 Idoia Martínez de LaPiscina1†
Idoia Martínez de LaPiscina1† Laura C. Hernández-Ramírez2†
Laura C. Hernández-Ramírez2† Nancy Portillo1,3
Nancy Portillo1,3 Ana L. Gómez-Gila4Inés Urrutia1Rosa Martínez-Salazar1Alejandro García-Castaño1Aníbal Aguayo1Itxaso Rica1,5Sonia Gaztambide1,6
Ana L. Gómez-Gila4Inés Urrutia1Rosa Martínez-Salazar1Alejandro García-Castaño1Aníbal Aguayo1Itxaso Rica1,5Sonia Gaztambide1,6 Fabio R. Faucz2
Fabio R. Faucz2 Margaret F. Keil2Maya B. Lodish2,7Martha Quezado8Nathan Pankratz9Prashant Chittiboina10John Lane9Denise M. Kay11James L. Mills12Luis Castaño1*
Margaret F. Keil2Maya B. Lodish2,7Martha Quezado8Nathan Pankratz9Prashant Chittiboina10John Lane9Denise M. Kay11James L. Mills12Luis Castaño1* Constantine A. Stratakis2 on behalf on the Spanish Corticotroph Adenomas Collaborative1
Constantine A. Stratakis2 on behalf on the Spanish Corticotroph Adenomas Collaborative1- 1Section on Endocrinology, Metabolism, Nutrition and Renal Diseases, Biocruces Bizkaia Health Research Institute, Cruces University Hospital, UPV/EHU, CIBERER, CIBERDEM, Barakaldo, Spain
- 2Section on Endocrinology and Genetics, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH), Bethesda, MD, United States
- 3Pediatric Endocrinology Service, Alto Deba Hospital, Arrasate, Spain
- 4Pediatric Endocrinology Service, Virgen del Rocío University Hospital, Sevilla, Spain
- 5Pediatric Endocrinology Service, Cruces University Hospital, Barakaldo, Spain
- 6Endocrinology Service, Cruces University Hospital, Barakaldo, Spain
- 7Division of Pediatric Endocrinology, Department of Pediatrics, Mission Hall, University of California, San Francisco, San Francisco, CA, United States
- 8Laboratory of Pathology, National Cancer Institute, National Institutes of Health (NIH), Bethesda, MD, United States
- 9Department of Laboratory Medicine and Pathology, University of Minnesota Medical School, Minneapolis, MN, United States
- 10Neurosurgery Unit for Pituitary and Inheritable Diseases, National Institute of Neurological Disorders and Stroke, Bethesda, MD, United States
- 11Newborn Screening Program, Wadsworth Center, New York State Department of Health, Albany, NY, United States
- 12Epidemiology Branch, Division of Intramural Population Health Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH), Bethesda, MD, United States
Context: The DICER1 syndrome is a multiple neoplasia disorder caused by germline mutations in the DICER1 gene. In DICER1 patients, aggressive congenital pituitary tumors lead to neonatal Cushing's disease (CD). The role of DICER1 in other corticotropinomas, however, remains unknown.
Objective: To perform a comprehensive screening for DICER1 variants in a large cohort of CD patients, and to analyze their possible contribution to the phenotype.
Design, setting, patients, and interventions: We included 192 CD cases: ten young-onset (age <30 years at diagnosis) patients were studied using a next generation sequencing panel, and 182 patients (170 pediatric and 12 adults) were screened via whole-exome sequencing. In seven cases, tumor samples were analyzed by Sanger sequencing.
Results: Rare germline DICER1 variants were found in seven pediatric patients with no other known disease-associated germline defects or somatic DICER1 second hits. By immunohistochemistry, DICER1 showed nuclear localization in 5/6 patients. Variant transmission from one of the parents was confirmed in 5/7 cases. One patient had a multinodular goiter; another had a family history of melanoma; no other patients had a history of neoplasms.
Conclusions: Our findings suggest that DICER1 gene variants may contribute to the pathogenesis of non-syndromic corticotropinomas. Clarifying whether DICER1 loss-of-function is disease-causative or a mere disease-modifier in this setting, requires further studies.
Clinical trial registration: ClinicalTrials.gov: NCT00001595.
Introduction
Multiple molecular mechanisms causing pituitary neuroendocrine tumors (PitNETs) have been identified, but the genetic defects underlying such neoplasms remain unknown for the majority of the sporadic cases. The most common somatic genetic defects in somatotropinomas and corticotropinomas are activating somatic mutations in the GNAS (guanine nucleotide-activating alpha-subunit) and USP8 (ubiquitin-specific protease 8) genes, respectively. On the other hand, 5–8% of PitNETs arise in a familial setting, either isolated (familial isolated pituitary adenoma) or as part of a syndrome affecting other organs, such as multiple endocrine neoplasia type 1 and type 4, McCune-Albright syndrome and Carney complex, and more rarely, within the DICER1 and succinate dehydrogenase-related syndromes (1).
The DICER1 gene (14q32.13) encodes a ubiquitously expressed enzyme containing two endoribonuclease III domains (RNase IIIa and IIIb), required for the cleavage of precursor molecules into mature microRNAs (2–4). Germline or mosaic loss-of-function (LOF) DICER1 mutations predispose to DICER1 syndrome or pleuropulmonary blastoma (PPB) familial tumor and dysplasia syndrome, an autosomal dominant condition causing PPB and multiple other tumors (5, 6). Pituitary blastoma (PitB) is a recently described component of this syndrome, causing severe neonatal Cushing's disease (CD) due to a malignant congenital pituitary tumor, with or without other associated tumors (7–9). Deleterious germline and somatic DICER1 variants have also been reported in various sporadically occurring neoplasms (10–12).
The clinical presentation of the DICER1 syndrome is quite variable, and PitB displays low penetrance (8). Growing evidence has demonstrated the role of miRNAs and long non-coding RNAs in pituitary tumorigenesis (13, 14). Nevertheless, the specific role of DICER1 in pituitary function and tumorigenesis remains unknown. Therefore, we have performed a thorough screening for DICER1 variants in CD patients using targeted next generation sequencing (NGS) and whole-exome sequencing (WES). We describe, for the first time, rare variants in the DICER1 gene in seven patients presenting with isolated corticotropinomas and CD arising during childhood.
Subjects and Methods
Patients and Samples
We studied 170 pediatric (≤18 years at disease onset) and 12 adult CD patients who are part of a large cohort recruited at the National Institutes of Health (NIH) Clinical Research Center between 1997 and 2018 under the research protocol 97-CH-0076 (ClinicalTrials.gov: NCT00001595). The Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional Review Board approved the study, and informed assent/consent was obtained from all the patients and their parents or guardians. Clinical data were obtained directly from the patients and/or from their medical records. For the patients and their parents, if available, DNA was extracted either from peripheral blood samples using the Maxwell 16 Blood DNA Purification Kit in a Maxwell 16 Instrument (Promega AS1015 and AS3050) or from saliva using the Oragene-Dx collection kit and the PrepIT-L2P DNA extraction kit (DNA Genotek OGD-500 and PT-L2P-45), according to the manufacturer's protocols. Additional studies in this cohort have been reported elsewhere (15–22).
We also studied ten young patients (age <30 years at diagnosis) with diagnosed familial or sporadic CD; these cases are part of a multicenter study performed at the Biocruces Bizkaia Health Research Institute and the Department of Endocrinology and Pediatric Endocrinology at the Cruces University Hospital (Barakaldo, Spain). Clinical data were provided by the clinicians responsible for these patients and written informed consent was obtained from all participants or their parents. The study was approved by the local Ethics committees. Extraction and purification of DNA from peripheral blood leukocytes was performed using the MagPurix Blood DNA Extraction Kit 200, according to the manufacturer's instructions (Zinexts Life Science Corp., Taiwan).
When possible, unstained tumor slides were obtained from the corresponding departments of Anatomical Pathology. After manual delimitation of the tumor area, DNA was extracted from unstained sections using the Pinpoint Slide DNA Isolation System (Zymo Research D3001).
Written, informed consent was obtained from the individuals and/or minors' legal guardian for the publication of any potentially identifiable images or data included in this article.
Design of a Targeted Gene Panel and Screening via Next-Generation Sequencing
A targeted panel for PitNETs was designed to include nine genes (AIP, CDKN1B, DICER1, GNAS, MEN1, PRKAR1A, SDHB, SDHC, and SDHD) associated with human pituitary tumors in online databases, including PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), Human Gene Mutation Database (HGMD, https://portal.biobase-international.com) and Online Mendelian Inheritance in Man (https://www.ncbi.nlm.nih.gov/omim). The design of the panel was performed by Ion AmpliSeq™ Designer (Thermo Fisher Scientific, USA) and oligonucleotides against exons, flanking intronic and untranslated (UTR) regions were included. Library preparation was done using the Ion Ampliseq Library Kit v2.0 (Thermo Fisher Scientific) according to manufacturer's instructions. Samples were then sequenced using the Personal Genome Machine system with an Ion 316™ Chip (Thermo Fisher Scientific). Base calling, read filtering, alignment to the reference human genome GRCh37/hg19, and variant calling were done using Ion Torrent Suites (Thermo Fisher Scientific). Further QC analysis, coverage analysis, and variant filtering were completed with the Ion Reporter Software (Thermo Fisher Scientific). Coverage depth and read quality were also evaluated with the Integrative Genomics Viewer (broadinstitute.org).
Germline DNA samples from the 182 patients studied at NIH were submitted for WES at the University of Minnesota Genomics Center (UMGC, 91 patients) or Novogene (91 patients) and analyzed at UMGC. Targeted capture libraries were generated using the Agilent QXT v5 + UTRs (UMGC) or SureSelect Human All Exon v6 (Novogene) kits. Samples were sequenced on an Illumina HiSeq 2000 platform producing 100 base pair paired-end reads (UMGC) or on an Illumina HiSeq 2500 platform producing 150 bp paired-end reads (Novogene), while tumor samples were sequenced on a HiSeq 2500 platform producing 1,265 bp paired-end reads. WES results for all samples were analyzed together. FASTQ files were processed using a Genome Analysis Toolkit (GATK) v3.7 based pipeline, including BWA-MEM v0.7.17 for alternate contig aware alignment to the hg38 reference genome (GRCh38_full_analysis_set_plus_decoy_hla.fa), Picard Tools v2.6.0 to mark duplicates (picard, retrieved from http://broadinstitute.github.io/picard/), and GATK for indel realignment, base quality recalibration, genotyping (HaplotypeCaller), variant quality score recalibration, and to split multiallelic sites (23, 24). ANNOVAR was used to determine the effect of coding variants using both the RefSeq and UCSC gene sets, including putative amino acid changes, distance to intron-exon boundary, the creation or removal of a stop-codon, and location within known non-coding RNAs (25). Non-synonymous variants were annotated based on their computationally predicted deleteriousness using information from dbNSFP. All variants were annotated for their presence and frequency in multiple variant collections (e.g., dbSNP, 1,000 Genomes, and internal WES datasets totaling over 10,000 samples). All the results were filtered to include only variants with a Phred-like score ≥30. The median number of on-target reads generated per sample was 451 million, resulting in a median target coverage of 60X (89% of targets covered at >20X). For DICER1, the first three exons (of the 29 total exons) represent 5′ untranslated region (5′ UTR) and had a median percent covered at 20X of 0.0%. Of the 26 coding exons in DICER1, 20 had a median percent covered at 20X of 95% or greater. Exons 7 (73.1%), 8 (41.9%), 9 (20.2%), 10 (90.0%), 28 (46.7%), and 29 (80.6%) each had a median percent covered at 20X of <95%.
In silico Analyses and Variant Classification
The impact of non-synonymous variants on protein structure and function was assessed in silico using Mutation Taster (http://www.mutationtaster.org/), MutPred (http://mutpred.mutdb.org/), Polyphen 2 (http://genetics.bwh.harvard.edu/pph2/), PROVEAN (Protein Variation Effect Analyzer) (http://provean.jcvi.org/index.php), SIFT (Sorting Intolerant from Tolerant, http://sift.jcvi.org/), SNPs and Go (http://snps.biofold.org/snps-and-go/snps-and-go.html), and VarSome (https://varsome.com). Variants were classified following the standards and guidelines from the American College of Medical Genetics and Genomics (26). We focused our study on coding variants with a minor allele frequency (MAF) <1% in gnomAD (http://gnomad.broadinstitute.org).
Sanger Sequencing
Variants of interest were confirmed by bidirectional direct sequencing using the BigDye Terminator 3.1 Cycle Sequencing Kit (Thermo Fisher Scientific 4337456) in a 3500xL Genetic Analyzer (Applied Biosystems, primer sequences, and conditions are available from the authors, on request). All variants were annotated according to the GenBank reference sequences NM_030621.4 and NP_085124.2. When available, DNA samples from the patients' parents were screened likewise. Since all the selected variants were heterozygous at the germline level, loss of heterozygosity (LOH) in the tumors was also investigated. In addition, the sequence encoding the RNAse IIIb domain (amino acids 1666–1824) of DICER1 was sequenced from tumor DNA samples. Previously reported clinical associations were searched in the databases ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), catalog of Somatic Mutations in Cancer (COSMIC, https://cancer.sanger.ac.uk/cosmic), and HGMD. In selected cases, the primers 5′-CTTCCACCCCTCCAACTCAT-3′ and 5′-TGGAGTTACTGTTGGCTTCCT-3′ were used to amplify and sequence a region of 146 bp covering the USP8 mutational hotspot in tumor DNA.
Immunohistochemistry
Immunohistochemical staining for DICER1 was performed using 1:200 polyclonal rabbit anti-DICER1 primary antibody (Sigma-Aldrich HPA000694), and amplification with 1:1,000 Biotin-SP AffiniPure Goat Anti-rabbit IgG and 1:500 peroxidase streptavidin (Jackson ImmunoResearch Laboratories 111-065-144 and 016-030-084, respectively). Samples were developed using ImmPACT DAB peroxidase (HRP) substrate (Vector SK-4105). The rest of the protocol has been detailed elsewhere (19). Images were acquired using a Keyence BZ-X710 microscope and processed with the BZ-X Analyzer software (Keyence).
Statistical Analyses
The Prism v8.2.1 software (GraphPad Software) was used for all statistical analyses. Parametric data are presented as mean ± standard deviation and non-parametric data are presented as median and interquartile range. Gene variant frequencies in the study population were compared with the frequencies reported in public databases using the chi-squared or the Fisher's exact test, as appropriate. Results were considered statistically significant when P < 0.05.
Results
Study Population
The Biocruces Bizkaia Health Research Institute included 10 probands (60% females), with mean age at diagnosis of 17.9 (7.5–29.0) years and median tumor size of 17.4 (9–20) mm. The NICHD cohort included 182 patients, 56.6% (n = 103) females and 43.4% (n = 79) males. The median age at first symptoms and diagnosis were 10 (8–13) and 12.8 (10.5–15.5) years among the pediatric patients, while adult patients had a mean age of 38.8 ± 9.5 at first symptoms and 42 ± 12.6 at diagnosis. The median maximum tumor diameter was 5.8 (4–8) mm; 84.1% (n = 153) of patients had a microadenoma, 12.1% (n = 22) had a macroadenoma, 2.7% (n = 5) had multiple adenomas, and two patients had no tumor identifiable by histopathology (CD was proven in these cases by means of bilateral inferior petrosal sinus sampling, BIPSS, and resolution of hypercortisolemia after surgery). Fifteen patients had familial presentation and the rest were apparently sporadic. Except for somatic genomic instability in Case 2 (detailed below), none of the patients with DICER1 variants reported here have other known germline PitNET-associated genetic defects.
Patients With DICER1 Gene Variants of Interest
We found seven germline heterozygous missense DICER1 gene variants of interest each in an unrelated patient presenting with non-syndromic, apparently sporadic CD during childhood (one patient from the Biocruces Bizkaia cohort and six from the NICHD cohort). The patients' clinical, biochemical, and histopathological data are summarized in Table 1, and representative MRI images are presented in Figure 1. The genetic characterization and previous reports of the variants are detailed in Table 2 and Figure 2. In addition to these infrequent variants, we found that other, more common DICER1 variants, were overrepresented in our cohort, compared with the general population, as presented in Supplemental Table 1.
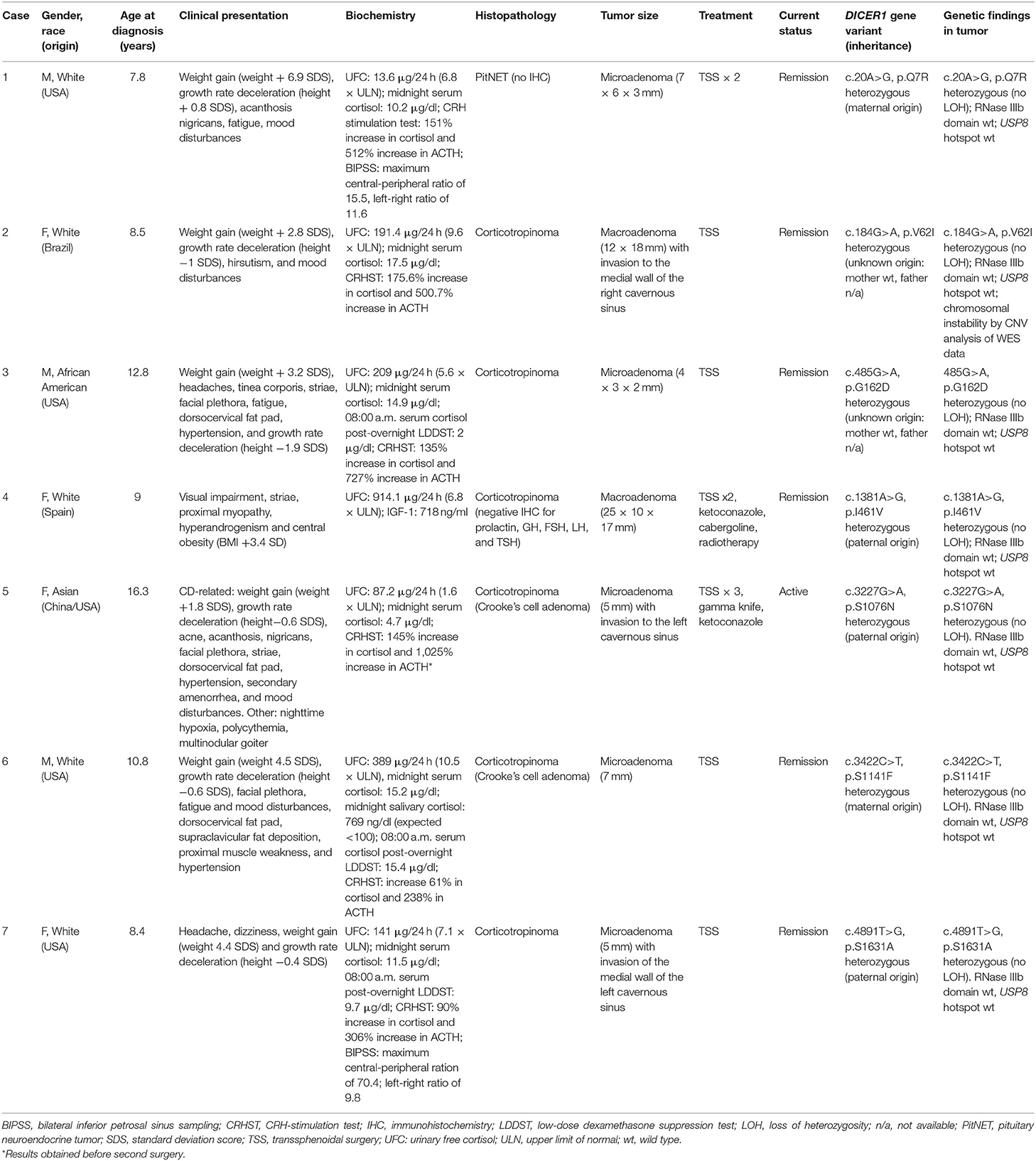
Table 1. Clinical and genetic characterization of patients harboring DICER1variants of interest.
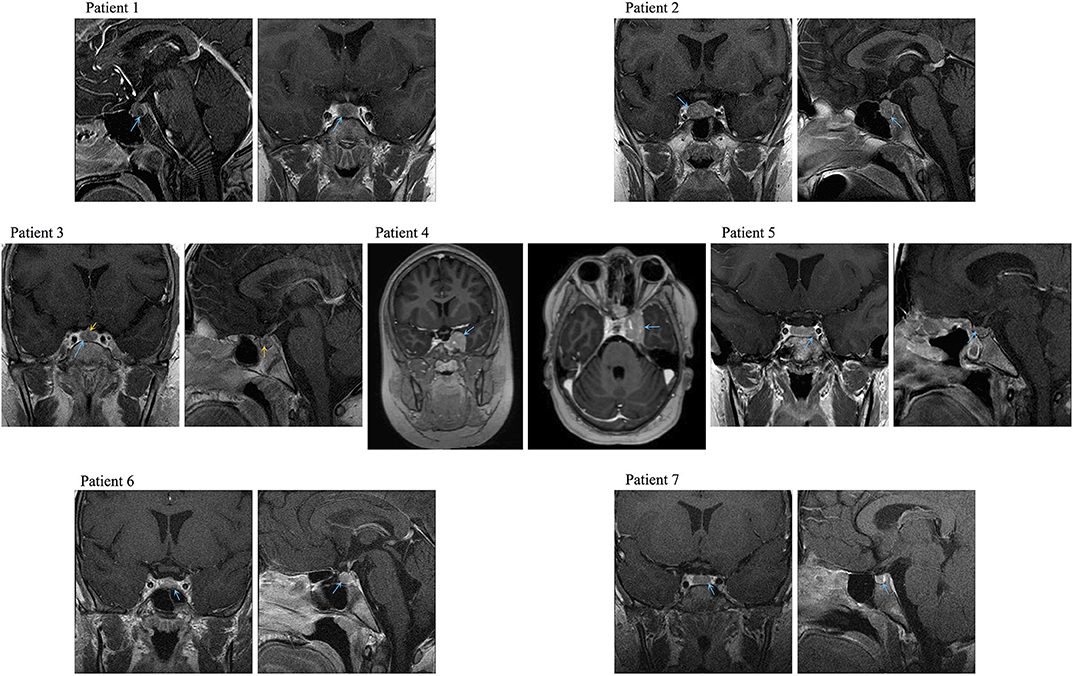
Figure 1. Representative preoperative MRI images of the PitNETs diagnosed in Cases 1–4, 6, and 7. Image for Case 5 was obtained after her first surgery (preoperative image not available). Blue arrows: tumors, yellow arrows: medial lobe cyst.
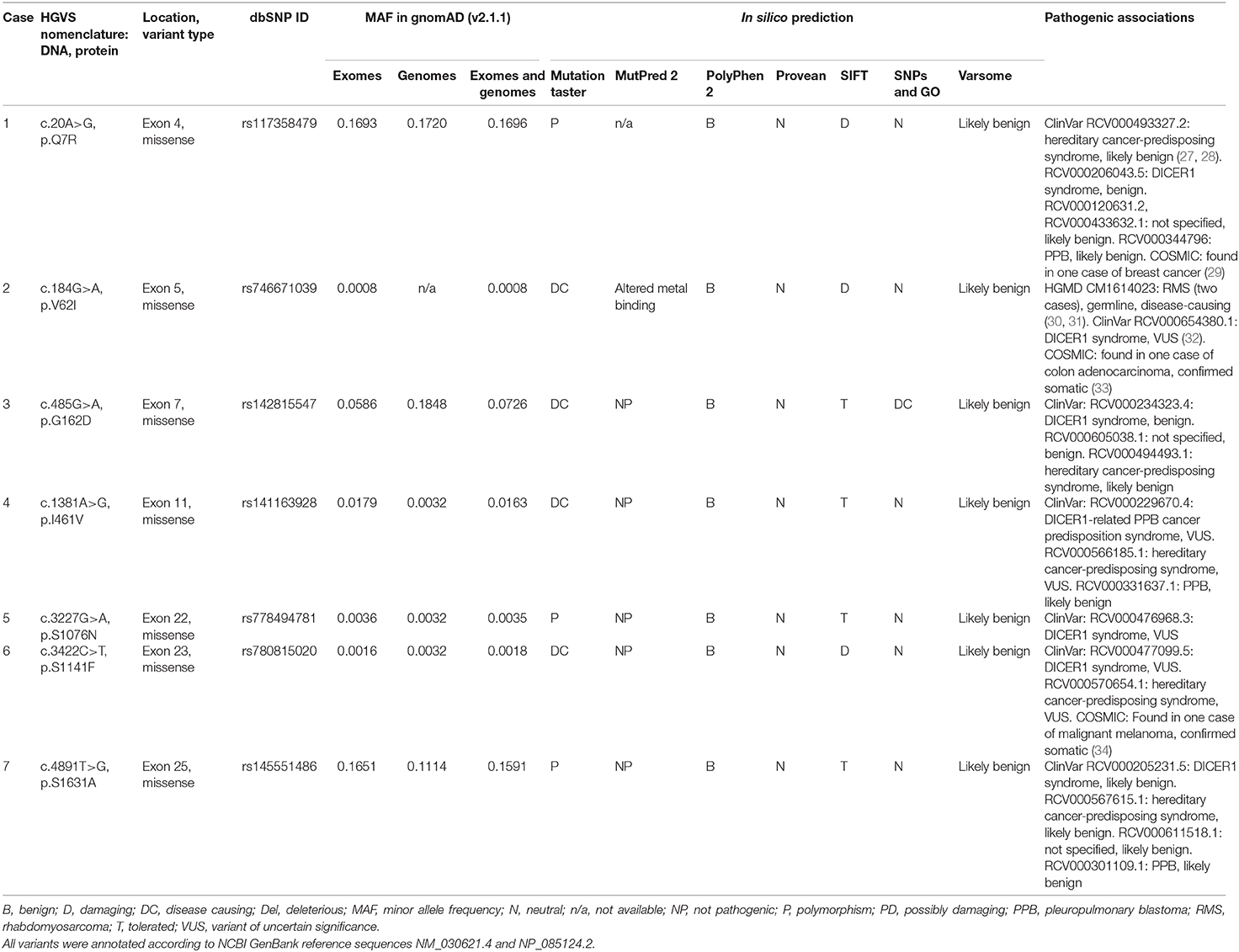
Table 2. DICER1 variants of interest identified among pediatric patients with pituitary neuroendocrine tumors.
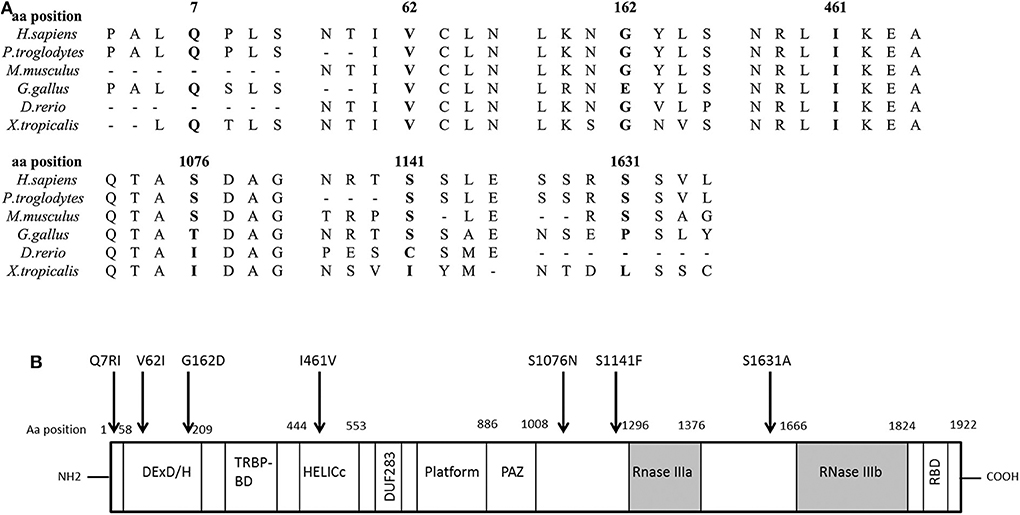
Figure 2. DICER1 protein comparison against different species and structure diagram of the protein. (A) Alignment of the DICER1 protein sequence across species. Position of the identified amino acid variants is presented in bold characters. Most of the identified variants are well conserved across species. (B) Scheme of the structure of the DICER1 protein and localization of studied variants. The main functional domains of DICER1 are the helicase domain including DEXD/H (DEAD box), TRBP-BD (trans-activation response RNA-binding protein-binding), HELICc (helicase conserved carboxy-terminal domain), and DUF283 (domain of unknown function), followed by a platform domain which separates the PAZ (Piwi/Argonaute, Zwille) domain from the two distinct RNase III domains (RNase IIIa and RNase IIIb) and the double-stranded RNA-binding domain (RBD). Localization of the DICER1 variants of interest described in this study is indicated by an arrow. Aa, amino acid.
Case 1
After a 1-year history of weight gain, reduced growth velocity, acanthosis nigricans, fatigue, and mood disturbances, this boy was diagnosed with CD at age 7.8 years. He had an elevated midnight serum cortisol, a positive CRH stimulation test (CRHST), and a possible pituitary microadenoma by magnetic resonance imaging (MRI); CD was confirmed by BIPSS. Remission was only achieved after two transsphenoidal surgeries and, in addition to a histologically confirmed PitNET, an intermediate lobe cyst was also resected. His family history is relevant for the diagnosis of skin melanoma in his mother, maternal grandfather, and a maternal great-uncle. The variant c.20A>G, p.Q7R (previously reported as a likely benign change in DICER1 syndrome patients and as a somatic change in a case of breast cancer) was detected in the patient, as well as in his mother and maternal grandfather.
Case 2
This female patient was diagnosed with CD at age 8.6 years, after having developed excessive weight gain, reduced growth velocity, hirsutism, and mood disturbances over the previous 1.5 years. She had elevated urinary free cortisol (UFC) and midnight serum cortisol, and her pituitary MRI revealed a macroadenoma. A corticotropinoma with invasion of the medial wall of the right cavernous sinus was resected, and the patient achieved remission. The variant identified in this case (c.184G>A, p.V62I), is extremely rare in the general population. It has been reported before as a germline variant probably associated with two cases of rhabdomyosarcoma, as a probable variant of uncertain significance (VUS) in DICER1 syndrome, and as a somatic change in colon adenocarcinoma.
Case 3
At age 9 years, this male patient developed excessive weight gain, headaches, tinea corporis, red striae, facial plethora, and fatigability. During his clinical evaluation, a dorsocervical fat pad, hypertension, and reduced growth velocity were also noted. He was diagnosed with CD at age 12.8 years, having elevated UFC and midnight serum cortisol, as well as a positive overnight low-dose dexamethasone suppression test (LDDST) and an MRI showing a microadenoma. The patient developed hypocortisolism after transsphenoidal surgery, and a corticotropinoma was histologically confirmed. He carried the variant c.485G>A, p.G162D, which has been reported before in patients with DICER1 syndrome as a benign or likely benign variant, and as a somatic change in a patient with Wilms tumor.
Case 4
At age 9 years, this female patient developed visual impairment, striae, proximal myopathy and clinical features of hyperandrogenism. Blood tests detected increased UFC and IGF-1, and the patient was diagnosed with CD. MRI showed a pituitary macroadenoma and transsphenoidal surgery was performed. Tissue immunohistochemistry was positive for ACTH. After surgery, the patient required treatment with ketoconazole and cabergoline, and 1 year later, a second surgery was performed due to persistent cortisol hypersecretion. At age 11 years, the patient presented with a second recurrence, which was successfully treated by radiotherapy. IGF-1 levels were within the normal range after the second surgery (127 ng/ml). At age 13 years, she presented with panhypopituitarism, obesity, and insulin resistance. The variant c.1381A>G, p.I461V in exon 11 of DICER1 was found in the patient and in her apparently healthy father.
Case 5
Around age 13 years, this female patient developed excessive weight gain with central fat distribution, reduced growth velocity, acne, acanthosis nigricans, facial plethora, striae, dorsocervical fat pad, hypertension, secondary amenorrhea, and mood disturbances. She had elevated UFC and midnight serum cortisol, and an MRI showing a microadenoma; a diagnosis of CD was established at age 16.1 years. During her first transsphenoidal surgery, invasion of the wall of the cavernous sinus was documented and histopathological examination confirmed a Crooke's cell corticotropinoma. A second transsphenoidal surgery resulted in remission; unfortunately, she presented with a new recurrence one year later and radiotherapy was then administered, followed by treatment with ketoconazole. The patient has a personal history of nighttime hypoxia, polycythemia, and multinodular goiter, but no family history of neoplasms. She carried the DICER1 variant c.3227G>A, p.S1076N, reported in ClinVar as a VUS for DICER1 syndrome, which she inherited from her apparently healthy father.
Case 6
At age 9 years, this previously healthy boy developed excessive weight gain with central fat distribution, growth rate deceleration, facial plethora, fatigability, and mood disturbances. During clinical examination, dorsocervical fat pad, supraclavicular fat deposition, proximal muscle weakness, and hypertension were also found. He was diagnosed with CD at age 10.8 years. Relevant findings during his diagnostic workup included combined dyslipidemia, as well as elevated UFC, unsuppressed midnight serum and salivary cortisol, positive LDDST, and CRHST and a pituitary microadenoma by MRI. Remission was achieved after transsphenoidal surgery and histopathological examination revealed a Crooke's cell corticotropinoma. The variant c.3422C>T, p.S1141F, detected in the patient and his mother, has been classified as a VUS in patients with DICER1 syndrome and has also been reported as a somatic change in one case of malignant melanoma.
Case 7
This female patient had a history of episodes of headache and dizziness starting at age 3 years. At age 5 she also developed, excessive weight gain with central fat distribution and growth rate deceleration. At age 8.4 the patient was diagnosed with CD, following biochemical tests showing elevated UFC and midnight serum cortisol and an MRI demonstrating a pituitary microadenoma. A corticotropinoma with invasion of the medial wall of the left cavernous sinus was resected, leading to disease remission. The patient carried the variant c.4891T>C, p.S1631A; this change was inherited from her father, who is affected with hypertension and hyperuricemia, but has no history of neoplasms. This variant has been reported as a likely benign change in patients with DICER1 syndrome and other associated phenotypes.
Genetic Screening and Immunohistochemistry in Corticotropinoma Tissues
At the somatic level, none of the cases presented LOH for the DICER1 germline variants of interest, and no additional variants were found in the gene region encoding the RNase IIIb domain (end of exon 25–28). None of these seven patients carried somatic hotspot USP8 mutations either. Interestingly, the corticotropinoma found in case 2 (previously published as PT282.03) displays somatic chromosomal instability, detected by WES copy number variation analysis, as we have recently reported (22).
DICER1 immunohistochemistry of the corticotropinomas (Figure 3), revealed predominantly nuclear but also cytoplasmic staining in most of the cells in Patients 1–3 and 5–6, in contrast with the purely cytoplasmic expression detected in PitNETs with no DICER1 variants of interest and in the normal pituitary gland. In contrast, tumor tissue from Patient 4 presented exclusively cytoplasmic staining.
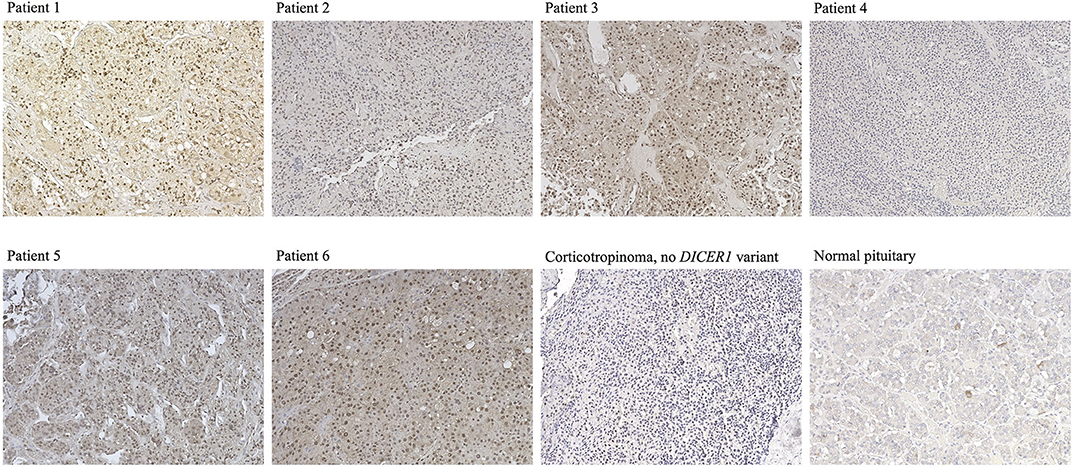
Figure 3. Immunostaining for DICER1 in PitNETs and normal pituitary. Representative images are presented for Cases 1–6 (no slides were available for staining for Case 7), as well as for a corticotropinoma from a patient with no DICER1 variants of interest and for a normal pituitary specimen. Magnification for all images: 20 ×.
Discussion
We hypothesized that DICER1 variants could play a role in the development and function of corticotroph cells, as well as in corticotroph tumorigenesis, based on the association of DICER1 LOF with congenital CD due to PitB. Supporting this hypothesis, we detected seven infrequent missense germline DICER1 variants in an equal number of pediatric patients with apparently sporadic corticotropinomas. In addition, we found an overrepresentation of other more common DICER1 variants in our cohort, compared with the general population. The study of a unique large cohort of young CD patients has allowed us to identify such rare genetic alterations, which might very infrequent in the general population of patients with PitNETs. We confirmed variant transmission from one of the parents in five of the cases of interest, but except for multinodular goiter in Case 5, no familial or personal history of any of the components of the DICER1 syndrome was found. There was, however, a history of familial melanoma in DICER1 variant carrier relatives of Case 1, and although not considered to be associated to the syndrome, somatic DICER1 defects have been described in melanoma cases. In the two cases where variant inheritance was not proven, only one of the parents was available for testing; thus, we could not determine whether those variants appeared de novo.
To date, the only pituitary lesion clearly associated with DICER1 mutations is PitB, an infrequent undifferentiated and aggressive subtype of PitNET first described by Scheithauer et al. (7, 35). Two main studies support this genetic association: a series of 13 cases which confirmed DICER1 mutations in 11 patients (nine proven germline), and a report of an infant with a germline frameshift deletion and a somatic missense variant causing aggressive CD (8, 9). In a recent report, a DICER1 mutation carrier was diagnosed with a microprolactinoma and papillary thyroid cancer with multinodular goiter; she had a daughter who died of ovarian cancer (36). Although the occurrence of a PitNET in this patient could be a coincidence, an association with the DICER1 defect cannot be ruled out.
All the variants identified in our patients have been classified previously as benign, likely benign, or VUS regarding their association with the DICER1 syndrome and other reported phenotypes, but no experimental data were found in the literature to validate their functional effects. They could potentially affect the tumor suppressor function of DICER1, although they are not clustered together in a specific protein domain (Figure 2). Performing the functional experiments required to analyze the impact of these variants was, however, beyond the scope of this manuscript. Causality has not been proven between DICER1 loss-of-function and PitNETs other than PitB, and our patients present a phenotype quite distinct from the DICER1 syndrome. Therefore, although non-syndromic corticotropinomas could be a new DICER1-associated phenotype, at the moment we cannot confidently determine whether the variants found in our patients are indeed causative or if they could act as disease modifiers.
DICER1 variants causing the DICER1 syndrome usually result in truncated proteins and are most often inherited, but appear de novo in 10–20% of cases (37). Although the majority of patients carry germline heterozygous variants, 10% of cases are due to mosaic defects, and most patients harbor second hits (somatic variants or, less frequently, LOH) (38). While germline variants can affect any region of the protein, reported somatic variants are localized in hotspots within the RNase IIIb domain and lead to reduced processing of mature 5′ miRNA strands, shifting the mature miRNA expression toward 3′-derived miRNAs, a potential oncogenic mechanism (39). In contrast, the variants found in our patients are non-truncating and lie outside the RNase IIIb domain, and no second hits were identified. Such findings were not completely unexpected, given the relatively benign phenotype of the tumors (none of the patients presented with pituitary blastoma or neonatal disease), and could indicate the presence of alternative causative genetic defects, meaning that DICER1 variants could be merely contributory in this setting. Interestingly, none of these CD cases harbored somatic USP8 mutations, which are rather frequent among sporadic CD cases, including in our cohort (17).
Another important observation from our study is the preferential nuclear localization of DICER1 by immunohistochemistry. Under physiological conditions, most of the functions of the DICER1 protein take place in the cytoplasm, but a small amount of the protein localizes to the nucleus, where it takes part in non-canonical RNAi processing, and DNA damage triggers its nuclear accumulation (40, 41). Therefore, the staining pattern observed could indicate either a response to previously induced DNA damage or an abnormal subcellular localization of defective DICER1 proteins. Unfortunately, inconsistencies in antibody performance for DICER1 immunohistochemistry have been reported in the literature (42). Analyzing a larger collection of tumors would be required to determine whether such findings are either indicative of DICER1 alterations, or a display of a physiological cellular response to tumorigenic stimuli. Of note, two patients had a histopathological diagnosis of Crooke's cell adenoma. Little is known about the molecular mechanisms underlying this relatively rare and particularly aggressive subtype of corticotropinoma, which represents 5.9% of the cases (n = 10) in our pediatric cohort (43). Whether DICER1 defects could be linked to this phenotype requires further investigations.
In conclusion, the identification of seven pediatric patients with corticotropinomas harboring heterozygous DICER1 variants revealed a possible role for DICER1 gene defects in corticotroph tumorigenesis. Further studies are required to assess the functional effect of these variants on miRNA processing, as well as to explain the specific role of DICER1 in the normal pituitary gland and in corticotroph tumorigenesis.
Data Availability Statement
All datasets presented in this study are included in the article/Supplementary Material.
Ethics Statement
Approval for this study was obtained from The Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional Review Board (research protocol 97-CH-0076 ClinicalTrials.gov: NCT00001595), and from Basque Clinical Research Ethics Committee (CEIC-E). Written informed assent or consent to participate in this study was provided by all the participants or legal guardians, as appropriate.
Author Contributions
IM, LH-R, LC, and CS contributed in the conception and study design, data were collected by IM, LH-R, NPo, AG-G, IU, RM-S, AG-C, AA, IR, SG, FF, MK, ML, MQ, NPa, PC, JL, DK, and JM. Data analysis and interpretation was carried out by IM, LH-R, IR, SG, JM, LC, and CS. IM and LH-R prepared the manuscript. All authors contributed to manuscript revision and approval. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Intramural Research Programs of Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and National Institute for Neurological Diseases and Stroke, National Institutes of Health, a grant from the Basque Department of Education (IT795-13), a grant from the Basque Department of Health (GV2018111082), the Merck Serono Research award from Fundación Salud 2000 (15-EP-004) and the José Igea 2018 grant, sponsored by Pfizer, from Fundación Sociedad Española de Endocrinología Pediátrica (SEEP).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was in part funded by the Intramural Research Program, NICHD, NIH. We would like to acknowledge Dr. Lyssikatos Charalampos and Ms. Maria de la Luz Sierra for their help with the collection and preparation of DNA and histopathological samples used in this study. Members of the Spanish Corticotroph Adenomas Collaborative Group include the following individuals and institutions: E Colino (Hospital San Rafael. Madrid), E García-García (Hospital Universitario Virgen del Rocío. Sevilla), JP López Siguero (Hospital Regional Universitario de Málaga. Málaga), F Moreno-Macián (Hospital Universitario y Politécnico de La Fe. Valencia), D Moure (Hospital Universitario Cruces, Barakaldo), C Villabona (Hospital Universitario Bellvitge. L'Hospitalet de Llobregat). In addition, we would like to thank the patients and their families for participating in our research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2020.00433/full#supplementary-material
References
1. Lim CT, Korbonits M. Update on the clinicopathology of pituitary adenomas. Endocr Pract. (2018) 24:473–88. doi: 10.4158/EP-2018-0034
2. Gurtan AM, Lu V, Bhutkar A, Sharp PA. In vivo structure-function analysis of human Dicer reveals directional processing of precursor miRNAs. RNA. (2012) 18:1116–22. doi: 10.1261/rna.032680.112
3. Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. (2014) 14:662–72. doi: 10.1038/nrc3802
4. Wilson RC, Tambe A, Kidwell MA, Noland CL, Schneider CP, Doudna JA. Dicer-TRBP complex formation ensures accurate mammalian microRNA biogenesis. Mol Cell. (2015) 57:397–407. doi: 10.1016/j.molcel.2014.11.030
5. Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. (2009) 325:965. doi: 10.1126/science.1174334
6. de Kock L, Wang YC, Revil T, Badescu D, Rivera B, Sabbaghian N, et al. High-sensitivity sequencing reveals multi-organ somatic mosaicism causing DICER1 syndrome. J Med Genet. (2016) 53:43–52. doi: 10.1136/jmedgenet-2015-103428
7. Scheithauer BW, Horvath E, Abel TW, Robital Y, Park SH, Osamura RY, et al. Pituitary blastoma: a unique embryonal tumor. Pituitary. (2012) 15:365–73. doi: 10.1007/s11102-011-0328-x
8. de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. (2014) 128:111–22. doi: 10.1007/s00401-014-1285-z
9. Sahakitrungruang T, Srichomthong C, Pornkunwilai S, Amornfa J, Shuangshoti S, Kulawonganunchai S, et al. Germline and somatic DICER1 mutations in a pituitary blastoma causing infantile-onset Cushing's disease. J Clin Endocrinol Metab. (2014) 99:E1487–92. doi: 10.1210/jc.2014-1016
10. Doros L, Schultz KA, Stewart DR, Bauer AJ, Williams G, Rossi CT, et al. DICER1-Related Disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews(R). Seattle, WA: University of Washington, Seattle (1993–2019).
11. Doros LA, Rossi CT, Yang J, Field A, Williams GM, Messinger Y, et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol. (2014) 27:1267–80. doi: 10.1038/modpathol.2013.242
12. Bandapalli OR, Paramasivam N, Giangiobbe S, Kumar A, Benisch W, Engert A, et al. Whole genome sequencing reveals DICER1 as a candidate predisposing gene in familial Hodgkin lymphoma. Int J Cancer. (2018) 143:2076–8. doi: 10.1002/ijc.31576
13. Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol. (2012) 48:R45–53. doi: 10.1530/JME-12-0008
14. Shi X, Tao B, He H, Sun Q, Fan C, Bian L, et al. MicroRNAs-based network: a novel therapeutic agent in pituitary adenoma. Med Hypotheses. (2012) 78:380–4. doi: 10.1016/j.mehy.2011.12.001
15. Stratakis CA, Tichomirowa MA, Boikos S, Azevedo MF, Lodish M, Martari M, et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin Genet. (2010) 78:457–63. doi: 10.1111/j.1399-0004.2010.01406.x
16. Trivellin G, Correa RR, Batsis M, Faucz FR, Chittiboina P, Bjelobaba I, et al. Screening for GPR101 defects in pediatric pituitary corticotropinomas. Endocr Relat Cancer. (2016) 23:357–65. doi: 10.1530/ERC-16-0091
17. Faucz FR, Tirosh A, Tatsi C, Berthon A, Hernández-Ramírez LC, Settas N, et al. Somatic USP8 gene mutations are a common cause of pediatric Cushing disease. J Clin Endocrinol Metab. (2017) 102:2836–43. doi: 10.1210/jc.2017-00161
18. Hernández-Ramírez LC, Tatsi C, Lodish MB, Faucz FR, Pankratz N, Chittiboina P, et al. Corticotropinoma as a component of carney complex. J Endocr Soc. (2017) 1:918–25. doi: 10.1210/js.2017-00231
19. Hernández-Ramírez LC, Gam R, Valdés N, Lodish MB, Pankratz N, Balsalobre A, et al. Loss-of-function mutations in the CABLES1 gene are a novel cause of Cushing's disease. Endocr Relat Cancer. (2017) 24:379–92. doi: 10.1530/ERC-17-0131
20. Makri A, Bonella MB, Keil MF, Hernández-Ramírez L, Paluch G, Tirosh A, et al. Children with MEN1 gene mutations may present first (and at a young age) with Cushing disease. Clin Endocrinol (Oxf). (2018) 89:437–43. doi: 10.1111/cen.13796
21. Cohen M, Persky R, Stegemann R, Hernández-Ramírez LC, Zeltser D, Lodish MB, et al. Germline USP8 mutation associated with pediatric cushing disease and other clinical features: a new syndrome. J Clin Endocrinol Metab. (2019) 104:4676–82. doi: 10.1210/jc.2019-00697
22. Tatsi C, Pankratz N, Lane J, Faucz FR, Hernández-Ramírez LC, Keil M, et al. Large genomic aberrations in corticotropinomas are associated with greater aggressiveness. J Clin Endocrinol Metab. (2019) 104:1792–801. doi: 10.1210/jc.2018-02164
23. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
24. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. (2013) 43:11.10.11-33. doi: 10.1002/0471250953.bi1110s43
25. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
26. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
27. Bodian DL, McCutcheon JN, Kothiyal P, Huddleston KC, Iyer RK, Vockley JG, et al. Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing. PLoS One. (2014) 9:e94554. doi: 10.1371/journal.pone.0094554
28. Slade I, Bacchelli C, Davies H, Murray A, Abbaszadeh F, Hanks S, et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet. (2011) 48:273–8. doi: 10.1136/jmg.2010.083790
29. Schneeweiss A, Park-Simon TW, Albanell J, Lassen U, Cortes J, Dieras V, et al. Phase Ib study evaluating safety and clinical activity of the anti-HER3 antibody lumretuzumab combined with the anti-HER2 antibody pertuzumab and paclitaxel in HER3-positive, HER2-low metastatic breast cancer. Invest New Drugs. (2018) 36:848–59. doi: 10.1007/s10637-018-0562-4
30. de Kock L, Foulkes WD. Sarcoma and germ-line DICER1 mutations. Lancet Oncol. (2016) 17:e470. doi: 10.1016/S1470-2045(16)30522-8
31. Kim J, Field A, Schultz KAP, Hill DA, Stewart DR. The prevalence of DICER1 pathogenic variation in population databases. Int J Cancer. (2017) 141:2030–6. doi: 10.1002/ijc.30907
32. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. (2015) 373:2336–46. doi: 10.1056/NEJMoa1508054
33. Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. (2014) 46:1264–6. doi: 10.1038/ng.3127
34. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. (2017) 23:703–13. doi: 10.1038/nm.4333
35. Scheithauer BW, Kovacs K, Horvath E, Kim DS, Osamura RY, Ketterling RP, et al. Pituitary blastoma. Acta Neuropathol. (2008) 116:657–66. doi: 10.1007/s00401-008-0388-9
36. Cotton E, Ray D. DICER1 mutation and pituitary prolactinoma. Endocrinol Diabetes Metab Case Rep. (2018) 2018:18-0087. doi: 10.1530/EDM-18-0087
37. Schultz KAP, Williams GM, Kamihara J, Stewart DR, Harris AK, Bauer AJ, et al. DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res. (2018) 24:2251–61. doi: 10.1158/1078-0432.CCR-17-3089
38. Brenneman M, Field A, Yang J, Williams G, Doros L, Rossi C, et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma/DICER1 syndrome: a unique variant of the two-hit tumor suppression model. F1000Res. (2015) 4:214. doi: 10.12688/f1000research.6746.2
39. Anglesio MS, Wang Y, Yang W, Senz J, Wan A, Heravi-Moussavi A, et al. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J Pathol. (2013) 229:400–9. doi: 10.1002/path.4135
40. White E, Schlackow M, Kamieniarz-Gdula K, Proudfoot NJ, Gullerova M. Human nuclear Dicer restricts the deleterious accumulation of endogenous double-stranded RNA. Nat Struct Mol Biol. (2014) 21:552–9. doi: 10.1038/nsmb.2827
41. Burger K, Gullerova M. Nuclear re-localization of Dicer in primary mouse embryonic fibroblast nuclei following DNA damage. PLoS Genet. (2018) 14:e1007151. doi: 10.1371/journal.pgen.1007151
42. Spoelstra NS, Cittelly DM, Christenson JL, Gordon MA, Elias A, Jedlicka P, et al. Dicer expression in estrogen receptor-positive versus triple-negative breast cancer: an antibody comparison. Hum Pathol. (2016) 56:40–51. doi: 10.1016/j.humpath.2016.05.014
Keywords: pituitary neuroendocrine tumor, Cushing's disease, DICER1, disease-modifying gene, corticotropinoma
Citation: Martínez de LaPiscina I, Hernández-Ramírez LC, Portillo N, Gómez-Gila AL, Urrutia I, Martínez-Salazar R, García-Castaño A, Aguayo A, Rica I, Gaztambide S, Faucz FR, Keil MF, Lodish MB, Quezado M, Pankratz N, Chittiboina P, Lane J, Kay DM, Mills JL, Castaño L and Stratakis CA (2020) Rare Germline DICER1 Variants in Pediatric Patients With Cushing's Disease: What Is Their Role? Front. Endocrinol. 11:433. doi: 10.3389/fendo.2020.00433
Received: 24 March 2020; Accepted: 02 June 2020;
Published: 03 July 2020.
Edited by:
Madhusmita Misra, Harvard Medical School and Massachusetts General Hospital, United StatesReviewed by:
Kris Ann Schultz, Children's Hospitals and Clinics of Minnesota, United StatesDouglas Stewart, National Cancer Institute (NCI), United States
Copyright © 2020 Martínez de LaPiscina, Hernández-Ramírez, Portillo, Gómez-Gila, Urrutia, Martínez-Salazar, García-Castaño, Aguayo, Rica, Gaztambide, Faucz, Keil, Lodish, Quezado, Pankratz, Chittiboina, Lane, Kay, Mills, Castaño and Stratakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis Castaño, bHVpc2FudG9uaW8uY2FzdGFub2dvbnphbGV6QG9zYWtpZGV0emEuZXVz
†These authors have contributed equally to this work