 Laura Van Moortel
Laura Van Moortel Kris Gevaert
Kris Gevaert Karolien De Bosscher
Karolien De Bosscher
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Endocrinol. , 24 September 2020
Sec. Molecular and Structural Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.559673
This article is part of the Research Topic Nuclear Receptors and Coregulators in Metabolism and Immunity View all 14 articles
Exogenous glucocorticoids are widely used in the clinic for the treatment of inflammatory disorders and hematological cancers. Unfortunately, their use is associated with debilitating side effects, including hyperglycemia, osteoporosis, mood swings, and weight gain. Despite the continued efforts of pharma as well as academia, the search for so-called selective glucocorticoid receptor modulators (SEGRMs), compounds with strong anti-inflammatory or anti-cancer properties but a reduced number or level of side effects, has had limited success so far. Although monoclonal antibody therapies have been successfully introduced for the treatment of certain disorders (such as anti-TNF for rheumatoid arthritis), glucocorticoids remain the first-in-line option for many other chronic diseases including asthma, multiple sclerosis, and multiple myeloma. This perspective offers our opinion on why a continued search for SEGRMs remains highly relevant in an era where small molecules are sometimes unrightfully considered old-fashioned. Besides a discussion on which bottlenecks and pitfalls might have been overlooked in the past, we elaborate on potential solutions and recent developments that may push future research in the right direction.
Glucocorticoids (GCs) are endogenous steroidal hormones involved in metabolism, stress, development, and immunity (1). They exert their effects by binding the glucocorticoid receptor (GR), a nuclear receptor (NR) consisting of an intrinsically disordered N-terminal domain (NTD), a central DNA binding domain (DBD), a hinge region (HR), and a C-terminal ligand-binding domain (LBD) (2). Upon ligand binding, GR typically translocates from the cytoplasm to the nucleus where it acts as a genuine transcription factor to regulate target gene expression via multiple mechanisms (Figure 1A), which are discussed in detail in (3). The discovery of the anti-inflammatory effects of endogenous GCs preceded the development of synthetic GCs, which are used to treat, among others, inflammatory disorders, and hematological cancers (4). Unfortunately, the therapeutic efficacy of such exogenous GCs is, particularly for systemic use, overshadowed by an unacceptably high number of undesired side effects such as hyperglycemia, osteoporosis, mood swings, and weight gain (5).
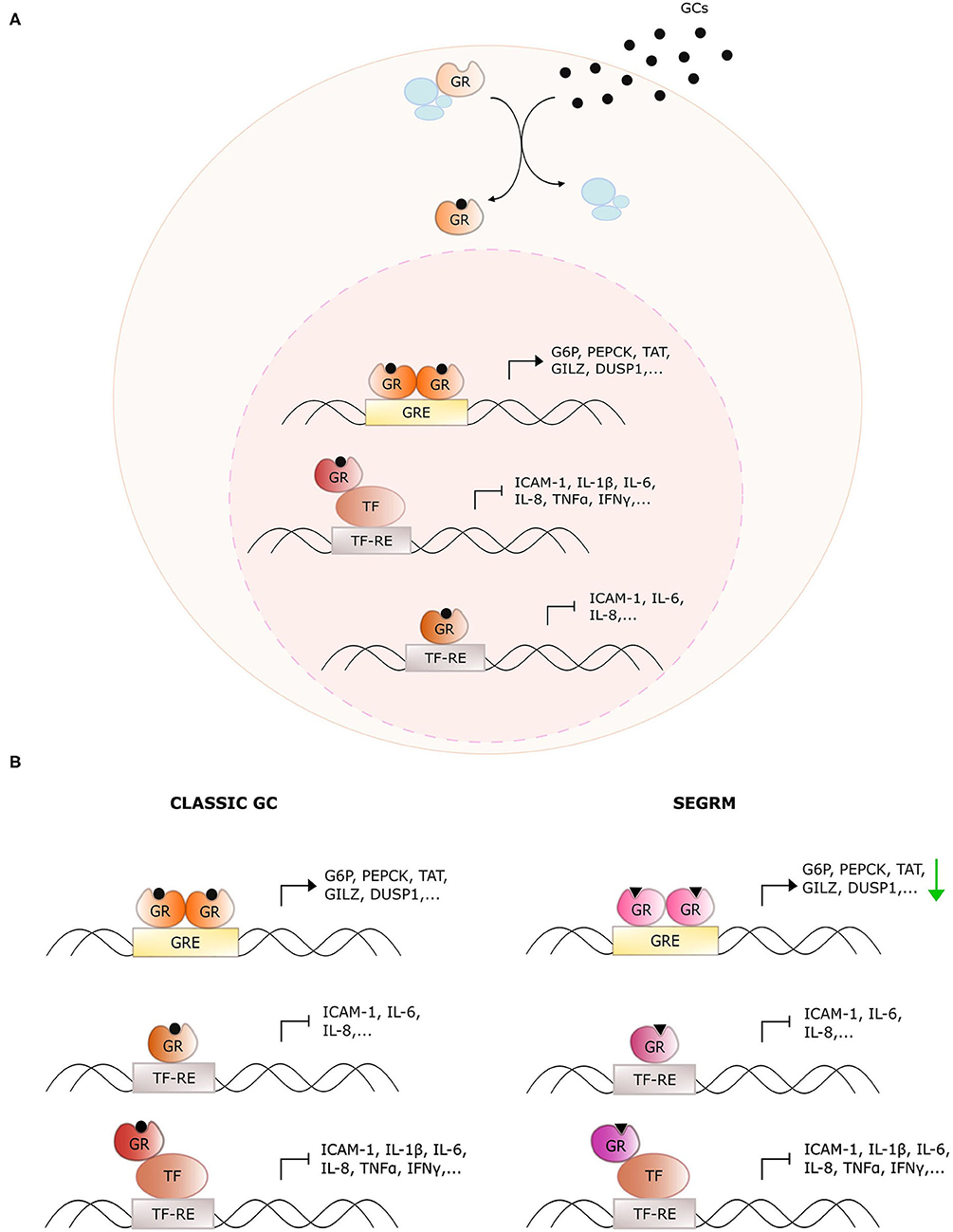
Figure 1. Overview of glucocorticoid receptor activity with classic glucocorticoids and selective GR modulators. (A) General action mechanism of the glucocorticoid receptor (GR). Glucocorticoids (GCs) diffuse through the cellular membrane and bind GR. The latter dissociates from its chaperone complex and migrates to the nucleus. There, it dimerizes and binds glucocorticoid response elements (GREs) to upregulate downstream target genes. Monomeric GR also undergoes protein-protein interactions with DNA-bound pro-inflammatory transcription factors (TFs) to downregulate their activity, or it binds directly to the TF response elements (TF-RE). (B) Distinct actions of classic GCs and selective GR modulators (SEGRMs). In contrast to classic GCs, SEGRMs are hypothesized to reduce GR's capacity to dimerize and therefore reduce GRE-mediated transcription. Interference with TF activity is driven via monomeric GR and therefore maintained with SEGRMs.
Some of these side effects stem from direct binding of homodimeric GR to pseudopalindromic glucocorticoid reponse elements (GREs) in the promoter regions of genes controlling key metabolic pathways (Figure 1). The resulting GRE-driven upregulation of tyrosine aminotransferase (TAT), glucose 6-phosphatase (G6P) and phosphoenolpyruvate carboxykinase (PEPCK) for instance leads to hyperglycemia (6). The suppression of nuclear factor (NF)-κB- and activator protein (AP)-1 activity on the other hand, is typically explained via protein-protein interactions with monomeric GR (called tethering) (7). Despite the controversies on the actual underlying mechanism (see further), the targeting of activities of pro-inflammatory transcription factors undoubtedly contributes substantially to the anti-inflammatory actions of GCs.
The discrepancy between monomer- and dimer-driven effects of GR was first suggested in 1994 with the demonstration that GR with a dimerization-disrupting mutation in the DBD (GRdim) is still able to repress AP-1-driven genes, while no longer able to induce GRE-mediated activation (8). Four years later, Reichardt et al. established that mice carrying this homozygous mutation were viable and healthy, in contrast to GR full knock-out mice (9), arguing for an equally viable mechanistic basis to separate beneficial from undesired effects. This was the starting point of the search for so-called dissociative or selective GR modulators (SEGRMs), GR ligands that can still repress inflammation via monomer-driven NF-κB, and AP-1 inhibition, while no longer inducing GRE-driven side effects (Figure 1B).
In the meantime, a few shades of gray have been added to the original black-and-white monomer-dimer paradigm. First of all, dimer-mediated gene activation also contributes to the anti-inflammatory effects of GCs via the upregulation of anti-inflammatory genes such as glucocorticoid-induced leucine zipper (GILZ) and dual specificity phosphatase (DUSP1) (10). This helps explaining why GRdim mice show increased sensitivity to acute inflammation such as septic shock (11). Secondly, it was shown in cellulo that Dex still promotes dimerization of the GRdim mutant (12). However, introducing an extra point mutation in the GR LBD almost completely disrupted dimerization and abrogated GRE-driven activity, but preserved the inhibition of NF-κB activity. Thirdly, monomeric GR was shown to bind directly to genomic NF-κB and AP-1 response elements, without the need for the transcription factor itself (13, 14). This finding challenges the original tethering hypothesis but still supports the notion that suppression of NF-κB and AP-1 activities does not require GR dimerization. Taken together, given the bodies of evidence on a large contribution of dimeric GR to particular side effects vs. the role of GR monomers to support anti-inflammatory actions in a chronic setting, the notion that compounds that favor signaling via monomeric GR can hold a therapeutic benefit against persistent inflammation, still stands.
The development of successful SEGRMs has proven to be a long and extremely bumpy road. Many compounds that showed promising initial results (listed in Table 1) never got past the pre-clinical stage or failed later on in clinical trials. It is well-known that the success rate for the development of any kind of small molecule drug from bench to clinic is very low, typically starting from 10,000 compounds to end up with one market-approved drug (44, 45). In addition, we believe that in the case of GR, multiple technical, and biological factors have been reducing the prospect to success even further. Fortunately, molecules are still being developed, trying to meet the hope of many patients who would benefit from GR modulators. For instance, AZD7495 (asthma, NCT03622112) and AZD9567 (rheumatoid arthritis, NCT03368235) are currently under evaluation in clinical trials.
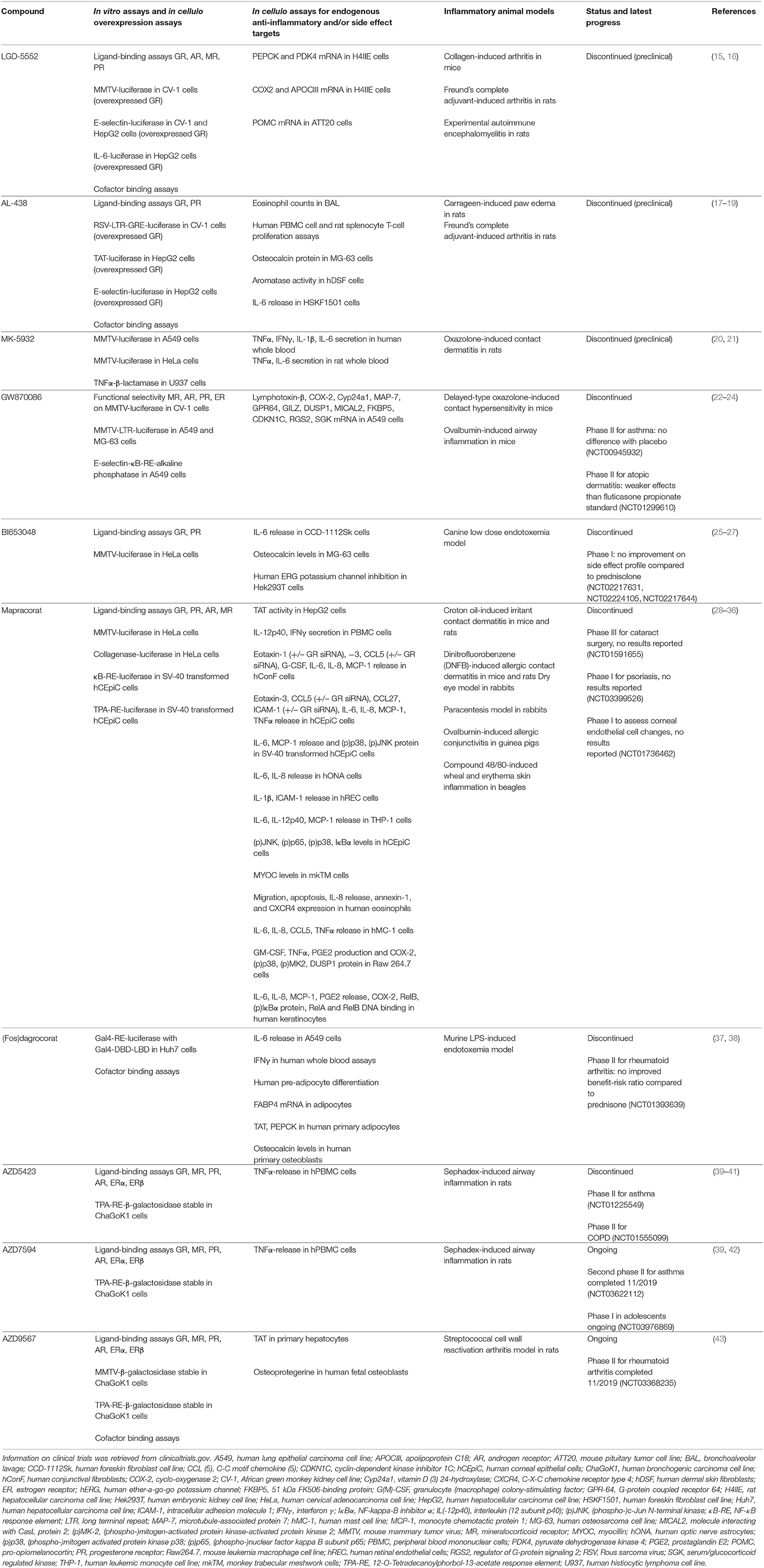
Table 1. Available pre-clinical data of SEGRMs.
This perspective offers our opinion as molecular biologists on the rationale why a continued search for SEGRMs still makes sense and bears significant relevance. We offer our view on a number of bottlenecks and pitfalls that might have hampered research progress in the past and elaborate on which new developments and insights could help overcome these issues.
The need for more selective GR ligands remains highly relevant. Although more targeted therapies have successfully been introduced, such as anti-tumor necrosis factor (TNF) for arthritic disorders and inflammatory bowel diseases (IBD), these therapies are not without limitations. For one, anti-TNF therapy has been associated with a 250% increase in the occurrence of tuberculosis (46). Furthermore, these therapies have been reported to trigger multiple sclerosis (MS) and other demyelinating conditions (47–50). This is in line with the reported disease worsening in patients with pre-existing MS in clinical trials for Lenercept and cA2, two types of anti-TNF therapy (51, 52). Beside such side effects, monoclonal therapies are generally very expensive, laying a huge burden on health care systems, which will only increase with aging populations in western countries. Their price also makes them unaffordable for most people in low income countries, which is particularly a problem for asthma, for which 80% of disease-related deaths occur in low to low-medium income countries (53).
GCs on the other hand are generally much cheaper and are still the first-line treatment for asthma, multiple sclerosis, and multiple myeloma among others (54–56). However, their side effects are a well-known problem and not necessarily limited to patients receiving oral or intravenous GCs. While topical skin treatments, especially with the newest generation glucocorticoids, impose a very low risk for systemic side effects (57–59), topical eye treatments, and inhaled GCs (IGCs) have both been associated with adrenal suppression (60, 61). This can lead to growth retardation in infants and children, who form a large cohort of the asthma patient population. The long-term use of high doses IGCs has also been associated with decreased bone mineral density in both children and adults (62–64). Although the benefits of ocular and IGCs usually outweigh the risks, patients would still benefit from GCs with lower risks for systemic side effects.
Taken together, the need for more selective GCs reaches further than systemic treatments and is also high for ocular and inhalation therapies.
Current tools for screening potential SEGRMs suffer from shortcomings and do not always capture the complexity of GR signaling. First of all, the lack of three-dimensional structures of full-length GR highly restricts our knowledge of GR's structure-activity relationship and decreases the predictive power of molecular modeling and docking studies. Additionally, all existing crystal structures of ligands in complex with GR's LBD were obtained upon the introduction of one or more mutations in this LBD. Although these mutations were predicted not to influence the LBD structure, this can never be claimed with absolute certainty. F602S for instance, one of the most commonly used GR mutations allowing growth of LBD crystals, causes chemical shift perturbations in LBD nuclear magnetic resonance spectra compared to wild-type LBD (65). Furthermore, GR is allosterically regulated through interactions with its corresponding response elements and cofactors (66), and more general also for other NR members, conformational changes in one NR domain can allosterically alter the conformation of another domain within the same NR molecule (67). Thus, most probably the conformation of the LBD studied in isolation is an incorrect reflection of this domain's conformation in the full-length protein.
Further, while high affinity and selectivity for GR can be captured using in vitro ligand-binding assays, confirmations in a cellular or in vivo context are sometimes lacking. This harbors an inherent risk to miss out on off-target effects of the compound in question. Therefore, the confirmation of GR dependency in a cellular and an in vivo context is still an important validation to make, for instance by testing compounds in wild-type vs. GR knock-out models.
Table 1 provides an overview of the assays typically carried out to characterize GR-mediated actions of a set of well-known SEGRMs. To our opinion, a lack of predictive power is one of the problems most difficult to solve, especially when moving from simplified assays to more complex biology. Direct GRE-driven activity, potentially leading to side effects, is almost universally monitored via reporters driven by a mouse mammary tumor virus (MMTV) promoter. Although a fast and straightforward and thus defendable method for initial compound characterization, a GRE-driven reporter assay can be a poor predictor for regulation of endogenous GRE-driven genes, as was also observed for MMTV (15, 22). The effects of GCs are highly gene-specific and GRE-driven activity can differ depending on the sequence of the GRE and the surrounding chromatin environment (68–70). The use of overexpressed GR should also be avoided in such assays, as this may lead to compound potencies and efficacies that are not necessarily representative for an endogenous context. Additionally, not all side effects are dimer-driven and are therefore not predictable via GRE-driven reporters. Mimicking the right gene- and context-specificity of GR activity remains one of the greatest challenges. Making a switch from reporters driven by minimal recombinant promoters to more physiologically relevant promoters could already offer some benefit. These promoters would ideally belong to genes that are confirmed mediators of underlying therapeutic - or side effects. Validation on a well-representative set of relevant endogenous target genes is even more important (see below, section Potential Solutions: The Way Forward).
Cell- and tissue-specificity of GC actions is another variable parameter. The MMTV-driven reporter for instance showed stronger upregulation by GW870086 in bone osteosarcoma cells compared to lung epithelial carcinoma cells (22). It thus remains essential to screen compounds in cell types that are the best proxies for the underlying therapeutic and/or side effects in vivo, for instance the use of hepatocytes to study effects on glucose and lipid metabolism, or the use of osteoblast or osteoclast cell lines for drugs that would be used in arthritis patients. Further, although characterization of compound activity in cellulo is essential, this will always be an oversimplification of the situation in a living organism. Therefore, validation of an improved therapeutic benefit depends on representative animal models. While this is readily implemented for anti-inflammatory effect scoring, concomitant testing of side effect parameters (such as glucose tolerance, insulin tolerance, cortisol levels, bone mineral density) presents a bottleneck, because a longer treatment protocol may be needed to surpass the thresholds of measurable results for these parameters or because of species differences (see below) (22, 43).
Lack of translatability from animal models to human patients is yet another hurdle to overcome. Differences in ligand activity between species can be an underlying cause, as observed for AL-438 and MK-5932, which both had stronger anti-inflammatory effects in rat vs. human blood (17, 20). While it would be recommended to perform initial cellular tests in human cells as much as possible, in vivo interspecies differences remain a hurdle in the entire field of drug discovery and are currently difficult to overcome. Another concern is when animal models used to study a particular disease insufficiently mimic the pathology observed in humans. A careful design and set-up of animal models remains key to study anti-inflammatory as well as side effects. If a well-known side effect (marker) of a classic GC in man is not observed in the animal model used, this model will obviously have no predictive power on (markers of) this particular side effect in patients and will therefore be unsuited to evaluate the improved benefit-risk ratio of SEGRMs over classic GCs. For instance, in a canine model of low dose endotoxemia used to investigate the anti-inflammatory and bone-sparing effects of BI653048, neither BI653048 nor prednisolone treatment affected osteocalcin levels (25). However, prednisolone does reduce bone mineral density in dogs and decreases bone formation markers in humans after 1 day (71, 72). Indeed, in a phase I clinical trial, BI653048, and prednisolone both caused decreased serum osteocalcin levels (26). Studies with other SEGRMs also concluded that osteocalcin levels in cellulo do not always reflect in vivo decreases in bone density (18, 26, 27, 37, 38), casting doubts on the value of osteocalcin as proxy for the in vivo reduction of bone mineral density.
Lastly, notwithstanding the notion that dissociating GCs may improve the benefit-risk ratio in chronic inflammatory disorders, a portion of the anti-inflammatory effects of GR does remain dimer-driven (73). Hence, the likelihood decreases for truly dissociating compounds to match the therapeutic efficacy of the strongest classic GCs. Taking into account that some side effects, such as osteoporosis, are at least partially mediated by monomeric GR (18), makes the quest to find a SEGRM that scores better on multiple side effects even more challenging.
Although pre-clinical characterization of compounds will never suffice to accurately predict their effects in patients, particular improvements on current screenings could increase the predictive power. First of all, reporter genes driven by physiological promoters relevant for the clinical context of the tested SEGRM should be preferred over artificial promoters. An example could be the use of the G6P- or PEPCK-promoters in liver cells to monitor hyperglycemia (74, 75), or a Runt-related transcription factor (Runx)2-driven promoter in osteoblasts or Smad-driven promoters in osteoclasts as markers for GC-induced osteoporosis (76, 77). A consistent and thorough screening of endogenous targets in a relevant human cellular context adds to importance. While monitoring GR activity in every targeted pathway for every compound is impossible to achieve, identification of reliable in cellulo biomarkers with a higher predictive power for species-independent in vivo anti-inflammatory and/or side effects would be a tremendous help. This requires a full understanding of the molecular mechanisms driving both anti-inflammatory and side effects in human tissues as well as in animal models. This is, particularly for side effects, not always the case. Continued efforts to unravel the underlying molecular mechanisms driving particular GC side effects are therefore crucial. However, some important side effect mediators have already been identified and could be suitable markers. Examples are muscle ring finger (MuRF)1, atrogin-1, and Krüppel-like factor (KLF)15 in muscle atrophy (78), regulated in development and DNA damage response (REDD)1 in skin (and muscle) atrophy (79, 80), and G6P and PEPCK in liver (74, 75). In bone, the upregulation of cleaved caspase 3 and −9 or the reduction of bone morphogenetic protein (BMP)2 and Runx2 activity are important predictors for reduction in osteoblast numbers (81, 82), while upregulation of receptor activator of nuclear factor-κB ligand (RANKL)-RANK signaling and cathepsin K activity are important markers for increased osteoclast differentiation and activity (respectively) (76, 83).
Reduction of publication bias toward “negative results” and joining forces between pharmaceutical companies and academic groups should push the current boundaries and drive research forward. At times, underlying reasons for discontinuation of (pre-)clinical research remain enigmatic. As one concrete example of many other examples that can be brought forward, results from three completed phase III clinical trials on the use of Mapracorat for post-operative treatment of cataract surgery (NCT01230125, NCT01591161, and NCT01591655) await publication, leaving fundamental scientists on the sideline wondering why Mapracorat was never market approved. More insights on where exactly discontinued SEGRMs failed, if those reasons are on the scientific level, will encourage academic labs with the right expertise to dig deeper into the underlying causes, and create feedback-knowledge that may flow back to industrial programs.
Even though fully dissociating SEGRMs might never reach the therapeutic efficacy of the most potent classic GCs, they can still offer relevant therapeutic benefit. Many inflammatory disorders are characterized by a disease course that alternates between periods of remission and exacerbation or relapse. SEGRMs might not trigger the full-on anti-inflammatory cascade that is required to suppress an exacerbation, but might be ideal for maintenance therapy. To maintain disease control, lower GC doses often suffice. SEGRMs could match the anti-inflammatory efficacy of the lower dose classic GCs while still showing a reduced side effect burden. Combination of classic GCs with SEGRMs or other therapeutic agents is another strategy to increase benefit-risk ratios. Combination of Dex with CpdA was for instance shown to increase anti-inflammatory effects while reducing GRE-driven signaling in cellulo (84). Finally, the development of compounds that do not bind the classic ligand-binding pocket but instead target the dimerization interface might be an interesting alternative strategy to disrupt GRE-driven signaling.
The intrinsically disordered nature of the GR NTD (85), has so far prohibited resolving a crystal structure of full-length GR. However, some smaller (however technically still challenging) advances could already lead to important new insights. Crystal structures of wild-type LBD in absence of stabilizing mutations would already give more confidence in the reliability of current docking approaches. Secondly, crystal structures of the DBD-hinge-LBD portion would not only lead to a better understanding of the structure-activity relationship of GR, but might pose extra advantages for molecular modeling or docking studies. Since efficient GR dimerization seems to require both DBD and LBD (12), crystal structures of at least the DBD-hinge-LBD portion of GR should improve predictions on those molecular entities that are truly dimer-disrupting.
Another important emerging strategy to find more efficacious GCs is to minimize exposure to non-inflamed tissues. IGCs can for instance be optimized to undergo rapid elimination once they enter the systemic circulation, a strategy that was applied for the development of AZD7594 (39). For systemic GCs, the use of liposomal formulations is showing very promising results. While these not only improve distribution to tissues that are anatomically difficult to reach (86–88), they can lower the side effects of systemic GCs by maximizing concentrations at the inflamed tissues while minimizing distribution to other tissues (89, 90).
While it may be utopia to try and develop compounds that alleviate all side effects, improved profiles for particular side effects may be achievable. Skin thinning and ocular hypertension are for instance among the most problematic side effects for topical and ocular GC treatments, respectively (5). For systemic treatments, liposomal formulations in combination with selective improvement of particular side effects may be a viable way forward. Liposomal SEGRMs that do not affect bone metabolism might for instance have an increased benefit-risk ratio over classic GCs for the treatment of arthritic disorders.
While there still seems a long road ahead toward SEGRMs with a real improved benefit-risk ratio, there is light at the end of the tunnel. The pipeline of SEGRM compounds under clinical evaluation is not empty and new insights from ongoing (or future) research is expected to lead to optimized screening tools with maximized predictive power. Additionally, strategies to limit exposure to off-targets tissues, such as liposomal formulations for systemic treatments, show promising results (86–90). Combination of these approaches with the identification of reliable markers to predict on-target side effects, (e.g., ocular hypertension in ocular treatment, osteoporosis in rheumatoid arthritis, skin thinning in topic applications) may be an effective and achievable leap forward.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
LVM wrote the manuscript with contributions from KG and KDB. LVM made the artwork with contributions from KG and KDB. All authors approved the final version.
LVM was supported by a strategic basic research fellowship of the Research Foundation-Flanders (FWO-Vlaanderen), grant number 1S14720N.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Tan C, Wahli W. A trilogy of glucocorticoid receptor actions. Proc Natl Acad Sci USA. (2016) 113:1115–7. doi: 10.1073/pnas.1524215113
2. Oakley R, Cidlowski J. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. (2013) 132:1033–44. doi: 10.1016/j.jaci.2013.09.007
3. Ramamoorthy S, Cidlowski JA. Corticosteroids. mechanisms of action in health and disease. Rheum Dis Clin N Am. (2016) 42:15–31. doi: 10.1016/j.rdc.2015.08.002
4. Caratti G, Matthews L, Poolman T, Kershaw S, Baxter M, Ray D. Glucocorticoid receptor function in health and disease. Clin Endocrinol. (2015) 83:441–8. doi: 10.1111/cen.12728
5. Schäcke H, Döcke W-D, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. (2002) 96:23–43. doi: 10.1016/S0163-7258(02)00297-8
6. Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck I, et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol. (2013) 380:41–54. doi: 10.1016/j.mce.2012.12.014
7. De Bosscher K, Haegeman G. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol. (2009) 23:281–91. doi: 10.1210/me.2008-0283
8. Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. (1994) 13:4087–95. doi: 10.1002/j.1460-2075.1994.tb06726.x
9. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. (1998) 93:531–41. doi: 10.1016/S0092-8674(00)81183-6
10. Clark A, Belvisi M. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther. (2012) 134:54–67. doi: 10.1016/j.pharmthera.2011.12.004
11. Kleiman A, Hübner S, Parkitna JMR, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. (2012) 26:722–9. doi: 10.1096/fj.11-192112
12. Presman DM, Ogara MF, Stortz M, Alvarez LD, Pooley JR, Schiltz RL, et al. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PLoS Biol. (2014) 12:e1001813. doi: 10.1371/journal.pbio.1001813
13. Weikum ER, de Vera IMS, Nwachukwu JC, Hudson WH, Nettles KW, Kojetin DJ, et al. Tethering not required : the glucocorticoid receptor binds directly to activator protein-1 recognition motifs to repress inflammatory genes. Nucleic Acids Res. (2017) 45:8596–608. doi: 10.1093/nar/gkx509
14. Hudson WH, de Vera IMS, Nwachukwu JC, Weikum ER, Herbst AG, Yang Q, et al. Cryptic glucocorticoid receptor-binding sites pervade genomic NF-κB response elements. Nat Commun. (2018) 9:1337. doi: 10.1038/s41467-018-03780-1
15. Miner JN, Ardecky B, Benbatoul K, Griffiths K, Larson CJ, Mais DE, et al. Antiinflammatory glucocorticoid receptor ligand with reduced side effects exhibits an altered protein-protein interaction profile. Proc Natl Acad Sci USA. (2007) 104:19244–9. doi: 10.1073/pnas.0705517104
16. López F, Ardecky R, Bebo B, Benbatoul K, de Grandpre L, Liu S, et al. LGD-5552, an antiinflammatory glucocorticoid receptor ligand with reduced side effects, in vivo. Endocrinology. (2008) 149:2080–89. doi: 10.1210/en.2007-1353
17. Kym PR, Kort ME, Coghlan MJ, Moore JL, Tang R, Ratajczyk JD, et al. Nonsteroidal selective glucocorticoid modulators: the effect of C-10 substitution on receptor selectivity and functional potency of 5-allyl-2,5-dihydro-2,2,4-trimethyl-lh-[1]benzopyrano[3,4-f]quinolines. J Med Chem. (2003) 46:1016–30. doi: 10.1021/jm020335m
18. Coghlan M, Jacobson P, Lane B, Nakane M, Lin CW, Elmore S, et al. A novel antiinflammatory maintains glucocorticoid efficacy with reduced side effects. Mol Endocrinol. (2003) 17:860–9. doi: 10.1210/me.2002-0355
19. Elmore SW, Miner JN, Jacobson PB, Wilcox DM, Lane BC, Coghlan MJ, et al. Nonsteroidal selective glucocorticoid modulators: the effect of C-5 alkyl substitution on the transcriptional activation/repression profile of 2,5-dihydro-10-methoxy-2,2,4-trimethyl-1H-[1]benzopyrano[3,4-f]quinolines. J Med Chem. (2001) 44:4481–91. doi: 10.1021/jm010367u
20. Brandish PE, Anderson K, Baltus GA, Bai C, Bungard CJ, Bunting P, et al. The preclinical efficacy, selectivity and pharmacologic profile of MK-5932, an insulin-sparing selective glucocorticoid receptor modulator. Eur J Pharmacol. (2014) 724:102–11. doi: 10.1016/j.ejphar.2013.12.031
21. Bungard CJ, Hartman GD, Manikowski JJ, Perkins JJ, Bai C, Brandish PE, et al. Discovery of selective glucocorticoid receptor modulator MK-5932. Bioorg Med Chem. (2011) 19:7374–86. doi: 10.1016/j.bmc.2011.10.054
22. Uings IJ, Needham D, Matthews J, Haase M, Austin R, Angell D, et al. Discovery of GW870086: a potent anti-inflammatory steroid with a unique pharmacological profile. Br J Pharmacol. (2013) 169:1389–403. doi: 10.1111/bph.12232
23. Bareille P, Hardes K, Donald AC. Efficacy and safety of once-daily GW870086 a novel selective glucocorticoid in mild-moderate asthmatics: a randomised, two-way crossover, controlled clinical trial. J Asthma. (2013) 50:1077–82. doi: 10.3109/02770903.2013.837480
24. Dölle S, Hielscher N, Bareille PJ, Hardes K, Robertson J, Worm M. Clinical efficacy and tolerability of a novel selective corticosteroid in atopic dermatitis - two randomised controlled trials. Skin Pharmacol Physiol. (2015) 28:159–66. doi: 10.1159/000367696
25. Bartko J, Derhaschnig U, Neels T, Nabozny GH, Harcken C, Leuschner J, et al. Selective glucocorticoid receptor modulation inhibits cytokine responses in a canine model of mild endotoxemia. Pharmacol Res. (2017) 125:215–23. doi: 10.1016/j.phrs.2017.09.006
26. Harcken C, Scholl P, Nabozny G, Thomson D, Bianchi D. Clinical profile of the functionally selective glucocorticoid receptor agonist BI 653048 in healthy male subjects. Expert Opin Investig Drugs. (2019) 28:489–96. doi: 10.1080/13543784.2019.1599859
27. Harcken C, Riether D, Liu P, Razavi H, Patel U, Lee T, et al. Optimization of drug-like properties of nonsteroidal glucocorticoid mimetics and identification of a clinical candidate. ACS Med Chem Lett. (2014) 5:1318–23. doi: 10.1021/ml500387y
28. Pfeffer BA, DeWitt CA, Salvador-Silva M, Cavet ME, López FJ, Ward KW. Reduced myocilin expression in cultured monkey trabecular meshwork cells induced by a selective glucocorticoid receptor agonist: comparison with steroids. Invest Ophthalmol Vis Sci. (2010) 51:437–46. doi: 10.1167/iovs.09-4202
29. Shafiee A, Bucolo C, Budzynski E, Ward KW, López FJ. In vivo ocular efficacy profile of mapracorat, a novel selective glucocorticoid receptor agonist, in rabbit models of ocular disease. Invest Ophthalmol Vis Sci. (2011) 52:1422–30. doi: 10.1167/iovs.10-5598
30. Schäcke H, Zollner T, Döcke W-D, Rehwinkel H, Jaroch S, Skuballa W, et al. Characterization of ZK 245186, a novel, selective glucocorticoid receptor agonist for the topical treatment of inflammatory skin diseases. Br J Pharmacol. (2009) 158:1088–1103. doi: 10.1111/j.1476-5381.2009.00238.x
31. Zhang J-Z, Cavet ME, vanDerMeid KR, Salvador-Silva M, López FJ, Ward KW. BOL-303242-X, a novel selective glucocorticoid receptor agonist, with full anti-inflammatory properties in human ocular cells. Mol Vis. (2009) 15:2606–16.
32. Baiula M, Spartà A, Bedini A, Carbonari G, Bucolo C, Ward KW, et al. Eosinophil as a cellular target of the ocular anti-allergic action of mapracorat, a novel selective glucocorticoid receptor agonist. Mol Vis. (2011) 17:3208–23.
33. Baiula M, Bedini A, Baldi J, Cavet ME, Govoni P, Spampinato S. Mapracorat, a selective glucocorticoid receptor agonist, causes apoptosis of eosinophils infiltrating the conjunctiva in late-phase experimental ocular allergy. Drug Des Devel Ther. (2014) 8:745–57. doi: 10.2147/DDDT.S62659
34. Cavet ME, Harrington KL, Ward KW, Zhang J-Z. Mapracorat, a novel selective glucocorticoid receptor agonist, inhibits hyperosmolar-induced cytokine release and MAPK pathways in human corneal epithelial cells. Mol Vis. (2010) 16:1791–800.
35. Spinelli SL, Xi X, McMillan DH, Woeller CF, Richardson ME, Cavet ME, et al. Mapracorat, a selective glucocorticoid receptor agonist, upregulates RelB, an anti-inflammatory nuclear factor-kappaB protein, in human ocular cells. Exp Eye Res. (2014) 127:290–8. doi: 10.1016/j.exer.2014.07.013
36. Vollmer TR, Stockhausen A, Zhang J-Z. Anti-inflammatory effects of mapracorat, a novel selective glucocorticoid receptor agonist, is partially mediated by MAP kinase phosphatase-1 (MKP-1). J Biol Chem. (2012) 287:35212–21. doi: 10.1074/jbc.M112.400671
37. Hu X, Du S, Tunca C, Braden T, Long K, Lee J, et al. The antagonists but not partial agonists of glucocorticoid receptor ligands show substantial side effect dissociation. Endocrinology. (2011) 152:3123–34. doi: 10.1210/en.2010-1447
38. Buttgereit F, Strand V, Lee EB, Simon-Campos A, McCabe D, Genet A, et al. Fosdagrocorat (PF-04171327) versus prednisone or placebo in rheumatoid arthritis: a randomised, double-blind, multicentre, phase IIb study. RMD Open. (2019) 5:e000889. doi: 10.1136/rmdopen-2018-000889
39. Hemmerling M, Nilsson S. Selective Nonsteroidal Glucocorticoid Receptor Modulators for the Inhaled Treatment of Pulmonary Diseases | Journal of Medicinal Chemistry (2017). Available online at: https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.7b01215 (accessed April 2, 2020).
40. Kuna P, Aurivillius M, Jorup C, Prothon S, Taib Z, Edsbäcker S. Efficacy and tolerability of an inhaled selective glucocorticoid receptor modulator – AZD5423 – in chronic obstructive pulmonary disease patients: Phase II study results. Basic Clin Pharmacol Toxicol. (2017) 121:279–89. doi: 10.1111/bcpt.12768
41. Gauvreau GM, Boulet L-P, Leigh R, Cockcroft DW, Killian KJ, Davis BE, et al. A nonsteroidal glucocorticoid receptor agonist inhibits allergen-induced late asthmatic responses. Am J Respir Crit Care Med. (2014) 191:161–7. doi: 10.1164/rccm.201404-0623OC
42. Brown MN, Fuhr R, Beier J, Su H-L, Chen Y, Forsman H, et al. Efficacy and safety of AZD7594, an inhaled non-steroidal selective glucocorticoid receptor modulator, in patients with asthma: a phase 2a randomized, double blind, placebo-controlled crossover trial. Respir Res. (2019) 20:37. doi: 10.1186/s12931-019-1000-7
43. Ripa L, Edman K, Dearman M, Edenro G, Hendrickx R, Ullah V, et al. Discovery of a novel oral glucocorticoid receptor modulator (AZD9567) with improved side effect profile. J Med Chem. (2018) 61:1785–99. doi: 10.1021/acs.jmedchem.7b01690
44. Brake K, Gumireddy A, Tiwari A, Chauhan H, Kumari D. In vivo studies for drug development via oral delivery: challenges, animal models and techniques. Pharm Anal Acta. (2017) 8:1–11. doi: 10.4172/2153-2435.1000560
45. Takebe T, Imai R, Ono S. The current status of drug discovery and development as originated in United States academia: the influence of industrial and academic collaboration on drug discovery and development. Clin Transl Sci. (2018) 11:597–606. doi: 10.1111/cts.12577
46. Minozzi S, Bonovas S, Lytras T, Pecoraro V, González-Lorenzo M, Bastiampillai AJ, et al. Risk of infections using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: a systematic review and meta-analysis. Expert Opin Drug Saf. (2016) 15:11–34. doi: 10.1080/14740338.2016.1240783
47. Kemanetzoglou E, Andreadou E. CNS demyelination with TNF-α blockers. Curr Neurol Neurosci Rep. (2017) 17:36. doi: 10.1007/s11910-017-0742-1
48. Dreyer L, Magyari M, Laursen B, Cordtz R, Sellebjerg F, Locht H. Risk of multiple sclerosis during tumour necrosis factor inhibitor treatment for arthritis: a population-based study from DANBIO and the Danish multiple sclerosis registry. Ann Rheum Dis. (2016) 75:785–6. doi: 10.1136/annrheumdis-2015-208490
49. Engel S, Luessi F, Mueller A, Schopf RE, Zipp F, Bittner S. PPMS onset upon adalimumab treatment extends the spectrum of anti-TNF-α therapy-associated demyelinating disorders. Ther Adv Neurol Disord. (2020) 13:1756286419895155. doi: 10.1177/1756286419895155
50. Honda Y, Otsuka A, Egawa G, Inoue Y, Kuzuya A, Takahashi R, et al. Multiple neurological abnormalities, including pontine hemorrhage, multiple sclerosis and aseptic meningitis, during anti-TNF-α therapy in psoriatic arthritis. Eur J Dermatol. (2015) 25:487–8. doi: 10.1684/ejd.2015.2558
51. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. Neurology. (2011) 77:1382. doi: 10.1212/01.wnl.0000406608.85830.82
52. van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, von Blomberg BME, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. (1996) 47:1531–4. doi: 10.1212/WNL.47.6.1531
53. WHO. Global Surveillance, Prevention and Control of Chronic Respiratory Diseases (2007). Available online at: https://www.who.int/respiratory/publications/global_surveillance/en/ (accessed April 23, 2020).
54. Nazareth T, Datar M, Yu T-C. Treatment effectiveness for resolution of multiple sclerosis relapse in a US health plan population. Neurol Ther. (2019) 8:383–95. doi: 10.1007/s40120-019-00156-5
55. Burwick N, Sharma S. Glucocorticoids in multiple myeloma: past, present, and future. Ann Hematol. (2019) 98:19–28. doi: 10.1007/s00277-018-3465-8
56. Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention. Global Initiative for Asthma - GINA. (2019). Available online at: https://ginasthma.org/gina-reports/ (accessed April 3, 2020).
57. Mooney E, Rademaker M, Dailey R, Daniel BS, Drummond C, Fischer G, et al. Adverse effects of topical corticosteroids in paediatric eczema: Australasian consensus statement. Australas J Dermatol. (2015) 56:241–51. doi: 10.1111/ajd.12313
58. Daniel BS, Orchard D. Ocular side–effects of topical corticosteroids: what a dermatologist needs to know. Australas J Dermatol. (2015) 56:164–9. doi: 10.1111/ajd.12292
59. Torrelo A. Methylprednisolone aceponate for atopic dermatitis. Int J Dermatol. (2017) 56:691–7. doi: 10.1111/ijd.13485
60. Bangsgaard R, Main KM, Boberg-Ans G, la Cour M, Forman JL, Haargaard B, et al. Adrenal suppression in infants treated with topical ocular glucocorticoids. Ophthalmology. (2018) 125:1638–43. doi: 10.1016/j.ophtha.2018.04.035
61. Hawcutt DB, Francis B, Carr DF, Jorgensen AL, Yin P, Wallin N, et al. Susceptibility to corticosteroid-induced adrenal suppression: a genome-wide association study. Lancet Respir Med. (2018) 6:442–50. doi: 10.1016/S2213-2600(18)30058-4
62. Gonzalez AV, Coulombe J, Ernst P, Suissa S. Long-term use of inhaled corticosteroids in COPD and the risk of fracture. Chest. (2018) 153:321–8. doi: 10.1016/j.chest.2017.07.002
63. Skoner DP. Inhaled corticosteroids: effects on growth and bone health. Ann Allergy Asthma Immunol. (2016) 117:595–600. doi: 10.1016/j.anai.2016.07.043
64. Wolfgram PM, Allen DB. Effects of inhaled corticosteroids on growth, bone metabolism, and adrenal function. Adv Pediatr. (2017) 64:331–45. doi: 10.1016/j.yapd.2017.03.006
65. Köhler C, Carlström G, Tångefjord S, Papavoine T, Lepistö M, Edman K, et al. Backbone 1H, 13C, and 15N resonance assignments of the ligand binding domain of the human wildtype glucocorticoid receptor and the F602S mutant variant. Biomol NMR Assign. (2018) 12:263–8. doi: 10.1007/s12104-018-9820-9
66. Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol. (2017) 18:159–74. doi: 10.1038/nrm.2016.152
67. Ricci CG, Silveira RL, Rivalta I, Batista VS, Skaf MS. Allosteric pathways in the PPARγ-RXRα nuclear receptor complex. Sci Rep. (2016) 6:19940. doi: 10.1038/srep19940
68. Meijsing S, Pufall M, So A, Bates D, Chen L, Yamamoto K. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. (2009) 324:407–10. doi: 10.1126/science.1164265
69. Telorac J, Prykhozhij S, Schöne S, Meierhofer D, Sauer S, Thomas-Chollier M, et al. Identification and characterization of DNA sequences that prevent glucocorticoid receptor binding to nearby response elements. Nucleic Acids Res. (2016) 44:6142–56. doi: 10.1093/nar/gkw203
70. Schöne S, Jurk M, Helabad M, Dror I, Lebars I, Kieffer B, et al. Sequences flanking the core-binding site modulate glucocorticoid receptor structure and activity. Nat Commun. (2016) 7:12621. doi: 10.1038/ncomms12621
71. Kauh E, Mixson L, Malice M-P, Mesens S, Ramael S, Burke J, et al. Prednisone affects inflammation, glucose tolerance, and bone turnover within hours of treatment in healthy individuals. Eur J Endocrinol. (2012) 166:459–67. doi: 10.1530/EJE-11-0751
72. Costa LAVS, Lopes BF, Lanis AB, Oliveira DCD, Giannotti JG, Costa FS. Bone demineralization in the lumbar spine of dogs submitted to prednisone therapy. J Vet Pharmacol Ther. (2010) 33:583–6. doi: 10.1111/j.1365-2885.2010.01174.x
73. Desmet S, De Bosscher K. Glucocorticoid receptors : finding the middle ground. J Clin Invest. (2017) 127:1136–45. doi: 10.1172/JCI88886
74. Imai E, Stromstedt PE, Quinn PG, Carlstedt-Duke J, Gustafsson JA, Granner DK. Characterization of a complex glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol. (1990) 10:4712–9. doi: 10.1128/MCB.10.9.4712
75. Schmoll D, Allan BB, Burchell A. Cloning and sequencing of the 5′ region of the human glucose-6-phosphatase gene: transcriptional regulation by cAMP, insulin and glucocorticoids in H4IIE hepatoma cells. FEBS Lett. (1996) 383:63–6. doi: 10.1016/0014-5793(96)00224-4
76. Fennen M, Pap T, Dankbar B. Smad-dependent mechanisms of inflammatory bone destruction. Arthritis Res Ther. (2016) 18:279. doi: 10.1186/s13075-016-1187-7
77. Morimoto E, Li M, Khalid AB, Krum SA, Chimge NO, Frenkel B. Glucocorticoids Hijack Runx2 to stimulate Wif1 for suppression of osteoblast growth and differentiation. J Cell Physiol. (2017) 232:145–53. doi: 10.1002/jcp.25399
78. Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda SI, et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. (2011) 13:170–82. doi: 10.1016/j.cmet.2011.01.001
79. Baida G, Bhalla P, Kirsanov K, Lesovaya E, Yakubovskaya M, Yuen K, et al. REDD1 functions at the crossroads between the therapeutic and adverse effects of topical glucocorticoids. EMBO Mol Med. (2015) 7:42–58. doi: 10.15252/emmm.201404601
80. Britto FA, Begue G, Rossano B, Docquier A, Vernus B, Sar C, et al. REDD1 deletion prevents dexamethasone-induced skeletal muscle atrophy. AJP Endocrinol Metab. (2014) 307:E983–93. doi: 10.1152/ajpendo.00234.2014
81. Lin H, Gao X, Chen G, Sun J, Chu J, Jing K, et al. Indole-3-carbinol as inhibitors of glucocorticoid-induced apoptosis in osteoblastic cells through blocking ROS-mediated Nrf2 pathway. Biochem Biophys Res Commun. (2015) 460:422–7. doi: 10.1016/j.bbrc.2015.03.049
82. Liu K, Jing Y, Zhang W, Fu X, Zhao H, Zhou X, et al. Silencing miR-106b accelerates osteogenesis of mesenchymal stem cells and rescues against glucocorticoid-induced osteoporosis by targeting BMP2. Bone. (2017) 97:130–8. doi: 10.1016/j.bone.2017.01.014
83. Lin NY, Chen CW, Kagwiria R, Liang R, Beyer C, Distler A, et al. Inactivation of autophagy ameliorates glucocorticoid-induced and ovariectomy-induced bone loss. Ann Rheum Dis. (2015) 75:1203–10. doi: 10.1136/annrheumdis-2015-207240
84. Desmet SJ, Bougarne N, Van Moortel L, de Cauwer L, Thommis J, Vuylsteke M, et al. Compound A influences gene regulation of the Dexamethasone- activated glucocorticoid receptor by alternative cofactor recruitment. Sci Rep. (2017) 7:e8063. doi: 10.1038/s41598-017-07941-y
85. Khan SH, McLaughlin WA, Kumar R. Site-specific phosphorylation regulates the structure and function of an intrinsically disordered domain of the glucocorticoid receptor. Sci Rep. (2017) 7:15440. doi: 10.1038/s41598-017-15549-5
86. Mamelle E, Kechai NE, Granger B, Sterkers O, Bochot A, Agnely F, et al. Effect of a liposomal hyaluronic acid gel loaded with dexamethasone in a guinea pig model after manual or motorized cochlear implantation. Eur Arch Otorhinolaryngol. (2017) 274:729–36. doi: 10.1007/s00405-016-4331-8
87. Altamirano-Vallejo JC, Navarro-Partida J, Gonzalez-De la Rosa A, Hsiao JH, Olguín-Gutierrez JS, Gonzalez-Villegas AC, et al. Characterization and pharmacokinetics of triamcinolone acetonide-loaded liposomes topical formulations for vitreoretinal drug delivery. J Ocul Pharmacol Ther. (2018) 34:416–25. doi: 10.1089/jop.2017.0099
88. Deshantri AK, Fens MH, Ruiter RWJ, Metselaar JM, Storm G, van Bloois L, et al. Liposomal dexamethasone inhibits tumor growth in an advanced human-mouse hybrid model of multiple myeloma. J Controlled Release. (2019) 296:232–40. doi: 10.1016/j.jconrel.2019.01.028
89. Meka RR, Venkatesha SH, Acharya B, Moudgil KD. Peptide-targeted liposomal delivery of dexamethasone for arthritis therapy. Nanomed. (2019) 14:1455–69. doi: 10.2217/nnm-2018-0501
Keywords: glucocorticoids, glucocorticoid receptor, selective GR modulators, drug discovery, inflammation, assay development, GR, SEGRM
Citation: Van Moortel L, Gevaert K and De Bosscher K (2020) Improved Glucocorticoid Receptor Ligands: Fantastic Beasts, but How to Find Them? Front. Endocrinol. 11:559673. doi: 10.3389/fendo.2020.559673
Received: 06 May 2020; Accepted: 26 August 2020;
Published: 24 September 2020.
Edited by:
Ines Pineda-Torra, University College London, United KingdomReviewed by:
Andrew Alt, University of Michigan, United StatesCopyright © 2020 Van Moortel, Gevaert and De Bosscher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karolien De Bosscher, a2Fyb2xpZW4uZGVib3NzY2hlckB2aWItdWdlbnQuYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.