 Bianca C. Field
Bianca C. Field Ruth Gordillo
Ruth Gordillo Philipp E. Scherer
Philipp E. Scherer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 02 October 2020
Sec. Diabetes: Molecular Mechanisms
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.569250
This article is part of the Research Topic The Role of Ceramides in Diabetes and Cardiovascular Disease View all 8 articles
Metabolic dysfunction is intertwined with the pathophysiology of both diabetes and cardiovascular disease. Recently, one particular lipid class has been shown to influence the development and sustainment of these diseases: ceramides. As a subtype of sphingolipids, these species are particularly central to many sphingolipid pathways. Increased levels of ceramides are known to correlate with impaired cardiovascular and metabolic health. Furthermore, the interaction between ceramides and adipokines, most notably adiponectin and leptin, appears to play a role in the pathophysiology of these conditions. Adiponectin appears to counteract the detrimental effects of elevated ceramides, largely through activation of the ceramidase activity of its receptors. Elevated ceramides appear to worsen leptin resistance, which is an important phenomenon in the pathophysiology of obesity and metabolic syndrome.
Sphingolipids are by far the most structurally diverse class of lipid molecules. The structural complexity of sphingolipids stems from variation in three components of these lipids: a long chain base, an amide-linked fatty acid, and a head group (20). They are signaling molecules involved in diverse cellular functions, from control of the cell cycle to degradation of plasma membrane proteins (21). They are involved in the most devastating inborn errors and degenerative diseases, including cancer, Alzheimer’s disease and dementia (22). In recent years, ceramides, a subgroup of sphingolipids, have received increased attention for their role in myriad pathophysiologic mechanisms, including those underlying cancer (23–25), inflammation (26), depression (27), and neurodegenerative disorders (28). Additionally, more recent studies have highlighted their increasingly important role in obesity and diabetes (9, 29–31) as well as cardiovascular disease (CVD) (32). Preventing aberrant ceramide deposition can prevent or ameliorate a variety of cardiometabolic pathologies, including insulin resistance, CVD (atherosclerosis, heart failure), and hepatic steatosis (33, 34), as well as mitochondrial dysfunction (Figure 1). Here, we will focus on ceramides’ role in diabetes and CVD, two of the leading causes of death in the United States (35) and worldwide (36).
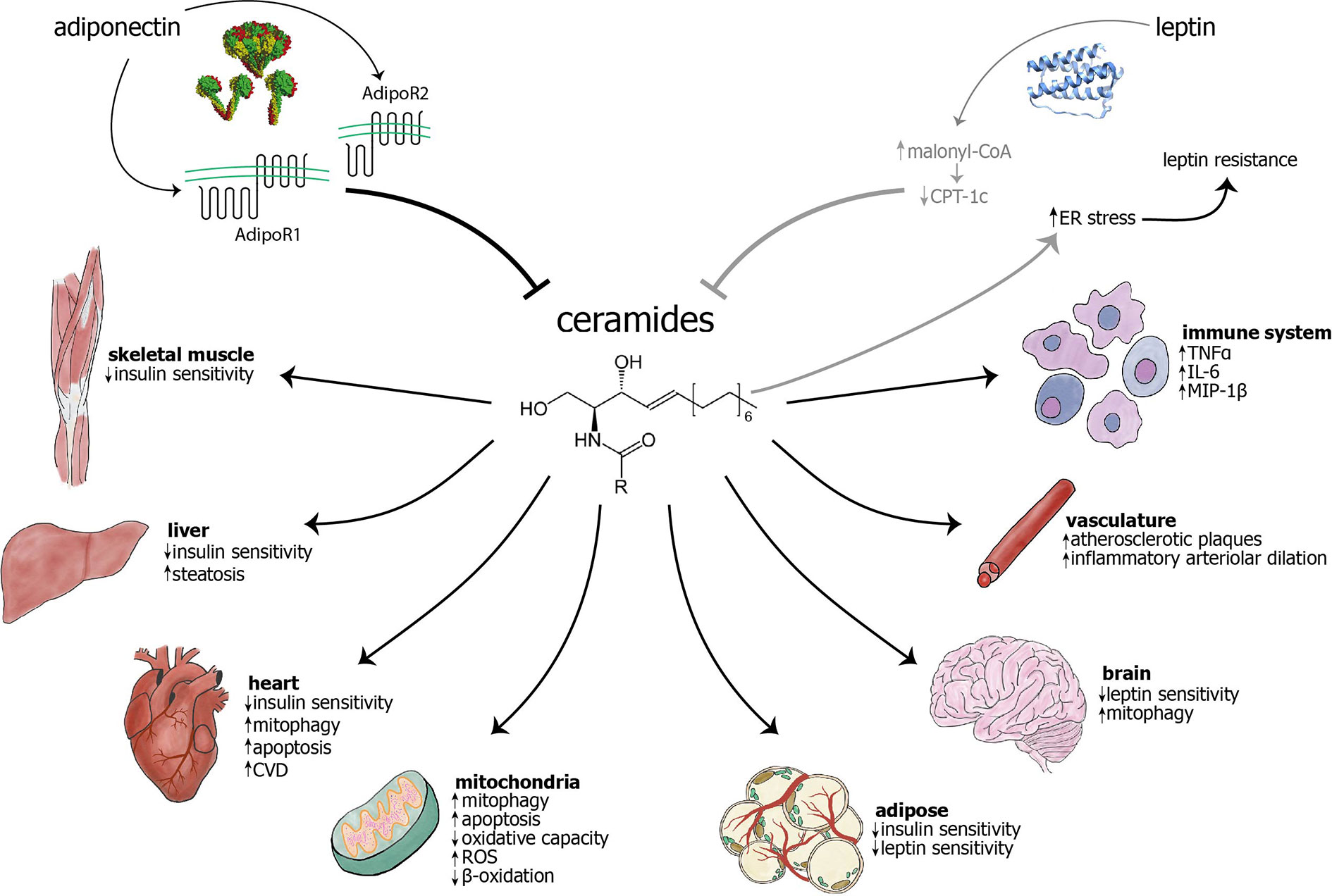
Figure 1 The adipokines adiponectin and leptin have largely inhibitory effects on ceramide levels, while ceramides are known to cause myriad effects in cellular components, different cell types, and organs. Adiponectin inhibits ceramides through the intrinsic ceramidase activity of its receptors (1), AdipoR1 and AdipoR2. The mechanism of leptin’s action on ceramides remains unknown, although it has been hypothesized that leptin may exert its influence through inhibition of carnitine palmitoyltransferase-1c (CPT-1c) by malonyl-CoA (2). Conversely, ceramide accumulation may cause ER stress and subsequent leptin resistance (3–6). The previous two relationships are visualized in gray to indicate that they have not been extensively studied and characterized. Ceramides are known to act on mitochondria and cause increased mitophagy, apoptosis, and generation of reactive oxygen species (ROS), as well as decreased β-oxidation and overall oxidative capacity. Ceramides have been shown to cause mitophagy in cardiomyocytes and neurons, among others (not shown) (7). Ceramides decrease insulin sensitivity in skeletal muscle, liver, heart, and adipose (8, 9). Their accumulation may cause decreased leptin sensitivity in the brain and adipose tissue. In the vasculature, ceramide accumulation leads to increased atherosclerotic plaques and inflammatory arteriolar dilation (10–13). They can also lead to increased cardiovascular disease (14, 15), hepatic steatosis (16), and increased inflammation in immune cells (17). As opposed to other targets, skin largely benefits from increased ceramides, with increased cell differentiation and wound healing, as well as decreased senescence (18, 19). AdipoR, adiponectin receptor; CPT-1c, carnitine palmitoyltranferase 1c; CVD, cardiovascular disease; ER, endoplasmic reticulum; IL-6, interleukin-6; MIP-1, macrophage inflammatory protein 1β; ROS, reactive oxygen species; TNFα, tumor necrosis factor α.
Ceramides constitute a simple subtype of sphingolipids composed of a variable-length fatty acid and an amino group (a sphingoid base, usually sphingosine). Their unique structure, highly hydrophobic with a small side chain, enable them to function well both as a structural membrane component and signaling molecule (20, 37). Under physiologic conditions, they range from short chain (C6:0) to very long chain (VLC) (C26:0), although longer chain ceramides are more commonly found in mammalian cells. Ceramide biosynthesis can occur through three distinct pathways: de novo, sphingomyelin hydrolysis, or sphingolipid salvage. De novo synthesis occurs in the endoplasmic reticulum (ER) and involves ceramide synthases (CerS) as well as serine palmitoyl-transferase (SPT), which catalyzes the rate-limiting step and is inhibited by the drug myriocin. In another pathway, sphingomyelinase (SMase) hydrolyzes sphingomyelin to form ceramide. Finally, in the salvage pathway, alkaline ceramidase (ACER), which is inhibited by the drug D-erythro-MAPP, and acid ceramidase (AC) work in the endolysosomal system to regenerate ceramide from sphingosine (38, 39).
Sphingomyelinases and ceramidases exist in three forms: acid, neutral, and alkaline. Their subtype depends on their subcellular localization, function, and optimal pH. Generally, acid SMases and ceramidases function in lysosomes, neutral enzymes along the plasma membrane, and alkaline enzymes in the Golgi or ER (40). Furthermore, CerSs, important in both de novo and salvage pathways, preferentially catalyze certain fatty acyl-CoA substrates in order to make different ceramide subtypes (39, 40). Because ceramides are the center of a web of sphingolipid biosynthesis reactions, it can be difficult to prove causation between particular metabolites and enzymes (38).
Dihydroceramides are the precursor metabolites of ceramides in the de novo sphingolipid biosynthesis pathway. CerS form dihydroceramides by condensing sphinganine and acyl-CoA, and they are a family of six isoforms (CerS1-6) that each display specificity for certain acyl-CoA chain lengths. Ceramides are formed from dihydroceramides by the action of the enzyme dihydroceramide desaturase (41, 42).
Ceramides are key to the structural integrity the plasma and mitochondrial membranes. While they are restricted to the membrane in which they are formed, they can flip-flop between membrane leaflets (43), although this movement occurs slowly (44). This incorporation can change membrane configuration and relative hydrophobicity of the plasma membrane-cytosol interface, changing the affinity for the membrane of a number of proteins, which in turn affect the activity of membrane enzymes (45).
Clinical studies show that circulating ceramides primarily originate in the liver, a major site of de novo ceramide synthesis (17, 46), although adipocytes may also contribute to circulating ceramides in some species (33). They circulate at a concentration of about 10μM and are primarily transported in circulation by lipoproteins (17, 46, 47), while exosomal transport may also significantly contribute (48). One study, using thin layer and high-performance liquid chromatography (TLC and HPLC, respectively), found that plasma ceramides are primarily (~80%) carried by very-low-density lipoprotein (VLDL) and low-density lipoprotein (LDL), with the remaining ceramide being carried by albumin (~15%) and high-density lipoprotein (HDL; ~5%) (46). Another study, using fast performance liquid chromatography (FPLC), separated lipoprotein fractions and found that ceramides were primarily carried by LDL (60.3 +/−6.7%) and to a lesser extent by HDL (24.1+/−7.4%), and VLDL (15.6+/−9.9%). However, ceramides comprised a small fraction of total lipids carried by these lipoproteins (LDL and VLDL ~0.6%; HDL ~0.1%). While the distribution of ceramide species carried by these lipoprotein classes were largely similar, LDL and VLDL carried more similar proportions of specific ceramide species (less C16:0, more C24:0), while HDL carried others (more C16:0, less C24:0) (47). Additionally, recent research has found that ceramide species are also transported in exosomes from adipocytes and endothelial cells (48).
Ceramides are a normal component of mitochondrial membranes, and they are necessary for normal mitochondrial function. Purified mitochondria and mitochondria-associated membranes have been shown to generate ceramides in vitro, and specific ceramide synthases (CerS2, CerS4, and CerS6) are present in the inner and outer mitochondrial membranes (49). Furthermore, normal biological mechanisms can be mediated by ceramides. For instance, CerS6-generated ceramides may mediate pruning of oligodendrocytes during normal brain development (50).
While ceramides are necessary for normal mitochondrial functions, accumulation of excess ceramides have been shown to induce mitochondrial dysfunction (51, 52). Increased mitochondrial ceramide content has been shown to have numerous deleterious effects, including inhibition of mitochondrial oxidative capacity, depletion of ATP, inhibition of the electron transport chain, and enhanced generation of reactive oxygen species (51, 53–55). Additionally, studies show that ceramides can alter or nearly eliminate the respiration-dependent mitochondrial membrane potential (51, 56, 57). They disrupt the normal structure of the inner and outer mitochondrial membranes, increasing their permeability independent of cytochrome c and electron transfer activity (51). One study in a mouse model of ischemia showed that ceramide accumulation leads to inhibition of the respiratory chain (58). In mouse models of diet-induced obesity (DIO), pharmacologic or genetic depletion of ceramides increases mitochondrial complex activity [complex IV in subcutaneous white adipose tissue (sWAT), complexes II and IV in brown adipose tissue (BAT)], as well as respiration in sWAT, BAT, and brain (59). Pharmacological depletion of ceramides also decreased the respiratory exchange ratio (RER) in models of DIO (60). Several ceramide lipid storage diseases, including Fabry disease and neuronal ceroid lipofuscinosis, are associated with dysfunction in the mitochondrial respiratory chain (7). Other sphingolipid storage diseases cause inhibition of mitochondrial biogenesis (61).
Beyond causing mitochondrial dysfunction, ceramides are known to regulate mitophagy (62). Accumulation of ceramide species in yeast led to increased mitochondrial fission and mitophagy (63). Similarly, in mouse and human cardiomyocytes, VLC ceramides produced by CerS2 led to increased mitophagy, along with increased oxidative stress and mitochondrial dysfunction (56). Ceramide-induced mitophagy can also help explain some pathophysiology and treatment mechanisms. For example, one study found that cigarette smoke caused mitophagy through increased levels of C16 ceramide, triggering necroptosis (64). Another study found that in glioblastoma cells, a plant agglutinin that is used to inhibit glioblastoma growth induced mitophagy-dependent cell death via increased ceramides (65).
Ceramides also have an established role in the induction of apoptosis (66), and their effects on mitochondria appear to be important in this process (52, 67). Apoptotic stimuli activate CerS in the mitochondrial membranes, indicating a role for ceramides in mitochondria-induced apoptosis (53). Mitochondria can function as sensors for cellular stress by monitoring local levels of specific lipids, including sphingolipids (45). This function facilitates the intrinsic apoptotic pathway. Formation of ceramide channels on the outer mitochondrial membrane leads to apoptosis, with ceramide species C16, C18, and C20 being most important in this process (52). Ceramides also work synergistically with the apoptotic protein Bax to induce mitochondrial outer membrane permeabilization in yeast (55). Furthermore, C2 ceramide was found to alter mitochondrial membrane potential and cause mitochondrial fragmentation, leading to apoptosis in cultured cardiomyocytes (57). Some interventions, including overexpression of Bcl-2 (68–71) and pharmacological inhibition of mitochondrial membrane depolarization (72, 73), have also prevented ceramide-induced apoptosis through prevention of mitochondrial dysfunction (74).
Ceramides may need to localize specifically to mitochondria to induce apoptosis. One group found that increased ceramides due to sphingomyelinase overexpression only induced apoptosis if the enzyme was targeted to mitochondria, but not if it was targeted to other cell compartments, including the inner plasma membrane, cytoplasm, ER, Golgi, and nucleus. Despite a resultant increase in whole-cell ceramide content, there was no induction of apoptosis when increased ceramides were targeted to these compartments (75). Furthermore, another group found that specific ceramide enzymes localize to the inner and outer membranes, respectively, suggesting that there exist distinct pools of ceramides (76).
Development of insulin resistance is a key step in the progression of metabolic syndrome, obesity, and diabetes, and it occurs when increasing levels of insulin are required to stimulate insulin-induced glucose uptake, particularly in myocytes and adipocytes. Studies have shown that plasma, adipose, and skeletal muscle ceramides are elevated in patients with obesity and type 2 diabetes mellitus (T2DM) (17, 77, 78), as well as in adipose, hepatic, skeletal muscle, and plasma samples from animal models of insulin resistance (17, 79–81). Moreover, ceramide levels in plasma, adipose, liver, and skeletal muscle negatively correlate with severity of insulin sensitivity in humans and in animal models (17, 77, 78, 81, 82). This is supported by the evidence that ceramides decrease the activity of Akt, which is a downstream effector of insulin signaling as well as of apoptosis and cell proliferation (24). Furthermore, adipose ceramide levels are higher in patients with increased hepatic lipid accumulation than in those with healthy livers (77). Reduced ceramide content prevents lipotoxicity induced by ceramide accumulation, preserving insulin and glucagon secretion (83). Ceramide bound to LDL also increases insulin resistance in skeletal myocytes and increases expression of inflammatory genes in macrophages, including IL-6 and TNFα (17). Clinical studies further prove that skeletal muscle ceramide content negatively correlates with insulin sensitivity (82, 84).
Relative amounts of particular ceramide species in insulin resistant states vary among sources. In one clinical study, skeletal muscle ceramides C16:0 and C18:3 were most strongly negatively correlated with insulin sensitivity (82). However, another clinical study showed that plasma C18:0 was most increased in obese T2DM participants over their healthy controls, but C24:0 was most abundant (78). More research is needed to clarify the relative impacts of specific ceramide species on insulin resistance.
Work by Summers and colleagues shows that ceramide production is an important factor in the development of insulin resistance through glucocorticoids and saturated fatty acids, but not through unsaturated fatty acids (16, 85). Glucocorticoids induce production of sphingolipid metabolites by promoting expression of the enzymes AC and glucosylceramide synthase (GCS), which produce ceramides through the salvage pathway. While Holland et al. did not measure or report an increase in acid sphingomyelinase activity, Cinque et al. demonstrated that dexamethasone, a glucocorticoid, induced thymocyte apoptosis by sequential activation of nuclear phosphoinositide specific phospholipase C-beta (PI-PLCβ) and acid sphingomyelinase (aSMase). Therefore, increased activity of both AC and aSMase, induced by glucocorticoids, could cause increased ceramide levels (86, 87). Saturated fatty acids promote ceramide accumulation in liver and skeletal muscle, as well as induction of insulin resistance (16). Conversely, reduction of ceramides has been shown to reverse the metabolic consequences of ceramide deposition in tissues. Induced overexpression of AC in mice, under the control of the adiponectin promoter, led to decreased ceramides in visceral and subcutaneous fat, liver, and serum, particularly ceramide species C16:0 and C18:0 (16, 88). After induction of AC expression, mice had reduced hepatic steatosis and improved insulin signaling and glucose metabolism, as shown by improvements in oral glucose tolerance tests and insulin tolerance tests, as well as increased glucose infusion rates and decreased hepatic glucose output during hyperinsulinemic-euglycemic clamps (88).
Ceramides’ induction of mitochondrial dysfunction appears to play a role in the development of metabolic syndrome and its associated conditions. Several studies specifically implicated ceramides produced by the enzyme ceramide synthase 6 (CerS6). One group found that ob/ob mice, which congenitally lack leptin expression, have elevated CerS6 in their liver, BAT, and sWAT, but not gonadal white adipose tissue (gWAT) (89), and another found that C16:0 ceramide from CerS6 inhibits mitochondrial beta-oxidation in BAT and liver (90). Another study found that in mice with high-fat diet (HFD)–induced weight gain, only CerS6 increased in mitochondria and mitochondria-associated membranes, not ceramide synthase 5 (CerS5). CerS6 also interacts with mitochondrial fission factor (MFF) to promote mitochondrial fission, and knockout of either of these proteins normalizes mitochondrial structure altered by HFD (91). In ob/ob and DIO mice, but not in db/db mice, which lack the leptin receptor, Raichur et al. found that these mice have elevated C16:0 ceramide in liver and plasma. They also found that these ablation of CerS6 and subsequent lowering of C16:0 ceramide by 50% in ob/ob and DIO mice corrected high blood glucose and insulin resistance, and also caused 25% weight loss without a change in food intake (89).
Evidence also points to roles for other CerS in metabolic dysfunction. In diabetic cardiomyopathy, ceramide synthase 2 (CerS2)- and CerS5-derived ceramides, including VLC ceramides, led to increased mitophagy and insulin resistance (56). In DIO mice, ceramide synthase 1 (CerS1) and C18:0 are elevated in skeletal muscle, and knockout of CerS1 globally as well as specifically from skeletal muscle improves insulin sensitivity through increased fibroblast growth factor 21 (FGF21) in muscle (92). The role of ceramides in metabolic syndrome will be discussed further in later sections examining the interaction of ceramides with adiponectin and leptin, respectively.
Ceramides have a well-established role in the development of CVDs, including atherosclerosis (15, 93), cardiomyopathy (15, 94, 95), heart failure (15, 96), myocardial infarction (14, 15), and stroke (14, 15). In particular, dihydroceramides, the precursors of ceramides in the de novo biosynthetic pathway of sphingolipids, are implicated in the development of CVD (15).
A substantial body of literature in rodent models of CVD indicates that ceramides are not only biomarkers of cardiovascular health, but also likely play a causative role in metabolic and CVD (8, 29, 94, 97–100). Studies in rodent models reveal that pharmacological inhibition of ceramide synthesis prevents heart failure secondary to ischemic cardiomyopathy and also prevents ventricular remodeling, fibrosis, and macrophage infiltration following myocardial infarction (94, 97–99). Moreover, such ceramide-lowering interventions also resolve many conditions underlying CVDs, including dyslipidemia, insulin resistance, hypertension, atherosclerosis, and hepatic steatosis (10–12, 16, 101–104). Manipulations of the de novo ceramide synthesis pathway further suggest that certain ceramide species are deleterious, whereas others are benign or beneficial (90, 92, 105, 106). Those containing the C16 or C18 acyl chain (90, 92, 105) and a double bond (i.e., ceramides, not dihydroceramides) (101) in the sphingolipid backbone are particularly harmful. Finally, studies in rodents reveal that ceramide degradation is a primary means by which adiponectin receptors, which have ceramidase activity (1), exert their antidiabetic, cardioprotective, and insulin-sensitizing actions (1, 107, 108). Cumulatively, these data identify ceramides as some of the more toxic metabolites that accumulate in metabolic distress.
Other authors have reported the existence of a strong correlation between CVD and increased dihydroceramide levels. Both dihydroceramides and ceramides correlate with the release of the inflammatory cytokine interleukin 6 (IL-6), but only dihydroceramides correlates with macrophage inflammatory protein 1β (MIP-1β) release (13, 15). Studies have also shown that dihydroceramide levels are elevated in patients with rheumatoid arthritis (109), patients with left ventricular assist devices (110) and hypertensive rats (13–15, 41, 111, 112).
Elevated levels of dihydroceramides have been found in atherosclerotic plaques (13) as well as in models of brain hypoxia (111, 112). What role this increase in dihydroceramides plays in plaque stability is still debatable, since the extracellular addition of dihydroceramides to human aortic smooth muscle cells did not cause apoptosis, whereas addition of ceramides did (13). After stroke, a key component of tissue injury results from ischemia and subsequent reperfusion, during which ceramides increase in mitochondria and the ER due to de novo synthesis (58, 74). Myocardial biopsies from patients with heart failure reveal increased levels of ceramide content. A common treatment for heart failure is implantation of a left ventricular assist device, which compensates for defects due to heart failure, leading to a reduction in mechanical load on the cardiac tissue. This intervention both lowered myocardial ceramide levels and improved whole-body and cardiac insulin sensitivity (113).
A meta-analysis of three prospective clinical studies in Finland, Switzerland, and Norway that followed patients with CVD found that ceramide levels, particularly certain ceramide ratios, were significantly positively correlated with cardiovascular death, independent of other lipid markers, including cholesterol, and C-reactive protein (114). It also found that ratios of long-chain ceramide species (C18:1/C16:0 and C18:1/C18:0) were more strongly correlated with negative cardiovascular outcomes than ratios of VLC ceramide species (C18:1/C24:0) were (114). Furthermore, ratios of ceramide species were shown to be more significant than gross changes (114, 115). Laaksonen et al. found that the ratios of (C18:1/C24:0)/(C18:1/C16:0) and (C18:1/C24:0)/(C18:1/C16:0) negatively correlate with coronary heart disease (CHD) and mortality. Another more recent study found that serum sphingolipids are markers of coronary artery disease, independent of cholesterol (100), further solidifying the strong correlation between these species and negative cardiac outcomes. Thus, plasma ceramide levels can be used as clinical biomarkers to predict risk of cardiovascular death.
Ceramides are also strongly implicated in mitochondrial dysfunction found in CVDs. In cultured cardiomyocytes, C2 ceramide was found to alter mitochondrial membrane potential and cause mitochondrial fragmentation, leading to apoptosis (57). Increased ceramides and resulting apoptosis were also found in cardiomyocytes of mouse models of various types of cardiomyopathy (94, 116). A mouse model of type 1 diabetic cardiomyopathy showed increased ceramide accumulation in myocardium, as well as diastolic dysfunction (117). In a mouse model of type 2 diabetic cardiomyopathy, CerS2- and CerS5-derived ceramides, including VLC ceramides, led to increased mitophagy and lipotoxic cardiomyocyte hypertrophy (56). In rats, mitochondria from reperfused cardiac tissue contained higher ceramide levels than controls did, specifically in the detergent-resistant portion of the outer mitochondrial membrane (118). This correlation was also found in human subjects with CVD. Myocardial tissue from heart failure patients revealed increased ceramide content as well as reduced PGC-1α expression, which indicates impaired oxidative respiration in mitochondria (113). Across species, from mice to rats to humans, ceramides are strongly linked to CVD through mitochondrial dysfunction.
Interestingly, specific diets may even play a role in ceramide-induced mitochondrial function and cardiomyopathy. Mice that developed diabetic cardiomyopathy due to HFD made from milk fat, rather than from lard, had higher levels of C14 ceramide in cardiac tissue. These mice on milk fat-based HFD also developed cardiac dysfunction more quickly than mice on a lard-based HFD. Treatment with myriocin reversed the previously abnormal electrocardiogram parameters in mice fed a milk fat-based HFD, and it also prevented development of left ventricular and cardiomyocyte hypertrophy (119). Cumulatively, these studies of animal models and patients strongly implicate ceramides in the development and maintenance of CVD.
Adiponectin is a prevalent adipokine that acts on many tissues and organs, including the heart, liver, and kidney (120–122). Its actions are correlated with better metabolic health, resulting in improved insulin sensitivity, reduced inflammation, and enhanced cell survival (122, 123). In congenitally leptin-deficient ob/ob mice, which have severe diabetes and metabolic dysfunction (124), adiponectin overexpression was able to reverse their phenotype, restoring normal glucose and insulin levels (125). This dramatic shift underscores the powerful, beneficial role of adiponectin in metabolic health. It has also been shown to improve both alcoholic and non-alcoholic hepatic steatosis (126). Adiponectin promotes expansion of adipose tissue in a benign manner, retaining metabolic health by preventing aberrant lipid accumulation, as occurs in the liver in hepatic steatosis (125, 127, 128).
After the discovery of adiponectin, the most common adiponectin receptors, AdipoR1 and AdipoR2, were cloned (129). They were later classified as part of the progestin and adiponectin receptor (PAQR) family (130). A fungal member of the PAQR family, Izh2p, was found to be inhibited by drugs affecting sphingolipid metabolism, including myriocin, which inhibits SPT and D-erythro-MAPP, which inhibits ACER (131). Later, discovery of the protein crystal structure and in vitro work established that AdipoRs are seven-transmembrane receptors with topology opposite that of GPCRs. These receptors have basal ceramidase activity that is increased by adiponectin binding. They have been shown to bind many anionic, but not neutral, phospholipids and sphingolipids, including species such as ceramide-1-phosphate and dihydroceramide-1-phosphate (132). Moreover, AdipoR2 was purified bound to a molecule of C18 free fatty acid, and a ceramide binding pose was identified by computer simulations. Using fluorophore-labeled C18:0, it was confirmed by fluorescence spectroscopy that ceramide binds AdipoR2 (1, 133). Adiponectin receptors can hydrolyze short ceramides and long ceramides, but C18:0 is hydrolyzed most efficiently. Adiponectin binding increases the basal AdipoR2 ceramidase activity 20-fold as shown by ultra-performance liquid chromatography mass spectrometry (UPLC-MS) (1). While this enzymatic activity is slow, other work has shown that intermembrane enzymes tend to be slower than their soluble counterparts (134). Due to the ceramidase activity of its receptors, adiponectin is hypothesized to have an important role in ceramide metabolism.
Adiponectin receptors have long been a potential target for amelioration of obesity-associated metabolic dysfunction (135), and its intrinsic ceramidase activity makes it a pharmacologic target for therapeutic lowering of ceramides as well. Kadowaki and colleagues developed a candidate drug for this purpose: an orally administered small-molecule agonist of adiponectin receptors 1 and 2 called AdipoRon that binds to both adiponectin receptors at low micromolar concentration (136). This compound has been shown to activate the ceramidase activity of adiponectin receptors, indicating its potential as a ceramide-lowering compound (137, 138).
Another protein, FGF21, has been implicated in the signaling of adiponectin and ceramides. This protein is known to have antidiabetic properties, as it causes weight loss and improved insulin sensitivity (139). Our group, as well as Lin and colleagues, found that adiponectin secretion in mice is increased by FGF21 (107, 140). We also found that increased FGF21, in mice and cell culture, lowered circulating ceramides, and that mice lacking FGF21 have impaired adiponectin production and increased circulating ceramides. Despite similar circulating FGF21 levels, mice lacking adiponectin and leptin did not respond to FGF21 as compared to ob/ob mice, which significantly lowered hepatic ceramides in response to FGF21. Furthermore, ceramides were increased in DIO mice (107). In another mouse model, an adipocyte-specific knockout of a serine palmitoyl transferase (SPT) subunit resulted in lower ceramide levels and increased FGF21, consistent with this proposed axis (59). In yet another model, depletion of ceramides from suppression of CerS1 globally and in skeletal muscle resulted in improved skeletal muscle insulin sensitivity that was mediated through FGF21 (92).
In female type 2 diabetic children and adolescents, as well as healthy controls, plasma adiponectin and plasma ceramide levels are inversely correlated. These obese diabetic patients had increased levels of ceramide species, including C18:0, C20:0, C22:0, and C24:1, as compared to their lean, non-diabetic counterparts (141). These findings were replicated in adult obese diabetic patients as compared to obese non-diabetic adult controls. Controlling for obesity, diagnosis of type 2 diabetes correlated with increased sphingolipids, dihydroceramides (total, dhC16:0, and dhC24:0), and ceramides (total and C16:0) (59). Dihydroceramides were elevated in patients up to nine years prior to their diabetes diagnosis, demonstrating the value of dihydroceramides as an early sign of metabolic dysfunction (142).
One study analyzed the metabolic health of women with polycystic ovarian syndrome (PCOS), a common gynecological condition linked to metabolic syndrome and insulin resistance (143). In this study, women with PCOS had 25% reduced whole-body insulin sensitivity and 40% lower circulating adiponectin levels. Importantly, their skeletal muscle showed a 300% increase in ceramide levels, further underlining the inverse relationship between circulating adiponectin and ceramide levels (144).
Numerous clinical studies have found correlation between ceramides and CVD. The Dallas Heart Study found that in plasma, adiponectin was inversely correlated with saturated fatty acid chain ceramides only, not unsaturated species. Furthermore, patients with unfavorable lipid profiles were more likely to have short chain ceramides, both saturated and monounsaturated. Importantly, total cholesterol and LDL, two long-accepted metrics of cardiovascular and metabolic health, were strongly positively correlated with increased circulating ceramides (145). In fact, one study found that circulating ceramides may be an early indicator of cardiovascular changes in healthy adults of all ages, and that their plasma levels were inversely correlated with adiponectin levels (115). They are also inversely correlated with the 6-minute walk test, a clinical diagnostic criterion that correlates with aerobic capacity (146). Another study showed that accumulation of ceramides led to arteriolar dilation by inflammatory mediators, while overexpression of ceramidase, exogenous adiponectin, or exogenous sphingosine-1-phosphate (a byproduct of ceramide degradation) led to healthy arteriolar dilation (147). This is important because dysfunction in small vessels often causes or contributes to chronic diseases and their complications, including diabetes mellitus and coronary artery disease (148–151).
The clinical studies detailed above collectively show that adiponectin and ceramides levels are negatively correlated, and they strongly suggest a causative link between adiponectin signaling and ceramide metabolism. However, they are unable to probe mechanisms more directly. To this end, studies conducted in cell culture and animal models provide greater insight into the relationship between ceramides and this adipokine.
This hypothesized relationship has been investigated with the use of transgenic mice and cells (152). In DIO and ob/ob mice, ceramides are elevated (107) and can be lowered by administration of exogenous adiponectin (108). When adiponectin was overexpressed in adipocytes of DIO mice, the animals remained insulin sensitive with low plasma ceramide levels, despite their increased adiposity. With this overexpression, mice even remained euglycemic when pancreatic beta cells were killed, impairing insulin secretion (108). Even indirect elevation of circulating adiponectin led to increased sphingosine and sphinganine in the liver, demonstrating increased ceramidase activity (153). Conversely, elimination of adiponectin production from adipocytes led to increased ceramide deposition in the liver (108). Specifically, ceramide species C16:0, C18:0, C24:0, and C24:1 were elevated, while ceramide breakdown products sphingosine and sphingosine-1-phosphate (S1P) were lowered (154). Blocking ceramide production with SPT inhibitor myriocin in DIO mice led to increased levels of circulating high molecular weight adiponectin (59). This inverse relationship between adiponectin and ceramides has also been shown in rats (155, 156) and dolphins.
Interestingly, a somewhat different relationship between adiponectin and ceramides is seen in the skin. In these cells, adiponectin plays a beneficial role, aiding in cell differentiation, wound healing, and cell senescence (18, 157–159). Hong et al. found that in response to adiponectin administration, ceramide levels, as well as sphingosine and S1P levels, increased in human epidermal keratinocytes. The authors hypothesized that ceramide levels are elevated either because adiponectin administration also led to increases in overall lipid synthesis through activation of nuclear hormone receptors, or because of inhibition of catabolic action by other ceramidases, which was not assessed (19).
As previously mentioned, adiponectin receptors 1 and 2 are thought to have ceramidase activity. AdipoR1 and AdipoR2 overexpression in adipocytes in gonadal, mesenteric, and subcutaneous adipose tissue (gWAT, mWAT, and sWAT, respectively) increased ceramidase activity (137). Conversely, cultured cells lacking these receptors had impaired ceramidase activity and increased lipid-induced cell death that could not be rescued by adiponectin administration (108). When these receptors are overexpressed in the liver, ceramidase activity increases (108). This hepatic overexpression also leads to lower ceramide species in mWAT and sWAT, indicating communication between these organs (137). In contrast, in their absence, mouse embryonic fibroblasts have impaired ceramidase activity (108). A synthetic adiponectin receptor agonist was developed that can stimulate AdipoR effects in the absence of adiponectin (136), and its injection into wildtype mice showed a comparable increase in ceramidase activity as compared to mice overexpressing AdipoR1 or AdipoR2 in hepatocytes (137).
Exogenous adiponectin administration is able to reverse the diabetic phenotype of ob/ob mice (125). Furthermore, reduction of ceramides has been shown to increase insulin sensitivity. However, the question remained whether specifically the ceramidase action of adiponectin receptors is required to improve insulin sensitivity in leptin-deficient mice. Holland et al. found that overexpression of AdipoR2, but not AC, was able to rescue glucose homeostasis in leptin-deficient mice. Adiponectin receptors lower a larger variety of ceramide species, including deoxyceramides, which are possibly more cytotoxic because of the limited pathways for metabolizing these alanine (rather than serine) derived sphingolipids, than does AC (137).
Leptin is another major adipokine that plays a prominent role in feeding and adipose homeostasis, as well as in reproduction, maintenance of bone mass, immunity, etc. (160, 161). Its release from adipose conveys a satiety signal to the hypothalamus, leading to lower food intake (162). The diabetic phenotype of ob/ob mice is ameliorated by administration of exogenous leptin. However, mice lacking the leptin receptor, known as db/db mice, do not respond to leptin as they cannot detect it (124, 163).
Leptin resistance is most extreme in the db/db mouse, but in mice and humans, it occurs in DIO as well (163–165). Hyperleptinemia has been shown to be necessary for the development of leptin resistance (166). These high levels of leptin have been shown to increase expression of SOCS3 in the hypothalamus, which blocks transduction of leptin signaling, causing leptin resistance (167, 168). Recently, our group has shown that reversing hyperleptinemia by reducing circulating leptin via genetic and pharmacological means restores hypothalamic sensitivity to leptin, leading to reduced weight gain and increased insulin sensitivity. This intervention leads to increased expression of LepR and POMC in the hypothalamus and subsequent improvement in metabolic phenotype (169, 170). Other research indicates that ceramide levels may play a role in modulating leptin sensitivity as well.
Several studies have directly linked ceramides to leptin resistance. One study showed that administration of myriocin, an inhibitor of de novo ceramide synthesis, to obese mice (both DIO and ob/ob) led to reduced levels of leptin expression in epididymal fat. Myriocin treatment had no effect on SOCS3 expression in leptin-deficient ob/ob mice, but it decreased SOCS3 expression in DIO mice. Together, these results indicate that lowering ceramide levels increases leptin sensitivity and that this increased sensitivity cannot occur in the absence of leptin signaling. Furthermore, administration of ceramides to 3T3-L1 adipocytes led to increased SOCS3 expression, further indicating that elevated ceramides induce leptin resistance (60). A recent study replicated this in rats, showing that administration of ceramides induced leptin resistance. Conversely, downregulation of acid sphingomyelinase, which produces ceramide, ameliorated leptin resistance in rats, as indicated by increased expression of leptin receptor and decreased expression of SOCS3 (171).
Several studies indicate that leptin can reduce ceramide levels, but its ability to do so may depend on leptin sensitivity. One study in Sprague-Dawley rats found that exercise-induced lowering of muscle ceramide content, which was initially elevated from HFD, precede any improvement in leptin or insulin sensitivity in skeletal muscle (172). From these data, the authors hypothesize that ceramides may be an important determinant of insulin sensitivity, and the same conclusion could be made regarding ceramides’ impact on leptin sensitivity as well. In another rat model, Wistar rats, leptin caused decreased total ceramide levels in gWAT, as well as decreased expression of genes encoding several ceramide synthesis enzymes, including SPT, LASS2, LASS4, SMPD1, and SMPD2 (173). In Zucker diabetic fatty (ZDF) rats, which have hyperleptinemia and leptin resistance, administration of leptin failed to block upregulation of SPT, resulting in increased ceramide content. However, with increased leptin sensitivity (conferred by delivery of leptin receptor cDNA to islet cells), leptin was able to block SPT mRNA expression (174). This indicates a relationship between leptin and ceramides that is dependent upon leptin sensitivity.
Just as leptin has been shown to reduce ceramides, some studies show that ceramides have a similar, reciprocal regulatory effect on leptin. Treatment of 3T3-L1 pre-adipocytes with ceramide-1-phosphate decreases leptin secretion (175). In DIO mice, which have hyperleptinemia and resultant leptin resistance, treatment with myriocin lead to lower circulating ceramides and decreased gene expression of leptin and SOCS3 in gWAT (60). Reduction of leptin levels has been shown to increase leptin sensitivity, and SOCS3 is known mediator of leptin resistance (167–169, 176). Therefore, reduced expression of leptin and SOCS3 secondary to ceramide reduction may enable increased leptin sensitivity.
However, the relationship between ceramides and leptin is still not entirely clear, as some studies have shown contradictory data. In both the DIO and ob/ob mouse models of obesity, which have high and nonexistent circulating levels of leptin respectively, total plasma and hepatic ceramide levels are increased (108, 177). These studies indicate that regardless of leptin levels, ceramides are increased in models of obesity and diabetes. In gonadal white adipose (gWAT) of leptin-deficient ob/ob mice, while C14 ceramide was increased, total ceramide levels as well as those of C18:1, C24:0, and C24:1 were decreased, showing that ceramides do not respond uniformly to the absence of leptin (177). In adipocytes, knocking out a subunit of SPT (leading to lowered adipose and circulating ceramides), led to lower levels of circulating leptin (59). Therefore, further research is necessary to clarify the relationship between leptin and ceramides, in states of both leptin sensitivity and leptin resistance.
Ceramide accumulation and leptin resistance can induce dysfunction in cardiovascular tissue as well. Leptin levels correlate with cardiovascular dysfunction in male patients, independent of degree of obesity and hypertension (178). One possible explanation of this correlation is demonstrated by one study, which showed that cardiac contractile dysfunction was exacerbated by accumulation of ceramides in cardiac muscle, which potentiated the contractile effects of leptin (179). Another study showed that decreasing ceramides in rat aortic endothelial cells, through downregulation of acid sphingomyelinase, improved leptin sensitivity (171). These studies suggest that ceramides contribute to leptin resistance in cardiovascular tissues.
Recent studies have shown that ceramide effects in the hypothalamus contribute to leptin resistance. It has been shown that palmitate, a lipid long known to cross the blood-brain barrier (180) and also induce inflammation (181) does so in the hypothalamus partially through ceramide synthesis (182). Mice subjected to intravenous emulsions of saturated fatty acids had ceramides accumulate in the hypothalamus, indicating a hypothalamic role for ceramide detection and regulation (108). Infusion of a ceramide analog directly into the hypothalamic arcuate nucleus, a brain region that regulates many neuroendocrine functions including feeding (183), of Sprague-Dawley rats suppressed leptin-induced anorectic effects, indicating an inverse relationship between ceramide levels and leptin sensitivity. Physiologic data supports this relationship as well, as fasting (a state of low leptin) increases ceramides, and refeeding (high leptin) decreases ceramides in the arcuate nucleus (2). Conversely, lowering hypothalamic ceramide levels through administration of myriocin increased leptin-induced anorectic effects. In the arcuate nucleus, mRNA expression of SPT, the enzyme driving de novo ceramide synthesis, was comparable to expression of AgRP and POMC, which are both crucial to the arcuate’s role in food intake (2). Such significant levels of expression of a key ceramide synthesis enzyme points to an important role for ceramides in this region.
Gao et al. propose that mechanistically, leptin reduces ceramide content through modulation of malonyl-CoA and carnitine palmitoyltransferase 1c (CPT-1c). They hypothesize that leptin induces its anorectic effects in part by increasing malonyl-CoA, which in turn inhibits CPT-1c, leading to decreased de novo ceramide synthesis. CPT-1c may act as a transporter of palmitoyl-CoA into the ER, enabling its use as a substrate for ceramide synthesis. Therefore, inhibition of CPT-1c through leptin-induced increases in malonyl-CoA would in turn lower ceramide levels (2). This group found further evidence of ceramide regulation by CPT-1c in hippocampal dendrites, bolstering evidence for this relationship between CPT-1c and ceramides (184).
Another study found that in Zucker rats, a strain known to have leptin resistance, an accumulation of ceramides in the mediobasal hypothalamus led to ER stress and lipotoxicity (185). ER stress has previously been shown to cause leptin resistance (3–5). The authors postulated that ceramides induce ER stress through prevention of proper protein folding. Relief of this ceramide-induced ER stress led to subsequent leptin sensitization, indicating bidirectional control of leptin sensitivity by ceramides (6). However, another study found that central administration of leptin caused decreased ceramide levels in the plasma membrane and Golgi, but not the ER of Wistar rat eWAT (173). Therefore, it is unclear whether ER stress is the true mediator of ceramides’ effect on leptin sensitivity.
Over the past few decades, studies have begun to show the significant impact of ceramides and adipokines on metabolic health, particularly in diabetes and CVD. Higher levels of ceramides correlate with low levels of adiponectin as well as with leptin and insulin resistance, indicating that these sphingolipids contribute to dysfunction in cardiovascular, hepatic, and adipose tissue. Cumulatively, these studies suggest that monitoring ceramide levels could allow better assessment of cardiovascular and metabolic disease progression and/or severity, and that ceramides are a possible target for future therapeutic intervention in cardiometabolic pathologies.
All authors contributed to the article and approved the submitted version.
The authors are supported by US National Institutes of Health (NIH) grants R01-DK55758, RC2-DK118620, P01-DK088761, R01-DK099110 and P01-AG051459 (PS). PS was also supported by an unrestricted research grant from the Novo Nordisk Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Vasiliauskaité-Brooks I, Sounier R, Rochaix P, Bellot G, Fortier M, Hoh F, et al. Structural insights into adiponectin receptors suggest ceramidase activity. Nature (2017) 544(7648):120–3. doi: 10.1038/nature21714
2. Gao S, Zhu G, Gao X, Wu D, Carrasco P, Casals N, et al. Important roles of brain-specific carnitine palmitoyltransferase and ceramide metabolism in leptin hypothalamic control of feeding. Proc Natl Acad Sci USA (2011) 108(23):9691–6. doi: 10.1073/pnas.1103267108
3. Hosoi T, Sasaki M, Miyahara T, Hashimoto C, Matsuo S, Yoshii M, et al. Endoplasmic reticulum stress induces leptin resistance. Mol Pharmacol (2008) 74(6):1610–9. doi: 10.1124/mol.108.050070
4. Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic Reticulum Stress Plays a Central Role in Development of Leptin Resistance. Cell Metab (2009) 9(1):35–51. doi: 10.1016/j.cmet.2008.12.004
5. Won JC, Jang P-G, Namkoong C, Koh EH, Kim SK, Park J-Y, et al. Central Administration of an Endoplasmic Reticulum Stress Inducer Inhibits the Anorexigenic Effects of Leptin and Insulin. Obesity (2009) 17(10):1861–5. doi: 10.1038/oby.2009.194
6. Contreras C, López M. Ceramide sensing in the hippocampus: The lipostatic theory and Ockham’s razor. Mol Metab (2014) 3(2):90–1. doi: 10.1016/j.molmet.2013.12.004
7. Kogot-Levin A, Saada A. Ceramide and the mitochondrial respiratory chain. Biochimie (2014) 100(1):88–94. doi: 10.1016/j.biochi.2013.07.027
8. Holland WL, Summers SA. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev (2008) 29(4):381–402. doi: 10.1210/er.2007-0025
9. Xia JY, Morley TS, Scherer PE. The adipokine/ceramide axis: Key aspects of insulin sensitization. Biochimie (2014) 96(1):130–9. doi: 10.1016/j.biochi.2013.08.013
10. Glaros EN, Kim WS, Quinn CM, Jessup W, Rye KA, Garner B. Myriocin slows the progression of established atherosclerotic lesions in apolipoprotein E gene knockout mice. J Lipid Res (2008) 49(2):324–31. doi: 10.1194/jlr.M700261-JLR200
11. Glaros EN, Kim WS, Wu BJ, Suarna C, Quinn CM, Rye K-A, et al. Inhibition of atherosclerosis by the serine palmitoyl transferase inhibitor myriocin is associated with reduced plasma glycosphingolipid concentration. Biochem Pharmacol (2007) 73(9):1340–6. doi: 10.1016/j.bcp.2006.12.023
12. Park TS, Rosebury W, Kindt EK, Kowala MC, Panek RL. Serine palmitoyltransferase inhibitor myriocin induces the regression of atherosclerotic plaques in hyperlipidemic ApoE-deficient mice. Pharmacol Res (2008) 58(1):45–51. doi: 10.1016/j.phrs.2008.06.005
13. Edsfeldt A, Dunér P, Ståhlman M, Mollet IG, Asciutto G, Grufman H, et al. Sphingolipids contribute to human atherosclerotic plaque inflammation. Arterioscler Thromb Vasc Biol (2016) 36(6):1132–40. doi: 10.1161/ATVBAHA.116.305675
14. Meeusen JW, Donato LJ, Bryant SC, Baudhuin LM, Berger PB, Jaffe AS. Plasma Ceramides. Arterioscler Thromb Vasc Biol (2018) 38(8):1933–9. doi: 10.1161/ATVBAHA.118.311199
15. Magaye RR, Savira F, Hua Y, Kelly DJ, Reid C, Flynn B, et al. The role of dihydrosphingolipids in disease. Cell Mol Life Sci (2019) 76(6):1107–34. doi: 10.1007/s00018-018-2984-8
16. Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of Ceramide Synthesis Ameliorates Glucocorticoid-, Saturated-Fat-, and Obesity-Induced Insulin Resistance. Cell Metab (2007) 5(3):167–79. doi: 10.1016/j.cmet.2007.01.002
17. Boon J, Hoy AJ, Stark R, Brown RD, Meex RC, Henstridge DC, et al. Ceramides contained in LDL are elevated in type 2 diabetes and promote inflammation and skeletal muscle insulin resistance. Diabetes (2013) 62(2):401–10. doi: 10.2337/db12-0686
18. Kawai K, Kageyama A, Tsumano T, Nishimoto S, Fukuda K, Yokoyama S, et al. Effects of adiponectin on growth and differentiation of human keratinocytes-Implication of impaired wound healing in diabetes. Biochem Biophys Res Commun (2008) 374(2):269–73. doi: 10.1016/j.bbrc.2008.07.045
19. Hong SP, Seo HS, Shin K-O, Park K, Park BC, Kim MH, et al. Adiponectin Enhances Human Keratinocyte Lipid Synthesis via SIRT1 and Nuclear Hormone Receptor Signaling. J Invest Dermatol (2019) 139(3):573–82. doi: 10.1016/j.jid.2018.08.032
20. Pruett ST, Bushnev A, Hagedorn K, Adiga M, Haynes CA, Sullards MC, et al. Biodiversity of sphingoid bases (“sphingosines”) and related amino alcohols. J Lipid Res (2008) 49(8):1621–39. doi: 10.1194/jlr.R800012-JLR200
21. Welsch CA, Roth LWA, Goetschy JF, Movva NR. Genetic, biochemical, and transcriptional responses of Saccharomyces cerevisiae to the novel immunomodulator FTY720 largely mimic those of the natural sphingolipid phytosphingosine. J Biol Chem (2004) 279(35):36720–31. doi: 10.1074/jbc.M406179200
22. Pralhada Rao R, Vaidyanathan N, Rengasamy M, Mammen Oommen A, Somaiya N, Jagannath MR. Sphingolipid Metabolic Pathway: An Overview of Major Roles Played in Human Diseases. J Lipids (2013) 2013:1–12. doi: 10.1155/2013/178910
23. Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Sci (80- ). (1993) 259(5102):1769–71. doi: 10.1126/science.8456305
24. Zhou H, Summers SA, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem (1998) 273(26):16568–75. doi: 10.1074/jbc.273.26.16568
25. Nganga R, Oleinik N, Ogretmen B. Mechanisms of Ceramide-Dependent Cancer Cell Death. Adv Cancer Res (2018) 140:1–25. doi: 10.1016/bs.acr.2018.04.007
26. Gomez-Muñoz A, Presa N, Gomez-Larrauri A, Rivera IG, Trueba M, Ordoñez M. Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog Lipid Res (2016) 61:51–62. doi: 10.1016/j.plipres.2015.09.002
27. Dinoff A, Herrmann N, Lanctôt KL. Ceramides and depression: A systematic review. J Affect Disord (2017) 213:35–43. doi: 10.1016/j.jad.2017.02.008
28. Wang G, Bieberich E. Sphingolipids in neurodegeneration (with focus on ceramide and S1P). Adv Biol Regul (2018) 70:51–64. doi: 10.1016/j.jbior.2018.09.013
29. Chaurasia B, Summers SA. Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol Metab (2015) 26(10):538–50. doi: 10.1016/j.tem.2015.07.006
30. Aburasayn H, Al Batran R, Ussher JR. Targeting ceramide metabolism in obesity. Am J Physiol Metab (2016) 311(2):E423–35. doi: 10.1152/ajpendo.00133.2016
31. Fang Z, Pyne S, Pyne NJ. Ceramide and sphingosine 1-phosphate in adipose dysfunction. Prog Lipid Res (2019) 74:145–59. doi: 10.1016/j.plipres.2019.04.001
32. Cogolludo A, Villamor E, Perez-Vizcaino F, Moreno L. Ceramide and Regulation of Vascular Tone. Int J Mol Sci (2019) 20(2):411. doi: 10.3390/ijms20020411
33. Apostolopoulou M, Gordillo R, Koliaki C, Gancheva S, Jelenik T, De Filippo E, et al. Specific Hepatic Sphingolipids Relate to Insulin Resistance, Oxidative Stress, and Inflammation in Nonalcoholic Steatohepatitis. Diabetes Care (2018) 41(June):1235–43. doi: 10.2337/dc17-1318
34. Gorden DL, Myers DS, Ivanova PT, Fahy E, Maurya MR, Gupta S, et al. Biomarkers of NAFLD progression: A lipidomics approach to an epidemic. J Lipid Res (2015) 56(3):722–36. doi: 10.1194/jlr.P056002
35. Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of Obesity Among Adults and Youth: United States, 2015-2016 (2017). Available at: https://www.cdc.gov/nchs/data/databriefs/db288.pdf (Accessed October 10, 2019).
36. World Health Organization G. Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000-2016 (2018). Available at: https://www.who.int/healthinfo/global_burden_disease/estimates/en/ (Accessed May 12, 2020).
37. Castro BM, Prieto M, Silva LC. Ceramide: A simple sphingolipid with unique biophysical properties. Prog Lipid Res (2014) 54(1):53–67. doi: 10.1016/j.plipres.2014.01.004
38. Hannun YA, Obeid LM. Many ceramides. J Biol Chem (2011) 286(32):27855–62. doi: 10.1074/jbc.R111.254359
39. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat Rev Mol Cell Biol (2008) 9(2):139–50. doi: 10.1038/nrm2329
40. Hernández-Corbacho MJ, Salama MF, Canals D, Senkal CE, Obeid LM. Sphingolipids in mitochondria. Biochim Biophys Acta - Mol Cell Biol Lipids (2017) 1862(1):56–68. doi: 10.1016/j.bbalip.2016.09.019
41. Liu A, Chu YJ, Wang X, Yu R, Jiang H, Li Y, et al. Serum Metabolomics Study Based on LC-MS and Antihypertensive Effect of Uncaria on Spontaneously Hypertensive Rats. Evidence-Based Complement Altern Med (2018) 2018:1–11. doi: 10.1155/2018/9281946
42. Levy M, Futerman AH. Mammalian ceramide synthases. IUBMB Life (2010) 62(5):347–56. doi: 10.1002/iub.319
43. López-Montero I, Rodriguez N, Cribier S, Pohl A, Vélez M, Devaux PF. Rapid transbilayer movement of ceramides in phospholipid vesicles and in human erythrocytes. J Biol Chem (2005) 280(27):25811–9. doi: 10.1074/jbc.M412052200
44. Simon CG, Holloway PW, Gear ARL. Exchange of C16-ceramide between phospholipid vesicles. Biochemistry (1999) 38(44):14676–82. doi: 10.1021/bi991537w
45. Tomassini B, Testi R. Mitochondria as sensors of sphingolipids. Biochimie (2002) 84(2-3):123–9. doi: 10.1016/S0300-9084(02)01377-9
46. Lightle S, Tosheva R, Lee A, Queen-Baker J, Boyanovsky B, Shedlofsky S, et al. Elevation of ceramide in serum lipoproteins during acute phase response in humans and mice: Role of serine-palmitoyl transferase. Arch Biochem Biophys (2003) 419(2):120–8. doi: 10.1016/j.abb.2003.08.031
47. Wiesner P, Leidl K, Boettcher A, Schmitz G, Liebisch G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J Lipid Res (2009) 50(3):574–85. doi: 10.1194/jlr.D800028-JLR200
48. Crewe C, Joffin N, Rutkowski JM, Kim M, Zhang F, Towler DA, et al. Endothelial-to-Adipocyte Extracellular Vesicle Axis Governed by Metabolic State. An Cell (2018) 175(3):695–708.e13. doi: 10.1016/j.cell.2018.09.005
49. Bionda C, Portoukalian J, Schmitt D, Rodriguez-Lafrasse C, Ardail D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem J (2004) 382(2):527–33. doi: 10.1042/BJ20031819
50. Novgorodov SA, Chudakova DA, Wheeler BW, Biewlawski J, Kindy MS, Obeid LM, et al. Developmentally regulated ceramide synthase 6 increases mitochondrial Ca2+ loading capacity and promotes apoptosis. J Biol Chem (2011) 286(6):4644–58. doi: 10.1074/jbc.M110.164392
51. Di Paola M, Cocco T, Lorusso M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry (2000) 39(22):6660–8. doi: 10.1021/bi9924415
52. Colombini M. Ceramide channels and mitochondrial outer membrane permeability. J Bioenerg Biomembr (2017) 49(1):57–64. doi: 10.1007/s10863-016-9646-z
53. Ueda N. Ceramide-Induced Apoptosis in Renal Tubular Cells: A Role of Mitochondria and Sphingosine-1-Phoshate. Int J Mol Sci (2015) 16(12):5076–124. doi: 10.3390/ijms16035076
54. Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem (1997) 272(39):24154–8. doi: 10.1074/jbc.272.39.24154
55. Ganesan V, Perera MN. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis (2010) 15:553–62. doi: 10.1007/s10495-009-0449-0
56. Law BA, Liao X, Moore KS, Southard A, Roddy P, Ji R, et al. Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J (2018) 32(3):1403–16. doi: 10.1096/fj.201700300R
57. Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res (2008) 77(2):387–97. doi: 10.1093/cvr/cvm029
58. Yu J, Novgorodov SA, Chudakova D, Zhu H, Bielawska A, Bielawski J, et al. JNK3 signaling pathway activates ceramide synthase leading to mitochondrial dysfunction. J Biol Chem (2007) 282(35):25940–9. doi: 10.1074/jbc.M701812200
59. Chaurasia B, Kaddai VA, Lancaster GI, Henstridge DC, Sriram S, Galam DLA, et al. Adipocyte Ceramides Regulate Subcutaneous Adipose Browning, Inflammation, and Metabolism. Cell Metab (2016) 24(6):820–34. doi: 10.1016/J.CMET.2016.10.002
60. Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, Samad F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol - Endocrinol Metab (2009) 297(1):E211–24. doi: 10.1152/ajpendo.91014.2008
61. Yambire KF, Fernandez-Mosquera L, Steinfeld R, Mühle C, Ikonen E, Milosevic I, et al. Mitochondrial biogenesis is transcriptionally repressed in lysosomal lipid storage diseases. Elife (2019) 8:1–29. doi: 10.7554/eLife.39598
62. Fugio LB, Coeli-Lacchini FB, Leopoldino AM. Sphingolipids and Mitochondrial Dynamic. Cells (2020) 9(3):581. doi: 10.3390/cells9030581
63. Teixeira V, Medeiros TC, Vilaça R, Pereira AT, Chaves SR, Côrte-Real M, et al. Ceramide signalling impinges on Sit4p and Hog1p to promote mitochondrial fission and mitophagy in Isc1p-deficient cells. Cell Signal (2015) 27(9):1840–9. doi: 10.1016/j.cellsig.2015.06.001
64. Mizumura K, Justice MJ, Schweitzer KS, Krishnan S, Bronova I, Berdyshev EV, et al. Sphingolipid regulation of lung epithelial cell mitophagy and necroptosis during cigarette smoke exposure. FASEB J (2018) 32(4):1880–90. doi: 10.1096/fj.201700571R
65. Panda PK, Naik PP, Meher BR, Das DN, Mukhopadhyay S, Praharaj PP, et al. PUMA dependent mitophagy by Abrus agglutinin contributes to apoptosis through ceramide generation. Biochim Biophys Acta - Mol Cell Res (2018) 1865(3):480–95. doi: 10.1016/j.bbamcr.2017.12.002
66. Dany M, Gencer S, Nganga R, Thomas RJ, Oleinik N, Baron KD, et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood (2016) 128(15):1944–58. doi: 10.1038/nrc.2017.96
67. Colombini M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim Biophys Acta - Bioenerg (2010) 1797(6-7):1239–44. doi: 10.1016/j.bbabio.2010.01.021
68. Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med (1995) 182(2):367–77. doi: 10.1084/jem.182.2.367
69. Petit PX, Gendron MC, Schrantz N, Métiviers D, Kroemer G, Maciorowska Z, et al. Oxidation of pyridine nucleotides during Fas- and ceramide-induced apoptosis in Jurkat cells: correlation with changes in mitochondria, glutathione depletion, intracellular acidification and caspase 3 activation. Biochem J (2001) 353(2):357–67. doi: 10.1042/0264-6021:3530357
70. Zhang I, Alter N, Reed JC, Borner C, Obeid LM, Hannun YA. Bcl-2 interrupts the ceramide-mediated pathway of cell death. Proc Natl Acad Sci U S A (1996) 93(11):5325–8. doi: 10.1073/pnas.93.11.5325
71. Geley S, Hartmann BL, Kofler R. Ceramides induce a form of apoptosis in human acute lymphoblastic leukemia cells that is inhibited by Bcl-2, but not by CrmA. FEBS Lett (1997) 400(1):15–8. doi: 10.1016/S0014-5793(96)01284-7
72. Stoica BA, Movsesyan VA, Lea IV PM, Faden AI. Ceramide-induced neuronal apoptosis is associated with dephosphorylation of Akt, BAD, FKHR, GSK-3β, and induction of the mitochondrial-dependent intrinsic caspase pathway. Mol Cell Neurosci (2003) 22(3):365–82. doi: 10.1016/S1044-7431(02)00028-3
73. Pastorino JG, Simbula G, Yamamoto K, Glascott PA, Rothman RJ, Farber JL. The Cytotoxicity of Tumor Necrosis Factor Depends on Induction of the Mitochondrial Permeability Transition. J Biol Chem (1996) 271(47):29792–8. doi: 10.1074/jbc.271.47.29792
74. Novgorodov SA, Gudz TI. Ceramide and mitochondria in ischemia/reperfusion. J Cardiovasc Pharmacol (2009) 53(3):198–208. doi: 10.1097/FJC.0b013e31819b52d5
75. Birbes H, Bawab SE, Hannun YA, Obeid LM. Selective hydrolysis of a mitochondrial pool of sphingomyelin induces apoptosis. FASEB J (2001) 15(14):2669–79. doi: 10.1096/fj.01-0539com
76. Novgorodov SA, Wu BX, Gudz TI, Bielawski J, Ovchinnikova TV, Hannun YA, et al. Novel pathway of ceramide production in mitochondria: Thioesterase and neutral ceramidase produce ceramide from sphingosine and acyl-CoA. J Biol Chem (2011) 286(28):25352–62. doi: 10.1074/jbc.M110.214866
77. Kolak M, Westerbacka J, Velagapudi VR, Wågsäter D, Yetukuri L, Makkonen J, et al. Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes (2007) 56(8):1960–8. doi: 10.2337/db07-0111
78. Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, DeFronzo RA, et al. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes (2009) 58(2):337–43. doi: 10.2337/db08-1228
79. Turinsky J, O’Sullivan DM, Bayly BP. 1,2-Diacylglycerol and Ceramide Levels in Insulin-resistant Tissues of the Rat in Vivo. J Biol Chem (1990) 265(28):16880–5.
80. Kim JK, Fillmore JJ, Chen Y, Yu C, Moore IK, Pypaert M, et al. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci USA (2001) 98(13):7522–7. doi: 10.1073/pnas.121164498
81. Brozinick JT, Hawkins E, Hoang Bui H, Kuo M-S, Tan B, Kievit P, et al. Plasma sphingolipids are biomarkers of metabolic syndrome in non-human primates maintained on a Western-style diet. Int J Obes (2013) 37:1064–70. doi: 10.1038/ijo.2012.191
82. Straczkowski M, Kowalska I, Nikolajuk A, Dzienis-Straczkowska S, Kinalska I, Baranowski M, et al. Relationship between Insulin Sensitivity and Sphingomyelin Signaling Pathway in Human Skeletal Muscle. Diabetes (2004) 53(5):1215–21. doi: 10.2337/diabetes.53.5.1215
83. Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discovery (2016) 15(9):639–60. doi: 10.1038/nrd.2016.75
84. Adams JM, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, et al. Ceramide Content Is Increased in Skeletal Muscle from Obese Insulin-Resistant Humans. Diabetes (2004) 53(1):25–31. doi: 10.2337/diabetes.53.1.25
85. Summers SA, Nelson DH. A role for sphingolipids in producing the common features of type 2 diabetes, metabolic syndrome X, and Cushing’s syndrome. Diabetes (2005) 54(3):591–602. doi: 10.2337/diabetes.54.3.591
86. Cifone MG, Migliorati G, Parroni R, Marchetti C, Millimaggi D, Santoni A, et al. Dexamethasone-induced thymocyte apoptosis: Apoptotic signal involves the sequential activation of phosphoinositide-specific phospholipase C, acidic sphingomyelinase, and caspases. Blood (1999) 93(7):2282–96. doi: 10.1182/blood.v93.7.2282
87. Cinque B, Fanini D, Di Marzio L, Palumbo P, La Torre C, Donato V, et al. Involvement of cPLA2 inhibition in dexamethasone-induced thymocyte apoptosis. Int J Immunopathol Pharmacol (2008) 21(3):539–51. doi: 10.1177/039463200802100307
88. Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, et al. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab (2015) 22(2):266–78. doi: 10.1016/j.cmet.2015.06.007
89. Raichur S, Brunner B, Bielohuby M, Hansen G, Pfenninger A, Wang B, et al. The role of C16:0 ceramide in the development of obesity and type 2 diabetes: CerS6 inhibition as a novel therapeutic approach. Mol Metab (2019) 21(January):36–50. doi: 10.1016/j.molmet.2018.12.008
90. Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab (2014) 20(4):678–86. doi: 10.1016/j.cmet.2014.08.002
91. Hammerschmidt P, Ostkotte D, Nolte H, Gerl MJ, Jais A, Brunner HL, et al. CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell (2019) 177(6):1536–1552.e23. doi: 10.1016/j.cell.2019.05.008
92. Turpin-Nolan SM, Hammerschmidt P, Chen W, Jais A, Timper K, Awazawa M, et al. CerS1-Derived C18:0 Ceramide in Skeletal Muscle Promotes Obesity-Induced Insulin Resistance. Cell Rep (2019) 26(1):1–10.e7. doi: 10.1016/j.celrep.2018.12.031
93. Bismuth J, Lin P, Yao Q, Chen C. Ceramide: A common pathway for atherosclerosis? Atherosclerosis (2008) 196(2):497–504. doi: 10.1016/j.atherosclerosis.2007.09.018
94. Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res (2008) 49(10):2101–12. doi: 10.1194/jlr.M800147-JLR200
95. Walls SM, Cammarato A, Chatfield DA, Ocorr K, Harris GL, Bodmer R. Ceramide-Protein Interactions Modulate Ceramide-Associated Lipotoxic Cardiomyopathy. Cell Rep (2018) 22(10):2702–15. doi: 10.1016/j.celrep.2018.02.034
96. Peterson LR, Xanthakis V, Duncan MS, Gross S, Friedrich N, Völzke H, et al. Ceramide remodeling and risk of cardiovascular events and mortality. J Am Heart Assoc (2018) 7(10):1–11. doi: 10.1161/JAHA.117.007931
97. Lee SY, Kim JR, Hu Y, Khan R, Kim S-J, Bharadwaj KG, et al. Cardiomyocyte specific deficiency of serine palmitoyltransferase subunit 2 reduces ceramide but leads to cardiac dysfunction. J Biol Chem (2012) 287(22):18429–39. doi: 10.1074/jbc.M111.296947
98. Park TS, Goldberg IJ. Sphingolipids, Lipotoxic Cardiomyopathy, and Cardiac Failure. Heart Fail Clin (2012) 8(4):633–41. doi: 10.1016/j.hfc.2012.06.003
99. Ussher JR, Koves TR, Cadete VJJ, Zhang L, Jaswal JS, Swyrd SJ, et al. Inhibition of De Novo Ceramide Synthesis Reverses Diet-Induced Insulin Resistance and Enhances Whole-Body Oxygen Consumption. Diabetes (2010) 59(10):2453–64. doi: 10.2337/db09-1293
100. Poss AM, Maschek JA, Cox JE, Hauner BJ, Hopkins PN, Hunt SC, et al. Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. J Clin Invest (2020) 130(3):1363–76. doi: 10.1172/JCI131838
101. Chaurasia B, Tippetts TS, Monibas RM, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Sci (80- ) (2019) 365(6451):386–92. doi: 10.1126/science.aav3722
102. Hojjati MR, Li Z, Zhou H, Tang S, Huan C, Ooi E, et al. Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J Biol Chem (2005) 280(11):10284–9. doi: 10.1074/jbc.M412348200
103. Park TS, Panek RL, Rekhter MD, Mueller SB, Rosebury WS, Robertson A, et al. Modulation of lipoprotein metabolism by inhibition of sphingomyelin synthesis in ApoE knockout mice. Atherosclerosis (2006) 189(2):264–72. doi: 10.1016/j.atherosclerosis.2005.12.029
104. Bharath LP, Ruan T, Li Y, Ravindran A, Wan X, Nhan JK, et al. Ceramide-initiated protein phosphatase 2A activation contributes to arterial dysfunction in vivo. Diabetes (2015) 64(11):3914–26. doi: 10.2337/db15-0244
105. Hla T, Kolesnick R. C16:0-ceramide signals insulin resistance. Cell Metab (2014) 20(5):703–5. doi: 10.1016/j.cmet.2014.10.017
106. Raichur S, Wang ST, Chan PW, Li Y, Chaing J, Chaurasia B, et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab (2014) 20(4):687–95. doi: 10.1016/j.cmet.2014.09.015
107. Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab (2013) 17(5):790–7. doi: 10.1016/j.cmet.2013.03.019
108. Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med (2011) 17(1):55–63. doi: 10.1038/nm.2277
109. Fang L, Mundra PA, Fan F, Galvin A, Weir JM, Wong G, et al. Plasma lipidomic profiling in patients with rheumatoid arthritis. Metabolomics (2016) 12(8):1–10. doi: 10.1007/s11306-016-1086-6
110. Fine B, Marx A, Topkara V, Gomez EA, Vunjak-Novakovic G, Colombo P. An Integrated Analysis of Metabolomics After Left Ventricular Assist Device Implantation. J Hear Lung Transplant (2017) 36(4):S93. doi: 10.1016/j.healun.2017.01.235
111. Testai FD, Kilkus JP, Berdyshev E, Gorshkova I, Natarajan V, Dawson G. Multiple sphingolipid abnormalities following cerebral microendothelial hypoxia. J Neurochem (2014) 131(4):530–40. doi: 10.1111/jnc.12836
112. Testai FD, Xu H-L, Kilkus J, Suryadevara V, Gorshkova I, Berdyshev E, et al. Changes in the metabolism of sphingolipids after subarachnoid hemorrhage. J Neurosci Res (2015) 93(5):796–805. doi: 10.1002/jnr.23542
113. Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation (2012) 125(23):2844–53. doi: 10.1161/CIRCULATIONAHA.111.060889
114. Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J (2016) 37:1967–76. doi: 10.1093/eurheartj/ehw148
115. Petrocelli JJ, McKenzie AI, Mahmassani ZS, Reidy PT, Stoddard GJ, Poss AM, et al. Ceramide Biomarkers Predictive of Cardiovascular Disease Risk Increase in Healthy Older Adults After Bed Rest. J Gerontol Biol Sci (2020) 75(9):1663–70. doi: 10.1093/gerona/glaa072
116. Chiu HC, Kovacs A, Ford DA, Hsu F-F, Garcia R, Herrero P, et al. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest (2001) 107(7):813–22. doi: 10.1172/JCI10947
117. Basu R, Oudit GY, Wang X, Zhang L, Ussher JR, Lopaschuk GD, et al. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am J Physiol - Hear Circ Physiol (2009) 297(6):2096–108. doi: 10.1152/ajpheart.00452.2009
118. Martínez-Abundis E, Correa F, Pavón N, Zazueta C. Bax distribution into mitochondrial detergent-resistant microdomains is related to ceramide and cholesterol content in postischemic hearts. FEBS J (2009) 276(19):5579–88. doi: 10.1111/j.1742-4658.2009.07239.x
119. Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, et al. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest (2012) 122(11):3919–30. doi: 10.1172/JCI63888
120. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem (1995) 270(45):26746–9. doi: 10.1074/jbc.270.45.26746
121. Turer AT, Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia (2012) 55(9):2319–26. doi: 10.1007/s00125-012-2598-x
122. Wang ZV, Scherer PE. Adiponectin, the past two decades. J Mol Cell Biol (2016) 8(2):93–100. doi: 10.1093/jmcb/mjw011
123. Scherer PE. The many secret lives of adipocytes: implications for diabetes. Diabetologia (2019) 62(2):223–32. doi: 10.1007/s00125-018-4777-x
124. Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chair BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Sci (80- ) (1995) 269(5223):543–6. doi: 10.1126/science.7624777
125. Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest (2007) 117(9):2621–37. doi: 10.1172/JCI31021
126. Xu A, Wang Y, Keshaw H, Xu LY, Lam KSL, Cooper GJS. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest (2003) 112(1):91–100. doi: 10.1172/jci17797
127. Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med (2012) 18(10):1539–51. doi: 10.1038/nm.2899
128. Scherer PE. The multifaceted roles of adipose tissue - Therapeutic targets for diabetes and beyond: The 2015 banting lecture. Diabetes (2016) 65(6):1452–61. doi: 10.2337/db16-0339
129. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature (2003) 423(6941):762–9. doi: 10.1038/nature01705
130. Tang YT, Hu T, Arterburn M, Boyle B, Bright JM, Emtage PC, et al. PAQR proteins: A novel membrane receptor family defined by an ancient 7-transmembrane pass motif. J Mol Evol (2005) 61(3):372–80. doi: 10.1007/s00239-004-0375-2
131. Villa NY, Kupchak BR, Garitaonandia I, Smith JL, Alonso E, Alford C, et al. Sphingolipids function as downstream effectors of a fungal PAQF. Mol Pharmacol (2009) 75(4):866–75. doi: 10.1124/mol.108.049809
132. Ye J, Bian X, Lim J, Medzhitov R. Adiponectin and related C1q/TNF-related proteins bind selectively to anionic phospholipids and sphingolipids. Proc Natl Acad Sci U S A (2019) 117(29):776690. doi: 10.1101/776690
133. Tanabe H, Fujii Y, Okada-Iwabu M, Iwabu M, Nakamura Y, Hosaka T, et al. Crystal structures of the human adiponectin receptors. Nature (2015) 520(7547):312–6. doi: 10.1038/nature14301
134. Langosch D, Scharnagl C, Steiner H, Lemberg MK. Understanding intramembrane proteolysis: From protein dynamics to reaction kinetics. Trends Biochem Sci (2015) 40(6):318–27. doi: 10.1016/j.tibs.2015.04.001
135. Holland WL, Scherer PE. Ronning after the adiponectin receptors. Science (2013) 342(6165):1460–1. doi: 10.1126/science.1249077
136. Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K-I, Matsuda K, et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature (2013) 503(7477):493–9. doi: 10.1038/nature12656
137. Holland WL, Xia JY, Johnson JA, Sun K, Pearson MJ, Sharma AX, et al. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol Metab (2017) 6(3):267–75. doi: 10.1016/j.molmet.2017.01.002
138. Choi SR, Lim JH, Kim MY, Kim EN, Kim Y, Choi BS, et al. Adiponectin receptor agonist AdipoRon decreased ceramide, and lipotoxicity, and ameliorated diabetic nephropathy. Metabolism (2018) 85:348–60. doi: 10.1016/j.metabol.2018.02.004
139. Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab (2015) 26(1):22–9. doi: 10.1016/j.tem.2014.10.002
140. Lin Z, Tian H, Lam KSL, Lin S, Hoo RCL, Konishi M, et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab (2013) 17(5):779–89. doi: 10.1016/j.cmet.2013.04.005
141. Lopez X, Goldfine AB, Holland WL, Gordillo R, Scherer PE. Plasma ceramides are elevated in female children and adolescents with type 2 diabetes. J Pediatr Endocrinol Metab (2013) 26(9–10):995–8. doi: 10.1515/jpem-2012-0407
142. Wigger L, Cruciani-Guglielmacci C, Nicolas A, Denom J, Fernandez N, Fumeron F, et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep (2017) 18(9):2269–79. doi: 10.1016/j.celrep.2017.02.019
143. De Groot PCM, Dekkers OM, Romijn JA, Dieben SWM, Helmerhorst FM. PCOS. coronary heart disease, stroke and the influence of obesity: a systematic review and meta-analysis. Hum Reprod Update (2011) 17(4):495–500. doi: 10.1093/humupd/dmr001
144. Hansen SL, Svendsen PF, Jeppesen JF, Hoeg LD, Andersen NR, Kristensen JM, et al. Molecular Mechanisms in Skeletal Muscle Underlying Insulin Resistance in Women Who Are Lean With Polycystic Ovary Syndrome. J Clin Endocrinol Metab (2019) 104(5):1841–54. doi: 10.1210/jc.2018-01771
145. Neeland IJ, Singh S, McGuire DK, Vega GL, Roddy T, Reilly DF, et al. Relation of plasma ceramides to visceral adiposity, insulin resistance and the development of type 2 diabetes mellitus: the Dallas Heart Study. Diabetologia (2018) 61(12):2570–9. doi: 10.1007/s00125-018-4720-1
146. Mänttäri A, Suni J, Sievänen H, Husu P, Vähä-Ypyä H, Valkeinen H, et al. Six-minute walk test: a tool for predicting maximal aerobic power (VO2max) in healthy adults. Clin Physiol Funct Imaging (2018) 38(6):1038–45. doi: 10.1111/cpf.12525
147. Schulz ME, Katunaric B, Hockenberry JC, Gutterman DD, Freed JK. Manipulation of the Sphingolipid Rheostat Influences the Mediator of Flow-Induced Dilation in the Human Microvasculature. J Am Heart Assoc (2019) 8(17):e013153. doi: 10.1161/JAHA.119.013153
148. Camici PG, Crea F. Coronary Microvascular Dysfunction. N Engl J Med (2007) 356(8):830–40. doi: 10.1056/NEJMra061889
149. Chao CYL, Cheing GLY. Microvascular dysfunction in diabetic foot disease and ulceration. Diabetes Metab Res Rev (2009) 25(7):604–14. doi: 10.1002/dmrr.1004
150. Karaca Ü, Schram MT, Houben AJHM, Muris DMJ, Stehouwer CDA. Microvascular dysfunction as a link between obesity, insulin resistance and hypertension. Diabetes Res Clin Pract (2014) 103(3):382–7. doi: 10.1016/j.diabres.2013.12.012
151. Stirban A. Microvascular Dysfunction in the Context of Diabetic Neuropathy. Curr Diabetes Rep (2014) 14(11):1–9. doi: 10.1007/s11892-014-0541-x
152. Wang ZV, Mu J, Schraw TD, Gautron L, Elmquist JK, Zhang BB, et al. PANIC-ATTAC: A mouse model for inducible and reversible β-cell ablation. Diabetes (2008) 57(8):2137–48. doi: 10.2337/db07-1631
153. Sharma AX, Quittner-Strom EB, Lee Y, Johnson JA, Martin SA, Yu X, et al. Glucagon Receptor Antagonism Improves Glucose Metabolism and Cardiac Function by Promoting AMP-Mediated Protein Kinase in Diabetic Mice. Cell Rep (2018) 22(7):1760–73. doi: 10.1016/j.celrep.2018.01.065
154. Xia JY, Sun K, Hepler C, Ghaben AL, Gupta RK, An YA, et al. Acute loss of adipose tissue-derived adiponectin triggers immediate metabolic deterioration in mice. Diabetologia (2018) 61(4):932–41. doi: 10.1007/s00125-017-4516-8
155. Blachnio-Zabielska AU, Hady HR, Markowski AR, Kurianiuk A, Karwowska A, Górski J, et al. Inhibition of Ceramide De Novo Synthesis Affects Adipocytokine Secretion and Improves Systemic and Adipose Tissue Insulin Sensitivity. Int J Mol Sci (2018) 19(12):3995. doi: 10.3390/ijms19123995
156. Sobolesky PM, Harrell TS, Parry C, Venn-Watson S, Janech MG. Feeding a modified fish diet to bottlenose dolphins leads to an increase in serum adiponectin and sphingolipids. Front Endocrinol (Lausanne) (2016) 7:33(APR):33. doi: 10.3389/fendo.2016.00033
157. Chang J, Li Y, Huang Y, Lam KSL, Hoo RLC, Wong WT, et al. Adiponectin prevents diabetic premature senescence of endothelial progenitor cells and promotes endothelial repair by suppressing the p38 MAP kinase/p16INK4A signaling pathway. Diabetes (2010) 59(11):2949–59. doi: 10.2337/db10-0582
158. Shibata S, Tada Y, Asano Y, Hau CS, Kato T, Saeki H, et al. Adiponectin Regulates Cutaneous Wound Healing by Promoting Keratinocyte Proliferation and Migration via the ERK Signaling Pathway. J Immunol (2012) 189(6):3231–41. doi: 10.4049/jimmunol.1101739
159. Jin T, Kim MJ, Heo WI, Park KY, Choi SY, Lee M-K, et al. Adiponectin corrects premature cellular senescence and normalizes antimicrobial peptide levels in senescent keratinocytes. Biochem Biophys Res Commun (2016) 477(4):678–84. doi: 10.1016/j.bbrc.2016.06.119
160. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 371:425–32. doi: 10.1038/372425a0
161. Friedman J. Leptin at 20: An overview. J Endocrinol (2014) 223(1):T1–8. doi: 10.1530/JOE-14-0405
162. Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, et al. Role of leptin in the neuroendocrine response to fasting. Nature (1996) 382:250–2. doi: 10.1038/382250a0
163. Cui H, López M, Rahmouni K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat Rev Endocrinol (2017) 13(6):338–51. doi: 10.1038/nrendo.2016.222
164. El-Haschimi K, Pierroz DD, Hileman SM, Bjørbæk C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest (2000) 105(12):1827–32. doi: 10.1172/JCI9842
165. Enriori PJ, Evans AE, Sinnayah P, Cowley MA. Leptin Resistance and Obesity. Obesity (2006) 14(S8):254S–8S. doi: 10.1038/oby.2006.319
166. Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia Is Required for the Development of Leptin Resistance. Stadler K, ed. PloS One (2010) 5(6):e11376. doi: 10.1371/journal.pone.0011376
167. Bjørbæk C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell (1998) 1(4):619–25. doi: 10.1016/S1097-2765(00)80062-3
168. Bjørbæk C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem (1999) 274(42):30059–65. doi: 10.1074/jbc.274.42.30059
169. Zhao S, Zhu Y, Schultz RD, Li N, He Z, Zhang Z, et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab (2019) 30(4):706–719.e6. doi: 10.1016/j.cmet.2019.08.005
170. Zhao S, Li N, Zhu Y, Straub L, Zhang Z, Wang M-Y, et al. Partial leptin deficiency confers resistance to diet-induced obesity in mice. Mol Metab (2020) 100995:1–12. doi: 10.1016/j.molmet.2020.100995
171. Cai B, Lu Y, Xu M. Acid sphingomyelinase downregulation alleviates vascular endothelial leptin resistance in rats. Acta Pharmacol Sin (2019) 41:650–60. doi: 10.1038/s41401-019-0328-3
172. Ritchie IRW, Gulli RA, Stefanyk LE, Harasim E, Chabowski A, Dyck DJ. Restoration of skeletal muscle leptin response does not precede the exercise-induced recovery of insulin-stimulated glucose uptake in high-fat-fed rats. Am J Physiol Integr Comp Physiol (2011) 300(2):R492–500. doi: 10.1152/ajpregu.00602.2010
173. Bonzón-Kulichenko E, Schwudke D, Gallardo N, Moltó E, Fernández-Agulló T, Shevchenko A, et al. Central Leptin Regulates Total Ceramide Content and Sterol Regulatory Element Binding Protein-1C Proteolytic Maturation in Rat White Adipose Tissue. Endocrinology (2009) 150(1):169–78. doi: 10.1210/en.2008-0505
174. Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, Unger RH. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem (1998) 273(49):32487–90. doi: 10.1074/jbc.273.49.32487
175. Ordoñez M, Presa N, Dominguez-Herrera A, Trueba M, Gomez-Muñoz A. Regulation of adipogenesis by ceramide 1-phosphate. Exp Cell Res (2018) 372(2):150–7. doi: 10.1016/j.yexcr.2018.09.021
176. Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab (2006) 17(9):365–71. doi: 10.1016/j.tem.2006.09.007
177. Samad F, Hester KD, Yang G, Hannun YA, Bielawski J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes (2006) 55(9):2579–87. doi: 10.2337/db06-0330
178. Paolisso G, Tagliamonte MR, Galderisi M, Zito GA, Petrocelli A, Carella C, et al. Plasma Leptin Level Is Associated With Myocardial Wall Thickness in Hypertensive Insulin-Resistant Men. Hypertension (1999) 34(5):1047–52. doi: 10.1161/01.HYP.34.5.1047
179. Ren J, Relling DP. Leptin-induced suppression of cardiomyocyte contraction is amplified by ceramide. Peptides (2006) 27(6):1415–9. doi: 10.1016/j.peptides.2005.11.022
180. Dhopeshwarkar GA, Mead JF. Uptake and transport of fatty acids into the brain and the role of the blood-brain barrier system. Adv Lipid Res (1973) 11:109–42. doi: 10.1016/b978-0-12-024911-4.50010-6
181. Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol - Endocrinol Metab (2009) 296(5):1003–12. doi: 10.1152/ajpendo.90377.2008
182. Sergi D, Morris AC, Kahn DE, McLean FH, Hay EA, Kubitz P, et al. Palmitic acid triggers inflammatory responses in N42 cultured hypothalamic cells partially via ceramide synthesis but not via TLR4. Nutr Neurosci (2020) 23(4):321–34. doi: 10.1080/1028415X.2018.1501533
183. Valassi E, Scacchi M, Cavagnini F. Neuroendocrine control of food intake. Nutr Metab Cardiovasc Dis (2008) 18(2):158–68. doi: 10.1016/j.numecd.2007.06.004
184. Carrasco P, Sahún I, McDonald J, Ramírez S, Jacas J, Gratacós E, et al. Ceramide levels regulated by carnitine palmitoyltransferase 1C control dendritic spine maturation and cognition. J Biol Chem (2012) 287(25):21224–32. doi: 10.1074/jbc.M111.337493
Keywords: leptin, adiponectin, adiponectin receptors, sphingolipids, ceramidase, adipokine
Citation: Field BC, Gordillo R and Scherer PE (2020) The Role of Ceramides in Diabetes and Cardiovascular Disease Regulation of Ceramides by Adipokines. Front. Endocrinol. 11:569250. doi: 10.3389/fendo.2020.569250
Received: 03 June 2020; Accepted: 09 September 2020;
Published: 02 October 2020.
Edited by:
Scott A. Summers, The University of Utah, United StatesReviewed by:
Gary Sweeney, York University, CanadaCopyright © 2020 Field, Gordillo and Scherer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philipp E. Scherer, UGhpbGlwcC5TY2hlcmVyQHV0c291dGh3ZXN0ZXJuLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.