 Cristiana Cipriani*
Cristiana Cipriani* Luciano Colangelo
Luciano Colangelo Rachele Santori
Rachele Santori Mario RenellaMonia Mastrantonio
Mario RenellaMonia Mastrantonio Salvatore MinisolaJessica Pepe
Salvatore MinisolaJessica Pepe- Department of Internal Medicine and Medical Disciplines, Sapienza University of Rome, Rome, Italy
The multiple endocrine functions of bone other than those related to mineral metabolism, such as regulation of insulin sensitivity, glucose homeostasis, and energy metabolism, have recently been discovered. In vitro and murine studies investigated the impact of several molecules derived from osteoblasts and osteocytes on glucose metabolism. In addition, the effect of glucose on bone cells suggested a mutual cross-talk between bone and glucose homeostasis. In humans, these mechanisms are the pivotal determinant of the skeletal fragility associated with both type 1 and type 2 diabetes. Metabolic abnormalities associated with diabetes, such as increase in adipose tissue, reduction of lean mass, effects of hyperglycemia per se, production of the advanced glycation end products, diabetes-associated chronic kidney disease, and perturbation of the calcium-PTH-vitamin D metabolism, are the main mechanisms involved. Finally, there have been multiple reports of antidiabetic drugs affecting the skeleton, with differences among basic and clinical research data, as well as of anti-osteoporosis medication influencing glucose metabolism. This review focuses on the aspects linking glucose and bone metabolism by offering insight into the most recent evidence in humans.
Introduction
Diabetes and osteoporosis are common chronic diseases with serious clinical complications. Pathophysiology of the two disorders and related complications is multifactorial, and several mechanisms are now fully recognized. In this context, research studies have actively investigated the interaction between bone and glucose metabolism. Experimental data have shown the significant detrimental effect that the perturbation of glucose metabolism has on the skeleton. Clinical studies confirmed these findings and contributed to the definition of the diabetes-associated bone disease. Moreover, studies on the endocrine function of the skeleton allowed the identification of mechanisms through which bone can modulate glucose homeostasis.
The paper reviews the most recent evidence on the mutual cross-talk between bone and glucose metabolism in humans.
How Glucose Metabolism Influences Bone
Type 2 diabetes mellitus (T2DM) is characterized by normal-high bone mineral density (BMD) and increased fracture risk (1, 2). Several bone-derived factors may be altered during perturbation of the glucose metabolism and are reported in Table 1. The main mechanisms involved in the pathogenesis of diabetes-associated skeletal disease are summarized in the following paragraphs.
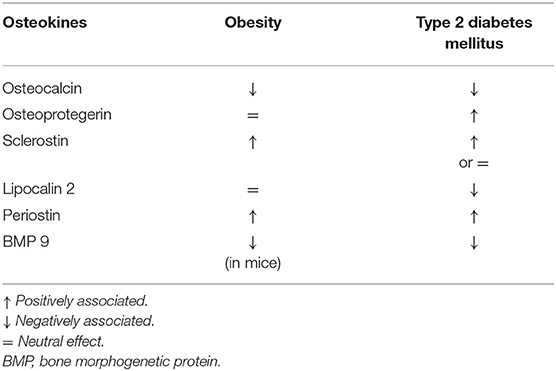
Table 1. Osteokines in human diseases where glucose metabolism is alterated.
Hyperglycemia and Adipokines
Hyperglycemia by itself has toxic effects on the differentiation of bone marrow mesenchymal cells (MSC) into adipocytes (3). In fact, high glucose levels stimulate the non-canonical Wnt/protein kinase C pathway (4) and upregulates the peroxisome proliferator-activated receptor gamma (PPARγ), resulting in increased adipogenesis and bone loss (5). Poor glycemic control in diabetes patients could therefore suppress some of the master genes, as Runx2, involved in osteoblastogenesis (6). Moreover, the increased adipogenesis in the bone marrow has a strong negative effect on bone health (7). Human studies in overweight postmenopausal women with T2DM have demonstrated the inverse association between bone marrow adipose tissue and BMD (5).
Loss of lean mass and increase in adipose tissue are other key mechanisms involved in diabetes-associated bone disease. Adipose tissue, and particularly visceral fat, produces several adipokines with different effects on bone metabolism (8–10). In experimental and human studies, adiponectin, visfatin, and omentin-1 had negative effects on bone, while leptin exerted positive actions (8–10). Additionally, the role of a specific adipo-myokine, irisin, has been recently described (11, 12). It is thought that irisin plays a positive effect on hyperglycemia by stimulating glucose uptake in muscle cells (13). As far as bone metabolism, irisin may promote osteogenic differentiation, increase in cortical bone mass and strength, and reduction in the number of osteoclasts (14). Irisin resistance has been postulated in diabetes, with a consequent theoretical loss of all these positive effects on bone mass and strength (12, 15).
Several studies have reported the association between metabolic syndrome and fragility fractures (16). Conditions that characterize the metabolic syndrome indeed induce perturbation in adipokines and cytokines secretion (9). These mechanisms, in association with the altered insulin signaling, might have an influence on bone metabolism, as documented by the reduction of bone formation markers (9). Components of the metabolic syndrome other than hyperglycemia and increased adipose tissue have been associated with poor skeletal health, as well. Increases in serum triglycerides levels have been negatively associated with BMD, particularly at the femoral neck, in postmenopausal women, and with increase in bone marrow fat in young men and women (17, 18). Finally, data have shown that arterial hypertension is associated with low BMD, mostly in relation to increases in urinary calcium excretion (19, 20).
Advanced Glycation End-Products
Hyperglycemia may act through non-enzymatic pathways and induce the formation of advanced glycation end-products (AGEs). AGEs have a detrimental effect on the skeleton, affecting the extracellular matrix and the vessels. Additionally, in vitro data demonstrated that high glucose levels and AGEs increase osteocytes expression of sclerostin, a negative regulator of bone formation (21). Pre-clinical observations were confirmed by clinical studies showing that sclerostin levels are higher in pre-diabetes subjects than in controls, and correlate with insulin resistance (22).
AGEs have a negative effect on bone quality, an aspect that is undetectable by dual X-ray absorptiometry. The increase in fracture risk in T2DM is indeed observed in the setting of normal BMD. Possible explanations for such a “paradox of BMD” are the high frequency of obesity in these patients, and the well-known positive association between high BMI and high BMD (23). Additionally, the role of insulin resistance, and consequent high insulin levels, has been postulated, even though some studies failed to find a positive association with BMD independently of BMI (24). On the contrary, altered bone quality has been demonstrated in patients with T2DM, as demonstrated by studies using the high-resolution peripheral quantitative computed tomography (HR-pQCT) (25). In particular, lower cortical volumetric BMD, thickness and cross-sectional area, and higher cortical porosity were observed, defining the concept of “relative deficit” at the cortical level as characteristic of the diabetic bone disease (25).
As hyperglycemia, AGEs, and microangiopathy are thought to exert negative effects on bone health, future research will define whether and how these mechanisms may be implicated in the deterioration of the cortical bone (25).
Systemic Mechanisms
There are several systemic mechanisms, whose stimulation is driven by altered glucose metabolism that may eventually affect bone metabolism.
Chronic kidney disease (CKD) is a common complication in diabetic patients. The intricacy between CKD and bone configures a specific metabolic disorder, the CKD-Mineral Bone Disorder (CKD-MBD), that plays an important role in the skeletal fragility associated with diabetes (26).
Disarrangement in the calcium-vitamin D-PTH axis contributes to bone loss in patients with diabetes. Poor glycemic control correlates with excessive urinary calcium loss, with subsequent stimulation of chronic PTH secretion and deleterious effects on the skeleton (27). Improvement in glucose control is associated with normalization of urinary calcium excretion (28) and may avoid stimulation of PTH secretion, with positive effects on BMD (29).
Bone Modulation of Glucose Metabolism
Osteokines are bone-derived factors that may modulate glucose homeostasis, as demonstrated in murine models. In particular, osteocalcin (OC), bone morphogenetic protein (BMP), and sclerostin (SOST) actively participate in energy metabolism, appetite, and browning of adipose tissue (30). Few experimental studies showed the possible involvement of the receptor activator of nuclear factor-kappaB ligand (RANKL), osteoprotegerin (OPG), lipocalin-2 (LCN2), and periostin in these pathways (30, 31). However, a small number of studies have been conducted to assess how these mechanisms could interplay in the modulation of glucose metabolism by the skeleton in patients with osteoporosis and/or diabetes. The major evidence is summarized in Table 2.
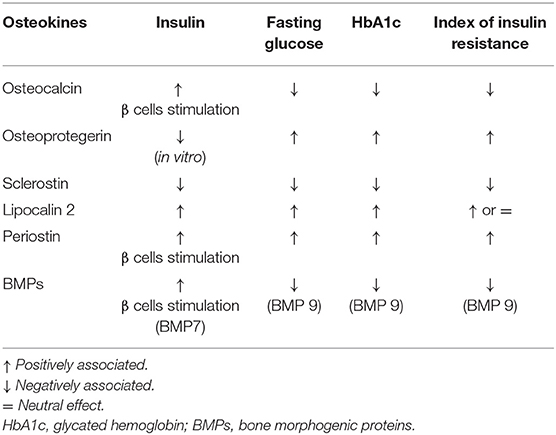
Table 2. Effects of osteokines on glucose metabolism in humans.
Osteocalcin
Osteocalcin is the most abundant osteoblast-specific, non-collagenous protein, and a key determinant of bone formation. The latest review on OC reported its role in glucose metabolism and adaptation to exercise, neuronal development, and male fertility (32) (Table 2). While this is true in mice, less evidence is available in humans. Observational studies reported that OC positively correlates with insulin sensitivity in T2DM patients and that high OC levels were associated with reduced risk of developing T2DM; other studies reported lower OC levels in diabetes and no association with the risk of T2DM (33, 34). Circulating OC comprises both the undercarboxylated (u) (the form that in mice influences glucose metabolism) and the carboxylated form. In clinical studies, the total level of OC is usually reported. Hence, such conflicting results may be ascribed to measurement bias in humans. In this context, interesting data were reported by Linossier et al. who assessed the effect of the acute increase in bone resorption on glucose metabolism in 12 healthy men exposed to microgravity (35). Authors observed that the onset of insulin-resistance was in response to increased bone resorption and concomitant to the increase of uOC (35).
Osteoprotegerin
Osteoprotegerin is a negative regulator of bone resorption through decreasing osteoclasts development. In vitro, OPG treatment of pancreatic ß cell lines decreased insulin release following glucose stimulation, thus preventing exhaustion of ß cells function (36, 37). Studies in postmenopausal women with normal and impaired fasting glucose levels showed that OPG levels are positively associated with insulin resistance index (38, 39). Additionally, OPG levels are higher in pre-diabetic and diabetic adults compared to those with normal glucose tolerance (40). Finally, Daniele et al. showed that OPG levels inversely correlate with the rate of insulin-mediated total body glucose disposal, while positively correlating with fasting endogenous glucose production and hepatic insulin resistance indexes (22).
RANKL
The RANKL is a well-known primary mediator of osteoclasts differentiation, but also regulates glucose metabolism. The inhibition of the RANKL signaling has been suggested to improve hepatic insulin sensitivity and to have a role in ß cells replication in mouse models (41). In patients with osteoporosis, inhibition of RANKL improves muscle strength and insulin sensitivity (42).
FGF23
FGF23, the key regulator of phosphate metabolism, has also been recently associated with fat metabolism (43). Data in 1,179 middle aged subjects showed that FGF23 levels were positively and independently associated with visceral obesity (44). Conversely, significant negative correlations between FGF23 levels and both fasting insulin and C-peptide levels were described in obese children and adolescents with hepatic steatosis (45).
Sclerostin
Slerostin is a well-known inhibitor of osteoblast differentiation acting through the Wnt signaling pathway. This pathway is also active in organs involved in glucose homeostasis, such as pancreas, adipose tissue, liver, and skeletal muscle (46). Clinical data showed negative correlations between sclerostin, insulin, and homeostasis model assessment of insulin resistance (HOMA-IR) in 55 children and adolescents with simple obesity (47). In a small cohort of girls with type 1 diabetes (T1DM), a negative association between serum sclerostin levels and glycated hemoglobin (HbA1c) was found (48). As far as adult patients, studies have shown that those with T1DM had either higher or comparable values of sclerostin compared to controls, while an increase in sclerostin levels is described in patients with T2DM (49–51). Finally, results from the Canadian Multicentre Osteoporosis Study (CaMos) showed that sclerostin levels are associated with fasting insulin levels and HOMA-IR, but not with the risk of incident T2DM (52).
Lipocalin-2
Lipocalin-2 has a prominent role in the pathological response of bone tissue to low mechanical forces in murine models (53). Recently, data in mice have shown that LCN2 crosses the blood-brain barrier and binds to specific neurons of the hypothalamus to control appetite (54). In humans, ROC curve analyses demonstrated that LCN-2 levels could discriminate between normal subjects and those with impaired glucose tolerance (IGT), as well as T2DM and IGT among obese women (55). In particular, significant positive correlation between LCN2 levels and fasting glucose and 2-h postprandial blood glucose, serum insulin, HbA1c, HOMA-IR was detected (55–57).
Periostin
Periostin is a matricellular protein derived from osteoblast and osteocytes. In mice, periostin is able to potentiate pancreatic β-cell regeneration, and is involved in the inhibition of sclerostin following skeletal mechanical loading (58, 59). In humans, high plasma periostin levels were observed in a study assessing 161 obese Chinese patients with T2DM (60). Periostin was strongly associated with triglyceride metabolism, chronic inflammation, and insulin resistance (60). In a cross-sectional study of 8,850 subjects aged 40 or older, periostin positively correlated with liver function, triglycerides levels, waist circumference, HOMA-IR, and fasting plasma insulin in overweight and obese subjects (61).
Bone Morphogenetic Proteins
Bone morphogenetic proteins control both osteoblasts and osteoclasts' function. These molecules are released from the bone matrix into circulation during bone resorption, thus may have effects on organs apart from the skeleton (30). Receptors of the BMPs were indeed found in multiple organs, such as the liver (62). Hence, regulation of bone turnover by BMPs may be coupled with their effect on organs related to glucose metabolism. Interestingly, in vitro data have reported the conversion of primary human pancreatic exocrine tissue into functional islet endocrine cells after exposure to BMP-7 (63).
In humans, circulating BMP-9 levels were found to be significantly higher in healthy subjects than in newly diagnosed T2DM patients and negatively correlated with HbA1c, fasting glucose, and HOMA-IR (64).
Anti-Diabetic Drugs That Interfere With Bone Metabolism
Anti-diabetic drugs may have detrimental, positive, and neutral effects on bone metabolism. In this context, experimental studies are often not corroborated by clinical data.
Metformin and Sulfonylureas
Metformin and sulfonylureas have no clinical significant effect on bone in humans. In pre-clinical studies, metformin activates differentiation of the mesenchymal stem cells toward the osteoblastic lineage while inhibiting adipogenesis and osteoclast differentiation (65–67). Clinical studies have shown inconsistent results on the effect of metformin on fracture risk. Some observational and retrospective studies and recent meta-analyses reported reduction in fracture occurrence in diabetic patients treated with metformin, while others did not observe any significant effect (68–72). Whether these results could be related to the overall low fracture risk of metformin users (72) or whether the use of metformin may have clinically significant protective effects on the skeleton needs to be addressed by future randomized prospective studies.
Pre-clinical data have shown the potential of sulfonylureas, particularly glimepiride, in stimulating bone formation (73, 74). Data in ovariectomized rats have shown that glimepiride could inhibit skeletal changes associated with menopause while stimulating bone formation (75). Clinical data have essentially reported a neutral effect of sulfonylureas on BMD and/or fractures (76–78). There are no clinical trials designed to assess fractures and/or falls as the primary endpoint in sulfonylureas users, in which the main fracture risk is hypoglycemia, particularly in older and frail individuals (78–80).
Insulin
Similarly to what is discussed for sulfonylureas, clinical effects of insulin on bone are mostly driven by the occurrence of hypoglycemia and consequent fracturing (81). The anabolic effects of insulin seen in experimental studies do not indeed translate into positive effects on bone health in humans (81). A recent population-based study of 58,853 newly diagnosed diabetic patients reported a 38% excess risk of major osteoporotic fractures in those treated with insulin (82).
Thiazolidinediones
Thiazolidinediones have been associated with reduction in BMD and increased incidence of fractures (83, 84). Rosiglitazone was associated with 6–20% increase in bone resorption and 4–13% reduction in bone formation markers in postmenopausal women, and with clinically significant bone loss (69, 83, 85–87). Similar data are available for pioglitazone (88, 89). Meta-analyses of studies have demonstrated that treatment with thiazolidinediones is associated with increased fracture risk in postmenopausal women, with a possible association to duration of therapy (73, 83). Main mechanisms include loss of the inhibitory effect of PPAR-γ on osteoclasts differentiation, increased production of sclerostin and DKK1 in osteocytes, and infiltration of adipocytes in the bone marrow (65).
Incretins
The presence of the GLP-1 receptor on the pre-osteoblasts and osteocytes surface was associated with the anabolic actions of liraglutide and exenatide in murine models (90–92). In humans, exenatide and liraglutide treatment were found to prevent bone loss associated with weight reduction, and a 16% increase in P1NP serum levels was observed in liraglutide-treated patients (93, 94). Results on the effect of GLP-1 agonists on fracture are inconclusive (65, 95), even though a recent meta-analysis suggests that treatment with liraglutide and lisixenatide were associated with decreased fracture events (96).
DPP4-Inhibitor
Suppression of bone resorption has been reported in association with DPP4-inhibitor sitagliptin in murine models, and only one study reported similar results in postmenopausal women (97, 98). While clinical studies showed a possible decrease in fracture risk in DPP4-inhibitors users, recent meta-analyses showed that this class of drug does not influence fracture risk when compared to placebo or other anti-diabetic agents (99–102).
Sodium-Glucose Cotransporter-2 Inhibitors
Possibly harmful effects on the skeleton from sodium-glucose cotransporter-2 inhibitors have been observed in the first clinical trials, but not confirmed by the subsequent data. Mechanisms such as weight loss, and increased urinary calcium excretion and PTH levels have been postulated (65). Data from the CANVAS study has reported a 4% incidence of fractures that was significantly higher vs. placebo (103). There was instead no difference in the incidence of fracture in the CANVAS-R study in the canaglifozin vs. placebo group, nor in the most recent CREDENCE trial (104, 105). Finally, the analysis of data from the CANVAS and the CANVAS-R trials comprising 10,142 participants concluded that data from the CANVAS trial could be related to chance or presumably to the presence of falls, whose prevalence was not specifically recorded in all studies (106).
As far as other agents of this class, pooled analysis of data in over 12,000 patients from placebo-controlled and head-to-head trials vs. glimepiride excluded the association of empaglifozin with fractures (77).
Anti-Osteoporosis Drugs That Interfere With Glucose Metabolism
Drugs currently used for treatment of osteoporosis modify bone turnover markers, which in turn could modify glucose metabolism. Reduction in bone turnover markers is observed during bisphosphonates and denosumab treatment (antiresorptive agents), while enhanced bone turnover is observed during PTH 1-34 and PTH 1-84 administration (anabolic agents).
Bisphosphonates
Registrative studies of alendronate, zoledronic acid, and denosumab did not report any significant effects of these drugs on incident diabetes or fasting glucose in postmenopausal women (107). Conversely, a retrospective population-based study conducted in Taiwan showed that the use of alendronate decreases the incidence of diabetes in subjects younger than 65 without dyslipidemia and hypertension (108). Reduction in the risk of diabetes was observed in another population-based retrospective study conducted in UK in individuals aged 60 and older with no baseline diabetes and more than 1 year of bisphosphonates exposure (109).
A recent randomized controlled trial (RCT) showed a significant decrease of 8.2 mg/dL in fasting glucose levels and of 0.2% in HbA1c levels in postmenopausal women with osteopenia and pre-diabetes treated with alendronate (110).
Denosumab
A post-hoc analysis of the FREEDOM trial showed no effect of denosumab on fasting glucose levels in postmenopausal women with diabetes and pre-diabetes. A modest decrease in fasting serum glucose (−6.8 mg/dL) was observed only in women with diabetes not using antidiabetic medications (111). A prospective study by Passeri et al. reported that a single dose of 60 mg of denosumab was not associated with changes in the glucose or insulin response to OGTT in non-diabetic postmenopausal women with osteoporosis (112). Authors observed a modest significant reduction in hepatic insulin resistance index only at 4 weeks (112). Similar results were observed in a subsequent study assessing the glucometabolic parameters and lipid profile in 48 non-diabetic osteoporotic postmenopausal women at 24 weeks after a single 60 mg denosumab dose (113). No significant changes were observed, with the exception of a significant reduction in insulin and HOMA-IR at 4 weeks (113).
Anabolic Agents
Anastasilakis et al. have shown that the intermittent administration of PTH 1-34 is followed by a subtle transient increase in calcium and PTH levels with no effect on glucose homeostasis (114).
As far as PTH 1-84 is concerned, a randomized, controlled, open-label trial involving 46 postmenopausal non-diabetic women with osteoporosis has shown that the hormone increases both OC and uOC, and decreases fasting plasma glucose (115). Interestingly, the effects of PTH 1-84 on OC and on uOC represented the mediator of more than the half (62%) at 12 months and almost half (48%) at 6 months, respectively, of the total effect of these hormones on fasting glucose (115).
Vitamin D and Calcium
Data from clinical studies showed that calcium and vitamin D supplementation might exert beneficial effect on glucose metabolism. However, different results have been observed among studies (116, 117). The most recent meta-analysis including 12 studies with 4,395 participants in the intervention arm and 4,551 in the control group demonstrated that calcium and vitamin D supplementation significantly reduce fasting glucose, HOMA-IR, and insulin levels (116).
Conclusions
Bone and glucose metabolism are strongly interrelated. Experimental studies have assessed the mechanisms of the mutual cross-talk between bone and glucose homeostasis, and allowed for a definition of diabetes-associated bone disease, for which a more proactive clinical evaluation and treatment is now recommended (118). Moreover, data on the endocrine actions of bone on glucose and energy metabolism have opened new and interesting insight into the possible risk of diabetes and obesity in patients with metabolic bone disease.
Future studies, particularly RCTs, will define the effect of anti-osteoporosis medications on glucose homeostasis, as well as how anti-diabetic agents could impact bone health.
Author Contributions
CC and JP contributed to ideation, drafting, and revising of the manuscript. LC, RS, MR, and MM contributed to the literature search and drafting of the manuscript. SM contributed to ideation and revising of the manuscript. All authors have revised and accepted the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hothersall EJ, Livingstone SJ, Looker HC, Ahmed SF, Cleland S, Leese GP, et al. Contemporary risk of hip fracture in type 1 and type 2 diabetes: a national registry study from Scotland. J Bone Miner Res. (2014) 29:1054–60. doi: 10.1002/jbmr.2118
2. Sellmeyer DE, Civitelli R, Hofbauer LC, Khosla S, Lecka-Czernik B, Schwartz AV. Skeletal metabolism, fracture risk, and fracture outcomes in type 1 and type 2 diabetes. Diabetes. (2016) 65:1757–66. doi: 10.2337/db16-0063
3. Zhuang H, Zhang X, Zhu C, Tang X, Yu F, Shang GW, et al. Molecular mechanisms of PPAR-gamma governing MSC osteogenic and adipogenic differentiation. Curr Stem Cell Res Ther. (2016) 11:255–64. doi: 10.2174/1574888X10666150531173309
4. Keats EC, Dominguez JM, 2nd, Grant MB, Khan ZA. Switch from canonical to non-canonical Wnt signaling mediates high glucose-induced adipogenesis. Stem Cells. (2014) 32:1649–60. doi: 10.1002/stem.1659
5. Fazeli PK, Horowitz MC, MacDougald OA, Scheller EL, Rodeheffer MS, Rosen CJ, et al. Marrow fat and bone–new perspectives. J Clin Endocrinol Metab. (2013) 98:935–45. doi: 10.1210/jc.2012-3634
6. Liu LF, Shen WJ, Zhang ZH, Wang LJ, Kraemer FB. Adipocytes decrease Runx2 expression in osteoblastic cells: roles of PPARgamma and adiponectin. J Cell Physiol. (2010) 225:837–45. doi: 10.1002/jcp.22291
7. Rharass T, Lucas S. Mechanisms in endocrinology: bone marrow adiposity and bone, a bad romance? Eur J Endocrinol. (2018) 179:R165–82. doi: 10.1530/EJE-18-0182
8. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. (2004) 89:2548–56. doi: 10.1210/jc.2004-0395
9. Pepe J, Cipriani C, Cilli M, Colangelo L, Minisola S. Adipokines and bone metabolism: an interplay to untangle. J Endocrinol Invest. (2016) 39:1359–61. doi: 10.1007/s40618-016-0549-y
10. Carnevale V, Romagnoli E, Del Fiacco R, Pepe J, Cipriani C, Piemonte S, et al. Relationship between bone metabolism and adipogenesis. J Endocrinol Invest. (2010) 33(7Suppl.):4–8.
11. Arhire LI, Mihalache L, Covasa M. Irisin: a hope in understanding and managing obesity and metabolic syndrome. Front Endocrinol. (2019) 10:524. doi: 10.3389/fendo.2019.00524
12. Osella AR, Colaianni G, Correale M, Pesole PL, Bruno I, Buongiorno C, et al. Irisin serum levels in metabolic syndrome patients treated with three different diets: a post-hoc analysis from a randomized controlled clinical trial. Nutrients. (2018) 10:E844. doi: 10.3390/nu10070844
13. Zhang Y, Li R, Meng Y, Li S, Donelan W, Zhao Y, et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes. (2014) 63:514–25. doi: 10.2337/db13-1106
14. Zhang Y, Xie C, Wang H, Foss RM, Clare M, George EV, et al. Irisin exerts dual effects on browning and adipogenesis of human white adipocytes. Am J Physiol Endocrinol Metab. (2016) 311:E530–41. doi: 10.1152/ajpendo.00094.2016
15. Moreno-Navarrete JM, Ortega F, Serrano M, Guerra E, Pardo G, Tinahones F, et al. Irisin is expressed and produced by human muscle and adipose tissue in association with obesity and insulin resistance. J Clin Endocrinol Metab. (2013) 98:E769–78. doi: 10.1210/jc.2012-2749
16. Muka T, Trajanoska K, Kiefte-de Jong JC, Oei L, Uitterlinden AG, Hofman A, et al. The association between metabolic syndrome, bone mineral density, hip bone geometry and fracture risk: the rotterdam study. PLoS ONE. (2015) 10:e0129116. doi: 10.1371/journal.pone.0129116
17. Kim HY, Choe JW, Kim HK, Bae SJ, Kim BJ, Lee SH, et al. Negative association between metabolic syndrome and bone mineral density in Koreans, especially in men. Calcified Tissue Int. (2010) 86:350–8. doi: 10.1007/s00223-010-9347-2
18. Bredella MA, Gill CM, Gerweck AV, Landa MG, Kumar V, Daley SM, et al. Ectopic and serum lipid levels are positively associated with bone marrow fat in obesity. Radiology. (2013) 269:534–41. doi: 10.1148/radiol.13130375
19. McFarlane SI. Bone metabolism and the cardiometabolic syndrome: pathophysiologic insights. J Cardiometab Syndr. (2006) 1:53–7. doi: 10.1111/j.0197-3118.2006.05457.x
20. Perez-Castrillon JL, Justo I, Silva J, Sanz A, Igea R, Escudero P, et al. Bone mass and bone modelling markers in hypertensive postmenopausal women. J Hum Hypertens. (2003) 17:107–10. doi: 10.1038/sj.jhh.1001520
21. Tanaka K, Yamaguchi T, Kanazawa I, Sugimoto T. Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte-like MLO-Y4-A2 cells. Biochem Biophys Res Commun. (2015) 461:193–9. doi: 10.1016/j.bbrc.2015.02.091
22. Daniele G, Winnier D, Mari A, Bruder J, Fourcaudot M, Pengou Z, et al. Sclerostin and insulin resistance in prediabetes: evidence of a cross talk between bone and glucose metabolism. Diabetes Care. (2015) 38:1509–17. doi: 10.2337/dc14-2989
23. Yang S, Shen X. Association and relative importance of multiple obesity measures with bone mineral density: the National Health and Nutrition Examination Survey 2005–2006. Arch Osteoporos. (2015) 10:14. doi: 10.1007/s11657-015-0219-2
24. Costantini S, Conte C. Bone health in diabetes and prediabetes. World J Diabet. (2019) 10:421–45. doi: 10.4239/wjd.v10.i8.421
25. Samelson EJ, Demissie S, Cupples LA, Zhang X, Xu H, Liu CT, et al. Diabetes and deficits in cortical bone density, microarchitecture, and bone size: framingham HR-pQCT study. J Bone Miner Res. (2018) 33:54–62. doi: 10.1002/jbmr.3240
26. Colangelo L, Biamonte F, Pepe J, Cipriani C, Minisola S. Understanding and managing secondary osteoporosis. Expert Rev Endocrinol Metab. (2019) 14:111–22. doi: 10.1080/17446651.2019.1575727
27. Schwartz AV. Diabetes mellitus: does it affect bone? Calcified Tissue Int. (2003) 73:515–9. doi: 10.1007/s00223-003-0023-7
28. Raskin P, Stevenson MR, Barilla DE, Pak CY. The hypercalciuria of diabetes mellitus: its amelioration with insulin. Clin Endocrinol. (1978) 9:329–35. doi: 10.1111/j.1365-2265.1978.tb02218.x
29. Gregorio F, Cristallini S, Santeusanio F, Filipponi P, Fumelli P. Osteopenia associated with non-insulin-dependent diabetes mellitus: what are the causes? Diabetes Res Clin Pract. (1994) 23:43–54. doi: 10.1016/0168-8227(94)90126-0
30. Bonnet N. Bone-derived factors: a new gateway to regulate glycemia. Calcified Tissue Int. (2017) 100:174–83. doi: 10.1007/s00223-016-0210-y
31. Mera P, Ferron M, Mosialou I. Regulation of energy metabolism by bone-derived hormones. Cold Spring Harb Perspect Med. (2018) 8:a031666. doi: 10.1101/cshperspect.a031666
32. Moser SC, van der Eerden BCJ. Osteocalcin-a versatile bone-derived hormone. Front Endocrinol. (2018) 9:794. doi: 10.3389/fendo.2018.00794
33. Liu Y, Liu X, J RL, Brock K, T CB-S, Teixeira-Pinto A. Relationship between serum osteocalcin/undercarboxylated osteocalcin and type 2 diabetes: a systematic review/meta-analysis study protocol. BMJ Open. (2019) 9:e023918. doi: 10.1136/bmjopen-2018-023918
34. Liu C, Wo J, Zhao Q, Wang Y, Wang B, Zhao W. Association between serum total osteocalcin level and type 2 diabetes mellitus: a systematic review and meta-analysis. Horm Metab Res. (2015) 47:813–9. doi: 10.1055/s-0035-1564134
35. Linossier MT, Amirova LE, Thomas M, Normand M, Bareille MP, Gauquelin-Koch G, et al. Effects of short-term dry immersion on bone remodeling markers, insulin and adipokines. PLoS ONE. (2017) 12:e0182970. doi: 10.1371/journal.pone.0182970
36. Kuroda Y, Maruyama K, Fujii H, Sugawara I, Ko SB, Yasuda H, et al. Osteoprotegerin regulates pancreatic beta-cell homeostasis upon microbial invasion. PLoS ONE. (2016) 11:e0146544. doi: 10.1371/journal.pone.0146544
37. Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocana A, Penninger JM, et al. Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-κB ligand pathway. Cell Metab. (2015) 22:77–85. doi: 10.1016/j.cmet.2015.05.021
38. Duan P, Yang M, Wei M, Liu J, Tu P. Serum osteoprotegerin is a potential biomarker of insulin resistance in chinese postmenopausal women with prediabetes and type 2 diabetes. Int J Endocrinol. (2017) 2017:8724869. doi: 10.1155/2017/8724869
39. Mashavi M, Menaged M, Shargorodsky M. Circulating osteoprotegerin in postmenopausal osteoporotic women: marker of impaired glucose regulation or impaired bone metabolism. Menopause. (2017) 24:1264–8. doi: 10.1097/GME.0000000000000914
40. Niu Y, Yang Z, Li X, Zhang W, Lu S, Zhang H, et al. Association of osteoprotegerin with impaired glucose regulation and microalbuminuria: the REACTION study. BMC Endocr Disord. (2015) 15:75. doi: 10.1186/s12902-015-0067-5
41. Kiechl S, Wittmann J, Giaccari A, Knoflach M, Willeit P, Bozec A, et al. Blockade of receptor activator of nuclear factor-κB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat Med. (2013) 19:358–63. doi: 10.1038/nm.3084
42. Bonnet N, Bourgoin L, Biver E, Douni E, Ferrari S. RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J Clin Invest. (2019) 129:3214–23. doi: 10.1172/JCI125915
43. Grethen E, Hill KM, Jones R, Cacucci BM, Gupta CE, Acton A, et al. Serum leptin, parathyroid hormone, 1,25-dihydroxyvitamin D, fibroblast growth factor 23, bone alkaline phosphatase, and sclerostin relationships in obesity. J Clin Endocrinol Metab. (2012) 97:1655–62. doi: 10.1210/jc.2011-2280
44. Xu Y, Ma X, Pan X, He X, Xiao Y, Bao Y. Correlations between serum concentration of three bone-derived factors and obesity and visceral fat accumulation in a cohort of middle aged men and women. Cardiovasc Diabetol. (2018) 17:143. doi: 10.1186/s12933-018-0786-9
45. Kutluturk Y, Akinci A, Ozerol IH, Yologlu S. The relationship between serum FGF-23 concentration and insulin resistance, prediabetes and dyslipidemia in obese children and adolescents. J Pediatr Endocrinol Metab. (2019) 32:707–14. doi: 10.1515/jpem-2018-0507
46. Elghazi L, Gould AP, Weiss AJ, Barker DJ, Callaghan J, Opland D, et al. Importance of β-Catenin in glucose and energy homeostasis. Sci Rep. (2012) 2:693. doi: 10.1038/srep00693
47. Wedrychowicz A, Sztefko K, Starzyk JB. Sclerostin and its association with insulin resistance in children and adolescents. Bone. (2019) 120:232–8. doi: 10.1016/j.bone.2018.07.021
48. Wedrychowicz A, Sztefko K, Starzyk JB. Sclerostin and its significance for children and adolescents with type 1 diabetes mellitus (T1D). Bone. (2019) 120:387–92. doi: 10.1016/j.bone.2018.08.007
49. Morales-Santana S, Garcia-Fontana B, Garcia-Martin A, Rozas-Moreno P, Garcia-Salcedo JA, Reyes-Garcia R, et al. Atherosclerotic disease in type 2 diabetes is associated with an increase in sclerostin levels. Diabetes Care. (2013) 36:1667–74. doi: 10.2337/dc12-1691
50. Garcia-Martin A, Rozas-Moreno P, Reyes-Garcia R, Morales-Santana S, Garcia-Fontana B, Garcia-Salcedo JA, et al. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. (2012) 97:234–41. doi: 10.1210/jc.2011-2186
51. Gennari L, Merlotti D, Valenti R, Ceccarelli E, Ruvio M, Pietrini MG, et al. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J Clin Endocrinol Metab. (2012) 97:1737–44. doi: 10.1210/jc.2011-2958
52. Yu OH, Richards B, Berger C, Josse RG, Leslie WD, Goltzman D, et al. The association between sclerostin and incident type 2 diabetes risk: a cohort study. Clin Endocrinol. (2017) 86:520–5. doi: 10.1111/cen.13300
53. Rucci N, Capulli M, Piperni SG, Cappariello A, Lau P, Frings-Meuthen P, et al. Lipocalin 2: a new mechanoresponding gene regulating bone homeostasis. J Bone Miner Res. (2015) 30:357–68. doi: 10.1002/jbmr.2341
54. Mosialou I, Shikhel S, Liu JM, Maurizi A, Luo N, He Z, et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature. (2017) 543:385–90. doi: 10.1038/nature21697
55. Rashad NM, El-Shal AS, Etewa RL, Wadea FM. Lipocalin-2 expression and serum levels as early predictors of type 2 diabetes mellitus in obese women. IUBMB Life. (2017) 69:88–97. doi: 10.1002/iub.1594
56. Liu X, Hamnvik OP, Petrou M, Gong H, Chamberland JP, Christophi CA, et al. Circulating lipocalin 2 is associated with body fat distribution at baseline but is not an independent predictor of insulin resistance: the prospective Cyprus Metabolism Study. Eur J Endocrinol. (2011) 165:805–12. doi: 10.1530/EJE-11-0660
57. Wallenius V, Elias E, Bergstrom GM, Zetterberg H, Behre CJ. The lipocalins retinol-binding protein-4, lipocalin-2 and lipocalin-type prostaglandin D2-synthase correlate with markers of inflammatory activity, alcohol intake and blood lipids, but not with insulin sensitivity in metabolically healthy 58-years-old Swedish men. Exp Clin Endocrinol Diabetes. (2011) 119:75–80. doi: 10.1055/s-0030-1265212
58. Smid JK, Faulkes S, Rudnicki MA. Periostin induces pancreatic regeneration. Endocrinology. (2015) 156:824–36. doi: 10.1210/en.2014-1637
59. Bonnet N, Standley KN, Bianchi EN, Stadelmann V, Foti M, Conway SJ, et al. The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J Biol Chem. (2009) 284:35939–50. doi: 10.1074/jbc.M109.060335
60. Luo Y, Qu H, Wang H, Wei H, Wu J, Duan Y, et al. Plasma periostin levels are increased in chinese subjects with obesity and type 2 diabetes and are positively correlated with glucose and lipid parameters. Mediators Inflamm. (2016) 2016:6423637. doi: 10.1155/2016/6423637
61. Yang Z, Zhang H, Niu Y, Zhang W, Zhu L, Li X, et al. Circulating periostin in relation to insulin resistance and non-alcoholic fatty liver disease among overweight and obese subjects. Sci Rep. (2016) 6:37886. doi: 10.1038/srep37886
62. Sugimoto H, Yang C, LeBleu VS, Soubasakos MA, Giraldo M, Zeisberg M, et al. BMP-7 functions as a novel hormone to facilitate liver regeneration. FASEB J. (2007) 21:256–64. doi: 10.1096/fj.06-6837com
63. Klein D, Alvarez-Cubela S, Lanzoni G, Vargas N, Prabakar KR, Boulina M, et al. BMP-7 induces adult human pancreatic exocrine-to-endocrine conversion. Diabetes. (2015) 64:4123–34. doi: 10.2337/db15-0688
64. Luo Y, Li L, Xu X, Wu T, Yang M, Zhang C, et al. Decreased circulating BMP-9 levels in patients with Type 2 diabetes is a signature of insulin resistance. Clin Sci. (2017) 131:239–46. doi: 10.1042/CS20160543
65. Kheniser KG, Polanco Santos CM, Kashyap SR. The effects of diabetes therapy on bone: a clinical perspective. J Diabetes Complications. (2018) 32:713–9. doi: 10.1016/j.jdiacomp.2018.04.005
66. Molinuevo MS, Schurman L, McCarthy AD, Cortizo AM, Tolosa MJ, Gangoiti MV, et al. Effect of metformin on bone marrow progenitor cell differentiation: in vivo and in vitro studies. J Bone Miner Res. (2010) 25:211–21. doi: 10.1359/jbmr.090732
67. Zhen D, Chen Y, Tang X. Metformin reverses the deleterious effects of high glucose on osteoblast function. J Diabetes Complications. (2010) 24:334–44. doi: 10.1016/j.jdiacomp.2009.05.002
68. Vestergaard P, Rejnmark L, Mosekilde L. Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk. Diabetologia. (2005) 48:1292–9. doi: 10.1007/s00125-005-1786-3
69. Stage TB, Christensen MH, Jorgensen NR, Beck-Nielsen H, Brosen K, Gram J, et al. Effects of metformin, rosiglitazone and insulin on bone metabolism in patients with type 2 diabetes. Bone. (2018) 112:35–41. doi: 10.1016/j.bone.2018.04.004
70. Salari-Moghaddam A, Sadeghi O, Keshteli AH, Larijani B, Esmaillzadeh A. Metformin use and risk of fracture: a systematic review and meta-analysis of observational studies. Osteoporos Int. (2019) 30:1167–73. doi: 10.1007/s00198-019-04948-1
71. Kanazawa I, Yamaguchi T, Yamamoto M, Sugimoto T. Relationship between treatments with insulin and oral hypoglycemic agents versus the presence of vertebral fractures in type 2 diabetes mellitus. J Bone Miner Metab. (2010) 28:554–60. doi: 10.1007/s00774-010-0160-9
72. Hidayat K, Du X, Wu MJ, Shi BM. The use of metformin, insulin, sulphonylureas, and thiazolidinediones and the risk of fracture: systematic review and meta-analysis of observational studies. Obes Rev. (2019) 20:1494–503. doi: 10.1111/obr.12885
73. Lecka-Czernik B. Diabetes, bone and glucose-lowering agents: basic biology. Diabetologia. (2017) 60:1163–9. doi: 10.1007/s00125-017-4269-4
74. Ma P, Gu B, Ma J, E L, Wu X, Cao J, et al. Glimepiride induces proliferation and differentiation of rat osteoblasts via the PI3-kinase/Akt pathway. Metabolism. (2010) 59:359–66. doi: 10.1016/j.metabol.2009.08.003
75. Fronczek-Sokol J, Pytlik M. Effect of glimepiride on the skeletal system of ovariectomized and non-ovariectomized rats. Pharmacol Rep. (2014) 66:412–7. doi: 10.1016/j.pharep.2013.12.013
76. Gilbert MP, Marre M, Holst JJ, Garber A, Baeres FM, Thomsen H, et al. Comparison of the long-term effects of liraglutide and glimepiride monotherapy on bone mineral density in patients with type 2 diabetes. Endocr Pract. (2016) 22:406–11. doi: 10.4158/EP15758.OR
77. Kohler S, Kaspers S, Salsali A, Zeller C, Woerle HJ. Analysis of fractures in patients with type 2 diabetes treated with empagliflozin in pooled data from placebo-controlled trials and a head-to-head study versus glimepiride. Diabetes Care. (2018) 41:1809–16. doi: 10.2337/dc17-1525
78. Lapane KL, Yang S, Brown MJ, Jawahar R, Pagliasotti C, Rajpathak S. Sulfonylureas and risk of falls and fractures: a systematic review. Drugs Aging. (2013) 30:527–47. doi: 10.1007/s40266-013-0081-0
79. Lapane KL, Jesdale BM, Dube CE, Pimentel CB, Rajpathak SN. Sulfonylureas and risk of falls and fractures among nursing home residents with type 2 diabetes mellitus. Diabetes Res Clin Pract. (2015) 109:411–9. doi: 10.1016/j.diabres.2015.05.009
80. Napoli N, Strotmeyer ES, Ensrud KE, Sellmeyer DE, Bauer DC, Hoffman AR, et al. Fracture risk in diabetic elderly men: the MrOS study. Diabetologia. (2014) 57:2057–65. doi: 10.1007/s00125-014-3289-6
81. Conte C, Epstein S, Napoli N. Insulin resistance and bone: a biological partnership. Acta Diabetol. (2018) 55:305–14. doi: 10.1007/s00592-018-1101-7
82. Losada-Grande E, Hawley S, Soldevila B, Martinez-Laguna D, Nogues X, Diez-Perez A, et al. Insulin use and excess fracture risk in patients with type 2 diabetes: a propensity-matched cohort analysis. Sci Rep. (2017) 7:3781. doi: 10.1038/s41598-017-03748-z
83. Zhu ZN, Jiang YF, Ding T. Risk of fracture with thiazolidinediones: an updated meta-analysis of randomized clinical trials. Bone. (2014) 68:115–23. doi: 10.1016/j.bone.2014.08.010
84. Dormuth CR, Carney G, Carleton B, Bassett K, Wright JM. Thiazolidinediones and fractures in men and women. Arch Intern Med. (2009) 169:1395–402. doi: 10.1001/archinternmed.2009.214
85. Berberoglu Z, Yazici AC, Demirag NG. Effects of rosiglitazone on bone mineral density and remodelling parameters in Postmenopausal diabetic women: a 2-years follow-up study. Clin Endocrinol. (2010) 73:305–12. doi: 10.1111/j.1365-2265.2010.03784.x
86. Zinman B, Haffner SM, Herman WH, Holman RR, Lachin JM, Kravitz BG, et al. Effect of rosiglitazone, metformin, and glyburide on bone biomarkers in patients with type 2 diabetes. J Clin Endocrinol Metab. (2010) 95:134–42. doi: 10.1210/jc.2009-0572
87. Bilezikian JP, Josse RG, Eastell R, Lewiecki EM, Miller CG, Wooddell M, et al. Rosiglitazone decreases bone mineral density and increases bone turnover in postmenopausal women with type 2 diabetes mellitus. J Clin Endocrinol Metab. (2013) 98:1519–28. doi: 10.1210/jc.2012-4018
88. Portillo-Sanchez P, Bril F, Lomonaco R, Barb D, Orsak B, Bruder JM, et al. Effect of pioglitazone on bone mineral density in patients with non-alcoholic steatohepatitis: a 36-month clinical trial. J Diabetes. (2019) 11:223–31. doi: 10.1111/1753-0407.12833
89. Pop LM, Lingvay I, Yuan Q, Li X, Adams-Huet B, Maalouf NM. Impact of pioglitazone on bone mineral density and bone marrow fat content. Osteoporos Int. (2017) 28:3261–9. doi: 10.1007/s00198-017-4164-3
90. Pereira M, Jeyabalan J, Jorgensen CS, Hopkinson M, Al-Jazzar A, Roux JP, et al. Chronic administration of Glucagon-like peptide-1 receptor agonists improves trabecular bone mass and architecture in ovariectomised mice. Bone. (2015) 81:459–67. doi: 10.1016/j.bone.2015.08.006
91. Wu X, Li S, Xue P, Li Y. Liraglutide, a glucagon-like peptide-1 receptor agonist, facilitates osteogenic proliferation and differentiation in MC3T3-E1 cells through phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT), extracellular signal-related kinase (ERK)1/2, and cAMP/protein kinase A (PKA) signaling pathways involving beta-catenin. Exp Cell Res. (2017) 360:281–91. doi: 10.1016/j.yexcr.2017.09.018
92. Mabilleau G, Pereira M, Chenu C. Novel skeletal effects of glucagon-like peptide-1 (GLP-1) receptor agonists. J Endocrinol. (2018) 236:R29–42. doi: 10.1530/JOE-17-0278
93. Bunck MC, Eliasson B, Corner A, Heine RJ, Shaginian RM, Taskinen MR, et al. Exenatide treatment did not affect bone mineral density despite body weight reduction in patients with type 2 diabetes. Diabetes Obes Metab. (2011) 13:374–7. doi: 10.1111/j.1463-1326.2010.01355.x
94. Iepsen EW, Lundgren JR, Hartmann B, Pedersen O, Hansen T, Jorgensen NR, et al. GLP-1 receptor agonist treatment increases bone formation and prevents bone loss in weight-reduced obese women. J Clin Endocrinol Metab. (2015) 100:2909–17. doi: 10.1210/jc.2015-1176
95. Jansen MA, Kiefte-de Jong JC, Gaillard R, Escher JC, Hofman A, Jaddoe VW, et al. Growth trajectories and bone mineral density in anti-tissue transglutaminase antibody-positive children: the Generation R Study. Clin Gastroenterol Hepatol. (2015) 13:913–20 e5. doi: 10.1016/j.cgh.2014.09.032
96. Cheng L, Hu Y, Li YY, Cao X, Bai N, Lu TT, et al. Glucagon-like peptide-1 receptor agonists and risk of bone fracture in patients with type 2 diabetes: a meta-analysis of randomized controlled trials. Diabetes Metab Res Rev. (2019) 35:e3168. doi: 10.1002/dmrr.3168
97. Glorie L, Behets GJ, Baerts L, De Meester I, D'Haese PC, Verhulst A. DPP IV inhibitor treatment attenuates bone loss and improves mechanical bone strength in male diabetic rats. Am J Physiol Endocrinol Metab. (2014) 307:E447–55. doi: 10.1152/ajpendo.00217.2014
98. Hegazy SK. Evaluation of the anti-osteoporotic effects of metformin and sitagliptin in postmenopausal diabetic women. J Bone Miner Metab. (2015) 33:207–12. doi: 10.1007/s00774-014-0581-y
99. Dombrowski S, Kostev K, Jacob L. Use of dipeptidyl peptidase-4 inhibitors and risk of bone fracture in patients with type 2 diabetes in Germany-A retrospective analysis of real-world data. Osteoporos Int. (2017) 28:2421–8. doi: 10.1007/s00198-017-4051-y
100. Driessen JH, de Vries F, van Onzenoort H, Harvey NC, Neef C, van den Bergh JP, et al. The use of incretins and fractures—a meta-analysis on population-based real life data. Br J Clin Pharmacol. (2017) 83:923–6. doi: 10.1111/bcp.13167
101. Yang Y, Zhao C, Liang J, Yu M, Qu X. Effect of dipeptidyl peptidase-4 inhibitors on bone metabolism and the possible underlying mechanisms. Front Pharmacol. (2017) 8:487. doi: 10.3389/fphar.2017.00487
102. Chen Q, Liu T, Zhou H, Peng H, Yan C. Risk of fractures associated with dipeptidyl peptidase-4 inhibitor treatment: a systematic review and meta-analysis of randomized controlled trials. Diabetes Ther. (2019) 10:1879–92. doi: 10.1007/s13300-019-0668-5
103. Watts NB, Bilezikian JP, Usiskin K, Edwards R, Desai M, Law G, et al. Effects of canagliflozin on fracture risk in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. (2016) 101:157–66. doi: 10.1210/jc.2015-3167
104. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
105. Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. (2019) 380:2295–306. doi: 10.1056/NEJMoa1811744
106. Zhou Z, Jardine M, Perkovic V, Matthews DR, Mahaffey KW, de Zeeuw D, et al. Canagliflozin and fracture risk in individuals with type 2 diabetes: results from the CANVAS program. Diabetologia. (2019) 62:1854–67. doi: 10.1007/s00125-019-4955-5
107. Schwartz AV, Schafer AL, Grey A, Vittinghoff E, Palermo L, Lui LY, et al. Effects of antiresorptive therapies on glucose metabolism: results from the FIT, HORIZON-PFT, and FREEDOM trials. J Bone Miner Res. (2013) 28:1348–54. doi: 10.1002/jbmr.1865
108. Chan DC, Yang RS, Ho CH, Tsai YS, Wang JJ, Tsai KT. The use of alendronate is associated with a decreased incidence of type 2 diabetes mellitus–a population-based cohort study in Taiwan. PLoS ONE. (2015) 10:e0123279. doi: 10.1371/journal.pone.0123279
109. Toulis KA, Nirantharakumar K, Ryan R, Marshall T, Hemming K. Bisphosphonates and glucose homeostasis: a population-based, retrospective cohort study. J Clin Endocrinol Metab. (2015) 100:1933–40. doi: 10.1210/jc.2014-3481
110. Karimi Fard M, Aminorroaya A, Kachuei A, Salamat MR, Hadi Alijanvand M, Aminorroaya Yamini S, et al. Alendronate improves fasting plasma glucose and insulin sensitivity, and decreases insulin resistance in prediabetic osteopenic postmenopausal women: a randomized triple-blind clinical trial. J Diabetes Investig. (2019) 10:731–7. doi: 10.1111/jdi.12944
111. Napoli N, Pannacciulli N, Vittinghoff E, Crittenden D, Yun J, Wang A, et al. Effect of denosumab on fasting glucose in women with diabetes or prediabetes from the FREEDOM trial. Diabetes Metab Res Rev. (2018) 34:e2991. doi: 10.1002/dmrr.2991
112. Passeri E, Benedini S, Costa E, Corbetta S. A single 60 mg dose of denosumab might improve hepatic insulin sensitivity in postmenopausal non-diabetic severe osteoporotic women. Int J Endocrinol. (2015) 2015:352858. doi: 10.1155/2015/352858
113. Lasco A, Morabito N, Basile G, Atteritano M, Gaudio A, Giorgianni GM, et al. Denosumab inhibition of RANKL and insulin resistance in postmenopausal women with osteoporosis. Calcified Tissue Int. (2016) 98:123–8. doi: 10.1007/s00223-015-0075-5
114. Anastasilakis AD, Efstathiadou Z, Plevraki E, Koukoulis GN, Slavakis A, Kita M, et al. Effect of exogenous intermittent recombinant human PTH 1–34 administration and chronic endogenous parathyroid hormone excess on glucose homeostasis and insulin sensitivity. Horm Metab Res. (2008) 40:702–7. doi: 10.1055/s-2008-1078729
115. D'Amelio P, Sassi F, Buondonno I, Spertino E, Tamone C, Piano S, et al. Effect of intermittent PTH treatment on plasma glucose in osteoporosis: a randomized trial. Bone. (2015) 76:177–84. doi: 10.1016/j.bone.2015.03.018
116. Asbaghi O, Khosroshahi MZ, Kashkooli S, Abbasnezhad A. Effect of calcium vitamin D co supplementation on insulin, insulin sensitivity, and glycemia: a systematic review and meta-analysis of randomized clinical trials. Horm Metab Res. (2019) 51:288–95. doi: 10.1055/a-0887-0205
117. Pittas AG, Dawson-Hughes B, Sheehan P, Ware JH, Knowler WC, Aroda VR, et al. Vitamin D supplementation and prevention of type 2 diabetes. N Engl J Med. (2019) 381:520–30. doi: 10.1056/NEJMoa1900906
Keywords: glucose, bone, metabolism, diabetes, osteoporosis, fracture
Citation: Cipriani C, Colangelo L, Santori R, Renella M, Mastrantonio M, Minisola S and Pepe J (2020) The Interplay Between Bone and Glucose Metabolism. Front. Endocrinol. 11:122. doi: 10.3389/fendo.2020.00122
Received: 02 October 2019; Accepted: 24 February 2020;
Published: 24 March 2020.
Edited by:
Amélie E. Coudert, Université Paris Diderot, FranceReviewed by:
Michaël R. Laurent, University Hospitals Leuven, BelgiumMelissa Orlandin Premaor, Federal University of Minas Gerais, Brazil
Copyright © 2020 Cipriani, Colangelo, Santori, Renella, Mastrantonio, Minisola and Pepe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristiana Cipriani, cristiana.cipriani@gmail.com