 Kwang Ho Kim
Kwang Ho Kim Chang Soo Kim
Chang Soo Kim- Clean Energy Research Center, Korea Institute of Science and Technology, Seoul, South Korea
Lignin is a major component of lignocellulosic biomass along with cellulose and hemicellulose, currently underutilized with the bulk of technical lignin being combusted for heat generation in plant. From the technoeconomic perspective, the success of a future biorefinery is highly dependent on lignin valorization. However, structural complexity, heterogeneity of lignin, and several undesirable reactions associated with the nature of lignin and process conditions obstruct the effective utilization of lignin. Lignin condensation has been a long-lasting problem from conventional to recent biorefineries, which makes the recovered lignin more difficult to decompose. Also, the undesirable condensation and repolymerization during the depolymerization of lignin is more problematic as this significantly limits its ability to be depolymerized to low molecular weight products at high yields. To solve this problem, which ultimately enables maximizing lignin utilization, there have been many efforts in various aspects. In this review, we focus on the undesirable reactions occurring during the fractionation and depolymerization process of lignin and introduce recent efforts to suppress or minimize those unwanted reactions. The aim of all these efforts is to maximize lignin conversion to low molecular weight products.
Introduction
Renewable biofuels and chemicals produced from lignocellulosic biomass have great potential to reduce our overdependence on fossil fuels and eventually mitigate climate change. However, there are still many technical barriers in developing economically feasible processes for converting biomass to fuels and chemicals (Himmel, 2009). Inherent recalcitrance of lignocellulosic biomass mainly associated with the presence of lignin is a major obstacle in developing a sustainable biorefinery. Lignin is the second most abundant natural polymer next to cellulose, accounting for 15–30 wt% of lignocellulosic biomass (Zakzeski et al., 2010). In addition to the existing pulp and paper industries, a significant amount of residual lignin is expected to be produced from the emerging lignocellulosic biorefineries. The availability of such biorefinery lignins further encourages the development of lignin valorization technologies, particularly for the production of high value-added chemicals (Zakzeski et al., 2012). Despite high-value opportunities, the technical lignin is simply burned to produce process heat in plants or sold as a natural ingredient of animal feed due to its natural heterogeneity and recalcitrance. From the industrial perspective, lignin valorization is essential for developing sustainable and advanced biorefineries in the future. In this respect, there have been substantial endeavors in developing an efficient and effective conversion process to produce value-added products from lignin because the development of such a conversion process is an important starting point for lignin valorization strategies (Ragauskas et al., 2014; Rahimi et al., 2014). However, due to the condensed and degraded nature of conventional technical lignins, it is challenging to develop effective depolymerization processes. Considering the fact that the yield of lignin-derived monomer is proportional to the frequency of inter-unit ether bonds (α-O-4 or β-O-4), preserving these cleavable linkages in lignin during the fractionation of biomass components is very important.
In addition to the effort to separate lignin as much as possible, researchers have put an enormous effort to develop an effective downstream depolymerization process for lignin valorization. Most conversion technologies developed recently use heat and metal catalysts to effectively break down lignin into low molecular weight aromatics. There are two important considerations for developing any processes, which are (1) maximizing the activity and stability of the catalyst under the reaction conditions and (2) controlling the condensation and repolymerization reactions of lignin that can form complex recondensed products (Xu et al., 2014). Taking the structural recalcitrance of lignin into account, an in-depth understanding of reaction mechanisms occurring during lignin depolymerization is important in maximizing value-added products. It is important to point out that the yields of target lignin products, liquid oil and low molecular weight phenolic products, are limited by the self-condensation and oligomerization reactions of the primary products (Roberts et al., 2011). Therefore, suppressing or preventing undesired reactions has been an important goal in lignin valorization.
Figure 1 shows the scheme of lignin depolymerization and repolymerization. There are competing reactions in the lignin depolymerization process. These reactions include recondensation to large molecular weight products or char between the primary products and stabilization to low molecular weight phenolic monomers. Although the competition is highly dependent on the reaction environment, the catalysts, and the quality of lignin, the undesirable reactions are very difficult to avoid. Thus, researchers have been exerting more efforts to minimize these unwanted secondary reactions in many aspects to maximize the value of lignin. This mini-review introduces the secondary reactions occurring during lignin extraction and the depolymerization process. Recent efforts to reduce or prevent undesirable reactions are also covered, providing insights toward further improvement of lignin valorization.
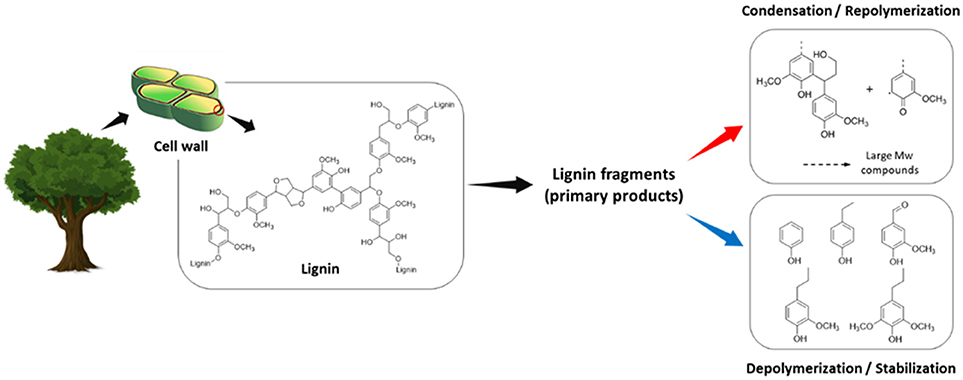
Figure 1. Lignin depolymerization and repolymerization scheme.
Lignin Condensation in the Pretreatment Process and New Fractionation Approach
The lignin condensation reaction has been a key problem at all stages of the process from the fractionation of biomass components to the depolymerization. The conventional fractionation process, namely biomass pretreatment, focuses on its effectiveness to remove lignin from biomass structure, generally employing acid or base catalysts. The resulting residual solid, mainly lignin, significantly undergoes irreversible repolymerization depending on the pretreatment conditions, which limits its ability to be depolymerized to low molecular weight products at high yields. Under acidic and neutral conditions, new intermolecular bonds (i.e., C–C bond) between lignin fragments form by the carbocation (Pielhop et al., 2015). Also, aryl-ether cleavage in Cα position in the presence of free Ar-OH group can lead to the formation of quinone methides, which further participate in cross-linking reactions (Grabber, 2005). In alkaline media, the reactive intermediates are mainly quinone methides. An addition reaction between phenolate ions and quinone methides results in the formation of C–C bonds, forming a lignin molecule with a larger size (Chakar and Ragauskas, 2004). In spite of their abundancy, the condensed lignin is very challenging to work with, even under harsh conditions with an effective catalyst and solvent system.
With the development of a recent biorefinery that integrates the conversion of carbohydrates and lignin, finding a suitable isolation process of lignin is becoming a primary research topic. As the proportion of β-O-4 linkages in lignin structure is the central factor for the yield and the nature of the monomeric products (Bouxin et al., 2015), a new lignin extraction method should be able to preserve the most frequent but readily cleavable aryl-ether linkages. As part of this effort, several fractionation processes have been developed and introduced, which utilize new types of solvents and catalysts. This includes ionic liquids (Shi et al., 2014, 2016; Sun et al., 2016; Xu et al., 2016; Dutta et al., 2017), deep eutectic solvents (Kim K. H. et al., 2018; Xu et al., 2018), γ-valerolactone (GVL; Luterbacher et al., 2014, 2015; Shuai et al., 2016b) and 2,5-furandicarboxylic acid (Weidener et al., 2018). Basically, the new solvent system demonstrated its applicability under mild processing conditions without strong acid or base catalysts, which minimizes carbocation- and quinone methides-induced condensation and repolymerization reactions. This could preserve the original structure of lignin with high aryl-ether linkages.
Undesirable Reactions During Lignin Depolymerization Process
In the liquefaction process of lignin, a substantial amount of solid residue is produced after reactions, which restricts the liquid oil yield. It is believed that formation of undesired solid products from intermediate products occurs significantly during the lignin depolymerization reactions (Bai et al., 2014). There have been many proposed reaction networks to explain these secondary condensation and repolymerization reactions. At high temperatures, the cleavage of abundant ether linkages (e.g., β-O-4 and α-O-4) in lignin can form highly reactive and unstable free radicals, with half-lives of < 10−3 s (Fossey et al., 1995). These radical species, in the early stage of the conversion process, can further react through rearrangement, abstraction and irreversible radical-radical coupling reactions to form large molecular weight products (Kim et al., 2017b).
As primary products, allyl- and vinyl-substituted intermediates (e.g., 4-vinylphenol, 2-methoxy-4-vinylphenol, isoeugenol, etc.) are produced from thermal lignin decomposition (Patwardhan et al., 2011; Ye et al., 2012). Because of their highly reactive nature toward polymerization and condensation reactions, rapid stabilization of reactive intermediates is required to prevent undesirable reactions, otherwise they readily repolymerize to form oligomers or even larger undesirable products (Hosoya et al., 2008; Bai et al., 2014; Kotake et al., 2014). Additionally, phenolic products with unsaturated carbonyl groups frequently found in lignin derivatives are prone to repolymerization (Shu et al., 2016).
Formaldehyde directly obtained from the cleavage of Cγ position at lignin side chains is also found to participate in repolymerization reactions between lignin decomposition products and char formation (Saisu et al., 2003; Huang et al., 2015). Considering the high reactivity of formaldehyde toward polymerization (e.g., phenol-formaldehyde resin), a cross-linking reaction between formaldehyde and phenolic fragments derived from lignin can readily occur resulting in the formation of large molecular weight products or char.
In addition to the lignin derived products or lignin fragments discussed above, some major functional groups in lignin are also key players in secondary reactions occurring in lignin decomposition. From the structural point of view, the Ar-OH group (free phenolic hydroxyl group) in lignin has gained much attention for its contribution to the secondary reactions due to its strong reactivity. The Ar-OH group is believed to be highly reactive toward electrophilic substitution reactions, and it can form reactive quinone methide intermediates depending on the reaction conditions (Kim et al., 2017a). All these species are responsible for secondary repolymerization. Furthermore, methoxyl groups have also been demonstrated to influence on char formation during lignin pyrolysis (Hosoya et al., 2009).
There are multiple condensation and repolymerization pathways that form larger molecules or char, which include (1) radical coupling, (2) vinyl condensation, (3) reactive fragments (e.g., formaldehyde) involving polymerization, and (4) reactive functional group (e.g., Ar-OH) induced repolymerization reactions. Apparently, the solid products or char produced in lignin depolymerization results from the combination of above reaction pathways (Nakamura et al., 2007).
Structural Modification of Lignin
Although the exact structure of lignin still remains unclear, improvements in modern analytical techniques have elucidated some important structural features of lignin. These features include functional groups and inter-unit linkages from their formation and reaction networks involved in lignin depolymerization. Abundant functional groups typically found in the lignocellulosic biomass include methoxyl (–OCH3), phenolic hydroxyl (Ar-OH), aliphatic hydroxyl, and carbonyl groups, depending on biomass species and extraction method (Chakar and Ragauskas, 2004). Among the different functional groups, the Ar-OH group has been of great interest to researchers since it is one of the most reactive functional groups in the lignin macromolecule (Kim et al., 2017a). Under both acidic and basic conditions, the primary products derived from lignin readily undergo facile addition and condensation reactions, forming high molecular weight products (Roberts et al., 2011). A recent study revealed that chemoselective masking of an Ar-OH group with a methyl group (Ar-OCH3) can dramatically reduce quinone methide-induced secondary condensation reactions up to 50% (Kim et al., 2017a). In-situ detection of free radical species, likely quinone methide radicals, using an electron paramagnetic resonance spectroscopy, elucidated that radical-induced reactions are involved in lignin depolymerization. If the Ar-OH is blocked by other groups, lignin cannot form quinone methide intermediates, which reduces repolymerization reactions. Even under pyrolysis conditions, selective methylation of Ar-OH enhances the production of aromatic hydrocarbon and inhibits secondary undesirable reactions (Kim J. Y et al., 2018). Ethanol is also found to be an effective reagent in suppressing char formation. In the lignin depolymerization with supercritical ethanol, ethanol not only acts as a hydrogen-donor solvent to stabilize the primary products, but it also forms O-alkylation of Ar-OH groups suppressing repolymerization reactions (Huang et al., 2014).
Another approach to enhance the yield of low molecular weight aromatic compounds is to oxidize lignin prior to the depolymerization reaction. A previous computational study indicated that the bond dissociation energy of the Cβ-O bond in β-O-4 linkages in lignin structure is substantially decreased (~14 kcal/mol) upon the oxidation of the Cα and Cγ (Kim et al., 2011). Thus, it has been hypothesized that the selective oxidation of lignin side chains could increase the cleavage of the most frequent β-O-4 linkages. Rahimi and coworkers focused on selective oxidation of the secondary benzylic alcohol to ketone at the Cα position using 4-acetamido-TEMPO, and the subsequent mild depolymerization with aqueous formic acid resulted in more than 60 wt% yield of phenolic monomers (Rahimi et al., 2013, 2014). Selective lignin oxidation enables the benzylic carbonyl group to polarize the C–H bond and lower the barrier for the rate-limiting elimination reaction during lignin depolymerization, producing more monoaromatic products. Another tests with model compounds demonstrated lignin degradation strategy by combining selective benzylic oxidation followed by catalytic C–O bond cleavage using zinc (Lancefield et al., 2015), NiMo sulfide (Zhang et al., 2017) or photocatalyst [Ir(ppy)2(dtbbpy)]PF6 (Nguyen et al., 2014).
More recently, a novel lignin extraction method that can prevent interunit C–C coupling while preserving aryl-ether (C–O) has been developed using formaldehyde (Shuai et al., 2016a). This effort aimed to strategically block the reactive benzylic positions with a protecting agent during lignin extraction. The addition of formaldehyde prevents lignin condensation reactions by forming 1,3-dioxane structures with –OH groups at the Cα and Cγ positions. The subsequent hydrogenolysis of structurally modified lignin results in near theoretical yields of monomers. Besides formaldehyde as a protecting agent, acetaldehyde and propionaldehyde were found to stabilize primary and secondary –OH groups at the lignin side chain (Lan et al., 2018). Hydrogenolysis of protected lignin significantly increased selectivity of final products (80% of 4-n-propanolsyringol).
It is also believed that the source of lignin (biomass origin) can affect the repolymerization reaction. For example, guaiacyl lignin (G unit) typically found in softwood lignin would be more susceptible to repolymerization and condensation reaction because it contains a greater proportion of unsubstituted C5 aromatic carbons. On the other hand, syringyl lignin (S unit), a main lignin unit in hardwood and herbaceous biomass contains relatively few condensed substructure, it would not undergo unwanted reactions significantly compared with G unit-rich lignins (Wariishi et al., 1991). New strategies including a selective substitution of open C5 aromatic carbons during the isolation of lignin can be considered before lignin depolymerization reactions.
The recent findings discussed above suggest that a slight structural modification of lignin can dramatically increase the valued products known as building block chemicals from lignin (Figure 2). Also this approach can be directed toward altering the biosynthesis process of lignin in the biomass cell wall to produce substrates that are more amenable to chemical depolymerization (Kim et al., 2017a).
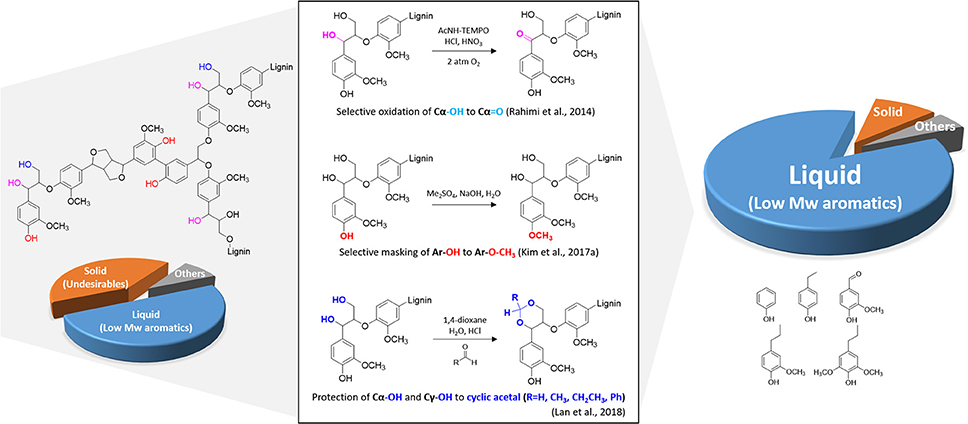
Figure 2. Chemoselective structural modification of lignin for maximizing the value.
Introducing Capping Agents
Researchers strategically introduced capping agents to stabilize the intermediates or reactive sites of lignin fragments. Boric acid has been used to protect the Ar-OH groups in the base-catalyzed lignin depolymerization (Roberts et al., 2011; Toledano et al., 2014a). Boric acid blocks addition and condensation reactions between the primary products by capping the Ar-OH groups with formation of boric esters (Roberts et al., 2011). The combination of base-catalyzed lignin breakdown and the boric acid as a protecting agent increases product yields (oil) to over 85 wt%. Based on the high reactivity of boric acid toward nucleophiles (Rietjens and Steenbergen, 2005), the addition of boric acid to lignin, a polymeric phenol, results in a strongly coordinated boric acid-lignin complex. This strong coordination can suppress the undesired reactions initiated from the reactive Ar-OH groups, although in-depth study is necessary to elucidate the detailed mechanisms.
Phenol has also been widely tested as a capping agent to stabilize phenolic moiety and formaldehyde. (Okuda et al., 2004; Yuan et al., 2010; Mahmood et al., 2013; Toledano et al., 2014a). Under the base-catalyzed lignin depolymerization reaction, the addition of phenol was able to reduce char and residual lignin, forming the production of a wide variety of phenolic compounds (Toledano et al., 2014a). Hydrolytic degradation of lignin under water-ethanol mixture in the presence of phenol as a capping agent reduced the char yield up to 70% (Yuan et al., 2010). However, it seems that the reaction conditions (i.e., reaction temperature, time and phenol to lignin ratio) need to be carefully controlled to maximize the conversion of lignin to low molecular weight products. Due to the highly reactive nature of phenol, it could lead to repolymerization reactions between the reactive sites of the combined phenol and the side chains of the lignin fragments, typically at higher temperature and longer reaction time.
A rapid quenching reactive radical species and unstable intermediates formed during depolymerization is another strategy to minimize the side reactions and produce low molecular weight phenols (Toledano et al., 2014b; Brittain et al., 2018). A hydrogenolytic approach of lignin with the assistance of an external hydrogen source and reducing catalysts is widely examined with an effort to quench the reactive intermediates and suppress the undesirable reactions. In principle, this approach employs hydrogens from molecular hydrogen or hydrogen donor solvents (e.g., ethanol, isopropyl alcohol, tetralin etc.) in the presence of metal catalysts (Ru, Pt, Pd, Ni, etc.). This strategy has been studied extensively and thoroughly reviewed elsewhere (Deuss and Barta, 2016; Sun et al., 2018).
Summary and Perspectives
Although there have been substantial efforts to identify the technical barriers for better utilization of lignins, the development of economically attractive processes is still challenging due to the complex nature of lignin. From the conventional pulp and paper industries to recent biorefineries, most of these processes significantly modify or damage lignin structure, which have restricted the yield of liquid oil and low molecular weight phenolic compounds to high level. Also, several undesirable reactions between the primary products to form large molecular weight oligomers or char during the lignin conversion process seem to be unavoidable. As discussed above, there are many contributing factors associated with inherent characteristics of lignin in this unwanted reaction network.
The history of lignin isolation and utilization traces back to the early 1900s, but researchers are still eagerly searching for effective lignin valorization methods. This highlights how lignin is definitely one of the most challenging materials to work with. To better utilize lignin, so that it brings benefits for future biorefineries, lignin valorization research has to be multidisciplinary. This multidisciplinary research may include computational modeling to calculate bond dissociation energy of linkages, plant science to engineer cell wall structures that are more amenable to chemical depolymerization, and chemistry along with process engineering to develop feedstock-agnostic biomass fractionation and depolymerization. Remembering the complex nature of lignin, there are still many unknown factors that affect the reaction network in lignin preparation and depolymerization, leaving opportunities for the scientific community to valorize lignin.
Author Contributions
KK organized and prepared all this manuscript; CK contributed for writing and reviewing the part of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by the Korea Institute of Science and Technology (KIST)—The University of British Columbia (UBC) Biorefinery on-site laboratory project.
References
Bai, X., Kim, K. H., Brown, R. C., Dalluge, E., Hutchinson, C., Lee, Y. J., et al. (2014). Formation of phenolic oligomers during fast pyrolysis of lignin. Fuel 128, 170–179. doi: 10.1016/j.fuel.2014.03.013
Bouxin, F. P., McVeigh, A., Tran, F., Westwood, N. J., Jarvis, M. C., and Jackson, S. D. (2015). Catalytic depolymerisation of isolated lignins to fine chemicals using a Pt/alumina catalyst: part 1-impact of the lignin structure. Green Chem. 17, 1235–1242. doi: 10.1039/C4GC01678E
Brittain, A. D., Chrisandina, N. J., Cooper, R. E., Buchanan, M., Cort, J. R., Olarte, M. V., et al. (2018). Quenching of reactive intermediates during mechanochemical depolymerization of lignin. Catal. Today 302, 180–189. doi: 10.1016/j.cattod.2017.04.066
Chakar, F. S., and Ragauskas, A. J. (2004). Review of current and future softwood kraft lignin process chemistry. Ind. Crops Prod. 20, 131–141. doi: 10.1016/j.indcrop.2004.04.016
Deuss, P. J., and Barta, K. (2016). From models to lignin: transition metal catalysis for selective bond cleavage reactions. Coord. Chem. Rev. 306, 510–532. doi: 10.1016/j.ccr.2015.02.004
Dutta, T., Isern, N. G., Sun, J., Wang, E., Hull, S., Cort, J. R., et al. (2017). Survey of lignin-structure changes and depolymerization during ionic liquid pretreatment. ACS Sust. Chem. Eng. 5, 10116–10127. doi: 10.1021/acssuschemeng.7b02123
Grabber, J. H. (2005). How do lignin composition, structure, and cross-linking affect degradability? A review of cell wall model studies. Crop Sci. 45, 820–831. doi: 10.2135/cropsci2004.0191
Himmel, M. E. (2009). Biomass Recalcitrance: Deconstructing the Plant Cell Wall for Bioenergy. Oxford, UK: Wiley-Blackwell.
Hosoya, T., Kawamoto, H., and Saka, S. (2008). Secondary reactions of lignin-derived primary tar components. J. Anal. Appl. Pyrolysis 83, 78–87. doi: 10.1016/j.jaap.2008.06.003
Hosoya, T., Kawamoto, H., and Saka, S. (2009). Role of methoxyl group in char formation from lignin-related compounds. J. Anal. Appl. Pyrolysis 84, 79–83. doi: 10.1016/j.jaap.2008.10.024
Huang, X., Korányi, T. I., Boot, M. D., and Hensen, E. J. (2014). Catalytic depolymerization of lignin in supercritical ethanol. ChemSusChem 7, 2276–2288. doi: 10.1002/cssc.201402094
Huang, X., Korányi, T. I., Boot, M. D., and Hensen, E. J. (2015). Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics. Green Chem. 17, 4941–4950. doi: 10.1039/C5GC01120E
Kim, J. Y., Heo, S., and Choi, J. W. (2018). Effects of phenolic hydroxyl functionality on lignin pyrolysis over zeolite catalyst. Fuel 232, 81–89. doi: 10.1016/j.fuel.2018.05.133
Kim, K. H., Dutta, T., Sun, J., Simmons, B., and Singh, S. (2018). Biomass pretreatment using deep eutectic solvents from lignin derived phenols. Green Chem. 20, 809–815. doi: 10.1039/C7GC03029K
Kim, K. H., Dutta, T., Walter, E. D., Isern, N. G., Cort, J. R., Simmons, B. A., et al. (2017a). Chemoselective methylation of phenolic hydroxyl group prevents quinone methide formation and repolymerization during lignin depolymerization. Acs Sust. Chem. Eng. 5, 3913–3919. doi: 10.1021/acssuschemeng.6b03102
Kim, K. H., Singh, S., Custodis, V., and van Bokhoven, J. (2017b). “Role of free radicals in fast pyrolysis,” in Fast Pyrolysis of Biomass (London, UK), 117–137.
Kim, S., Chmely, S. C., Nimos, M. R., Bomble, Y. J., Foust, T. D., Paton, R. S., et al. (2011). Computational study of bond dissociation enthalpies for a large range of native and modified lignins. J. Phys. Chem. Lett. 2, 2846–2852. doi: 10.1021/jz201182w
Kotake, T., Kawamoto, H., and Saka, S. (2014). Mechanisms for the formation of monomers and oligomers during the pyrolysis of a softwood lignin. J. Anal. Appl. Pyrolysis 105, 309–316. doi: 10.1016/j.jaap.2013.11.018
Lan, W., Amiri, M. T., Hunston, C. M., and Luterbacher, J. S. (2018). Protection group effects during alpha, gamma-diol lignin stabilization promote high-selectivity monomer production. Angew. Chem. Int. Edn. 57, 1356–1360. doi: 10.1002/anie.201710838
Lancefield, C. S., Ojo, O. S., Tran, F., and Westwood, N. J. (2015). Isolation of functionalized phenolic monomers through selective oxidation and C-O bond cleavage of the beta-O-4 linkages in lignin. Angew. Chem. Int. Edn. 54, 258–262. doi: 10.1002/anie.201409408
Luterbacher, J. S., Azarpira, A., Motagamwala, A. H., Lu, F., Ralph, J., and Dumesic, J. A. (2015). Lignin monomer production integrated into the γ-valerolactone sugar platform. Energy Env. Sci. 8, 2657–2663. doi: 10.1039/C5EE01322D
Luterbacher, J. S., Rand, J. M., Alonso, D. M., Han, J., Youngquist, J. T., Maravelias, C. T., et al. (2014). Nonenzymatic sugar production from biomass using biomass-derived γ-valerolactone. Science 343, 277–280. doi: 10.1126/science.1246748
Mahmood, N., Yuan, Z. S., Schmidt, J., and Xu, C. B. (2013). Production of polyols via direct hydrolysis of kraft lignin: effect of process parameters. Bioresour. Technol. 139, 13–20. doi: 10.1016/j.biortech.2013.03.199
Nakamura, T., Kawamoto, H., and Saka, S. (2007). Condensation reactions of some lignin related compounds at relatively low pyrolysis temperature. J. Wood Chem. Technol. 27, 121–133. doi: 10.1080/02773810701515143
Nguyen, J. D., Matsuura, B. S., and Stephenson, C. R. J. (2014). A Photochemical strategy for lignin degradation at room temperature. J. Am. Chem. Soc. 136, 1218–1221. doi: 10.1021/ja4113462
Okuda, K., Umetsu, M., Takami, S., and Adschiri, T. (2004). Disassembly of lignin and chemical recovery–rapid depolymerization of lignin without char formation in water-phenol mixtures. Fuel Process. Technol. 85, 803–813. doi: 10.1016/j.fuproc.2003.11.027
Patwardhan, P. R., Brown, R. C., and Shanks, B. H. (2011). Understanding the fast pyrolysis of lignin. ChemSusChem 4, 1629–1636. doi: 10.1002/cssc.201100133
Pielhop, T., Larrazabal, G. O., Studer, M. H., Brethauer, S., Seidel, C. M., and von Rohr, P. R. (2015). Lignin repolymerisation in spruce autohydrolysis pretreatment increases cellulase deactivation. Green Chem. 17, 3521–3532. doi: 10.1039/C4GC02381A
Ragauskas, A. J., Beckham, G. T., Biddy, M. J., Chandra, R., Chen, F., Davis, M. F., et al. (2014). Lignin valorization: improving lignin processing in the biorefinery. Science 344:1246843. doi: 10.1126/science.1246843
Rahimi, A., Azarpira, A., Kim, H., Ralph, J., and Stahl, S. S. (2013). Chemoselective metal-free aerobic alcohol oxidation in lignin. J. Am. Chem. Soc. 135, 6415–6418. doi: 10.1021/ja401793n
Rahimi, A., Ulbrich, A., Coon, J. J., and Stahl, S. S. (2014). Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 515, 249. doi: 10.1038/nature13867
Rietjens, M., and Steenbergen, P. A. (2005). Crosslinking mechanism of boric acid with diols revisited. Eur. J. Inorg. Chem. 2005, 1162–1174. doi: 10.1002/ejic.200400674
Roberts, V. M., Stein, V., Reiner, T., Lemonidou, A., Li, X. B., and Lercher, J. A. (2011). Towards quantitative catalytic lignin depolymerization. Chem. Eur. J. 17, 5939–5948. doi: 10.1002/chem.201002438
Saisu, M., Sato, T., Watanabe, M., Adschiri, T., and Arai, K. (2003). Conversion of lignin with supercritical water–phenol mixtures. Energy Fuels 17, 922–928. doi: 10.1021/ef0202844
Shi, J., Balamurugan, K., Parthasarathi, R., Sathitsuksanoh, N., Zhang, S., Stavila, V., et al. (2014). Understanding the role of water during ionic liquid pretreatment of lignocellulose: co-solvent or anti-solvent? Green Chem. 16, 3830–3840. doi: 10.1039/C4GC00373J
Shi, J., Pattathil, S., Parthasarathi, R., Anderson, N. A., Im Kim, J., Venketachalam, S., et al. (2016). Impact of engineered lignin composition on biomass recalcitrance and ionic liquid pretreatment efficiency. Green Chem. 18, 4884–4895. doi: 10.1039/C6GC01193D
Shu, R., Long, J., Xu, Y., Ma, L., Zhang, Q., Wang, T., et al. (2016). Investigation on the structural effect of lignin during the hydrogenolysis process. Bioresour. Technol. 200, 14–22. doi: 10.1016/j.biortech.2015.09.112
Shuai, L., Amiri, M. T., Questell-Santiago, Y. M., Heroguel, F., Li, Y. D., Kim, H., et al. (2016a). Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 354, 329–333. doi: 10.1126/science.aaf7810
Shuai, L., Questell-Santiago, Y. M., and Luterbacher, J. S. (2016b). A mild biomass pretreatment using γ-valerolactone for concentrated sugar production. Green Chem. 18, 937–943. doi: 10.1039/C5GC02489G
Sun, J., Konda, N. M., Shi, J., Parthasarathi, R., Dutta, T., Xu, F., et al. (2016). CO2 enabled process integration for the production of cellulosic ethanol using bionic liquids. Energy Environ. Sci. 9, 2822–2834. doi: 10.1039/C6EE00913A
Sun, Z. H., Fridrich, B., de Santi, A., Elangovan, S., and Barta, K. (2018). Bright side of lignin depolymerization: toward new platform chemicals. Chem. Rev. 118, 614–678. doi: 10.1021/acs.chemrev.7b00588
Toledano, A., Serrano, L., and Labidi, J. (2014a). Improving base catalyzed lignin depolymerization by avoiding lignin repolymerization. Fuel 116, 617–624. doi: 10.1016/j.fuel.2013.08.071
Toledano, A., Serrano, L., Pineda, A., Romero, A. A., Luque, R., and Labidi, J. (2014b). Microwave-assisted depolymerisation of organosolv lignin via mild hydrogen-free hydrogenolysis: catalyst screening. Appl. Catal. B Environ. 145, 43–55. doi: 10.1016/j.apcatb.2012.10.015
Wariishi, H., Valli, K., and Gold, M. H. (1991). In vitro depolymerization of lignin by manganese peroxidase of Phanerochaete chrysosporium. Biochem. Biophys. Res. Commun. 176, 269–275. doi: 10.1016/0006-291X(91)90919-X
Weidener, D., Klose, H., Leitner, W., Schurr, U., Usadel, B., Domínguez de María, P., et al. (2018). One-step lignocellulose fractionation using 2,5-furandicarboxylic acid as biogenic and recyclable catalyst. ChemSusChem. 11:2051–2056. doi: 10.1002/cssc.201800653
Xu, C. P., Arancon, R. A. D., Labidi, J., and Luque, R. (2014). Lignin depolymerisation strategies: towards valuable chemicals and fuels. Chem. Soc. Rev. 43, 7485–7500. doi: 10.1039/C4CS00235K
Xu, F., Sun, J., Konda, N. M., Shi, J., Dutta, T., Scown, C. D., et al. (2016). Transforming biomass conversion with ionic liquids: process intensification and the development of a high-gravity, one-pot process for the production of cellulosic ethanol. Energy Environ. Sci. 9, 1042–1049. doi: 10.1039/C5EE02940F
Xu, F., Sun, J., Wehrs, M., Kim, K. H., Rau, S. S., Chan, A. M., et al. (2018). Biocompatible choline-based deep eutectic solvents enable one-pot production of cellulosic ethanol. ACS Sust. Chem. Eng. 6, 8914–8919. doi: 10.1021/acssuschemeng.8b01271
Ye, Y., Fan, J., and Chang, J. (2012). Effect of reaction conditions on hydrothermal degradation of cornstalk lignin. J. Anal. Appl. Pyrolysis 94, 190–195. doi: 10.1016/j.jaap.2011.12.005
Yuan, Z. S., Cheng, S. N., Leitch, M., and Xu, C. B. (2010). Hydrolytic degradation of alkaline lignin in hot-compressed water and ethanol. Bioresour. Technol. 101, 9308–9313. doi: 10.1016/j.biortech.2010.06.140
Zakzeski, J., Bruijnincx, P. C., Jongerius, A. L., and Weckhuysen, B. M. (2010). The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 110, 3552–3599. doi: 10.1021/cr900354u
Zakzeski, J., Jongerius, A. L., Bruijnincx, P. C., and Weckhuysen, B. M. (2012). Catalytic lignin valorization process for the production of aromatic chemicals and hydrogen. ChemSusChem 5, 1602–1609. doi: 10.1002/cssc.201100699
Keywords: lignocellulosic biomass, fractionation, condensation, repolymerization, lignin modification, capping agent
Citation: Kim KH and Kim CS (2018) Recent Efforts to Prevent Undesirable Reactions From Fractionation to Depolymerization of Lignin: Toward Maximizing the Value From Lignin. Front. Energy Res. 6:92. doi: 10.3389/fenrg.2018.00092
Received: 11 July 2018; Accepted: 24 August 2018;
Published: 11 September 2018.
Edited by:
Arthur Jonas Ragauskas, University of Tennessee, Knoxville, United StatesReviewed by:
Somnath Shinde, Sandia National Laboratories (SNL), United StatesGopalakrishnan Kumar, University of Stavanger, Norway
Copyright © 2018 Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kwang Ho Kim, a3dhbmdob2tpbUBraXN0LnJlLmty