
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 12 May 2020
Sec. Cellular Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00281
This article is part of the Research Topic Update on the Endocrinology of Myocardial Aging/Heart Failure View all 7 articles
 Carmine Bruno1,2†
Carmine Bruno1,2† Andrea Silvestrini2,3*†
Andrea Silvestrini2,3*† Rodolfo Calarco1,2Angela M. R. Favuzzi1,2Edoardo Vergani1,2Maria Anna Nicolazzi1,2Claudia d'Abate1,2Elisabetta Meucci2,3
Rodolfo Calarco1,2Angela M. R. Favuzzi1,2Edoardo Vergani1,2Maria Anna Nicolazzi1,2Claudia d'Abate1,2Elisabetta Meucci2,3 Alvaro Mordente2,3Raffaele Landolfi1,2
Alvaro Mordente2,3Raffaele Landolfi1,2 Antonio Mancini1,2*
Antonio Mancini1,2*Purpose: In heart failure with reduced ejection fraction, catabolic mechanisms have a strong negative impact on mortality and morbidity. The relationship between anabolic hormonal deficiency and heart failure with preserved ejection fraction (HFpEF) has still been poorly investigated. On the other hand, oxidative stress is recognized as a player in the pathogenesis of HFpEF. Therefore, we performed a cohort study in HFpEF aimed to (1) define the multi-hormonal deficiency prevalence in HFpEF patients; (2) investigate the relationships between hormonal deficiencies and echocardiographic indexes; (3) explore the modulatory activity of anabolic hormones on antioxidant systems.
Methods: 84 patients with diagnosis of HFpEF were enrolled in the study. Plasma levels of N-terminal pro-brain natriuretic peptide, fasting glucose, insulin, lipid pattern, insulin-like growth factor-1, dehydroepiandrosterone-sulfate (DHEA-S), total testosterone (T, only in male subjects) were evaluated. Hormonal deficiencies were defined according to T.O.S.C.A. multi-centric study, as previously published. An echocardiographic evaluation was performed. Plasma total antioxidant capacity (TAC) was measured using the system metmyoglobin –H2O2 and the chromogen ABTS, whose radical form is spectroscopically revealed; latency time (LAG) in the appearance of ABTS• is proportional to antioxidants in sample.
Results: Multiple deficiencies were discovered. DHEA-S deficiency in 87% of patients, IGF-1 in 67% of patients, T in 42%. Patients with DHEA-S deficiency showed lower levels of TAC expressed by LAG (mean ± SEM 91.25 ± 9.34 vs. 75.22 ± 4.38 s; p < 0.05). No differences between TAC in patients with or without IGF-1 deficiency were found. A trend toward high level of TAC in patients without hormonal deficiencies compared with patients with one or multiple deficiencies was found. Regarding echocardiographic parameters, Left Atrial and Left Atrial Volume Index were significantly higher in patients with low IGF-1 values (mean ± SD 90.84 ± 3.86 vs. 72.83 ± 3.78 mL; 51.03 ± 2.33 vs. 40.56 ± 2.46 mL/m2, respectively; p < 0.05).
Conclusions: Our study showed high prevalence of anabolic deficiencies in HFpEF. DHEA-S seems to influence antioxidant levels; IGF-1 deficiency was associated with alteration in parameters of myocardial structure and dysfunction. These data suggest a role of anabolic hormones in the complex pathophysiological mechanisms of HFpEF and could represent the basis for longitudinal studies and investigations on possible benefits of replacement therapy.
Heart Failure (HF) is a clinical syndrome characterized by typical signs and symptoms (dyspnea, fluid retention, fatigue, and exercise intolerance) associated with reduced cardiac output and/or elevated intracardiac pressures at rest or during stress. Classically, HF is classified according to left ventricle ejection fraction (LVEF) in two main variants: HF with reduced ejection fraction (HFrEF) characterized by LFEF <40%, and HF with preserved ejection fraction (HFpEF) when LVEF ≥ 50% (1).
These two variants of HF, while sharing common clinical features, have been associated with different pathophysiological models (2) and therapeutic efficacy of medications on mortality (3).
About HFpEF pathophysiology, diastolic dysfunction is the main mechanism involved, but several other alterations, such as left atrial dysfunction, right ventricular dysfunction, pulmonary hypertension, and increased vascular stiffness, have been identified, which contribute to HFpEF (4–7). Advanced age and comorbidities are the leading risk factors for HFpEF. According to a recent proposal, comorbidities play a pivotal role in leading a systemic proinflammatory status that is responsible, via oxidative stress at microvascular level, of functional and structural myocardial alteration typical of HFpEF (8). Among comorbidities, the most important and widely prevalent in HFpEF patients are represented by obesity, diabetes, iron deficiency, chronic obstructive pulmonary syndrome (COPD), hypertension, and renal failure. The clinical course of comorbidities significantly influences HFpEF prognosis (9–11) and many interventions, both pharmacological and non-pharmacological, directed to improve comorbidities have been associated to better clinical outcomes in patients with HFpEF (12).
Regarding HFpEF therapies, many efforts have been made to find effective pharmacological approaches; conventional HF drugs, including ACE inhibitors, sartans, and β1-antagonists (13–15), did not show improvements in HFpEF morbidity and mortality; about mineralocorticoids receptor antagonists (MRI) and neprilisin-inhibitors, post-hoc analyses of randomized trials suggest the need of accurate stratification of HFpEF patients, in order to implement “tailor-made” therapeutic strategies (16, 17). In this direction, pharmacogenetics could represent a promising field of investigations (18).
On the other hand, anabolic hormones, insulin-like growth factor-1 (IGF-1), dehydroepiandrosterone-sulfate (DHEA-S), testosterone (T), have an important role on cardiac morphology and function (19–21); Anabolic hormones deficiency, in which the so called “cardiac cachexia” represents the end-stage, demonstrated detrimental impact on disease progression and mortality in heart failure with reduced ejection fraction (HFrEF) (22–24).
The relationship between anabolic hormone deficiency and HFpEF has been poorly investigated. A study of Salzano et al. (25) showed a lower impact of anabolic drive deficiencies in HFpEF than HFrEF, although about half of the HFpEF patients demonstrated single or multiple hormonal deficiency. These data were partially in agreement with our preliminary study on prevalence of anabolic deficiencies in HFpEF (26).
No data were reported about the impact of hormonal deficiencies on oxidative stress parameters; therefore, we performed an observational cross-sectional study to quantify anabolic hormonal deficiency prevalence, and to investigate the relationships between anabolic alterations, echocardiographic parameters, and antioxidants levels, with the aim to gain insight into pathophysiology of HFpEF.
Chronic HFpEF patients with NYHA functional class I–III, admitted to the Department of Internal Medicine of the “Fondazione Policlinico Universitario A. Gemelli IRCCS” between April 2016 and May 2019 were recruited. The HFpEF diagnosis was based on current European Society of Cardiology guidelines (1). Patients with symptoms and signs of HF, elevated natriuretic peptides levels, N-terminal proBNP > 125 pg/mL and LVEF > 50%) and echocardiographic evidence of diastolic dysfunction were considered. The echocardiographic criteria of diastolic dysfunction were defined as left atrial volume index (LAVI) > 34 mL/m2, left ventricular mass index left atrial volume index (LAVI) > 34 mL/m or a left ventricular mass index (LVMI) ≥ 115 g/m2 for males, and ≥95 g/m2 for females. Doppler parameters were a ratio of transmitral early filling velocity to tissue early diastolic mitral annular velocity (E/e′) ≥ 13 and a mean e′ septal and lateral wall <9 cm/s.
Patients with acute HF, NYHA class IV, end stage renal disease, liver cirrhosis, neoplastic or autoimmune diseases were excluded, as well as patients with known endocrinopathies, taking hormonal replacement therapy or previous/current amiodarone treatment.
Information about physiological and medical history, including the main risk factors for cardiovascular disease and pharmacological therapy, were acquired. Standard medical therapy, including loop diuretics, angiotensin converting enzyme inhibitors (ACEi), angiotensin II receptor blockers (ARB), and beta-blockers (BBs) had to be stable for at least 2 months. We investigated the prevalence of the following comorbidities: arterial hypertension, diabetes mellitus (DM), chronic obstructive pulmonary disease (COPD), renal failure, anemia, atrial fibrillation, peripheral artery disease, or coronary artery disease.
Body weight was measured in light clothes with an electronic scale (Seca 910; Seca, Ham- burg, Germany) to the nearest 0.1 kg and height was measured with a stadiometer (Seca 220 tele-scopic measured rod; Seca, Hamburg, Germany) to the nearest 0.1 cm. Body Mass Index (BMI) was calculated with the formula Body weight (kg)/[height (m)]2. All patients signed written informed consent, according to the declaration of Helsinki. The study was approved by the Local Ethics Committee.
In all patients, venous blood samples were collected in lithium-heparin tubes in the morning after an overnight fast and after a supine rest of at least 15 min, in order to evaluate plasma levels of N-terminal pro-brain natriuretic peptide (NT-proBNP), plasma fasting glucose, IGF-1, DHEA-S. In male subjects, total testosterone (T) was also assayed. All samples were centrifuged within 2 h after collection and separate plasma aliquots were stored at −80°C until assayed.
A complete echocardiography evaluation was performed (Affiniti 70c, Echocardiography Philips, Philips s.p.a. Milan, Italy), calculating the following parameters: left ventricular ejection fraction (LVEF), left ventricular end-diastolic volume (LVEDV), left ventricular end-systolic volume (LVESV), septal thickness (IVS), posterior wall thickness (PW), peak E-wave velocity (E), peak A-wave velocity (A), E/A ratio, pulsed- wave TDI E′ velocity (E′), E/e′ ratio, deceleration time (DT), left atrial volume (LAV), indexed left atrial volume (LAVI), systolic pulmonary artery pressure (SPAP), tricuspid annular plane systolic excursion (TAPSE), right ventricular mid cavity diameter value (RVEDV), TAPSE/SPAP ratio, and tricuspidal peak velocity (TPV).
NT-proBNP plasma concentrations, DHEA-S, T, and IGF-1 were measured using immunochemiluminometric assays on a Roche Modular E170 analyzer (Roche Diagnostics, Indianapolis, IN, USA). The intra-assay and inter-assay CV for all hormones were, <5.0 and <7.0%, respectively. Echocardiographic and hormonal parameters were obtained in a stable clinical condition.
Our laboratory considered the following ranges as normal: NT-proBNP (>126 pg/mL), DHEA-S (800–3,500 ng/mL), and T (2.5–8.4 ng/mL). Values equal or below the lower normal limit of normal were defined as deficiency. IGF-1 deficiency was defined according to T.O.S.C.A. registry criteria (i.e., 122 ng/mL for age range >55 years, 109 ng/mL for range 55–64 years, 102 ng/mL for range 65–74, 99 ng/mL for age >75 years), referring to the 33th percentile of a male population with Chronic Heart Failure due to age-related variations (27).
Plasma total antioxidant capacity (TAC) was evaluated using the method developed by Rice-Evans and Miller (28). The method is based on inhibition, determined by antioxidants, of the absorbance of the radical cation 2,2′-azinobis (3-ethylbenzothiazoline-6 sulfonate) (ABTS•+) formed by interaction between ABTS (150 μM) and ferrylmyoglobin radical species, generated by activation of metmyoglobin (2.5 μM) with H2O2 (75 μM). Aliquots of the frozen plasma were thawed at room temperature and 10 μL of the samples were tested immediately. The manual procedure was used with only minor modifications, as previously described (29). The reaction was started directly in cuvette through H2O2 addition after 1 min equilibration of all other reagents and followed for 10 min, monitoring at 734 nm, typical of the spectroscopically detectable ABTS•+. The presence of chain-breaking antioxidants induces a lag time (the “Lag phase”) in the accumulation of ABTS•+ proportional to antioxidants concentration and expressed as length of such Lag phase (s). This assay mainly measures non-protein and non-enzymatic antioxidants that are primarily extra-cellular chain-breaking antioxidants, such as ascorbate, urate, and glutathione. Trolox, a water-soluble tocopherol analog, was used as a reference standard. Absorbance was measured with a Hewlett-Packard 8450A UV/Vis spectrophotometer (Palo Alto, CA) equipped with a cuvette stirring apparatus and a constant temperature cell holder. Measurements of pH were made with a PHM84 Research pH meter (Radiometer, Copenhagen, Denmark); the electrode response was corrected for temperature. Intraassay CV was <8%.
Patients with normal values and hormonal deficiencies were identified for each single measured hormonal parameter. Mean and Standard Error of the Mean (SEM) were used to describe quantitative variables, absolute and relative frequencies for qualitative variables. Student T-Test was used to evaluate the differences in echocardiographic parameters, NT-proBNP values, BMI, and age between patients divided in groups according to the presence or absence of hormone deficiency (in particular, IGF-1 and testosterone deficiencies).
Mann-Whitney U-Test was used to compare the same parameters between patients with or without DHEA-S deficiency. In order to investigate the associations between hormonal deficiency and comorbidities, Fisher's exact or Chi-squared test was used. A value of p < 0.05 was considered statistically significant and the analysis was performed using Prism 6.
Our cohort of patients consisted of 84 patients, 36 men, and 48 women, aged between 59 and 98 years (mean ± SEM: 80.31 ± 0.94 years). Table 1 showed the demographic and clinical features of the entire cohort.
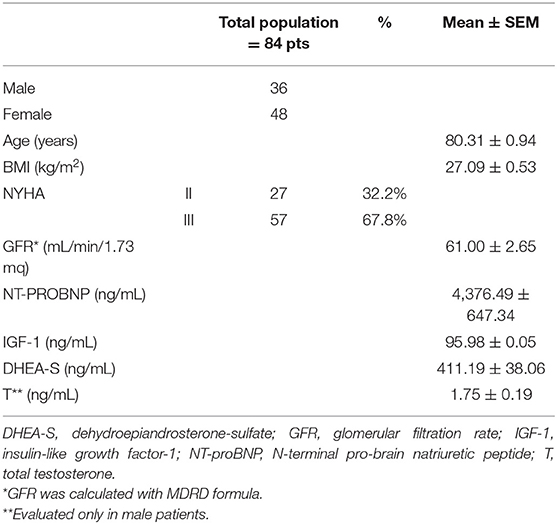
Table 1. Demographic, clinical features, and hormonal levels of the cohort.
Table 2 showed prevalence of comorbidities.
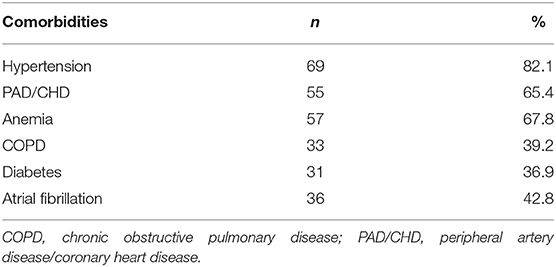
Table 2. Prevalence of comorbidities.
Figure 1 showed the prevalence of hormone deficiencies. DHEA-S deficiency represent the most prevalent anabolic hormone deficiency.
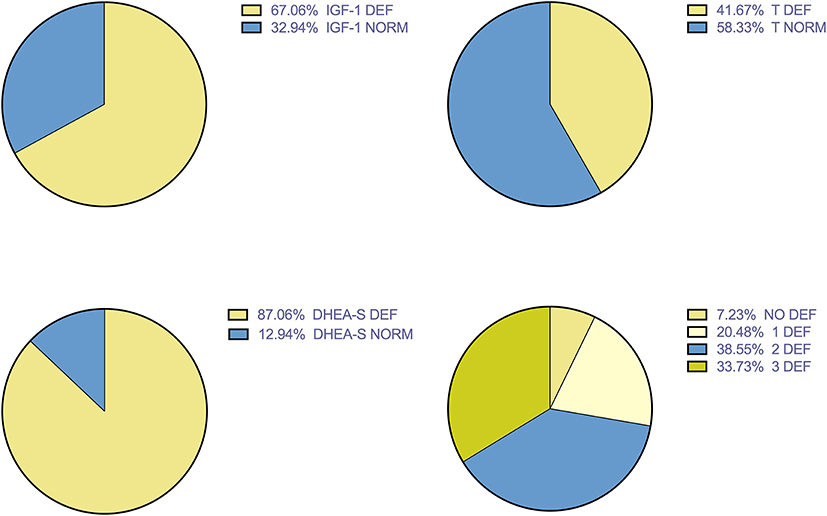
Figure 1. Prevalence of hormonal deficiencies.
Figure 2 showed differences in TAC, expressed by LAG, in group of patients with or without each hormone deficiency. As it could be noticed, higher LAG levels were observed in patients with DHEA-S deficiency compared with patients with normal hormonal values.
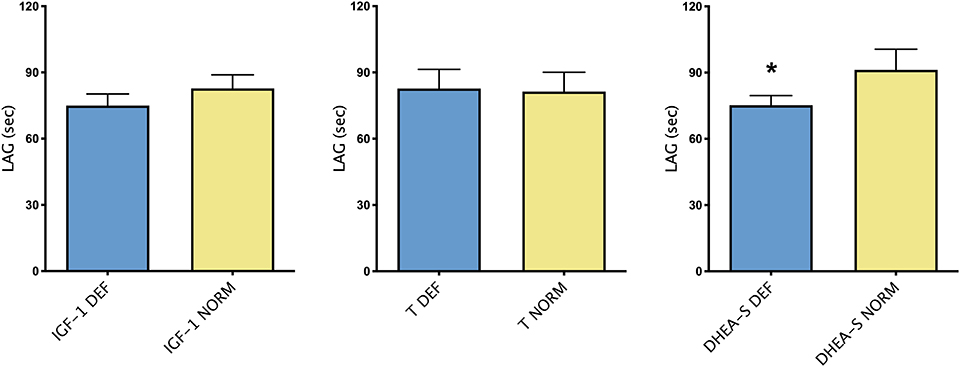
Figure 2. Mean ± SEM of TAC, expressed by LAG (s), in patients divided according to the presence/absence of each hormonal deficiency. *p < 0.05.
Figure 3 showed mean ± SEM LAG in patients divided by the number of hormonal deficiencies observed. Although not significant, patients without hormonal deficiencies presented a trend toward higher levels of LAG compared with patients with one or more hormonal deficiencies.
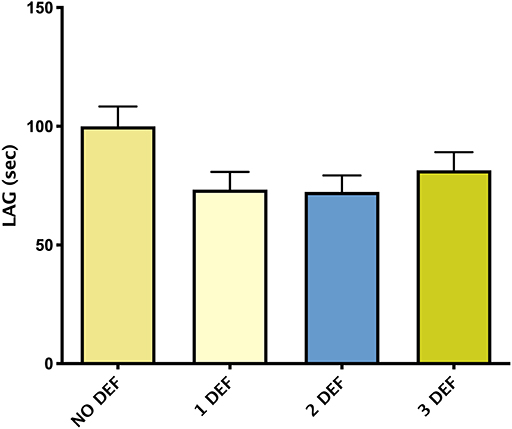
Figure 3. Mean ± SEM of TAC, expressed by LAG (s), in patients divided according to the number of hormonal deficiencies observed.
Table 3 showed echocardiographic parameters in patients with or without IGF-1 deficiency. Patients with IGF-1 deficiency showed higher LAV and LAVI. About prevalence of comorbidities, no statistically significant differences were found between the two groups.
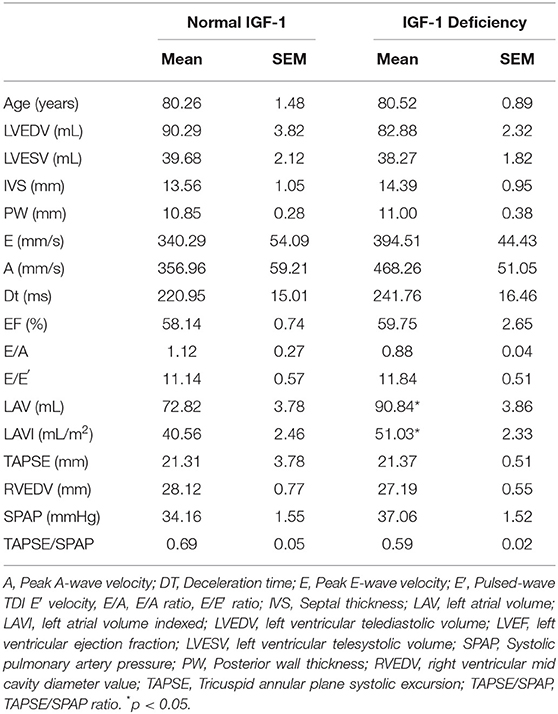
Table 3. Echocardiographic parameters in patients with or without IGF-1 deficiency.
No statistically significant differences in echocardiographic parameters and in prevalence of comorbidities were found in patients with or without T and DHEA-S deficiency.
Our data showed high prevalence of anabolic hormones deficiencies in HFpEF. This confirms previous preliminary report of our group (26), underlying DHEA-S deficiency as the most prevalent. They also partially agree with the study of Salzano et al. (25), in which demographic and clinical features of their cohort of patients were slightly different (better NYHA class, lower mean age). However, our data are not simply related to the age of patients since no significant differences were observed between patients with normal or low levels of the studied hormones.
Furthermore, it is matter of discussion if alterations in anabolic drive observed in HF patients represent a cause or a consequence of the clinical syndrome. Our data, showing a reduced TAC in patients with DHEA-S deficiency, are in favor of a role of hormonal deficiency in influencing HFpEF pathogenesis and/or pathophysiology. In fact, over the past several decades, clinical and experimental studies (30–33) have provided substantial evidence that oxidative stress (OS), defined as an excessive production of reactive radical species of oxygen (ROS) compared to antioxidant defenses, is enhanced in heart failure (HF). ROS in the heart are involved in the lowering of contractile function, myocardial growth and hypertrophy, extracellular matrix remodeling, fibroblast proliferation, and definitely in the breakthrough and progression of the disease (34). Mechanisms involved have been previously reviewed (35). Oxidative stress is considered a main player in pathogenesis of HF, but with different pathways in the two models of HF. In fact, as recently proposed, while in HFrEF the process starts with primary ischemic or oxidative damage of cardiomyocytes, in HFpEF a cascade of events is increased by the systemic pro-inflammatory state related to multiple co-morbidities (36). For instance, diabetic patients showed a dysregulation of the innate and adaptive immune systems in myocardial tissue (37, 38). In obesity and insulin resistance visceral adipose tissue shows a higher CD8+:CD4+ T-cell ratio and macrophage M1 polarization (39), that represent a chronic pro-inflammatory response that, via cytokines activation, contributes to HFpEF progression (40). Cardiac macrophages—and related oxidative stress—are responsible of inducing cardiomyocyte apoptosis and interstitial fibrosis in the HFpEF heart (40). Moreover, the resultant endothelial damage leads to microvascular coronary alterations, and, ultimately, to myocardial dysfunction (8).
Finally, a new important field is the differential response to medical therapy, suggested by recent studies about pharmacogenetics, in the view of personalized medicine; a great importance could be exerted by genetic variations of G-protein coupled receptors (GPCRs) which have been studied in some models of heart failure, in particular variants of the β adrenergic receptors have been linked to a worse clinical course of HF (41, 42). Some other classes of drugs could be also indirectly involved, such as polymorphism of AT1R associated with hypertension (43) and dopamine receptor D2 variations linked to obesity (44).
In this complex interplay between inflammation, oxidative stress and individual response to drugs, anabolic hormones impairment could be another mosaic tile. DHEA-S, in fact, is involved in the regulation of OS (45), and also in HF (35). According to literature, DHEA-S can exert reciprocal effects (pro-oxidant or antioxidant) depending on tissue specificity and hormonal levels (46, 47). DHEA-S treatment may reduce lipogenesis and weight gain in rats (48), delay atherosclerosis in rabbits (49), increase insulin secretion and sensitivity in rats (50, 51), and it may reduce cardiac fibrosis in diabetic rats (52). Various investigations showed a potential role of DHEA in antioxidants modulation. Yorek et al. (53) demonstrated that DHEA reduces plasma oxidative stress markers production in arterioles of diabetic rats, such as thiobarbituric acid-reactive substances (TBARS) and superoxide anion, while Aragno et al. (54) observed ROS reduction in hearts of DHEA-treated diabetic rats. It also narrows oxidative stress-induced skeletal muscle damage in diabetic rats (55). In vivo and in vitro studies showed that DHEA-S counteracts lipid peroxidation (56, 57). Moreover, OS parameters in plasma and in peripheral blood mononuclear cells in diabetic subjects are significantly decreased by DHEA treatment (58).
GH/IGF-1 axis alteration is another factor with high prevalence in our cohort, with an impact on left atrium parameters, considered important indexes of myocardial dysfunction. Although GH is also a regulator of oxidative balance as previously showed, we did not find any influence on TAC in cohort object of our study. It is well-known that IGF-1 is a key factor in cardiomyocytes trophism and contractility. A higher IGFBP/IGF-1 ratio was associated with increased E/e′ ratio, NT-proBNP levels and left atrial enlargement (59). IGF-1 decrease or IGFBP-7 increases were associated with a poor clinical course triggering fibrosis mechanism via increased collagen deposition in the myocardium, one of the mechanisms involved in structural abnormalities leading to diastolic dysfunction. Elevation of soluble suppression of tumorigenicity-2 (sST2), a known collagen synthesis marker from myocardial fibroblast (60) are in favor of this hypothesis. Other links between IGF-1 deficit and diastolic dysfunction were described. Patients with growth hormone and IGF-1 deficiencies exhibit endothelial dysfunction, reduced nitric oxide (NO) production, and high peripheral vascular resistance. GH replacement therapy normalizes NO production and ameliorates peripheral resistance (61). Reduced NO production could contribute to worsening of ventricular compliance in HFpEF patients.
As antioxidants levels are concerned, we observed a trend toward higher levels of TAC in patients with normal hormonal milieu compared with patients with multiple hormonal deficiencies, although not significant. This pattern is not overlapping with one observed in HFrEF as previously described by our group (62); this could reflect a different mechanism underlying oxidative stress related, as above discussed, to a primitive myocardial damage in HFrEF (i.e., ischemic heart disease), and to systemic comorbidities in HFpEF.
Nevertheless, some potential limitations of the present study can be identified. Firstly, the cohort of patients studied although greater than previous reports, needs to be further enlarged. Secondly, TAC as measure of OS should be coupled with other parameters of oxidative damage; finally, our study was focused on IGF-1 levels without evaluating GH secretion via dynamic tests. Therefore, further investigations should be performed to evaluate prevalence of GH deficiency in HFpEF.
Our study, extending previous observation, confirmed a high prevalence of anabolic hormone deficiencies in patients with HFpEF, with particular relevance of DHEA-S. It seems to be related to antioxidants modulation, with higher levels of TAC in patients without DHEA-S deficiency; considering the role of OS in HFpEF pathogenesis, our data could suggest a pathogenetic involvement of this hormone.
Another anabolic key factor could be IGF-1; patients with low IGF-1levels presented left atrial enlargement that is considered one of main diastolic dysfunction criteria.
In conclusion, HFpEF seems to be a complex model with a reciprocal interaction between comorbidities, including anabolic deficiencies, and myocardial function. Oxidative stress could represent the trait union between the three components of the systemic picture, typical of HFpEF.
Therapeutically consequences of these observations remain to be further investigated.
The datasets generated for this study are available on request to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics commitee of Fondazione Policlinico Universiario A Gemelli IRCSS, Largo A Gemelli 00168, Rome, Italy. The patients/participants provided their written informed consent to participate in this study.
CB, AS, AF, and AMa were involved in conception and design of the study. CB, EV, and AS wrote the manuscript. EV, CB, RC, MN, and Cd'A were involved in acquisition and analysis of data. RL, EM, AMa, and AMo were involved in supervision. All authors were involved in critical discussion and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2016) 37:2129–200. doi: 10.5603/KP.2016.0141
2. Silvestrini A, Bruno C, Vergani E, Venuti A, Favuzzi AMR, Guidi F, et al. Circulating irisin levels in heart failure with preserved or reduced ejection fraction: a pilot study. PLoS One. (2019) 14:e0210320. doi: 10.1371/journal.pone.0210320
3. Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. (2011) 32:670–9. doi: 10.1093/eurheartj/ehq426
4. Brubaker PH, Kitzman DW. Prevalence and management of chronotropic incompetence in heart failure. Curr Cardiol Rep. (2007) 9:229–35. doi: 10.1007/BF02938355
5. Lam CSP, Roger VL, Rodeheffer RJ, Borlaug BA, Enders FT, Redfield MM. Pulmonary hypertension in heart failure with preserved ejection fraction. A community-based study. J Am Coll Cardiol. (2009) 53:1119–26. doi: 10.1016/j.jacc.2008.11.051
6. Borlaug BA, Kass DA. Ventricular-vascular interaction in heart failure. Cardiol Clin. (2011) 29:447–59. doi: 10.1016/j.ccl.2011.06.004
7. Phan TT, Shivu GN, Abozguia K, Davies C, Nassimizadeh M, Jimenez D, et al. Impaired heart rate recovery and chronotropic incompetence in patients with heart failure with preserved ejection fraction. Circ Hear Fail. (2010) 3:29–34. doi: 10.1161/CIRCHEARTFAILURE.109.877720
8. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/j.jacc.2013.02.092
9. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2017) 14:591–602. doi: 10.1038/nrcardio.2017.65
10. Ergatoudes C, Schaufelberger M, Andersson B, Pivodic A, Dahlström U, Fu M. Non-cardiac comorbidities and mortality in patients with heart failure with reduced vs. preserved ejection fraction: a study using the Swedish Heart Failure Registry. Clin Res Cardiol. (2019) 108:1025–33. doi: 10.1007/s00392-019-01430-0
11. Riedel O, Ohlmeier C, Enders D, Elsässer A, Vizcaya D, Michel A, et al. The contribution of comorbidities to mortality in hospitalized patients with heart failure. Clin Res Cardiol. (2018) 107:487–97. doi: 10.1007/s00392-018-1210-x
12. Wintrich J, Kindermann I, Ukena C, Selejan S, Werner C, Maack C, et al. Therapeutic approaches in heart failure with preserved ejection fraction: past, present, and future. Clin Res Cardiol. (2020) doi: 10.1007/s00392-020-01633-w. [Epub ahead of print].
13. Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. (2008) 359:2456–67. doi: 10.1056/NEJMoa0805450
14. Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJV, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-preserved trial. Lancet. (2003) 362:777–81. doi: 10.1016/S0140-6736(03)14285-7
15. Cleland JGF, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. (2006) 27:2338–45. doi: 10.1093/eurheartj/ehl250
16. Solomon SD, Vaduganathan ML, Claggett B, Packer M, Zile M, Swedberg K, et al. Sacubitril/valsartan across the spectrum of ejection fraction in heart failure. Circulation. (2020) 141:352–61. doi: 10.1161/CIRCULATIONAHA.119.044586
17. McMurray JJV, Jackson AM, Lam CSP, Redfield MM, Anand IS, Ge J, et al. Effects of sacubitril-valsartan versus valsartan in women compared with men with heart failure and preserved ejection fraction: insights from PARAGON-HF. Circulation. (2020) 141:338–51. doi: 10.1161/CIRCULATIONAHA.119.044491
18. Thompson MD, Cole DE, Capra V, Siminovitch KA, Rovati GE, Burnham WM, et al. Pharmacogenetics of the G protein-coupled receptors. Methods Mol Biol. (2014) 1175:189–242. doi: 10.1007/978-1-4939-0956-8_9
19. Sacca L, Cittadini A, Fazio S. Growth hormone and the heart. Endocr Rev. (1994) 15:555–73. doi: 10.1210/edrv-15-5-555
20. Savineau JP, Marthan R, Dumas De La Roque E. Role of DHEA in cardiovascular diseases. Biochem Pharmacol. (2013) 85:718–26. doi: 10.1016/j.bcp.2012.12.004
21. Ayaz O, Howlett SE. Testosterone modulates cardiac contraction and calcium homeostasis: cellular and molecular mechanisms. Biol Sex Differ. (2015) 6:9. doi: 10.1186/s13293-015-0027-9
22. Anker SD, Chua TP, Ponikowski P, Harrington D, Swan JW, Kox WJ, et al. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation. (1997) 96:526–34. doi: 10.1161/01.CIR.96.2.526
23. Berry C, Clark AL. Catabolism in chronic heart failure. Eur Heart J. (2000) 21:521–32. doi: 10.1053/euhj.1999.1882
24. Jankowska EA, Biel B, Majda J, Szklarska A, Lopuszanska M, Medras M, et al. Anabolic deficiency in men with chronic heart failure: prevalence and detrimental impact on survival. Circulation. (2006) 114:1829–37. doi: 10.1161/CIRCULATIONAHA.106.649426
25. Salzano A, Marra AM, Ferrara F, Arcopinto M, Bobbio E, Valente P, et al. Multiple hormone deficiency syndrome in heart failure with preserved ejection fraction. Int J Cardiol. (2016) 225:1–3. doi: 10.1016/j.ijcard.2016.09.085
26. Favuzzi AMR, Venuti A, Bruno C, Nicolazzi MA, Fuorlo M, Dajko M, et al. Hormonal deficiencies in heart failure with preserved ejection fraction: prevalence and impact on diastolic dysfunction: a pilot study. Eur Rev Med Pharmacol Sci. (2020) 24:352–61. doi: 10.26355/eurrev_202001_19933
27. Bossone E, Arcopinto M, Iacoviello M, Triggiani V, Cacciatore F, Maiello C, et al. Multiple hormonal and metabolic deficiency syndrome in chronic heart failure: rationale, design, and demographic characteristics of the T.O.S.CA. Registry. Intern Emerg Med. (2018) 13:661–71. doi: 10.1007/s11739-018-1844-8
28. Rice-Evans C, Miller NJ. Total antioxidant status in plasma and body fluids. Methods Enzymol. (1994) 234:279–93. doi: 10.1016/0076-6879(94)34095-1
29. Mancini A, Leone E, Festa R, Grande G, Di Donna V, De Marinis L, et al. Evaluation of antioxidant systems (coenzyme Q10 and total antioxidant capacity) in morbid obesity before and after biliopancreatic diversion. Metabolism. (2008) 57:1384–9. doi: 10.1016/j.metabol.2008.05.007
30. Belch JJF, Bridges AB, Scott N, Chopra M. Oxygen free radicals and congestive heart failure. Br Heart J. (1991) 65:245–8. doi: 10.1136/hrt.65.5.245
31. Hill MF, Singal PK. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol. (1996) 148:291–300.
32. Hill MF, Singal PK. Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation. (1997) 96:2414–20. doi: 10.1161/01.CIR.96.7.2414
33. Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A. Elevated levels of 8-iso-prostaglandin F2α in pericardial fluid of patients with heart failure. Circulation. (1998) 97:1536–9. doi: 10.1161/01.CIR.97.16.1536
34. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301:H2181–90. doi: 10.1152/ajpheart.00554.2011
35. Mancini A, Vergani E, Bruno C, Olivieri G, Di Segni C, Silvestrini A, et al. Oxidative stress as a possible mechanism underlying multi-hormonal deficiency in chronic heart failure. Eur Rev Med Pharmacol Sci. (2018) 22:3936–61. doi: 10.26355/eurrev_201806_15279
36. Frantz S, Falcao-Pires I, Balligand JL, Bauersachs J, Brutsaert D, Ciccarelli M, et al. The innate immune system in chronic cardiomyopathy: a European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur J Heart Fail. (2018) 20:445–59. doi: 10.1002/ejhf.1138
37. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. (2015) 116:1022–33. doi: 10.1161/CIRCRESAHA.116.303697
38. Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res. (2015) 116:354–67. doi: 10.1161/CIRCRESAHA.116.304072
39. Sell H, Habich C, Eckel J. Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol. (2012) 8:709–16. doi: 10.1038/nrendo.2012.114
40. Jia G, Habibi J, Bostick BP, Ma L, Demarco VG, Aroor AR, et al. Uric acid promotes left ventricular diastolic dysfunction in mice fed a western diet. Hypertension. (2015) 65:531–9. doi: 10.1161/HYPERTENSIONAHA.114.04737
41. Liggett SB. Polymorphisms of β-adrenergic receptors in heart failure. Am J Med. (2004) 117:525–7. doi: 10.1016/j.amjmed.2004.07.039
42. Liggett SB, Wagoner LE, Craft LL, Hornung RW, Hoit BD, McIntosh TC, et al. The Ile164 β2-adrenergic receptor polymorphism adversely affects the outcome of congestive heart failure. J Clin Invest. (1998) 102:1534–1539. doi: 10.1172/JCI4059
43. Bonnardeaux A, Davies E, Jeunemaitre X, Féry I, Charru A, Clauser E, et al. Angiotensin II type 1 receptor gene polymorphisms in human essential hypertension. Hypertension. (1994) 24:63–9. doi: 10.1161/01.HYP.24.1.63
44. Comings DE, Gade R, MacMurray JP, Muhleman D, Peters WR. Genetic variants of the human obesity (OB) gene: association with body mass index in young women, psychiatric symptoms, and interaction with the dopamine D2 receptor (DRD2) gene. Mol Psychiatry. (1996) 1:325–35.
45. Mancini A, Festa R, Di Donna V, Leone E, Littarru GP, Silvestrini A, et al. Hormones and antioxidant systems: role of pituitary and pituitary-dependent axes. J Endocrinol Invest. (2010) 33:422–33. doi: 10.1007/BF03346615
46. Jacob MHVM, Janner D, da R, Belló-Klein A, Llesuy SF, Ribeiro MFM. Dehydroepiandrosterone modulates antioxidant enzymes and Akt signaling in healthy Wistar rat hearts. J Steroid Biochem Mol Biol. (2008) 112:138–44. doi: 10.1016/j.jsbmb.2008.09.008
47. Çelebi F, Yilmaz I, Aksoy H, Gümüş M, Taysi S, Oren D. Dehydroepiandrosterone prevents oxidative injury in obstructive jaundice in rats. J Int Med Res. (2004) 32:400–5. doi: 10.1177/147323000403200408
48. Han DH, Hansen PA, Chen MM, Holloszy JO. DHEA treatment reduces fat accumulation and protects against insulin resistance in male rats. J Gerontol A Biol Sci Med Sci. (1998) 53:B19–B24. doi: 10.1093/gerona/53A.1.B19
49. Hayashi T, Esaki T, Muto E, Kano H, Asai Y, Thakur NK, et al. Dehydroepiandrosterone retards atherosclerosis formation through its conversion to estrogen: the possible role of nitric oxide. Arterioscler Thromb Vasc Biol. (2000) 20:782–92. doi: 10.1161/01.ATV.20.3.782
50. Campbell CSG, Caperuto LC, Hirata AE, Araujo EP, Velloso LA, Saad MJ, et al. The phosphatidylinositol/AKT/atypical PKC pathway is involved in the improved insulin sensitivity by DHEA in muscle and liver of rats in vivo. Life Sci. (2004) 76:57–70. doi: 10.1016/j.lfs.2004.06.017
51. Medina MC, Souza LC, Caperuto LC, Anhê GF, Amanso AM, Teixeira VPA, et al. Dehydroepiandrosterone increases β-cell mass and improves the glucose-induced insulin secretion by pancreatic islets from aged rats. FEBS Lett. (2006) 580:285–90. doi: 10.1016/j.febslet.2005.12.014
52. Aragno M, Mastrocola R, Alloatti G, Vercellinatto I, Bardini P, Geuna S, et al. Oxidative stress triggers cardiac fibrosis in the heart of diabetic rats. Endocrinology. (2008) 149:380–8. doi: 10.1210/en.2007-0877
53. Yorek MA, Coppey LJ, Gellett JS, Davidson EP, Bing X, Lund DD, et al. Effect of treatment of diabetic rats with dehydroepiandrosterone on vascular and neural function. Am J Physiol Endocrinol Metab. (2002) 283:E1067–75. doi: 10.1152/ajpendo.00173.2002
54. Aragno M, Mastrocola R, Medana C, Catalano MG, Vercellinatto I, Danni O, et al. Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes. Endocrinology. (2006) 147:5967–74. doi: 10.1210/en.2006-0728
55. Aragno M, Mastrocola R, Catalano MG, Brignardello E, Danni O, Boccuzzi G. Oxidative stress impairs skeletal muscle repair in diabetic rats. Diabetes. (2004) 53:1082–8. doi: 10.2337/diabetes.53.4.1082
56. Boccuzzi G, Aragno M, Seccia M, Brignardello E, Tamagno E, Albano E, et al. Protective effect of dehydroepiandrosterone against copper induced lipid peroxidation in the rat. Free Radic Biol Med. (1997) 22:1289–94. doi: 10.1016/S0891-5849(96)00543-6
57. Khalil A, Lehoux JG, Wagner RJ, Lesur O, Cruz S, Dupont E, et al. Dehydroepiandrosterone protects low density lipoproteins against peroxidation by free radicals produced by gamma-radiolysis of ethanol-water mixtures. Atherosclerosis. (1998) 136:99–107. doi: 10.1016/S0021-9150(97)00194-9
58. Brignardello E, Runzo C, Aragno M, Catalano MG, Cassader M, Perin PC, et al. Dehydroepiandrosterone administration counteracts oxidative imbalance and advanced glycation end product formation in type 2 diabetic patients. Diabetes Care. (2007) 30:2922–7. doi: 10.2337/dc07-1110
59. Barroso MC, Kramer F, Greene SJ, Scheyer D, Köhler T, Karoff M, et al. Serum insulin-like growth factor-1 and its binding protein-7: potential novel biomarkers for heart failure with preserved ejection fraction. BMC Cardiovasc Disord. (2016) 16:199. doi: 10.1186/s12872-016-0376-2
60. Zile MR, Baicu CF. Biomarkers of diastolic dysfunction and myocardial fibrosis: application to heart failure with a preserved ejection fraction. J Cardiovasc Transl Res. (2013) 6:501–15. doi: 10.1007/s12265-013-9472-1
61. Caicedo D, Díaz O, Devesa P, Devesa J. Growth hormone (GH) and cardiovascular system. Int J Mol Sci. (2018) 19:290. doi: 10.3390/ijms19010290
Keywords: heart failure, cardiovascular endocrinology, antioxidants, hormones, myocardial dysfunctions
Citation: Bruno C, Silvestrini A, Calarco R, Favuzzi AMR, Vergani E, Nicolazzi MA, d'Abate C, Meucci E, Mordente A, Landolfi R and Mancini A (2020) Anabolic Hormones Deficiencies in Heart Failure With Preserved Ejection Fraction: Prevalence and Impact on Antioxidants Levels and Myocardial Dysfunction. Front. Endocrinol. 11:281. doi: 10.3389/fendo.2020.00281
Received: 31 December 2019; Accepted: 15 April 2020;
Published: 12 May 2020.
Edited by:
Yang Yang, Northwest University, ChinaReviewed by:
Miles Douglas Thompson, University of California, San Diego, United StatesCopyright © 2020 Bruno, Silvestrini, Calarco, Favuzzi, Vergani, Nicolazzi, d'Abate, Meucci, Mordente, Landolfi and Mancini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Silvestrini, YW5kcmVhLnNpbHZlc3RyaW5pQHVuaWNhdHQuaXQ=; Antonio Mancini, YW50b25pby5tYW5jaW5pQHVuaWNhdHQuaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.