 Haipeng Pang
Haipeng Pang Shuoming Luo
Shuoming Luo Gan Huang
Gan Huang Ying Xia
Ying Xia Zhiguo Xie
Zhiguo Xie Zhiguang Zhou
Zhiguang Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 12 March 2020
Sec. Diabetes: Molecular Mechanisms
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00119
T1DM (type 1 diabetes mellitus), which results from the irreversible elimination of beta-cells mediated by autoreactive T cells, is defined as an autoimmune disease. It is widely accepted that T1DM is caused by a combination of genetic and environmental factors, but the precise underlying molecular mechanisms are still unknown. To date, more than 50 genetic risk regions contributing to the pathogenesis of T1DM have been identified by GWAS (genome-wide association studies). Notably, more than 60% of the identified candidate genes are expressed in islets and beta-cells, which makes it plausible that these genes act at the beta-cell level and play a key role in the pathogenesis of T1DM. In this review, we focus on the current status of candidate genes that act at the beta-cell level by regulating the innate immune response and antiviral activity, affecting susceptibility to proapoptotic stimuli and influencing the pancreatic beta-cell phenotype.
The autoimmune disease T1DM (type 1 diabetes mellitus) is characterized by the selective destruction of insulin-producing pancreatic beta-cells by autoreactive T cells, absolute insulin deficiency and subsequent hyperglycemia (1). Both genetic and environmental factors are important in the pathogenesis of T1DM; specifically, environmental factors, such as viral infection and the gut microbiome, may act as triggers that induce the onset of diabetes in individuals with a genetically susceptible background (2–6). However, the precise pathogenic mechanisms have not been established. A more complete understanding of the roles and consequences of risk-associated variants would be beneficial for applying targeted genomic approaches to prevent T1DM.
GWAS (genome-wide association studies) have identified more than 50 genetic risk regions associated with T1DM, but most of these regions comprise several genes, and the risk-conferring variants and genes remain to be defined (7, 8). Of note, more than 60% of these candidate genes are expressed in islets and beta-cells (Table 1), indicating that their roles in the onset and development of T1DM may be at the beta-cell level (13).

Table 1. Candidate T1DM genes expressed in islets.
The results of many studies imply that these candidate genes act at the beta-cell level and contribute to the pathogenesis of T1DM mainly by regulating the innate immune response and antiviral activity, affecting susceptibility to proapoptotic stimuli and influencing pancreatic beta-cell phenotypes (Figure 1) (8, 14). Existing evidence shows that innate immunity is involved in the early induction and amplification of the autoimmune process in pancreatic islets (15, 16). Of all the innate immune responses, the type 1 IFN (interferon) signaling pathway plays a particularly important role in the pathogenesis of T1DM (17). Proinflammatory cytokines and chemokines can suppress beta-cell function, evoke apoptosis and maintain insulitis, which causes the progressive loss of beta-cells (15). Pancreatic beta-cell apoptosis has been viewed as the final and most critical step in the progression of T1DM. If the dying beta-cells are not efficiently eliminated, they become the most significant source of autoantigens, which can worsen insulitis and autoimmunity (18, 19). Beta-cell phenotypes are mainly related to residual function, mass, neogenesis, proliferation and so-called beta-cell suicide (1). Intriguingly, patients with long-standing T1DM have residual insulin-positive beta-cells and exhibit endogenous insulin production (20, 21). Therefore, it will be beneficial to reveal the mechanisms of specific candidate genes that act at the beta-cell level to induce beta-cell dysfunction or death in order to identify new therapeutic targets to treat and cure T1DM.
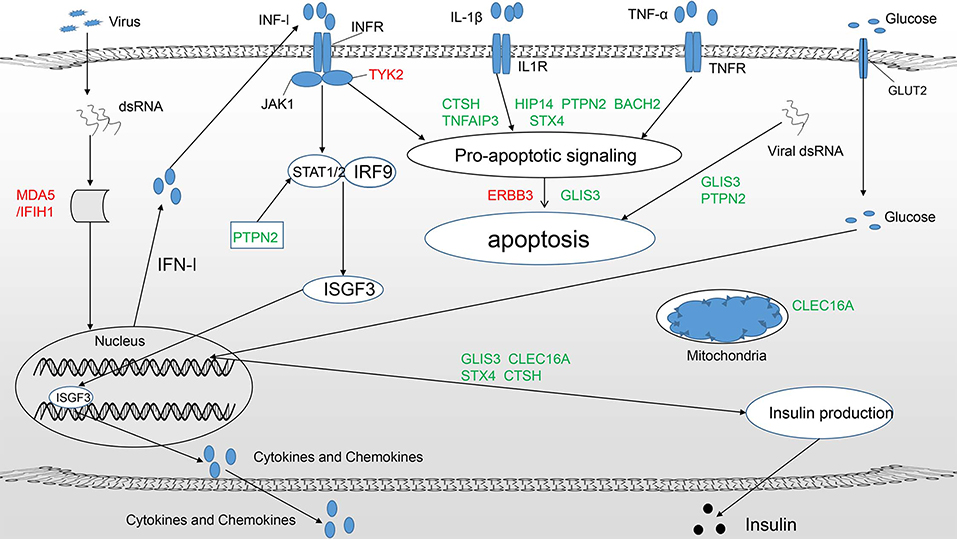
Figure 1. T1DM candidate genes acting at the beta-cell level mainly play a role in three pathways (11): (1) Regulate the innate immune response and pathways important for antiviral activity, such as the type 1 IFN signaling pathway (IFIH1, TYK2, PTPN2). (2) Modulate susceptibility to proapoptotic stimuli (BACH2, TNFAIP3, ERBB3, HIP14, STX4, CTSH, PTPN2). (3) Affect beta-cell phenotypes, primarily insulin production (GLIS3). Candidate genes in green and red represent protective and predisposing candidates, respectively. Some candidate genes clearly participate in more than one pathway.
This review will briefly introduce innate immunity, beta-cell apoptosis and beta-cell phenotypes in patients with T1DM. We focus on the relationship between innate immunity, beta-cell apoptosis, beta-cell phenotypes and T1DM. Later in the review, previous and the most recent findings on T1DM candidate genes acting at the beta-cell level are discussed.
As the front line of the immune system, innate immunity plays an important role in eradicating invading pathogens and initiating the adaptive immune response. Humans can detect environmental pathogens through interactions between innate PRRs (pattern recognition receptors), including RLRs (RIG-I-like receptors), TLRs (Toll-like receptors) and NLRs (nucleotide oligomerization domain-like receptors), and PAMPs (pathogen-associated molecular patterns), which are highly conserved structures shared among large groups of microorganisms (22, 23). The recognition of pathogens by PRRs induces a series of innate immune responses, including the production and release of proinflammatory chemokines and cytokines, such as IFNs, IL-1 (interleukin-1), and TNF-α (tumor necrosis factor-α) (22). A moderate innate immune response protects the body against further injury; however, an excessive response can be detrimental in individuals with a predisposing genetic background because of their increased risk of developing autoimmune diseases, such as T1DM (24, 25).
The induction and development of T1DM involve extremely complicated interactions between pancreatic beta-cells and the immune system, which doubtlessly include innate immunity (26). Among all innate immune responses, the type 1 IFN signaling pathway is especially important for beta-cell damage, as demonstrated by pathway analysis (13). A large body of evidence has confirmed the connection between type 1 IFNs and T1DM in both human and animal model studies. It was originally reported that chronic hepatitis patients treated with IFN-α occasionally develop T1DM, indicating a relationship between IFNs and T1DM (27). This finding was further confirmed by the fact that IFN expression levels were elevated in the pancreas of patients recently diagnosed with T1DM (28). Furthermore, self-neutralizing antibodies targeting IFN-α were associated with protection against T1DM in patents with APS1 (autoimmune polyglandular syndrome type 1) (29). It has also been reported that overactivation of the type 1 IFN signaling pathway occurs prior to the appearance of T1DM-associated antibodies, which highlights the role of type 1 IFN as a potentially precipitating factor in the early phase of T1DM (30, 31). These discoveries have been supported by animal experiments in which transgenic mouse models that overexpress IFN-α in beta-cells were shown to develop hypoinsulinemic diabetes, and self-reactive antibodies against IFN-α and its receptors prevented the development of inflammation and diabetes (32, 33).
The mechanisms underlying type 1 IFN-induced T1DM can be divided into two groups (34). In the first group, the non-immunologic mechanisms include the ER (endoplasmic reticulum) stress-mediated impairment of insulin production and the induction of beta-cell apoptosis via the mitochondrial pathway (35, 36). For mechanisms in the second group, type 1 IFN is central to activating innate immunity and adaptive immune responses. The type 1 IFN signaling pathway promotes the production of proinflammatory mediators and the recruitment of innate immune cells, including macrophages, monocytes, NK (natural killer) cells, and DCs (dendritic cells), which can cause and maintain insulitis in a genetically predisposed background (17). Additionally, the IFN-α-triggered overexpression of MHC-| (major histocompatibility complex class I) can evoke more efficient self-antigen presentation and render beta-cells more easily attacked by autoreactive immune cells (35). Under this circumstance, the adaptive immune response is amplified, resulting in the attack of beta-cells by CD8+ T cells.
Apoptosis, also termed programmed cell death, is characterized by cell shrinkage, chromatin condensation, DNA and protein cleavage, and the formation of apoptotic bodies accompanied by almost no inflammatory response. This physiological process, which can be divided into the mitochondrial pathway and the death receptor pathway, maintains homeostasis and benefits the organism by eliminating unneeded cells. Both apoptosis pathways function by activating cysteine proteases called caspases.
It is widely accepted that the loss of pancreatic beta-cells due to apoptosis is the significant and final step in the pathogenesis of T1DM (37). The process of apoptosis is closely connected to the innate immune response (Figure 2). For example, the enhanced apoptosis of beta-cells and defective apoptotic cell clearance lead to the leakage of cellular content and exposure of autoantigens, which amplify insulitis and autoimmunity; moreover, DNA accumulation from cellular apoptosis can cause excessive type 1 IFN production. In another example, proinflammatory agents produced by leukocytes, such as IL-1β, IFN-γ, TNF-α, and other soluble mediators, can induce beta-cell apoptosis (19, 38). All these cytokines can activate cytosolic signal transduction pathways that regulate the apoptosis of affected beta-cells. For instance, IL-1β and TNF-α function by activating the NF-κB (nuclear factor-κB), and MAPK (mitogen-activated protein kinase) pathways, and IFN-γ mainly exerts activity via the JAK (Janus kinase)-STAT (signal transducers and activators of transcription) pathway.

Figure 2. Innate immunity (especially the type 1 IFN signaling pathway) and cytokine-induced apoptosis together contribute to beta-cell death. (1) When PAMPs are bound by PRRs, including the cytosolic receptors RIG-1 and MDA5 and endosomal TLRs, the interactions can promote the synthesis and secretion of type 1 IFNs. IFN-α/β bind their receptor IFNAR and induce the production of cytokines and chemokines that can cause and worsen insulitis and apoptosis. (2) The signaling pathways underlying cytokine-induced apoptosis mainly include (i) JAK/STAT signaling induced by IFN-γ binding to its receptor IFNGR and (ii) NF-κB and MAPK signaling induced by IL-β/IL-R and TNF-α/TNFR.
After activation, NF-κB translocates to the nucleus and regulates iNOS (inducible nitric oxide synthase) gene expression. Previous evidence shows that NF-κB inhibition prevents cytokine-induced beta-cell apoptosis in vitro and in vivo and exerts a protective effect against diabetes induced by multiple low-dose treatments with streptozotocin in mice (39–41). After phosphorylation by JAK2, activated STAT1 translocates to the nucleus and regulates the expression of many genes. STAT1 deletion prevents cytokine-induced beta-cell death and diabetes induced by multiple low-dose treatments with streptozotocin in mice (42–44), and STAT1 can also regulate caspase expression (45). The MAPK family includes ERK (extracellular signal-regulated kinase), p38 and JNK (c-Jun N-terminal kinase). The downstream protein kinases and transcription factors, including ATF-2 (activating transcription factor 2), AP-1 (activator protein 1), and c-Jun, modify nuclear gene expression, and AP-1 may be the major transcription factor promoting MAPK-associated apoptosis (46, 47). Additionally, cytokine-mediated signal transduction pathways can interact with each other through MAPKs (47).
The beta-cell phenotypes of patients with T1DM mainly relate to beta-cell suicide and the function and mass of residual beta-cells (1). Beta-cell suicide is a consequence of MHC-| overexpression and increased ER stress (1). Overexpression of MHC-| can render insulin-producing beta-cells more sensitive to attack by cytotoxic T lymphocytes, and ER stress is associated with alterations in mRNA splicing and the production of abnormal proteins, which may serve as immunogenic antigens (9, 48). As T1DM develops, most pancreatic beta-cells are lost. However, some studies have identified a substantial number of residual beta-cells in patients with T1DM at diagnosis (8). Furthermore, patients with long-standing T1DM, even more than 50 years, retain identifiable residual beta-cells (21, 49). Moreover, in non-diabetogenic in vivo culture, impaired islets from T1DM patients can regain the ability to secrete insulin (50). All these findings demonstrate that the loss of beta-cell function results from both decreased beta-cell mass and decreased function. These studies provide insight into the development of new therapeutic interventions aimed at preserving and augmenting residual beta-cells.
To date, more than 50 candidate regions associated with T1DM have been identified by GWAS (7). Some candidate genes are potentially involved in other inflammatory and autoimmune diseases in addition to T1DM, suggesting that they could be key regulators of abnormal autoimmune responses. Previous studies have focused primarily on genes affecting the immune system, such as HLA, CTCL4, and PTPN22. However, gene function at the beta-cell level is receiving increasing attention (Table 1). Genes with such activity exert influence by regulating the innate immune response and antiviral activity (IFIH1, TYK2), influencing susceptibility to proapoptotic stimuli (HIP14, BACH2), and affecting the beta-cell phenotype (GLIS3) (Figure 1). Of note, some genes play a role in both the immune system and at the beta-cell level, such as HLA, INS, and BACH2, which implies that interactions between the abnormal immune system and pancreatic islet beta-cells contribute to the development of T1DM.
IFIH1 (interferon induced helicase C domain 1), which is located on human chromosome 2q24.2, was identified by GWAS as a candidate gene conferring risk to T1DM; it is expressed in human pancreatic beta-cells and encodes MDA (melanoma differentiation-associated protein 5), a cytoplasmic sensor that recognizes dsRNA, a byproduct of viral replication (51, 52). The interaction between MDA and dsRNA leads to a cascade of antiviral responses, such as the synthesis and secretion of type 1 IFNs (53). IFIH1 promotes cytokine and chemokine production when induced by enterovirus infection or PIC (polyinosinic-polycytidylic acid) (54, 55). Knockout of MDA5 in INS-1E cells and primary beta-cells decreases PIC-induced cytokines and chemokines, which indicates that IFIH1 modulates the local release of inflammatory mediators at the pancreatic beta-cell level (54). Moreover, in NOD mice, partial loss of MDA (MDA+/−) reduces the incidence of spontaneous diabetes, and complete MDA5 deficiency (MDA−/−) fully protects against spontaneous diabetes compared with wild-type (WT) MDA5 status (MDA+/+) (51). Various SNPs (single nucleotide polymorphisms) have been found to confer either susceptibility to or protection against T1DM (56). Among all these mutations, the gain-of-function missense mutation A946T in IFIH1 (rs1990760) has been confirmed to be associated with T1DM as well as several other autoimmune diseases in several independent studies (57–59). IFIH1 A946T confers increased basal and ligand-triggered type 1 IFN expression, and transgenic mice with the A946T risk allele exhibit increased basal type 1 IFN expression (60). Additionally, several studies showed that the IFIH1 A946T risk allele exerts its effect via the IFN-β-mediated response rather than through IFN-α, and IFN-β can promote persistent LCMV (lymphocytic choriomeningitis virus) infection, which causes enduring beta-cell damage (61–65). Intriguingly, a previous study indicated an association between the SNP rs1990760 and seasonal variation in the onset of T1DM and found that the predisposing gene was more likely to be associated with the onset of T1DM in summer (66). This finding may be explained by the theory that T1DM is caused by environmental factors in individuals with a genetically susceptible background. In contrast, two rare protective loss-of-function mutations in IFIH1, rs35667974 (I923V) and rs35744605 (E627X), are associated with potent inhibition of PIC-stimulated IFN-β production (67). In summary, IFIH1 may play an important role in the pathogenesis of T1DM by regulating the innate immune response, especially the type 1 IFN signaling pathway; as the downregulation of IFIH1 may have a positive effect on preventing the onset of T1DM in the initial phase, it may become a useful strategy for preventing T1DM in the future.
Located on human chromosome 19p13.2, TYK2 (tyrosine kinase 2) is a T1DM-associated candidate gene encoding a tyrosine kinase belonging to the JAK family that interacts with the cytoplasmic part of INFAR and plays a role in the type 1 IFN signaling pathway (68). Several SNPs within TYK2 are associated with autoimmune and inflammatory diseases, such as T1DM, RA (rheumatoid arthritis), SLE (systemic lupus erythematosus), MS (multiple sclerosis), and IBD (inflammatory bowel disease) (69–71). A SNP within TYK2 (rs2304256) that causes a missense mutation leading to decreased function has been associated with a decreased risk of developing T1DM (71). Human beta-cells with TYK2 knockout display lower PIC-induced JAK-STAT pathway activation; lower IFN-α, CXCL10, and MHC-| expression; and greater prevention of PIC-induced apoptosis (72). However, mice with lower expression of TYK2, caused by either TYK2 gene knockout or the presence of mutants with reduced TYK2 promoter activity, leading to decreased expression, are more sensitive to virus-induced diabetes, accompanied by higher virus titers and type 1 IFN levels, than mice with WT TYK2 (73). These findings demonstrate that WT TYK2 is crucial for maintaining the appropriate activation of the type 1 IFN signaling pathway. Differences in tissues and species may partially account for the opposing outcomes, but the exact mechanism by which the expression level of this gene regulates T1DM susceptibility remains to be explored. Regardless, there is no doubt that TYK2 can alter the inflammatory response toward beta-cells and may be a promising antidiabetic target.
PTPN2 (protein tyrosine phosphatase, non-receptor type 2), which is located on human chromosome 18p11, is expressed in human islet cells and exerts negative feedback on the JAK-STAT signaling pathway by dephosphorylating JAKs and STATs (8, 74, 75). In addition to the JAK-STAT signaling pathway, ERK, EGFR (epidermal growth factor receptor) and IRβ (insulin receptor β) are also regulated by PTPN2 (76–78). PTPN2 expression can be upregulated by proinflammatory cytokines and PIC, and PTPN2 knockout in INS-1E cells and primary beta-cells exacerbates PIC-induced apoptosis and proinflammatory cytokine production via the upregulation of STATs (54, 74). In another study, PTPN2 knockout in INS-1E cells, primary rat beta-cells and human beta-cells increased apoptosis by activating JNK, Bim (BH3-only protein) and the intrinsic apoptotic pathway. All these findings show that decreased PTPN2 expression sensitizes beta-cells to apoptosis induced by danger signals, and SNPs within PTPN2 that evoke decreased expression or function may increase the risk of T1DM (8). In addition to apoptosis, insulin secretion is also potentially affected by PTPN2; a previous study found that PTPN2 knockout in mice affected beta-cell function and led to decreased insulin secretion (79).
Of note, all three candidate genes mentioned above participate in regulating the type 1 IFN signaling pathway, and a common trait of risk-conferring variants is the promotion of excessive activation of the inflammatory response, leading to an increased risk of T1DM (13). The evidence not only emphasizes the importance of the type 1 IFN signaling pathway in the pathogenesis of T1DM but also provides a potential treatment strategy, namely, moderately downregulating the expression of type 1 IFNs by using targeted genomic approaches.
Located on human chromosome 6q15, BACH2 (BTB and CNC homology 1, basic leucine zipper transcription factor 2) was traditionally thought to function at the immune system level but has been shown to be expressed in pancreatic beta-cells as well and to be upregulated by proinflammatory cytokines (80). BACH2 knockout in human and mouse beta-cells increases cytokine-induced beta-cell apoptosis via the upregulation of JUN1, BIM and the intrinsic apoptotic pathway; in contrast, BACH2 overexpression has a protective effect on beta-cell apoptosis (80). Moreover, inhibition of BACH2 downregulates PTPN2 expression (80). Although the exact mechanism is still unknown, this finding supports the hypothesis that the network formed by T1DM candidate risk genes renders beta-cells hyper-responsive to danger signals. A recent study found that a BACH2 risk allele (rs3757247) might contribute to the development of insulin-triggered T1DM by affecting the immune response (81). The finding that the BACH2 gene functions at both the immune system and beta-cell levels suggests interplay between these two systems and implies an intricate network underlying T1DM pathogenesis.
TNFAIP3 (TNF-induced protein 3), which is located on human chromosome 6q23, has been identified by GWAS as a candidate gene associated with the onset and pathogenesis of T1DM and other autoimmune diseases, such as RA, IBD and psoriasis (82, 83). The TNFAIP3 gene encodes the zinc finger protein A20, a cytoplasmic ubiquitin-editing protein that is upregulated by cytokines in INS-1E cells and primary mouse islets (83). TNFAIP3 knockout increases INS-1E cell apoptosis induced by proinflammatory cytokines; in contrast, overexpression of this gene decreases apoptosis (84). A20 exerts function via multiple pathways: it negatively regulates NF-κB activation and NO production, inhibits JNK activation, upregulates Akt (a protein controlling beta-cell survival) and subsequently downregulates the intrinsic apoptotic pathway (84). These functions highlight the multiple antiapoptotic effects of A20 in beta-cells (8). In addition to influencing apoptosis, TNFAIP3 also affects beta-cell function by regulating the expression level of ZnT8, which is essential for insulin production and secretion, as determined by experiments showing that TNFAIP3 overexpression protects ZnT8 from cytokine-induced downregulation (85). Furthermore, a SNP in the non-coding region of TNFAIP3 (rs2327832) is associated with lower C-peptide and higher HbA1c (hemoglobin A1c) levels, which indicates reduced beta-cell function and impaired glycemic control in children with recent onset of T1DM (84). Although further investigation in different cohorts is needed, this finding provides evidence that A20 influences beta-cell death and function. Another recent study indicated that islet allografts with A20 upregulation show increased survival via NF-κB inhibition, AP-1 reporter activation and CXCL10 transcription (86), which sheds some light on the possibility of reducing immunosuppression therapies after islet transplantation and increasing the success rate of this operation.
Located on human chromosome 12q13.2, the ERBB3 (erb-b2 receptor tyrosine kinase 3) gene is known for its role in cancer. The ERBB3 gene encodes a protein in the EGFR family that functions as a heterodimer with other EGFR family members (87). The SNP rs2292239, located in intron 7 of ERBB3, is associated with T1DM, residual beta-cell function and metabolic control (88–90). Previous studies focused on this gene reported that it confers a risk for T1DM by modulating APC function to exert immunoregulatory effects (91). A later study demonstrated that ERBB3 also affects beta-cell apoptosis (89). ERBB3 knockdown decreases basal and cytokine-induced apoptosis, but ERBB3 expression is downregulated by proinflammatory cytokines, indicating that this gene may participate in negative regulation by cytokines (89). Thus, further investigation is needed to resolve the contradiction that the ERBB3 gene is downregulated by proinflammatory cytokines but increases cytokine-induced apoptosis. Additionally, this contradiction may suggest that additional unknown mechanisms affect beta-cell death.
The HIP14 (huntingtin-interacting protein 14) gene located on human chromosome 12 encodes a palmitoyl transferase that is highly expressed in the brain (92). HIP14 was identified as a T1DM candidate protein by in silico phenome-interactome network analysis (12). HIP14 is expressed in pancreatic islets, with predominant expression in beta-cells (12). Unlike PTPN2, BACH2, and TNFAIP3, which are upregulated by proinflammatory cytokines, HIP14 is downregulated by cytokines (12). HIP14 is thought to participate in T1DM development through interactions with two proteins physically associated with T1DM, HTT (huntingtin protein), and GAD65 (glutamate decarboxylase 65) (12). HIP14 knockout leads to increased apoptosis, whereas HIP14 overexpression results in decreased apoptosis due to reduced NF-κB activation (12). Another study indicated that caspase 6, which plays an important role in apoptosis, can be inhibited by the palmitoyl transferase activity of HIP14 in the mouse brain (93). However, further investigation is needed to clarify whether this effect also occurs in pancreatic beta-cells. In addition to apoptosis, insulin release is also affected by the palmitoyl transferase activity of HIP14 (12). The knockout of HIP14 and overexpression of mutant HIP14 lacking the palmitoyl transferase domain lead to decreased insulin release, indicating that the palmitoyl transferase activity of HIP14 participates in insulin secretion (12). In summary, HIP14 may contribute to the development of T1DM by regulating beta-cell apoptosis and insulin secretion, but more evidence is required to determine whether it is a candidate risk gene of T1DM.
Located on human chromosome 16, the STX4 (syntaxin 4) gene is situated within the T1DM susceptibility region, and similar to HIP14, the Stx4 protein encoded by STX4 was identified as a T1DM candidate protein by in silico phenome-interactome network analysis (12, 94). STX4, which localizes to the plasma membrane, is associated with insulin secretion (94). STX4 overexpression restricted to pancreatic beta-cells increases the capacity for insulin secretion, promotes glucose tolerance and protects STZ-treated mice from developing diabetes (94). Furthermore, increased STX4 expression can downregulate the expression of chemokine genes associated with inflammation and the apoptosis of pancreatic islets, such as CXCL9, CXCL10, and CXCL11 (94). Additionally, increased STX4 expression leads to decreased apoptosis by decreasing the translocation and activation of NF-κB (94). In conclusion, STX4 can influence both insulin secretion and beta-cell apoptosis, and it may be a novel target for the treatment of T1DM.
CLEC16A (c-type lectin domain family 16, member A), which is located on human chromosome 16p13, encodes a membrane-associated endosomal protein that has been associated with T1DM, MS, primary adrenal insufficiency and other inflammatory and autoimmune diseases (95–97). CLEC16A plays a role in mitochondrial autophagy (mitophagy), a process to eliminate unhealthy mitochondria that is essential for maintaining beta-cell function, glucose homeostasis and GSIS (glucose-stimulated insulin secretion); inhibition of the CLEC16A-related pathway impairs beta-cell oxygen consumption and insulin secretion (98). A ubiquitin-dependent tripartite composed of CLEC16A, NRDP1, and USP18 was reported to act as a regulator of beta-cell mitophagy (99). A previous study found that pancreas-specific CLEC16A deficiency led to impaired glucose tolerance, ER stress and GSIS in mice, and a SNP in the CLEC16A gene (rs12708716) associated with reduced expression resulted in impaired beta-cell function in humans (98). A recent finding indicated that risk variants within CLEC16A might lead to insulin-triggered T1DM due to less efficient negative selection in the thymus (81). These findings shed light on how mitophagy maintains and promotes beta-cell function and suggest the candidate gene CLEC16A as a new potential therapeutic target for T1DM.
DEXI, which is located in the same region as the CLEC16A gene, encodes a dexamethasone-induced protein of unknown function that is highly expressed in human pancreatic islets; this gene has been implicated in the pathogenesis of T1DM and other autoimmune diseases, such as MS (95, 100). According to gene expression analysis, SNPs within the CLEC16A gene modulate the expression level of DEXI, suggesting DEXI as a potential candidate gene related to T1DM (101). A previous study found that DEXI knockout led to the decreased activation of STAT1 and production of proinflammatory chemokines, such as CCL5, CXCL9, and CXCL1, in PIC-treated INS-1E cells, and DEXI overexpression has the opposite results (100); moreover, DEXI was shown to modulate IFN-β transcription (100). Based on these findings, the researchers concluded that DEXI might participate in the pathogenesis of T1DM by regulating the type 1 IFN signaling pathway (100). However, another recent study found that DEXI knockout did not alter the frequency of diabetes or influence the protective effect afforded by CLEC16A knockout in NOD mice (102). These researchers concluded that CLEC16A, rather than DEXI, is a causal gene of T1DM within human chromosome region 16p13 (102). Different cell types and species may partly explain the opposite conclusions, but the precise underlying mechanisms remain to be further investigated.
Located on human chromosome 15q25.1, the CTSH (cathepsin H) gene encodes a lysosomal cysteine protease that is expressed in human pancreatic islet beta-cells and downregulated upon exposure to proinflammatory cytokines. Overexpression of CTSH leads to decreased cytokine-induced apoptosis by decreasing the activation of the JNK1/2 and p38 pathways and the production of proapoptotic factors, including c-Myc, Bim and DP5 (death protein 5), in insulin-producing INS-1 cells (103). In addition to its effects on beta-cell apoptosis, CTSH overexpression also resulted in increased insulin accumulation in the medium and higher Ins2 levels. In line with this finding, CTSH(-/-) mice have lower plasma insulin levels than WT mice (103). These facts indicate the antiapoptotic effects of CTSH and its ability to enhance beta-cell function. A SNP in CTSH (rs3825932) associated with lower expression affects disease progression in children with newly diagnosed T1DM in an allele dose-dependent manner, characterized by the requirement for a higher daily insulin dose and a lower chance of remission (103). This variant also influences beta-cell function in healthy adults (103). However, another SNP (rs2289702) in low LD (linkage disequilibrium) with rs3825932 was recently discovered to have an adverse effect; rs2289702 correlates with decreased CTSH expression and plays a protective role in T1DM (104). The researchers speculated that increased CTSH expression might lead to an excessive innate immune response, thus increasing the risk of T1DM, based on the fact that CTSH can increase the activation of TLR3, a protein expressed in human islets, via cleavage of the N-terminus (104). Further investigation is needed to clarify whether this gene has protective properties and to elucidate its underlying mechanisms.
GLIS3 (Gli-similar 3), which is located on human chromosome 9p24.2, encodes a transcription factor in the zinc finger family; this gene was identified by GWAS as a candidate gene for both T1DM and T2DM (82, 105). It plays an important role in the development and generation of beta-cells by maintaining mature beta-cell mass and function and INS gene expression (106, 107). Loss-of-function mutations within GLIS3 lead to a rare syndrome mainly characterized by neonatal diabetes and congenital hypothyroidism in humans, and in accordance with this, GLIS3(−/−) mice develop neonatal diabetes caused by impaired pancreatic beta-cell generation and insulin production (108, 109). Additionally, GLIS3 knockout increases basal and proinflammatory cytokine-induced apoptosis by promoting the formation of a proapoptotic splice variant of BIM (110). These findings indicate that GLIS3 protects against T1DM by maintaining beta-cell function and mass and by exerting antiapoptotic effects. It is conceivable that we can prevent the onset of T1DM by enhancing GLIS3 in the future.
As evident from the above discussion, candidate genes acting at the beta-cell level play important roles in the onset and development of T1DM and, together with genes acting at the immune system, constitute the complete pathogenic network. There is a critical need to elucidate the exact underlying mechanisms of these genes to fully understand T1DM. Additionally, advances in this field will provide new therapeutic strategies for T1DM, i.e., avenues to moderately downregulate the innate immune response and cytokine-induced apoptosis and to strengthen residual beta-cell function and viability by using genetic engineering techniques.
T1DM is a multifactorial autoimmune disease, and its precise mechanisms are still unknown. However, it is widely accepted that a combination of environmental and genetic factors contributes to the onset and pathogenesis of T1DM. Candidate genes identified by GWAS influence not only the immune system but also pancreatic islet beta-cells. Some studies have revealed that risk genes act at the beta-cell level mainly through modulating the innate immune system, antiviral activity, and beta-cell apoptosis and phenotypes, and understanding potential pathogenic mechanisms will be helpful in the development of new treatments. However, T1DM is an extremely complex and heterogeneous disease, and these characteristics may be attributed to different genetic backgrounds and environmental components. To develop a more precise predictive model and more effective treatment and prevention measures, it is necessary to fully elucidate the pathogenic network of T1DM. A scoring system for quantifying the genetic and environmental elements may help considerably. To reach this ambitious goal, we propose roughly dividing the process into the following steps. First, screen candidate risk genes for T1DM and establish a pathogenic network comprising genetic and environmental elements. Next, assign these elements a value according to importance in conferring risk for T1DM and build a formula based on epidemiological information. Finally, assess the susceptibility of developing T1DM using the novel formula, and take individualized prevention measures in the predisposed population. Since the pathogenic mechanisms are not fully understood, there is still a long way to go to achieve this goal.
HP searched references, wrote the first draft of the paper and revised the text. SL, GH, and YX critically revised the text and provided substantial scientific contributions. ZZ and ZX proposed the project and revised the manuscript. All the authors approved the final version of the manuscript.
This work was supported by the National Natural Science Foundation of China (81873634, 81400783), the National Key R&D Program of China (2016YFC1305000, 2016YFC1305001, 2018YFC1315603), and the Science and Technology Major Project of Hunan Province (2017SK1020).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet. (2018) 391:2449–62. doi: 10.1016/S0140-6736(18)31320-5
2. Xie Z, Chang C, Zhou Z. Molecular mechanisms in autoimmune type 1 diabetes: a critical review. Clin Rev Allergy Immunol. (2014) 47:174–92. doi: 10.1007/s12016-014-8422-2
3. Zheng P, Li Z, Zhou Z. Gut microbiome in type 1 diabetes: a comprehensive review. Diabetes Metab Res Rev. (2018) 34:e3043. doi: 10.1002/dmrr.3043
4. Wang Z, Xie Z, Lu Q, Chang C, Zhou Z. Beyond genetics: what causes type 1 diabetes. Clin Rev Allergy Immunol. (2017) 52:273–86. doi: 10.1007/s12016-016-8592-1
5. Xia Y, Xie Z, Huang G, Zhou Z. Incidence and trend of type 1 diabetes and the underlying environmental determinants. Diabetes Metab Res Rev. (2019) 35:e3075. doi: 10.1002/dmrr.3075
6. Esposito S, Toni G, Tascini G, Santi E, Berioli MG, Principi N. Environmental factors associated with type 1 diabetes. Front Endocrinol. (2019) 10:592. doi: 10.3389/fendo.2019.00592
7. Johnson MB, Cerosaletti K, Flanagan SE, Buckner JH. Genetic mechanisms highlight shared pathways for the pathogenesis of polygenic type 1 diabetes and monogenic autoimmune diabetes. Curr Diab Rep. (2019) 19:20. doi: 10.1007/s11892-019-1141-6
8. Storling J, Pociot F. Type 1 diabetes candidate genes linked to pancreatic islet cell inflammation and beta-cell apoptosis. Genes. (2017) 8:72. 8:72. doi: 10.3390/genes8020072
9. Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. (2012) 8:e1002552. doi: 10.1371/journal.pgen.1002552
10. Nica AC, Ongen H, Irminger JC, Bosco D, Berney T, Antonarakis SE, et al. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. (2013) 23:1554–62. doi: 10.1101/gr.150706.112
11. Floyel T, Kaur S, Pociot F. Genes affecting β-cell function in type 1 diabetes. Curr Diab Rep. (2015) 15:97. doi: 10.1007/s11892-015-0655-9
12. Berchtold LA, Storling ZM, Ortis F, Lage K, Bang-Berthelsen C, Bergholdt R, et al. Huntingtin-interacting protein 14 is a type 1 diabetes candidate protein regulating insulin secretion and beta-cell apoptosis. Proc Natl Acad Sci USA. (2011) 108:E681–8. doi: 10.1073/pnas.1104384108
13. Santin I, Eizirik DL. Candidate genes for type 1 diabetes modulate pancreatic islet inflammation and β-cell apoptosis. Diabetes Obes Metab. (2013) 15(Suppl.3):71–81. doi: 10.1111/dom.12162
14. Santin I, Dos Santos RS, Eizirik DL. Pancreatic beta cell survival and signaling pathways: effects of type 1 diabetes-associated genetic variants. Methods Mol Biol. (2016) 1433:21–54. doi: 10.1007/7651_2015_291
15. Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. (2009) 5:219–26. doi: 10.1038/nrendo.2009.21
16. Pino SC, Kruger AJ, Bortell R. The role of innate immune pathways in type 1 diabetes pathogenesis. Curr Opin Endocrinol Diabetes Obes. (2010) 17:126–30. doi: 10.1097/MED.0b013e3283372819
17. Qaisar N, Jurczyk A, Wang JP. Potential role of type I interferon in the pathogenic process leading to type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. (2018) 25:94–100. doi: 10.1097/MED.0000000000000399
18. Lee MS, Chang I, Kim S. Death effectors of β-cell apoptosis in type 1 diabetes. Mol Genet Metab. (2004) 83:82–92. doi: 10.1016/j.ymgme.2004.08.002
19. Vives-Pi M, Rodriguez-Fernandez S, Pujol-Autonell I. How apoptotic β-cells direct immune response to tolerance or to autoimmune diabetes: a review. Apoptosis. (2015) 20:263–72. doi: 10.1007/s10495-015-1090-8
20. Gianani R, Campbell-Thompson M, Sarkar SA, Wasserfall C, Pugliese A, Solis JM, et al. Dimorphic histopathology of long-standing childhood-onset diabetes. Diabetologia. (2010) 53:690–8. doi: 10.1007/s00125-009-1642-y
21. Keenan HA, Sun JK, Levine J, Doria A, Aiello LP, Eisenbarth G, et al. Residual insulin production and pancreatic ss-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes. (2010) 59:2846–53. doi: 10.2337/db10-0676
22. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
23. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73. doi: 10.1128/CMR.00046-08
24. Bach JF, Bendelac A, Brenner MB, Cantor H, De Libero G, Kronenberg M, et al. The role of innate immunity in autoimmunity. J Exp Med. (2004) 200:1527–31. doi: 10.1084/jem.20042110
25. Beyan H, Buckley LR, Yousaf N, Londei M, Leslie RD. A role for innate immunity in type 1 diabetes? Diabetes Metab Res Rev. (2003) 19:89–100. doi: 10.1002/dmrr.341
26. Huang J, Xiao Y, Xu A, Zhou Z. Neutrophils in type 1 diabetes. J Diabetes Investig. (2016) 7:652–63. doi: 10.1111/jdi.12469
27. Fabris P, Betterle C, Floreani A, Greggio NA, de Lazzari F, Naccarato R, et al. Development of type 1 diabetes mellitus during interferon alfa therapy for chronic HCV hepatitis. Lancet. (1992) 340:548. doi: 10.1016/0140-6736(92)91744-S
28. Huang X, Yuang J, Goddard A, Foulis A, James RF, Lernmark A, et al. Interferon expression in the pancreases of patients with type I diabetes. Diabetes. (1995) 44:658–64. doi: 10.2337/diab.44.6.658
29. Stewart TA. Neutralizing interferon alpha as a therapeutic approach to autoimmune diseases. Cytokine Growth Factor Rev. (2003) 14:139–54. 14:139–54. doi: 10.1016/S1359-6101(02)00088-6
30. Ferreira RC, Guo H, Coulson RM, Smyth DJ, Pekalski ML, Burren OS, et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes. (2014) 63:2538–50. doi: 10.2337/db13-1777
31. Kallionpaa H, Elo LL, Laajala E, Mykkanen J, Ricano-Ponce I, Vaarma M, et al. Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes. (2014) 63:2402–14. doi: 10.2337/db13-1775
32. Stewart TA, Hultgren B, Huang X, Pitts-Meek S, Hully J, MacLachlan NJ. Induction of type I diabetes by interferon-alpha in transgenic mice. Science. (1993) 260:1942–6. doi: 10.1126/science.8100367
33. Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA. (2008) 105:12439–44. doi: 10.1073/pnas.0806439105
34. Lombardi A, Tsomos E, Hammerstad SS, Tomer Y. Interferon alpha: the key trigger of type 1 diabetes. J Autoimmun. (2018) 94:7–15. doi: 10.1016/j.jaut.2018.08.003
35. Marroqui L, Dos Santos RS, Op de Beeck A, Coomans de Brachene A, Marselli L, Marchetti P, et al. Interferon-α mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia. (2017) 60:656–67. doi: 10.1007/s00125-016-4201-3
36. Lombardi A, Tomer Y. Interferon alpha impairs insulin production in human beta cells via endoplasmic reticulum stress. J Autoimmun. (2017) 80:48–55. doi: 10.1016/j.jaut.2017.02.002
37. Quan W, Jo EK, Lee MS. Role of pancreatic β-cell death and inflammation in diabetes. Diabetes Obes Metab. (2013) 15(Suppl.3):141–51. doi: 10.1111/dom.12153
38. Keating SE, Baran M, Bowie AG. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. (2011) 32:574–81. doi: 10.1016/j.it.2011.08.004
39. Gilmore TD. The Rel/NF-kappaB signal transduction pathway: introduction. Oncogene. (1999) 18:6842–4. doi: 10.1038/sj.onc.1203237
40. Eldor R, Yeffet A, Baum K, Doviner V, Amar D, Ben-Neriah Y, et al. Conditional and specific NF-kappaB blockade protects pancreatic beta cells from diabetogenic agents. Proc Natl Acad Sci USA. (2006) 103:5072–7. doi: 10.1073/pnas.0508166103
41. Ortis F, Pirot P, Naamane N, Kreins AY, Rasschaert J, Moore F, et al. Induction of nuclear factor-kappaB and its downstream genes by TNF-α and IL-1β has a pro-apoptotic role in pancreatic beta cells. Diabetologia. (2008) 51:1213–25. doi: 10.1007/s00125-008-0999-7
42. Tau G, Rothman P. Biologic functions of the IFN-gamma receptors. Allergy. (1999) 54:1233–51. doi: 10.1034/j.1398-9995.1999.00099.x
43. Callewaert HI, Gysemans CA, Ladriere L, D'Hertog W, Hagenbrock J, Overbergh L, et al. Deletion of STAT-1 pancreatic islets protects against streptozotocin-induced diabetes and early graft failure but not against late rejection. Diabetes. (2007) 56:2169–73. doi: 10.2337/db07-0052
44. Gysemans CA, Ladriere L, Callewaert H, Rasschaert J, Flamez D, Levy DE, et al. Disruption of the gamma-interferon signaling pathway at the level of signal transducer and activator of transcription-1 prevents immune destruction of beta-cells. Diabetes. (2005) 54:2396–403. doi: 10.2337/diabetes.54.8.2396
45. Stephanou A, Brar BK, Scarabelli TM, Jonassen AK, Yellon DM, Marber MS, et al. Ischemia-induced STAT-1 expression and activation play a critical role in cardiomyocyte apoptosis. J Biol Chem. (2000) 275:10002–8. doi: 10.1074/jbc.275.14.10002
46. Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. J Clin Immunol. (1999) 19:350–64. doi: 10.1023/A:1020546615229
47. Eizirik DL, Mandrup-Poulsen T. A choice of death–the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia. (2001) 44:2115–33. doi: 10.1007/s001250100021
48. Richardson SJ, Rodriguez-Calvo T, Gerling IC, Mathews CE, Kaddis JS, Russell MA, et al. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia. (2016) 59:2448–58. doi: 10.1007/s00125-016-4067-4
49. Lam CJ, Jacobson DR, Rankin MM, Cox AR, Kushner JA. β cells persist in T1D pancreata without evidence of ongoing beta-cell turnover or neogenesis. J Clin Endocrinol Metab. (2017) 102:2647–59. doi: 10.1210/jc.2016-3806
50. Krogvold L, Skog O, Sundstrom G, Edwin B, Buanes T, Hanssen KF, et al. Function of isolated pancreatic islets from patients at onset of type 1 diabetes: insulin secretion can be restored after some days in a non-diabetogenic environment in vitro: results from the DiViD study. Diabetes. (2015) 64:2506–12. doi: 10.2337/db14-1911
51. Downes K, Pekalski M, Angus KL, Hardy M, Nutland S, Smyth DJ, et al. Reduced expression of IFIH1 is protective for type 1 diabetes. PLoS One. (2010) 5:e12646. doi: 10.1371/journal.pone.0012646
52. Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. (2009) 324:387–9. doi: 10.1126/science.1167728
53. Morse ZJ, Horwitz MS. Innate viral receptor signaling determines type 1 diabetes onset. Front Endocrinol. (2017) 8:249. doi: 10.3389/fendo.2017.00249
54. Colli ML, Moore F, Gurzov EN, Ortis F, Eizirik DL. MDA5 and PTPN2, two candidate genes for type 1 diabetes, modify pancreatic beta-cell responses to the viral by-product double-stranded RNA. Hum Mol Genet. (2010) 19:135–46. doi: 10.1093/hmg/ddp474
55. Skog O, Korsgren O, Frisk G. Modulation of innate immunity in human pancreatic islets infected with enterovirus in vitro. J Med Virol. (2011) 83:658–64. doi: 10.1002/jmv.21924
56. Looney BM, Xia CQ, Concannon P, Ostrov DA, Clare-Salzler MJ. Effects of type 1 diabetes-associated IFIH1 polymorphisms on MDA5 function and expression. Curr Diab Rep. (2015) 15:96. doi: 10.1007/s11892-015-0656-8
57. Jermendy A, Szatmari I, Laine AP, Lukacs K, Horvath KH, Korner A, et al. The interferon-induced helicase IFIH1 Ala946Thr polymorphism is associated with type 1 diabetes in both the high-incidence Finnish and the medium-incidence Hungarian populations. Diabetologia. (2010) 53:98–102. doi: 10.1007/s00125-009-1561-y
58. Qu HQ, Marchand L, Grabs R, Polychronakos C. The association between the IFIH1 locus and type 1 diabetes. Diabetologia. (2008) 51:473–5. doi: 10.1007/s00125-007-0895-6
59. Zurawek M, Fichna M, Fichna P, Skowronska B, Dzikiewicz-Krawczyk A, Januszkiewicz D, et al. Cumulative effect of IFIH1 variants and increased gene expression associated with type 1 diabetes. Diabetes Res Clin Pract. (2015) 107:259–66. doi: 10.1016/j.diabres.2014.11.008
60. Gorman JA, Hundhausen C, Errett JS, Stone AE, Allenspach EJ, Ge Y, et al. The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat Immunol. (2017) 18:744–52. doi: 10.1038/ni.3766
61. Cheng G, Wang LC, Fridlender ZG, Cheng GS, Chen B, Mangalmurti NS, et al. Pharmacologic activation of the innate immune system to prevent respiratory viral infections. Am J Respir Cell Mol Biol. (2011) 45:480–8. doi: 10.1165/rcmb.2010-0288OC
62. Molineros JE, Maiti AK, Sun C, Looger LL, Han S, Kim-Howard X, et al. Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS Genet. (2013) 9:e1003222. doi: 10.1371/journal.pgen.1003222
63. Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, et al. Blockade of interferon Beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe. (2015) 17:653–61. doi: 10.1016/j.chom.2015.04.005
64. Robinson T, Kariuki SN, Franek BS, Kumabe M, Kumar AA, Badaracco M, et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-alpha and serologic autoimmunity in lupus patients. J Immunol. (2011) 187:1298–303. doi: 10.4049/jimmunol.1100857
65. Sheikh F, Dickensheets H, Gamero AM, Vogel SN, Donnelly RP. An essential role for IFN-beta in the induction of IFN-stimulated gene expression by LPS in macrophages. J Leukoc Biol. (2014) 96:591–600. doi: 10.1189/jlb.2A0414-191R
66. Jermendy A, Szatmari I, Korner A, Szabo AJ, Toth-Heyn P, Hermann R. Association between interferon-induced helicase (IFIH1) rs1990760 polymorphism and seasonal variation in the onset of type 1 diabetes mellitus. Pediatr Diabetes. (2018) 19:300–4. doi: 10.1111/pedi.12569
67. Chistiakov DA, Voronova NV, Savost'Anov KV, Turakulov RI. Loss-of-function mutations E6 27X and I923V of IFIH1 are associated with lower poly(I:C)-induced interferon-beta production in peripheral blood mononuclear cells of type 1 diabetes patients. Hum Immunol. (2010) 71:1128–34. doi: 10.1016/j.humimm.2010.08.005
68. Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell. (1992) 70:313–22. doi: 10.1016/0092-8674(92)90105-L
69. Bergholdt R, Brorsson C, Palleja A, Berchtold LA, Floyel T, Bang-Berthelsen CH, et al. Identification of novel type 1 diabetes candidate genes by integrating genome-wide association data, protein-protein interactions, and human pancreatic islet gene expression. Diabetes. (2012) 61:954–62. doi: 10.2337/db11-1263
70. Tao JH, Zou YF, Feng XL, Li J, Wang F, Pan FM, et al. Meta-analysis of TYK2 gene polymorphisms association with susceptibility to autoimmune and inflammatory diseases. Mol Biol Rep. (2011) 38:4663–72. doi: 10.1007/s11033-010-0601-5
71. Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet. (2010) 42:68–71. doi: 10.1038/ng.493
72. Marroqui L, Dos Santos RS, Floyel T, Grieco FA, Santin I, Op de Beeck A, et al. TYK2, a Candidate gene for type 1 diabetes, modulates apoptosis and the innate immune response in human pancreatic beta-cells. Diabetes. (2015) 64:3808–17. doi: 10.2337/db15-0362
73. Izumi K, Mine K, Inoue Y, Teshima M, Ogawa S, Kai Y, et al. Reduced Tyk2 gene expression in beta-cells due to natural mutation determines susceptibility to virus-induced diabetes. Nat Commun. (2015) 6:6748. doi: 10.1038/ncomms7748
74. Moore F, Colli ML, Cnop M, Esteve MI, Cardozo AK, Cunha DA, et al. PTPN2, a candidate gene for type 1 diabetes, modulates interferon-gamma-induced pancreatic beta-cell apoptosis. Diabetes. (2009) 58:1283–91. doi: 10.2337/db08-1510
75. Zhu W, Mustelin T, David M. Arginine methylation of STAT1 regulates its dephosphorylation by T cell protein tyrosine phosphatase. J Biol Chem. (2002) 277:35787–90. doi: 10.1074/jbc.C200346200
76. Galic S, Klingler-Hoffmann M, Fodero-Tavoletti MT, Puryer MA, Meng TC, Tonks NK, et al. Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol Cell Biol. (2003) 23:2096–108. doi: 10.1128/MCB.23.6.2096-2108.2003
77. Tiganis T, Bennett AM, Ravichandran KS, Tonks NK. Epidermal growth factor receptor and the adaptor protein p52Shc are specific substrates of T-cell protein tyrosine phosphatase. Mol Cell Biol. (1998) 18:1622–34. doi: 10.1128/MCB.18.3.1622
78. Walchli S, Curchod ML, Gobert RP, Arkinstall S, Hooft van Huijsduijnen R. Identification of tyrosine phosphatases that dephosphorylate the insulin receptor. A brute force approach based on “substrate-trapping” mutants. J Biol Chem. (2000) 275:9792–6. doi: 10.1074/jbc.275.13.9792
79. Xi Y, Liu S, Bettaieb A, Matsuo K, Matsuo I, Hosein E, et al. Pancreatic T cell protein-tyrosine phosphatase deficiency affects beta cell function in mice. Diabetologia. (2015) 58:122–31. doi: 10.1007/s00125-014-3413-7
80. Marroqui L, Santin I, Dos Santos RS, Marselli L, Marchetti P, Eizirik DL. BACH2, a candidate risk gene for type 1 diabetes, regulates apoptosis in pancreatic beta-cells via JNK1 modulation and crosstalk with the candidate gene PTPN2. Diabetes. (2014) 63:2516–27. doi: 10.2337/db13-1443
81. Onuma H, Kawamura R, Tabara Y, Yamashita M, Ohashi J, Kawasaki E, et al. Variants in the BACH2 and CLEC16A gene might be associated with susceptibility to insulin-triggered type 1 diabetes. J Diabetes Investig. (2019) 10:1447–53. doi: 10.1111/jdi.13057
82. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. (2009) 41:703–7. doi: 10.1038/ng.381
83. Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol. (2014) 35:22–31. doi: 10.1016/j.it.2013.10.005
84. Fukaya M, Brorsson CA, Meyerovich K, Catrysse L, Delaroche D, Vanzela EC, et al. A20 inhibits β-cell apoptosis by multiple mechanisms and predicts residual beta-cell function in type 1 diabetes. Mol Endocrinol. (2016) 30:48–61. doi: 10.1210/me.2015-1176
85. Cheng L, Zhang D, Chen B. Tumor necrosis factor alpha-induced protein-3 protects zinc transporter 8 against proinflammatory cytokine-induced downregulation. Exp Ther Med. (2016) 12:1509–14. doi: 10.3892/etm.2016.3457
86. Zammit NW, Walters SN, Seeberger KL, O'Connell PJ, Korbutt GS, Grey ST. A20 is an immune tolerance factor that can determine islet transplant outcomes. JCI Insight. 2019. 2019. doi: 10.1101/770834
87. Ma J, Lyu H, Huang J, Liu B. Targeting of erbB3 receptor to overcome resistance in cancer treatment. Mol Cancer. (2014) 13:105. doi: 10.1186/1476-4598-13-105
88. Keene KL, Quinlan AR, Hou X, Hall IM, Mychaleckyj JC, Onengut-Gumuscu S, et al. Evidence for two independent associations with type 1 diabetes at the 12q13 locus. Genes Immun. (2012) 13:66–70. doi: 10.1038/gene.2011.56
89. Saffen D. The genetic architecture of autism spectrum disorders (ASDs) and the potential importance of common regulatory genetic variants. Sci China Life Sci. (2015) 58:968–75. doi: 10.1007/s11427-012-4336-5
90. Wang D, Pan G. The association between rs2292239 polymorphism in ERBB3 gene and type 1 diabetes: a meta-analysis. Biomed Res Int. (2019) 2019:7689642. doi: 10.1155/2019/7689642
91. Wang H, Jin Y, Reddy MV, Podolsky R, Liu S, Yang P, et al. Genetically dependent ERBB3 expression modulates antigen presenting cell function and type 1 diabetes risk. PLoS ONE. (2010) 5:e11789. doi: 10.1371/journal.pone.0011789
92. Singaraja RR, Hadano S, Metzler M, Givan S, Wellington CL, Warby S, et al. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet. (2002) 11:2815–28. doi: 10.1093/hmg/11.23.2815
93. Skotte NH, Sanders SS, Singaraja RR, Ehrnhoefer DE, Vaid K, Qiu X, et al. Palmitoylation of caspase-6 by HIP14 regulates its activation. Cell Death Differ. (2017) 24:433–44. doi: 10.1038/cdd.2016.139
94. Oh E, Ahn M, Afelik S, Becker TC, Roep BO, Thurmond DC. Syntaxin 4 expression in pancreatic β-cells promotes islet function and protects functional β-cell mass. Diabetes. (2018) 67:2626–39. doi: 10.2337/db18-0259
95. Gingerich MA, Sidarala V, Soleimanpour SA. Clarifying the function of genes at the chromosome 16p13 locus in type 1 diabetes: CLEC16A and DEXI. Genes Immun. (2019). doi: 10.1038/s41435-019-0087-7. [Epub ahead of print].
96. Nischwitz S, Cepok S, Kroner A, Wolf C, Knop M, Muller-Sarnowski F, et al. More CLEC16A gene variants associated with multiple sclerosis. Acta Neurol Scand. (2011) 123:400–6. doi: 10.1111/j.1600-0404.2010.01421.x
97. Skinningsrud B, Husebye ES, Pearce SH, McDonald DO, Brandal K, Wolff AB, et al. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J Clin Endocrinol Metab. (2008) 93:3310–7. doi: 10.1210/jc.2008-0821
98. Soleimanpour SA, Gupta A, Bakay M, Ferrari AM, Groff DN, Fadista J, et al. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell. (2014) 157:1577–90. doi: 10.1016/j.cell.2014.05.016
99. Pearson G, Chai B, Vozheiko T, Liu X, Kandarpa M, Piper RC, et al. Clec16a, Nrdp1, and USP8 form a ubiquitin-dependent tripartite complex that regulates β-cell mitophagy. Diabetes. (2018) 67:265–77. doi: 10.2337/db17-0321
100. Dos Santos RS, Marroqui L, Velayos T, Olazagoitia-Garmendia A, Jauregi-Miguel A, Castellanos-Rubio A, et al. DEXI, a candidate gene for type 1 diabetes, modulates rat and human pancreatic beta cell inflammation via regulation of the type I IFN/STAT signalling pathway. Diabetologia. (2019) 62:459–72. doi: 10.1007/s00125-018-4782-0
101. Davison LJ, Wallace C, Cooper JD, Cope NF, Wilson NK, Smyth DJ, et al. Long-range DNA looping and gene expression analyses identify DEXI as an autoimmune disease candidate gene. Hum Mol Genet. (2012) 21:322–33. doi: 10.1093/hmg/ddr468
102. Nieves-Bonilla JM, Kiaf B, Schuster C, Kissler S. The type 1 diabetes candidate gene Dexi does not affect disease risk in the non-obese diabetic mouse model. Genes Immun. (2019) 21:1–7. doi: 10.1038/s41435-019-0083-y
103. Floyel T, Brorsson C, Nielsen LB, Miani M, Bang-Berthelsen CH, Friedrichsen M, et al. CTSH regulates β-cell function and disease progression in newly diagnosed type 1 diabetes patients. Proc Natl Acad Sci USA. (2014) 111:10305–10. doi: 10.1073/pnas.1402571111
104. Inshaw JRJ, Cutler AJ, Crouch DJM, Wicker LS, Todd JA. Genetic variants predisposing most strongly to type 1 diabetes diagnosed under age 7 years lie near candidate genes that function in the immune system and in pancreatic β-cells. Diabetes Care. (2019) 43:169–77. doi: 10.2337/dc19-0803
105. Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. (2011) 44:67–72. doi: 10.1038/ng.1019
106. Kang HS, Kim YS, ZeRuth G, Beak JY, Gerrish K, Kilic G, et al. Transcription factor Glis3, a novel critical player in the regulation of pancreatic β-cell development and insulin gene expression. Mol Cell Biol. (2009) 29:6366–79. doi: 10.1128/MCB.01259-09
107. Yang Y, Chang BH, Chan L. Sustained expression of the transcription factor GLIS3 is required for normal beta cell function in adults. EMBO Mol Med. (2013) 5:92–104. doi: 10.1002/emmm.201201398
108. Senee V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. (2006) 38:682–7. doi: 10.1038/ng1802
109. Watanabe N, Hiramatsu K, Miyamoto R, Yasuda K, Suzuki N, Oshima N, et al. A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett. (2009) 583:2108–13. doi: 10.1016/j.febslet.2009.05.039
110. Nogueira TC, Paula FM, Villate O, Colli ML, Moura RF, Cunha DA, et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. (2013) 9:e1003532. doi: 10.1371/journal.pgen.1003532
Keywords: T1DM, GWAS, pancreatic beta-cell, candidate gene, apoptosis, innate immunity, beta-cell phenotype
Citation: Pang H, Luo S, Huang G, Xia Y, Xie Z and Zhou Z (2020) Advances in Knowledge of Candidate Genes Acting at the Beta-Cell Level in the Pathogenesis of T1DM. Front. Endocrinol. 11:119. doi: 10.3389/fendo.2020.00119
Received: 16 January 2020; Accepted: 24 February 2020;
Published: 12 March 2020.
Edited by:
Sangeeta Dhawan, City of Hope National Medical Center, United StatesReviewed by:
Senta Georgia, Children's Hospital of Los Angeles, United StatesCopyright © 2020 Pang, Luo, Huang, Xia, Xie and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiguo Xie, eGllemhpZ3VvQGNzdS5lZHUuY24=; Zhiguang Zhou, emhvdXpoaWd1YW5nQGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.