 Jes Sloth Mathiesen1,2*
Jes Sloth Mathiesen1,2* Søren Grønlund Nielsen1Åse Krogh Rasmussen3Katalin Kiss4Karin Wadt5Anne Pernille Hermann6Morten Frost Nielsen6Stine Rosenkilde Larsen7
Søren Grønlund Nielsen1Åse Krogh Rasmussen3Katalin Kiss4Karin Wadt5Anne Pernille Hermann6Morten Frost Nielsen6Stine Rosenkilde Larsen7 Klaus Brusgaard8
Klaus Brusgaard8 Anja Lisbeth Frederiksen9
Anja Lisbeth Frederiksen9 Christian Godballe1
Christian Godballe1 Maria Rossing10
Maria Rossing10- 1Department of ORL Head and Neck Surgery and Audiology, Odense University Hospital, Odense, Denmark
- 2Department of Clinical Research, University of Southern Denmark, Odense, Denmark
- 3Department of Medical Endocrinology, Copenhagen University Hospital, Copenhagen, Denmark
- 4Department of Pathology, Copenhagen University Hospital, Copenhagen, Denmark
- 5Department of Clinical Genetics, Copenhagen University Hospital, Copenhagen, Denmark
- 6Department of Endocrinology, Odense University Hospital, Odense, Denmark
- 7Department of Pathology, Odense University Hospital, Odense, Denmark
- 8Department of Clinical Genetics, Odense University Hospital, Odense, Denmark
- 9Department of Clinical Genetics, Aalborg University Hospital, Aalborg, Denmark
- 10Center for Genomic Medicine, Copenhagen University Hospital, Copenhagen, Denmark
Background: Previous studies have suggested that the variability in age of onset and aggressiveness of medullary thyroid carcinoma (MTC) in patients with multiple endocrine neoplasia type 2A (MEN 2A) carrying the same REarranged during Transfection (RET) mutation may be caused by additional RET germline variants or somatic variants.
Methods: This study was a retrospective case comparison study of all MEN 2A index patients (n = 2) with the RET L790F germline mutation in Denmark. Whole blood and MTC tissue were analyzed for RET germline variants and other somatic variants (>500), respectively.
Results: Patient 1 presented with MTC (T1aN1bM0) at age 14 years, while patient 2 presented with MTC (T1bN0M0) at age 70 years. No germline RET germline variants nor other variants were found to explain this MTC variability.
Conclusions: We could not confirm the previously reported finding of a somatic RET variant as likely responsible for the early onset and aggressiveness of MTC in a RET germline mutation carrier. Also, we found no RET germline variants that could explain the MTC variability among our index patients. We did, however, identify a somatic FLT3 R387Q variant with an unknown potential as genetic modifier. Further large-scale studies are needed to investigate genetic modifiers in RET L790F carriers.
Introduction
Multiple endocrine neoplasia (MEN) 2 is an autosomal dominant inherited cancer syndrome subdivided into MEN 2A and MEN 2B (1, 2). With a point prevalence of 13–24 and 1–2 per million, MEN 2A and MEN 2B are rare (3–6). The syndromes, however, are accompanied by major implications as virtually all patients develop medullary thyroid carcinoma (MTC) while variable proportions develop pheochromocytoma, primary hyperparathyroidism, cutaneous lichen amyloidosis, Hirschsprung disease, ganglioneuromatosis of the aerodigestive tract, facial, ophthalmologic, and skeletal abnormalities (1).
MEN 2A and 2B are caused by germline mutations of the REarranged during Transfection (RET) proto-oncogene (7–11). Despite the recognition of strong genotype-phenotype correlations (12, 13), several studies have observed a substantial inter- and intra-familial variability in the age of onset and aggressiveness of MTC among carriers of the same mutation (14–21). Some authors have suggested that certain RET germline variants (IVS1-126G>T, IVS8+82A>G; 85-86 insC, G691S, L769L, S836S, S904S) in combination with a RET germline mutation may modify the age of onset and aggressiveness of MTC in MEN 2 patients (22–26). The issue, however, is controversial (25–30). Others have suggested that the variability may be caused by a somatic variant acting as the “second hit” in Knudson's two hit model (16, 31, 32). To our knowledge, no such studies exist for MEN 2A patients carrying the RET L790F germline mutation.
Consequently, we conducted the first study of RET germline variants and somatic variants in L790F carriers, aiming to explain the MTC variability among index patients in Denmark.
Methods
Study Design and Participants
This investigation is a retrospective case study of all MEN 2A index patients (n = 2) with the RET L790F germline mutation in Denmark.
Data Sources
Patients were identified from the extended nationwide Danish RET cohort 1994–2017 (33, 34), and data were drawn from the Danish MEN 2A cohort 1901–2014, the Danish MTC cohort 1997–2014, and medical records (4, 35).
Genetic Testing
Testing for RET germline variants were performed using Next Generation Sequencing as previously described (33). Formalin-fixed paraffin-embedded tumoir tissue from MTC primary tumor and metastases were tested for somatic RET variants as reported earlier (36). Testing for somatic variants in oncogenes was done by targeted exome sequencing; In short, genomic DNA (200 ng) was fragmented to 300 bp adaptor ligation using KAPA HTP Library Preparation Kit (Roche) and enrichment of exome using SureSelectXT Clinical Research Exome kit (Agilent). Paired-end sequencing (2 × 100 or 2 × 150 bp) was performed to gain an average coverage of 50–100 × using the NextSeq500 platforms (Illumina). Raw sequencing data were processed using CASAVA-1.8.2 and reads were aligned to the human reference genome (hg19/GRCh37) using CLC Biomedical Genomics Workbench (Qiagen). Variant calling >5% frequency and filtering for 523 cancer relevant genes, including RET, H-RAS, K-RAS, and N-RAS using Ingenuity Variant Analysis (Qiagen). For this study, “mutation” denotes a disease-causing (pathogenic) sequence change, while “variant” denotes any sequence change, whether pathogenic or not.
The investigation was approved by the Danish Health Authority (3-3013-395/3) and the Danish Data Protection Agency (18/17801). The Regional Committees on Health Research Ethics for Southern Denmark found further review was not liable to notification. Informed consent was obtained from the cases for the publication of any potentially identifiable images or data included in this article. The data that support the findings of this study are available from the corresponding author upon request.
Results
Patient 1
A 14-year-old female of Lebanese origin, presented with B-symptoms and multiple enlarged lymph nodes on the right side of the neck. Fine-needle aspiration suggested plasma cells or cells with thyroid or paraganglioma origin. A lymph node biopsy was taken and histology showed MTC metastasis. After this, basal calcitonin was measured to 18.3 μg/L (<0.1 μg/L). The patient underwent a total thyroidectomy and right-sided neck dissection. Histology displayed multifocal MTC (largest tumor 7 mm) with metastases to 17 of 20 removed lymph nodes and a normal parathyroid gland. The final TNM-stage was T1aN1bM0. Two months postoperatively basal calcitonin was 19.0 μg/L (<0.1 μg/L) and adjuvant radiotherapy was given. RET testing revealed the L790F (c.2370G>T) germline mutation. There was no family history of MEN 2A. The father was tested negative for RET mutations, while the mother was positive for the L790F mutation. The latter, however, declined further work up. Fourteen years later basal calcitonin had risen to 59,280 ng/L (<7.3 ng/L). Distant metastases were cytologically verified in mediastinal lymph nodes and suspected in the neck, lungs, mamma, liver, and in proximity to the transverse colon. The patient was initiated on Vandetanib, a tyrosine kinase inhibitor. Thirty months after initiation, the patient had basal calcitonin of 80 ng/L and apart from the suspected liver metastasis that was assessed without change there were no radiological evidence of metastases on CT scan. Neither pheochromocytoma nor primary hyperparathyroidism has been diagnosed during the 16 years of follow-up. Table 1 shows clinical characteristics and results of genetic analyses for patient 1.
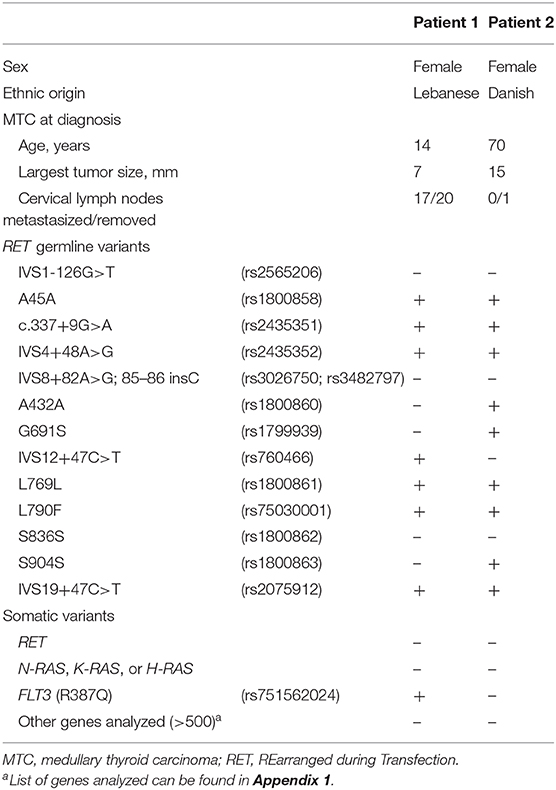
Table 1. Clinical and genetic characteristics of two index patients with the RET L790F germline mutation.
Patient 2
A 70-year-old female, presented with a right-sided thyroid tumor, but was otherwise asymptomatic. Fine-needle aspiration showed follicular neoplasm of unknown malignant potential. The medical history was unremarkable apart from a right-sided ductal carcinoma in situ treated with lumpectomy and radiotherapy 18 months earlier. The patient underwent a left-sided hemithyroidectomy without prior measurement of serum calcitonin, as MTC was unsuspected. Histology revealed multifocal MTC with the largest tumor size of 15 mm. At this point, basal serum calcitonin was 28 ng/L (<7.3 ng/L) and family history revealed that the mother and aunt had undergone thyroid operations for unknown causes. The patient underwent right-sided completion thyroidectomy. Histology showed multifocal MTC with several tumors measuring 1–3 mm. Also, a removed lymph node was without metastasis. The final TNM-stage was T1bN0M0. A RET test identified the L790F (c.2370G>T) germline mutation. Subsequently, normal tissue from the deceased mother was also tested positive for the L790F mutation. Basal serum calcitonin was immeasurable and no biochemical signs of pheochromocytoma or primary hyperparathyroidism have been present after 1 year of follow-up. Clinical characteristics and results of genetic analyses for patient 2 can be seen in Table 1.
Genetic Characteristics
The only RET germline variant and the only somatic variant exclusively identified in patient 1 were IVS12+47C>T and FLT3 R387Q (c.1160G>A), respectively. The RET variants exclusively found in patient 2 were A432A, G691S, and S904S.
Discussion
Despite thorough investigations, this study found no RET germline variants or other somatic variants that could convincingly explain the substantial variability in MTC phenotype seen in our two L790F index patients.
Patient 1 presented with MTC and lymph node metastases at 14 years old, several years earlier than the reported median age (57 years) for MTC presentation in L790F index patients (37). The only RET germline variant exclusively identified in this patient was IVS12+47C>T. While other RET intron variants (IVS1-126G>T, IVS8+82A>G; 85-86 insC) have been associated to earlier age at MTC onset and the presence of lymph node metastases at diagnosis (26), no similar association has been reported for the RET IVS12+47C>T variant. The variant is classified as benign in the ClinVar database according to the criteria from the American College of Medical Genetics and Genomics (38, 39). To the best of our knowledge, no functional studies of this variant exist.
The only somatic variant exclusively found in patient 1 was FLT3 R387Q (c.1160G>A). Fms-like tyrosine kinase 3 ligand (FL) is the cytokine ligand of FLT3, which is considered an important growth and differentiation factor. FLT3 is commonly found in hematopoietic malignancies. In most cases, this is due to activating mutations in the FLT3 gene but a significant number of leukemias are also characterized by a higher than normal expression level of un-mutated, wild-type FLT3, thus emphasizing the importance of FLT3 signaling perturbations in malignant transformation. FL interacts with FLT3 at the EF-loop of the homodimeric FLT3. R387 is situated in the D4 domain of FLT3 interacting with FL (Figure 1) and is part of the interface of D4 taking part in homodimerization. R387 is part of the FG-loop situated next to the highly conserved EF-loop constituting the tyrosine corner. In the majority of other tyrosine kinase receptors the homodimerization is achieved by hydrogen bonds between argenines and glutamines of the FG-Loop. FLT3 show sequence dissimilarity in this particular region of the D4 domain region to other tyrosine kinase receptors. It can be hypothesized that R387Q could be part of a hydrogen binding motif with the opposing Glutamine 365 or 366 in the FLT3 partner. Thus, we cannot exclude a possible genetic modifying role for the R387Q variant.
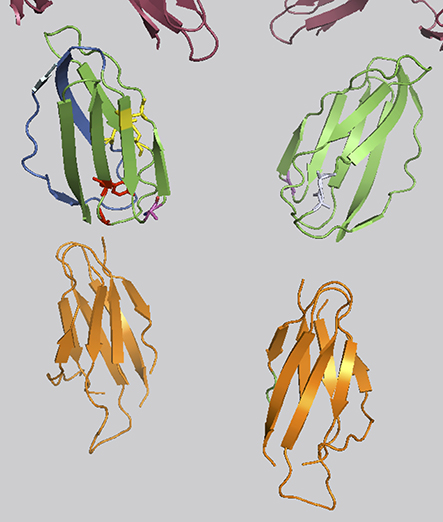
Figure 1. FLT3: FL interaction. D4 interface taking part in FL binding shown in marine blue. Conserved disulfid bridges of the FLT3 Homotype interacting D4 domains in yellow. The highly conserved Tyrosin of the EF-Loop is shown in red. D5 domains in orange. The variation R387 in magenta. Glutamines opposing R387 involved in homodimerisation shown in light grey. The upper part of the figure (in raspberry) represents part of the D1, D2 and D3 domains.
Patient 2 presented relatively late with an intrathyroidal MTC at 70 years old. Variants exclusively found in this patient were A432A, G691S, and S904S. Of these variants, the G691S and S904S variant and their association to MTC have been investigated in several MEN 2A studies (22, 23, 25–27, 29). A study of 104 MEN 2A patients from Spain suggested that G691S and S904S could be related to early appearance of symptoms in an overall MEN 2A group and in a group with codon 634 mutations only (22). Similarly, a recent Italian study observed an earlier age at MTC diagnosis in patients carrying the G691S variant and the S891A germline mutation compared to those only carrying the S891A mutation (23). However, in the present study, we could not confirm a genetic modifier effect of the G691S and S904S variant when comparing our two L790F index patients. This is in accordance with other investigations that studied the G691S and S904S variant in MEN 2A patients carrying mutations of codon 533, 618, and 634 (25–27, 29).
The last variant exclusively found in patient 2 was A432A. An international collaborative study of 384 MEN 2A patients from four different European populations suggested a protective effect of the A432A variant with a 50% decreased risk of developing pheochromocytoma and/or primary hyperparathyroidism (P = 0.03) (27). Such an effect cannot be ruled out for patient 2, as she has not presented with other MEN 2A manifestations despite a relatively advanced age at MTC diagnosis. However, the penetrance of pheochromocytoma and primary hyperparathyroidism in L790F carriers has been reported as exceedingly low (20). Although pheochromocytoma often is reported in carriers of codon 634 mutations (40–45), there have also been reports of this manifestation in L790F carriers (20, 46–48). Opposed to the phenotype of our two patients, having only MTC and no pheochromocytoma, a 44-year-old L790F carrier has been reported to present with bilateral pheochromocytoma and no evidence of MTC. The report underlines the need for MEN 2 screening in all patients with pheochromocytoma and the need for lifelong screening for pheochromocytoma in all MEN 2 patients (46, 49).
We found no somatic variants in RET, H-RAS, K-RAS, and N-RAS in neither of our MEN 2A index patients. This is consistent with some studies (50–53), but in contrast to others (54, 55). None of these studies correlated the absence or presence of somatic variants to the age at diagnosis and aggressiveness of MTC. This was, however, done in a French-Italian collaborative study of V804L carriers (16). Despite V804L carriers usually displaying a late onset and an indolent course of C-cell disease, the authors reported of an index patient, who presented with a thyroid tumor (25 mm), clinical lymph node involvement, and elevated basal calcitonin (1,750 pg/ml) at 12 years of age. A somatic M918T mutation was detected in the patient's tumor. The authors hypothesized that the two mutations were likely responsible for both the early clinical onset and the aggressiveness of the tumor (16). The lack of somatic RET variants in both our index patients suggests that a similar hypothesis may not be valid for L790F carriers. However, future large-scale studies are needed to adequately address the question. Such studies are also needed to provide more information of useful genotype-phenotype correlations for better prognostication in L790 carries. The present investigation is limited by the lack of functional studies although they can be misleading from time to other (36, 56).
As patient 1 was of Lebanese origin (immigrated to Denmark at four years of age) and patient 2 of Danish origin, influence of ethnic, and geographical factors cannot be completely ruled out. Even though other studies have found geographical differences in pheochromocytoma of MEN 2A and survival of MEN 2B patients, the issue remains hypothetical (57, 58).
Conclusions
We could not confirm the previously reported finding of a somatic RET variant as likely responsible for the early onset and aggressiveness of MTC in a RET germline mutation carrier. Also, we found no RET germline variants that could explain the MTC variability among our index patients. We did, however, identify a somatic FLT3 R387Q variant with an unknown potential as genetic modifier. Further large-scale studies are needed to investigate genetic modifiers in RET L790F carriers.
Data Availability Statement
Due to anonymity of the two cases reported in the article, we would prefer not to deposit the sequencing data in public community-supported repository. However, the data are available upon reasonable request to the authors.
Ethics Statement
Written informed consent was obtained from the cases for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JM conceived the study, collected data, and drafted the manuscript. MR contributed with genetic analyses, data collection, and manuscript drafting. KK and SL contributed with collection of tumor tissue and manuscript drafting. KB contributed with interpretation of variants and manuscript drafting. SN, ÅR, KW, AH, MN, AF, and CG contributed with data collection and manuscript drafting.
Funding
This work was supported by the University of Southern Denmark, the Region of Southern Denmark, Odense University Hospital, Copenhagen University Hospital, the Danish Cancer Society, the Danish Cancer Research Foundation and the A. P. Moller Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2020.00251/full#supplementary-material
References
1. Wells SA Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. (2015) 25:567–610. doi: 10.1089/thy.2014.0335
2. Castinetti F, Waguespack SG, Machens A, Uchino S, Hasse-Lazar K, Sanso G, et al. Natural history, treatment, and long-term follow up of patients with multiple endocrine neoplasia type 2B: an international, multicentre, retrospective study. Lancet Diabetes Endocrinol. (2019) 7:213–20. doi: 10.1016/S2213-8587(18)30336-X
3. Opsahl EM, Brauckhoff M, Schlichting E, Helset K, Svartberg J, Brauckhoff K, et al. A nationwide study of multiple endocrine neoplasia type 2A in Norway: predictive and prognostic factors for the clinical course of medullary thyroid carcinoma. Thyroid. (2016) 26:1225–38. doi: 10.1089/thy.2015.0673
4. Mathiesen JS, Kroustrup JP, Vestergaard P, Stochholm K, Poulsen PL, et al. Incidence and prevalence of multiple endocrine neoplasia 2A in Denmark 1901-2014: a nationwide study. Clin Epidemiol. (2018) 10:1479–87. doi: 10.2147/CLEP.S174606
5. Znaczko A, Donnelly DE, Morrison PJ. Epidemiology, clinical features, and genetics of multiple endocrine neoplasia type 2B in a complete population. Oncologist. (2014) 19:1284–6. doi: 10.1634/theoncologist.2014-0277
6. Mathiesen JS, Kroustrup JP, Vestergaard P, Madsen M, Stochholm K, Poulsen PL, et al. Incidence and prevalence of multiple endocrine neoplasia 2B in Denmark: a nationwide study. Endocr Relat Cancer. (2017) 24:L39–L42. doi: 10.1530/ERC-17-0122
7. Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. (1993) 2:851–6. doi: 10.1093/hmg/2.7.851
8. Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. (1993) 363:458–60. doi: 10.1038/363458a0
9. Carlson KM, Dou S, Chi D, Scavarda N, Toshima K, Jackson CE, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci U S A. (1994) 91:1579–83. doi: 10.1073/pnas.91.4.1579
10. Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. (1994) 367:375–6. doi: 10.1038/367375a0
11. Eng C, Smith DP, Mulligan LM, Nagai MA, Healey CS, Ponder MA, et al. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet. (1994) 3:237–41. doi: 10.1093/hmg/3.2.237
12. Mulligan LM, Marsh DJ, Robinson BG, Schuffenecker I, Zedenius J, Lips CJ, et al. Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the International RET Mutation Consortium. J Intern Med. (1995) 238:343–6. doi: 10.1111/j.1365-2796.1995.tb01208.x
13. Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, et al. The relationship between pecific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. (1996) 276:1575–9. doi: 10.1001/jama.276.19.1575
14. Long KL, Etzel C, Rich T, Hyde S, Perrier ND, Graham PH, et al. All in the family? Analyzing the impact of family history in addition to genotype on medullary thyroid carcinoma aggressiveness in MEN2A patients. Fam Cancer. (2017) 16:283–9. doi: 10.1007/s10689-016-9948-7
15. Lindskog S, Nilsson O, Jansson S, Nilsson B, Illerskog AC, Ysander L, et al. Phenotypic expression of a family with multiple endocrine neoplasia type 2A due to a RET mutation at codon 618. Br J Surg. (2004) 91:713–8. doi: 10.1002/bjs.4457
16. Lombardo F, Baudin E, Chiefari E, Arturi F, Bardet S, Caillou B, et al. Familial medullary thyroid carcinoma: clinical variability and low aggressiveness associated with RET mutation at codon 804. J Clin Endocrinol Metab. (2002) 87:1674–80. doi: 10.1210/jcem.87.4.8403
17. Decker RA. Long-term follow-up of a large North American kindred with multiple endocrine neoplasia type 2A. Surgery. (1992) 112:1066–72; discussion 1072–3.
18. Feldman GL, Edmonds MW, Ainsworth PJ, Schuffenecker I, Lenoir GM, Saxe AW, et al. Variable expressivity of familial medullary thyroid carcinoma (FMTC) due to a RET V804M (GTG–>ATG) mutation. Surgery. (2000) 128:93–8. doi: 10.1067/msy.2000.107103
19. Martins-Costa MC, Cunha LL, Lindsey SC, Camacho CP, Dotto RP, Furuzawa GK, et al. M918V RET mutation causes familial medullary thyroid carcinoma: study of 8 affected kindreds. Endocr Relat Cancer. (2016) 23:909–20. doi: 10.1530/ERC-16-0141
20. Bihan H, Murat A, Fysekidis M, Al-Salameh A, Schwartz C, Baudin E, et al. The clinical spectrum of RET proto-oncogene mutations in codon 790. Eur J Endocrinol. (2013) 169:271–6. doi: 10.1530/EJE-13-0050
21. Kaldrymides P, Mytakidis N, Anagnostopoulos T, Vassiliou M, Tertipi A, Zahariou M, et al. A rare RET gene exon 8 mutation is found in two Greek kindreds with familial medullary thyroid carcinoma: implications for screening. Clin Endocrinol. (2006) 64:561–6. doi: 10.1111/j.1365-2265.2006.02509.x
22. Robledo M, Gil L, Pollan M, Cebrian A, Ruiz S, Azanedo M, et al. Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Res. (2003) 63:1814–7. Available online at: https://cancerres.aacrjournals.org/content/63/8/1814.article-info
23. Colombo C, Minna E, Rizzetti MG, Romeo P, Lecis D, Persani L, et al. The modifier role of RET-G691S polymorphism in hereditary medullary thyroid carcinoma: functional characterization and expression/penetrance studies. Orphanet J Rare Dis. (2015) 10:25. doi: 10.1186/s13023-015-0231-z
24. Magalhaes PK, de Castro M, Elias LL, Soares EG, Maciel LM. Polymorphisms in the RET proto-oncogene and the phenotypic presentation of familial medullary thyroid carcinoma. Thyroid. (2004) 14:848–52. doi: 10.1089/1050725042451239
25. Siqueira DR, Romitti M, da Rocha AP, Ceolin L, Meotti C, Estivalet A, et al. The RET polymorphic allele S836S is associated with early metastatic disease in patients with hereditary or sporadic medullary thyroid carcinoma. Endocr Relat Cancer. (2010) 17:953–63. doi: 10.1677/ERC-09-0312
26. Tamanaha R, Camacho CP, Pereira AC, da Silva AM, Maciel RM, Cerutti JM. Evaluation of RET polymorphisms in a six-generation family with G533C RET mutation: specific RET variants may modulate age at onset and clinical presentation. Clin Endocrinol. (2009) 71:56–64. doi: 10.1111/j.1365-2265.2008.03491.x
27. Lesueur F, Cebrian A, Robledo M, Niccoli-Sire P, Svensson KA, Pinson S, et al. Polymorphisms in RET and its coreceptors and ligands as genetic modifiers of multiple endocrine neoplasia type 2A. Cancer Res. (2006) 66:1177–80. doi: 10.1158/0008-5472.CAN-05-2995
28. Weber F, Eng C. Editorial: germline variants within RET: clinical utility or scientific playtoy? J Clin Endocrinol Metab. (2005) 90:6334–6. doi: 10.1210/jc.2005-2030
29. Du ZF, Li PF, Zhao JQ, Cao ZL, Li F, Ma JM, et al. Genetic diagnosis of a Chinese multiple endocrine neoplasia type 2A family through whole genome sequencing. J Biosci. (2017) 42:209–18. doi: 10.1007/s12038-017-9686-5
30. Fugazzola L, De Leo S, Perrino M. The optimal range of RET mutations to be tested: European comments to the guidelines of the American Thyroid Association. Thyroid Res. (2013) 6(Suppl 1):S8. doi: 10.1186/1756-6614-6-S1-S8
31. Machens A, Dralle H. Multiple endocrine neoplasia type 2: achievements and current challenges. Clinics (Sao Paulo). (2012) 67(Suppl 1):113–8. doi: 10.6061/clinics/2012(Sup01)19
32. Machens A, Dralle H. Long-term outcome after DNA-based prophylactic neck surgery in children at risk of hereditary medullary thyroid cancer. Best Pract Res Clin Endocrinol Metab. (2019) 33:101274. doi: 10.1016/j.beem.2019.04.008
33. Mathiesen JS, Kroustrup JP, Vestergaard P, Stochholm K, Poulsen PL, Rasmussen AK, et al. Distribution of RET mutations in multiple endocrine neoplasia 2 in Denmark 1994-2014: a nationwide study. Thyroid. (2017) 27:215–223. doi: 10.1089/thy.2016.0411
34. Hoxbroe Michaelsen S, Ornstrup MJ, Poulsen MM, Bennedbaek FN, Gaustadnes M, Rossing M, et al. Long-term follow-up of RET Y791F carriers in Denmark 1994-2017: a national cohort study. J Surg Oncol. (2019) 119:687–93. doi: 10.1002/jso.25371
35. Mathiesen JS, Kroustrup JP, Vestergaard P, Stochholm K, Poulsen PL, Rasmussen AK, et al. Survival and long-term biochemical cure in medullary thyroid carcinoma in Denmark 1997-2014: a nationwide study. Thyroid. (2019) 29:368–77. doi: 10.1089/thy.2018.0564
36. Mathiesen JS, Hansen TVO, Rasmussen AK, Hjortshoj TD, Kiss K, Larsen SR, et al. Novel somatic RET mutation questioning the causality of the RET I852M germline sequence variant in multiple endocrine neoplasia 2A. Thyroid. (2017) 27:1103–4. doi: 10.1089/thy.2017.0131
37. Frank-Raue K, Machens A, Scheuba C, Niederle B, Dralle H, Raue F. Difference in development of medullary thyroid carcinoma among carriers of RET mutations in codons 790 and 791. Clin Endocrinol. (2008) 69:259–63. doi: 10.1111/j.1365-2265.2008.03215.x
38. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
39. Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. (2016) 44:D862–D8. doi: 10.1093/nar/gkv1222
40. Accardo G, Conzo G, Esposito D, Gambardella C, Mazzella M, Castaldo F, et al. Genetics of medullary thyroid cancer: an overview. Int J Surg. (2017) 41(Suppl 1):S2–S6. doi: 10.1016/j.ijsu.2017.02.064
41. Castinetti F, Qi XP, Walz MK, Maia AL, Sanso G, Peczkowska M, et al. Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: an international retrospective population-based study. Lancet Oncol. (2014) 15:648–55. doi: 10.1530/endoabs.37.S12.3
42. Imai T, Uchino S, Okamoto T, Suzuki S, Kosugi S, Kikumori T, et al. High penetrance of pheochromocytoma in multiple endocrine neoplasia 2 caused by germ line RET codon 634 mutation in Japanese patients. Eur J Endocrinol. (2013) 168:683–7. doi: 10.1530/EJE-12-1106
43. Conzo G, Circelli L, Pasquali D, Sinisi A, Sabatino L, Accardo G, et al. Lessons to be learned from the clinical management of a MEN 2A patient bearing a novel 634/640/700 mutation of the RET proto-oncogene. Clin Endocrinol. (2012) 77:934–6. doi: 10.1111/j.1365-2265.2012.04412.x
44. Martinelli P, Maruotti GM, Pasquali D, Paladini D, Agangi A, Rippa E, et al. Genetic prenatal RET testing and pregnancy management of multiple endocrine neoplasia Type II A (MEN2A): a case report. J Endocrinol Invest. (2004) 27:357–60. doi: 10.1007/BF03351062
45. Tessitore A, Sinisi AA, Pasquali D, Cardone M, Vitale D, Bellastella A, et al. A novel case of multiple endocrine neoplasia type 2A associated with two de novo mutations of the RET protooncogene. J Clin Endocrinol Metab. (1999) 84:3522–7. doi: 10.1210/jc.84.10.3522
46. Min JW, Park YJ, Kim HJ, Chang MC. Bilateral adrenal pheochromocytoma with a germline L790F mutation in the RET oncogene. J Korean Surg Soc. (2012) 82:185–9. doi: 10.4174/jkss.2012.82.3.185
47. Berndt I, Reuter M, Saller B, Frank-Raue K, Groth P, Grussendorf M, et al. A new hot spot for mutations in the ret protooncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab. (1998) 83:770–4. doi: 10.1210/jc.83.3.770
48. Rodien P, Jeunemaitre X, Dumont C, Beldjord C, Plouin PF. Genetic alterations of the RET proto-oncogene in familial and sporadic pheochromocytomas. Horm Res. (1997) 47:263–8. doi: 10.1159/000185474
49. Neumann HP, Berger DP, Sigmund G, Blum U, Schmidt D, Parmer RJ, et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med. (1993) 329:1531–8. doi: 10.1056/NEJM199311183292103
50. Eng C, Mulligan LM, Smith DP, Healey CS, Frilling A, Raue F, et al. Mutation of the RET protooncogene in sporadic medullary thyroid carcinoma. Genes Chromosomes Cancer. (1995) 12:209–12. doi: 10.1002/gcc.2870120308
51. Komminoth P, Kunz EK, Matias-Guiu X, Hiort O, Christiansen G, Colomer A, et al. Analysis of RET protooncogene point mutations distinguishes heritable from nonheritable medullary thyroid carcinomas. Cancer. (1995) 76:479–89.
52. Zedenius J, Wallin G, Hamberger B, Nordenskjold M, Weber G, Larsson C. Somatic and MEN 2A de novo mutations identified in the RET proto-oncogene by screening of sporadic MTC:s. Hum Mol Genet. (1994) 3:1259–62. doi: 10.1093/hmg/3.8.1259
53. Blaugrund JE, Johns MM Jr., Eby YJ, Ball DW, Baylin SB, Hruban RH, et al. RET proto-oncogene mutations in inherited and sporadic medullary thyroid cancer. Hum Mol Genet. (1994) 3:1895–7. doi: 10.1093/hmg/3.10.1895
54. Eng C, Mulligan LM, Healey CS, Houghton C, Frilling A, Raue F, et al. Heterogeneous mutation of the RET proto-oncogene in subpopulations of medullary thyroid carcinoma. Cancer Res. (1996) 56:2167–70.
55. Marsh DJ, Andrew SD, Eng C, Learoyd DL, Capes AG, Pojer R, et al. Germline and somatic mutations in an oncogene: RET mutations in inherited medullary thyroid carcinoma. Cancer Res. (1996) 56:1241–1243.
56. Machens A, Spitschak A, Lorenz K, Putzer BM, Dralle H. Germline RET sequence variation I852M and occult medullary thyroid cancer: harmless polymorphism or causative mutation? Clin Endocrinol. (2011) 75:801–5. doi: 10.1111/j.1365-2265.2011.04158.x
57. Yoshimoto K, Iwahana H, Itakura M. Relatively good prognosis of multiple endocrine neoplasia type 2B in Japanese: review of cases in Japan and analysis of genetic changes in tumors. Endocr J. (1993) 40:649–57. doi: 10.1507/endocrj.40.649
Keywords: multiple endocrine neoplasia type 2, medullary thyroid carcinoma, REarranged during Transfection, L790F, variability, gene variants, FLT3 R387Q
Citation: Mathiesen JS, Nielsen SG, Rasmussen ÅK, Kiss K, Wadt K, Hermann AP, Nielsen MF, Larsen SR, Brusgaard K, Frederiksen AL, Godballe C and Rossing M (2020) Variability in Medullary Thyroid Carcinoma in RET L790F Carriers: A Case Comparison Study of Index Patients. Front. Endocrinol. 11:251. doi: 10.3389/fendo.2020.00251
Received: 23 January 2020; Accepted: 06 April 2020;
Published: 28 April 2020.
Edited by:
Wen Zhou, Case Western Reserve University, United StatesReviewed by:
Maria Alevizaki, National and Kapodistrian University of Athens, GreeceDaniela Pasquali, University of Campania Luigi Vanvitelli, Italy
Copyright © 2020 Mathiesen, Nielsen, Rasmussen, Kiss, Wadt, Hermann, Nielsen, Larsen, Brusgaard, Frederiksen, Godballe and Rossing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jes Sloth Mathiesen, amVzX21hdGhpZXNlbkB5YWhvby5kaw==