden17
Brian Hayden17 Berry van der Hoorn5Philippe Jarne13J. Iwan Jones18
Berry van der Hoorn5Philippe Jarne13J. Iwan Jones18 Battle Karimi1Francois Keck7Martyn Kelly19
Battle Karimi1Francois Keck7Martyn Kelly19 Ineke E. Knot20
Ineke E. Knot20 Louie Krol5,6Francois Massol21,22
Louie Krol5,6Francois Massol21,22 Wendy A. Monk2,23John Murphy18
Wendy A. Monk2,23John Murphy18 Jan Pawlowski11
Jan Pawlowski11 Timothée Poisot24Teresita M. Porter16,25
Timothée Poisot24Teresita M. Porter16,25 Kate C. Randall14Emma Ransome26
Kate C. Randall14Emma Ransome26 Virginie Ravigné27Alan Raybould28,29,30Stephane Robin31
Virginie Ravigné27Alan Raybould28,29,30Stephane Robin31 Maarten Schrama5,6
Maarten Schrama5,6 Bertrand Schatz13Alireza Tamaddoni-Nezhad32
Bertrand Schatz13Alireza Tamaddoni-Nezhad32 Krijn B. Trimbos6
Krijn B. Trimbos6 Corinne Vacher33
Corinne Vacher33 Valentin Vasselon34
Valentin Vasselon34 Susie Wood35Guy Woodward26
Susie Wood35Guy Woodward26 David A. Bohan1*
David A. Bohan1*- 1Agroécologie, AgroSup Dijon, INRA, Université Bourgogne, Université Bourgogne Franche-Comté, Dijon, France
- 2Environment and Climate Change Canada @ Canadian Rivers Institute, Department of Biology, University of New Brunswick, NB, Canada
- 3Centre for Environmental Genomics Applications, St. John's, NL, Canada
- 4Department of Natural Resources Management, Texas Tech University, Lubbock, TX, United States
- 5Naturalis Biodiversity Center, Leiden, Netherlands
- 6Institute of Environmental Sciences, Leiden University, Leiden, Netherlands
- 7CARRTEL, USMB, INRA, Thonon-les-Bains, France
- 8School of Natural Sciences, Bangor University, Bangor, United Kingdom
- 9Lancaster Environment Centre, Lancaster University, Lancaster, United Kingdom
- 10UMR 1349 IGEPP, INRA, Université de Rennes 1, Agrocampus Ouest Rennes, Domaine de la Motte, Le Rheu, France
- 11Department of Genetics and Evolution, University of Geneva, Science III, Geneva, Switzerland
- 12Canadian Rivers Institute, Biology, Forestry and Environmental Management, University of New Brunswick, Fredericton, NB, Canada
- 13CEFE UMR 5175, CNRS—Université de Montpellier - Université Paul-Valéry Montpellier–IRD—EPHE, Montpellier, France
- 14School of Biological Sciences, University of Essex, Colchester, United Kingdom
- 15Département de Biologie, Université de Sherbrooke, Sherbrooke, Canada
- 16Centre for Biodiversity Genomics and Department of Integrative Biology, University of Guelph, Guelph, ON, Canada
- 17Stable Isotopes in Nature Laboratory, Canadian Rivers Institute, University of New Brunswick, Fredericton, NB, Canada
- 18School of Biological and Chemical Sciences, Queen Mary University of London, London, United Kingdom
- 19Bowburn Consultancy, Durham, United Kingdom
- 20Institute of Biodiversity and Ecosystem Dynamics, University of Amsterdam, Amsterdam, Netherlands
- 21Univ. Lille, CNRS, UMR 8198—Evo-Eco-Paleo, SPICI Group, Lille, France
- 22Univ. Lille, CNRS, Inserm, CHU Lille, Institut Pasteur de Lille, U1019–UMR 8204–CIIL–Center for Infection and Immunity of Lille, Lille, France
- 23Faculty of Forestry and Environmental Management, University of New Brunswick, Fredericton, NB, Canada
- 24Département de sciences biologiques, Université de Montréal, Montreal, QC, Canada
- 25Great Lakes Forestry Centre, Natural Resources Canada, Sault Ste. Marie, ON, Canada
- 26Department of Life Sciences, Silwood Park Campus, Imperial College London, London, United Kingdom
- 27CIRAD, UMR PVBMT, Saint-Pierre, France
- 28Syngenta Crop Protection AG, Basel, Switzerland
- 29School of Social and Political Science, The University of Edinburgh, Edinburgh, United Kingdom
- 30The Royal (Dick) School of Veterinary Studies, The University of Edinburgh, Global Academy of Agriculture and Food Security, Edinburgh, United Kingdom
- 31Laboratoire MMIP—UMR INRA 518/AgroParisTech, Paris, France
- 32Department of Computer Science, University of Surrey, Guildford, United Kingdom
- 33BIOGECO, INRA, Univ. Bordeaux, Pessac, France
- 34Agence Française pour la Biodiversité, Pôle R&D ECLA, Évian-les-Bains, France
- 35Cawthron Institute, Nelson, New Zealand
Classical biomonitoring techniques have focused primarily on measures linked to various biodiversity metrics and indicator species. Next-generation biomonitoring (NGB) describes a suite of tools and approaches that allow the examination of a broader spectrum of organizational levels—from genes to entire ecosystems. Here, we frame 10 key questions that we envisage will drive the field of NGB over the next decade. While not exhaustive, this list covers most of the key challenges facing NGB, and provides the basis of the next steps for research and implementation in this field. These questions have been grouped into current- and outlook-related categories, corresponding to the organization of this paper.
Introduction
Classical biomonitoring techniques (Table 1) have focused primarily on measures linked to various biodiversity metrics (e.g., species richness, beta diversity; Li et al., 2010; Gutiérrez-Cánovas et al., 2019) and indicator species (but see Vandewalle et al., 2010; Culhane et al., 2014; Saito et al., 2015 for other approaches). Next-generation biomonitoring (NGB) describes a suite of tools and approaches that allow the examination of a broader spectrum of organizational levels—from genes to entire ecosystems. A more holistic vision of evaluating ecological structure and change has long been a goal of ecology, but only recently have the tools emerged to bring it toward fruition. In this issue of Frontiers in Ecology & Evolution, which explores the research topic, “A Next Generation of Biomonitoring to Detect Global Ecosystem Change,” we explore this complementary suite of new tools that could be forged into a global approach to biomonitoring. In this overview paper, we attempt to synthesize opinion on the key issues that are necessary to address en route to this next generation of biomonitoring tools. We focus on a key subset of these tools—those based on DNA metabarcoding as a new standard methodology for multiple taxonomic identifications—for which the number of papers published has increased exponentially since 2010 (Figure 1).
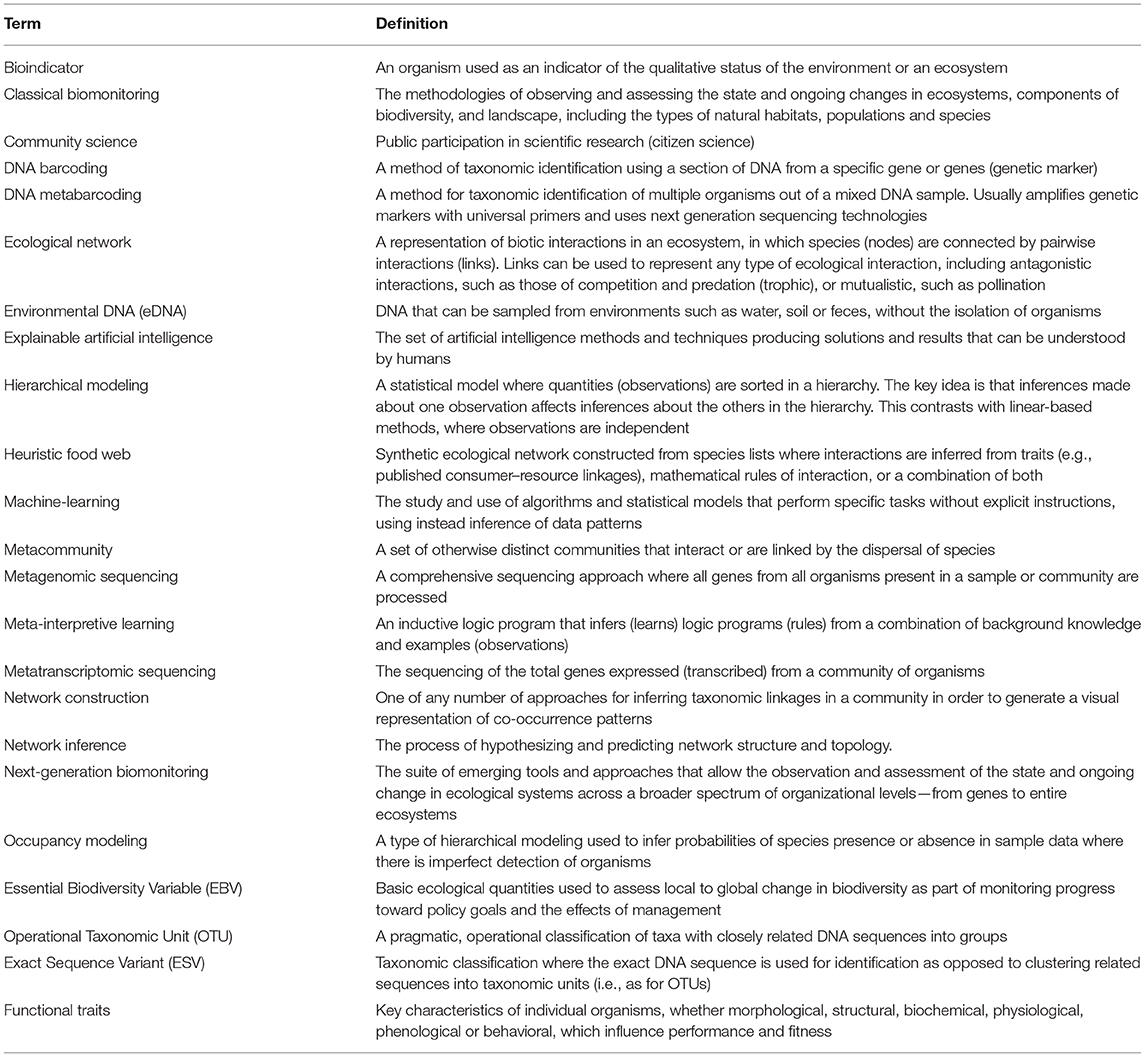
Table 1. Glossary of terms as used in this paper.
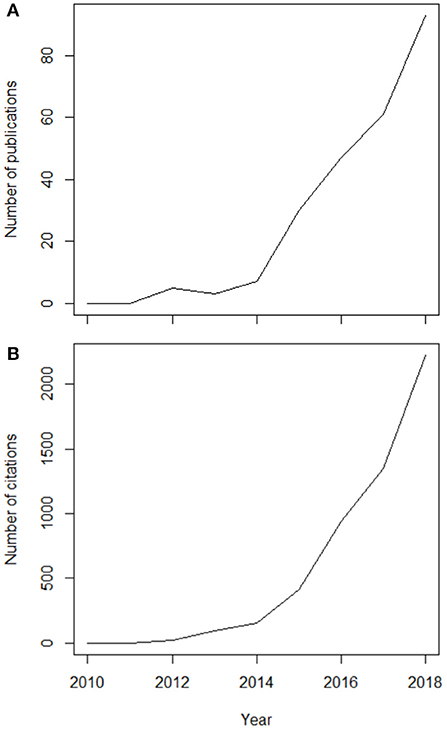
Figure 1. Exponential rise in the number of published, peer-reviewed articles (A) and the number of citations of these articles (B) about next-generation biomonitoring. Figures depict data obtained through a systematic query of the Web of Science database using the Boolean search: “*monitoring” AND “*DNA” AND “metabarcoding”.
DNA metabarcoding generates massive amounts of data on taxonomic units (e.g., operational taxonomic units, OTUs, or exact sequence variants, ESVs; Callahan et al., 2017) rapidly, and these can be linked increasingly to functional attributes (Douglas et al., 2018; Makiola et al., 2019). DNA metabarcoding is highly complementary to whole metagenomic and metatranscriptomic sequencing (Knight et al., 2018), existing sources of ecological information (Cordier et al., 2018; Derocles et al., 2018) and classical biomonitoring approaches (Deiner et al., 2017); in all cases, adding genomic and/or ecological information to the rich taxonomic lists afforded by DNA metabarcoding would allow deeper exploration of ecological or biodiversity patterns. This would move biomonitoring closer to being able to extract both structural and functional attributes from the same multispecies sample (Keck et al., 2017; Cordier et al., 2019). By merging DNA metabarcoding with ecological information and machine learning approaches, NGB extends modern analytical potential beyond the classical morphological identification of bioindicator species. For instance, taxonomic lists from DNA metabarcodes can identify anthropogenic drivers behind community change and infer networks of possible ecological interactions and associated ecosystem properties (Bohan et al., 2017; Compson et al., 2018). While challenges to constructing these networks from NGB data remain (e.g., Barner et al., 2018; Freilich et al., 2018; Deagle et al., 2019), this overview paper discusses some promising ways of overcoming these limitations, including using trait filters developed from published literature and methods of inferring interactions (e.g., machine learning), and these ideas are developed in more depth in the associated manuscripts of this special issue. Indeed, the ultimate aim of NGB is to deliver this more integrated view of natural ecosystems at a fraction of the time and cost of classical approaches (Baird and Hajibabaei, 2012; Keck et al., 2017; Leese et al., 2018; Cordier et al., 2019). Building this large-scale monitoring poses many challenging questions, from the practical and logistical to the political and philosophical.
Here, we frame and describe the interplay of ten key questions that we envisage will drive the field over the next decade (Figure 2). Questions 1–7 address issues that are of current importance, and pertain to the scope of NGB. Questions 8–10 are questions of outlook and opportunity, exploring where the field might be going. This list emerged as an overview of the current Frontiers special issue on the research topic: “A Next Generation of Biomonitoring to Detect Global Ecosystem Change.” While not exhaustive it covers most of the key challenges facing NGB, and provides the basis of the next steps for research and implementation in this field.
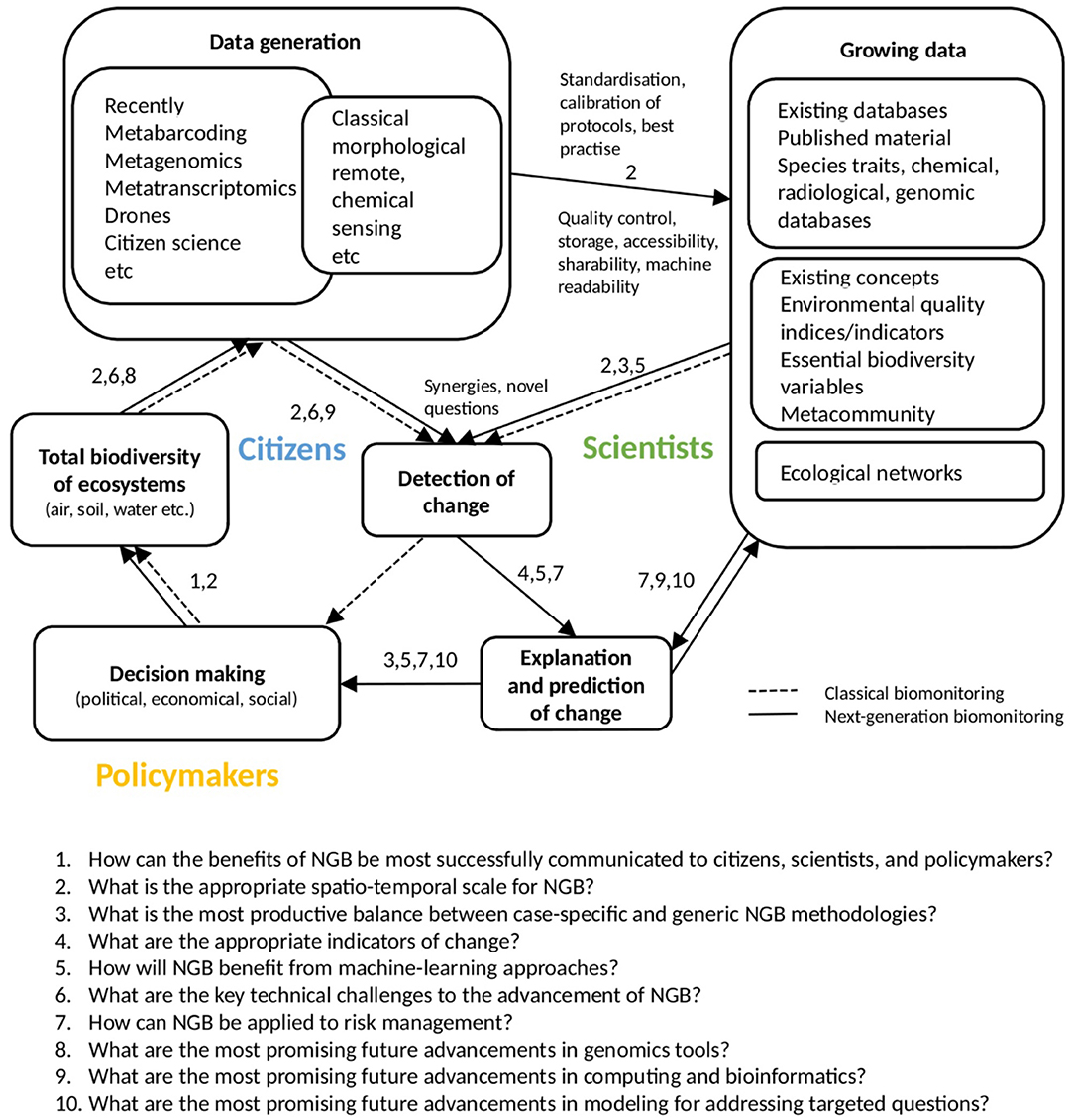
Figure 2. Diagrammatic representation of the interplay between the Key questions for next-generation biomonitoring presented in this paper. Next-generation biomonitoring (NGB) is based on a holistic view of ecosystems through integrating new technologies and exploring synergies with existing data sources. For its realization, it will be necessary to both automate many bioassessment processes and separate the steps of biodiversity detection and explanation of ecosystem change.
Current Questions
How Can the Benefits of NGB Be Most Successfully Communicated to Citizens, Scientists, and Policymakers?
Managing issues of human health, food production and security, and the intertwined environmental issues of biodiversity and ecosystem services necessitates biomonitoring (Bush et al., 2019a; Schmidt-Traub et al., 2019). Information about the status of these issues, such as changes in the frequency of human (Jones et al., 2008) and crop diseases (Savary et al., 2019), insect declines (Hallmann et al., 2017), and losses of species of flowering plants (Carvell et al., 2006) are expected to lead to profound changes in human behavior and appreciation of the environment (Schröter et al., 2017). However, the vision of a broader scale evaluation of ecosystem change, and the benefits this will bring to citizens, scientists, and policymakers, needs to be clearly communicated if wide adoption of NGB approaches is to be realized.
There are three clear benefits of NGB. First, as is argued across the papers of this Issue, NGB has the potential to provide a more holistic method of assessment than classical biomonitoring, affording improved decision-making and management of issues that affect citizens' quality of life. Second, while NGB will provide methods for detailing the complexity of ecosystems, it will also use methods, such as ecological networks, which render this complexity comprehensible, communicating to citizens the richness of their local ecosystem and responses to change (Pocock et al., 2016). Third, NGB can foster citizen participation and buy-in to biomonitoring if it underpins evidence-based decision-making (Hodgetts et al., 2018), and projects with high public participation or strong community science components can produce tangible change in management (Schröter et al., 2017). Portable DNA sequencing instruments allow individuals with relatively little training to generate data; for example, Quick et al. (2016) used this approach to develop a tool to monitor the 2015 Ebola outbreak in Central Africa with a 24 h response time. Similar kits are being developed for use by members of the public to monitor local plant and human disease prevalence and the status of pests in agricultural fields and waterways.
For policy, NGB will not only achieve what classical biomonitoring currently does, such as by reporting on agreed classic indicator species or assemblages, but will also allow the inference and prediction of higher level ecosystem properties (Evans et al., 2016; Compson et al., 2019). In principle, NGB could facilitate remedial decision-making, allowing its accompanying management to be trialed before it is implemented. NGB has the potential to enable monitoring of changes in ecosystem structure and function in something close to real time, because large elements of biomonitoring can be automated, reducing the latencies and biases in human-dependent biomonitoring (Quick et al., 2016; Bohan et al., 2017), bringing science one step closer to the vision of biomonitoring any ecosystem in any biome of the globe. Large coverage would also help to avoid some of the “shocks” associated with the loss or the sharp decline of keystone species and major ecosystem processes long after a tipping point has occurred (Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services, IPBES1). Finally, the generality of NGB enables fusing of multiple areas of biomonitoring that are currently distinct and managed separately. Monitoring of disease, invasions, climate, and land-use change could be undertaken simultaneously, greatly reducing the cost of NGB by pooling resources and sharing expenses. This would, in turn, increase the amount of biomonitoring that might be done, increasing its efficiency.
Despite these potential benefits, which have become apparent, adoption of the latest methodologies into management and decision-making processes has been slow, often hindered by miscommunication between research/scientists and management/policy partners (Darling and Mahon, 2011). Nevertheless, the number of initiatives for a global-scale biomonitoring of biodiversity that maximize cooperation and communication between scientists, policymakers and citizens is increasing. These include the development of new indicators, such as Essential Biodiversity Variables (EBVs, Kissling et al., 2018), ontologies for global biomonitoring (Global Infrastructures for Supporting Biodiversity, GLOBIS-B), storage and linking of data-sets (Global Biodiversity Information Facility, GBIF2), and routes into global scale policy (Group on Earth Observations Biodiversity Observation Network, GEO BON3; Global Earth Observation System of Services, GEOSS4). Scientists working on NGB should participate actively in these efforts. For example, the EU Co-Operation in Science and Technology (COST) action DNAqua-Net5 gathers scientists in order to improve biomonitoring of aquatic ecosystems (Leese et al., 2018; Pawlowski et al., 2018), and has a working group dedicated to the discussion of regulatory and policy frameworks where scientists and stakeholders work collaboratively (Hering et al., 2018). The Interreg European Regional Development Fund project Synergie transfrontalière pour la bio-surveillance et la préservation des écosystèmes Aquatiques (SYNAQUA6) shares a similar aim to gather panels of stakeholders to design scenarios for future NGB implementation for freshwater ecosystem biomonitoring in France and Switzerland (Lefrançois et al., 2018). The benefits of NGB should, in turn, leverage new policy, providing a better fit into the current regulatory and policy frameworks for the more “complex” metrics and indicators of ecosystem structure, function, and services. The role of science and scientists should be to critically appraise the development of the NGB approach and, in so doing, advocate for the benefits of NGB and establish a dialogue between relevant biomonitoring scientists, citizens, industry end-users, and policymakers.
What Is the Appropriate Spatio-Temporal Scale for NGB?
A recurrent message of the papers in this issue is that the scales of biomonitoring, both in terms of spatial extent and temporal frequency of sampling, need to be greatly enlarged if we are to appropriately monitor and assess the risks to ecosystems (see Ovaskainen et al., 2019), identify and evaluate the core drivers of ecosystem dynamics and stability, and make decisions for their management. This will require solutions to some of the practical framework problems that limit the scales of the current generation of biomonitoring approaches, including socio-economic, political, and local management issues.
Current biomonitoring is heavily skewed toward terrestrial Europe, North America, Australia, and New Zealand (Cavallo et al., 2019; McGee et al., 2019). This is due, in part, to a lack of expertise in biomonitoring and interpretation in many countries, a global shortage of finance, as well as a limited acceptance of conventional methods. One avenue that might contribute to a solution, besides better communication of NGB (see Question 1), is to simplify the biomonitoring process into component steps. NGB would consist of two essential steps: (1) sample collection and the detection of ecosystem change; and then, only where change is detected, (2) explanation and prediction. Such separation would greatly reduce the need for expertise in all parts of the globe. Automated and high-throughput sampling and detection of change would take place at large temporal and spatial scales, including parts of the globe with poor coverage at present (using field technicians, citizen scientists, or drones), with the expertise to explain any detected change being outsourced to regional centers of excellence, much as already exists for the World Health Organization Regional Offices and the networks of experts they support (WHO7). The two-stage process would also lower costs for a given scale of coverage, thus making better use of the available finance. A challenging framework question will be what the definition of “change” is, which may vary between different countries and regions. Having the necessary, near-real-time assessments of change is something that is currently only achievable using the NGB approach.
While it is clear that scalability and reusability of global biomonitoring data are necessary to answer large-scale ecological management questions, this can only be achieved where the steps of sampling and detection of change are automated and standardized, making data machine-readable so that information from different systems is comparable and shareable, and can be integrated with other, existing sources of environmental and ecological information (Poisot et al., 2016, 2019). Automating the process of taxa identification, network construction and inference, and comparison to reference states will require considerable technological development (Bohan et al., 2017; Lausch et al., 2018).
Environmental DNA (eDNA) describes genomic materials shed from organisms into their environment that represent the “template” for NGB analysis. eDNA data quality can be influenced by almost every step in the taxa identification workflow (Zinger et al., 2019), from sample collection (Dickie et al., 2018), DNA extraction (Lear et al., 2018), choice of gene or target region, selection of Taq polymerase, polymerase chain reaction (PCR) cycling protocol, primer, choice of sequencing platform, bioinformatic pipelines (Deiner et al., 2017; Makiola et al., 2018; Bush et al., 2019a; Pauvert et al., 2019), and taxonomic reference databases utilized (Porter and Hajibabaei, 2018). These potential challenges compound with the myriad context-specific influences on the ecology of eDNA, such as abiotic and biotic influences on eDNA production, degradation, and transport in the environment (Barnes and Turner, 2016). Standardization or calibration of sampling protocols and other methods in the workflow can improve reproducibility by allowing compilation and comparison of data from across studies (Dickie et al., 2018). Such standardization can be attractive for the majority of users, being both cheap and efficient, even where their research needs differ, as has been successfully demonstrated in The Earth Microbiome Project8 (Thompson et al., 2017) and the Global ARMS (Autonomous Reef Monitoring Structures) Program9 (Ransome et al., 2017).
To tap the full potential of biomonitoring data, it will be necessary to improve curation and access to the rich reference datasets that have already been generated. Due in part to specific institutional regulations, there is a lot of genetic reference material that is only available to researchers within certain institutions. Since molecular-based identifications are heavily dependent on the quality and completeness of the reference databases, this research field will collectively benefit from incentives to curate and upload reference sequences to publicly available databases. Ensuring that these datasets are available in a usable format to interested researchers across the globe represents a major challenge to the field, but one which must be met in order to address global changes in biodiversity and species distribution (Poisot et al., 2016, 2019; Desjardins-Proulx et al., 2019). The definition of the ontologies that will allow NGB data to be machine-read and automated, assuring quality control and the integration of metadata from biomonitoring and associated disciplines, has begun but requires large-scale adoption across fields to be useful.
Knowledge from existing sources (e.g., remote sensing, chemical screening, trait databases) could be integrated into NGB via machine-readable ontologies to generate data synergies and explore novel ecological questions (Bohan et al., 2017; Lausch et al., 2018). For example, this approach could be used to supplement DNA taxa lists with functional trait information for the development of more advanced, predictive heuristic network models (sensu, Compson et al., 2018), while simultaneously creating new—and supplementing existing—databases of taxonomic traits, such as organismal body size or trophic linkages (Kissling et al., 2018). Since the integration of multiple traits and bioindicators holds one of the biggest potential synergies, a possible answer to this question could be working with other initiatives, such as GLOBIS-B, GEO BON, GBIF (Canhos et al., 2015), and the Aquatic eDNA Atlas Project10, as noted in Question 1, toward a common, decentralized, global biodiversity data platform.
What Is the Most Productive Balance Between Case-Specific and Generic NGB Methodologies?
One promise of NGB is to provide general biomonitoring methodologies and comparisons across potentially any ecosystem, including those currently poorly studied or unknown. The search for rationalized, common approaches has begun in certain disciplines, including in aquatic environments (Goldberg et al., 2016), but as the field matures more general guides or approaches may be achievable. Ecosystems are ecologically distinct, but each has unique scales of operation that should be reflected in the spatial scales, frequencies, and replication of sampling. The scales of application of biomonitoring are currently constrained by the methodology used, with most survey methods designed to assess local taxonomic groups of interest. This leads to methodological heterogeneity across regions (Borja et al., 2009; Birk et al., 2012), encumbering efforts to scale up to regional or national levels (Voulvoulis et al., 2017).
Two approaches might be adopted to standardize NGB methodologies. The first would be to sample at the finest spatial resolution possible—at high frequency, in any or all ecosystems across the globe—to store copious amounts of data and to invest in the computational hardware and bioinformatics to detect, forecast, and monitor change. This approach would produce datasets that are both close to complete and an invaluable monitoring and ecological resource, with as yet unforeseen benefits, but the data would come at the cost of collection and curation that may not warrant the increase in efficacy, especially where the detection of system change or changing processes does not require such high-resolution data. However, with plummeting costs this approach will likely be increasingly feasible in the future.
The alternative approach would build upon generic expectations of the rate and temporal dynamics of change in order to identify the required frequency of sampling. The spatial scales of sample independence and representation might then be identified across examples of the ecosystem, indicating appropriate levels of replication to assure detection, with an appropriate power, of given levels of acceptable change. Ma et al. (2018a) described generic, multiscale approaches adopted from the theory of networks to examine temporal and spatial variation. These approaches treat network structure as essentially being independent of the taxa involved in the networks, and use network profiling, null models, and multilayer networks to make statements about the expected level of change that is and is not acceptable in pure network structural terms. This information can then be fed into ecological modeling and robust forecasting studies.
A standardized but general methodology for sampling would maximize scalability, interpretability, and impact of NGB. It is unlikely, however, that the specification of sampling would conveniently lead to a common set of results for all ecosystems to be examined. Rather, any generality that might exist would likely be limited to some combination of the biome being sampled (i.e., air, soil, water), and the organizational (i.e., regional and local networks, communities, species, populations, individuals, or genes) and taxonomic levels. Generality may only be delivered by an ecological understanding of ecosystem structure, probably facilitated by network approaches.
What Are the Appropriate Indicators of Change?
To move biomonitoring forward, science and policy need to explore how: (1) NGB information could lead to new indicators for metacommunities; (2) novel indicators build upon and contribute to existing indicators and frameworks (e.g., Tapolczai et al., 2019); and (3) spatio-temporal metacommunity scales influence the interpretation of these novel indicators. The indicator concept proposes that the ecological state of an ecosystem can be evaluated by observing a particular taxon or taxonomic group or function (De Cáceres and Legendre, 2009). Taxon-free indicator metrics, such as Indices of Biotic Integrity (IBI), are appealing to environmental practitioners and policymakers because they distill a lot of information down to a simple metric that, in principle, can be compared across systems. However, their simplicity is likely the reason why such metrics may be misused in practice (Seegert, 2000). Further, while indicator species or IBIs might be useful at local spatial scales, they are not applicable across the many habitats, ecosystems, or biomes (Angermeier et al., 2000) that can be monitored using next-generation methods. Pairing molecular-based approaches with machine learning for NGB can potentially recover orders of magnitude more information in biomonitoring data, thus eliminating many of the constraints that hindered the development of biomonitoring indicators we use today. For example, building ecological networks from this recovered data might be used to analyze whole-network properties with ecosystem functions and services (Evans et al., 2016), providing a mechanistic link between network structural change and ecological functions. There certainly is a lot of work to be done to explore and develop these higher-level, network indicators, as well as to determine which network properties will be useful for predicting ecosystem consequences to environmental change. Once developed, however, these tools should provide immediate added-value to the taxonomic lists generated by NGB, as well as to the classical, biomonitoring approaches, especially considering the cost effectiveness of routine, open-source pipelines for the rapid calculation of such (e.g., ecological network) indicators.
Scaling up from a local- to a large-scale approach should furthermore incorporate recent advances in metacommunity ecology into biomonitoring, in order to make sense of the connections that exist among communities across landscapes. Leibold and Chase (2017) expounded the compelling argument that we should combine previously competing concepts of community assembly, such as neutral theory, species sorting, patch dynamics, and mass effects into a single, overarching theory. Ecosystem biomonitoring is strongly rooted in local observation and a normative interpretation, yet it often fails to take into account spatio-temporal variability and connections among sampled localities, arguably leading to over-interpretation of local-scale deviations from a putative “normal state” (Baattrup-Pedersen et al., 2017). We may also underestimate the influence of metacommunity effects on the drivers of local dynamics and, consequently, biomonitoring observations. The scale-limited spatio-temporal scope of biomonitoring studies also carries a serious risk of missing large-scale phenomena that could have potentially devastating impacts, such as biological invasions (Kamenova et al., 2017) or global declines in insects (Hallmann et al., 2017) that went largely unnoticed in policy for nearly 30 years (IPBES1). DNA-based approaches offer a potential avenue to address this challenge, and we should seize the opportunity to both develop NGB methods by further refinement and testing and promote these methods to policymakers, citing their many benefits.
How Will NGB Benefit From Machine-Learning Approaches?
Statistical methods for extracting information from data represent some of the basic tools that ecologists wield. Standard statistics are used to explore the covariation between dependent and independent variables and to test hypotheses of interaction. Machine-learning approaches work analogously, exploring the probabilistic or logical correlations across matrices of species data. Machine learning of networks has been successfully applied to classical, macro-ecological sample data (e.g., Bohan et al., 2011) and to evaluate ecosystem responses to changed management (Ma et al., 2018b). In contrast, the reconstruction of microbial networks or the inference of networks and trophic links from DNA data has proven to be more difficult (Barner et al., 2018; Freilich et al., 2018; Deagle et al., 2019), with results that appear to depend upon a combination of the machine learning technique and the data used. No one algorithm will work best for every problem, mirroring the “no free lunch” theorem of Wolpert and Macready (1997). The rhetorical question, “How will NGB benefit from machine-learning approaches?,” is one that we can answer only by continual work to further develop and integrate ever better learning approaches into ecology and biomonitoring.
Because NGB represents an emerging field, it is useful to look at examples where machine learning and metabarcoding have been successfully combined. Naïve Bayesian and random forest classifiers have been used to make taxonomic assignments from metabarcodes, produce statistical measures of confidence, and reduce rates of false positive identifications (Wang et al., 2007). Supervised machine learning has been used to classify environmental samples in a meta-analysis of microbial community samples collected by hundreds of researchers for the Earth Microbiome Project (Thompson et al., 2017). Recently, eDNA datasets have been analyzed using supervised machine learning to predict the status of aquatic ecosystems (Cordier et al., 2018). The combination of taxonomy-free molecular data and machine-learning techniques outperformed biomonitoring methods based on the screening of known indicator species by classic metabarcoding (Cordier et al., 2018).
Moving toward the reconstruction of networks of explicit interactions is a logical next step that would afford an ecological explanation of change. Such ecological network reconstruction would require the incorporation of background knowledge or information, for example, about species traits or existing interactions (Tamaddoni-Nezhad et al., 2013, 2015). Taxon interaction knowledge can be text-mined from direct observations recorded in the literature, or inferred from published trait information, and, when used to reconstruct interaction networks such as food webs, offer the potential to generate new biomonitoring metrics derived from network properties (Compson et al., 2018). Recent results suggest that, in the absence of background information, model-free inference of network structure is also feasible using information from the overall network structure and those interactions that are known (Stock et al., 2017). Hypotheses or explicit models for how species interact can also be incorporated into machine learning as background knowledge (Tamaddoni-Nezhad et al., 2013, 2015). As symbolic representations of interactions, these hypotheses and models have the benefit of rendering the machine-learning output human-comprehensible and explainable for decision-making and prediction (Muggleton et al., 2018). The challenge for this model-based approach is that we have relatively few symbolic descriptions of species interactions for organisms, especially in understudied biomes. While there are rules for trophic interactions between macro-organisms, for example, based upon body- or gape-size (Jonsson et al., 2018), there are few such rules for microorganisms. The generation of hypotheses for potentially new mechanisms of interaction in understudied systems could also be supported by artificial intelligence: first, using text mining to recover information about taxa and functions that is not readily accessible from reference databases like Global Biotic Interactions (GloBI) or the United States Geological Survey (USGS) traits database; and then by employing machine learning, such as Meta-Interpretive Learning (Tamaddoni-Nezhad et al., 2015), to hypothesize interaction rules that explain the text-mined information and metabarcoding data.
Considerable amounts of this kind of information exist in literature databases such as Google Scholar, Academic Search Premier, and Web of Science. Unfortunately, the publishing rights to these data are often difficult for scientists to disentangle, and the various text-mining exercises that have been conducted have been treated as hacking attacks, which are resisted. Until these publishing rights are relaxed, such as is proposed in Europe (Enserink, 2018), populating many ecosystems with biological and functional information will remain a limitation.
What Are the Key Technical Challenges to the Advancement of NGB?
NGB aims to detect and explain changes in the total biodiversity of ecosystems to understand and predict the ecological structure of ecosystems. This requires that NGB methods generate accurate data for the presence, absence, and abundance of taxa. Uncertainty in the detection of a taxon, as false negatives or positives, can lead to erroneous conclusions with consequences that could impair biomonitoring and decision making. As noted in Question 3, detection uncertainty can arise from multiple sources, such as sampling, laboratory, and bioinformatics, and these have been extensively reviewed elsewhere (e.g., Deiner et al., 2017; Knight et al., 2018; Larsson et al., 2018; Lear et al., 2018; Porter and Hajibabaei, 2018; Zinger et al., 2019). Work to reduce rates of false negatives and positives in DNA metabarcoding data is an active field of research, and progress has been made through using occupancy modeling (Ficetola et al., 2015, 2016) and probability distribution modeling for tag jumping and contamination issues (Larsson et al., 2018).
The next logical step is to ask whether DNA concentrations in the environment relate to organismal abundance or biomass. The question is intuitive, in the sense that a greater abundance or biomass of organisms should, in principle, produce a higher concentration of DNA, but as with detection uncertainty DNA concentration is determined by many other factors. Studies have demonstrated that the relative abundance of an organism between samples can relate to eDNA concentrations (Takahara et al., 2012; Thomas et al., 2016; Piñol et al., 2019). However, the leap from relative abundance to absolute abundance (or anything close) has been confounded by multiple effects, including an inability to distinguish between live and dead biomass, the observation that different age classes of the same organism release DNA at different rates into the environment (Maruyama et al., 2014), and an increased awareness of the complex environmental interactions of eDNA, relating to its origin, state, transport, and fate (Cristescu and Hebert, 2018). How to treat read count data is critical now that microbiome datasets are understood to be compositional in nature and sensitive to library size and several other biases (Gloor et al., 2017). For NGB it is clear that we need to establish how DNA technologies relate to absolute organismal abundance and how we can minimize methodological biases through best practices (e.g., Knight et al., 2018). However, the debate about the confidence to be invested in metabarcoding data will likely continue until we attain technical advances, such as PCR-free sequencing systems, curated and complete reference databases, and modeling that can explain and correct for errors.
How Can NGB Be Applied to Risk Management?
With further development of NGB, multiple lines of evidence and data will need to be combined in real time to provide managers with cost-effective tools needed to make robust decisions and mitigate impacts on the natural environment. To incorporate these multiple sources of information and move beyond purely descriptive models of ecosystem structure and change, such as eDNA-derived lists of taxa and co-occurrence networks, it will be necessary both to develop explanatory and predictive models of ecosystem function and services, and to test, explore, and understand these models, possibly using developments in text-mining (Compson et al., 2018) and Explainable Artificial Intelligence (Miller, 2019; Rudin, 2019).
As the “universe of observation” (Bush et al., 2019b) expands toward a more integrative ecosystem approach, driven by the growing capacity of molecular and analytical methods, it remains unclear what amount of information will be needed to make good management decisions. For example, how much do we benefit if we incorporate all possible data, or do we just add noise? The application of DNA-isolation from bulk environmental samples or mixed communities coupled with high throughput sequencing and automated taxonomic assignment removes many of the taxonomic constraints currently hindering biomonitoring, particularly for multiple trophic groups and otherwise cryptic groups of organisms (Hug et al., 2016). Increasing taxonomic resolution and greater sampling intensity expands the number of observed biological units. This greater volume of information will also require a parallel expansion of our abilities to interpret biodiversity changes.
Artificial intelligence, in the form of machine learning algorithms such as Meta-interpretive Learning, can help process these large amounts of information and aid in hypothesizing explanatory models of interaction that humans can comprehend and machines can read symbolically (Tamaddoni-Nezhad et al., 2015). The explanations used in biomonitoring will evolve from existing concepts of ecosystem indicators and indices that do not attempt to explain the reason for changes in ecosystems (Derocles et al., 2018) toward models that provide a holistic view of ecological change, such as EBVs (Jetz et al., 2019); models that provide an understanding of the underlying mechanisms behind ecosystem functions; and models that recognize the complex and dynamic nature of ecosystems, including all trophic levels and their interactions. This evolution of biomonitoring, moving from a descriptive toward a predictive risk management tool, based on new hypotheses and models, will have the greatest impact on decision and policy making, which will in turn feed-back to biomonitoring.
Outlook Questions
To this point, the questions posed have focused on contemporary issues about the framework of NGB, as well as technical and conceptual challenges to implementing NGB (Figure 2). We also foresee rapid advancement in this field beyond what is needed to establish NGB as a biomonitoring approach, facilitating exploration of new frontiers of science and providing solutions to some of the problems we have outlined in this article. These are related, in large part, to rapid developments in computing and genomics. Specifically, we believe that three areas of advancement in biodiversity assessment and analytical capacity will drastically improve NGB: (1) advances in genomics tools that will lead to greater sequencing capacity, providing unprecedented recovery of information from DNA (Question 8); (2) advances in computing, bioinformatics, and open-source pipelines (Question 9); and, (3) improved models that will allow for more targeted use by practitioners interested in adopting NGB approaches (Question 10).
What Are the Most Promising Future Advancements in Genomics Tools?
Many widely used, next-generation sequencing technologies have attained greater sequencing depth (i.e., the product of the number of reads and the read length standardized to the genome length) despite using shorter read lengths by exponentially increasing the amount of sequences generated (Sims et al., 2014). We anticipate a next-next-generation revolution that will achieve whole genome sequencing for entire communities, with enough sequencing depth to provide information about individual sequence variation necessary to begin exploration of evolutionary and functional questions in conjunction with NGB. Already, technologies are emerging that provide orders of magnitude more sequencing depth than current platforms. For example, a single flow cell of Illumina's Novaseq platform can generate ~700 times greater sequencing depth than is typically available, allowing for the detection of dramatically more diversity, even at coarse taxonomic levels; standardizing sequencing depth using patterned flow cells further improves sequencing performance by preventing the merging of neighboring sequences (Singer et al., 2019). Eventually, as such platforms advance, shotgun sequencing will become the norm, and the need for PCR will be circumvented, eliminating many of the issues currently associated with sequencing and subsequent data processing. Such advances in sequencing capacity and error reduction will translate to higher detection probabilities, greater coverage of species, and better assessments of abundance and rare or endangered species in all systems, including those that are remote and difficult to access or under-studied. Additionally, we foresee three new frontiers of science that the added information from new sequencing technologies will enable us to explore.
First, greater sequencing depth across a larger complement of the community will make it possible to construct robust phylogenetic trees for entire communities, which will help advance NGB method development by providing better phylogenetic information for improving ecological information and prediction. The practice of metaphylogenetics is currently limited by short sequence read lengths (i.e., normally ~150–300 base pairs). Furthermore, PCR and primer choice can greatly influence the resultant community (Hajibabaei et al., 2019), leading to coarse and unresolved phylogenetic trees. With greater sequencing depth, these limitations will become a thing of the past, allowing for more robust phylogenetic analysis. Community assembly can only be understood in the context of species' evolutionary histories, and such an advance in phylogenetic community ecology will not only be crucial for advancement of ecological theory, but also improve the current standards of biodiversity assessment, allowing for a more holistic exploration of rare or unknown taxa in hyperdiverse, poorly studied ecosystems (Papadopoulou et al., 2015).
Second, future platforms will improve sequencing depth per individual such that it will soon be possible to assess intraspecific genetic variation in an assemblage. To date, studies of population genetics have been limited by primer development for target organisms, focusing on no more than a few taxa at a time in order to answer very specific questions. For example, mitochondrial metagenomics approaches that combine shotgun sequencing and DNA metabarcoding allow for read mapping that may provide the quantitative information on intraspecific genetic variation needed to assess population genetic structure (reviewed in Crampton-Platt et al., 2016). In combination with DNA metabarcoding (sensu, Elbrecht et al., 2018), these approaches would then make it possible to assess the genetic structure of any taxa of interest in the community, and enable practitioners to ask questions about the entire metacommunity and test macroecological theory (e.g., species-genetic diversity correlations).
Third, enhanced sequencing depth will allow for a wider exploration of functional genes in environmental samples. This would make it possible to map functional genes to taxa for entire communities of organisms, linking communities and networks with broad-scale ecosystem assessment of function. Recent efforts have attempted to utilize machine learning to link genus-level predictions of function in microbial communities, for example by using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt), for inferential assessments of function and hypothesis generation (Douglas et al., 2018). With more sequence data and better inferential methodologies, machine learning in biomonitoring will progress. Concurrent efforts to expand and annotate functional gene databases (e.g., Kyoto Encyclopedia of Genes and Genomes, KEGG11) are facilitating the mapping of genes to function across a wide range of biodiversity, bringing incredible added value to projects using the greater sequencing depth afforded by newer sequencing platforms. As these efforts advance, not only will metacommunity and ecosystem theory be advanced by linking structure to function at multiple scales of observation, but potentially transformative changes in biomonitoring and biodiversity assessment will occur, as functional profiles could have greater discriminatory power for detecting change compared to taxonomic profiles, especially in cases where taxonomic profiles are highly variable.
What Are the Most Promising Future Advancements in Computing and Bioinformatics?
With unprecedented data generation, NGB practitioners will be confronted with the enormous task of dealing with an overwhelming amount of information (Keck et al., 2017). Advances in computing and bioinformatics are required to maximize the use of this biodiversity information. Much work still needs to be done to test for and correct errors that inherently emerge from bioinformatics approaches (reviewed in Olson et al., 2017). One solution is to quantitatively assess genome assembly by incorporating evolutionary expectations of gene content, using single copy orthologs (Seppey et al., 2019). These problems of genome assembly and amplification bias will eventually be eliminated as whole-genome sequencing approaches are adopted, but this will, in turn, require even more sophisticated bioinformatics tools (e.g., NanoPack, De Coster et al., 2018).
Another area that will benefit greatly from advances in computing and bioinformatics is database generation, maintenance, and expansion. Existing taxonomic, trait, and functional gene databases (e.g., GenBank, GloBI, KEGG) are incomplete, and the task of updating and expanding these databases is daunting. Artificial intelligence could also be used to advance data discovery (Gonzalez et al., 2016; Compson et al., 2018). Text-mining pipelines, for example, currently make use of open-source, artificial intelligence tools (e.g., OrganismTagger: Naderi et al., 2011). The consequent improvements that these tools will make to taxonomic and functional databases will lead to further advancements of biomonitoring tools, such as cloud-based, rapid ecological network and food web construction, driving a virtuous cycle where more robust datasets lead to improved models.
The promise of these advancements will only be met, however, via improvements in data accessibility, data discoverability, and development of data standards. These will likely emerge from consortiums developing ontologies for genomics and other data (reviewed by Levy and Myers, 2016), as noted in Questions 1 and 3. More work needs to be done, in particular, to develop, peer-review, and publish open-source tools for bioinformatics pipelines (Mangul et al., 2019). Without parallel improvements in tool archival and version control, the improvements that should follow will be inconsistent, reducing their utility and widespread adoption. This work would likely be facilitated by open-source archival services (e.g., GitHub or SourceForge) or package managers (e.g., Bioconda, Grüning et al., 2018).
What Are the Most Promising Future Advancements in Modeling for Addressing Targeted Questions?
While genomic and technological advancements will affect the field of biodiversity assessment, advances in modeling will specifically help end-users, including regulators and resource managers, using NGB approaches. For example, as the costs of sample and bioinformatic processing reduce, more sophisticated hierarchical occupancy models could be applied to repeated sampling data to quantify detection probabilities and inform practitioners about the sampling effort required to answer system-specific questions. These models, which can account for multiple categorical factors influencing a response variable, can accommodate samples of repeated presence-absence data to provide estimates of occurrence and detection probabilities of species and communities, enabling to account for false negatives due to imperfect detection (Campos-Cerqueira and Aide, 2016; Steenweg et al., 2016), a limitation that is seldom considered in bioassessment studies (McClenaghan et al., 2019). Occupancy modeling could also provide a way past the critical limitation of current DNA metabarcoding—that of obtaining absolute abundance information. Applied hierarchical occupancy modeling has been used to address questions related to the detection and abundance of species (Kery and Andrew Royle, 2015), and future genomic and technical advancements will broaden the application of these models via the generation of larger datasets covering wider ranges and along more gradients of environmental change. Hierarchical occupancy models will enable further leveraging of these more robust datasets by incorporating variation in the pathway from sample collection to sequencing and bioinformatics. Detection probabilities, for example, can be built into Bayesian hierarchical models to detect probabilities associated with different primers, sequencing approaches, and other steps along the sampling-to-sequencing pathway (Doi et al., 2019), providing NGB practitioners with better experiments that make more efficient use of resources (Lugg et al., 2018).
As the field of NGB evolves, we foresee synergistic advancements from merging occupancy-modeling and machine-learning approaches with additional layers of information coded in DNA, recovered by improved sequencing technologies and greater sequencing depth. Incorporating relative read abundance information into occupancy models could be used to assess the abundance of functional gene classes in environmental samples. Shotgun sequencing will also remove the constraints and biases of PCR amplification of DNA, leading to better estimates of sample abundance and biomass (Bista et al., 2018). Much of this information could be incorporated into ecological networks and heuristic food webs to estimate interaction strengths and calculate probabilities of interaction (Morales-Castilla et al., 2015). Finally, with increases in occupancy and food web model sophistication, and as more data are generated that capitalize on these approaches, there will be increasing volumes of high-quality information to feed into machine learning algorithms, leading to more predictive modeling of diverse ecosystems and an unprecedented opportunity for NGB practitioners to anticipate change and prevent ecosystem impairment in real time.
Author Contributions
AM conceived, contributed and led the writing of the paper. DAB conceived, contributed and wrote the paper. ZC conceived, contributed and wrote the paper. All other authors contributed and wrote the paper.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
ABo and JP would like to acknowledge funding from the FEDER and Swiss Confederation through the SYNAQUA project. ABo, FK, VV, MK, JP, and TC would like to acknowledge fruitful discussions in the framework of the COST action DNAqua-Net (CA15219).
AD, KR, AT-N, and GW would like to acknowledge funding support from the UK Natural Environment Research Council (NERC-NE/M02086X/1 and NE/M020843/1).
DAB, AM, EC, PD, FM, SR, VR, and CV would like to acknowledge the financial support of the French Agence Nationale de la Recherche project NGB (ANR-17-CE32-011).
DAB, AM, SR, and CV would like to acknowledge the support of the Consortium Biocontrole, which provides funding for the BCMicrobiome project.
TMP would like to acknowledge funding from the Canadian government through the Genomics Research and Development Initiative, Ecobiomics project.
Footnotes
1. ^www.ipbes.net/assessment-reports (accessed May 30, 2019).
2. ^www.gbif.org (accessed May 30, 2019).
3. ^www.geobon.org (accessed May 30, 2019).
4. ^http://www.earthobservations.org/ (accessed May 30, 2019).
5. ^www.dnaqua.net (accessed May 30, 2019)
6. ^www.interreg-francesuisse.eu/beneficiaire/synaqua-synergie-transfrontaliere-pour-la-bio-surveillance-et-la-preservation-des-ecosystemes-aquatiques (accessed May 30, 2019)
7. ^www.who.int/ (accessed May 30, 2019).
8. ^www.earthmicrobiome.org (accessed May 30, 2019)
9. ^www.oceanarms.org (accessed May 30, 2019)
10. ^www.fs.fed.us/rm/boise/AWAE/projects/the-aquatic-eDNAtlas-project.html (accessed May 30, 2019).
11. ^www.genome.jp/kegg (accessed May 30, 2019).
References
Angermeier, P. L., Smogor, R. A., and Stauffer, J. R. (2000). Regional frameworks and candidate metrics for assessing biotic integrity in mid-Atlantic highland streams. Trans. Am. Fish. Soc. 129, 962–981. doi: 10.1577/1548-8659(2000)129<0962:RFACMF>2.3.CO;2
Baattrup-Pedersen, A., Emma Göthe Riis, T., Andersen, D. K., and Larsen, S. E. (2017). A new paradigm for biomonitoring: an example building on the danish stream plant index. Methods Ecol. Evol. 8, 297–307. doi: 10.1111/2041-210X.12676
Baird, D. J., and Hajibabaei, M. (2012). Biomonitoring 2.0: a new paradigm in ecosystem assessment made possible by next-generation DNA sequencing. Mol. Ecol. 21, 2039–2044. doi: 10.1111/j.1365-294X.2012.05519.x
Barner, A., Coblentz, K., Hacker, S., and Menge, B. (2018). Fundamental contradictions among observational and experimental estimates of non-trophic species interactions. Ecology 99, 557–566. doi: 10.1002/ecy.2133
Barnes, M. A., and Turner, C. R. (2016). The ecology of environmental DNA and implications for conservation genetics. Conserv. Genet. 17, 1–17. doi: 10.1007/s10592-015-0775-4
Birk, S., Bonne, W., Borja, A., Brucet, S., Courrat, A., Poikane, S., et al. (2012). Three hundred ways to assess Europe's surface waters: an almost complete overview of biological methods to implement the water framework directive. Ecol. Indic. 18, 31–41. doi: 10.1016/j.ecolind.2011.10.009
Bista, I., Carvalho, G. R., Tang, M., Walsh, K., Zhou, X., Hajibabaei, M., et al. (2018). Performance of amplicon and shotgun sequencing for accurate biomass estimation in invertebrate community samples. Mol. Ecol. Resour. 18, 1020–1034. doi: 10.1111/1755-0998.12888
Bohan, D. A., Caron-Lormier, G., Muggleton, S., Raybould, A., and Tamaddoni-Nezhad, A. (2011). Automated discovery of food webs from ecological data using logic-based machine learning. PLoS ONE 6:e29028. doi: 10.1371/journal.pone.0029028
Bohan, D. A., Vacher, C., Tamaddoni-Nezhad, A., Raybould, A., Dumbrell, A. J., and Woodward, G. (2017). Next-generation global biomonitoring: large-scale, automated reconstruction of ecological networks. Trends Ecol. Evol. 32, 477–487. doi: 10.1016/j.tree.2017.03.001
Borja, A., Miles, A., Occhipinti-Ambrogi, A., and Berg, T. (2009). Current status of macroinvertebrate methods used for assessing the quality of European marine waters: implementing the water framework directive. Hydrobiologia 633, 181–196. doi: 10.1007/s10750-009-9881-y
Bush, A., Compson, Z., Monk, W., Porter, T. M., Steeves, R., Emilson, E., et al. (2019a). Studying ecosystems with DNA metabarcoding: lessons from biomonitoring of aquatic macroinvertebrates. Front. Ecol. Evol. 7:434. doi: 10.1101/578591
Bush, A., Compson, Z., Monk, W., Porter, T. M., Steeves, R., Emilson, E., et al. (2019b). Studying ecosystems with DNA metabarcoding: lessons from aquatic biomonitoring. bioRxiv 578591. doi: 10.3389/fevo.2019.00434
Callahan, B. J., McMurdie, P. J., and Holmes, S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643. doi: 10.1038/ismej.2017.119
Campos-Cerqueira, M., and Aide, T. M. (2016). Improving distribution data of threatened species by combining acoustic monitoring and occupancy modelling. Methods Ecol. Evol. 7, 1340–1348. doi: 10.1111/2041-210X.12599
Canhos, D. A. L., Sousa-Baena, M. S., de Souza, S., Maia, L. C., Stehmann, J. R., Canhos, V. P., et al. (2015). The importance of biodiversity e-infrastructures for megadiverse countries. PLoS Biol. 13:e1002204. doi: 10.1371/journal.pbio.1002204
Carvell, C., Roy, D. B., Smart, S. M., Pywell, R. F., Preston, C. D., and Goulson, D. (2006). Declines in forage availability for bumblebees at a national scale. Biol. Conserv. 132, 481–489. doi: 10.1016/j.biocon.2006.05.008
Cavallo, M., Borja, Á., Elliott, M., Quintino, V., and Touza, J. (2019). Impediments to achieving integrated marine management across borders: the case of the EU marine strategy framework directive. Mar. Policy 103, 68–73. doi: 10.1016/j.marpol.2019.02.033
Compson, Z. G., Monk, W. A., Curry, C. J., Gravel, D., Bush, A., Baker, C. J. O., et al. (2018). Linking DNA metabarcoding and text mining to create network-based biomonitoring tools: a case study on boreal wetland macroinvertebrate communities. Adv. Ecol. Res. 59, 33–74. doi: 10.1016/bs.aecr.2018.09.001
Compson, Z. G., Monk, W. A., Hayden, B., Bush, A., O'Malley, Z., Hajibabaei, M., et al. (2019). Network-based biomonitoring: exploring freshwater food webs with stable isotope analysis and DNA metabarcoding. Front. Ecol. Evol. 7:395. doi: 10.3389/fevo.2019.00395
Cordier, T., Forster, D., Dufresne, Y., Martins, C. I. M., Stoeck, T., and Pawlowski, J. (2018). Supervised machine learning outperforms taxonomy-based environmental DNA metabarcoding applied to biomonitoring. Mol. Ecol. Resour. 18, 1381–1391. doi: 10.1111/1755-0998.12926
Cordier, T., Lanzén, A., Apothéloz-Perret-Gentil, L., Stoeck, T., and Pawlowski, J. (2019). Embracing environmental genomics and machine learning for routine biomonitoring. Trends Microbiol. 27, 387–397. doi: 10.1016/j.tim.2018.10.012
Crampton-Platt, A., Yu, D. W., Zhou, X., and Vogler, A. P. (2016). Mitochondrial metagenomics: letting the genes out of the bottle. GigaScience 5:15. doi: 10.1186/s13742-016-0120-y
Cristescu, M. E., and Hebert, P. D. N. (2018). Uses and misuses of environmental DNA in biodiversity science and conservation. Annu. Rev. Ecol. Evol. Syst. 49, 209–230. doi: 10.1146/annurev-ecolsys-110617-062306
Culhane, F. E., Briers, R. A., Tett, P., and Fernandes, T. F. (2014). Structural and functional indices show similar performance in marine ecosystem quality assessment. Ecol. Indic. 43, 271–280. doi: 10.1016/j.ecolind.2014.03.009
Darling, J. A., and Mahon, A. R. (2011). From molecules to management: adopting DNA-based methods for monitoring biological invasions in aquatic environments. Environ. Res. 111, 978–988. doi: 10.1016/j.envres.2011.02.001
De Cáceres, M., and Legendre, P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Deagle, B., Thomas, A., McInnes, J., Clarke, L., Vesterinen, E., Clare, E., et al. (2019). Counting with DNA in metabarcoding studies: how should we convert sequence reads to dietary data? Mol. Ecol. 28, 391–406. doi: 10.1111/mec.14734
Deiner, K., Bik, H. M., Mächler, E., Seymour, M., Lacoursière-Roussel, A., Altermatt, F., et al. (2017). Environmental DNA metabarcoding: transforming how we survey animal and plant communities. Mol. Ecol. 26, 5872–5895. doi: 10.1111/mec.14350
Derocles, S. A. P., Bohan, D. A., Dumbrell, A. J., Kitson, J. J. N., Massol, F., Pauvert, C., et al. (2018). Biomonitoring for the 21st century: integrating next-generation sequencing into ecological network analysis. Adv. Ecol. Res. 58, 1–62. doi: 10.1016/bs.aecr.2017.12.001
Desjardins-Proulx, P., Poisot, T., and Gravel, D. (2019). Artificial intelligence for ecological and evolutionary synthesis. Front. Ecol. Evol. 7:402. doi: 10.3389/fevo.2019.00402
Dickie, I. A., Boyer, S., Buckley, H. L., Duncan, R. P., Gardner, P. P., Hogg, I. D., et al. (2018). Towards robust and repeatable sampling methods in eDNA-based studies. Mol. Ecol. Resour. 18, 940–952. doi: 10.1111/1755-0998.12907
Doi, H., Fukaya, K., Oka, S.-I., Sato, K., Kondoh, M., and Miya, M. (2019). Evaluation of detection probabilities at the water-filtering and initial PCR steps in environmental DNA metabarcoding using a multispecies site occupancy model. Sci. Rep. 9:3581. doi: 10.1038/s41598-019-40233-1
Douglas, G. M., Beiko, R. G., and Langille, M. G. I. (2018). Predicting the functional potential of the microbiome from marker genes using PICRUSt. Methods Mol. Biol. 1849, 169–177. doi: 10.1007/978-1-4939-8728-3_11
Elbrecht, V., Vamos, E. E., Steinke, D., and Leese, F. (2018). Estimating intraspecific genetic diversity from community DNA metabarcoding data. PeerJ 6:e4644. doi: 10.7717/peerj.4644
Enserink, M. (2018). European funders seek to end reign of paywalled journals. Science 361, 957–958. doi: 10.1126/science.361.6406.957
Evans, D. M., Kitson, J. J. N., Lunt, D. H., Straw, N. A., and Pocock, M. J. O. (2016). Merging DNA metabarcoding and ecological network analysis to understand and build resilient terrestrial ecosystems. Funct. Ecol. 30, 1904–1916. doi: 10.1111/1365-2435.12659
Ficetola, G. F., Pansu, J., Bonin, A., Coissac, E., Giguet-Covex, C., De Barba, M., et al. (2015). Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol. Ecol. Resour. 15, 543–556. doi: 10.1111/1755-0998.12338
Ficetola, G. F., Taberlet, P., and Coissac, E. (2016). How to limit false positives in environmental DNA and metabarcoding? Mol. Ecol. Resour. 16, 604–607. doi: 10.1111/1755-0998.12508
Freilich, M., Wieters, E., Broitman, B., Marquet, P., and Navarrete, S. (2018). Species co-occurrence networks: can they reveal trophic and non-trophic interactions in ecological communities? Ecology 99, 690–699. doi: 10.1002/ecy.2142
Gloor, G. B., Macklaim, J. M., Pawlowsky-Glahn, V., and Egozcue, J. J. (2017). Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 8:2224. doi: 10.3389/fmicb.2017.02224
Goldberg, C. S., Turner, C. R., Deiner, K., Klymus, K. E., Thomsen, P. F., Murphy, M. A., et al. (2016). Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 7, 1299–1307. doi: 10.1111/2041-210X.12595
Gonzalez, G. H., Tahsin, T., Goodale, B. C., Greene, A. C., and Greene, C. S. (2016). Recent advances and emerging applications in text and data mining for biomedical discovery. Brief. Bioinform. 17, 33–42. doi: 10.1093/bib/bbv087
Grüning, B., Dale, R., Sjödin, A., Chapman, B. A., Rowe, J., Tomkins-Tinch, C. H., et al. (2018). Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods 15, 475–476. doi: 10.1038/s41592-018-0046-7
Gutiérrez-Cánovas, C., Arribas, P., Naselli-Flores, L., Bennas, N., Finocchiaro, M., Millán, A., et al. (2019). Evaluating anthropogenic impacts on naturally stressed ecosystems: revisiting river classifications and biomonitoring metrics along salinity gradients. Sci. Total Environ. 658, 912–921. doi: 10.1016/j.scitotenv.2018.12.253
Hajibabaei, M., Porter, T. M., Wright, M., and Rudar, J. (2019). COI metabarcoding primer choice affects richness and recovery of indicator taxa in freshwater systems. PLoS ONE.14:e0220953. doi: 10.1371/journal.pone.0220953
Hallmann, C. A., Sorg, M., Jongejans, E., Siepel, H., Hofland, N., Schwan, H., et al. (2017). More than 75 percent decline over 27 years in total flying insect biomass in protected areas. PLoS ONE 12:e0185809. doi: 10.1371/journal.pone.0185809
Hering, D., Borja, A., Jones, J. I., Pont, D., Boets, P., Bouchez, A., et al. (2018). Implementation options for DNA-based identification into ecological status assessment under the European Water Framework Directive. Water Res. 138, 192–205. doi: 10.1016/j.watres.2018.03.003
Hodgetts, T., Grenyer, R., Greenhough, B., McLeod, C., Dwyer, A., and Lorimer, J. (2018). The microbiome and its publics: a participatory approach for engaging publics with the microbiome and its implications for health and hygiene. EMBO Rep. 19:e45786. doi: 10.15252/embr.201845786
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1:16048. doi: 10.1038/nmicrobiol.2016.48
Jetz, W., McGeoch, M. A., Guralnick, R., Ferrier, S., Beck, J., Costello, M. J., et al. (2019). Essential biodiversity variables for mapping and monitoring species populations. Nat. Ecol. Evol. 3, 539–551. doi: 10.1038/s41559-019-0826-1
Jones, K. E., Patel, N. G., Levy, M. A., Storeygard, A., Balk, D., Gittleman, J. L., et al. (2008). Global trends in emerging infectious diseases. Nature 451, 990–993. doi: 10.1038/nature06536
Jonsson, T., Kaartinen, R., Jonsson, M., and Bommarco, R. (2018). Predictive power of food web models based on body size decreases with trophic complexity. Ecol. Lett. 21, 702–712. doi: 10.1111/ele.12938
Kamenova, S., Bartley, T. J., Bohan, D. A., Boutain, J. R., Colautti, R. I., Domaizon, I., et al. (2017). Invasions toolkit. Adv. Ecol. Res. 56, 85–182. doi: 10.1016/bs.aecr.2016.10.009
Keck, F., Vasselon, V., Tapolczai, K., Rimet, F., and Bouchez, A. (2017). Freshwater biomonitoring in the Information Age. Front. Ecol. Environ. 15, 266–274. doi: 10.1002/fee.1490
Kery, M., and Andrew Royle, J. (2015). Applied Hierarchical Modeling in Ecology: Analysis of Distribution, Abundance and Species Richness in R and BUGS: Volume 1:Prelude and Static Models. London: Academic Press. doi: 10.1016/B978-0-12-801378-6.00001-1
Kissling, W. D., Walls, R., Bowser, A., Jones, M. O., Kattge, J., Agosti, D., et al. (2018). Towards global data products of essential biodiversity variables on species traits. Nat. Ecol. Evol. 2, 1531–1540. doi: 10.1038/s41559-018-0667-3
Knight, R., Vrbanac, A., Taylor, B. C., Aksenov, A., Callewaert, C., Debelius, J., et al. (2018). Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422. doi: 10.1038/s41579-018-0029-9
Larsson, A. J. M., Stanley, G., Sinha, R., Weissman, I. L., and Sandberg, R. (2018). Computational correction of index switching in multiplexed sequencing libraries. Nat. Methods 15, 305–307. doi: 10.1038/nmeth.4666
Lausch, A., Borg, E., Bumberger, J., Dietrich, P., Heurich, M., Huth, A., et al. (2018). Understanding forest health with remote sensing, part III: requirements for a scalable multi-source forest health monitoring network based on data science approaches. Remote Sens. 10:1120. doi: 10.3390/rs10071120
Lear, G., Dickie, I., Banks, J., Boyer, S., Buckley, H., Buckley, T., et al. (2018). Methods for the extraction, storage, amplification and sequencing of DNA from environmental samples. N. Z. J. Ecol. 42:10. doi: 10.20417/nzjecol.42.9
Leese, F., Bouchez, A., Abarenkov, K., Altermatt, F., Borja, Á., Bruce, K., et al. (2018). Why we need sustainable networks bridging countries, disciplines, cultures and generations for aquatic biomonitoring 2.0: a perspective derived from the DNAqua-net COST action. Adv. Ecol. Res. 58, 63–99. doi: 10.1016/bs.aecr.2018.01.001
Lefrançois, E., Apothéloz-Perret-Gentil, L., Blancher, P., Botreau, S., Chardon, C., Crepin, L., et al. (2018). Development and implementation of eco-genomic tools for aquatic ecosystem biomonitoring: the SYNAQUA French-Swiss program. Environ. Sci. Pollut. Res. Int. 25, 33858–33866. doi: 10.1007/s11356-018-2172-2
Leibold, M. A., and Chase, J. M. (2017). Metacommunity Ecology. Princeton, NJ: Princeton University Press. doi: 10.2307/j.ctt1wf4d24
Levy, S. E., and Myers, R. M. (2016). Advancements in next-generation sequencing. Annu. Rev. Genomics Hum. Genet. 17, 95–115. doi: 10.1146/annurev-genom-083115-022413
Li, L., Zheng, B., and Liu, L. (2010). Biomonitoring and bioindicators used for river ecosystems: definitions, approaches and trends. Procedia environ. Sci. 2, 1510–1524. doi: 10.1016/j.proenv.2010.10.164
Lugg, W. H., Griffiths, J., van Rooyen, A. R., Weeks, A. R., and Tingley, R. (2018). Optimal survey designs for environmental DNA sampling. Methods Ecol. Evol. 9, 1049–1059. doi: 10.1111/2041-210X.12951
Ma, A., Bohan, D. A., Canard, E., Derocles, S. A. P., Gray, C., Lu, X., et al. (2018a). A replicated network approach to “Big Data” in ecology. Adv. Ecol. Res. 59, 225–264. doi: 10.1016/bs.aecr.2018.04.001
Ma, A., Lu, X., Gray, C., Raybould, A., Tamaddoni-Nezhad, A., Woodward, G., et al. (2018b). Ecological networks reveal resilience of agro-ecosystems to changes in farming management. Nat. Ecol. Evol. 3, 260–264. doi: 10.1038/s41559-018-0757-2
Makiola, A., Dickie, I. A., Holdaway, R. J., Wood, J. R., Orwin, K. H., and Glare, T. R. (2019). Land use is a determinant of plant pathogen alpha-but not beta-diversity. Mol. Ecol 28, 3786–3789. doi: 10.1111/mec.15177
Makiola, A., Dickie, I. A., Holdaway, R. J., Wood, J. R., Orwin, K. H., Lee, C. K., et al. (2018). Biases in the metabarcoding of plant pathogens using rust fungi as a model system. MicrobiologyOpen 8:e00780. doi: 10.1002/mbo3.780
Mangul, S., Martin, L. S., Eskin, E., and Blekhman, R. (2019). Improving the usability and archival stability of bioinformatics software. Genome Biol. 20:47. doi: 10.1186/s13059-019-1649-8
Maruyama, A., Nakamura, K., Yamanaka, H., Kondoh, M., and Minamoto, T. (2014). The release rate of environmental dna from juvenile and adult fish. PLoS ONE 9:e114639. doi: 10.1371/journal.pone.0114639
McClenaghan, B., Compson, Z. G., and Hajibabaei, M. (2019). Validating metabarcoding-based biodiversity assessments with multi-species occupancy models: a case study using coastal marine eDNA. bioRxiv 797852. doi: 10.1101/797852
McGee, K. M., Robinson, C., and Hajibabaei, M. (2019). Gaps in DNA-based biomonitoring across the globe. Front. Ecolo. Evol. 7:337. doi: 10.3389/fevo.2019.00337
Miller, T. (2019). Explanation in artificial intelligence: insights from the social sciences. Artif. Intell. 267, 1–38. doi: 10.1016/j.artint.2018.07.007
Morales-Castilla, I., Matias, M. G., Gravel, D., and Araújo, M. B. (2015). Inferring biotic interactions from proxies. Trends Ecol. Evol. 30, 347–356. doi: 10.1016/j.tree.2015.03.014
Muggleton, S. H., Schmid, U., Zeller, C., Tamaddoni-Nezhad, A., and Besold, T. (2018). Ultra-strong machine learning: comprehensibility of programs learned with ILP. Mach. Learn. 107, 1119–1140. doi: 10.1007/s10994-018-5707-3
Naderi, N., Kappler, T., Baker, C. J. O., and Witte, R. (2011). OrganismTagger: detection, normalization and grounding of organism entities in biomedical documents. Bioinformatics 27, 2721–2729. doi: 10.1093/bioinformatics/btr452
Olson, N. D., Treangen, T. J., Hill, C. M., Cepeda-Espinoza, V., Ghurye, J., Koren, S., et al. (2017). Metagenomic assembly through the lens of validation: recent advances in assessing and improving the quality of genomes assembled from metagenomes. Brief. Bioinform. 20, 1140–1150. doi: 10.1093/bib/bbx098
Ovaskainen, O., Abrego, N., Somervuo, P., Palorinne, I., Hardwick, B., Pitkänen, J.-M., et al. (2019). Monitoring fungal communities with the Global Spore Sampling Project. Front. Ecol. Evol. 7:511. doi: 10.3389/fevo.2019.00511
Papadopoulou, A., Taberlet, P., and Zinger, L. (2015). Metagenome skimming for phylogenetic community ecology: a new era in biodiversity research. Mol. Ecol. 24, 3515–3517. doi: 10.1111/mec.13263
Pauvert, C., Buée, M., Laval, V., Edel-Hermann, V., Fauchery, L., Gautier, A., et al. (2019). Bioinformatics matters: the accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline. Fungal Ecol. 41, 23–33. doi: 10.1016/j.funeco.2019.03.005
Pawlowski, J., Kelly-Quinn, M., Altermatt, F., Apothéloz-Perret-Gentil, L., Beja, P., Boggero, A., et al. (2018). The future of biotic indices in the ecogenomic era: integrating (e)DNA metabarcoding in biological assessment of aquatic ecosystems. Sci. Total Environ. 637–638, 1295–1310. doi: 10.1016/j.scitotenv.2018.05.002
Piñol, J., Senar, M. A., and Symondson, W. O. C. (2019). The choice of universal primers and the characteristics of the species mixture determine when DNA metabarcoding can be quantitative. Mol. Ecol. 28, 407–419. doi: 10.1111/mec.14776
Pocock, M. J. O., Evans, D. M., Fontaine, C., Harvey, M., Julliard, R., McLaughlin, Ó., et al. (2016). The visualisation of ecological networks, and their use as a tool for engagement, advocacy and management. Adv. Ecol. Res. 54, 41–85. doi: 10.1016/bs.aecr.2015.10.006
Poisot, T., Baiser, B., Dunne, J. A., Kéfi, S., Massol, F., Mouquet, N., et al. (2016). Mangal - making ecological network analysis simple. Ecography 39, 384–390. doi: 10.1111/ecog.00976
Poisot, T., Bruneau, A., Gonzalez, A., Gravel, D., and Peres-Neto, P. (2019). Ecological data should not be so hard to find and reuse. Trends Ecol. Evol. 34, 494–496. doi: 10.1016/j.tree.2019.04.005
Porter, T. M., and Hajibabaei, M. (2018). Scaling up: a guide to high-throughput genomic approaches for biodiversity analysis. Mol. Ecol. 27, 313–338. doi: 10.1111/mec.14478
Quick, J., Loman, N. J., Duraffour, S., Simpson, J. T., Severi, E., Cowley, L., et al. (2016). Real-time, portable genome sequencing for Ebola surveillance. Nature 530, 228–232. doi: 10.1038/nature16996
Ransome, E., Geller, J. B., Timmers, M., Leray, M., Mahardini, A., Sembiring, A., et al. (2017). The importance of standardization for biodiversity comparisons: a case study using autonomous reef monitoring structures (ARMS) and metabarcoding to measure cryptic diversity on Mo'orea coral reefs, French Polynesia. PLoS ONE 12:e0175066. doi: 10.1371/journal.pone.0175066
Rudin, C. (2019). Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 1, 206–215. doi: 10.1038/s42256-019-0048-x
Saito, V. S., Siqueira, T., and Fonseca-Gessner, A. A. (2015). Should phylogenetic and functional diversity metrics compose macroinvertebrate multimetric indices for stream biomonitoring? Hydrobiologia 745, 167–179. doi: 10.1007/s10750-014-2102-3
Savary, S., Willocquet, L., Pethybridge, S. J., Esker, P., McRoberts, N., and Nelson, A. (2019). The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 3, 430–439. doi: 10.1038/s41559-018-0793-y
Schmidt-Traub, G., Obersteiner, M., and Mosnier, A. (2019). Fix the broken food system in three steps. Nature 569, 181–183. doi: 10.1038/d41586-019-01420-2
Schröter, M., Kraemer, R., Mantel, M., Kabisch, N., Hecker, S., Richter, A., et al. (2017). Citizen science for assessing ecosystem services: status, challenges and opportunities. Ecosyst. Serv. 28, 80–94. doi: 10.1016/j.ecoser.2017.09.017
Seegert, G. (2000). The development, use, and misuse of biocriteria with an emphasis on the index of biotic integrity. Environ. Sci. Policy 3, 51–58. doi: 10.1016/S1462-9011(00)00027-7
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245. doi: 10.1007/978-1-4939-9173-0_14
Sims, D., Sudbery, I., Ilott, N. E., Heger, A., and Ponting, C. P. (2014). Sequencing depth and coverage: key considerations in genomic analyses. Nat. Rev. Genet. 15, 121–132. doi: 10.1038/nrg3642
Singer, G. A. C., Fahner, N. A., Barnes, J. G., McCarthy, A., and Hajibabaei, M. (2019). Comprehensive biodiversity analysis via ultra-deep patterned flow cell technology: a case study of eDNA metabarcoding seawater. Sci. Rep. 9:5991. doi: 10.1038/s41598-019-42455-9
Steenweg, R., Whittington, J., Hebblewhite, M., Forshner, A., Johnston, B., Petersen, D., et al. (2016). Camera-based occupancy monitoring at large scales: Power to detect trends in grizzly bears across the Canadian Rockies. Biol. Conserv. 201, 192–200. doi: 10.1016/j.biocon.2016.06.020
Stock, M., Poisot, T., Waegeman, W., and De Baets, B. (2017). Linear filtering reveals false negatives in species interaction data. Sci. Rep. 7:45908. doi: 10.1038/srep45908
Takahara, T., Minamoto, T., Yamanaka, H., Doi, H., and Kawabata, Z. (2012). Estimation of fish biomass using environmental DNA. PLoS ONE 7:e35868. doi: 10.1371/journal.pone.0035868
Tamaddoni-Nezhad, A, Bohan, D, Raybould, A, and Muggleton, S. (2015). “Towards machine learning of predictive models from ecological data,” in Inductive Logic Programming. Lecture Notes in Computer Science, Vol. 9046, eds J. Davis and J. Ramon (Cham: Springer). doi: 10.1007/978-3-319-23708-4_11
Tamaddoni-Nezhad, A., Milani, G. A., Raybould, A., Muggleton, S., and Bohan, D. A. (2013). Construction and validation of food webs using logic-based machine learning and text mining. Adv. Ecol. Res. 49, 225–289. doi: 10.1016/B978-0-12-420002-9.00004-4
Tapolczai, K., Keck, F., Bouchez, A., Rimet, F., Kahlert, M., Vasselon, V., et al. (2019). Diatom DNA metabarcoding for biomonitoring : strategies to avoid major taxonomical and bioinformatical biases limiting molecular indices capacities. Front. Ecol. Evol. 7:409. doi: 10.3389/fevo.2019.00409
Thomas, A. C., Deagle, B. E., Paige Eveson, J., Harsch, C. H., and Trites, A. W. (2016). Quantitative DNA metabarcoding: improved estimates of species proportional biomass using correction factors derived from control material. Mol. Ecol. Resour. 16, 714–726. doi: 10.1111/1755-0998.12490
Thompson, L. R., Sanders, J. G., McDonald, D., Amir, A., Ladau, J., Locey, K. J., et al. (2017). A communal catalogue reveals earth's multiscale microbial diversity. Nature 551, 457–463. doi: 10.1038/nature24621
Vandewalle, M., De Bello, F., Berg, M. P., Bolger, T., Doledec, S., Dubs, F., et al. (2010). Functional traits as indicators of biodiversity response to land use changes across ecosystems and organisms. Biodivers. Conserv. 19, 2921–2947. doi: 10.1007/s10531-010-9798-9
Voulvoulis, N., Arpon, K. D., and Giakoumis, T. (2017). The EU water framework directive: from great expectations to problems with implementation. Sci. Total Environ. 575, 358–366. doi: 10.1016/j.scitotenv.2016.09.228
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wolpert, D. H., and Macready, W. G. (1997). No free lunch theorems for optimization. IEEE Trans. Evol. Comput. 1, 67–82. doi: 10.1109/4235.585893
Keywords: eDNA, metabarcoding, biodiversity assessment, artificial intelligence, ecological networks
Citation: Makiola A, Compson ZG, Baird DJ, Barnes MA, Boerlijst SP, Bouchez A, Brennan G, Bush A, Canard E, Cordier T, Creer S, Curry RA, David P, Dumbrell AJ, Gravel D, Hajibabaei M, Hayden B, van der Hoorn B, Jarne P, Jones JI, Karimi B, Keck F, Kelly M, Knot IE, Krol L, Massol F, Monk WA, Murphy J, Pawlowski J, Poisot T, Porter TM, Randall KC, Ransome E, Ravigné V, Raybould A, Robin S, Schrama M, Schatz B, Tamaddoni-Nezhad A, Trimbos KB, Vacher C, Vasselon V, Wood S, Woodward G and Bohan DA (2020) Key Questions for Next-Generation Biomonitoring. Front. Environ. Sci. 7:197. doi: 10.3389/fenvs.2019.00197
Received: 06 June 2019; Accepted: 04 December 2019;
Published: 09 January 2020.
Edited by:
Marco Casazza, Università degli Studi di Napoli Parthenope, ItalyReviewed by:
Erin Grey, Governors State University, United StatesShuisen Chen, Guangzhou Institute of Geography, China
Copyright © 2020 Makiola, Compson, Baird, Barnes, Boerlijst, Bouchez, Brennan, Bush, Canard, Cordier, Creer, Curry, David, Dumbrell, Gravel, Hajibabaei, Hayden, van der Hoorn, Jarne, Jones, Karimi, Keck, Kelly, Knot, Krol, Massol, Monk, Murphy, Pawlowski, Poisot, Porter, Randall, Ransome, Ravigné, Raybould, Robin, Schrama, Schatz, Tamaddoni-Nezhad, Trimbos, Vacher, Vasselon, Wood, Woodward and Bohan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Bohan, ZGF2aWQuYm9oYW5AaW5yYS5mcg==