 Weitong Xu
Weitong Xu Tingting Zhao†
Tingting Zhao†- Lab for Aging Research, State Key Laboratory of Biotherapy, National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, Chengdu, China
It is widely recognized that chemical, physical, and biological factors can singly or synergistically evoke the excessive production of oxidative stress in pulmonary tissue that followed by pulmonary lesions and pneumonia. In addition, metabolic and endocrine disorder-induced diseases such as diabetes and obesity often expressed higher susceptibility to pulmonary infections, and presented severe symptoms which increasing the mortality rate. Therefore, the connection between the lesion of the lungs and the metabolic/endocrine disorders is an interesting and essential issue to be addressed. Studies have noticed a similar pathological feature in both infectious pneumonia and metabolic disease-intercurrent pulmonary lesions, that is, from the view of molecular pathology, the accumulation of excessive reactive oxygen species (ROS) in pulmonary tissue accompanying with activated pro-inflammatory signals. Meanwhile, Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and nuclear factor erythroid-2-related factor 2 (Nrf2) signaling plays important role in metabolic/endocrine homeostasis and infection response, and it's closely associated with the anti-oxidative capacity of the body. For this reason, this review will start from the summary upon the implication of ROS accumulation, and to discuss how AMPK-Nrf2 signaling contributes to maintaining the metabolic/endocrine homeostasis and attenuates the susceptibility of pulmonary infections.
Introduction
Pneumonia is one of the most common clinical problems in human beings, characterized by the inflammation in the terminal airway, alveoli, and lung mesenchyme, usually caused by pathogen infection such as bacteria, viruses, fungi, and mycoplasma (1). If uncontrolled, pulmonary inflammation will progress to serious respiratory dysfunction and mortality especially in child and elderly people (2–4). Although amounts of antibiotics have been applied, the mortality rate of pneumonia has increased due to the rising drug resistance.
The outcome of pneumonia is closely associated with the individual's basic diseases. For example, patients with chronic obstructive pulmonary disease who smoke or inhale irritant gas for a long time have a high risk of pneumonia due to the irreversible decline in pulmonary function (5–8). Clinical data also have presented that pneumonia is a threat globally in obesity and diabetes with increased incidence and severity of disease (9).
The way that underlying the promotion of metabolic/endocrine disorders on the occurrence of pneumonia is not fully understood yet, but increasing evidence implies that one of the culprits is the excessive and persistent reactive oxygen species (ROS) accumulation evoked by inflammation response, which is originally executed by immune cells recruited in lungs but consequently implicated to other cells there. As known, when acting properly, inflammation response in organ and tissue is an innate response upon insults, because ROS produced by this response is the killer of various pathogens. However, if acting too intensively, inflammation response will cause excessive ROS accumulation and tissue damage in local, sometimes even cytokine storm in the whole body (10, 11), In other words, the rational ROS accumulation plays as the weapon against pathogens, but excessive ROS accumulation becomes injurer of the body itself. Therefore, keeping the intensity of inflammation and ROS accumulation at the proper level is a crucial task for pneumonia protection, including metabolic/endocrine disorder-implicated pneumonia protection. It is worthy to mention, moreover, excepting excessive ROS accumulation from acute inflammatory response, high ROS burden also exists in the organs of individuals suffering from some metabolic/endocrine disorders, such as diabetes and obesity (12–14). Generally, the chronic disease-induced ROS burden is not powerful enough to wipe out pathogens, but its consistent existent can bring about the damages in cells and tissues. Of importance, these damages provide more probabilities for pathogen invasion in the lungs, which can progress into pneumonia (15, 16).
Fortunately, innate cellular antioxidant systems also exist, acting for the blockage of over-intensive inflammation response and excessive ROS accumulation, therefore being beneficial to damage limitation and disease suppression. As the key transcription factor mediating anti-oxidant response element (ARE) associated anti-oxidative signals, nuclear factor erythroid-2-related factor 2 (Nrf2) plays an important role in eliminating ROS formed by oxidative stress (17, 18). Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) involves even more cellular antioxidant pathways and contributes significantly to inflammation defense (19, 20). Interestingly, recent studies report that Nrf2 and AMPK are functionally connected and play a cascade-like effect (21). Given AMPK closely links to a diverse metabolic axis in the body, it shall be reasonable to have a better understanding of the implication of oxidative stress and AMPK-Nrf2 anti-oxidative signaling in inflammatory pathogenesis, and to further explore the therapeutic potential of AMPK-Nrf2 signaling on metabolic and endocrine disorder-impacted pulmonary diseases.
We have two intentions by means of this review: to summarize the contributions of oxidative stress, either pulmonary lesions related ROS accumulation and metabolic/endocrine disorders-related ROS accumulation, to pneumonia susceptibility, and to discuss how AMPK-Nrf2 signaling contributes to maintain the metabolic and endocrine homeostasis that is the fundamental status of the body to resist pulmonary infection, to alleviate pulmonary lesions, and to prevent pneumonia.
ROS Accumulation and Pneumonia Susceptibility
Excessive ROS Production Caused by Pulmonary Lesions
Respiratory system exchanges with the external environment. Therefore, the lungs are easily exposed to harmful substances in the air, such as flue gas and various oxidants. Under detrimental environments, the lungs suffer mild or severe oxidative stress which results in ROS accumulation (22). ROS are the byproducts of electron transfer reactions in cells, including hydroxyl (HO•), superoxide anion (O2•−), singlet oxygen (1O2), hydrogen peroxide (H2O2), and other intermediates (23). It is worth noting that the carbonyl compounds are regarded as a new kinds of ROS, like acrolein (ACR) and methyl vinyl ketone (MVK) (24, 25).
The ROS production following pulmonary lesions results in either acute ROS increase or chronic ROS accumulation. The infection of pathogens like bacteria, viruses, and fungi, often evoke acute ROS increase. When pathogen infection occurs, immune cells infiltrate into pulmonary mesenchyme between alveolar cells and interstitial cells to respond via releasing ROS and inflammatory factors. The interaction between the pathogen and the host's immune response is intricate, and the latter sometimes acts traumatic for cells, and tissue that causes damages in the lung (26). Viewing from the molecular level, excessive ROS can react quickly and nonspecifically with the chemical groups in DNA, lipids, and proteins, resulting in cellular metabolism aberrations, severe extracellular matrix remodeling, increased mucus secretion, abnormal apoptosis and pulmonary fibrosis (10, 27, 28). Moreover, pulmonary acute ROS elevation can increase the hazard and risk of secondary lung infections, for instance, children with a history of respiratory diseases have an increased risk of reinfection pneumonia (29–31). In summary, oxidative stress is an upstream cause of pulmonary lesion, and inflammation resulted from pulmonary lesion versa aggravates oxidative stress.
As to the states of chronic ROS accumulation in the lungs, two situations are well-known. One is the sustained exposure to irritant gas that directly stimulates the upper respiratory tract, leading the injury of epithelial cells and the filtration of immune cells, then the oxidative response in the lungs including the production of large amount of ROS (32, 33). This situation is generally caused by long-term smoking, industrial pollution from the combustion of coal and petroleum, and other fuels in the atmosphere and raised dust or respirable particulate matter due to urban environmental pollution. There is also evidence that irons in the air can increase the concentration of ROS in the surface fluid of the lung epithelial cells (34). Another is chronic respiratory diseases-related ROS accumulation. Traditionally, these diseases are limited to chronic respiratory diseases, such as chronic obstructive pulmonary disease (COPD), bronchiectasis, interstitial lung disease, chronic pulmonary heart disease, and pulmonary fibrosis. As indicated by studies, a variety of immune cells in the lungs with these diseases suffer from significant oxidative stress. For example, long-term smokers and COPD patients have higher activities of alveolar macrophages, with elevated cationic protein level in eosinophil and cell surface adhesion factor and increased expression of peroxidase (MPO) in neutrophil (35, 36). Since macrophages and neutrophils are the main sources of ROS in inflammatory tissue (37), these changes occurred in cells bring about the uncontrolled ROS production and accumulation further in the lungs, together with significantly release proinflammatory mediators, an array of cytokines and metalloproteinases that participate in tissue damage (38). Moreover, it has known wildly that, disease-induced ROS elevation can continually regulate the location, modification, and complex formation of nuclear proteins, thus affecting the status of chromatin and the function of transcriptional machinery. For example, ROS can effectively promote the recruitment of RNA polymerase to the promoters of the genes encoding NF-κB- and AP)-1-responsive pro-inflammatory (39). On the other several mutations in antioxidant genes, such as GSTP1, glutathione S-transferase (GST) M1, superoxide dismutase 3 (SOD3) and microsomal epoxy hydrolase (EPHX1) is associated with the severity of COPD infection, and the function of the proteins encoded by these genes was impaired, therefore breaking the steady-state of the oxidation-reduction system and increasing the susceptibility to bacterial infections (40).
Excessive ROS Production Caused by Endocrine/Metabolic Disorders
Despite respiratory diseases, the connection between unbalanced endocrine and metabolic circumstance and ROS accumulation starts to enter our view in the recent. ROS burden exists generally in the organs of individuals suffering from metabolic disorders, such as diabetes, obesity, and disturbances of water and electrolyte balance (14, 16). In obesity, excessive fat storage releases high level of free fatty acid (FFA) in plasma, that promotes the elevated generation of O2•− from the mitochondrial electron transport chain, by inhibiting the translocation of adenine nucleotides (41). ROS can also derive from the activation of NADPH oxidase (NOX). In fact, hyperglycemia, hyperlipidemia, and angiotensin II all activate NOX, which acts to produce ROS molecules (42, 43), that remarkedly affects the status of the tissues such as adipose tissue, muscles, and liver (42, 43). What matters is that the excessive ROS can inhibit the biogenesis and dynamics of mitochondria, which in turn disrupts the homeostasis between the production and elimination of ROS. Actually, this mechanism has been revealed by studies involving both high-fat mouse models and obese patients (44, 45). For instance, in an animal study, obesity was observed to increase NOX activity but reduce the activity of antioxidant enzymes such as SOD, catalase, and glutathione in white fat tissue (46). Moreover, Importantly, as reported by Tanaka et al. (47), chronic exposure of pancreatic islets to high concentrations of glucose is toxic to beta cells, but this toxicity can be antagonized by antioxidants, N-acetyl-L-cysteine or aminoguanidine. Calcium dysregulation syndrome due to high sodium diet, low calcium diet, or insufficient vitamin D is also a type of endocrine/metabolic disorders, impaired Ca2+ homeostasis leads to mitochondrial dysfunction, which is characterized by changes in alterations in ATP synthesis and NADP(H) oxidation, resulting in an excessive ROS production and cell apoptosis (48). At the same time, Ca2+ disorders will also induce endoplasmic reticulum (ER) stress, which will cause the activation of ROS-related protein Nox4 resident on the ER (49), further lead to the accumulation of ROS.
Studies have shown that obesity and diabetes are potentially infectious with the respiratory system, such as tuberculosis, influenza and pneumococcal, staphylococcal, and opportunistic pathogens (9). Diabetes is often accompanied by the impaired innate immune system and the adaptive immune system (50, 51), one reason for Ros to clear obstacles. Moreover, leptin resistance affects the regulation of ROS in obesity which leptin increases may also regulate the accumulation of ROS in the immune response (52).
ROS Increase Susceptibility to Pathogens
The implication of ROS to tissues largely depends on their quantities and action time, so they are a kind of double-edged sword. As mentioned in the introduction, physiologic level and action of ROS are positive for the body, contributing to antimicrobial ability. wound healing and so on (53), partially by playing roles in lymphocytes recruitment and phagocytic ability improvement (54, 55). However, if the reaction become drastic and persistent, ROS will eventually promote pathogens infections that cause tissue damage.
Excessive ROS production break immune response, either innate and adoptive. For instance, in diabetes and obesity individuals, over produced ROS influence antibody response and CD4+/CD8 ratio of T cells, also weaken the functions of the natural killer cells and the migration/phagocytosis of neutrophils (56–58). Moreover, oxidative stress is thought to drive impaired phagocytosis of alveolar macrophages, that closely relates to the repeated bacterial and viral infections in respiratory airways. As ROS molecules, carbonyl groups produced by various oxidative stresses, can directly cause macrophage cytoskeletal instability and, thus impair the macrophage-mediated bacterial phagocytosis (25). The interaction between carbonyl-modified extracellular matrix proteins and macrophages also inhibits the phagocytosis of macrophages (59). Consistent findings from in vivo study are there. For example, alveolar macrophages activation and antibacterial activity parallelly reduced in diabetic patients, accompanying the inability to deal with the challenge of M. tuberculosis (60). In COPD patients, experimental rhinovirus infection leads to elevated systemic and airway infectious inflammation (61, 62). The specific mechanism is still unclear, but some people believe that the internalization of the virus triggers the production of cellular hydrogen peroxide, and the NOX2-dependent ROS production inhibits the antiviral signaling network by modifying the Toll-like receptor 7 (63). Notably, ROS-caused damage of respiratory muscle cells has negative impact on the mechanical function of the lungs, which is important for breathing and gas exchange, therefore being another force to disturb the health of the lungs and to increase the susceptibility of this organ to pathogen infection (64).
On the other hand, ROS accumulation creates an environment suitable for pathogen settlement. One of the bacterial survival strategies depends on the ability to form biofilms and establish a special community. It has been reported recently that ROS accumulation can increase the variability of biofilm formation and induce the antibiotic resistance of Pseudomonas aeruginosa (65). Clinical evidence from patients with diabetes and cardiovascular diseases, collected more recently, have confirmed this notion (66). It is suggested that, when biofilm changed, the host is difficult to neutralize bacterial pathogens, which leads the colonization of bacteria becomes easier in the airway epithelial cells, herein resulting in infectious pneumonia, even severe acute respiratory distress syndrome (ARDs) and sepsis (67).
One more point that needs to be mention is that the accumulation of ROS can accelerate cellular aging, so-called senescence, at least partially through the prompted secretion of proinflammatory cytokines and proteases, companied with the oxidative response and produced by inflammatory cells. Importantly, studies have shown that Keratin 10, laminin receptor, and PAFR are highly expressed in senescent cells, and these proteins are ligands for pneumonia-associated bacteria (68). Oxidative stress-induced senescence is connecting with the raise of inflammation response. This notion comes from the study demonstrating that Oxidative stress and senescence are significantly responsible for the over-activated expression of pro-inflammatory cytokines (69). Interestingly, with high ROS-induced intracellular DNA damage, the innate anti-viral pathway, cGAS-STING, in these cells did not elevate the expression of the IFN1 family genes, while that will be observed in normal cells. It suggests a possibility that the endogenous ROS accumulation probably can weaken the power of the innate immune pathway, at least in certain case, toward DNA virus infection. The recent emergence of SARS-CoV-2 related coronavirus COVID-19 and influenza B virus has made respiratory viruses an important pathogen for infectious pneumonia (70, 71). A recent meta-analysis has shown that the most common comorbidities of COVID-19 clinical symptoms are hypertension and diabetes, followed by cardiovascular diseases and respiratory diseases (72). Statistics also show that patients with diabetes and influenza have more COVID-19 infections (73, 74). This result reveals that the high ROS environment of the body caused by various diseases increases the risk of viral infection. This persistent pandemic threat makes the identification and development of new treatment strategies, especially the treatment of infectious pneumonia, an urgent matter.
ROS Clearance Alleviates Pathogen Susceptibility
The body relies on an antioxidant system to remove ROS and maintain redox homeostasis. According to their molecular structure and mechanism of action, antioxidants in organisms can be divided into: (1) antioxidants involved in ROS captures, such as glutathione (GSH), vitamin C and E; (2) Enzyme active substances participated in the antioxidant pathway, including superoxide dismutase (SOD), catalase, glutathione oxidase (GPx), and glutathione reductase (GR) (75). For example, GSH can be combined with hydrogen peroxide (H2O2) and is oxidized to oxidized glutathione (GSSG), in turn, GSSG can be reduced to GSH by glutathione reductase and re-participate in body metabolism. In addition, H2O2 also can be converted to water by mitochondrial glutathione peroxidase (GPx) (76, 77). Clinical studies have shown that taking high doses of vitamin C can alleviate endothelial tissue damage caused by ROS production from ischemia and reduce the risk of bacterial infection in patients after ischemia/reperfusion (78). Moreover, vitamin C supplementation may prevent community-acquired pneumonia, but the prophylactic use of vitamin C to prevent pneumonia in the general population deserves more researches and evidences (79). Another research in the acute exacerbation of chronic obstructive pulmonary disease (AECOPD), use of GPx mimic Ebselen and Apocynin (a NOX2 oxidase inhibitor) can effectively reduce the excess ROS produced and reduces inflammation caused by influenza A (80).
Recent studies have shown that intervention of ROS clearance by external drugs can effectively prevent pathogen infection. For instance, N-acetyl-L-cysteine (NAC) is a commonly antioxidant which has been used in some antiviral models. NAC can inhibit the replication of related viruses, such as HIV (81, 82), Influenza A (83, 84), RSV (85), PCV2 (86), alleviate susceptibility of viruses. Resveratrol as an antioxidant also has antibacterial activity against Staphylococcus aureus, Bacillus cereus, and others (87–89). A 2012 study showed that combined use of resveratrol and antibiotics has a greater antibacterial effect than single-use (90). But, researchers reported that resveratrol combined with levofloxacin inhibits the antibacterial activity, levofloxacin perform bactericidal effect through ROS activation while resveratrol inhibits it (91). The mechanism of resveratrol eliminates ROS for antibacterial is not clear, but resveratrol's own strong antioxidant properties and its ability to inhibit the diffusion of biofilms (92, 93) have given the possibility of an investigation. Besides, it has also been reported that antioxidants have the same molecular structure as some antibacterial drugs, such as vanillin, resveratrol, curcumin, may be a possibility for a new antibacterial agent to address antibiotic resistance (94).
For a long time, people have generally believed that ROS is a weapon for the immune system against pathogens. However, the use of antioxidants also is one of the treatments for infections. We can speculate that the long-accumulated ROS and the ROS of the immune response are different. This difference may exist in production, regulatory mechanisms, chemical composition, and others, which needs further exploration.
The Role of AMPK-Nrf2 Pathway in Pneumonia
AMPK—an Upstream Regulator of Antioxidative Response
AMPK is a serine/threonine kinase in all eukaryotic tissues and organs, can be activated by various stimuli that affected cell metabolism, including hypoxemia, alimentary deficiency, exercise, and many hormones and substances. The ratio of AMP to ATP mainly regulated AMPK, The activation of AMPK requires the binding of AMP and ADP to the γ subunit and the β subunit myristoylation to promote phosphorylation at Thr172 (95). If the AMP content is excessive (3–5 folds), the myristoylation β subunit is not required to directly activate AMPK (96, 97). In addition, Ca2+/calmodulin-dependent protein kinaseβ (CaMKKβ), kinase serine/threonine kinase 11 (LKB1), and transforming growth factor-β-activated kinase 1 also can phosphorylation of Thr172 to activate AMPK (95). In addition, the activation of AMPK also closes the anabolic pathway, like a catabolic switch (98). The biological functions of AMPK mainly include: stimulate fatty acid oxidation of liver and skeletal muscle, promote muscle glucose uptake, inhibit the production of triglyceride, cholesterol, adipose, inhibit lipolysis of adipocytes, and regulate pancreatic insulin secretion (95, 99, 100). Overall, AMPK is a key balancer of energy supply and demand and a key integrator of important pathways such as redox, inflammatory production, and autophagy in cells.
Most previous studies have shown that metabolic disorders can seriously affect AMPK activity. For example, in diabetic patients, the AMPK activity of glomerular epithelial cells in a high glucose environment is significantly reduced, and the glomerular epithelial cells are significantly proliferated and hypertrophic (101). Similarly, in pancreatic β cells, the high glucose environment significantly reduced AMPK activity. However, it is worth noting that activating AMPK activity in pancreatic β-cells can inhibit glucose-stimulated insulin release, and that AMPK activation can promote the increase of ROS levels in β-cells, while inhibiting the expression of glucokinase, and ultimately induce β-cell apoptosis (102). In type 2 diabetes, the AMPK to its downstream glucose transporter-4 (GLUT4) signaling pathway is impaired in patients' skeletal muscles. The LKB1-AMPK-GLUT4 signaling pathway is an important signaling pathway that promotes skeletal muscle glucose uptake, transport, and utilization (103, 104). In skeletal muscle of obese individuals, the uptake rate of long-chain fatty acids (LCFA) was significantly higher than normal (105), and the plasma membrane protein FAT/CD36, which plays a key role in LCFA transport, was found to be highly expressed in the myotubes of obese patients (106). More importantly, activating AMPK with the drug AICAR can induce all LCFAs to transfer to the plasma membrane (107). Studies of other signaling cascade pathways have shown that inhibition of extracellular signal receptor kinase (ERK1/2) can prevent FAT/CD36 translocation (108, 109), and stimulation of protein kinase C can induce the shift of FAT/CD36. The internal Ca2+ concentration is related to signal transport, and subsequent further research proves that CaMKK located upstream of AMPK mainly affects LCFA transport (110). Because glucagon activates AMPK and insulin inhibits AMPK, this indicates that AMPK participates in the endocrine feedback loop and can respond to feedback regulation of hormone (such as insulin) release (111), which is necessary to respond to nutrient and energy stress. But hormones have opposite effects on AMPK activity in the central and peripheral circuits (112). Therefore, we can reasonably speculate that excessively transported LCFA can also inhibit AMPK activity.
Several studies have shown that activation of AMPK can inhibit oxidative stress triggered by different lesions, hyperglycemia, and hyperlipidemia. For example, in hyperglycemic conditions, AMPK activation induces the expression of mitochondrial uncoupling protein 2 (UCP-2), which significantly reduces the production of superoxide free radicals in mitochondrial of hyperglycemic endothelial cells (113). On the other hand, there is emerging evidence that AMPK can inhibit the activation of the NF-κB system (a key class of immune stress-related nuclear transcription factors) through its signaling network, thereby blocking the transcription of innate immunity and inflammation-related factors (114).
Previous studies have shown that the AMPK-FOXO3-Thioredoxin (Trx) is an antioxidant pathway (115). Trx is a disulfide reductase which protects cellular proteins from cysteine oxidation. Under physiological conditions, Trx binds to thioredoxin interacting protein (Txnip) and functions are inhibited (116). When it comes to oxidative stress, the complex between Trx and Txnip dissociates, then Txnip bind to the inflammatory hormone receptor Nod-like receptor protein 3 (NLRP3) receptor, triggering the expression of a variety of downstream inflammatory factors (117). Meanwhile, Trx regulated by AMPK can play an antioxidant role under oxidative stress situation. Experiments have shown that the injection of AICAR in ApoE−/− mice induced by high-fat increased Trx expression of the aortic wall, accompanied with ROS decreased (116) Moreover, AMPK downstream protein mTOR, a nutrient and growth factor sensing complex, by phosphorylating eukaryotic translation initiation factor 4E (eIF4E) and ribosomal protein S6 kinase (S6K) to control protein synthesis (118, 119). Protein anabolism is positively correlated with ROS production (120). The activation of AMPK can inhibit the activity of mTOR1, reducing anabolism-related ROS accumulation.
Furthermore, the PPAR proteins family, as ligand-dependent transcriptional regulators, are closely linked to antioxidants. Researches have shown that PPARδ is downstream of AMPK through enhance antioxidant capacity to protect endothelial function from ER stress and oxidative stress (121), such as increase Sirt1 activity and NO production (121, 122). Previous studies have shown that PPARγ in elderly dermal fibroblasts (HDF) reduces the expression of pro-inflammatory cytokines (123). Moreover, Ligand-activated PPARδ also blocked angiotensin II (Ang II), inhibiting ROS production (124).
Nrf2—a Key Antioxidant Transcription Factor
The activation of the Nrf2-antioxidant response element (ARE) signaling pathway is the main antioxidant means of the cell. This pathway regulates protein produce to eliminate active oxidants by group binding or enzyme reaction. A variety of chronic diseases, including neurodegenerative diseases, metabolic diseases, and cardiovascular diseases, are closely related to Nrf2 and serve as potential therapeutic targets (125). In normal conditions without stress, Nrf2 is retained in the cytoplasm, mainly because Nrf2 binds to the E3 ubiquitin ligase substrate adaptor [Kelch-like ECH-related protein 1 (Keap1)], which makes Nrf2 fast degrade by ubiquitination.
Keap1 is highly sensitive to oxidants and electrophiles. Under the oxidative stress, The cysteine residue of Keap1 combined with oxidants and be modified, thereby releasing Nrf2 that binds to Keap1, resulting in the accumulation of Nrf2 in the nucleus and activation of transcription of the ARE-related genes, which coding the major detoxication enzymes, including Heme Oxygenase-1 (HO-1), NADPH quinone oxidoreductase 1 (NQO1), and GSH redox system-related enzymes, such as γ-glutamyl cysteine synthetase (γ-GCS) subunit GCLC, glutathione peroxidase (GPx), glutathione reductase (GSR), and glutathione-S-transferase (GSTs), prevent oxidative damage by increasing the synthesis of reduced GSH, free radicals are scavenged and the cell redox balance is maintained (126–129).
The loss or inactivation of Nrf2 can induce a variety of inflammatory-related diseases. In experimental diabetic mouse models, Nrf2 deficiency increases oxidative stress and induces ROS accumulation, leading to early heart damage and cardiovascular dysfunction (mainly increased cardiac fibrosis). Treatment with Nrf2 inducers (such as sulforaphane and myricetin) can effectively protect the mouse heart from damage caused by high ROS (130). In humans, the transcriptional activity of Nrf2 affects the occurrence of pulmonary diseases. For example, the transcriptional characteristics of Nrf2 in alveolar macrophages of smoking-induced emphysema patients are lower than normal (131). Moreover, Nrf2−/− mice exhibit enhanced susceptibility to cigarette smoke-induced emphysema (132).
AMPK Mediates Antioxidative Cascade by Activating Nrf2
Recent studies have shown that AMPK can directly phosphorylate with Nrf2. In cells, there is phenomenon display that activation of AMPK leads to migration and aggregation of transcription factor Nrf2 into the nucleus. AMPK phosphorylates Nrf2 located in the Ser558 residue (Ser550 in mice) in the normal nuclear export signal, a result confirmed from an in vitro kinase and peptide competition assays (21), and the nuclear import effect of Nrf2 is lost in the case of the Ser550A mutation, even under AMPK activation. Moreover, it has been reported that AMPK inhibits the activity of glycogen synthase kinase-3 (GSK3β) (133). Specific activation of GSK3β can significantly reduce nuclear Nrf2 levels. GSK3β inhibits Nrf2 activity through phosphorylation and nuclear exclusion (134). Therefore, it can be considered that AMPK indirectly reduces the level of Nrf2 in the nucleus by inhibiting the activity of GSK3β.
Numerous pharmacological experiments have also shown that activating the AMPK-Nrf2 pathway can effectively alleviate many inflammatory-mediated diseases. Compounds HP-1c and NBP, which are used in brain inflammation, can prevents cerebral ischemia, reduces neurotoxicity, and active angiotensin II receptor blockers (ARBs) to establish neuroprotective effects on neurodegenerative diseases (135) by activating AMPK. In addition, AMPK activating also promotes the conversion of microglia from pro-inflammatory M1 to anti-inflammatory M2, thereby reducing the inflammatory response in brain (136). It is known that ARBs and NBP can restore and activate Nrf2 expression, either cause AMPK activation to alleviate inflammation (136), therefore it can be speculated that there may be a crosstalk between AMPK and Nrf2. About other activation pathways of AMPK to Nrf2, previous studies have shown that the cysteine residues (C151) in Keap1 were essential for inhibiting nuclear translocation of Nrf2 (137). Moreover, some studies suggested that some compounds, such as TBHQ, sulforaphane, and quercetin, could modify the cysteine residue in Keap1 (138), also activate AMPK-related pathways.
AMPK-Nrf2 Activation Ameliorates Pneumonia
There are many pieces of evidences confirm that activation of the AMPK-Nrf2 pathway can alleviate pneumonia-related symptoms. As an agonist of AMPK, BBR down-regulates the expression of pro-inflammatory genes in the lungs of mice injected with LPS and up-regulates the expression of Nrf2 targeting genes. Besides, this effect of BBR is impaired in Nrf2 gene-deficient mice. Furthermore, in an Nrf2-depleted mice model, BBR prolonged the survival time of endotoxin-stimulated mice, and its effect on plasma redox regulation was greatly attenuated (139).
Enhancing the activity of Nrf2 can play the same role. Isothiocyanate thiocyanate (SFN) activation of Nrf2 restores bacterial recognition and phagocytosis, enhances the ability of alveolar macrophages to clear lung bacteria, and reduces lung inflammation in wild-type mice exposed to cigarette smoke, while mice with knockout Nrf2 are not (140). Syndecan 4, a transmembrane proteoglycan, could increase the number of bronchiolar progenitor cells that have subsided due to inflammation by dedifferentiation of pulmonary club cells into bronchiolar progenitor cells, and its mechanism is mainly through regulating Nrf2 activity (141, 142). In recent years, it has been proved that the normal function of Nrf2 is one of the key factors to ensure the homeostasis of airway epithelial health. Keap1-Nrf2 system is helpful for the lineage-specific differentiation of stem and progenitor cells, which play an important role in the stable operation of various types of cells in the lung (143).
Activation of the AMPK-Nrf2 pathway can also prevent or relieve pneumonia indirectly by alleviate the symptoms of obesity and diabetes. Surveys have shown that the mortality and morbidity of pneumococcal pneumonia and influenza in obese and diabetic patients are generally higher (9), and that the mortality of pneumonia-related surgery in diabetic patients is higher (144). Therefore, it is reasonable to believe that obesity or diabetes caused by metabolic disorders inhibits AMPK-Nrf2 pathway activity, just as free fatty acid accumulation and high glucose environment are accompanied with oxidative stress damage and ROS accumulation (145). The use of drugs to activate AMPK, including AICAR (146), metformin (147), TZDs (148), berberine (149), and resveratrol (150), can activate skeletal muscle AMPK, improve insulin sensitivity, improve T2D symptoms, and promote lipolysis. It has also been reported that in T2D model mice treated with cassia seed extract, the mRNA and protein expression levels of P-LKB1, P-AMPKa12, AMPKa2, P-AMPKa2, GLUT4 are increased, which means the glucose transport pathway is restored, results in glutathione levels increased and ROS levels decreased significantly (151). However, the detailed molecular mechanism of mediation of insulin sensitization directly through AMPK-Nrf2-ROS is unclear. To summarize, the activation of AMPK-Nrf2 reduces the accumulation of ROS in the lungs, especially in the case of severe oxidative stress in body tissues such as obese or diabetic patients. We can use this new mechanism to discover new treatment strategies to prevent and improve the occurrence of pneumonia.
Conclusion and Perspectives
Pulmonary lesions and metabolic disorders can cause oxidative stress and accumulation of ROS, which further remodeling the pulmonary microenvironment. These damages result in the impaired immune system and increases colonization of pathogens, to raise the susceptibility of pneumonia. The new molecular mechanism reveals that AMPK-Nrf2 has an antioxidant cascade effect, and the regulation of the AMPK-Nrf2 pathway to eliminate pulmonary ROS can prevent or improve pneumonia. As shown in the figure (see Figure 1), the use of AMPK activators has been increased for the treatment of various inflammations. However, for many chronic inflammations, especially in the lungs constantly exposed to high levels of oxidative stress, the body's antioxidant activity is crucial. Current research has revealed that Nrf2 is located downstream of AMPK and has a direct effect on the inflammatory response. In addition, promoting the phosphorylation of Nrf2 through AMPK by pharmacological or genetic methods to alleviate the accumulation of excess ROS in the body may be a potential therapeutic strategy. Therefore, the regulation of the activity of the AMPK-Nrf2 pathway has great research prospects for the treatment of pneumonia.
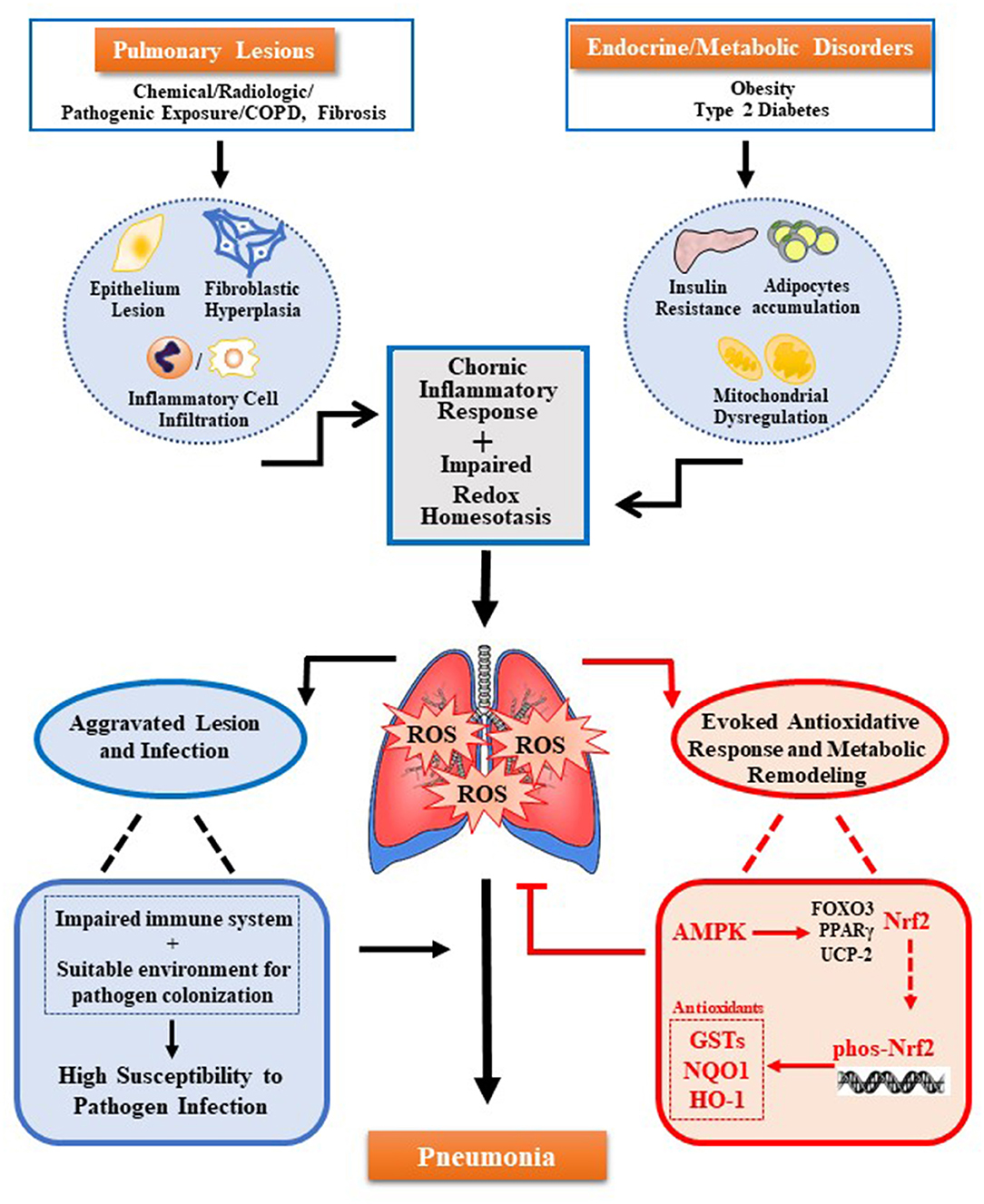
Figure 1. Multiple oxidative stress factors induce pneumonia and AMPK-Nrf2 pathway inhibits pulmonary ROS accumulation. Pulmonary ROS accumulation mainly caused by pulmonary lesions and endocrine/metabolic disorders. Trigger factors include chronic inflammation response induced by damage of pulmonary cells (alveolar epithelial cells, alveolar cells, lung fibroblasts, and others) and infiltration of immune cells in the lungs, as well as the impaired redox homeostasis induced by metabolic disease-related insulin resistance, adipocytes accumulation, and mitochondrial dysfunction. Accumulated ROS can cause damage to the immune system and colonization of pathogens, both of which increase pulmonary susceptibility and cause pneumonia. Meanwhile, ROS can also trigger several antioxidant pathways regulated by AMPK. Among them, the antioxidant effect of AMPK-Nrf2-AREs plays a key role. The activation of this pathway can inhibit the accumulation of ROS in the lungs to prevent or alleviate pneumonia.
Author Contributions
WX and TZ mainly performed the review writing and revision. HX guided the writing planning and manuscript preparation. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Grant No. Z2018B04), National Natural Science Foundation of China (Grant No. 81771511), and National Key Research and Development Program of China (Grant No. 2018YFC2000400).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mackenzie GJP. The definition and classification of pneumonia. Pneumonia (Nathan). (2016) 8:14. doi: 10.1186/s41479-016-0012-z
2. Matera MG, Calzetta L, Cazzola M. Oxidation pathway and exacerbations in COPD: the role of NAC. Exp Rev Respir Med. (2016) 10:89–97. doi: 10.1586/17476348.2016.1121105
3. Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy. (2018) 14:221–32. doi: 10.1080/15548627.2017.1389823
4. Hara H, Kuwano K, Araya J. Mitochondrial quality control in COPD and IPF. Cells. (2018) 7:86. doi: 10.3390/cells7080086
5. Hooven TA, Polin RA. Pneumonia. Sem Fetal Neonatal Med. (2017) 22:206–13. doi: 10.1016/j.siny.2017.03.002
6. Mandell LA. Community-acquired pneumonia: an overview. Postgrad Med. (2015) 127:607–15. doi: 10.1080/00325481.2015.1074030
7. Torres A, Blasi F, Dartois N, Akova M. Which individuals are at increased risk of pneumococcal disease and why? Impact of COPD, asthma, smoking, diabetes, and/or chronic heart disease on community-acquired pneumonia and invasive pneumococcal disease. Thorax. (2015) 70:984–9. doi: 10.1136/thoraxjnl-2015-206780
8. Almirall J, Blanquer J, Bello S. Community-acquired pneumonia among smokers. Arch Bronconeumol. (2014) 50:250–4. doi: 10.1016/j.arbr.2013.11.004
9. Fisher-Hoch SP, Mathews CE, McCormick JB. Obesity, diabetes and pneumonia: the menacing interface of non-communicable and infectious diseases. Trop Med Int Health. (2013) 18:1510–9. doi: 10.1111/tmi.12206
10. Poljsak B, Šuput D, Milisav I. Achieving the balance between ROS and antioxidants : when to use the synthetic antioxidants. Oxid Med Cell Longev. (2013) 2013:956792. doi: 10.1155/2013/956792
11. Agita A, Alsagaff MT. Inflammation, immunity, and hypertension. Acta Med Indones. (2017) 49:158–165. doi: 10.1161/hypertensionaha.110.163576
12. Newsholme P, Cruzat VF, Keane KN, Carlessi R, de Bittencourt PI Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J. (2016) 473: 4527–50. doi: 10.1042/BCJ20160503C
13. Niemann B, Rohrbach S, Miller MR, Newby DE, Fuster V, Kovacic JC. Oxidative stress and cardiovascular risk: obesity, diabetes, smoking, and pollution: part 3 of a 3-part series. J Am Coll Cardiol. (2017) 70:230–51. doi: 10.1016/j.jacc.2017.05.043
14. Rani V, Deep G, Singh RK, Palle K, Yadav UC. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. (2016) 148:183–93. doi: 10.1016/j.lfs.2016.02.002
15. Feillet-Coudray C, Fouret G, Vigor C, Bonafos B, Jover B, Blachnio-Zabielska A, et al. Long-term measures of dyslipidemia, inflammation, and oxidative stress in rats fed a high-fat/high-fructose diet. Lipids. (2019) 54:81–97. doi: 10.1002/lipd.12128
16. Wang Y, Fouret G, Bonafos B, Blachnio-Zabielska A, Leroy T, Crouzier D, et al. Long-term follow-up of muscle lipid accumulation, mitochondrial activity and oxidative stress and their relationship with impaired glucose homeostasis in high fat high fructose diet-fed rats. J Nutr Biochem. (2019) 64:182–97. doi: 10.1016/j.jnutbio.2018.10.021
17. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev. (2016) 2016:4350965. doi: 10.1155/2016/4350965
18. Sajadimajd S, Khazaei M. Oxidative stress and cancer: the role of Nrf2. Curr Cancer Drug Targets. (2018) 18:538–57. doi: 10.2174/1568009617666171002144228
19. Liu MY, Zhang MJ, Xie MJ. [Molecular mechanisms of anti-inflammatory action of AMPK]. Sheng Li Xue Bao. (2018) 70:329–34. doi: 10.13294/j.aps.2018.0020
20. Peng XW, Zhou HH, Dai J, Zhang L. [Advances on the anti-inflammatory and protective effect of AMPK activators]. Sheng Li Xue Bao. (2019) 71:319–26. doi: 10.13294/j.aps.2018.0094
21. Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol. (2016) 36:1931–42. doi: 10.1128/MCB.00118-16
22. Chung KF, Marwick JA. Molecular mechanisms of oxidative stress in airways and lungs with reference to asthma and chronic obstructive pulmonary disease. Ann N Y Acad Sci. (2010) 1203:85–91. doi: 10.1111/j.1749-6632.2010.05600.x
23. Ivanov AV, Valuev-Elliston VT, Tyurina DA, Ivanova ON, Kochetkov SN, Bartosch B, et al. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget. (2017) 8:3895–932. doi: 10.18632/oncotarget.13904
24. Horinouchi T, Higashi T, Mazaki Y, Miwa S. Carbonyl compounds in the gas phase of cigarette mainstream smoke and their pharmacological properties. Biol Pharm Bull. (2016) 39:909–14. doi: 10.1248/bpb.b16-00025
25. Kirkham PA, Spooner G, Ffoulkes-Jones C, Calvez R. Cigarette smoke triggers macrophage adhesion and activation: role of lipid peroxidation products and scavenger receptor. Free Radic Biol Med. (2003) 35:697–710. doi: 10.1016/S0891-5849(03)00390-3
26. Quinton LJ, Mizgerd JP. Dynamics of lung defense in pneumonia: resistance, resilience, and remodeling. Annu Rev Physiol. (2015) 77:407–30. doi: 10.1146/annurev-physiol-021014-071937
27. Yang J, Zhao X, Tang M, Li L, Lei Y, Cheng P, et al. The role of ROS and subsequent DNA-damage response in PUMA-induced apoptosis of ovarian cancer cells. Oncotarget. (2017) 8:23492–506. doi: 10.18632/oncotarget.15626
28. Labuschagne CF, Brenkman AB. Current methods in quantifying ROS and oxidative damage in Caenorhabditis elegans and other model organism of aging. Ageing Res Rev. (2013) 12:918–30. doi: 10.1016/j.arr.2013.09.003
29. Branche AR, Falsey AR. Parainfluenza virus infection. Sem Respir Crit Care Med. (2016) 37:538–54. doi: 10.1055/s-0036-1584798
30. Panda S, Mohakud NK, Pena L, Kumar S. Human metapneumovirus: review of an important respiratory pathogen. Int J Infect Dis. (2014) 25:45–52. doi: 10.1016/j.ijid.2014.03.1394
31. Esposito S, Blasi F, Arosio C, Fioravanti L, Fagetti L, Droghetti R, et al. Importance of acute Mycoplasma pneumoniae and Chlamydia pneumoniae infections in children with wheezing. Eur Respir J. (2000) 16:1142–6. doi: 10.1034/j.1399-3003.2000.16f21.x
32. Böhm F, Edge R, Burke M, Truscott TG. Dietary uptake of lycopene protects human cells from singlet oxygen and nitrogen dioxide - ROS components from cigarette smoke. J Photochem Photobiol B. (2001) 64:176–8. doi: 10.1016/S1011-1344(01)00221-4
33. Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. (2006) 28:219–42. doi: 10.1183/09031936.06.00053805
34. Mateos F, Brock JH, Pérez-Arellano JL. Iron metabolism in the lower respiratory tract. Thorax. (1998) 53:594–600. doi: 10.1136/thx.53.7.594
35. Fiorini G, Crespi S, Rinaldi M, Oberti E, Vigorelli R, Palmieri GJB, et al. Serum ECP and MPO are increased during exacerbations of chronic bronchitis with airway obstruction. Biomed Pharmacother. (2000) 54:274–8. doi: 10.1016/S0753-3322(00)80071-2
36. Noguera A, Busquets X, Sauleda J, Villaverde JM, Macnee W, Agustí AG, et al. Expression of adhesion molecules and G proteins in circulating neutrophils in chronic obstructive pulmonary disease. Am J Respir. (1998) 158:1664–8. doi: 10.1164/ajrccm.158.5.9712092
37. Rahman I, Macnee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med. (1996) 21:669–681. doi: 10.1016/0891-5849(96)00155-4
38. Rahman I, Morrison D, Donaldson K, Macnee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med. (1996) 154:1055–60. doi: 10.1164/ajrccm.154.4.8887607
39. Cruz CM, Rinna A, Forman HJ, Ventura ALM, Persechini PM, Ojcius DM. ATP activates an ros-dependent oxidative stress response and secretion of pro-inflammatory cytokines in macrophages. J Biol Chem. (2006) 282:2871–9. doi: 10.1074/jbc.M608083200
40. Postma DS, Marjan K, Marike HB, Koppelman GH. Asthma and chronic obstructive pulmonary disease: common genes, common environments? Am J Respir Crit Care Med. (2011) 183:1588–94. doi: 10.1164/rccm.201011-1796PP
41. Bakker SJ, Ijzerman RG, Teerlink T, Westerhoff HV, Gans RO, Heine RJ. Cytosolic triglycerides and oxidative stress in central obesity: the missing link between excessive atherosclerosis, endothelial dysfunction, and β-cell failure? Atherosclerosis. (2000) 148:17–21. doi: 10.1016/S0021-9150(99)00329-9
42. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes. (2003) 52:1–8. doi: 10.2337/diabetes.52.1.1
43. Morgan D, Oliveira-Emilio HR, Keane D, Hirata AE, Santos Da Rocha M, Bordin S, et al. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia. (2007) 50:359–69. doi: 10.1007/s00125-006-0462-6
44. Lee HY, Lee JS, Alves T, Ladiges W, Rabinovitch PS, Jurczak MJ, et al. Mitochondrial targeted catalase protects against high-fat diet-induced muscle insulin resistance by decreasing intramuscular lipid accumulation. Diabetes. (2017) 66:db161334. doi: 10.2337/db16-1334
45. Chen D, Li X, Zhang L, Zhu M, Gao L. A high-fat diet impairs mitochondrial biogenesis, mitochondrial dynamics, and the respiratory chain complex in rat myocardial tissues. J Cell Biochem. (2018) 119:9602. doi: 10.1002/jcb.27068
46. Shigetada F, Takuya F, Michio S, Masanori I, Yukio Y, Yoshimitsu N, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. (2004) 114:1752–1761. doi: 10.1172/JCI21625
47. Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A. (1999) 96:10857–62. doi: 10.1073/pnas.96.19.10857
48. Krabbendam IE, Honrath B, Culmsee C, Dolga AM. Mitochondrial Ca2+-activated K+ channels and their role in cell life and death pathways. Cell Calc. (2018) 69:101–11. doi: 10.1016/j.ceca.2017.07.005
49. Zeeshan HM, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci. (2016) 17:327. doi: 10.3390/ijms17030327
50. Jean-Philippe B, Mustapha M, Claire L, Ji KM, Martine C, Hubert V, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. (2006) 17:4–12.
51. Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nat Rev Immunol. (2011) 11:81. doi: 10.1038/nri2922
52. Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol. (2000) 68:437–446. doi: 10.1089/10799900050163299
53. Dharmaraja AT. Role of reactive oxygen species (ROS) in therapeutics and drug resistance in cancer and bacteria. J Med Chem. (2017) 60:3221–40. doi: 10.1021/acs.jmedchem.6b01243
54. Pinegin B, Vorobjeva N, Pashenkov M, Chernyak B. The role of mitochondrial ROS in antibacterial immunity. J Cell Physiol. (2018) 233:3745–54. doi: 10.1002/jcp.26117
55. Dunnill C, Patton T, Brennan J, Barrett J, Dryden M, Cooke J, et al. Reactive oxygen species (ROS) and wound healing: the functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process. Int Wound J. (2017) 14:89–96. doi: 10.1111/iwj.12557
56. Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabet Care. (2004) 27:813–23. doi: 10.2337/diacare.27.3.813
57. Sawant JM. Biochemical changes in polymorphonuclear leucocytes in diabetic patients. J Postgrad Med. (1993) 39:183.
58. Krol E, Agueel R, Banue S, Smogorzewski M, Kumar D, Massry S, et al. Amlodipine reverses the elevation in [Ca2+]i and the impairment of phagocytosis in PMNLs of NIDDM patients. Kidney Int. (2003) 64:2188–95. doi: 10.1046/j.1523-1755.2003.00311.x
59. Mullick AE, Mcdonald JM, Melkonian G, Talbot P, Pinkerton KE, Rutledge JC. Reactive carbonyls from tobacco smoke increase arterial endothelial layer injury. Am J Physiol Heart Circ Physiol. (2002) 283:591–7. doi: 10.1152/ajpheart.01046.2001
60. Wang CH, Yu CT, Lin HC, Liu CY, Kuo HP. Hypodense alveolar macrophages in patients with diabetes mellitus and active pulmonary tuberculosis. Tuber Lung Dis. (1999) 79:235–42. doi: 10.1054/tuld.1998.0167
61. Wijnhoven HJ, Heunks LM, Geraedts MC, Hafmans T, Viña JR, Dekhuijzen PNR. Oxidative and nitrosative stress in the diaphragm of patients with COPD. Int J Chron Obstruct Pulmon Dis. (2006) 1:173–9. doi: 10.2147/copd.2006.1.2.173
62. Gunawardana N, Finney L, Johnston SL, Mallia P. Experimental rhinovirus infection in COPD: implications for antiviral therapies. Antiviral Res. (2014) 102:95–105. doi: 10.1016/j.antiviral.2013.12.006
63. Mallia P, Message SD, Contoli M, Gray KK, Telcian A, Laza-Stanca, et al. Neutrophil adhesion molecules in experimental rhinovirus infection in COPD. Respir Res. (2013) 14:72. doi: 10.1186/1465-9921-14-72
64. Falagas ME, Athanasoulia AP, Peppas G, Karageorgopoulos DE. Effect of body mass index on the outcome of infections: a systematic review. Obes Rev. (2010) 10:280–9. doi: 10.1111/j.1467-789X.2008.00546.x
65. Song LC, Ding Y, Liu Y, Cai Z, Zhou J, Swarup, et al. Reactive oxygen species drive evolution of pro-biofilm variants in pathogens by modulating cyclic-di-GMP levels. Open Biol. (2016) 6:160162. doi: 10.1098/rsob.160162
66. Aragon-Martinez OH, Martinez-Morales F, Isiordia-Espinoza MA, Luque CD, Zapata Morales JR, Gonzalez-Rivera ML. Bacterial resistance and failure of clinical cure could be produced by oxidative stress in patients with diabetes or cardiovascular diseases during fluoroquinolone therapy. Med Hypotheses. (2017) 103:32–4. doi: 10.1016/j.mehy.2017.04.004
67. Jung HS, Kang BJ, Ra SW, Seo KW, Jegal Y, Jun JB, et al. Elucidation of bacterial pneumonia-causing pathogens in patients with respiratory viral infection. Tuberc Respir Dis (Seoul). (2017) 80:358–67. doi: 10.4046/trd.2017.0044
68. Shivshankar P, Boyd AR, Le SC, Yeh IT, Orihuela CJ. Cellular senescence increases expression of bacterial ligands in the lungs and is positively correlated with increased susceptibility to pneumococcal pneumonia. Aging Cell. (2011) 10:798–806. doi: 10.1111/j.1474-9726.2011.00720.x
69. Han X, Chen H, Gong H, Tang X, Huang N, Xu W, et al. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress-induced senescence. J Biol Chem. (2020) 295:4451–63. doi: 10.1074/jbc.RA119.010734
70. Baden LR, Rubin EJ. Covid-19 - the search for effective therapy. N Engl J Med. (2020) 382:1851–2. doi: 10.1056/NEJMe2005477
71. Koutsakos M, Nguyen TH, Barclay WS, Kedzierska K. Knowns and unknowns of influenza B viruses. Future Microbiol. (2016) 11:119–35. doi: 10.2217/fmb.15.120
72. Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, et al. Prevalence of comorbidities in the novel Wuhan coronavirus (COVID-19) infection: a systematic review and meta-analysis. Int J Infect Dis. (2020) 94:91–5. doi: 10.1016/j.ijid.2020.03.017
73. Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. (2020) 8:e21. doi: 10.1016/S2213-2600(20)30116-8
74. Maddaloni E, Buzzetti R. Covid-19 and diabetes mellitus: unveiling the interaction of two pandemics. Diabetes Metab Res Rev. (2020) e33213321. doi: 10.1002/dmrr.3321
75. Apostolova N, Victor VM. Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications. Antioxid Redox Signal. (2015) 22:686–729. doi: 10.1089/ars.2014.5952
76. Flohé L. The fairytale of the GSSG/GSH redox potential. Biochim Biophys Acta. (2013) 1830:3139–42. doi: 10.1016/j.bbagen.2012.10.020
77. Calabrese G, Morgan B, Riemer J. Mitochondrial glutathione: regulation and functions. Antioxid Redox Signal. (2017) 27:1162–77. doi: 10.1089/ars.2017.7121
78. Oudemans-van Straaten HM, Spoelstra-de Man AM, de Waard MC. Vitamin C revisited. Crit Care. (2014) 18:460. doi: 10.1186/s13054-014-0460-x
79. Hemilä H, Louhiala P. Vitamin C for preventing and treating pneumonia. Cochrane Database Syst Rev. (2013) Cd005532. doi: 10.1002/14651858.CD005532.pub3
80. Oostwoud LC, Gunasinghe P, Seow HJ, Ye JM, Selemidis S, Bozinovski S, et al. Apocynin and ebselen reduce influenza A virus-induced lung inflammation in cigarette smoke-exposed mice. Sci Rep. (2016) 6:20983. doi: 10.1038/srep20983
81. Roederer M, Staal FJ, Raju PA, Ela SW, Herzenberg LA, Herzenberg LA. Cytokine-stimulated human immunodeficiency virus replication is inhibited by N-acetyl-L-cysteine. Proc Natl Acad Sci U S A. (1990) 87:4884–8. doi: 10.1073/pnas.87.12.4884
82. Staal FJ, Roederer M, Herzenberg LA, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci U S A. (1990) 87:9943–7. doi: 10.1073/pnas.87.24.9943
83. Geiler J, Michaelis M, Naczk P, Leutz A, Langer K, Doerr HW, et al. N-acetyl-L-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549cells infected with highly pathogenic H5N1 influenza A virus. Biochem Pharmacol. (2010) 79:413–20. doi: 10.1016/j.bcp.2009.08.025
84. Kesic MJ, Simmons SO, Bauer R, Jaspers I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic Biol Med. (2011) 51:444–53. doi: 10.1016/j.freeradbiomed.2011.04.027
85. Mata M, Morcillo E, Gimeno C, Cortijo J. N-acetyl-L-cysteine (NAC) inhibit mucin synthesis and pro-inflammatory mediators in alveolar type II epithelial cells infected with influenza virus A and B and with respiratory syncytial virus (RSV). Biochem Pharmacol. (2011) 82:548–5. doi: 10.1016/j.bcp.2011.05.014
86. Chen X, Ren F, Hesketh J, Shi X, Li J, Gan F, et al. Reactive oxygen species regulate the replication of porcine circovirus type 2 via NF-κB pathway. Virology. (2012) 426:66–72. doi: 10.1016/j.virol.2012.01.023
87. Nøhr-Meldgaard K, Ovsepian A, Ingmer H, Vestergaard M. Resveratrol enhances the efficacy of aminoglycosides against Staphylococcus aureus. Int J Antimicrob Agents. (2018) 52:390–6. doi: 10.1016/j.ijantimicag.2018.06.005
88. Ma DSL, Tan LT, Chan KG, Yap WH, Pusparajah P, Chuah LH, et al. Resveratrol-potential antibacterial agent against foodborne pathogens. Front Pharmacol. (2018) 9:102. doi: 10.3389/fphar.2018.00102
89. Vestergaard M, Ingmer H. Antibacterial and antifungal properties of resveratrol. Int J Antimicrob Agents. (2019) 53:716–23. doi: 10.1016/j.ijantimicag.2019.02.015
90. Kumar SN, Siji JV, Nambisan B, Mohandas C. Activity and synergistic interactions of stilbenes and antibiotic combinations against bacteria in vitro. World J Microbiol Biotechnol. (2012) 28:3143–50. doi: 10.1007/s11274-012-1124-0
91. Tosato MG, Schilardi PL, de Mele MFL, Thomas AH, Miñán A, Lorente C. Resveratrol enhancement Staphylococcus aureus survival under levofloxacin and photodynamic treatments. Int J Antimicrob Agents. (2018) 51:255–9. doi: 10.1016/j.ijantimicag.2017.10.006
92. Ferreira S, Domingues F. The antimicrobial action of resveratrol against Listeria monocytogenes in food-based models and its antibiofilm properties. J Sci Food Agric. (2016) 96:4531–5. doi: 10.1002/jsfa.7669
93. Lee JH, Cho HS, Joo SW, Chandra Regmi S, Kim JA, Ryu CM, et al. Diverse plant extracts and trans-resveratrol inhibit biofilm formation and swarming of Escherichia coli O157:H7. Biofouling. (2013) 29:1189–203. doi: 10.1080/08927014.2013.832223
94. Martelli G, Giacomini D. Antibacterial and antioxidant activities for natural and synthetic dual-active compounds. Eur J Med Chem. (2018) 158:91–105. doi: 10.1016/j.ejmech.2018.09.009
95. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
96. Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. (2011) 472:230–3. doi: 10.1038/nature09932
97. Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. (2011) 332:1433–5. doi: 10.1126/science.1200094
98. Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. (2007) 403:139–48. doi: 10.1042/BJ20061520
99. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. (2009) 89:1025–78. doi: 10.1152/physrev.00011.2008
100. Cantó C, Gerharthines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. (2009) 458:1056–60. doi: 10.1038/nature07813
101. Rajani R, Pastor-Soler NM, Hallows KR. Role of AMP-activated protein kinase in kidney tubular transport, metabolism, and disease. Curr Opin Nephrol Hypertens. (2017) 26:375–83. doi: 10.1097/MNH.0000000000000349
102. Kim WH, Lee JW, Suh YH, Lee HJ, Lee SH, Oh YK, et al. AICAR potentiates ROS production induced by chronic high glucose: roles of AMPK in pancreatic β-cell apoptosis. Cell Signal. (2007) 19:791–805. doi: 10.1016/j.cellsig.2006.10.004
103. Shen Y, Honma N, Kobayashi K, Jia LN, Hosono T, Shindo K, et al. Cinnamon extract enhances glucose uptake in 3T3-L1 adipocytes and C2C12 myocytes by inducing LKB1-AMP-activated protein kinase signaling. PLoS One. (2014) 9:e87894. doi: 10.1371/journal.pone.0087894
104. Zhou X, Shentu P, Xu Y. Spatiotemporal regulators for insulin-stimulated GLUT4 vesicle exocytosis. J Diabet Res. (2017) 2017:1683678. doi: 10.1155/2017/1683678
105. Habets DD, Coumans WA, El Hasnaoui M, Zarrinpashneh E, Bertrand L, Viollet B, et al. Crucial role for LKB1 to AMPKα2 axis in the regulation of CD36-mediated long-chain fatty acid uptake into cardiomyocytes. Biochim Biophys Acta. (2009) 1791:212–9. doi: 10.1016/j.bbalip.2008.12.009
106. García-Martínez C, Marotta M, Moore-Carrasco R, Guitart M, Camps M, Busquets S, et al. Impact on fatty acid metabolism and differential localization of FATP1 and FAT/CD36 proteins delivered in cultured human muscle cells. Am J Physiol Cell Physiol. (2005) 288:C1264–72. doi: 10.1152/ajpcell.00271.2004
107. Chabowski A, Coort SL, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, et al. The subcellular compartmentation of fatty acid transporters is regulated differently by insulin and by AICAR. FEBS Lett. (2005) 579:2428–32. doi: 10.1016/j.febslet.2004.11.118
108. Turcotte LP, Raney MA, Todd MK. ERK1/2 inhibition prevents contraction-induced increase in plasma membrane FAT/CD36 content and FA uptake in rodent muscle. Acta Physiol Scand. (2005) 184:131–9. doi: 10.1111/j.1365-201X.2005.01445.x
109. Luiken JJ, Coort SL, Koonen DP, van der Horst DJ, Bonen A, Zorzano A, et al. Regulation of cardiac long-chain fatty acid and glucose uptake by translocation of substrate transporters. Pflugers Arch. (2004) 448:1–15. doi: 10.1007/s00424-003-1199-4
110. Jeppesen J, Albers PH, Rose AJ, Birk JB, Schjerling P, Dzamko N, et al. Contraction-induced skeletal muscle FAT/CD36 trafficking and FA uptake is AMPK independent. J Lipid Res. (2011) 52:699–711. doi: 10.1194/jlr.M007138
111. Liu Q, Gauthier MS, Sun L, Ruderman N, Lodish H. Activation of AMP-activated protein kinase signaling pathway by adiponectin and insulin in mouse adipocytes: requirement of acyl-CoA synthetases FATP1 and Acsl1 and association with an elevation in AMP/ATP ratio. FASEB J. (2010) 24:4229–39. doi: 10.1096/fj.10-159723
112. Sun G, Tarasov AI, McGinty J, McDonald A, da Silva Xavier G, Gorman T, et al. Ablation of AMP-activated protein kinase α1 and α2 from mouse pancreatic β cells and RIP2.Cre neurons suppresses insulin release in vivo. Diabetologia. (2010) 53:924–36. doi: 10.1007/s00125-010-1692-1
113. Xie Z, Zhang J, Wu J, Viollet B, Zou MH. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes. (2008) 57:3222–30. doi: 10.2337/db08-0610
114. Guo Y, Zhang Y, Hong K, Luo F, Gu Q, Lu N, et al. AMPK inhibition blocks ROS-NFκB signaling and attenuates endotoxemia-induced liver injury. PLoS One. (2014) 9:e86881. doi: 10.1371/journal.pone.0086881
115. Li XN, Song J, Zhang L, LeMaire SA, Hou X, Zhang C, et al. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes. (2009) 58:2246–57. doi: 10.2337/db08-1512
116. Ogata FT, Batista WL, Sartori A, Gesteira TF, Masutani H, Arai RJ, et al. Nitrosative/oxidative stress conditions regulate thioredoxin-interacting protein (TXNIP) expression and thioredoxin-1 (TRX-1) nuclear localization. PLoS One. (2013) 8:e84588. doi: 10.1371/journal.pone.0084588
117. Hou Y, Wang Y, He Q, Li L, Xie H, Zhao Y, et al. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav Brain Res. (2018) 336:32–9. doi: 10.1016/j.bbr.2017.06.027
118. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. (2015) 22:377–88. doi: 10.1038/cdd.2014.150
119. Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol. (2015) 33:55–66. doi: 10.1016/j.ceb.2014.12.001
120. Schieke SM, Phillips D, McCoy JP Jr, Aponte AM, Shen RF, Balaban RS, et al. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. (2006) 281:27643–652. doi: 10.1074/jbc.M603536200
121. Jeong AY, Min YL, Sang HL, Park JH, Han HJ. PPARδ agonist-mediated ROS stimulates mouse embryonic stem cell proliferation through cooperation of p38 MAPK and Wnt/β-catenin. Cell Cycle. (2009) 8:611–9. doi: 10.4161/cc.8.4.7752
122. Atsuko N, Yoko W, Yasuko K, Satoru M. Link between PI3K/AKT/PTEN pathway and NOX proteinin diseases. Aging Dis. (2014) 5:203–11. doi: 10.14336/AD.2014.0500203
123. Lee YH, Lee NH, Bhattarai G, Yun JS, Kim TI, Jhee EC, et al. PPARγ inhibits inflammatory reaction in oxidative stress induced human diploid fibloblast. Cell Biochem Funct. (2010) 28:490–6. doi: 10.1002/cbf.1681
124. Lee H, Ham SA, Kim MY, Kim JH, Paek KS, Kang ES, et al. Activation of PPARδ counteracts angiotensin II-induced ROS generation by inhibiting rac1 translocation in vascular smooth muscle cells. Free Radic Res. (2012) 46:912–9. doi: 10.3109/10715762.2012.687448
125. Singh S, Vrishni S, Singh BK, Rahman I, Kakkar P. Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radiac Res. (2010) 44:1267–88. doi: 10.3109/10715762.2010.507670
126. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. (2007) 47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046
127. Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. (2005) 7:385–94. doi: 10.1089/ars.2005.7.385
128. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. (2009) 284:13291–5. doi: 10.1074/jbc.R900010200
129. Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. (2006) 38:769–89. doi: 10.1080/03602530600971974
130. Jiang T, Huang Z, Lin Y, Zhang Z, Fang D, Zhang DD. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes. (2010) 59:850–60. doi: 10.2337/db09-1342
131. Lv X, Li Y, Xiao Q, Li D. Daphnetin activates the Nrf2-dependent antioxidant response to prevent arsenic-induced oxidative insult in human lung epithelial cells. Chem Biol Interact. (2019) 302:93–100. doi: 10.1016/j.cbi.2019.02.001
132. Zheng G, Ren H, Li H, Li X, Dong T, Xu S, et al. Lycium barbarum polysaccharide reduces hyperoxic acute lung injury in mice through Nrf2 pathway. Biomed Pharmacother. (2019) 111:733–9. doi: 10.1016/j.biopha.2018.12.073
133. Song HC, Kim YW, Sang GK. AMPK-mediated GSK3β inhibition by isoliquiritigenin contributes to protecting mitochondria against iron-catalyzed oxidative stress. Biochem Pharmacol. (2010) 79:1352–62. doi: 10.1016/j.bcp.2009.12.011
134. Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. (2006) 281:14841–51. doi: 10.1074/jbc.M513737200
135. Saavedra JM. Angiotensin II AT1 receptor blockers as treatments for inflammatory brain disorders. Clin Sci (Lond). (2012) 123:567–90. doi: 10.1042/CS20120078
136. Wang Y, Huang Y, Xu Y, Ruan W, Wang H, Zhang Y et al. A dual AMPK/Nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxid Redox Signal. (2018) 28:141–63. doi: 10.1089/ars.2017.7003
137. Zhang DD, Hannink MJM, Biology C. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. (2003) 23:8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003
138. Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, et al. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. (2008) 28:2758–70. doi: 10.1128/MCB.01704-07
139. Mo C, Wang L, Zhang J, Numazawa S, Tang H, Tang X, et al. The crosstalk between Nrf2 and AMPK signal pathways is important for the anti-inflammatory effect of berberine in LPS-stimulated macrophages and endotoxin-shocked mice. Antioxid Redox Signal. (2014) 20:574–88. doi: 10.1089/ars.2012.5116
140. Hu R, Hebbar V, Kim BR, Chen C, Winnik B, Buckley B, et al. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther. (2004) 310:263–71. doi: 10.1124/jpet.103.064261
141. Tram Thu V, Reine TM, Amanda S, Jenssen TG, Kolset SO. Syndecan-4 is a major syndecan in primary human endothelial cells in vitro, modulated by inflammatory stimuli and involved in wound healing. J Histochem Cytochem. (2015) 63:280–92. doi: 10.1369/0022155415568995
142. Kon S, Ikesue M, Kimura C, Aoki M, Nakayama Y, Saito Y, et al. Syndecan-4 protects against osteopontin-mediated acute hepatic injury by masking functional domains of osteopontin. J Exp Med. (2008) 205:25–33. doi: 10.1084/jem.20071324
143. Kärkkäinen V, Pomeshchik Y, Savchenko E, Dhungana H, Kurronen A, Lehtonen S, et al. Nrf2 regulates neurogenesis and protects neural progenitor cells against Aβ toxicity. Stem Cells. (2014) 32:1904–16. doi: 10.1002/stem.1666
144. Ma CM, Liu Q, Li ML, Ji MJ, Zhang JD, Zhang BH, et al. The effects of type 2 diabetes and postoperative pneumonia on the mortality in inpatients with surgery. Diabet Metab Syndr Obes. (2019) 12:2507–13. doi: 10.2147/DMSO.S232039
145. Karam BS, Chavez-Moreno A, Koh W, Akar JG, Akar FG. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc Diabetol. (2017) 16:120. doi: 10.1186/s12933-017-0604-9
146. Pold R, Jensen LS, Jessen N, Buhl ES, Schmitz O, Flyvbjerg A, et al. Long-term AICAR administration and exercise prevents diabetes in ZDF rats. Diabetes. (2005) 54:928–34. doi: 10.2337/diabetes.54.4.928
147. Musi N, Goodyear LJ. Targeting the AMP-activated protein kinase for the treatment of type 2 diabetes. Curr Drug Targets Immune Endocr Metabol Disord. (2002) 2:119–27. doi: 10.2174/1568008023340730
148. Lessard SJ, Chen ZP, Watt MJ, Hashem M, Reid JJ, Febbraio MA, et al. Chronic rosiglitazone treatment restores AMPKα2 activity in insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab. (2006) 290:E251–7. doi: 10.1152/ajpendo.00096.2005
149. Turner N, Li JY, Gosby A, To SW, Cheng Z, Miyoshi H, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes. (2008) 57:1414–8. doi: 10.2337/db07-1552
150. Timmers S, Konings E, Bilet L, Houtkooper RH, van de Weijer T, Goossens GH, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. (2011) 14:612–22. doi: 10.1016/j.cmet.2011.10.002
151. Wang QY, Tong AH, Pan YY, Zhang XD, Ding WY, Xiong W. The effect of cassia seed extract on the regulation of the LKB1-AMPK-GLUT4 signaling pathway in the skeletal muscle of diabetic rats to improve the insulin sensitivity of the skeletal muscle. Diabetol Metab Syndr. (2019) 11:108. doi: 10.1186/s13098-019-0504-0
Keywords: AMPK, Nrf2, pneumonia, oxidative stress, diabetes, obesity
Citation: Xu W, Zhao T and Xiao H (2020) The Implication of Oxidative Stress and AMPK-Nrf2 Antioxidative Signaling in Pneumonia Pathogenesis. Front. Endocrinol. 11:400. doi: 10.3389/fendo.2020.00400
Received: 19 October 2019; Accepted: 18 May 2020;
Published: 17 June 2020.
Edited by:
Liang-Jun Yan, University of North Texas Health Science Center, United StatesReviewed by:
Marcel Bonay, INSERM U1179 Handicap neuromusculaire: Physiopathologie, Biothérapie et Pharmacologie appliquées (END-ICAP), FranceMarianna Jung, University of North Texas Health Science Center, United States
Copyright © 2020 Xu, Zhao and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hengyi Xiao, aGVuZ3lpeEBzY3UuZWR1LmNu
†These authors have contributed equally to this work