 Petra Loid1,2,3*
Petra Loid1,2,3* Taina Mustila4,5
Taina Mustila4,5 Riikka E. Mäkitie2,3,6Heli Viljakainen2,7
Riikka E. Mäkitie2,3,6Heli Viljakainen2,7 Anders Kämpe8Päivi Tossavainen9Marita Lipsanen-Nyman1,3Minna Pekkinen1,2,3
Anders Kämpe8Päivi Tossavainen9Marita Lipsanen-Nyman1,3Minna Pekkinen1,2,3 Outi Mäkitie1,2,3,8
Outi Mäkitie1,2,3,8- 1Children's Hospital, Helsinki University Hospital, Helsinki, Finland
- 2Folkhälsan Research Center, Genetics Research Program, Helsinki, Finland
- 3Research Program for Clinical and Molecular Metabolism, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 4Department of Pediatrics, Seinäjoki Central Hospital, Seinäjoki, Finland
- 5City of Turku, Welfare Division, Preventive Healthcare, Turku, Finland
- 6Molecular Endocrinology Laboratory, Department of Medicine, Hammersmith Campus, Imperial College London, London, United Kingdom
- 7The Department of Food and Nutrition, University of Helsinki, Helsinki, Finland
- 8Department of Molecular Medicine and Surgery, Karolinska Institutet, and Department of Clinical Genetics, Karolinska University Hospital, Stockholm, Sweden
- 9Department of Children and Adolescents, PEDEGO Research Unit, University of Oulu, Oulu, Finland
Context: The hypothalamic circuit has an essential role in the regulation of appetite and energy expenditure. Pathogenic variants in genes involved in the hypothalamic leptin–melanocortin pathway, including melanocortin-4-receptor (MC4R), have been associated with monogenic obesity.
Objective: To determine the rate and spectrum of rare variants in genes involved in melanocortin pathway or hypothalamic development in patients with severe early-onset obesity (height-adjusted weight >60% before age 10 years).
Methods: We used a custom-made targeted exome sequencing panel to assess peripheral blood DNA samples for rare (minor allele frequency <0.5%), pathogenic/likely pathogenic variants in 24 genes related to the hypothalamic circuit in 92 subjects (51% males, median age 13.7 years) with early-onset severe obesity (median body mass index (BMI) Z-score + 4.0).
Results: We identified a novel frameshift deletion in MC4R (p.V103Afs5*) in two unrelated patients and a previously reported MC4R variant (p.T112M) in one patient. In addition, we identified rare heterozygous missense variants in ADCY3 (p.G1110R), MYT1L (p.R807Q), ISL1 (p.I347F), LRP2 (p.R2479I, and p.N3315S) and a hemizygous missense variant in GRPR (p.L87M) (each in one patient), possibly contributing to the obesity phenotype in these patients. Altogether 8 % (7/92) of the subjects had rare pathogenic/likely pathogenic variants in the studied genes.
Conclusions: Rare genetic variants within the hypothalamic circuit are prevalent and contribute to the development of severe early-onset obesity. Targeted exome sequencing is useful in identifying affected subjects. Further studies are needed to evaluate the variants' clinical significance and to define optimal treatment.
Introduction
Obesity is a complex disease with various contributing environmental and genetic factors. To uncover the principal genetic factors behind obesity, several advanced research strategies have been used, including twin studies, genome-wide association studies (GWASs), chromosomal microarray analyses and next generation sequencing (1). For example, common single nucleotide polymorphisms (SNPs) in multiple genes have been shown to modulate the risk of obesity, although each of these have only a minor effect on body mass index (BMI) variation (2, 3). We and others have also shown that rare copy number variants (CNVs) are enriched in patients with early-onset severe obesity (4–6). Recently, it has also been suggested that obesity could be a consequence of rare genetic variants with strong effect (7, 8). Despite these advancements, the genetic causes and underlying molecular mechanisms behind obesity are still inadequately understood.
Mutations in genes linked to the leptin-melanocortin signaling in the hypothalamus are associated with monogenic obesity. These genes include i.e., LEP, LEPR, POMC, PCSK1, MC4R, MC3R, SH2B1, NTRK2, MRAP2, and TUB (1). The discovery of these monogenic mutations has increased our understanding of the mechanisms controlling energy balance. Single-gene mutations are estimated to account for ~7% of cases of non-syndromic severe obesity (1). Of them, MC4R mutations are the most frequent with over 300 reported variants (9). The prevalence of pathogenic variants in MC4R in Finnish patients with early-onset obesity was reported to be 1.8% (10), and in other populations 1.5–5.8% (7, 11–14). Patients with loss-of-function MC4R mutations typically present with severe childhood obesity, hyperphagia, hyperinsulinemia, and increased linear growth (11, 15). MC4R is a G-protein-coupled receptor (GPCR), which activates the stimulatory G protein (Gs), increases cAMP production and activates protein kinase A (PKA), leading to decreased food intake and increased energy expenditure (16). Lately, it has been suggested that in addition to Gs activation the MC4R downstream signaling includes several other pathways potentially important for weight regulation and that more patients than previously assumed may have functionally relevant genetic variants in MC4R (16, 17).
SIM1 and BDNF are transcription and neurotropic factors that control the neuronal differentiation of the hypothalamus and mutations in genes encoding these proteins have been associated with obesity (1). Recently, new obesity related genes involved in hypothalamic development or melanocortin pathway have been identified, such as ADCY3, MYT1L, POU3F2, GRPR, and LRP2 (1, 5, 18–22).
In recent years, advances have been made in the development of targeted therapy for patients with rare genetic disorders. Setmelanotide, a MC4R agonist, has shown promising results in promoting weight loss in patients with genetic loss-of-function defects in POMC, LEPR, and MC4R (9, 23, 24). Genetic testing in patients with severe obesity is therefore important for the patients and their families, as it allows early detection of those at increased risk, optimal targeting of preventive measures and helps to identify patients likely to benefit from pharmacological treatment.
This study evaluated the prevalence and nature of genetic variants within the melanocortin pathway in patients affected by severe early-onset obesity. We have chosen to focus on the most severe end of the spectrum of obesity, namely on forms presenting already in early childhood, as they are more likely to be explained by genetic causes. We evaluated a Finnish cohort of 92 patients with severe childhood obesity and aimed to determine the rate and spectrum of rare pathogenic/likely pathogenic genetic variants in 24 genes linked to the melanocortin pathway or hypothalamic development.
Materials and Methods
Study Subjects
This study included altogether 92 children and adolescents with severe early-onset childhood obesity, as defined according to the Finnish growth standards (25) as height-adjusted weight >60% before 10 years of age. All children were born to non-consanguineous parents. The patients were recruited at Children's Hospital at Helsinki University Hospital, Seinäjoki Central Hospital and Department of Children and Adolescents at Oulu University Hospital in Finland during years 2011–2017. All participating patients had been followed up by a pediatrician and patients diagnosed with an underlying endocrine or genetic disorder were excluded from the study (e.g., Prader–Willi syndrome, hypercortisolism, and hypothyroidism). Ethical approvals for the study were obtained from the research ethics committees of the Hospital District of Helsinki and Uusimaa, the Pirkanmaa Hospital District and the Northern Ostrobothnia Hospital District. Informed written consents were obtained from all the participants or from one of their guardians (for subjects aged <18 years).
Hospital records were reviewed for clinical data. Anthropometric measurements including height, weight and waist circumference were measured during a study visit. BMI was calculated as weight divided by square height (kg/m2). Sex-and age-specific BMI Z-scores were derived based on the World Health Organization reference values (www.who.int/childgrowth/standards). Blood samples were collected for genetic studies from all participants and from available first-degree family members (parents and siblings). Heights and weights were collected from the participating family members.
Genes in the Panel
We performed a literature search to identify genes related to the melanocortin pathway and the development of the hypothalamus (1, 5, 12, 18, 19, 21, 26–33). The 24 genes included in the panel were: (1) genes in which variants have previously been reported to cause or associate with obesity (ADCY3, BDNF, CPE, GRPR, LEP, LEPR, LRP2, MC3R, MC4R, MRAP2, MYT1L, NPY, NTRK2, PCSK1, POMC, SH2B1, SIM1, TUB) and (2) genes previously reported in animal models/linkage analysis/CNV studies to be involved in the melanocortin pathway or development of hypothalamus (ARNT2, ISL1, NEUROG3, OTP, OXT, POU3F2). Summary of the genes in the panel are presented in Table 1 and Figure 1.
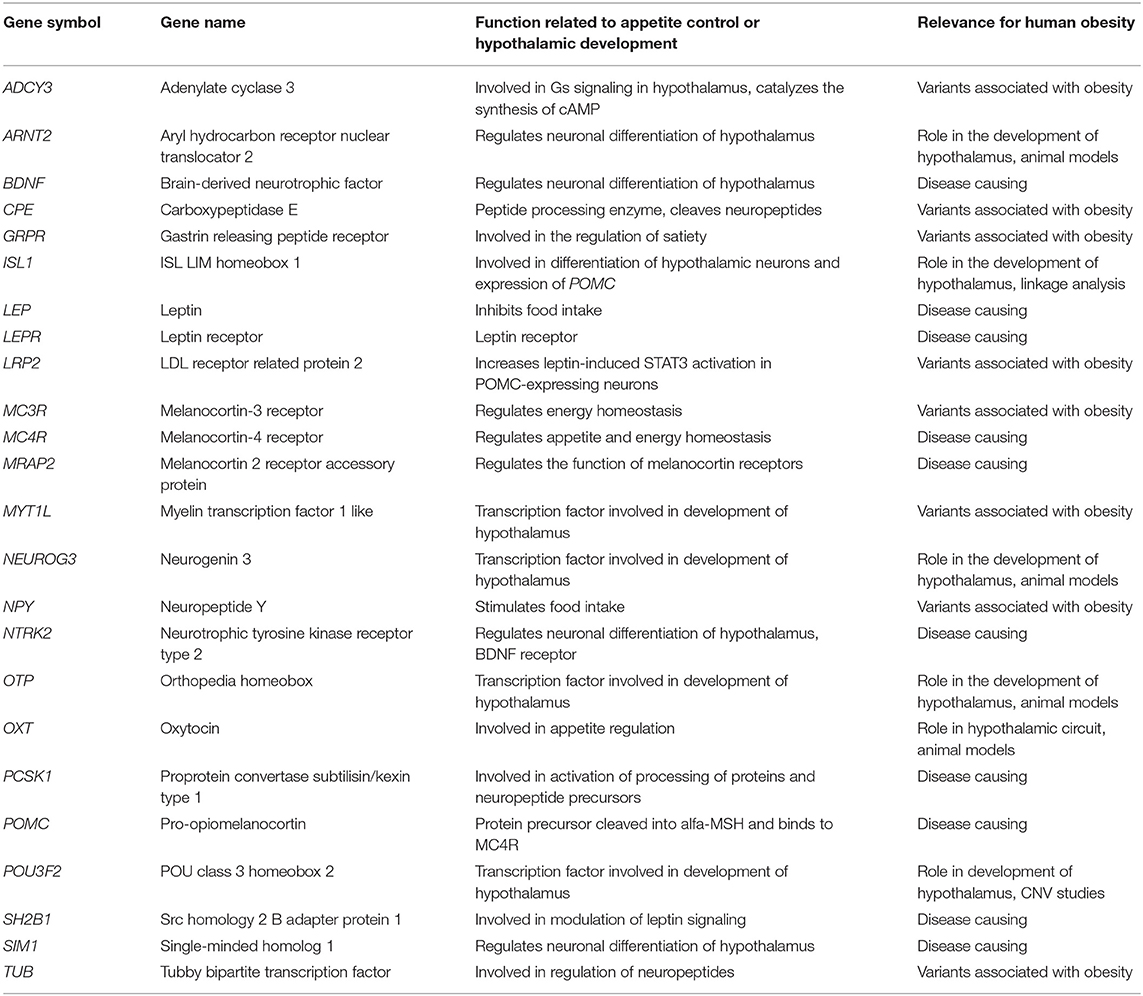
Table 1. List of the obesity related genes included in the panel.
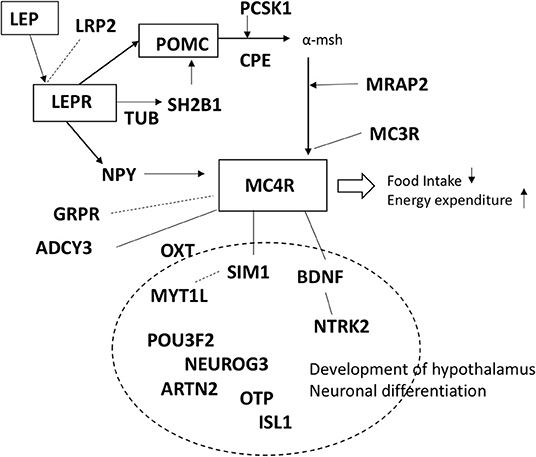
Figure 1. Schematic presentation of the genes included in the panel.
Genetic Analysis
Genomic DNA was isolated from obtained peripheral blood samples according to standard procedures. The probes for targeted exome sequencing were designed using SeqCap EZ Choice Library and NimbleDesign (Roche NimbleGen, United States). DNA capture and Next generation sequencing were performed at Oxford Genomics Center. Sequence reads were aligned to the reference human genome GRCh37. Variants were called using software Platypus software (version 0.8.1) and functionally annotated using Ensembl Variant Effector Predictor (VEP). Variant filtering was performed using VarAFT (version 2.13) (34). Non-synonymous exonic and splice site variants were considered. Variants with allele frequency higher than 0.5% (in Exome Aggregation Consortium (http://exac.broadinstitute.org), the Genome Aggregation Database (http://gnomad.broadinstitute.org), 1,000 Genomes Project (http://www.internationalgenome.org) and the Sequencing Initiative Suomi project (SISu) (http://sisuproject.fi) were filtered out. Possible pathogenicity of the variants was assessed using different variant prediction databases (Polyphen-2, SIFT, Combined Annotation Dependent Depletion (CADD) and MutationTaster2). We considered rare variants with allele frequency <0.5% and variants predicted to be pathogenic by several in silico prediction tools, including a CADD score >20, as likely pathogenic.
Sanger sequencing was performed to confirm the findings and to analyze parental and siblings' samples to determine inheritance pattern. Primers were designed using Primer3 software and PCR was performed using DreamTaqTM DNA Polymerase (Thermo Fischer Scientific) according to standard protocol. Chromatograms were analyzed with Sequencer v5.0 software. Primer sequences are available upon request.
Statistical Analyses
Results are reported as medians (interquartile range, IQR). Statistical analyses were performed with SPSS Statistical package (version 25.0.0.1).
Results
Characteristics of the Study Cohort
The study included a total of 92 subjects (51% males). At the time of the study visit their median age was 13.7 years (IQR 10.6–16.8 years) and median BMI Z-score +4.0 (IQR + 3.4 to + 4.9). The study subjects fulfilled our inclusion criteria (height-adjusted weight >60 %) at the median age of 6 years (IQR 4.5–7.0 years).
Targeted Exome Sequencing
On average 99.3% of bases in the coding exons were covered by at least 100 reads and the mean read depth of coverage was 755X. We found rare pathogenic/likely pathogenic variants in 7/92 (8%) of the study subjects and one rare variant of uncertain significance in one patient. The rare variants were in six of the 24 genes in the panel, with variants in the MC4R gene being the most common finding (3/92). The genetic and clinical features of the patients with the rare variants are presented below and summarized in Table 2. The prediction values of the identified variants are presented in Table 3.
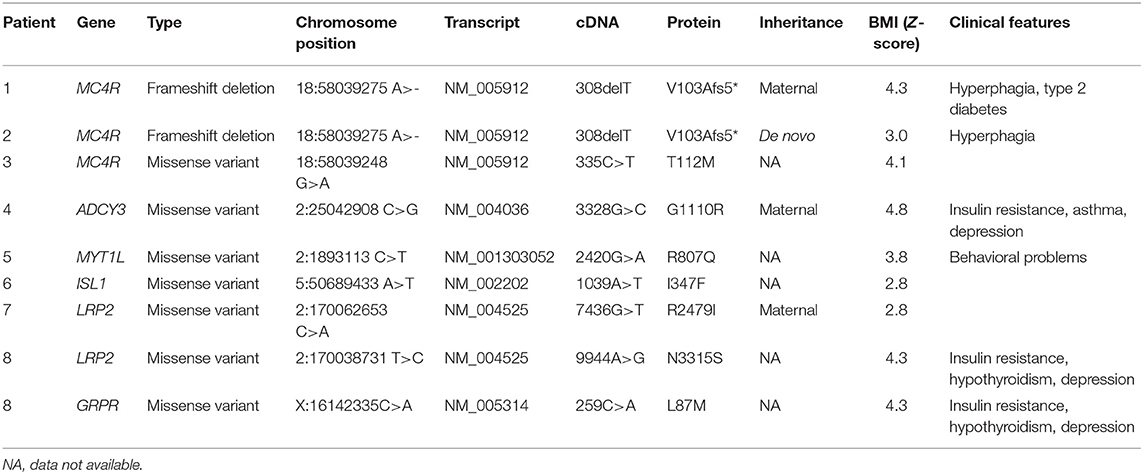
Table 2. Genetic and clinical characteristics of the patients with rare genetic variants (MAF < 0.5%), reference human genome GRCh37.
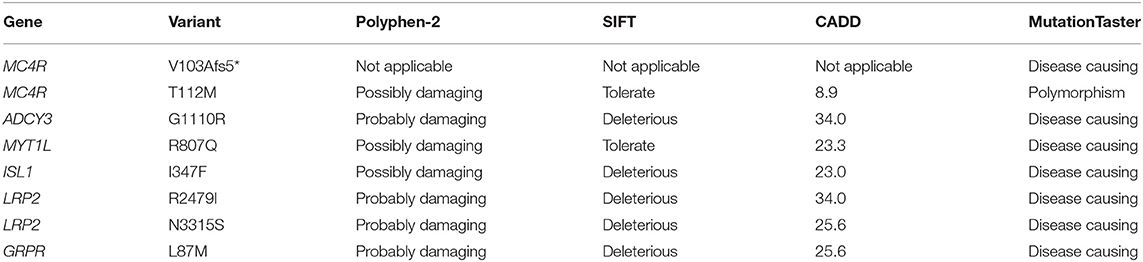
Table 3. Prediction values of the rare genetic variants detected in the obesity cohort.
Rare Exonic Variants and Associated Phenotypes
MC4R Variants
In MC4R (NM_005912) we identified altogether 2 variants. The first was a novel heterozygous frameshift deletion c.308delT present in two unrelated patients. The frameshift variant was confirmed by Sanger sequencing (Figure 2). The deletion causes a frameshift at amino acid 103 (p.V103Afs5*) and introduces four new amino acids resulting in a stop codon in the region of the second transmembrane domain, leading to a significantly truncated protein of only 107 amino acids compared to the normal 332-amino-acid MC4R (Figure 3). The variant is absent in all databases (ExAC, gnomAD, 1,000 Genomes, SISu). MutationTaster2 predicted the deletion as disease-causing.
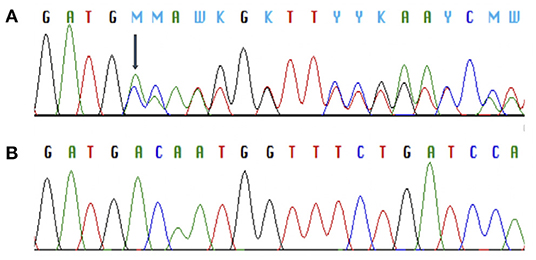
Figure 2. Chromatograms of Sanger sequence analysis of MC4R showing a heterozygous frameshift deletion (p.V103Afs5*) in two index patients with monogenic obesity (A) and the normal MC4R sequence (B).
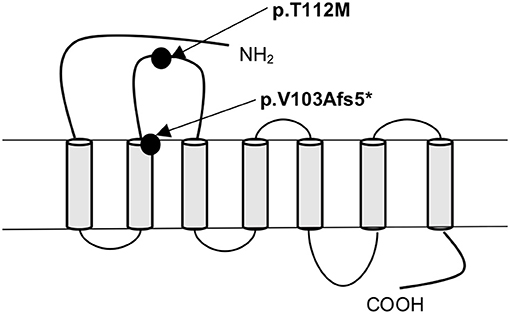
Figure 3. Schematic presentation of the MC4R protein and the location of the frameshift variant (p.V103Afs5*) and the missense variant (p.T112M) identified in patients with obesity.
The pedigrees of the two unrelated families with the MC4R frameshift variant are presented in Figure 4.
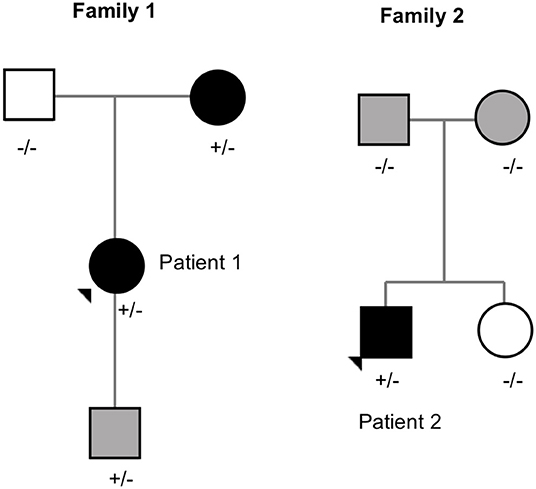
Figure 4. Pedigrees of the families with MC4R frameshift variant (p.V103Afs5*). Square = male, circle = female, +/– heterozygote carrier, –/– wildtype. Black square/circle = obesity, gray square/circle = overweight, white square/circle = normal weight.
Patient 1 is a 20-year-old female with normal birth measurements: weight 3.6 kg and length 48 cm. She presented with severe obesity already at the age of 1 year with weight 17.8 kg and height 81.5 cm (BMI Z-score + 5.8). Presently at age 20 years, her weight is 121 kg, height 170.5 cm (BMI 44) and waist circumference 133 cm. She also has type 2 diabetes and hyperphagia. Her mother, heterozygous for the same mutation, presented with obesity in childhood and overweight during adulthood. The mutation-negative father had normal weight during childhood. The index patient's 2.5-year-old son also carries the same frameshift deletion. He presented with overweight at age 9 months with a BMI Z-score of + 2.9. Currently, his BMI Z-score is +1.8 and he requires close monitoring for pronounced hyperphagia.
Patient 2 is an 18-year-old male with BMI 35 (weight 106 kg and height 173.5 cm) and waist circumference 110 cm. His birth measurements were normal (3.4 kg and 50.5 cm) and he presented with obesity at the age of 3 years measuring 18.5 kg and 93.5 cm (BMI Z-score +3.9). He also presented with hyperphagia. The variant was de novo as his overweight parents and normal weight sister did not have the variant.
The other identified MC4R variant is a previously reported heterozygous missense variant, c.335C>T (p.T112M) (Figure 3) in Patient 3; a 12-year-old boy with normal birth measurements (3.5 kg and 50 cm). He presented with obesity at age 4 years measuring 29.2 kg and 114.3 cm (BMI Z-score +4.5). Presently at the age of 12 years, his weight is 114.4 kg and height 177.1 cm (BMI Z-score +4.1). He had accelerated linear growth with growth velocity of 12 cm/year at the age of 5 years, leading to height-for-age Z-score +4.1 at age 12 years. His mother's height was 163 cm and father's 183 cm. The patient's mother with obesity did not harbor the variant. No DNA was available from the other family members. The allele frequency of the variant is 0.09% in all populations in gnomAD, 0.2% in the Finnish population in gnomAD and 0.18% in SISu.
ADCY3 Missense Variant
We identified a heterozygous missense variant in ADCY3 (c.3328G>C, p.G1110R) in Patient 4; a 16-year-old female with a BMI Z-score of +4.8, insulin resistance, asthma and depression. She had normal birth measurements (3.25 kg and 51 cm). She presented with obesity already at the age of 1.5 years measuring 16.3 kg and 86.4 cm (BMI Z-score + 3.6). The patient's mother (BMI 35) also carried the same variant. The father's DNA was not available. According to gnomAD, the variant is found in 1/3474 in the Finnish population.
MYT1L Missense Variant
We detected a heterozygous missense variant in MYT1L (c.2420G>A, p.R807Q) in Patient 5; a 12-year-old girl with a BMI Z-score of + 3.8. She presented with aggressive behavior and reading problems. The variant was not found in her normal-weight mother; DNA of the normal-weight father was not available. The allele frequency of the variant is 0.01% in all populations in gnomAD, 0.1% in the Finnish population in gnomAD and 0.1% in SISu.
ISL1 Missense Variant
We found a heterozygous missense variant in ISL1 (c.1039A>T, p.I347F) in Patient 6; a 9-year-old girl with a BMI Z-score of + 2.8. The patient had normal development and no endocrinological abnormalities. Her mother (BMI 39) was also a carrier of the variant but her normal-weight father was not. The variant is found in 5/25038 (all heterozygotes) in the Finnish population in gnomAD and the allele frequency is 0.03 % in SISu.
LRP2 Missense Variants
We identified two heterozygous missense variants in LRP2. The first variant (c.7436G>T, p.R2479I) was found in Patient 7, a 19-year-old boy with a BMI Z-score of + 2.8, and in his mother (BMI 39). The variant is not found in any of the above-mentioned databases.
The second variant in LRP2 (c.9944A>G, p.N3315S) was detected in Patient 8; a 16-year-old boy with a BMI Z-score of + 4.3, insulin resistance, fatty liver, hypothyroidism and severe depression. The patient had an overweight mother and a normal-weight father, but parental DNAs were unavailable. The variant is not found in the Finnish population in gnomAD or in SISu, but it is found in 2/24956 (both heterozygotes) in African population in gnomAD.
GRPR Missense Variant
In Patient 8, harboring an LRP2 variant (p.N3315S), we also identified a hemizygous missense variant (c.259C>A, p.L87M) in GRPR. This variant is present with allele frequency 0.02% in the Finnish population in gnomAD and SISu.
Discussion
Recent advances in genetic methodology have enabled identification of novel genetic factors that may contribute to obesity. Such discoveries have increased our understanding of the significance of various components involved in the hypothalamic circuit regulating appetite and energy expenditure. By using targeted exome sequencing, we identified several rare exonic variants in genes related to the leptin-melanocortin pathway and hypothalamic development in a large cohort of Finnish patients with severe childhood-onset obesity. Altogether 7 of the 92 studied subjects (8%) harbored at least one rare and pathogenic/likely pathogenic variant in the studied 24 genes, suggesting that a significant proportion of severe childhood-onset obesity could be related to genetic defects within the hypothalamic circuit.
The prevalence of monogenic obesity varies with ethnicity and consanguinity. Close consanguinity is rare in the Finnish population (35) and there were no consanguineous families in our cohort. We did not detect any pathogenic variants in LEPR, LEP, POMC, and PCSK1 genes. Patients with these gene defects often have very early and rapid weight gain and associated endocrinopathies. It is therefore possible, that such patients may have been identified earlier by pediatricians and thus excluded from our study focusing on obesity of unknown etiology.
The prevalence of pathogenic variants in MC4R in our cohort was 2% (2/92), which is comparable to previous reports (7, 10–14). The patients in our study with the novel MC4R frameshift deletion had severe childhood obesity and hyperphagia. The frameshift deletion is considered pathogenic as it leads to a significantly truncated protein and its functional consequences on the receptor function are likely to be considerable.
Additionally, we detected a previously identified rare heterozygous missense variant (p.T112M) in MC4R. Our patient presented with childhood obesity, markedly increased linear growth and tall stature during childhood. MC4R deficiency has been associated with accelerated linear growth and increased final height (15). In contrast, we did not observe increased final height or linear growth in the patients with the MC4R frameshift variant. The missense variant is not shown to be pathogenic according to prediction tools and the variant has previously been reported as having normal Gs signaling according to in vitro functional studies (10, 36). However, the variant has been described as a biased signaling variant that leads to decreased ERK1/2 activation. MC4R variants that alter the ERK1/2 signaling have been suggested to be involved in regulation of food intake (16, 17). We considered this variant to be of uncertain significance as further studies are needed to explore the impact of non-Gs signaling MC4R variants on weight regulation.
ADCY3 encodes adenylyl cyclase 3, which localizes at the cilia of paraventricular nucleus in the hypothalamus and is involved in the Gs signaling. Previous studies have shown that Adcy3 heterozygous null mice present with obesity, insulin resistance and increased adiposity (37). A glucagon-like peptide-1 analog, liraglutide, has been shown to increase the expression of hepatic Adcy3 in obese and diabetic mice and the Adcy3 activation may partly account for the reduction of blood glucose levels and bodyweight (38). Furthermore, previous studies have identified homozygous mutations in ADCY3 in children with severe obesity (33), and an ADCY3 variant has been associated with increased risk of obesity and type 2 diabetes in homozygous individuals and to a lesser degree in heterozygous carriers (18). The patient in our study was heterozygous for a non-synonymous ADCY3 variant with a high CADD score. She had severe obesity and insulin resistance and her mother, harboring the same variant, also presented with obesity. The ADCY3 variant was predicted disease causing and is likely to play a role in the obesity and insulin resistance in our patient.
MYT1L is expressed in the brain and involved in the development of neuroendocrine hypothalamus. Several MYT1L mutations have been identified in patients with obesity, intellectual disability, developmental delay, autistic features and behavioral problems (19, 20, 39). Loss of MYT1L in zebrafish was related to altered hypothalamic development and loss of oxytocin (OXT) in the neuroendocrine preoptic area. MYT1L was demonstrated to be part of the melanocortin pathway, functioning downstream of SIM1, and OXT was recognized as a potential treatment target (19). We have previously described a Finnish patient with a frameshift deletion in MYT1L presenting with obesity, intellectual disability and developmental delay (20). The patient in this study with the presumed de novo MYT1L variant had severe obesity, aggressive behavior and learning difficulties. Although our patient did not present with marked developmental delay or intellectual disability, her aggressive behavior, learning difficulties and the early-onset obesity are likely to be related to the identified MYT1L variant.
The Insulin gene enhance protein ISL-1, encoded by ISL1, is a transcription factor involved in many developmental processes. Mice and zebrafish experiments have shown that Isl1 is expressed in the developing brain, regulates the differentiation of hypothalamic neurons and is important for the expression of Pomc in the hypothalamus. Inactivation of Isl1 in Pomc neurons reduces the expression of Pomc and leads to obesity (32, 40). A linkage analysis in French families with morbid obesity showed linkage between ISL1 and BMI and leptin (41). Variants in ISL1 have previously been associated with congenital heart defects (42), but to our knowledge, no mutations in ISL1 have been reported in monogenic obesity. The ISL1 missense variant found in our study segregated with the obesity phenotype in the family and was predicted disease causing. The role of ISL1 in the development of obesity and the functional relevance of the variant require further studies.
Low-density lipoprotein receptor 2 gene (LRP2), encodes an endocytic receptor, LRP2 or megalin. LRP2 has many ligands in various tissues. LRP2 increases leptin-induced STAT3 activation in POMC-expressing neurons in hypothalamus and inhibits food intake. STAT3 signaling is reduced in the absence of functional LRP2 (22). LRP2 has been associated with monogenic obesity (1, 22). Here we identified two patients with rare heterozygous variants in LRP2; one novel missense variant (p.R2479I), and another rare missense variant (p.N3315S), neither one has previously been found in the Finnish population. Both LRP2 variants were predicted disease-causing. More studies are required to explore the functional significance of these LRP2 variants.
GRPR, encoding gastrin-releasing peptide receptor, is an X-linked 7-transmembrane G-protein coupled receptor that activates the phospholipase C signaling pathway. GRPR is expressed in several tissues, including the brain and is involved in the regulation of satiety. A previous study identified in five patients with obesity heterozygous or hemizygous mutations in GRPR that were predicted functionally relevant (5). The hemizygous GRPR variant (p.L87M) identified in our study was also detected in a boy with severe obesity by Serra-Juhe et al. (5). In their co-expression analysis GRPR showed clear enrichment of co-expression with genes from the melanocortin pathway (5). Our patient presented with early-onset obesity, insulin resistance, fatty liver, hypothyroidism, and severe depression. Interestingly, GRPR has also been implicated in psychiatric disorders like depression (43), suggesting that the hemizygous GRPR variant contributes to the severe depression seen in our patient. In addition, our patient also harbors a rare variant (p.N3315S) in the LRP2 gene and it therefore remains uncertain how each of the two variants contribute to the phenotype.
Next generation sequencing is an important tool in identifying genetic causes in monogenic obesity. Targeted exome sequencing provides several advantages compared to whole-exome sequencing; it is cost-effective, has high quality with high read depth and coverage, data interpretation is easier as the number of genes is limited, and chance of incidental findings is minimal. Its weakness is the limited number of genes included in panel. Because of this methodological limitation we may have missed variants in novel genes that were not included in the gene panel but still contribute to hypothalamic function. Another limitation in our study is the lack of available family members for genetic testing in some instances and therefore interpretation of the pathogenicity of the identified variants was more challenging. Further, our study did not include any functional evaluations of the genetic findings and therefore the contribution of some of the variants to the obesity development remains uncertain. However, we only considered rare exonic variants with allele frequency <0.5% and variants predicted to be pathogenic by several in silico prediction tools, including a high CADD score as likely pathogenic.
In conclusion, we report rare pathogenic/likely pathogenic variants in genes involved in hypothalamic circuit in 7/92 (8%) of patients with early-onset obesity. Our results support the role of hypothalamic melanocortin pathway in the development of obesity. Further studies are required to explore the clinical relevance of the rare genetic variants. Novel therapies targeting specific genetic defects are under development and may offer future treatment modalities in patients with monogenic obesity.
Data Availability Statement
Data cannot be shared publicly because the data consists of sensitive patient data. More specifically the data consists of individual clinical data and individual genotypes for young children. Data are available from the Helsinki University Hospital's Institutional Data Access/Ethics Committee for researchers who meet the criteria for access to confidential data. Data availability contacts: Outi Mäkitie, b3V0aS5tYWtpdGllJiN4MDAwNDA7aGVsc2lua2kuZmk=.
Author Contributions
PL, TM, HV, ML-N, MP, and OM: study design. PL, TM, HV, PT, and ML-N: data collection. PL, AK, MP, and OM: data analysis and data interpretation. PL, RM, AK, MP, and OM: drafting of manuscript. All authors contributed to manuscript revision and approved the final version.
Funding
This study was financially supported by the Academy of Finland; Sigrid Jusélius Foundation; Foundation for Pediatric Research; Folkhälsan Research Foundation; Päivikki ja Sakari Sohlberg Foundation; Stiftelsen Dorothea Olivia, Karl Walter och Jarl Walter Perkléns minne; Emil Aaltonen Foundation; Swedish Research Council; Novo Nordisk Foundation; University of Helsinki through the Doctoral Program in Clinical Research; Helsinki University Hospital research funds.
Conflict of Interest
OM declares consultancy to Kyowa Kirin, Alexion, and Sandoz.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the patients and their family members for participating in the study. We thank RN Päivi Turunen for help with data collection.
References
1. da Fonseca ACP, Mastronardi C, Johar A, Arcos-Burgos M, Paz-Filho G. Genetics of non-syndromic childhood obesity and the use of high-throughput DNA sequencing technologies. J Diabetes Complicat. (2017) 31:1549–61. doi: 10.1016/j.jdiacomp.2017.04.026
2. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. (2015) 518:197–206. doi: 10.1038/nature14177
3. Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet. (2018) 27:3641–9. doi: 10.1093/hmg/ddy271
4. Pettersson M, Viljakainen H, Loid P, Mustila T, Pekkinen M, Armenio M, et al. Copy number variants are enriched in individuals with early-onset obesity and highlight novel pathogenic pathways. J Clin Endocrinol Metab. (2017) 102:3029–39. doi: 10.1210/jc.2017-00565
5. Serra-Juhe C, Martos-Moreno GA, Bou de Pieri F, Flores R, Gonzalez JR, Rodriguez-Santiago B, et al. Novel genes involved in severe early-onset obesity revealed by rare copy number and sequence variants. PLoS Genet. (2017) 13:e1006657. doi: 10.1371/journal.pgen.1006657
6. Wheeler E, Huang N, Bochukova EG, Keogh JM, Lindsay S, Garg S, et al. Genome-wide SNP and CNV analysis identifies common and low-frequency variants associated with severe early-onset obesity. Nat Genet. (2013) 45:513–7. doi: 10.1038/ng.2607
7. Serra-Juhe C, Martos-Moreno GA, Bou de Pieri F, Flores R, Chowen JA, Perez-Jurado LA, et al. Heterozygous rare genetic variants in non-syndromic early-onset obesity. Int J Obes. (2019). doi: 10.1038/s41366-019-0357-5. [Epub ahead of print].
8. Hendricks AE, Bochukova EG, Marenne G, Keogh JM, Atanassova N, Bounds R, et al. Rare variant analysis of human and rodent obesity genes in individuals with severe childhood obesity. Sci Rep. (2017) 7:4394. doi: 10.1038/s41598-017-03054-8
9. Collet TH, Dubern B, Mokrosinski J, Connors H, Keogh JM, Mendes de Oliveira E, et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (setmelanotide) in MC4R deficiency. Mol Metab. (2017) 6:1321–9. doi: 10.1016/j.molmet.2017.06.015
10. Valli-Jaakola K, Lipsanen-Nyman M, Oksanen L, Hollenberg AN, Kontula K, Bjorbaek C, et al. Identification and characterization of melanocortin-4 receptor gene mutations in morbidly obese finnish children and adults. J Clin Endocrinol Metab. (2004) 89:940–5. doi: 10.1210/jc.2003-031182
11. Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. (2003) 348:1085–95. doi: 10.1056/NEJMoa022050
12. Kleinendorst L, Massink MPG, Cooiman MI, Savas M, van der Baan-Slootweg OH, Roelants RJ, et al. Genetic obesity: next-generation sequencing results of 1230 patients with obesity. J Med Genet. (2018) 55:578–86. doi: 10.1136/jmedgenet-2018-105315
13. Akinci A, Turkkahraman D, Tekedereli I, Ozer L, Evren B, Sahin I, et al. Novel mutations in obesity-related genes in turkish children with non-syndromic early onset severe obesity: a multicentre study. J Clin Res Pediatr Endocrinol. (2019) 11:341–9. doi: 10.4274/jcrpe.galenos.2019.2019.0021
14. De Rosa MC, Chesi A, McCormack S, Zhou J, Weaver B, McDonald M, et al. Characterization of rare variants in MC4R in African American and latino children with severe early-onset obesity. J Clin Endocrinol Metab. (2019) 104:2961–70. doi: 10.1210/jc.2018-02657
15. Martinelli CE, Keogh JM, Greenfield JR, Henning E, van der Klaauw AA, Blackwood A, et al. Obesity due to melanocortin 4 receptor (MC4R) deficiency is associated with increased linear growth and final height, fasting hyperinsulinemia, and incompletely suppressed growth hormone secretion. J Clin Endocrinol Metab. (2011) 96:E181–8. doi: 10.1210/jc.2010-1369
16. Kuhnen P, Krude H, Biebermann H. Melanocortin-4 receptor signalling: importance for weight regulation and obesity treatment. Trends Mol Med. (2019) 25:136–48. doi: 10.1016/j.molmed.2018.12.002
17. He S, Tao YX. Defect in MAPK signaling as a cause for monogenic obesity caused by inactivating mutations in the melanocortin-4 receptor gene. Int J Biol Sci. (2014) 10:1128–37. doi: 10.7150/ijbs.10359
18. Grarup N, Moltke I, Andersen MK, Dalby M, Vitting-Seerup K, Kern T, et al. Loss-of-function variants in ADCY3 increase risk of obesity and type 2 diabetes. Nat Genet. (2018) 50:172–4. doi: 10.1038/s41588-017-0022-7
19. Blanchet P, Bebin M, Bruet S, Cooper GM, Thompson ML, Duban-Bedu B, et al. MYT1L mutations cause intellectual disability and variable obesity by dysregulating gene expression and development of the neuroendocrine hypothalamus. PLoS Genet. (2017) 13:e1006957. doi: 10.1371/journal.pgen.1006957
20. Loid P, Makitie R, Costantini A, Viljakainen H, Pekkinen M, Makitie O. A novel MYT1L mutation in a patient with severe early-onset obesity and intellectual disability. Am J Med Genet A. (2018) 176:1972–5. doi: 10.1002/ajmg.a.40370
21. Kasher PR, Schertz KE, Thomas M, Jackson A, Annunziata S, Ballesta-Martinez MJ, et al. Small 6q16.1 deletions encompassing POU3F2 cause susceptibility to obesity and variable developmental delay with intellectual disability. Am J Hum Genet. (2016) 98:363–72. doi: 10.1016/j.ajhg.2015.12.014
22. Paz-Filho G, Boguszewski MC, Mastronardi CA, Patel HR, Johar AS, Chuah A, et al. Whole exome sequencing of extreme morbid obesity patients: translational implications for obesity and related disorders. Genes. (2014) 5:709–25. doi: 10.3390/genes5030709
23. Kuhnen P, Clement K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med. (2016) 375:240–6. doi: 10.1056/NEJMoa1512693
24. Clement K, Biebermann H, Farooqi IS, Van der Ploeg L, Wolters B, Poitou C, et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat Med. (2018) 24:551–5. doi: 10.1038/s41591-018-0015-9
25. Saari A, Sankilampi U, Hannila ML, Kiviniemi V, Kesseli K, Dunkel L. New finnish growth references for children and adolescents aged 0 to 20 years: length/height-for-age, weight-for-length/height, and body mass index-for-age. Ann Med. (2011) 43:235–48. doi: 10.3109/07853890.2010.515603
26. Alsters SI, Goldstone AP, Buxton JL, Zekavati A, Sosinsky A, Yiorkas AM, et al. Truncating Homozygous mutation of carboxypeptidase E (CPE) in a morbidly obese female with type 2 diabetes mellitus, intellectual disability and hypogonadotrophic hypogonadism. PLoS ONE. (2015) 10:e0131417. doi: 10.1371/journal.pone.0131417
27. Ramachandrappa S, Raimondo A, Cali AM, Keogh JM, Henning E, Saeed S, et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J Clin Invest. (2013) 123:3042–50. doi: 10.1172/JCI68016
28. Pelling M, Anthwal N, McNay D, Gradwohl G, Leiter AB, Guillemot F, et al. Differential requirements for neurogenin 3 in the development of POMC and NPY neurons in the hypothalamus. Dev Biol. (2011) 349:406–16. doi: 10.1016/j.ydbio.2010.11.007
29. Anthwal N, Pelling M, Claxton S, Mellitzer G, Collin C, Kessaris N, et al. Conditional deletion of neurogenin-3 using Nkx2.1iCre results in a mouse model for the central control of feeding, activity and obesity. Dis Model Mech. (2013) 6:1133–45. doi: 10.1242/dmm.011916
30. Moir L, Bochukova EG, Dumbell R, Banks G, Bains RS, Nolan PM, et al. Disruption of the homeodomain transcription factor orthopedia homeobox (Otp) is associated with obesity and anxiety. Mol Metab. (2017) 6:1419–28. doi: 10.1016/j.molmet.2017.08.006
31. Camerino C. Low sympathetic tone and obese phenotype in oxytocin-deficient mice. Obesity. (2009) 17:980–4. doi: 10.1038/oby.2009.12
32. Nasif S, de Souza FS, Gonzalez LE, Yamashita M, Orquera DP, Low MJ, et al. Islet 1 specifies the identity of hypothalamic melanocortin neurons and is critical for normal food intake and adiposity in adulthood. Proc Natl Acad Sci USA. (2015) 112:E1861–70. doi: 10.1073/pnas.1500672112
33. Saeed S, Bonnefond A, Tamanini F, Mirza MU, Manzoor J, Janjua QM, et al. Loss-of-function mutations in ADCY3 cause monogenic severe obesity. Nat Genet. (2018) 50:175–9. doi: 10.1038/s41588-017-0023-6
34. Desvignes JP, Bartoli M, Delague V, Krahn M, Miltgen M, Beroud C, et al. VarAFT: a variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. (2018) 46:W545-w53. doi: 10.1093/nar/gky471
35. Norio R. Finnish disease heritage I: characteristics, causes, background. Hum Genet. (2003) 112:441–56. doi: 10.1007/s00439-002-0875-3
36. Tao YX, Segaloff DL. Functional analyses of melanocortin-4 receptor mutations identified from patients with binge eating disorder and nonobese or obese subjects. J Clin Endocrinol Metab. (2005) 90:5632–8. doi: 10.1210/jc.2005-0519
37. Tong T, Shen Y, Lee HW, Yu R, Park T. Adenylyl cyclase 3 haploinsufficiency confers susceptibility to diet-induced obesity and insulin resistance in mice. Sci Rep. (2016) 6:34179. doi: 10.1038/srep34179
38. Liang Y, Li Z, Liang S, Li Y, Yang L, Lu M, et al. Hepatic adenylate cyclase 3 is upregulated by liraglutide and subsequently plays a protective role in insulin resistance and obesity. Nutr Diabetes. (2016) 6:e191. doi: 10.1038/nutd.2015.37
39. Al Tuwaijri A, Alfadhel M. MYT1L mutation in a patient causes intellectual disability and early onset of obesity: a case report and review of the literature. J Pediatr Endocrinol Metab. (2019) 32:409–13. doi: 10.1515/jpem-2018-0505
40. Alvarez-Bolado G. Development of neuroendocrine neurons in the mammalian hypothalamus. Cell Tissue Res. (2019) 375:23–39. doi: 10.1007/s00441-018-2859-1
41. Clement K, Dina C, Basdevant A, Chastang N, Pelloux V, Lahlou N, et al. A sib-pair analysis study of 15 candidate genes in French families with morbid obesity: indication for linkage with islet 1 locus on chromosome 5q. Diabetes. (1999) 48:398–402. doi: 10.2337/diabetes.48.2.398
42. Ma L, Wang J, Li L, Qiao Q, Di RM, Li XM, et al. ISL1 loss-of-function mutation contributes to congenital heart defects. Heart Vessels. (2019) 34:658–68. doi: 10.1007/s00380-018-1289-z
Keywords: childhood obesity, hypothalamus, appetite regulation, hyperphagia, MC4R
Citation: Loid P, Mustila T, Mäkitie RE, Viljakainen H, Kämpe A, Tossavainen P, Lipsanen-Nyman M, Pekkinen M and Mäkitie O (2020) Rare Variants in Genes Linked to Appetite Control and Hypothalamic Development in Early-Onset Severe Obesity. Front. Endocrinol. 11:81. doi: 10.3389/fendo.2020.00081
Received: 16 November 2019; Accepted: 07 February 2020;
Published: 21 February 2020.
Edited by:
Paolo Marzullo, Università degli Studi del Piemonte Orientale, ItalyReviewed by:
Vibha Singhal, Massachusetts General Hospital and Harvard Medical School, United StatesBeatrice Dubern, Hôpital Armand Trousseau, France
Copyright © 2020 Loid, Mustila, Mäkitie, Viljakainen, Kämpe, Tossavainen, Lipsanen-Nyman, Pekkinen and Mäkitie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petra Loid, cGV0cmEubG9pZCYjeDAwMDQwO2hlbHNpbmtpLmZp