 Jinlyung Choi1
Jinlyung Choi1 Elizabeth Bach2,3
Elizabeth Bach2,3 Jaejin Lee1Jared Flater1Shane Dooley1
Jaejin Lee1Jared Flater1Shane Dooley1 Adina Howe1*†
Adina Howe1*† Kirsten S. Hofmockel2,4*†
Kirsten S. Hofmockel2,4*†- 1Department of Agricultural and Biosystems Engineering, Iowa State University, Ames, IA, United States
- 2Department of Ecology, Evolution, and Organismal Biology, Iowa State University, Ames, IA, United States
- 3Department of Biology and School of Global Environmental Sustainability, Colorado State University, Fort Collins, CO, United States
- 4Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA, United States
Microbial mechanisms controlling cellulose degradation in soil habitats remains a critical knowledge gap in understanding and modeling terrestrial carbon-cycling. We investigated land management and soil micro-habitat influences on soil bacterial communities and distribution of cellulose-degrading enzyme genes in three bioenergy cropping systems (corn, prairie, and fertilized prairie). Within the soil, aggregates have been examined as potential micro- habitats with specific characteristics influencing resource partitioning and regulation, thus we also investigated genes associated with cellulose degradation within soil aggregate fractions from the fertilized prairie system. Soil bacterial communities and carbon-cycling gene presence varied across land management and soil microhabitats. Examination of genes specifically involved in cellulose-degradation pathways showed high levels of redundancy across the bioenergy cropping systems, but medium macroaggregates (1,000–2,000 μm) supported greater cellulose-degrading enzyme gene abundance than other aggregate fractions and whole soil. In medium aggregates, the enriched cellulose-degrading genes were most similar to genes previously observed in Actinobacteria. These findings represent gentic potential only, and our previous work on the same samples found elevated cellulase exo-enzyme activity in microaggregates. These contrasting results emphasize the importance of measuring community, functional genes, and metabolic potentials in a coordinated manner. Together, these data indicate that location within the soil matrix matters. Overall, our results indicate that soil aggregate environments are hot-spots that select for organisms with functional attributes like cellulose degradation, and future work should further explore micro-environmental factors that affect realized C-cycling processes.
Introduction
Cellulose comprises 40–60% of plant residues (Lynd et al., 2002), contributing more than 70 × 109 Mg of carbon (C) annually to the global C budget (Paul, 2014). Cellulose is a simple polymer that forms insoluble crystalline microfibrils that are highly resistant to enzymatic hydrolysis (Béguin and Aubert, 1994). Therefore, a suite of enzymes is involved in depolymerizing cellulose into molecules that can be assimilated by soil microbes (Lynd et al., 2002; Figure 1A). Generally cellulose degraders are not able to produce the full suite of enzymes involved in breaking down cellulose; therefore, it is considered a community process. Simultaneously, across and within phyla, multiple genes encode for similar enzymatic processes, contributing to functional redundancy within a microbial community. For example, many polysaccharide degraders harbor multiple glycoside hydrolases, promoting the coordinated activity among multiple enzymes (Wilson, 2011; Berlemont and Martiny, 2015). Further, many organisms have the ability to degrade oligosaccharides, but few lineages have been identified with enhanced potential for complex carbohydrate decomposition (Berlemont and Martiny, 2015; Berlemont, 2017). In fact, observations of complex carbohydrate deconstruction is limited to only a few lineages of potential polysaccharide degraders, while the majority of opportunistic microbes participate indirectly by maintaining low oligosaccharide concentrations to prevent enzyme inhibition (Xu et al., 2013; Berlemont and Martiny, 2016).
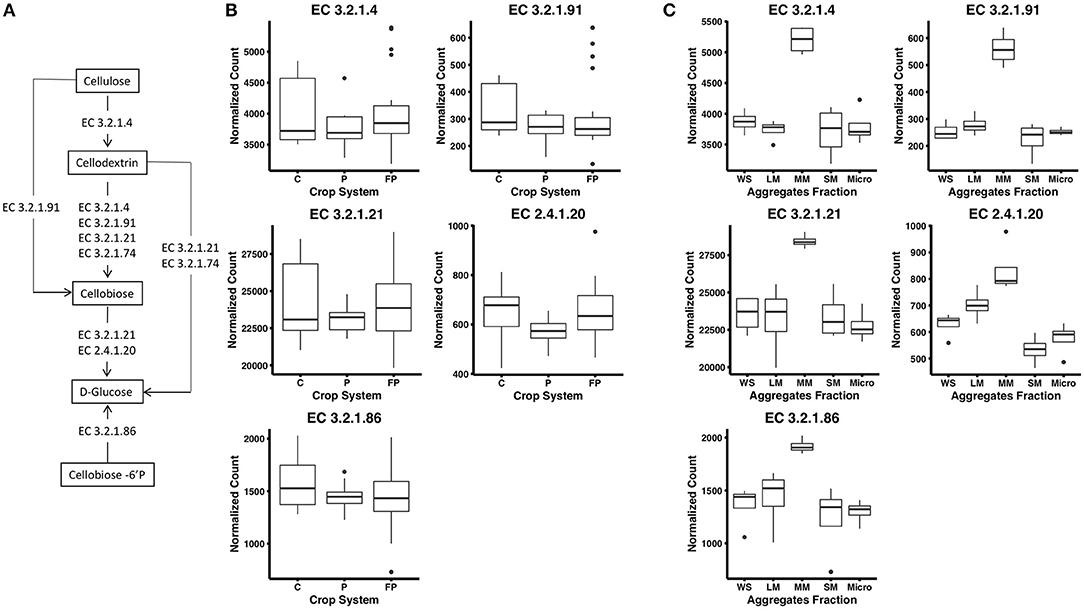
Figure 1. (A) Cellulose decomposition enzymes from the KEGG database. Breaking down cellulose microfibrils requires endoglucanases that randomly attack the cellulose chain (EC 3.2.1.4) and exoglucanases including cellodextrinases (EC 3.2.1.74) that cleave from the end of the cellulose chains releasing cellobiose. Cellobiohydrolases (EC 3.2.1.91) are endoglucanases that can degrade cellulose or cellodextrin to cellobiose. Finally, β-glucosidases (EC 3.2.1.21) cellobiose phosphorylase (2.4.1.20) and 6-phospho-β-glucosidase (EC 3.2.1.86) release glucose from cellobiose and other soluble cellodextrins. (B) Abundance of reads encoding for KEGG enzymes in corn (C), unfertilized prairie (P), and fertilized prairie (FP) metagenomes. (C) Abundance of reads encoding for KEGG enzymes in aggregates from fertilized prairie (FP) metagenomes. WS, whole soil; LM, large; MM, medium; SM, small; Micro, micro aggregates. Normalized counts estimated with DeSeq2 package.
Recent studies considering large-scale patterns in microbial biogeography reveal that different locations harbor microorganisms that differ in genotypic composition (Langenheder and Prosser, 2008; Hanson et al., 2012). Yet few studies have considered the influence of the soil matrix itself, which contains micro-scale environments with stable soil structures that differ in size and breadth of resources (Tisdall and Oades, 1982; Six et al., 2001). This heterogeneity in resource availability can influence community assembly and the abundance and expression of genes contributing to a specific function (Salles et al., 2009). Within the soil matrix, soil aggregates have been examined as potential microbial habitats (or community niche; Leibold, 1995), where the level of complementarity in resource niche partitioning regulates the relative abundance of functional genes with in the soil matrix. Consequently, much of what we know of soil-associated carbohydrate decomposition has stemmed from the isolation of soil microbes that have been characterized with enhanced complex carbohydrate decomposition or potential enzyme measurements of cellobiohydrolase and β-glucosidases to understand how cellulose decomposition varies across ecosystems (Sinsabaugh et al., 2008) and land management regimes (Bowles et al., 2014).
Some studies have measured the potential activity of extracellular enzymes and taxonomic diversity among soil aggregate fractions (Marx et al., 2005; Allison and Jastrow, 2006; Bailey et al., 2012; Kim et al., 2015). In a comparison of individual soil aggregates, relative abundance of bacterial family Chitinophagaceae was greater in aggregates with high β-glucosidase activity (Bailey et al., 2013), suggesting that community membership of a functional group at the micro-scale may play a role in the potential for soil carbon cycling. Further evidence for variation among soil aggregates was observed in a mollisol under corn, prairie, and fertilized prairie bioenergy cropping systems in central Iowa, where increased plant inputs in the prairies correlated with increased potential cellobiohydrolase enzyme activity and microbial biomass carbon pools (Bach and Hofmockel, 2015, 2016). Within all three bioenergy systems, total carbon was greatest in soil macroaggregate fractions in contrast to greater potential cellobiohydrolase activity in microaggregates (<250 μm) (Bach and Hofmockel, 2016). Further exploration of the soil microbial communities in these aggregates revealed that the bacterial (16S rRNA) and fungal (ITS) communities showed greater microbial diversity and distinct microbial communities in microaggregate fractions (Bach et al., 2018), evidence of the role of microbial membership within aggregate fractions to observed differences in enzyme activity potentials.
To identify genes encoding enzymes involved in cellulose degradation and the phylogenetic distribution of their bacterial hosts across cropping systems and aggregate scales, we performed full metagenomic sequencing on whole soil from three bioenergy systems (i.e., corn, prairie, and fertilized prairie) and five soil aggregate fractions from a fertilized prairie system. The fertilized prairie soils were strategically selected to examine aggregate-scale differences because our previous work showed it supports greater extracellular enzyme activity and microbial biomass than the corn and unfertilized prairie systems (Bach and Hofmockel, 2015). We hypothesize that cellulose degradation is enhanced in prairie systems due to enriched abundance of genes encoding for key cellulose-degrading enzymes, particularly more energetically expensive endocellulases, and support more phyla contributing those genes. Further, we hypothesize that cellulose degrading genes are not evenly distributed among micro-habitats, or aggregate fractions, due to differences in the distribution of carbon among aggregates. Specifically, we expected to see more genes encoding cellulose-degrading enzymes in microaggregates, where we previously observed increased cellobiohydrolase activity (Bach and Hofmockel, 2016).
Materials and Methods
Soil Sampling and Sequencing
Soil samples were collected at the Comparison of Biofuel Systems (COBS) research site at Iowa State University (Ames, IA). Replicated plots growing continuous corn (C, corn planted every year without rotation), prairie (P, prairie without fertilizer), and fertilized prairie (FP, prairie with fertilizer, 84 Kg N ha−1) were studied. The study site is a randomized complete block design with four replicate blocks containing each treatment (n = 12). Three replicate soil samples (5.5 cm diameter, 10 cm deep) were collected from each plot in July, 2012. Replicate cores from each plot were pooled and physically sieved into five different aggregate sizes after collection [whole soil, large (>2,000 μm), medium (1,000–2,000 μm), small (250–1,000 μm), and micro aggregate (<250 μm)] as previously described (Bach and Hofmockel, 2014). Among the resulting 60 samples (5 fractions × 4 replicates × 3 blocks), a total of 44 samples were selected for DNA extraction for this study, including whole soil from all plots and all fractions from fertilized prairie. DNA was extracted using PowerSoil®-htp DNA Isolation Kit (MoBio, Carlsbad, CA, United States) as previously described (Bach et al., 2018). Paired-end metagenome libraries (read length 100 bp) were prepared and sequenced using HiSeq at Argonne National Laboratory. All 44 metagenomes are publicly available at the MG-RAST Metagenomics Analysis Server (https://www.mg-rast.org/mgmain.html?mgpage=project&project=mgp13620), Supplementary Table 1). In total, this study represents a total of 3.1 billion reads (average 71 million reads per sample).
Analysis of Metagenomes
Sequences were aligned to the KEGG prokaryote and eukaryote protein database (obtained January 17, 2017) using Diamond (v0.7.9.58), requiring a minimum E-value threshold of 0.001. The best-matching amino acid sequence with these criteria was selected as the KEGG protein annotation. Count data for the abundance of genes encoding for enzymes was analyzed with DESeq2 (version 1.16.1). Taxonomy associated with metagenomic reads was assigned by nucleotide homology to genes encoding enzymes in the KEGG database, requiring 97% similarity and a minimum E-value of 1e-5 (BLAST+, version 2.2.30). For estimation of taxonomy, abundances were calculated as total reads associated with an annotation normalized by the total number of reads in each metagenome. To characterize cellulose degradation, a subset of KEGG enzymes associated with cellulose decomposition were selected within the starch and sucrose metabolism pathway (Kanehisa et al., 2017), including endoglucanases (EC 3.2.1.4), β-glucosidases (EC 3.2.1.21), 6-phospho-β-glucosidase (EC 3.2.1.86), cellobiose phosphorylase (EC 2.4.1.20), cellobiohydrolases (EC 3.2.1.91) (Figure 1A). Co-occurrence analysis was used to understand interactions of genes associated with cellulose decomposition. Spearman correlation coefficient values were calculated using co-occurrence software FastCoOccur (version 0.0.1) (https://github.com/germs-lab/FastCoOccur), which is based on methods previously described (Williams et al., 2014). Nodes representing genes associated with KEGG enzymes were considered significant if the Spearman's rank correlation coefficient, ρ > 0.7 or smaller than −0.7 and the P < 0.01. Visualization of networks was performed using the Force Atlas layout of Gephi software (version 0.9.2).
Statistical Analyses
All statistical analysis was performed in R v.3.4.1. Phyloseq (version 1.20.0) (McMurdie and Holmes, 2013) was used to for metagenome comparisons. All NMDS ordinations were plotted based on Bray-Curtis distances, and significance between treatments was determined based on permutational multivariate analysis of variance (PERMANOVA) using Adonis function in vegan package (version 2.4-4) (Oksanen et al., 2017). Cellulose decomposition genes that were significantly different between treatments were determined using DESeq2 (version 1.16.1) (Love et al., 2014), requiring p < 0.01. All analysis associated with this study are available at https://github.com/germs-lab/cobs-study.
Results
Genes Associated With Cellulose Decomposition Vary in Abundances Between Corn and Prairies
For each soil sample, a metagenome library was sequenced, and extracellular enzyme activity was measured. Specifically, carbon-degrading enzymes β- glucosidase and cellobiohydrolase were measured, and we observed greater potential activity of both in fertilized and unfertilized prairie crops compared with corn (Bach and Hofmockel, 2015, 2016). To better understand the genes associated with the observed differences in enzyme potentials, we sequenced metagenomes from the three crops (Howe et al., 2016). The microbial community membership between the corn, prairie, and fertilized prairie were compared based on the 16S rRNA gene distribution in soil metagenomes. We observed significant differences between 16S rRNA genes identified in corn and both prairie metagenomes (prairie vs corn: p = 0.0021, corn vs fertilized prairie: p = 0.0001) and between prairie and fertilized prairie (p = 0.0173; Supplementary Figure 1), suggesting distinct microbial membership among microbiomes. Comparing the distribution of all KEGG genes in the three cropping systems, we observed significant differences between corn and both prairie crop systems (prairie vs corn: p = 0.0098, corn vs fertilized prairie: p = 0.0007), but no significant differences between unfertilized and fertilized prairie metagenomes (p = 0.4; Supplementary Figure 2).
To investigate cellulose degradation in these cropping systems, we selected genes associated with the starch and sucrose metabolism pathway (KEGG map00500) and five cellulose degrading enzymes (Figure 1A). These enzymes were selected because of their consistent detection across soil metagenomes. Overall, genes associated with these cellulose degradation enzymes comprised only a small fraction of the soil metagenomes, ~0.5% of total reads. Among these, genes associated with β-glucosidases (EC 3.2.1.21) were the most abundant, with an average abundance of 23,903 reads per total number of reads in fertilized prairie soil metagenomes (average, normalized), followed by endoglucanases (EC 3.2.1.4; 3,954 average reads), 6-phospho-β-glucosidase (EC 3.2.1.86; 1,481 average reads) and cellobiose phosphorylase (EC 2.4.1.20; 629 average reads) (Figures 1B,C). Cellulose degrading enzyme genes observed in low abundance were cellobiohydrolases (EC 3.2.1.91; 306 average reads) (Figure 1B). In comparison, the range of abundances of reads associated with any genes associated with a KEGG EC was 0 to 254,355, with an average of 933. Overall, the relative abundance of each cellulase gene was similar in all three cropping systems, suggesting that these genes are generally conserved within the same soil type across land management differences (Supplementary Figure 3). We identified the taxonomy associated with these cellulase genes in the three cropping systems, observing that similar bacteria are associated with genetic potential for cellulose decomposition in these soils (Supplementary Figure 4). Overall, 25 phyla were associated with the 1,214 genes encoding the six enzymes associated with cellulose degradation (Figure 2). In all soils, we identified that Actinobacteria and Proteobacteria comprise the majority of phyla associated with endoglucanases (EC 3.2.1.4) and β-glucosidases (EC 3.2.1.21), while mainly Proteobacteria are associated with genes encoding for 6-phospho-β-glucosidase (EC 3.2.1.86). Cellobiohydrolases (EC 3.2.1.91) was associated with mainly Actinobacteria and Ascomycota (Figure 2).
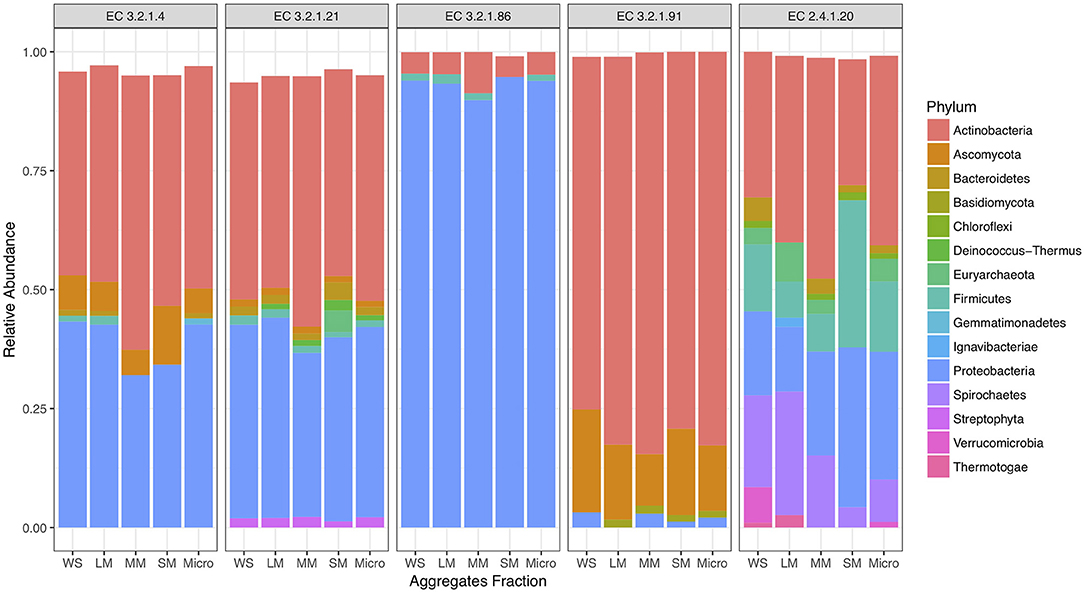
Figure 2. Proportion of taxonomy associated with cellulose decomposition enzymes including endoglucanases (EC 3.2.1.4), β-glucosidases (EC 3.2.1.21), 6-phospho-β-glucosidase (EC 3.2.1.86), cellobiose phosphorylase (EC 2.4.1.20), and cellobiohydrolases (EC 3.2.1.91) in fertilized prairie aggregate metagenomes.
Genes Associated With Cellulose Decomposition Vary in Abundances in Aggregates of Fertilized Prairie Metagenomes
To examine distribution of cellulase gene abundance within the soil matrix, four aggregate fractions and whole soil metagenomes were compared from the fertilized prairie treatment. Laboratory measures of extracellular enzyme activity across these same aggregate samples found increased cellobiohydrolase activity in microaggregates (<250 μm) compared with other aggregate fractions (Bach and Hofmockel, 2016). To identify functional differences in aggregates, we compared KEGG genes in fertilized prairie metagenomes. In contrast to 16S rRNA gene distributions (Supplementary Figure 5, p-value between 0.28 and 0.79), we observed significant differences in gene abundances associated with KEGG enzymes (Supplementary Figure 6, medium vs other aggregates: p < 0.05; micro vs large: p = 0.035; among other aggregate: p > 0.05). For KEGG genes specific to cellulose degradation (Figure 1A), we observed consistent contributions of genes among aggregate fractions (Supplementary Figure 7). Further, the abundance of these genes in micro, small, and large aggregates were similar to whole soils (Figure 1C); however, in contrast to other aggregates, medium aggregates were observed to consistently be enriched for genes associated with enzymes in the cellulose degradation pathway (p < 0.05, Figure 1C). Specifically, we observed that genes encoding for enzymes cleaving cellobiose (EC 3.2.1.91) and subsequently D-glucose (EC 3.2.1.21, EC 2.4.1.20) were significantly enriched in medium aggregates compared to other soil fractions. Further, genes encoding for enzymes associated with extracellular cellulose degradation, specifically extracellular cellobiose (EC 3.2.1.86), were also observed in significantly greater abundances in medium aggregate soils (Figure 1C). This contrasts with our direct measures of extracellular cellobiose degradation, which were elevated in microaggregates (Bach and Hofmockel, 2016).
To further characterize the gene abundance differences we observed in medium aggregates, we identified the taxonomy of cellulose degradation genes observed to be significantly different in medium aggregates. In these aggregates, we observed a significantly greater abundance of genes associated with Actinobacteria in four cellulose decomposition enzymes [endoglucanases (EC 3.2.1.4) and β-glucosidases (EC 3.2.1.21), 6-phospho-β-glucosidase (EC 3.2.1.86), cellobiohydrolases (EC 3.2.1.91), p < 0.01; Figure 3]. Overall, Actinobacteria comprised on average 7.5% of the taxonomy observed in soil (based on 16S rRNA gene abundances), suggesting despite its low presence in bulk soils, it may play an important role in medium aggregates. In endoglucanases (EC 3.2.1.4) and β-glucosidases (EC 3.2.1.21), genes associated with Streptophyta and Basidiomycota were observed at significantly greater abundance in medium aggregates (p < 0.01), suggesting that fungal organisms may play important roles in decomposing cellulose in the soil (Figure 3).
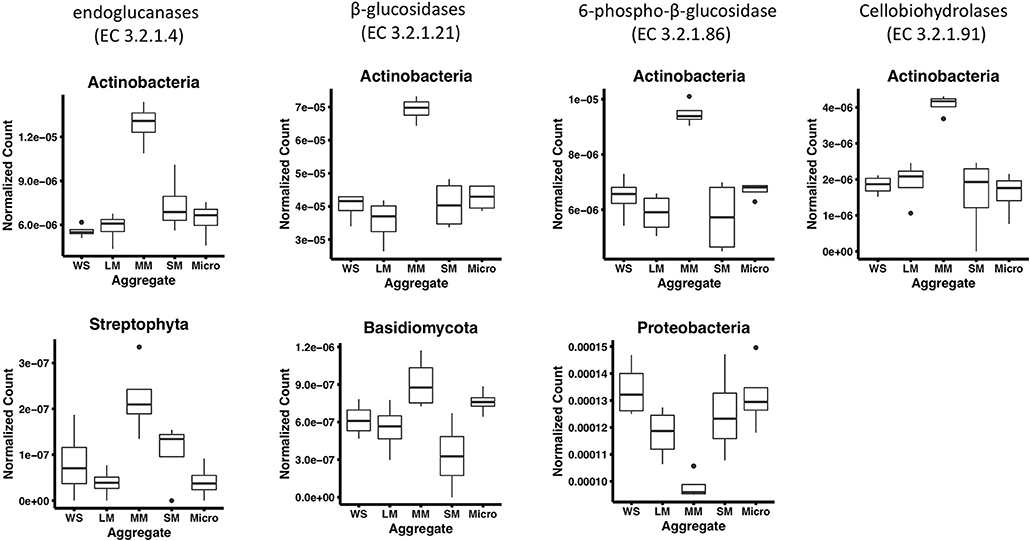
Figure 3. Phyla that significantly differ among aggregates associated with cellulose decomposition enzymes in fertilized prairie aggregate metagenomes (p < 0.01, ANOVA). Cellobiose phosphorylase (EC 2.4.1.20) is exclude since no significantly different phyla were observed.
Genes Potentially Interacting With Cellulose Enzymes in Medium Aggregates in Fertilized Prairie Metagenomes
Broadening out beyond only cellulose decomposition enzymes, we examined the 3,546 genes within the KEGG database and found that 346 genes were observed to be significantly different between medium aggregates and whole soil. There were similar numbers of genes observed to be more or less prevalent in medium aggregates. A total of 162 genes (associated with 146 KEGG pathways) were observed enriched in medium aggregates relative to other aggregates and whole soils (Supplementary Figure 8A). In contrast, 184 genes (associated with 204 KEGG pathways), were less abundant in medium aggregates (Supplementary Figure 8C). Genes associated with similar KEGG pathways were observed as both increased and decreased in medium aggregates. Similar results were observed in carbohydrate metabolism associated genes (Supplementary Figures 8B,D). In other words, we could not identify specific non-cellulose processing pathway genes that were enriched in medium aggregates. Consequently, to better understand the genes unique to medium aggregates, we next evaluated genes of enzymes that may interact with enzymes associated with cellulose degradation.
We performed a co-occurrence network analysis on all genes within metagenomes that were correlated to genes encoding enzymes in the cellulose degradation pathway (Figure 1). In total, we identified 192 and 121 KEGG genes that were either positively or negatively correlated with cellulase genes, respectively (Figure 4). We next evaluated whether these KEGG enzyme genes were significantly different (increased or decreased, p < 0.01) within medium aggregate metagenomes compared to other aggregates and bulk soil. A total of 54 positively-correlated KEGG genes were observed to be enriched in medium aggregate compared with whole soil (Figure 4A, green nodes, Supplementary Table 2A), and 42 negatively-correlated genes decreased abundances in medium aggregates (Figure 4B, red nodes, Supplementary Table 2B). EC class 4 (Lyases) were not observed in 54 positively-correlated genes that were enriched in medium aggregates. However all six EC classes were observed in negatively-correlated relationships with decreased abundance in medium aggregates. Among EC class 3 (Hydrolases), six of glycosidase class (EC 3.2.1.-) were observed in positive-correlations, but not observed among the negative-correlations.
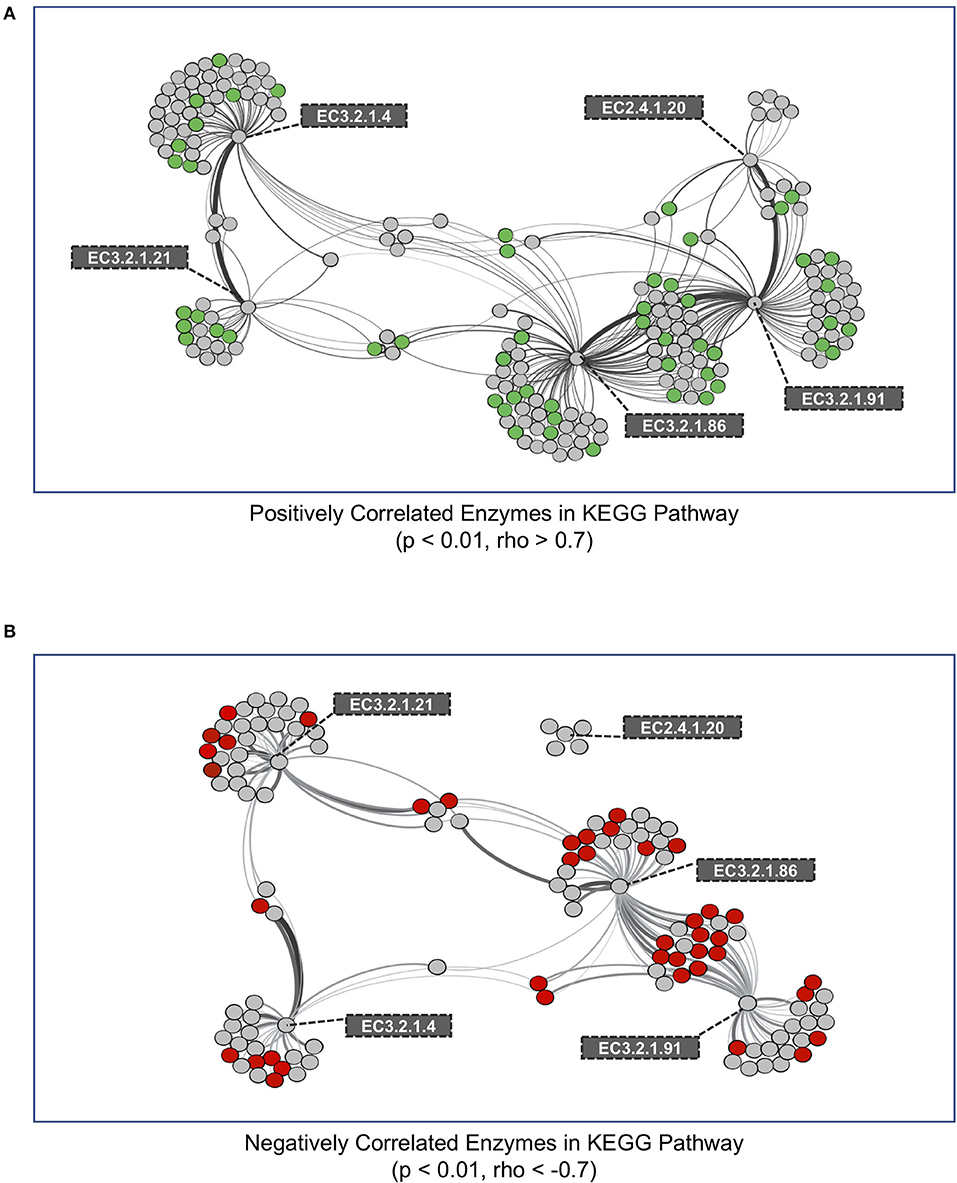
Figure 4. Co-occurrence network of KEGG enzymes with significant interactions among cellulose decomposition enzymes. (A) Positively correlated KEGG enzymes with an enzyme in the cellulose decomposition pathway. Green nodes represent KEGG enzymes that are enriched in medium aggregates compared to whole soil. (B) Negatively correlated KEGG enzymes with an enzyme in the cellulose decomposition pathway. Red nodes represent KEGG enzymes that are observed in lower abundances in medium aggregate compared to whole soil. Thickness of edges represent the strength of correlation between nodes.
Discussion
Microbial mechanisms controlling cellulose degradation in soil habitats remains a critical knowledge gap in understanding and modeling terrestrial C-cycling. Our metagenomic investigation of mollisols under three bioenergy cropping systems in central Iowa demonstrates that soil bacterial communities and KEGG associated genes vary across land management and soil microhabitats. Examination of genes specifically involved in cellulose-degradation pathways showed high levels of redundancy across the bioenergy cropping systems, but medium macroaggregates (1,000–2,000 μm) supported greater cellulose-degrading enzyme gene abundance than other aggregate fractions and whole soil. In contrast, direct measures of potential cellobiohydrolase activity in these same soil samples were observed to be elevated in microaggregate fractions (Bach and Hofmockel, 2016). Hence, realized C-cycling functions like cellulose degradation do not necessarily reflect observed differences in microbial genetic potential (Frossard et al., 2012). Similar pattern is observed in higher level of taxonomic group (Supplementary Figure 9). Together, these data indicate that genetic potential and realized cellulase activity are redundant across bacterial taxa that respond to changes in land management but are sensitive to changes in soil microbial habitat.
Bacterial Communities and Cellulose Degradation Potential Differ Under Land Management
At an ecosystem scale, bacterial communities responded to stark differences in root input quantity and quality among the three bioenergy cropping systems. Corn systems had the least root biomass, fertilized prairie produced 8 times more root biomass than corn in 2011, and unfertilized prairie produced more than twice as much root biomass as unfertilized prairie (Dietzel et al., 2015). In addition, corn roots had a greater cellulose index than either prairie treatment, and unfertilized prairie roots had greater hemicellulose index, driven by greater concentrations of arabinose, galactose, glucose, and xylose (Rivas et al., 2014). These differences in root inputs likely contributed to our observed increased extracellular enzyme activity in both prairie systems compared with corn (Bach and Hofmockel, 2015) as well as the differences in the bacterial communities and KEGG C-cycling gene profiles among the three systems observed in this study (Supplementary Figure 1). However, we found genes associated with cellulose degradation pathways were relatively rare in the full metagenomes, only ~0.5% of total reads and were similarly distributed among the three cropping systems.
Across the cropping systems, there are several reasons the capacity for cellulose degradation may be highly conserved. First, these prairie plantings were only 4 years old at the time of sampling and had been managed as corn fields for many decades before the planting, so we may be observing a legacy effect on cellulose degradation capacity (Kulmatiski and Beard, 2011). Reciprocal transplant of organic matter among agricultural and natural systems detected management system effects on microbial consumption of organic matter, implying land use and organic matter inputs influence carbon decomposition (Hunting et al., 2017). Secondly, we did observe taxonomic shifts in bacterial communities among the systems, supporting several others studies showing functional redundancy in bacterial and fungal communities (Fierer et al., 2013; Talbot et al., 2014). Given cellulose-degrading enzyme genes were such a small proportion of the total metagenome, it is likely these genes are hosted by a small portion of the total bacterial community, and that portion may not be driving taxonomic shifts in the full community. Small changes in the phyla housing β-glucosidases (EC 3.2.1.21), endoglucanases (EC 3.2.1.4), 6-phospho-β-glucosidase (EC 3.2.1.86), cellobiose phosphorylase (EC 2.4.1.20), and cellobiohydrolases (EC 3.2.1.91) indicate similar levels of functional genes were found overall in all the cropping systems, but were contributed by different bacterial community members (Supplementary Figure 4). Differences in cellulose input levels may also lead to differences in gene expression and/or enzyme efficiency. It is also possible that other soil organisms such as fungi, nematodes, collembolan, and/or earthworms may be the primary consumers of plant inputs, performing the initial stages of cellulose decomposition within their guts. To better understand micro-scale processes that may influence ecosystem-scale shifts in community and cellulose decomposition, we investigated microbial metagenomes from within soil aggregates.
Medium Macroaggregates Enriched in Genes Related to Cellulose Degradation
We identified medium macroaggregates as a “hot-spot” for genetic potential for cellulose degradation. Previous work on these same samples indicated microaggregates, not medium macroaggregates support elevated C-cycling enzyme activity and distinct bacterial or fungal communities(Bach and Hofmockel, 2016; Bach et al., 2018). Other studies have also found contrasting results. Allison and Jastrow (2006) also found elevated cellulase activity within microaggregates, but Kim et al. (2015) found no relationship between extracellular enzyme activity rates and microbial community composition within soil aggregates. Investigating enzyme activity within individual macroaggregates (<1,000 μm diameter), revealed higher cellulase activity in small volume macroaggregates, and microbial communities within aggregates with high β-glucosidase activity did not differ in overall microbial diversity and richness, but did differ in relative abundance of Chitinophagaceae family of bacteria (Bailey et al., 2013).
In this study, enrichment of genes encoding cellulose decomposing enzymes were predominantly associated with Actinobacteria and Proteobacteria. Previous field based experiments have noted the importance of Actinobacteria and Proteobacteria for predicting the activities of cellobiohydrolase activity (Trivedi et al., 2016). Phylogenetic investigations also support a predominance of cellulolytic capabilities among the order Actinomycetales, noting the trait-based advantage of filamentous morphology that preferentially enables penetration of cellulosic substrates within heterogeneous environments (Lynd et al., 2002). Enrichment of cellulose degrading Actinobacteria within the medium aggregates support the concept of niche differentiation, where sources of cellulose, such as plant residues, may accumulate in macroaggregates (Six et al., 2000, 2002) creating separate habitats that harbor functionally distinct communities. Here we build upon this understanding of macroaggregates along with previous evidence from the COBS field experiment by demonstrating microsite differences in cellulose degrading communities, genetic potential, and enzyme activity across soil aggregate fractions within whole soil.
Cellulose degradation is a community process involving multiple enzymes that cleave cellulose molecules from the end (exoglucanases) and within (endocellulases) of the polymer (Figure 1A). Endocellulases are critical to decomposition, but energetically expensive, because cellodextrin cannot be assimilated intact, due to the large size. Yet breaking the interior bonds within a cellulose chain is essential for generating multiple fragments that can be cleaved into assimilable substrates. Our results indicate that despite the strong influence of endocellulases on cellulose decomposition, and in turn ecosystem functioning, these genes are rare, and may provide a keystone function (Chapin et al., 2000; Crowther et al., 2013). Endocellulases are much less prevalent relative to β-glucosidases, which are abundant and broadly distributed among taxa. In general, we observed that the proportion of phyla associated with cellulose degradation was consistent among soil aggregate fractions throughout the pathway of genes, including Actinobacteria, and Proteobacteria with contributions from Basidiomycota and Ascomycota.
While we observed that aggregate-specific dynamics resulted in the enrichment of genes associated with cellulose decomposition, we could not identify these trends for specific metabolic pathways. This result emphasizes the complexity of organic matter decomposition pathways in soil and the difficulty to unraveling microbial multifunctionality. Breaking down cellulose microfibrils requires endoglucanases that randomly attack the cellulose chain, but do not necessarily produce assimilable substrates (EC 3.2.1.4). Subsequently exoglucanases including cellodextrinases (EC 3.2.1.74) cleave from the end of the cellulose chains releasing cellobiose. Cellobiohydrolases (EC 3.2.1.91) are endoglucanases that can degrade cellulose or cellodextrin to cellobiose. Finally, β-glucosidases (EC 3.2.1.21) cellobiose phosphorylase (2.4.1.20) and 6-phospho-β-glucosidase (EC 3.2.1.86) release glucose from cellobiose and other soluble cellodextrins (Schimz et al., 1983; Singh and Hayashi, 1995; Lin et al., 2012; Montella et al., 2017). In addition to genes encoding for these enzymes, we found many genes that positively or negatively correlated with cellulose decomposition genes. However, the vast majority of these genes are not studied in association with cellulose decomposition. Our findings suggest hypotheses for future researcher aimed at understanding the genetic mechanisms underpinning microbial decomposition of cellulose in soil. Our results indicate that this metabolism is accomplished with diverse microbes with similar functions operating distinctively depending on their microenvironment.
Conclusions
Cellulose-degradation is an important, yet complex process involving multiple pathways and microbial species. Both natural and human-induced alterations can therefore constrain this process in numerous ways. Our deep exploration of soil metagenomes showed that bacterial communities were larger and more diverse in prairie plantings, across all aggregate sizes, and more diverse in microaggregates, regardless of land management. Presence of cellulose-degradation pathways were similar across land management regimes, but were modestly enriched in medium macroaggregate habitats. This finding contrasts our previous work on the same samples, which found elevated cellulase exo-enzyme activity in microaggregates, emphasizing the importance of considering the potential nature of many standard soil measures. One consistent point in these data is that spatial structuring within the soil matrix differentiates the genetic and enzymatic potential as well as the distribution of organisms within the soil. Soil aggregate environments have substrate hot-spots that select for organisms with functional attributes. To identify the mechanisms driving realized functions in-situ, future work will continue to incorporate molecular information and substrate inputs that captures the pools and fluxes of metabolites and enzymes expressed by organisms under field conditions.
Author Contributions
JC, EB, AH, and KH wrote the manuscript. EB performed the experiment. JC, JL, JF, and SD performed analysis. AH and KH designed the experiment and overall project.
Funding
This work was supported by U.S. Department of Energy (Award Number SC0010775). Additional sequencing technical support provided by DOE Genomic Sciences and Sarah Owens at Argonne National Laboratory. Folker Meyer of Argonne National Laboratory provided support for data archiving through MG-RAST.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sarah Hargreaves, Ryan Williams, Fan Yang, Giselle Narvaez-Rivera, Becka Luzbetack, Austin Putz, Eric Asbe, and Elyssa McFarland for help with laboratory and field work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2018.00107/full#supplementary-material
Supplementary Figure 1. NMDS of 16S rRNA genes from corn (C), unfertilized prairie (P), and fertilized prairie (FP) metagenomes.
Supplementary Figure 2. NMDS of KEGG genes associated with corn (C), unfertilized prairie (P), and fertilized prairie (FP) metagenomes.
Supplementary Figure 3. Proportion of KEGG enzyme associated with cellulose decomposition enzymes including endoglucanases (EC 3.2.1.4), β-glucosidases (EC 3.2.1.21), 6-phospho-β-glucosidase (EC 3.2.1.86), cellobiose phosphorylase (EC 2.4.1.20), and cellobiohydrolases (EC 3.2.1.91) in three bioenergy crops (CC, corn; P, prairie; FP, fertilized prairie).
Supplementary Figure 4. Proportion of taxonomy associated with cellulose decomposition enzymes including endoglucanases (EC 3.2.1.4), β-glucosidases (EC 3.2.1.21), 6-phospho-β-glucosidase (EC 3.2.1.86), cellobiose phosphorylase (EC 2.4.1.20), and cellobiohydrolases (EC 3.2.1.91) in three bioenergy crops (CC, corn; P, prairie; FP, fertilized prairie). Phylum comprising <1% of the total community abundance were removed.
Supplementary Figure 5. NMDS of 16S rRNA genes in metagenomes from aggregate fractions and whole soil of fertilized prairie metagenomes. (WS, whole soil; LM, large; MM, medium; SM, small; MM, micro aggregates).
Supplementary Figure 6. NMDS of KEGG genes in aggregate fractions and whole soil of fertilized prairie metagenomes. (WS, whole soil; LM, large; MM, medium; SM, small; MM, micro aggregates).
Supplementary Figure 7. Proportion of KEGG enzyme associated with cellulose decomposition enzymes including endoglucanases (EC 3.2.1.4), β-glucosidases (EC 3.2.1.21), 6-phospho-β-glucosidase (EC 3.2.1.86), cellobiose phosphorylase (EC 2.4.1.20), and cellobiohydrolases (EC 3.2.1.91) in aggregate fractions and whole soil. (WS, whole soil; LM, large; MM, medium; SM, small; MM, micro aggregates).
Supplementary Figure 8. All enzymes significantly increased (A) or decreased (B) in abundance in medium aggregates relative to other soil metagenomes. Enzymes associated with carbohydrate metabolism increased (C) or decreased (D) in abundance in medium aggregates relative to other soil metagenomes. Z-score represents distribution of a numerical measurement of a value's relationship to the mean.
Supplementary Figure 9. Proportion of taxonomy associated with cellulose decomposition enzymes including endoglucanases (EC 3.2.1.4), β-glucosidases (EC 3.2.1.21), 6-phospho-β-glucosidase (EC 3.2.1.86), cellobiose phosphorylase (EC 2.4.1.20), and cellobiohydrolases (EC 3.2.1.91) in aggregate fractions and whole soil. (WS, whole soil; LM, large; MM, medium; SM, small; MM, micro aggregates). Phylum comprising <1% of the total community abundance were removed.
Supplementary Table 1. Summary of metagenomes used in this study. Aggregate fractions included whole soil (WS), large (>2,000 μm, LM), medium (1,000–2,000 μm, MM), small (250–1,000 μm, SM), and micro aggregate (<250 μm, micro) sieved fractions.
Supplementary Table 2. All enzymes significantly positively and negatively correlated with cellulose decomposition enzymes in medium aggregates.
References
Allison, S. D., and Jastrow, J. D. (2006). Activities of extracellular enzymes in physically isolated fractions of restored grassland soils. Soil Biol. Biochem. 38, 3245–3256. doi: 10.1016/j.soilbio.2006.04.011
Bach, E. M., and Hofmockel, K. S. (2014). Soil aggregate isolation method affects measures of intra-aggregate extracellular enzyme activity. Soil Biol. Biochem. 69, 54–62. doi: 10.1016/j.soilbio.2013.10.033
Bach, E. M., and Hofmockel, K. S. (2015). Coupled carbon and nitrogen inputs increase microbial biomass and activity in prairie bioenergy systems. Ecosystems 18, 417–427. doi: 10.1007/s10021-014-9835-8
Bach, E. M., and Hofmockel, K. S. (2016). A time for every season: soil aggregate turnover stimulates decomposition and reduces carbon loss in grasslands managed for bioenergy. GCB Bioenergy 8, 588–599. doi: 10.1111/gcbb.12267
Bach, E. M., Williams, R. J., Hargreaves, S. K., Yang, F., and Hofmockel, K. S. (2018). Greatest soil microbial diversity found in micro-habitats. Soil Biol. Biochem. 118, 217–226. doi: 10.1016/j.soilbio.2017.12.018
Bailey, V. L., Bilskis, C. L., Fansler, S. J., McCue, L. A., Smith, J. L., and Konopka, A. (2012). Measurements of microbial community activities in individual soil macroaggregates. Soil Biol. Biochem. 48, 192–195. doi: 10.1016/j.soilbio.2012.01.004
Bailey, V. L., Fansler, S. J., Stegen, J. C., and McCue, L. A. (2013). Linking microbial community structure to β-glucosidic function in soil aggregates. ISME J. 7, 2044–2053. doi: 10.1038/ismej.2013.87
Béguin, P., and Aubert, J. P. (1994). The biological degradation of cellulose. FEMS Microbiol. Rev. 13, 25–58. doi: 10.1111/j.1574-6976.1994.tb00033.x
Berlemont, R. (2017). Distribution and diversity of enzymes for polysaccharide degradation in fungi. Sci. Rep. 7:222. doi: 10.1038/s41598-017-00258-w
Berlemont, R., and Martiny, A. C. (2015). Genomic potential for polysaccharide deconstruction in bacteria. Appl. Environ. Microbiol. 81, 1513–1519. doi: 10.1128/AEM.03718-14
Berlemont, R., and Martiny, A. C. (2016). Glycoside hydrolases across environmental microbial communities. PLoS Comput. Biol. 12:e1005300. doi: 10.1371/journal.pcbi.1005300
Bowles, T. M., Acosta-Martínez, V., Calderón, F., and Jackson, L. E. (2014). Soil enzyme activities, microbial communities, and carbon and nitrogen availability in organic agroecosystems across an intensively-managed agricultural landscape. Soil Biol. Biochem. 68, 252–262. doi: 10.1016/j.soilbio.2013.10.004
Chapin, F. S., Zavaleta, E. S., Eviner, V. T., Naylor, R. L., Vitousek, P. M., Reynolds, H. L., et al. (2000). Consequences of changing biodiversity. Nature 405, 234–242. doi: 10.1038/35012241
Crowther, T. W., Stanton, D. W. G., Thomas, S. M., A'Bear, A. D., Hiscox, J., Jones, T. H., et al. (2013). Top-down control of soil fungal community composition by a globally distributed keystone consumer. Ecology 94, 2518–2528. doi: 10.1890/13-0197.1
Dietzel, R., Jarchow, M. E., and Liebman, M. (2015). Above- and belowground growth, biomass, and nitrogen use in maize and reconstructed prairie cropping systems. Crop Sci. 55, 910–923. doi: 10.2135/cropsci2014.08.0572
Fierer, N., Ladau, J., Clemente, J. C., Leff, J. W., Owens, S. M., Pollard, K. S., et al. (2013). Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 342, 621–624. doi: 10.1126/science.1243768
Frossard, A., Gerull, L., Mutz, M., and Gessner, M. O. (2012). Disconnect of microbial structure and function: enzyme activities and bacterial communities in nascent stream corridors. ISME J. 6, 680–691. doi: 10.1038/ismej.2011.134
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C., and Martiny, J. B. H. (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Publ. Group 10, 497–506. doi: 10.1038/nrmicro2795
Howe, A., Yang, F., Williams, R. J., Meyer, F., and Hofmockel, K. S. (2016). Identification of the core set of carbon-associated genes in a bioenergy grassland soil. PLoS ONE 11:e0166578. doi: 10.1371/journal.pone.0166578
Hunting, E. R., Barmentlo, S. H., Schrama, M., van Bodegom, P. M., Zhai, Y., and Vijver, M. G. (2017). Agricultural constraints on microbial resource use and niche breadth in drainage ditches. PeerJ. 5:e4175. doi: 10.7717/peerj.4175
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucl. Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kim, H., Nunan, N., Dechesne, A., Juarez, S., and Grundmann, G. (2015). The spatial distribution of exoenzyme activities across the soil micro-landscape, as measured in micro- and macro-aggregates, and ecosystem processes. Soil Biol. Biochem. 91, 258–267. doi: 10.1016/j.soilbio.2015.08.042
Kulmatiski, A., and Beard, K. H. (2011). Long-term plant growth legacies overwhelm short-term plant growth effects on soil microbial community structure. Soil Biol. Biochem. 43, 823–830. doi: 10.1016/j.soilbio.2010.12.018
Langenheder, S., and Prosser, J. I. (2008). Resource availability influences the diversity of a functional group of heterotrophic soil bacteria. Environ. Microbiol. 10, 2245–2256. doi: 10.1111/j.1462-2920.2008.01647.x
Leibold, M. A. (1995). The niche concept revisited: mechanistic models and community context. Ecology 76, 1371–1382. doi: 10.2307/1938141
Lin, L., Kan, X., Yan, H., and Wang, D. (2012). Characterization of extracellular cellulose-degrading enzymes from Bacillus thuringiensis strains. Electron. J. Biotechnol. 15. doi: 10.2225/vol15-issue3-fulltext-1
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lynd, L. R., Weimer, P. J., van Zyl, W. H., and Pretorius, I. S. (2002). Microbial cellulose utilization: fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 66, 506–577. doi: 10.1128/MMBR.66.3.506-577.2002
Marx, M. C., Kandeler, E., Wood, M., Wermbter, N., and Jarvis, S. C. (2005). Exploring the enzymatic landscape: distribution and kinetics of hydrolytic enzymes in soil particle-size fractions. Soil Biol. Biochem. 37, 35–48. doi: 10.1016/j.soilbio.2004.05.024
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
Montella, S., Ventorino, V., Lombard, V., Henrissat, B., Pepe, O., and Faraco, V. (2017). Discovery of genes coding for carbohydrate-active enzyme by metagenomic analysis of lignocellulosic biomasses. Sci. Rep. 7:42623. doi: 10.1038/srep42623
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2017). Vegan: Community Ecology Package. R package.
Paul, E. A. (2014). Soil Microbiology, Ecology and Biochemistry. London, UK; San Diego, CA; Waltham, MA; Oxford, UK: Academic Press.
Rivas, F. A., Tabatabai, M. A., Olk, D. C., and Thompson, M. L. (2014). Kinetics of short-term carbon mineralization in roots of biofuel crops in soils. Biol. Fertil. Soils 50, 527–535. doi: 10.1007/s00374-013-0870-y
Salles, J. F., Poly, F., Schmid, B., and Le Roux, X. (2009). Community niche predicts the functioning of denitrifying bacterial assemblages. Ecology 90, 3324–3332. doi: 10.1890/09-0188.1
Schimz, K. L., Broll, B., and John, B. (1983). Cellobiose phosphorylase (EC 2.4.1.20) of cellulomonas: occurrence, induction, and its role in cellobiose metabolism. Arch. Microbiol. 135, 241–249. doi: 10.1007/BF00413475
Singh, A., and Hayashi, K. (1995). Microbial cellulases: protein architecture, molecular properties, and biosynthesis. Adv. Appl. Microbiol. 40, 1–44. doi: 10.1016/S0065-2164(08)70362-9
Sinsabaugh, R. L., Lauber, C. L., Weintraub, M. N., Ahmed, B., Allison, S. D., Crenshaw, C., et al. (2008). Stoichiometry of soil enzyme activity at global scale. Ecol. Lett. 11, 1252–1264. doi: 10.1111/j.1461-0248.2008.01245.x
Six, J., Conant, R. T., Paul, E. A., and Paustian, K. (2002). Stabilization mechanisms of soil organic matter: implications for C-saturation of soils. Plant Soil 241, 155–176. doi: 10.1023/A:1016125726789
Six, J., Elliott, E. T., and Paustian, K. (2000). Soil macroaggregate turnover and microaggregate formation: a mechanism for C sequestration under no-tillage agriculture. Soil Biol. Biochem. 32, 2099–2103. doi: 10.1016/S0038-0717(00)00179-6
Six, J., Guggenberger, G., Paustian, K., Haumaier, L., Elliott, E. T., and Zech, W. (2001). Sources and composition of soil organic matter fractions between and within soil aggregates. Eur. J. Soil Sci. 52, 607–618. doi: 10.1046/j.1365-2389.2001.00406.x
Talbot, J. M., Bruns, T. D., Taylor, J. W., Smith, D. P., Branco, S., Glassman, S. I., et al. (2014). Endemism and functional convergence across the North American soil mycobiome. Proc. Natl. Acad. Sci. U.S.A. 111, 6341–6346. doi: 10.1073/pnas.1402584111
Tisdall, J. M., and Oades, J. M. (1982). Organic matter and water-stable aggregates in soils. Eur. J. Soil Sci. 33, 141–163. doi: 10.1111/j.1365-2389.1982.tb01755.x
Trivedi, P., Delgado-Baquerizo, M., Anderson, I. C., and Singh, B. K. (2016). Response of soil properties and microbial communities to agriculture: implications for primary productivity and soil health indicators. Front. Plant Sci. 7:990. doi: 10.3389/fpls.2016.00990
Williams, R. J., Howe, A., and Hofmockel, K. S. (2014). Demonstrating microbial co-occurrence pattern analyses within and between ecosystems. Front. Microbiol. 5:358. doi: 10.3389/fmicb.2014.00358
Wilson, D. B. (2011). Microbial diversity of cellulose hydrolysis. Curr. Opin. Microbiol. 14, 259–263. doi: 10.1016/j.mib.2011.04.004
Keywords: microbiome, carbon cycling, metagenomes, aggregates, prairie, bioenergy
Citation: Choi J, Bach E, Lee J, Flater J, Dooley S, Howe A and Hofmockel KS (2018) Spatial Structuring of Cellulase Gene Abundance and Activity in Soil. Front. Environ. Sci. 6:107. doi: 10.3389/fenvs.2018.00107
Received: 02 March 2018; Accepted: 06 September 2018;
Published: 02 October 2018.
Edited by:
Wilfred Otten, Cranfield University, United KingdomReviewed by:
Ellard Roy Hunting, Woods Hole Oceanographic Institution, United StatesHannes Schmidt, Universität Wien, Austria
Copyright © 2018 Choi, Bach, Lee, Flater, Dooley, Howe and Hofmockel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kirsten S. Hofmockel, a2lyc3Rlbi5ob2Ztb2NrZWxAcG5ubC5nb3Y=
Adina Howe, YWRpbmFAaWFzdGF0ZS5lZHU=
†These authors have contributed equally to this work