 Stine M. Præstholm
Stine M. Præstholm Catarina M. Correia
Catarina M. Correia Lars Grøntved
Lars Grøntved- Department of Biochemistry and Molecular Biology, University of Southern Denmark, Odense, Denmark
Glucocorticoids (GCs) and the glucocorticoid receptor (GR) are important regulators of development, inflammation, stress response and metabolism, demonstrated in various diseases including Addison's disease, Cushing's syndrome and by the many side effects of prolonged clinical administration of GCs. These conditions include severe metabolic challenges in key metabolic organs like the liver. In the liver, GR is known to regulate the transcription of key enzymes in glucose and lipid metabolism and contribute to the regulation of circadian-expressed genes. Insights to the modes of GR regulation and the underlying functional mechanisms are key for understanding diseases and for the development of improved clinical uses of GCs. The activity and function of GR is regulated at numerous levels including ligand availability, interaction with heat shock protein (HSP) complexes, expression of GR isoforms and posttranslational modifications. Moreover, recent genomics studies show functional interaction with multiple transcription factors (TF) and coregulators in complex transcriptional networks controlling cell type-specific gene expression by GCs. In this review we describe the different regulatory steps important for GR activity and discuss how different TF interaction partners of GR selectively control hepatic gene transcription and metabolism.
Introduction
Any living organism must adapt and respond to the surrounding environment to maintain its existence. For multicellular organisms such as mammals, this includes daily transitions between different physiological conditions including sleep/awake, fasted/fed, and physical inactivity/activity. Moreover, occasional response to environmental changes such as confinement, predator stress, extreme temperatures, inflammation and prolonged lack of food is critical for survival. Glucocorticoids (GCs) serve as important endocrine signaling molecules controlling many molecular signaling pathways that enable cells in the organism to respond to different extrinsic cues. This is particularly evident for cellular responses in the arousal state including the transitions mentioned above. Importantly, pathophysiological conditions leading to dysfunctional GC signaling have dramatic effects on many important biological functions including development, inflammatory response, reproduction, cognitive function, anxiety, circadian entrainment, cardiovascular regulation and cellular metabolism in a tissue-specific manner (1). For example, uncontrolled GC secretion observed in Cushing's syndrome leads to metabolic complications such as type 2 diabetes and osteoporosis, which are also observed in situations of prolonged treatment with GCs. In contrast, conditions of low GC production, seen in Addison's disease, are associated with muscle weakness, low blood pressure and weight loss (2).
Glucocorticoids exert their actions primarily by binding to the glucocorticoid receptor (GR or Nr3c1), which is expressed in most cells in mammals. Yet, GCs have highly tissue/cell-specific effects regulated by multiple mechanisms. As a DNA-binding transcription factor (TF), GR is primarily involved in the control of gene expression, with transcription of GR target genes in a given cell being controlled by three overall mechanisms (Figure 1). First, activity of GR is directly correlated with the amount of GC molecules available in the cell. This is controlled by adrenal GC synthesis and local availability of GCs in the cell. Second, expression of active GR in the nucleus determines the molecular response to GCs. This is regulated by GR turnover (synthesis and breakdown), expression of different GR isoforms, posttranslational modifications (PTMs) and nuclear translocation. Third, genomic action of GR is controlled by cell type-specific accessibility of GR response elements (GRE) in the genome in synergy with cell-specific TFs, coregulators and regulatory RNAs. In this review we will discuss all three regulatory aspects of GR signaling with a specific focus on GR interaction with the genome. We will primarily refer to studies from mouse liver tissue to discuss recent insights to hepatic gene regulatory networks and metabolism controlled by GCs. This will specifically be related to the hepatic transcriptional response to the circadian rhythm, feeding and fasting.
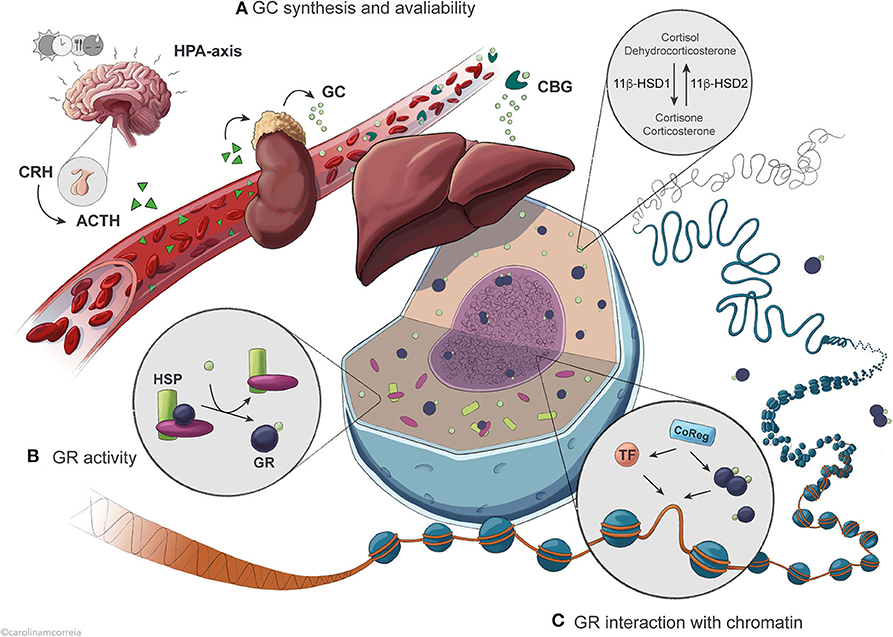
Figure 1. Overview of the regulatory levels affecting GR activity in the control of hepatic transcription. (A) Circadian and ultradian synthesis of GCs is controlled by the HPA axis in response to external stimuli including feeding, stress, light and circadian timekeepers. Availability of active GCs is further influenced by binding to the serum protein CBG and by intracellular conversion catalyzed by the enzyme 11β-HSD1/2. (B) Once in the cell, GCs are bound by the GR with an affinity that is conditioned by association with chaperone complexes containing HSPs, expression of specific GR isoforms and GR protein turnover. (C) GR exerts its action after translocation to the nucleus, where it binds GRE sequences in the DNA to regulate transcription of target genes as a result of dynamic interaction with different TFs and coregulators.
Regulation of Glucocorticoid Secretion and Availability in the Cell
Glucocorticoids (cortisol in humans; corticosterone in rodents) are steroid hormones secreted circadianly by the adrenal cortex. Their daily levels peak immediately before the active phase (early morning for humans; early evening for rodents) in anticipation of a waking state, but also in quick response to external stimuli such as stress, hypoglycemia and exercise (3, 4). The hypothalamic-pituitary-adrenal (HPA) axis controls and maintains GC secretion into the bloodstream (Figure 1A). The hypothalamus produces corticosteroid-releasing hormone (CRH), stimulating the pituitary gland to secrete adrenocorticotropic hormone (ACTH), which in turn promotes GC secretion by the adrenal gland (5). As many other hormones, including growth hormone and insulin (6, 7), GCs are secreted in an ultradian pattern with pulsatile secretion once every 60 to 90 min, as a result of feedback and feedforward mechanisms between ACTH, CRH and GC secretion keeping GC levels in a physiological range (4, 8, 9). The circadian secretion of GCs results partly from oscillations in ACTH secretion, but mostly from varying adrenal sensitivity to ACTH (3–5). In the blood, GCs circulate in association with corticosteroid-binding globulin (CBG) or, to a lower extent, albumin, and only a small fraction remains unbound in most vertebrates. As only free GCs diffuse into the target cells, CBG modulates GC bioavailability (10–12). Disruption of CBG expression in mice leads to reduced total serum GC (13) and as CBG and albumin are synthesized by the liver, it is possible that hepatic regulation of GC-binding proteins modulates the levels of available GC.
Additionally, non-adrenal production of cortisol has been described in visceral adipose tissue and liver via the conversion of inert cortisone catalyzed by the enzyme 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) in humans (dehydrocorticosterone to corticosterone in rodents), and reversely by 11β-HSD2 (14, 15) (Figure 1A). Liver activity of this enzyme is particularly relevant to the whole-body non-adrenal production of cortisol; however, HPA axis feedback mechanisms likely blunt any systemic effects (15). Therefore, activity of 11β-HSD1 mostly contributes to locally maintaining intracellular levels of active GCs in the liver and visceral adipose tissue, fine-tuning the highly variable GC levels. This enzyme thus regulates the availability of receptor-active GCs in the cell, modulating access to GR and amplifying GC effects (16–18). In mice, absence of 11β-HSD1 leads to an inability to produce active GCs from the inert form, resulting in compensatory activation of the HPA axis, increased basal corticosterone levels and failure to fully elicit a hepatic gluconeogenic response to fasting, similarly to absence or impairment of GR (19). Dysregulation of 11β-HSD1 expression and activity is associated with apparent hypercortisolemia, disrupted metabolism and HPA axis function, obesity, type 2 diabetes and metabolic syndrome; however, the specific contribution of the enzyme to these processes is still controversial (16, 18).
Circadian Control of Glucocorticoid Levels
The circadian synthesis and secretion of GCs by the adrenal glands is controlled by both the local molecular clock and the central clock in the suprachiasmatic nucleus (SCN) via a sympathetic neuronal pathway, and can be blunted by stress stimuli (3, 4). The SCN is important for GC rhythmicity, as it regulates the hypothalamic-hypophysial portions of the HPA axis affecting CRH secretion (20–22). During light-induced HPA axis-independent GC secretion, the SCN directly activates the adrenal glands via the adrenal sympathetic nerves, suggesting that GCs can act as SCN-gated mediators of the light stimuli to entrain metabolic-responsive peripheral clocks (5). The ubiquity of GR expression and the marked circadian secretion of GCs imply that these are efficient SCN-driven synchronizers of peripheral clocks and, specifically in the liver, are fundamental for the circadian expression of metabolic genes, even with contribution from other hormonal signals and entrainment factors (3, 4, 23). However, GCs do not affect the central clock, since GR is not expressed in the SCN (3, 23).
Unlike the SCN, the phase of peripheral clocks can be modulated by feeding, and even uncoupled from the SCN (3). As a metabolic organ, the liver is particularly responsive to feeding patterns, which can lead to desynchronization of its peripheral clock from the central clock (24, 25), an entrainment partly mediated by GCs (26–29). The interplay between eating behavior and GCs can be observed during day-restricted feeding of mice (opposite to their normal feeding pattern), leading to secretion of GCs with two distinct peaks instead of a single one, with one being feeding-responsive (before feeding time, in the early morning) and the other light-entrained (before the normal active period, in the early evening) (3, 4, 27, 30). Misalignment also occurs as a result of the disruption of normal activity patterns due to jet lag, shift work, sleep disorders or social jet lag, and associates with the development of metabolic disorders, such as diet-induced obesity and non-alcoholic fatty liver disease (31).
GR Structure, Splice Variants and PTMs in the Modulation of GR Activity
The effects of GCs are mediated by GR through its three functional domains: a hydrophobic C-terminal ligand-binding domain (LBD) containing a ligand-dependent trans-activation portion (τ2, or AF2), a zinc-finger DNA-binding domain (DBD) located adjacently, and an N-terminal trans-activation domain (τ1, or AF1) (32–34). There is extensive alternative splicing and translation of human GR, impacting cell-specific GC actions. Alternative splicing originates multiple isoforms varying primarily in the DBD and the C-terminal LBD/AF2, while multiple translational start sites give rise to GR proteins with different lengths of the AF1 domain. The expression of some GR isoforms is evolutionarily conserved, but while many have shown biological relevance in humans (35), isoforms in rodents are less characterized. In humans and rodents, GRα (referred to simply as GR henceforth) is considered the canonical GR isoform that mediates most actions of GCs and is the primary isoform expressed in most tissues. Alternative splicing of the GR primary transcript in humans and rodents can give rise to additional GR isoforms, including GRβ, which has a truncated C-terminus, resulting in an inactive AF2, with compromised ability to bind GCs. Thus, GRβ is considered dominant negative (36, 37). Although expressed to a lower level than GRα, GRβ is considered a functional TF in a number of tissues, including the liver (36, 38). Additional isoforms include the widely expressed GRγ, which exhibits similar affinities to both GCs and DNA as GRα, but has a compromised transactivation potential and is associated with GC resistance. Expression of GR is also affected by the activity of miRNA molecules that bind to the 3′ UTR of GR transcripts, affecting their stability and preventing their translation (37). Additionally, lncRNAs such as Gas5 repress ligand-activated GR activity by binding to its DBD as a decoy GRE in starvation conditions, leading to suppression of GC-stimulated mRNA expression of key gluconeogenic enzymes G6Pase and Pck1 during fasting (39).
In addition to the coregulatory function of specific GR isoforms, the activity of hormone-bound GR in different tissues can be modulated by specific sets of PTMs (40). For example, upon hormone binding, ligand-selective phosphorylation of the GR affects GR-mediated transcriptional activity and recruitment of coregulators, and is thus involved in directing and modulating GR action as a repressor or activator, namely via crosstalk from other signaling pathways such as in GSK3β-mediated phosphorylation (40–45). The relevance of PTMs on the GR protein and their effects on GR function are also illustrated by the protein-protein interactions between clock components and GR leading to suppression of GR activity via acetylation of a lysine residue by the CLOCK protein, potentiated by the presence of BMAL1 (46). Additionally, modifications such as GC-dependent phosphorylation reduce GR stability and half-life by tagging it for ubiquitination and subsequent degradation, and also influence its subcellular localization (37, 43, 44, 47–49). Other PTMs affecting GR function include SUMOylation, which reduces protein stability and regulates transcriptional activity, as well as nitrosylation and oxidation, both associated with reduction of GC-binding (37).
Regulation of GR Translocation to the Nucleus
Inactive GR is located in the cytoplasm, monomerically associated with a multimeric chaperone complex important for GR stability, folding and translocation (Figure 1B). The maturation of the complex involves a stepwise ATP-dependent assembly from the initial GR-HSP70-HSP40 complex, to the recruitment of HSP90 and Hop facilitating the assembly of a final high GC affinity complex consisting of GR, HSP90, p23, and FKBP51 (50). Circulating GCs enter the cells via diffusion across the cell membrane and interact with GR. Upon ligand-binding, a FKBP51-FKBP52 switch exposes the GR nuclear localization signals, which are recognized by importins and nucleoporins, facilitating the translocation of activated GR through a nuclear pore via microtubules (50, 51). Disruption of FKBP52 leads to reduced expression of GR target genes in the liver and augmented hepatic steatosis as a result of diet-induced obesity (52), also observed in liver-specific GR knock out (L-GRKO) mice (26), demonstrating a functional role of the multimeric chaperone complex for hepatic GR function. In general, the subcellular location of GR follows the diurnal GC concentration (53). However, both ligand-bound and unbound GR shuttle dynamically between the nucleus and the cytoplasm with a variable rate, consequently regulating GR activity. Aberrantly high cytosolic pH and chemical stress can lead to dissociation of HSP90 and increased nuclear import of GR. GR nuclear translocation can also be regulated by context-specific PTMs, e.g., phosphorylation of GR by kinases like MAPKs, CDK, and GSK3 (50). In the liver, factors including HDAC6 and REV-ERBα have been found to affect GR translocation, thus affecting GR activity (53, 54).
Genomic Actions of GR: General Concepts
Following nuclear translocation, GR accumulates at specific gene regulatory regions (e.g., enhancers) depending on the DNA sequence, occupancy of other TFs, organization of nucleosomes and higher order chromatin structures (Figure 1C). GR residence time at specific regions of chromatin lasts seconds, whereas freely diffusing unbound GR occupies chromatin in milliseconds (55) (Figure 2A). This enables GR to efficiently probe tens of thousands of putative enhancers within a short time frame and initiate transcription of hundreds of genes within minutes of activation by hormone (56). Also, the dynamic nature of chromatin interaction is shared by transcriptional coregulators known to interact with GR (57), both likely playing an important kinetic role in GC-regulated gene expression, including the duration and frequency of transcriptional bursting (58). As a result of the pulsatile secretion pattern, GC concentration in the serum is highly dynamic, allowing a rapid transcriptional response that can be translated into a fast biochemical response (59). For example, transcriptional bursting has been linked to a fast-acting metabolic switch in hepatic glucose metabolism, where expression of gluconeogenic genes such as G6pc and Pck1 is rapidly decreased in response to feeding (60), the latter being regulated by GCs (61, 62).
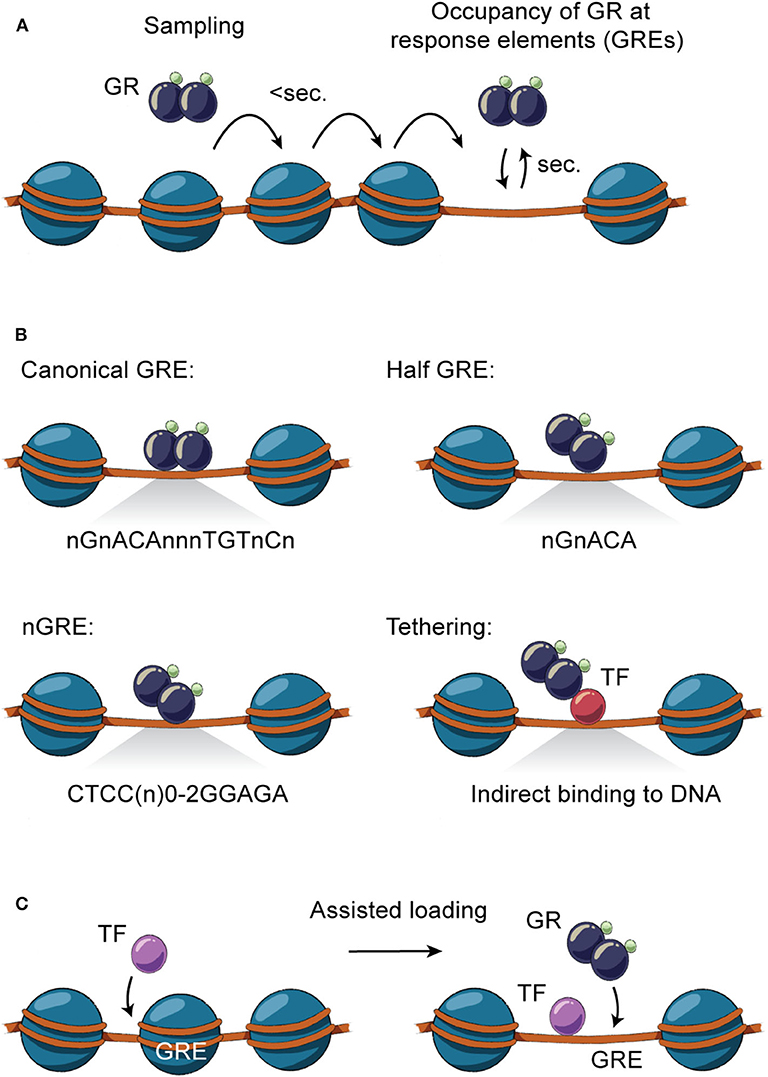
Figure 2. Direct and indirect GR-DNA interactions. (A) GR interacts dynamically with DNA. Freely diffusing GR occupies chromatin with residence time in milliseconds, whereas GR binding at specific regions of chromatin is measured in the order of seconds. (B) GR interacts directly with DNA by binding to canonical GRE (nGnACAnnnTGTnCn), half-sites (nGnACA) and nGRE (CTCC(n)0−2GGAGA) or indirectly by tethering to DNA-bound TFs by protein-protein interactions. (C) TFs can assist the loading of GR, or vice versa, by facilitating an accessible chromatin environment at the regulatory site.
Direct and Indirect GR Interaction With the DNA Template
Genomic occupancy of GR is facilitated by direct GR binding to GREs on DNA as a monomer, homodimer or tetramer (63) (Figure 2B), with the tetrameric structure being suggested as the final active form of GR (64). GR binds directly to the canonical DNA motif consisting of inverted repeats separated by 3 bps (nGnACAnnnTGTnCn) or to half-sites of these inverted repeats (nGnACA) (63, 65) and degenerate versions of these (66). In addition, GR can bind other inverted repeats separated by 0-2 bps (CTCC(n)0−2GGAGA) (67, 68), termed negative GREs (nGRE). Besides binding directly to DNA, GR can occupy enhancers by tethering to DNA-bound TFs by protein-protein interactions (63, 65).
Binding of GR to canonical DNA motifs as homodimers and tetramers is generally associated with GC-mediated transactivation (63, 69–71). Also, studies suggest that GR association with GR half-sites is linked to active gene expression (63, 65). Once GR is associated with enhancers, GC-induced transactivation involves recruitment of transcriptional coactivators to facilitate chromatin remodeling, histone hyperacetylation and mediator recruitment which leads to recruitment and/or increased activity of RNA polymerase II at juxtaposed gene promoters (56, 72–74). In contrast, GC-mediated transrepression has been widely discussed, and hence several different models have been presented, including direct binding of GR to nGRE motifs, interaction with DNA sequences bound by other TFs, tethered GR binding to transactivating TFs, redistribution of monomeric GR, sequestering of transactivating coregulators and/or GR-regulated expression of negative modulators of transcription (75). Even though nGREs have been associated with transcriptional repression (67, 68, 76), their role has been debated (63, 72, 74). For example, recent studies found no enrichment of nGREs at enhancers juxtaposed to GC-repressed genes (74). In contrast to enhancers induced by GC, repressed enhancers show marginal canonical GR binding motifs, suggesting that GR binds other DNA motifs (77) or tethers to other TFs (78). This type of GR interaction with DNA is generally believed to be mediated by monomeric GR, based on structural studies of the GR DBD and mice expressing a mutant GR (GRdim) unable to achieve DBD dimerization (69, 76–79). Although mice expressing GRdim indeed show reduced GR transactivation ability in the liver and maintain transrepressive activity (70), studies have suggested that GRdim forms dimers in the nucleus through another dimerization surface of the LBD (80). This suggests that binding to GR half-sites or other DNA motifs may be mediated by GR dimers, where possibly only one part of the dimer binds directly to DNA (Figure 2B). Cistromic analysis of GR and GRdim in the liver and in macrophages suggests extensive GR binding to chromatin through GR half-sites, which in many cases colocalizes with lineage-determining TFs driving cell-specific gene transcription (63, 70). Accordingly, GC treatment has been suggested to induce pronounced GR redistribution from GR half-sites to canonical GREs leading to reduced transcription of genes controlled by lineage-specific TFs (63). Introducing a mutation that completely disrupts direct GR binding to DNA (GRΔZn) leads to a perinatal lethal phenotype similar to knock out of GR, emphasizing an essential function of direct binding to DNA. Interestingly, studies of mouse embryonic fibroblasts isolated from GRΔZn mice show that direct GR-DNA interaction is essential for both transcriptional activation and repression by GCs, arguing that tethering is not a dominant mechanism for GR transrepression (81). Thus, genomic action of GR is primarily mediated by multimeric or monomeric actions involving direct interaction with the DNA template.
GR Interaction With Chromatin
GR binding to DNA is not solely dependent on the DNA sequence of the GRE. As GR binding sites are part of enhancer regions organized in higher order chromatin structures, occupancy of GR to specific regions of the genome is determined by a number of interdependent factors. This includes selective chromatin accessibility, epigenetic modifications of the histones, and the presence of other signal-dependent TFs, lineage-determining TFs and transcriptional coregulators (56). In the mouse liver, GR binds at least 11,000 distinct regions which are primarily located in intronic and intergenic distal regions (26, 61, 63, 72, 82). The vast majority of the GR binding sites are accessible prior to GC stimulation (pre-accessible chromatin) and only some are de novo remodeled following GR recruitment (72). Similar findings are observed for other cell types (56, 83, 84), demonstrating that selective GR occupancy of chromatin is largely determined by the accessibility of GREs. This pre-programmed chromatin landscape is shaped by cell-specific TFs and interacting coregulators that facilitate an accessible chromatin environment thereby assisting the loading of other TFs to the chromatin (discussed below; Figure 2C) (85). Accordingly, when comparing the liver cistrome across a number of well-described GC-responsive cell types, more than 80% of GR binding sites are unique to the liver and only 0.5% of the binding sites in the liver are shared with other cell types (72). This correlates with the findings that GR-occupied enhancers active in one cell type are inaccessible and nucleosomal in another cell type (73). GR has also been found to facilitate binding of other TFs to enhancers in the liver by establishment of accessible chromatin (72). In fact, binding of GR to genomic regions with different levels of chromatin accessibility has been linked to the type and strength of the GRE motif, with weaker motifs being found at nucleosome-depleted enhancers, compared to more nucleosomal dense sites (73).
Control of Gene Transcription by Recruitment of Coregulators and Chromatin Remodeling
Upon GR binding to chromatin, the local nucleosome-sparse region expands and the accessibility of the chromatin is further increased trough recruitment of chromatin remodeling complexes such as SWI/SNF and additional TFs (86–88). In addition, GR facilitates recruitment of widely expressed coactivators including histone acetyl transferases CBP, P300, GRIP1, PCAF and SRC-2 and components of the Mediator complex such as MED1 and MED14 (56, 66, 73, 89, 90). Moreover, other important GR coactivators have been identified in the liver, including CRTC2 (91), SIRT1, PGC-1α (92), ASCOM complex (93) and SETDB2 (94). On the other hand, GR has been found to interact with corepressors including SMRT (95), HDAC1 (96), CtBP (97), SMAD6-HDAC3 (98), CRY1 (99) and recently TAZ (100), although these interactions are not necessarily associated with transcriptional repression. The wide variety of coregulator interactions allows transcriptional fine-tuning of specific genes in a given cell in a concerted response to cellular signals and circulating GC levels.
Local recruitment of GR and associated coregulators to specific enhancers is translated to a transactivation potential by assembly into higher order enhancer-enhancer and enhancer-promoter condensates (101), facilitating localized increased concentration of the transcriptional machinery (102). Interestingly, interaction between promoters and enhancers occupied by GR is mostly established prior to GC stimulation (103, 104), suggesting that GC treatment does not necessarily lead to new chromosomal interactions but rather increases existing interactions between GR-occupied enhancers and GC-regulated target genes (74). Importantly, availability of GCs has been shown to be central for this differential interaction, suggesting that rapid regulation of gene transcription in response to changes in GC levels not only involves dynamic loading of GR and coregulators on the genome but also differential regulation of enhancer-promoter interaction (103).
GR Operates in Transcriptional Networks to Control Hepatic Gene Expression
The general GR working model described above illustrates that cell-specific GR actions are orchestrated by auxiliary lineage-determining and signal-dependent TFs. As any given cell expresses multiple cell-specific TFs that shape the accessible chromatin landscape, it is evident that GR-GC action in a given cell is controlled by signaling pathways regulating the activity and expression of these TFs. For example, the liver receives a variety of context-dependent signals controlling specific signaling pathways including circadian cues, insulin, glucagon, growth hormone and free fatty acids, that collectively shape and are shaped by the GC response in hepatocytes. These different signals are integrated in spatial and temporal TF signaling networks that regulate and fine-tune the hepatic transcriptional response. GR interaction with different TFs and the importance of these interactions for transcriptional regulation have been investigated for decades (105). Recently, several key genome-wide studies in mouse liver tissue have demonstrated that GR interacts with a large repertoire of TFs and that these interactions are diverse, bidirectional, dynamic and highly context- and cell-specific (Table 1). The interactions between GR and TFs can be classified as direct or indirect. Direct interactions cover protein-protein interactions or concurrent and co-localized binding to regulatory sites in the chromatin (Figure 3A), impacting coregulator recruitment, and consequently enhancer activity (Figure 3B). Indirect interactions involve TF cascades, where the expression of one TF regulates the expression of another TF (Figure 3C).
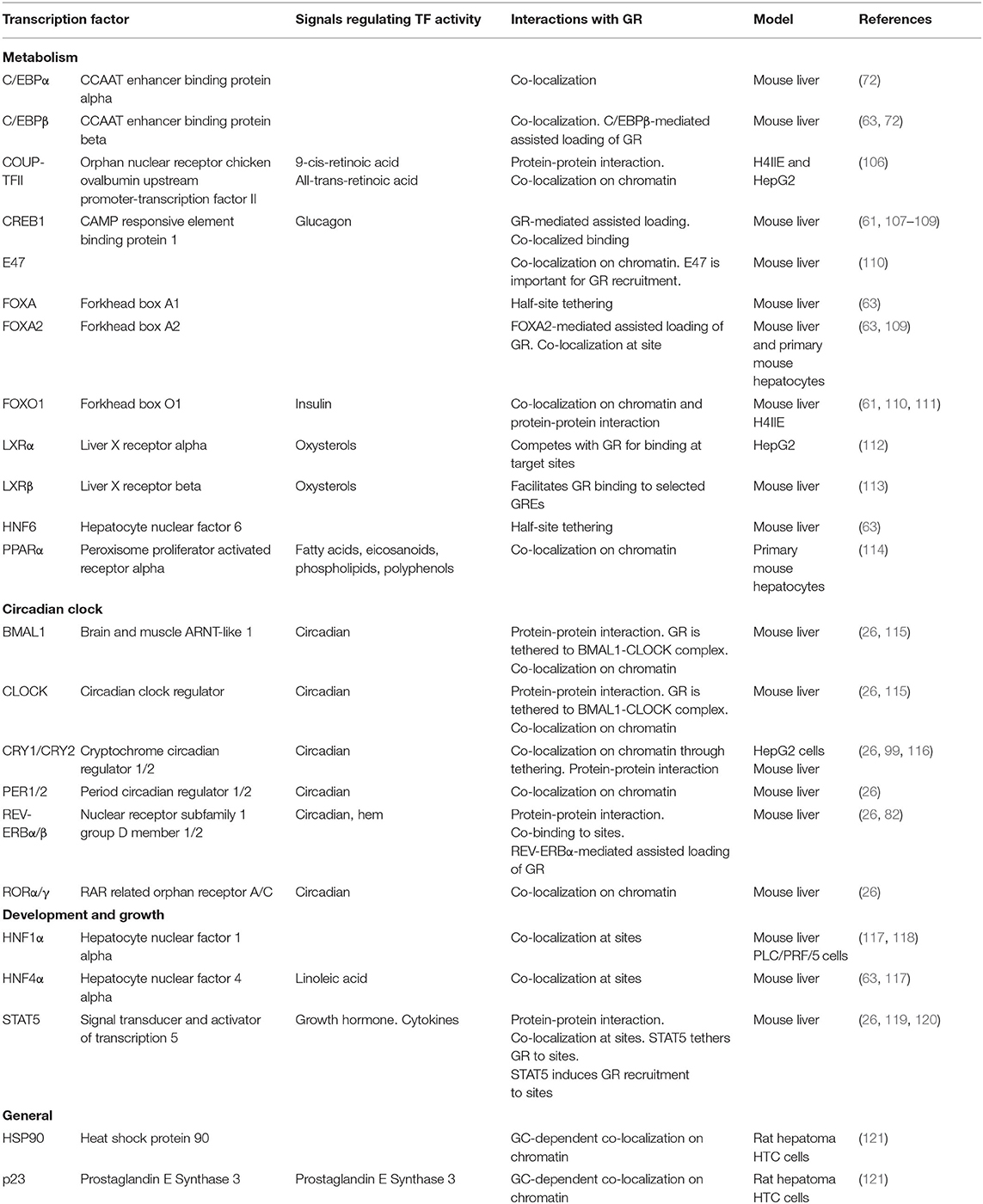
Table 1. Examples of hepatocyte expressed transcription factors interacting with GR on chromatin.
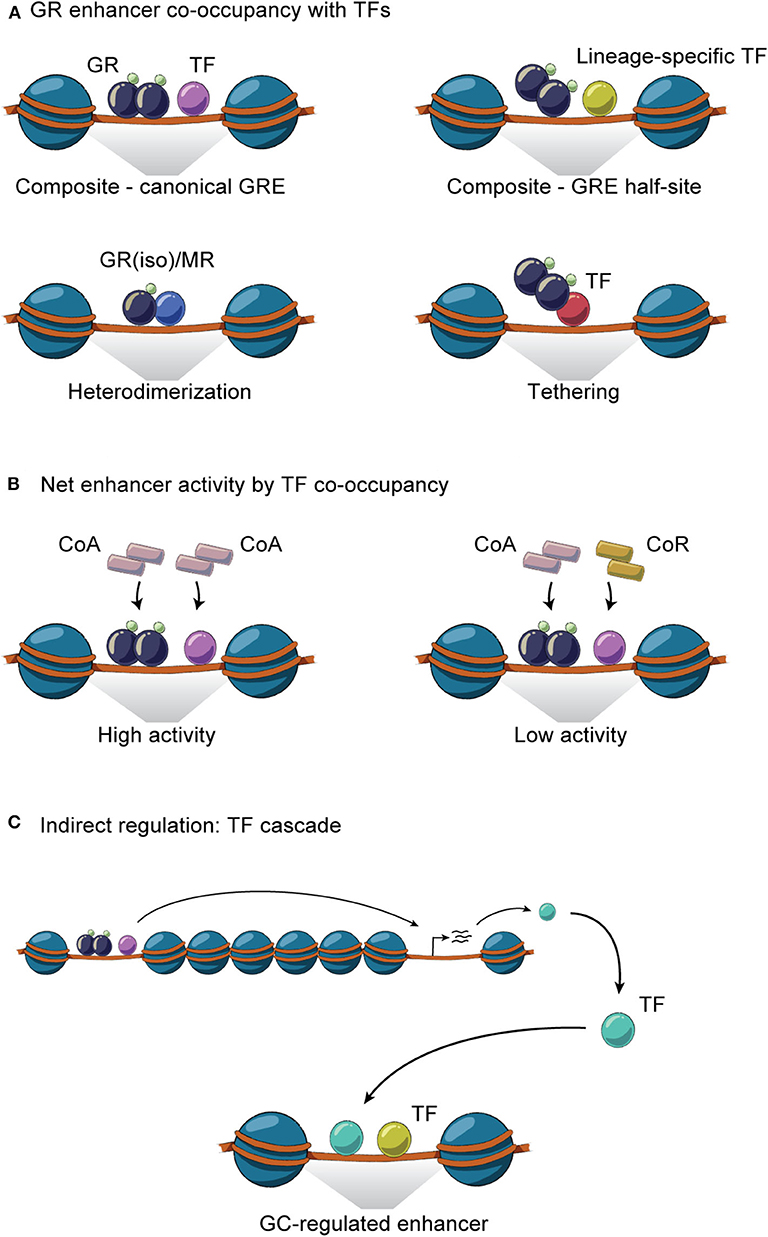
Figure 3. GR interaction with TFs on chromatin. (A) GR and TFs co-occupy enhancers through homodimeric or monomeric GR binding together with TFs at composite sites, by heterodimerization and through tethering. (B) GR- and TF-mediated recruitment of coactivators (CoA) and/or corepressors (CoR) to co-occupied regulatory sites controls the net enhancer activity. (C) Indirect GR-TF interaction involves TF cascades, where the expression of GR regulates the expression of TF or vice versa.
Composite TF Interactions and Assisted Loading
At composite sites, GR binds to GREs and can functionally interact with other TFs bound to a neighboring site in the same regulatory region, co-operatively regulating enhancer activity. These binding sites can be overlapping or closely located on the DNA strand and involve GREs, half GREs and/or nGREs (Figure 3A). Many liver-expressed TFs have been found or suggested to co-occupy GR binding sites (Table 1). ChIP-seq experiments have confirmed the composite binding of CREB1, FOXO1, FOXA, HNF4α, HNF6, C/EBPα, C/EBPβ, PPARα, E47, STAT5, and REV-ERBα at several GR-occupied enhancers (26, 61, 63, 72, 82, 107, 108, 110, 114). In the liver, ChIP-seq data suggests that GR binds GRE half-sites together with lineage-determining TFs including HNF4α, C/EBPβ, HNF6, and FOXA (63, 72). In addition, AP-1 and SP1 motifs have been found to be enriched at GR binding sites (122) and the AhR binding site contains a GRE (123), suggesting that these TFs could work together with GR at specific sites to regulate transcription (124). However, further investigations are needed to determine the relevance of AP-1, SP1, and AhR on GR activity in the liver.
Several confirmed composite GR-TF interactions have been found to impact GR activity and hepatic metabolism, including C/EBPβ, E47, STAT5, and LXRβ, which are required for GR recruitment to specific sites (26, 72, 110, 113), in accordance with the model for assisted loading. For example, GR and E47 co-occupy many promoters and enhancers, working in synergy to regulate GC-induced metabolic genes. Studies using liver-specific E47 knock-out mice emphasize the importance of E47 in the recruitment of GR, FOXO1, and the mediator complex to composite sites. This cooperation affects glucose, fatty acid and lipid metabolism, which is demonstrated by E47 knock-out mice being protected from GC-induced hyperglycemia, dyslipidemia and hepatic steatosis (110). Another example is the C/EBP-facilitated assisted loading of GR. C/EBP has been found to occupy and prime the majority of GR target sites in the liver, making the chromatin accessible for GR binding. Disruption of C/EBP binding attenuates GR recruitment and GR-induced chromatin remodeling at composite sites (72). The concept of assisted loading is also found reversely, with GR assisting the loading of TFs including C/EBP and CREB1 at a subset of sites (Figure 2C) (72, 107). For example, GR-mediated assisted loading of CREB1 at a subset of CREB1 target enhancers doubles the number of CREB1 bound sites and increases chromatin accessibility, eventually leading to increased hepatic glucose production during fasting (107).
Protein-Protein Interactions: Heterodimerization and Tethering at Chromatin
As mentioned above, multiple GR isoforms can be generated from the primary transcript and protein processing. Thus, GRα/β heterodimers can be formed on chromatin, impacting the activity of occupied enhancers (125–127) (Figure 3A). In fact, GRβ has been shown to have metabolic relevance in the liver. For example, feeding induces GRβ expression within 7 h, likely in response to insulin (36). This is supported by observations that hepatic GRβ expression increases in diet-induced obese mice (128). Overexpression of GRβ in mouse liver reduces expression of known GRα target genes such as Pck1 and Ppara, associated with disrupted gluconeogenesis and increased hepatic lipid accumulation and inflammation, respectively (128, 129). Moreover, the GRβ-mediated increase in lipid accumulation is also seen in L-GRKO mice (26, 130), suggesting that GRβ may function as a negative regulator of GRα in hepatic fatty acid metabolism. Importantly, GRβ expression in a GRα-negative background leads to expression of a specific set of genes not regulated in the presence of GRα (129), suggesting that GRα and GRβ regulate each other's activities by mechanisms involving accessibility to chromatin, cooperation with TFs and coregulators and indirect regulation of enhancer activity (Figures 3A–C). Likewise, GR has been found to form a heterodimer with the mineralocorticoid receptor (MR) (34) in a number of different tissues and cells, including the hippocampus and mammary cells. Here, the GR-MR complex binds to GREs and regulates gene expression (131, 132). Although a GR-MR complex has not, to our knowledge, been shown to be functional in the liver, it has been suggested that GR-MR could regulate hepatic expression of G6Pase (133) (Figure 3B). However, further investigations are needed.
Besides heterodimerization on DNA, GR has been suggested to form other protein-protein interactions on chromatin which tether GR to enhancers independently of its DBD. This includes interaction with COUP-TFII, STAT5, PPARα and the molecular clock components BMAL1, CLOCK, and REV-ERBα, influencing GR activity and hepatic metabolism (82, 106, 115, 116, 119). For example, COUP-TFII protein interaction with GR is important for GC-induced promoter activity and hepatic Pck1 gene expression (106). Also, GR is suggested to be recruited to a subset of sites via tethering to DNA-bound PPARα to regulate metabolic genes in the liver including Pdk4 (114). Moreover, GR tethering to the BMAL1-CLOCK complex is suggested to repress hepatic Rev-erbα expression (115), demonstrating how GR and the molecular circadian clock interconnect to regulate shared gene programs.
Controlling Enhancer Activity by Co-occupancy of Multiple TFs
The transcriptional effect of multiple TF interactions at enhancers can be evaluated by looking at the expression of juxtaposed target genes or at localized histone acetylation and mediator recruitment. In the case of TF cooperation at individual enhancers, activation of several TFs will result in synergistic effects on enhancer activity and gene expression. In contrast, TFs working independently at the shared enhancer would result in gene expression corresponding to a sum of the contribution from each TF. For example, composite GR-PPARα sites have been found to synergistically affect the expression of fatty acid oxidation and ketogenic genes while GR-CREB1 sites synergistically regulate gluconeogenic genes (107). Likewise, synergistic and additive regulation has been reported for genes controlled by GR and FOXO1 in co-occupancy (61). These cooperative effects likely reflect increased recruitment of coactivators to a given set of enhancers involved in transcription of a specific gene (Figure 3B).
In contrast to the synergistic action of composite GR-TF binding sites to increase enhancer activity, several studies have suggested negative regulation between GR and TFs occupied at a given enhancer. Such negative regulation can be understood as a competition between the TFs for a given DNA sequence. For example, in the liver, LXRα binds GREs together with its heterodimerization partner RXRα, thereby potentially competing with GR for binding to the same sites leading to differential regulation of genes involved in glucose metabolism (112). Another example is the GR isoform competition model, which seeks to explain how dominant negative GRβ functions as a negative regulator for GRα at some sites. Similarly, GR has been suggested to compete with AP1 at AP1 motifs with embedded GR half-sites (77). However, these competitional models do not agree with the dynamic nature of GR and most other TFs as these factors bind transiently to chromatin with residence times in a matter of seconds (55, 134), possibly allowing multiple factors to interact with the same site (135). Thus, GR-TF competition at composite sites is likely not a competition for the same response element. Instead, the negative regulation likely reflects the different coregulators recruited to the response element. Composite binding of different TFs recruiting coregulators of opposite activity or competition between limited amount of avaliable coregulators for binding to the specific TFs would balance the transcriptional response. For example, corepressors and coactivators have been suggested to bind GR in equilibrium, balancing GR activity (136), which has also been suggested for other nuclear receptors in the liver, including the thyroid hormone receptor (137).
Regulating TF Networks by GR
The direct interaction between GR and other key TFs on chromatin in the liver can take different forms, as described above, to jointly regulate hepatic gene expression. However, indirect GR-TF interactions involving TF cascades are equally important, though more challenging to investigate, with several potential interaction steps (Figure 3C). Important indirect pathways have been studied in the liver. For example, GR binds GREs near core clock genes to induce transcription of Per1, Bmal1, Cry1, Dbp (138, 139). This in turn controls a range of circadian-regulated genes. In regards to energy metabolism, GR interacts with several key factors in TF cascades connecting and impacting different signaling pathways. For example, glucagon-mediated activation of CREB1 induces the transcription of YY1, which then induces the transcription of GR. This interaction cascade is important in hepatic gluconeogenesis (140). Moreover, GR induces the transcription of Klf9, which has been linked to the downstream induction of PGC1α expression and of hepatic gluconeogenic genes (141). GR interaction with PGC1α has furthermore been suggested to regulate mitochondrial oxidative phosphorylation (142). Additionally, GR induces the transcription of PPARα upon long-term fasting, initiating hepatic fatty acid oxidation and the ketogenic gene program (107).
GR Regulatory Networks Impact Multiple Aspects of Hepatic Metabolism
The emerging studies in complex gene-regulatory networks controlled by GR and controlling GR activity emphasize the importance of the context-dependent action of GCs in tissues like the liver. Accordingly, genetic disruption of GR in the liver impacts a range of metabolic pathways leading to dysregulated glucagon synthesis, lipid metabolism, gluconeogenesis, urea metabolism and bile acid synthesis and uptake (26, 143–146). For example, L-GRKO mice and GRdim mice show dysregulated glucose, fatty acid and bile acid metabolism (26, 144, 146). Reduced expression of key gluconeogenic genes including Pck1, G6Pc, and Pfkfb3 in L-GRKO mice is linked to fasting hypoglycemia (26, 144–146), and around half of newborn albumin-alpha-fetoprotein-driven L-GRKO mice die within 48 h after birth, possibly due to hypoglycemia (120, 146). L-GRKO mice are more sensitive to insulin than WT littermates and liver glycogen content in L-GRKO mice is reduced (145). These effects of L-GRKO on glucose metabolism could in part be explained by the interaction with TFs such as CREB1, FOXO1, FOXA2, PPARα, E47, STAT5, LXRα, LXRβ, and circadian regulators (26, 61, 82, 107, 109, 110, 112–114) (Figure 4). Yet, the effects of L-GRKO on glucose metabolism seem to be partially compensated by increased gluconeogenesis in the kidney (145) and by a shifted hormonal balance involving reduced plasma concentration of insulin and increased glucagon levels, compared to WT mice (146).
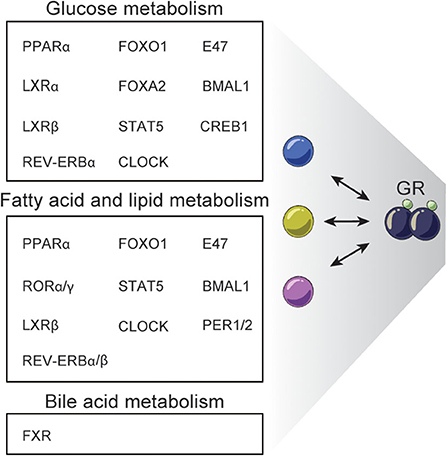
Figure 4. Examples of TFs interacting with GR regulating hepatic metabolism. GR interacts with different TFs to regulate specific processes in hepatic glucose, fatty acid, lipid, and bile acid metabolism.
Hepatic GR disruption also leads to decreased fat mass (145) and lower plasma triglyceride levels (26, 146), while free fatty acid plasma levels are similar in fasted and fed L-GRKO mice and WT mice (146). Recently, L-GRKO mice were reported to accumulate triglycerides in the liver and to develop hepatic steatosis (26), although this is controversial (130), but may be explained by the promoter controlling CRE expression. Many TFs have been found to work together with GR to regulate fatty acid and lipid metabolism including STAT5, PPARα, FOXO1, E47, LXRβ, CLOCK, REV-ERBα/β, CRY, BMAL1, RORα/γ, and PER1/2 (26, 82, 110, 111, 113, 114) (Figure 4).
Finally, disruption of hepatic GR function leads to dysregulated systemic bile acid homeostasis. Specifically, mice with hepatic GR knock down by shRNA have a reduced amount of bile acid in the gallbladder, elevated serum bile acid levels, impaired bile acid uptake/transport and are more susceptible to develop gallstones when fed on cholesterol-rich diet. Moreover, these mice do not undergo the normal changes in bile acid levels in the serum, liver and intestines in the fast-refeeding transition (144). GRdim mice fed a lithogenic diet have elevated fasting serum bile acid levels and decreased gallbladder bile acid volume. These effects have been associated with interaction between GR and FXR, a key TF regulating bile acid metabolism (97, 144). GR deficiency reduces the expression of the classical FXR-target gene Shp encoding the SHP repressor, leading to increased expression of the rate-limiting enzymes in bile acid synthesis Cyp7a1 and Cyp8b1 (144). Additionally, dex-induced GR recruits the co-repressor CtBP to block FXR activity at shared sites related to bile acid gene metabolism, e.g. Shp promoter (97) (Figure 4).
Examples of Key Hepatic Gene Regulatory Networks Controlled by GR
GR Crosstalk With FOXO1
The daily change from the inactive fasting phase to the active feeding phase requires a major transcriptional reprogramming of the liver. This is particularly relevant at the transition between the unfed and fed states, which takes place around zeitgeber time (ZT) 12 (i.e., 6 p.m.) in nocturnal animals such as mice. The interaction between GR and the insulin-regulated TF FOXO1 is involved in driving this transcriptional transition. Pre-prandial high GC and low insulin levels are associated with GR and FOXO1 binding to chromatin, respectively, and regulation of target genes. In fact, in this fasted state, more than half of all FOXO1 binding sites are co-occupied with GR regulating gene expression. Conversely, the post-prandial increased insulin and reduced GC lead to reduced FOXO1 and GR occupancy, respectively, and reduced transcriptional regulatory activity. Importantly, more than 80% of feeding-repressed genes in the liver are associated with a nearby enhancer bound by GR, FOXO1 or both (61). One example of a metabolic gene coregulated by GR and FOXO1 in the liver is Angptl4, associated with the regulation of glucose and lipid metabolism. In a fasted state, GR and FOXO1 bind a specific GRE and forkhead box transcription factor response element (FRE), respectively, located in the regulatory region of Angptl4. GCs induce, while insulin abolishes, the occupancy of both factors at the region (111). Besides the direct interaction between GR and FOXO1 at enhancers in the liver, GR has been found to induce the expression of Foxo1 gene in the liver and in this way indirectly regulate target genes (147). Furthermore, FOXO1 binding has been found at the promoter of GR, suggesting that the indirect interaction is bidirectional (148).
GR Crosstalk With PPARα
Like GR, PPARα is important for the hepatic response to fasting. The role of PPARα in regulating metabolism and inflammation as well as the importance of crosstalk between PPARα and other TFs, including GR, have been covered in detail in previous reviews (1, 149). The GR-PPARα interactions in the liver include co-localization to chromatin and coregulation of genes involved in lipid and glucose metabolism (150). More specifically, in co-ligand treatment of primary murine hepatocytes, 13% of GR peaks are co-bound with PPARα (114). Furthermore, other studies have found that, during fasting, GR and PPARα have a synergistic effect on genes involved in ketogenesis and fatty acid oxidation; however, the GR-PPARα interaction has been suggested to be indirect as GR induces the expression of PPARα and time-course experiments show a gradual effect of GR on PPARα activity (107).
GR Crosstalk With STAT5
STAT5 is activated by the growth hormone through the growth hormone receptor-JAK2 signaling pathway and by cytokine signaling. In the liver, STAT5 is known to regulate genes involved in body growth, cell cycle, lipid, bile acid, drug and steroid metabolism (151). The STAT5 and GR signaling pathways are connected as exemplified by the reduced body size in mice with inactivated hepatic GR showing impaired growth hormone signaling (120). Furthermore, the importance of STAT5 and GR signaling is demonstrated in liver-specific STAT5 and STAT5 GR double mutant mice exhibiting hepatic steatosis and, for the double mutant, also hepatic carcinoma (130). The STAT5 and GR crosstalk at multiple levels. STAT5 and GR form protein-protein interactions in hepatocytes, which have been found to be important for postnatal growth and maturation-related gene expression. Mice expressing a point mutation in the GR DBD (GRdim mice), previously suggested to reduce GR DNA-binding and GR dimerization (69, 79), have an unaltered ability to interact with STAT5 (120). These GRdim mice have normal body size, suggesting that the joint GR-STAT5 regulation of growth genes happens through tethering of GR to the STAT5 bound sites or through half GREs in conjunction with STAT5 binding sites (119, 120). However, as mentioned above, more recent studies have found that GRdim is able to dimerize and bind DNA (80), suggesting a reassessment of GR and STAT5 interaction type at shared sites.
Recently, it has been shown that high-fat diet feeding of mice leads to reprogramming of the hepatic GR cistrome primarily during the active feeding phase. Many sites with high-fat diet-induced increased GR recruitment are associated with increased STAT5 co-occupancy. These co-occupied sites showed increased enhancer activity and were associated with genes involved in fatty acid, lipid and glucose metabolism. Hepatocyte-specific STAT5 and GR KO mice demonstrated that STAT5 facilitated the recruitment of GR at gained sites, whereas GR had no effect on STAT5 recruitment. It is still unknown whether the increased STAT5 activity in obese mice is a response to altered growth hormone or cytokine signaling or if it originates from nutritional adaptations in the chromatin landscape (26).
GR Crosstalk With Molecular Clock Components
In the liver, the effect of exogenous GCs on gene regulation is highly dependent on the time of administration. For example, in mice about eight times more genes are differentially regulated by GCs at daytime compared to nighttime. Pathway analysis shows a strong time-dependent regulation of genes in glucose and lipid metabolism (82), which has also been observed in studies looking at endogenous GC effects (26). Hence, timing of GC administration according to the endogenous GC levels has shown positive effects. Administration of GCs at ZT12, as opposed to ZT0, leads to less hepatic lipid accumulation and behavioral changes. This time-differential effect of GC is suggested to be caused by a disrupted circadian regulation of GC-target genes with administration at ZT0, which is supposedly more critical compared to an over-activation of GR at ZT12 (152).
This diurnal oscillation of GC action stems from cooperativity and multiple interactions between GR and the molecular clock components in the liver. For example, GR and central clock components including BMAL1, CLOCK, REV-ERBα/β, PER1, PER2, CRY1, CRY2, and RORα/γ co-occupy different genes involved in clock function and in metabolism (26, 82, 99). The cooperativity also involves different physical interactions between GR and clock factors on the chromatin level, regulating the expression of other clock factors and metabolic genes (see Table 1). For example, GR physically interacts with CRY1/2 in a GC-induced manner and, in the post-prandial phase, CRY1/2 represses GR activity on e.g. the expression of Pck1. CRY1/2 deficient mice have constitutively high GC levels and exhibit glucose intolerance, suggesting reduced suppression of HPA axis and increased GR activity in the liver (99).
It has been long known that GC and GC-activated GR influence the expression and circadian phase-shifts of several clock factors, including Per1, Dbp and Cry1 (23, 30, 139). In fact, GR is recruited to the promoters and enhancers of all central clock genes, suggesting a gene regulatory function of GR (26). Reversely, molecular clock elements also affect GR function, as exemplified by the previously mentioned binding of REV-ERBα to HSP90 (53) and the acetylation of GR by CLOCK (46), both leading to suppression of GR action.
The interaction between GR and members of the molecular clock and its influence on hepatic metabolism can be further exemplified focusing on a single molecular factor. REV-ERBα is one of the key transcriptional repressors in the molecular transcriptional clock, contributing to the characteristic circadian expression in many tissues, including the brain and metabolic tissues like the liver, muscle, pancreas and adipose tissue. In the liver, REV-ERBα is involved in the daily regulation of glucose and lipid metabolism (153). REV-ERBα represses clock genes by binding to RevDR2/RORE DNA elements and recruiting the corepressor complex NCoR-HDAC3. On the other hand, REV-ERBα regulates many metabolic genes by tethering to cell-type specific TFs. Hepatic REV-ERBα tethers to e.g., HNF6 and recruits HDAC3 for active repression of lipogenic genes (154, 155). GR has been found to interact with REV-ERBα on different levels. REV-ERBα interacts physically with GR and, together with HNF4α and HNF6, binds regulatory regions controlling gene expression in mouse liver. REV-ERBα was found to be important for efficient GR recruitment to chromatin during the day, presumably by maintaining histone acetylation at binding sites (82). Moreover, indirect interactions between GR and REV-ERBα have also been observed. REV-ERBα inhibits GR protein expression and nuclear localization (53), and GR inhibits REV-ERBα RNA expression (156) by forming a complex with CLOCK and BMAL1, where GR may be tethered to the regulatory site of the REV-ERBα gene (115).
Perspectives in Disease and Clinical Use of GCs
Glucocorticoids have immunosuppressant and anti-inflammatory properties, making them an effective treatment for allergies, inflammatory and autoimmune diseases. The anti-inflammatory effects mediated by GR are conducted by the immune cells, with the macrophages having a particularly important role in the repression of inflammatory genes [reviewed in (157)]. However, by administering GCs systemically, there is a risk of eliciting undesirable side effects on other tissues and cellular processes, such as hepatic metabolism, which is highly impacted by GR regulation. In this review, we described the multiple layers of regulation of GR function, from the control of hormonal availability to the modulation of GR expression at both mRNA and protein levels, as well as PTMs and interactions with different proteins and TFs affecting the transcriptional activity of GR. In depth knowledge of the multifaced control of GR activity provides a unique opportunity to tailor GC treatment and prevent metabolic-related side effects.
One strategy could involve administration of different GR ligands affecting interacting coregulators to modulate transcriptional regulation by GR (8). Another strategy could be to selectively activate or inhibit specific and relevant GR-mediated regulatory pathways, where treatments involving a combination of different TF ligands could have potential. For example, co-administration of GC and LXR agonists attenuates the transcriptional activity of GR on a subset of genes in glucose and lipid metabolism, suggesting co-treatment with LXRβ agonists might reduce metabolic side effects in patients with autoimmune or inflammatory diseases (112). However, the function of LXRs on GR target sites is debated (113), and the mechanisms behind the positive and negative effects of LXRs on GR should be elucidated. Also, the antagonistic effect of activated PPARα on GR-mediated transcription of metabolic genes to circumvent GC side effects seems promising (150), with potentials and challenges recently discussed in another review (1). Additionally, the natural ultradian GC release and subsequent dynamic activation of GR contrasts with the constant exposure to GCs during pharmacological therapies. The development of new synthetic GCs and pulsatory administration strategies could potentially minimize side effects by mimicking physiology (58, 59, 103). Finally, pharmacological chronotherapy involving GCs seems promising in several inflammatory disorders, with outcomes improving when GC administration is consistently timed (4). This timed GC-administration has been shown to be beneficial in, for example, patients with rheumatoid arthritis (158).
Concluding Remarks
The multifaceted regulation of GC action and GR activity discussed in this review highlights the complexity of transcriptional regulation by ligand-dependent TFs. The cooperation with signal-dependent and lineage-specific TFs makes GC-dependent gene regulation very responsive to environmental cues and is thus essential to understand for future optimized usage of GCs in the clinic. Specifically, a deeper understanding of the regulatory mechanisms underlying GR action would be fundamental for future development of safer and more effective therapies for disorders where GC secretion and signaling is involved. The recent genomics studies into the GR interactome show promise in the elucidation of the complex GR-TF networks and could contribute to a shift toward future tailored pharmacological strategies including spatio-temporal drug delivery and personalized medicine.
Author Contributions
SP and CC wrote the manuscript with supervision from LG. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from the SDU2020 initiative, the Novo Nordisk Foundation and Independent Research Fund Denmark (Sapere Aude: DFF-Starting Grant).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Illustration of Figure 1 and elements used for Figures 2, 3 was created by scientific illustrator CC. We would like to acknowledge Claes S. Wassmann for critical review and comments.
References
1. Vandewalle J, Luypaert A, de Bosscher K, Libert C. Therapeutic mechanisms of glucocorticoids. Trends Endocrinol Metab. (2018) 29:42–54. doi: 10.1016/j.tem.2017.10.010
2. Rose AJ, Vegiopoulos A, Herzig S. Role of glucocorticoids and the glucocorticoid receptor in metabolism: insights from genetic manipulations. J Steroid Biochem Mol Biol. (2010) 122:10–20. doi: 10.1016/j.jsbmb.2010.02.010
3. Challet E. Keeping circadian time with hormones. Diabetes Obes Metab. (2015) 17:76–83. doi: 10.1111/dom.12516
4. Spencer RL, Chun LE, Hartsock MJ, Woodruff ER. Glucocorticoid hormones are both a major circadian signal and major stress signal: how this shared signal contributes to a dynamic relationship between the circadian and stress systems. Front Neuroendocrinol. (2018) 49:52–71. doi: 10.1016/j.yfrne.2017.12.005
5. Ishida A, Mutoh T, Ueyama T, Bando H, Masubuchi S, Nakahara D, et al. Light activates the adrenal gland: timing of gene expression and glucocorticoid release. Cell Metab. (2005) 2:297–307. doi: 10.1016/j.cmet.2005.09.009
6. Bertram R, Satin LS, Sherman AS. Closing in on the mechanisms of pulsatile insulin secretion. Diabetes. (2018) 67:351–9. doi: 10.2337/dbi17-0004
7. Giustina A, Veldhuis JD. Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev. (1998) 19:717–97. doi: 10.1210/er.19.6.717
8. Biddie SC, Conway-Campbell BL, Lightman SL. Dynamic regulation of glucocorticoid signalling in health and disease. Rheumatology. (2012) 51:403–12. doi: 10.1093/rheumatology/ker215
9. Lightman SL, Birnie MT, Conway-Campbell BL. Dynamics of ACTH and cortisol secretion and implications for disease. Endocr Rev. (2020) 41:470–90. doi: 10.1210/endrev/bnaa002
10. Perogamvros I, Kayahara M, Trainer PJ, Ray DW. Serum regulates cortisol bioactivity by corticosteroid-binding globulin-dependent and independent mechanisms, as revealed by combined bioassay and physicochemical assay approaches. Clin Endocrinol. (2011) 75:31–8. doi: 10.1111/j.1365-2265.2011.04003.x
11. Desantis LM, Delehanty B, Weir JT, Boonstra R. Mediating free glucocorticoid levels in the blood of vertebrates: are corticosteroid-binding proteins always necessary? Funct Ecol. (2013) 27:107–19. doi: 10.1111/1365-2435.12038
12. Breuner CW, Orchinik M. Plasma binding proteins as mediators of corticosteroid action in vertebrates. J Endocrinol. (2002) 175:99–112. doi: 10.1677/joe.0.1750099
13. Richard EM, Helbling JC, Tridon C, Desmedt A, Minni AM, Cador M, et al. Plasma transcortin influences endocrine and behavioral stress responses in mice. Endocrinology. (2010) 151:649–59. doi: 10.1210/en.2009-0862
14. Basu R, Singh RJ, Basu A, Chittilapilly EG, Johnson CM, Toffolo G, et al. Splanchnic cortisol production occurs in humans. Diabetes. (2004) 53:2051. doi: 10.2337/diabetes.53.8.2051
15. Stimson RH, Andersson J, Andrew R, Redhead DN, Karpe F, Hayes PC, et al. Cortisol release from adipose tissue by 11β-hydroxysteroid dehydrogenase type 1 in humans. Diabetes. (2009) 58:46–53. doi: 10.2337/db08-0969
16. Maser E, Völker B, Friebertshäuser J. 11β-hydroxysteroid dehydrogenase type 1 from human liver: dimerization and enzyme cooperativity support its postulated role as glucocorticoid reductase. Biochemistry. (2002) 41:2459–65. doi: 10.1021/bi015803t
17. Andrew R, Westerbacka J, Wahren J, Yki-Järvinen H, Walker BR. The contribution of visceral adipose tissue to splanchnic cortisol production in healthy humans. Diabetes. (2005) 54:1364. doi: 10.2337/diabetes.54.5.1364
18. Dammann C, Stapelfeld C, Maser E. Expression and activity of the cortisol-activating enzyme 11 beta-hydroxysteroid dehydrogenase type 1 is tissue and species-specific. Chem Biol Interact. (2019) 303:57–61. doi: 10.1016/j.cbi.2019.02.018
19. Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, et al. 11 beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA. (1997) 94:14924–9. doi: 10.1073/pnas.94.26.14924
20. Moore RY, Eichler VB. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res. (1972) 42:201–6. doi: 10.1016/0006-8993(72)90054-6
21. Focke CMB, Iremonger KJ. Rhythmicity matters: circadian and ultradian patterns of HPA axis activity. Mol Cell Endocrinol. (2020) 501:110652. doi: 10.1016/j.mce.2019.110652
22. Neumann AM, Schmidt CX, Brockmann RM, Oster H. Circadian regulation of endocrine systems. Auton Neurosci Basic Clin. (2019) 216:1–8. doi: 10.1016/j.autneu.2018.10.001
23. Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, et al. Resetting of circadian time peripheral tissues by glucocorticoid signaling. Science. (2000) 289:2344–7. doi: 10.1126/science.289.5488.2344
24. Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. (2000) 14:2950–61. doi: 10.1101/gad.183500
25. Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M. Entrainment of the circadian clock in the liver by feeding. Science. (2001) 291:490–3. doi: 10.1126/science.291.5503.490
26. Quagliarini F, Mir AA, Balazs K, Wierer M, Dyar KA, Jouffe C, et al. Cistromic reprogramming of the diurnal glucocorticoid hormone response by high-fat diet. Mol Cell. (2019) 76:531–45.e5. doi: 10.1016/j.molcel.2019.10.007
27. Silver R, Balsam P. Oscillators entrained by food and the emergence of anticipatory timing behaviors. Sleep Biol Rhythms. (2010) 8:120–36. doi: 10.1111/j.1479-8425.2010.00438.x
28. Sheward WJ, Maywood ES, French KL, Horn JM, Hastings MH, Seckl JR, et al. Entrainment to feeding but not to light: circadian phenotype of VPAC(2) receptor-null mice. J Neurosci. (2007) 27:4351–8. doi: 10.1523/JNEUROSCI.4843-06.2007
29. Sasaki H, Hattori Y, Ikeda Y, Kamagata M, Iwami S, Yasuda S, et al. Phase shifts in circadian peripheral clocks caused by exercise are dependent on the feeding schedule in PER2::LUC mice. Chronobiol Int. (2016) 33:849–62. doi: 10.3109/07420528.2016.1171775
30. Le Minh N, Damiola F, Tronche F, Schutz G, Schibler U. Glucocorticoid hormones inhibit food-induced phase-shifting of peripheral circadian oscillators. Embo J. (2001) 20:7128–36. doi: 10.1093/emboj/20.24.7128
31. Tahara Y, Shibata S. Entrainment of the mouse circadian clock: effects of stress, exercise, and nutrition. Free Radic Biol Med. (2018) 119:129–38. doi: 10.1016/j.freeradbiomed.2017.12.026
32. Carlstedt-Duke J, Strömstedt PE, Wrange O, Bergman T, Gustafsson JA, Jörnvall H. Domain structure of the glucocorticoid receptor protein. Proc Natl Acad Sci USA. (1987) 84:4437–40. doi: 10.1073/pnas.84.13.4437
33. Hollenberg SM, Evans RM. Multiple and cooperative trans-activation domains of the human glucocorticoid receptor. Cell. (1988) 55:899–906. doi: 10.1016/0092-8674(88)90145-6
34. Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. (2002) 110:93–105. doi: 10.1016/S0092-8674(02)00817-6
35. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. (2013) 132:1033–44. doi: 10.1016/j.jaci.2013.09.007
36. Hinds TD Jr, Ramakrishnan S, Cash HA, Stechschulte LA, Heinrich G, Najjar SM, et al. Discovery of glucocorticoid receptor-beta in mice with a role in metabolism. Mol Endocrinol. (2010) 24:1715–27. doi: 10.1210/me.2009-0411
37. Vandevyver S, Dejager L, Libert C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr Rev. (2014) 35:671–93. doi: 10.1210/er.2014-1010
38. DuBois DC, Sukumaran S, Jusko WJ, Almon RR. Evidence for a glucocorticoid receptor beta splice variant in the rat and its physiological regulation in liver. Steroids. (2013) 78:312–20. doi: 10.1016/j.steroids.2012.11.014
39. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA Gas5 is a growth arrest– and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. (2010) 3:ra8. doi: 10.1126/scisignal.2000568
40. Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3 beta-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. (2008) 28:7309–22. doi: 10.1128/MCB.00808-08
41. Orti E, Mendel DB, Smith LI, Munck A. Agonist-dependent phosphorylation and nuclear dephosphorylation of glucocorticoid receptors in intact-cellS. J Biol Chem. (1989) 264:9728–31.
42. Orti E, Mendel DB, Smith LI, Bodwell JE, Munck A. A dynamic-model of glucocorticoid receptor phosphorylation and cycling in intact-cells. J Steroid Biochem Mol Biol. (1989) 34:85–96. doi: 10.1016/0022-4731(89)90069-1
43. Webster JC, Jewell CM, Bodwell JE, Munck A, Sar M, Cidlowski JA. Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J Biol Chem. (1997) 272:9287–93. doi: 10.1074/jbc.272.14.9287
44. Galliher-Beckley AJ, Cidlowski JA. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. (2009) 61:979–86. doi: 10.1002/iub.245
45. Avenant C, Ronacher K, Stubsrud E, Louw A, Hapgood JP. Role of ligand-dependent GR phosphorylation and half-life in determination of ligand-specific transcriptional activity. Mol Cell Endocrinol. (2010) 327:72–88. doi: 10.1016/j.mce.2010.06.007
46. Nader N, Chrousos GP, Kino T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. Faseb J. (2009) 23:1572–83. doi: 10.1096/fj.08-117697
47. Wilkinson L, Verhoog N, Louw A. Novel role for receptor dimerization in post-translational processing and turnover of the GR alpha. Sci Rep. (2018) 8:17. doi: 10.1038/s41598-018-32440-z
48. Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. (2001) 276:42714–21. doi: 10.1074/jbc.M106033200
49. Wallace AD, Cao Y, Chandramouleeswaran S, Cidlowski JA. Lysine 419 targets human glucocorticoid receptor for proteasomal degradation. Steroids. (2010) 75:1016–23. doi: 10.1016/j.steroids.2010.06.015
50. Vandevyver S, Dejager L, Libert C. On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic. (2012) 13:364–74. doi: 10.1111/j.1600-0854.2011.01288.x
51. Timmermans S, Souffriau J, Libert C. A general introduction to glucocorticoid biology. Front Immunol. (2019) 10:1545. doi: 10.3389/fimmu.2019.01545
52. Warrier M, Hinds TD, Ledford KJ, Cash HA, Patel PR, Bowman TA, et al. Susceptibility to diet-induced hepatic steatosis and glucocorticoid resistance in FK506-Binding protein 52-deficient mice. Endocrinology. (2010) 151:3225–36. doi: 10.1210/en.2009-1158
53. Okabe T, Chavan R, Fonseca Costa SS, Brenna A, Ripperger JA, Albrecht U. REV-ERBalpha influences the stability and nuclear localization of the glucocorticoid receptor. J Cell Sci. (2016) 129:4143–54. doi: 10.1242/jcs.190959
54. Winkler R, Benz V, Clemenz M, Bloch M, Foryst-Ludwig A, Wardat S, et al. Histone deacetylase 6 (HDAC6) is an essential modifier of glucocorticoid-induced hepatic gluconeogenesis. Diabetes. (2012) 61:513–23. doi: 10.2337/db11-0313
55. Keizer VIP, Coppola S, Houtsmuller AB, Geverts B, van Royen ME, Schmidt T, et al. Repetitive switching between DNA-binding modes enables target finding by the glucocorticoid receptor. J Cell Sci. (2019) 132:jcs.217455. doi: 10.1242/jcs.217455
56. McDowell IC, Barrera A, D'Ippolito AM, Vockley CM, Hong LK, Leichter SM, et al. Glucocorticoid receptor recruits to enhancers and drives activation by motif-directed binding. Genome Res. (2018) 28:1272–84. doi: 10.1101/gr.233346.117
57. Paakinaho V, Presman DM, Ball DA, Johnson TA, Schiltz RL, Levitt P, et al. Single-molecule analysis of steroid receptor and cofactor action in living cells. Nat Commun. (2017) 8:15896. doi: 10.1038/ncomms15896
58. Stavreva DA, Garcia DA, Fettweis G, Gudla PR, Zaki GF, Soni V, et al. Transcriptional bursting and co-bursting regulation by steroid hormone release pattern and transcription factor mobility. Mol Cell. (2019) 75:1161–77.e11. doi: 10.1016/j.molcel.2019.06.042
59. Stavreva DA, Wiench M, John S, Conway-Campbell BL, McKenna MA, Pooley JR, et al. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol. (2009) 11:1093–102. doi: 10.1038/ncb1922
60. Bahar Halpern K, Tanami S, Landen S, Chapal M, Szlak L, Hutzler A, et al. Bursty gene expression in the intact mammalian liver. Mol Cell. (2015) 58:147–56. doi: 10.1016/j.molcel.2015.01.027
61. Kalvisa A, Siersbaek MS, Praestholm SM, Christensen LJL, Nielsen R, Stohr O, et al. Insulin signaling and reduced glucocorticoid receptor activity attenuate postprandial gene expression in liver. PLos Biol. (2018) 16:e2006249. doi: 10.1371/journal.pbio.2006249
62. Cassuto H, Kochan K, Chakravarty K, Cohen H, Blum B, Olswang Y, et al. Glucocorticoids regulate transcription of the gene for phosphoenolpyruvate carboxykinase in the liver via an extended glucocorticoid regulatory unit. J Biol Chem. (2005) 280:33873–84. doi: 10.1074/jbc.M504119200
63. Lim HW, Uhlenhaut NH, Rauch A, Weiner J, Hubner S, Hubner N, et al. Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res. (2015) 25:836–44. doi: 10.1101/gr.188581.114
64. Presman DM, Ganguly S, Schiltz RL, Johnson TA, Karpova TS, Hager GL. DNA binding triggers tetramerization of the glucocorticoid receptor in live cells. Proc Natl Acad Sci USA. (2016) 113:8236–41. doi: 10.1073/pnas.1606774113
65. Schiller BJ, Chodankar R, Watson LC, Stallcup MR, Yamamoto KR. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol. (2014) 15:418. doi: 10.1186/s13059-014-0418-y
66. Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol. (2017) 18:159–74. doi: 10.1038/nrm.2016.152
67. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. (2011) 145:224–41. doi: 10.1016/j.cell.2011.03.027
68. Hudson WH, Kossmann BR, de Vera IM, Chuo SW, Weikum ER, Eick GN, et al. Distal substitutions drive divergent DNA specificity among paralogous transcription factors through subdivision of conformational space. Proc Natl Acad Sci USA. (2016) 113:326–31. doi: 10.1073/pnas.1518960113
69. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. (1998) 93:531–41. doi: 10.1016/S0092-8674(00)81183-6
70. Frijters R, Fleuren W, Toonen EJ, Tuckermann JP, Reichardt HM, van der Maaden H, et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genomics. (2010) 11:359. doi: 10.1186/1471-2164-11-359
71. Petta I, Dejager L, Ballegeer M, Lievens S, Tavernier J, De Bosscher K, et al. The interactome of the glucocorticoid receptor and its influence on the actions of glucocorticoids in combatting inflammatory and infectious diseases. Microbiol Mol Biol Rev. (2016) 80:495–522. doi: 10.1128/MMBR.00064-15
72. Grøntved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, et al. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. Embo J. (2013) 32:1568–83. doi: 10.1038/emboj.2013.106
73. Johnson TA, Chereji RV, Stavreva DA, Morris SA, Hager GL, Clark DJ. Conventional and pioneer modes of glucocorticoid receptor interaction with enhancer chromatin in vivo. Nucleic Acids Res. (2018) 46:203–14. doi: 10.1093/nar/gkx1044
74. D'Ippolito AM, McDowell IC, Barrera A, Hong LK, Leichter SM, Bartelt LC, et al. Pre-established chromatin interactions mediate the genomic response to glucocorticoids. Cell Syst. (2018) 7:146–60.e7. doi: 10.1016/j.cels.2018.06.007
75. Desmet SJ, de Bosscher K. Glucocorticoid receptors: finding the middle ground. J Clin Invest. (2017) 127:1136–45. doi: 10.1172/JCI88886
76. Hudson WH, Youn C, Ortlund EA. The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol. (2013) 20:53–8. doi: 10.1038/nsmb.2456
77. Weikum ER, de Vera IMS, Nwachukwu JC, Hudson WH, Nettles KW, Kojetin DJ, et al. Tethering not required: the glucocorticoid receptor binds directly to activator protein-1 recognition motifs to repress inflammatory genes. Nucleic Acids Res. (2017) 45:8596–608. doi: 10.1093/nar/gkx509
78. Johnson GD, Barrera A, McDowell IC, D'Ippolito AM, Majoros WH, Vockley CM, et al. Human genome-wide measurement of drug-responsive regulatory activity. Nat Commun. (2018) 9:5317. doi: 10.1038/s41467-018-07607-x
79. Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. Embo J. (1994) 13:4087–95. doi: 10.1002/j.1460-2075.1994.tb06726.x
80. Presman DM, Ogara MF, Stortz M, Alvarez LD, Pooley JR, Schiltz RL, et al. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PLoS Biol. (2014) 12:e1001813. doi: 10.1371/journal.pbio.1001813
81. Escoter-Torres L, Greulich F, Quagliarini F, Wierer M, Uhlenhaut NH. Anti-inflammatory functions of the glucocorticoid receptor require DNA binding. Nucleic Acids Res. (2020) 48:8393–407. doi: 10.1093/nar/gkaa565
82. Caratti G, Iqbal M, Hunter L, Kim D, Wang P, Vonslow RM, et al. REVERBa couples the circadian clock to hepatic glucocorticoid action. J Clin Invest. (2018) 128:4454–71. doi: 10.1172/JCI96138
83. John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. (2011) 43:264–8. doi: 10.1038/ng.759
84. Love MI, Huska MR, Jurk M, Schopflin R, Starick SR, Schwahn K, et al. Role of the chromatin landscape and sequence in determining cell type-specific genomic glucocorticoid receptor binding and gene regulation. Nucleic Acids Res. (2017) 45:1805–19. doi: 10.1093/nar/gkw1163
85. Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet. (2014) 15:69–81. doi: 10.1038/nrg3623
86. Burd CJ, Ward JM, Crusselle-Davis VJ, Kissling GE, Phadke D, Shah RR, et al. Analysis of chromatin dynamics during glucocorticoid receptor activation. Mol Cell Biol. (2012) 32:1805–17. doi: 10.1128/MCB.06206-11
87. Nagaich AK, Walker DA, Wolford R, Hager GL. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol Cell. (2004) 14:163–74. doi: 10.1016/S1097-2765(04)00178-9
88. Muratcioglu S, Presman DM, Pooley JR, Grontved L, Hager GL, Nussinov R, et al. Structural modeling of GR interactions with the SWI/SNF chromatin remodeling complex and C/EBP. Biophys J. (2015) 109:1227–39. doi: 10.1016/j.bpj.2015.06.044
89. Bartlett AA, Lapp HE, Hunter RG. Epigenetic mechanisms of the glucocorticoid receptor. Trends Endocrinol Metab. (2019) 30:807–18. doi: 10.1016/j.tem.2019.07.003
90. Chen W, Roeder RG. The mediator subunit MED1/TRAP220 is required for optimal glucocorticoid receptor-mediated transcription activation. Nucleic Acids Res. (2007) 35:6161–9. doi: 10.1093/nar/gkm661
91. Hill MJ, Suzuki S, Segars JH, Kino T. CRTC2 is a coactivator of gr and couples gr and creb in the regulation of hepatic gluconeogenesis. Mol Endocrinol. (2016) 30:104–17. doi: 10.1210/me.2015-1237
92. Suzuki S, Iben JR, Coon SL, Kino T. SIRT1 is a transcriptional enhancer of the glucocorticoid receptor acting independently to its deacetylase activity. Mol Cell Endocrinol. (2018) 461:178–87. doi: 10.1016/j.mce.2017.09.012
93. Ananthanarayanan M, Li Y, Surapureddi S, Balasubramaniyan N, Ahn J, Goldstein JA, et al. Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is downregulated in cholestasis. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G771–81. doi: 10.1152/ajpgi.00499.2010
94. Roqueta-Rivera M, Esquejo RM, Phelan PE, Sandor K, Daniel B, Foufelle F, et al. SETDB2 links glucocorticoid to lipid metabolism through insig2a regulation. Cell Metab. (2016) 24:474–84. doi: 10.1016/j.cmet.2016.07.025
95. Ki SH, Cho IJ, Choi DW, Kim SG. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol Cell Biol. (2005) 25:4150–65. doi: 10.1128/MCB.25.10.4150-4165.2005
96. Qiu Y, Zhao Y, Becker M, John S, Parekh BS, Huang S, et al. HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Mol Cell. (2006) 22:669–79. doi: 10.1016/j.molcel.2006.04.019
97. Lu Y, Zhang Z, Xiong X, Wang X, Li J, Shi G, et al. Glucocorticoids promote hepatic cholestasis in mice by inhibiting the transcriptional activity of the farnesoid X receptor. Gastroenterology. (2012) 143:1630–40.e8. doi: 10.1053/j.gastro.2012.08.029
98. Ichijo T, Voutetakis A, Cotrim AP, Bhattachryya N, Fujii M, Chrousos GP, et al. The Smad6-histone deacetylase 3 complex silences the transcriptional activity of the glucocorticoid receptor: potential clinical implications. J Biol Chem. (2005) 280:42067–77. doi: 10.1074/jbc.M509338200
99. Lamia KA, Papp SJ, Yu RT, Barish GD, Uhlenhaut NH, Jonker JW, et al. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. (2011) 480:552–6. doi: 10.1038/nature10700
100. Xu S, Liu Y, Hu R, Wang M, Stöhr O, Xiong Y, et al. TAZ inhibits GR and coordinates hepatic glucose homeostasis in normal physiologic states. bioRxiv. (2020). doi: 10.1101/2020.05.12.091264
101. Stortz M, Pecci A, Presman DM, Levi V. Unraveling the molecular interactions involved in phase separation of glucocorticoid receptor. BMC Biol. (2020) 18:59. doi: 10.1186/s12915-020-00788-2
102. Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA. A phase separation model for transcriptional control. Cell. (2017) 169:13–23. doi: 10.1016/j.cell.2017.02.007
103. Stavreva DA, Coulon A, Baek S, Sung MH, John S, Stixova L, et al. Dynamics of chromatin accessibility and long-range interactions in response to glucocorticoid pulsing. Genome Res. (2015) 25:845–57. doi: 10.1101/gr.184168.114
104. Hakim O, Sung MH, Voss TC, Splinter E, John S, Sabo PJ, et al. Diverse gene reprogramming events occur in the same spatial clusters of distal regulatory elements. Genome Res. (2011) 21:697–706. doi: 10.1101/gr.111153.110
105. Schule R, Muller M, Kaltschmidt C, Renkawitz R. Many transcription factors interact synergistically with steroid receptors. Science. (1988) 242:1418–20. doi: 10.1126/science.3201230
106. De Martino MU, Bhattachryya N, Alesci S, Ichijo T, Chrousos GP, Kino T. The glucocorticoid receptor and the orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II interact with and mutually affect each other's transcriptional activities: implications for intermediary metabolism. Mol Endocrinol. (2004) 18:820–33. doi: 10.1210/me.2003-0341
107. Goldstein I, Baek S, Presman DM, Paakinaho V, Swinstead EE, Hager GL. Transcription factor assisted loading and enhancer dynamics dictate the hepatic fasting response. Genome Res. (2017) 27:427–39. doi: 10.1101/gr.212175.116
108. Everett LJ, Le Lay J, Lukovac S, Bernstein D, Steger DJ, Lazar MA, et al. Integrative genomic analysis of CREB defines a critical role for transcription factor networks in mediating the fed/fasted switch in liver. BMC Genomics. (2013) 14:337. doi: 10.1186/1471-2164-14-337
109. Zhang L, Rubins NE, Ahima RS, Greenbaum LE, Kaestner KH. Foxa2 integrates the transcriptional response of the hepatocyte to fasting. Cell Metab. (2005) 2:141–8. doi: 10.1016/j.cmet.2005.07.002
110. Hemmer MC, Wierer M, Schachtrup K, Downes M, Hubner N, Evans RM, et al. E47 modulates hepatic glucocorticoid action. Nat Commun. (2019) 10:306. doi: 10.1038/s41467-018-08196-5
111. Kuo T, Chen TC, Yan S, Foo F, Ching C, McQueen A, et al. Repression of glucocorticoid-stimulated angiopoietin-like 4 gene transcription by insulin. J Lipid Res. (2014) 55:919–28. doi: 10.1194/jlr.M047860
112. Nader N, Ng SS, Wang Y, Abel BS, Chrousos GP, Kino T. Liver x receptors regulate the transcriptional activity of the glucocorticoid receptor: implications for the carbohydrate metabolism. PLoS ONE. (2012) 7:e26751. doi: 10.1371/journal.pone.0026751
113. Patel R, Patel M, Tsai R, Lin V, Bookout AL, Zhang Y, et al. LXRbeta is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J Clin Invest. (2011) 121:431–41. doi: 10.1172/JCI41681
114. Ratman D, Mylka V, Bougarne N, Pawlak M, Caron S, Hennuyer N, et al. Chromatin recruitment of activated AMPK drives fasting response genes co-controlled by GR and PPARalpha. Nucleic Acids Res. (2016) 44:10539–53. doi: 10.1093/nar/gkw742
115. Murayama Y, Yahagi N, Takeuchi Y, Aita Y, Mehrazad Saber Z, Wada N, et al. Glucocorticoid receptor suppresses gene expression of Rev-erbalpha (Nr1d1) through interaction with the CLOCK complex. FEBS Lett. (2019) 593:423–32. doi: 10.1002/1873-3468.13328
116. Kriebs A, Jordan SD, Soto E, Henriksson E, Sandate CR, Vaughan ME, et al. Circadian repressors CRY1 and CRY2 broadly interact with nuclear receptors and modulate transcriptional activity. Proc Natl Acad Sci USA. (2017) 114:8776–81. doi: 10.1073/pnas.1704955114
117. Bailly A, Briançon N, Weiss MC. Characterization of glucocorticoid receptor and hepatocyte nuclear factor 4α (HNF4α) binding to the hnf4α gene in the liver. Biochimie. (2009) 91:1095–103. doi: 10.1016/j.biochi.2009.06.009
118. Arenas F, Hervias I, Úriz M, Joplin R, Prieto J, Medina JF. Combination of ursodeoxycholic acid and glucocorticoids upregulates the AE2 alternate promoter in human liver cells. J Clin Invest. (2008) 118:695–709. doi: 10.1172/JCI33156
119. Engblom D, Kornfeld JW, Schwake L, Tronche F, Reimann A, Beug H, et al. Direct glucocorticoid receptor-Stat5 interaction in hepatocytes controls body size and maturation-related gene expression. Genes Dev. (2007) 21:1157–62. doi: 10.1101/gad.426007
120. Tronche F, Opherk C, Moriggl R, Kellendonk C, Reimann A, Schwake L, et al. Glucocorticoid receptor function in hepatocytes is essential to promote postnatal body growth. Genes Dev. (2004) 18:492–7. doi: 10.1101/gad.284704
121. Freeman BC, Yamamoto KR. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science. (2002) 296:2232. doi: 10.1126/science.1073051
122. Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. (2011) 43:145–55. doi: 10.1016/j.molcel.2011.06.016
123. Sato S, Shirakawa H, Tomita S, Tohkin M, Gonzalez FJ, Komai M. The aryl hydrocarbon receptor and glucocorticoid receptor interact to activate human metallothionein 2A. Toxicol Appl Pharmacol. (2013) 273:90–9. doi: 10.1016/j.taap.2013.08.017
124. Wang SH, Liang CT, Liu YW, Huang MC, Huang SC, Hong WF, et al. Crosstalk between activated forms of the aryl hydrocarbon receptor and glucocorticoid receptor. Toxicology. (2009) 262:87–97. doi: 10.1016/j.tox.2009.03.020
125. Fruchter O, Kino T, Zoumakis E, Alesci S, De Martino M, Chrousos G, et al. The human glucocorticoid receptor (GR) isoform {beta} differentially suppresses GR{alpha}-induced transactivation stimulated by synthetic glucocorticoids. J Clin Endocrinol Metab. (2005) 90:3505–9. doi: 10.1210/jc.2004-1646
126. Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. (1995) 95:2435–41. doi: 10.1172/JCI117943
127. Scheschowitsch K, Leite JA, Assreuy J. New insights in glucocorticoid receptor signaling-more than just a ligand-binding receptor. Front Endocrinol. (2017) 8:16. doi: 10.3389/fendo.2017.00016
128. Marino JS, Stechschulte LA, Stec DE, Nestor-Kalinoski A, Coleman S, Hinds TD Jr. Glucocorticoid receptor beta induces hepatic steatosis by augmenting inflammation and inhibition of the peroxisome proliferator-activated receptor (PPAR) alpha. J Biol Chem. (2016) 291:25776–88. doi: 10.1074/jbc.M116.752311
129. He B, Cruz-Topete D, Oakley RH, Xiao X, Cidlowski JA. Human glucocorticoid receptor beta regulates gluconeogenesis and inflammation in mouse liver. Mol Cell Biol. (2015) 36:714–30. doi: 10.1128/MCB.00908-15
130. Mueller KM, Kornfeld JW, Friedbichler K, Blaas L, Egger G, Esterbauer H, et al. Impairment of hepatic growth hormone and glucocorticoid receptor signaling causes steatosis and hepatocellular carcinoma in mice. Hepatology. (2011) 54:1398–409. doi: 10.1002/hep.24509
131. Pooley JR, Rivers CA, Kilcooley MT, Paul SN, Cavga AD, Kershaw YM, et al. Beyond the heterodimer model for mineralocorticoid and glucocorticoid receptor interactions in nuclei and at DNA. PLoS ONE. (2020) 15:e0227520. doi: 10.1371/journal.pone.0227520
132. Mifsud KR, Reul JM. Acute stress enhances heterodimerization and binding of corticosteroid receptors at glucocorticoid target genes in the hippocampus. Proc Natl Acad Sci USA. (2016) 113:11336–41. doi: 10.1073/pnas.1605246113
133. Liu G, Grifman M, Keily B, Chatterton JE, Staal FW, Li QX. Mineralocorticoid receptor is involved in the regulation of genes responsible for hepatic glucose production. Biochem Biophys Res Commun. (2006) 342:1291–6. doi: 10.1016/j.bbrc.2006.02.065
134. Phair RD, Scaffidi P, Elbi C, Vecerova J, Dey A, Ozato K, et al. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol Cell Biol. (2004) 24:6393–402. doi: 10.1128/MCB.24.14.6393-6402.2004
135. Voss TC, Schiltz RL, Sung MH, Yen PM, Stamatoyannopoulos JA, Biddie SC, et al. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell. (2011) 146:544–54. doi: 10.1016/j.cell.2011.07.006
136. Wang Q, Blackford JA Jr, Song LN, Huang Y, Cho S, Simons SS Jr. Equilibrium interactions of corepressors and coactivators with agonist and antagonist complexes of glucocorticoid receptors. Mol Endocrinol. (2004) 18:1376–95. doi: 10.1210/me.2003-0421
137. Præstholm SM, Siersbæk MS, Nielsen R, Zhu X, Hollenberg AN, Cheng S-y, et al. Multiple mechanisms regulate H3 acetylation of enhancers in response to thyroid hormone. PLOS Genetics. (2020) 16:e1008770. doi: 10.1371/journal.pgen.1008770
138. Reddy AB, Maywood ES, Karp NA, King VM, Inoue Y, Gonzalez FJ, et al. Glucocorticoid signaling synchronizes the liver circadian transcriptome. Hepatology. (2007) 45:1478–88. doi: 10.1002/hep.21571
139. Yamamoto T, Nakahata Y, Tanaka M, Yoshida M, Soma H, Shinohara K, et al. Acute physical stress elevates mouse period1 mRNA expression in mouse peripheral tissues via a glucocorticoid-responsive element. J Biol Chem. (2005) 280:42036–43. doi: 10.1074/jbc.M509600200
140. Lu Y, Xiong X, Wang X, Zhang Z, Li J, Shi G, et al. Yin Yang 1 promotes hepatic gluconeogenesis through upregulation of glucocorticoid receptor. Diabetes. (2013) 62:1064–73. doi: 10.2337/db12-0744
141. Cui A, Fan H, Zhang Y, Zhang Y, Niu D, Liu S, et al. Dexamethasone-induced Kruppel-like factor 9 expression promotes hepatic gluconeogenesis and hyperglycemia. J Clin Invest. (2019) 129:2266–78. doi: 10.1172/JCI66062
142. Li R, Jia Y, Pan S, Li X, Song H, Zhao R. Glucocorticoid receptor mediates the effect of high-fat diet on mitochondrial oxidative phosphorylation in mouse liver. DNA Cell Biol. (2016) 35:51–8. doi: 10.1089/dna.2015.2932
143. Okun JG, Conway S, Schmidt KV, Schumacher J, Wang X, de Guia R, et al. Molecular regulation of urea cycle function by the liver glucocorticoid receptor. Mol Metab. (2015) 4:732–40. doi: 10.1016/j.molmet.2015.07.006
144. Rose AJ, Berriel Diaz M, Reimann A, Klement J, Walcher T, Krones-Herzig A, et al. Molecular control of systemic bile acid homeostasis by the liver glucocorticoid receptor. Cell Metab. (2011) 14:123–30. doi: 10.1016/j.cmet.2011.04.010
145. Bose SK, Hutson I, Harris CA. Hepatic glucocorticoid receptor plays a greater role than adipose GR in metabolic syndrome despite renal compensation. Endocrinology. (2016) 157:4943–60. doi: 10.1210/en.2016-1615
146. Opherk C, Tronche F, Kellendonk C, Kohlmüller D, Schulze A, Schmid W, et al. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol. (2004) 18:1346–53. doi: 10.1210/me.2003-0283
147. Langlet F, Haeusler RA, Linden D, Ericson E, Norris T, Johansson A, et al. Selective inhibition of FOXO1 activator/repressor balance modulates hepatic glucose handling. Cell. (2017) 171:824–35.e18. doi: 10.1016/j.cell.2017.09.045
148. Chen J, Zhang Z, Wang N, Guo M, Chi X, Pan Y, et al. Role of HDAC9-FoxO1 axis in the transcriptional program associated with hepatic gluconeogenesis. Sci Rep. (2017) 7:6102. doi: 10.1038/s41598-017-06328-3
149. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular actions of PPARalpha in lipid metabolism and inflammation. Endocr Rev. (2018) 39:760–802. doi: 10.1210/er.2018-00064
150. Bougarne N, Paumelle R, Caron S, Hennuyer N, Mansouri R, Gervois P, et al. PPARalpha blocks glucocorticoid receptor alpha-mediated transactivation but cooperates with the activated glucocorticoid receptor alpha for transrepression on NF-kappaB. Proc Natl Acad Sci USA. (2009) 106:7397–402. doi: 10.1073/pnas.0806742106
151. Baik M, Yu JH, Hennighausen L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann N Y Acad Sci. (2011) 1229:29–37. doi: 10.1111/j.1749-6632.2011.06100.x
152. Wu T, Jiang J, Yang L, Li H, Zhang W, Chen Y, et al. Timing of glucocorticoid administration determines severity of lipid metabolism and behavioral effects in rats. Chronobiol Int. (2017) 34:78–92. doi: 10.1080/07420528.2016.1238831
153. Delezie J, Dumont S, Dardente H, Oudart H, Grechez-Cassiau A, Klosen P, et al. The nuclear receptor REV-ERBalpha is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. (2012) 26:3321–35. doi: 10.1096/fj.12-208751
154. Zhang Y, Fang B, Emmett MJ, Damle M, Sun Z, Feng D, et al. GENE REGULATION. Discrete functions of nuclear receptor Rev-erbalpha couple metabolism to the clock. Science. (2015) 348:1488–92. doi: 10.1126/science.aab3021
155. Zhang Y, Fang B, Damle M, Guan D, Li Z, Kim YH, et al. HNF6 and Rev-erbalpha integrate hepatic lipid metabolism by overlapping and distinct transcriptional mechanisms. Genes Dev. (2016) 30:1636–44. doi: 10.1101/gad.281972.116
156. Torra IP, Tsibulsky V, Delaunay F, Saladin R, Laudet V, Fruchart JC, et al. Circadian and glucocorticoid regulation of Rev-erbalpha expression in liver. Endocrinology. (2000) 141:3799–806. doi: 10.1210/endo.141.10.7708
157. Greulich F, Hemmer MC, Rollins DA, Rogatsky I, Uhlenhaut NH. There goes the neighborhood: assembly of transcriptional complexes during the regulation of metabolism and inflammation by the glucocorticoid receptor. Steroids. (2016) 114:7–15. doi: 10.1016/j.steroids.2016.05.003
158. Buttgereit F, Doering G, Schaeffler A, Witte S, Sierakowski S, Gromnica-Ihle E, et al. Efficacy of modified-release versus standard prednisone to reduce duration of morning stiffness of the joints in rheumatoid arthritis (CAPRA-1): a double-blind, randomised controlled trial. Lancet. (2008) 371:205–14. doi: 10.1016/S0140-6736(08)60132-4
Keywords: Glucocorticoid receptor, chromatin, transcription, metabolism, liver
Citation: Præstholm SM, Correia CM and Grøntved L (2020) Multifaceted Control of GR Signaling and Its Impact on Hepatic Transcriptional Networks and Metabolism. Front. Endocrinol. 11:572981. doi: 10.3389/fendo.2020.572981
Received: 15 June 2020; Accepted: 03 September 2020;
Published: 08 October 2020.
Edited by:
Nicolas Venteclef, Sorbonne Universités, FranceReviewed by:
Andrew C. B. Cato, Karlsruhe Institute of Technology (KIT), GermanyDino Moras, INSERM U964 Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), France
Copyright © 2020 Præstholm, Correia and Grøntved. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lars Grøntved, bGFyc2dyQGJtYi5zZHUuZGs=
†These authors have contributed equally to this work