 Hsuan Lung
Hsuan Lung Edward C. Hsiao
Edward C. Hsiao Kelly L. Wentworth
Kelly L. Wentworth- 1Division of Endocrinology and Metabolism and the Institute for Human Genetics, Department of Medicine, University of California, San Francisco, San Francisco, CA, United States
- 2Oral and Craniofacial Sciences Graduate Program, School of Dentistry, University of California, San Francisco, San Francisco, CA, United States
- 3Department of Dentistry, Chang Gung Memorial Hospital and College of Medicine, Chang Gung University, Kaohsiung, Taiwan
- 4Division of Endocrinology and Metabolism, Zuckerberg San Francisco General Hospital, University of California, San Francisco, San Francisco, CA, United States
The Gs G-protein coupled receptor pathway is a critical regulator of normal bone formation and function. The Gs pathway increases intracellular cAMP levels by ultimately acting on adenylate cyclase. McCune-Albright Syndrome (MAS) and fibrous dysplasia (FD) of the bone are two proto-typical conditions that result from increased cellular Gs signaling activity. Both are caused by somatic activating mutations in the GNAS gene that encodes for the Gsα subunit. FD bone lesions are particularly difficult to treat because of their variability and because of the lack of effective medical therapies. In this review, we briefly discuss the key clinical presentations of FD/MAS. We also review the current status of mouse models that target the Gs GPCR signaling pathway and human cellular models for FD/MAS. These powerful tools and our improving clinical knowledge will allow further elucidation of the roles of GPCR signaling in FD/MS pathogenesis, and facilitate the development of novel therapies for these medically significant conditions.
Introduction
Musculoskeletal disorders such as skeletal dysplasias are a significant health problem affecting both children and adults. A variety of cellular pathways, including G-protein coupled receptors (GPCRs) and their ligands, have been identified as key regulators of osteoblast formation and function—two critical steps in normal bone formation and homeostasis. The human GPCR family includes over 340 non-olfactory and 400 olfactory receptors (1, 2), making it the largest class of receptors in the human genome. GPCRs mediate a wide variety of biological processes and are activated by multiple types of extracellular signals, ranging from photons and ions to small molecules to peptides. The diversity of GPCRs and their responses to small molecules has made them major targets for over 40% of modern pharmaceuticals (3).
GPCRs signal through a select number of canonical pathways (4): among these, the Gs and Gi pathways increase or decrease intracellular cAMP levels, respectively, by acting on adenylate cyclase, while the Gq pathway increases intracellular calcium by activating phospholipase C.
McCune-Albright Syndrome (MAS) is a proto-typical disease caused by activating mutations in the GNAS locus, encoding the Gsα protein (5). MAS is a mosaic genetic disease characterized by the classic triad of polyostotic fibrous dysplasia (FD) of the bone, café-au-lait skin hyperpigmentation, and peripheral precocious puberty. Patients with MAS may have other endocrinopathies, including Cushing's syndrome, hyperthyroidism, acromegaly, and solid organ malignancies of the breast, thyroid, and pancreas. FD/MAS is caused by an acquired somatic mutation in GNAS, the gene that encodes the alpha subunit of the stimulatory G-protein (Gsα), leading to constitutive activation of Gs signaling in affected cells. This mutation occurs post-zygotically, resulting in tissue mosaicism, and is not inherited through the germline. As a result of this mosaicism, the clinical disease spectrum of FD ranges from single bone involvement to multi-organ involvement. The most common cause is a missense mutation at either position c.602G>A (p.R201H) or c.601C>T (p.R201C). This mutation results in an amino acid substitution in the GTP hydrolase domain of the Gsα protein, inhibiting the intrinsic GTPase activity, and leading to persistently elevated intracellular cAMP levels (Figure 1).
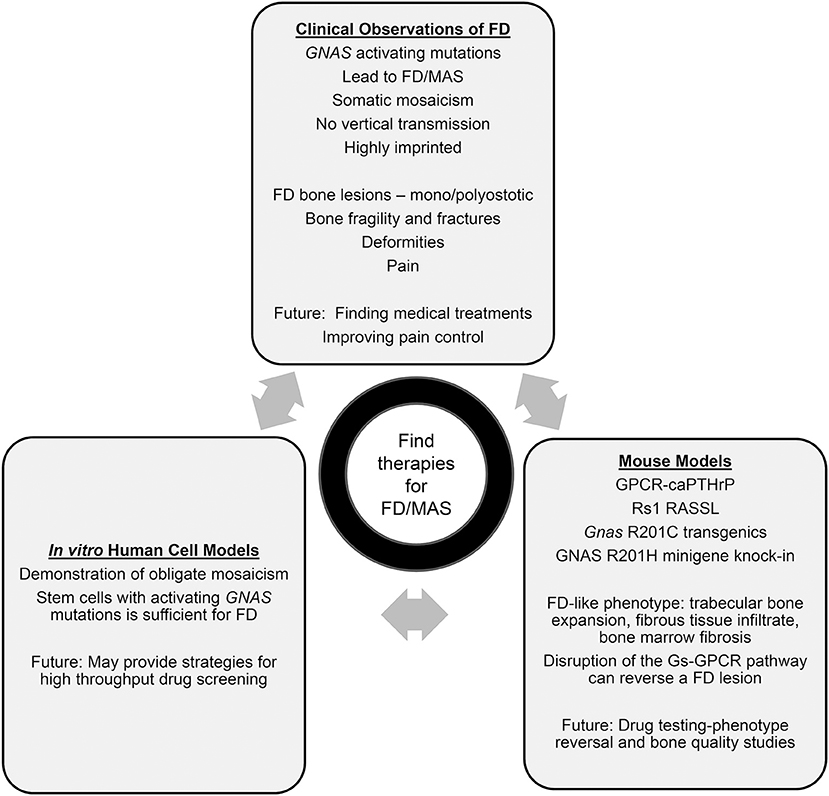
Figure 1. Summary of human, mouse, and clinical findings in FD.
Clinical Presentation of Fibrous Dysplasia
A major clinical feature of MAS is FD of the bone, where expansile bone lesions cause fragility, malformations, and pain (Figure 2). FD also occurs without MAS, and is a common congenital skeletal dysplasia that can affect one bone (monoostotic) or multiple bones (polyostotic) (5). FD is arguably the most significant medical complication of MAS, since no effective treatments are available to manage the bone complications. In addition, the broad clinical spectrum of FD/MAS and the mosaic nature of the disease leads to variability in the radiographic presentation, making FD challenging to accurately diagnose.
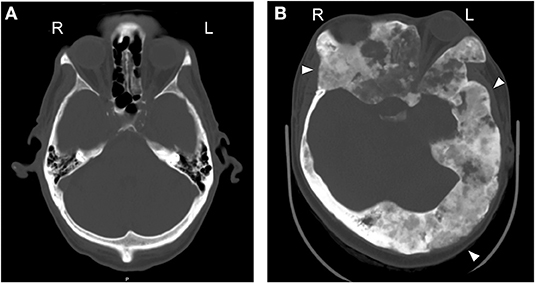
Figure 2. Fibrous dysplasia commonly affects the skull and is mosaic. Axial CT across the orbit. (A) A normal CT scan of the craniofacial bones, from a skeletally normal 61 y.o. male. (B) A 29 y.o. female with craniofacial FD. Note the asymmetry in the skull [ground glass expanded lesions (arrowheads) and displaced left globe].
In 2019, the FD/MAS International Consortium put forth a position statement to help guide clinicians in the diagnosis and management of FD/MAS (6). The first step in diagnosing FD/MAS is to perform a complete skeletal and extra-skeletal evaluation to determine the extent of the disease. MAS can be diagnosed if a patient has FD and at least one extraskeletal manifestation (café-au-lait skin hyperpigmentation, peripheral precocious puberty, thyroid lesions consistent with FD/MAS, growth hormone excess, neonatal hypercortisolism) or the absence of skeletal involvement but 2 extra-skeletal manifestations (6). An accurate diagnosis of FD/MAS can usually be made after a complete physical examination, combined with biochemical, hormonal, and radiologic evaluation of the skeletal, dermatologic, and endocrine systems. Biopsy of FD lesions is needed only if there is uncertainty about the diagnosis (i.e., atypical radiologic features) or concern for underlying malignancy (6). In these situations, affected tissue can be tested for the presence of a GNAS activating mutation, with the understanding that false negatives may occur if the sample has a low mutational burden. Peripheral blood is usually not sufficient for diagnosis due to the mosaicism of the disease. Next-generation sequencing is associated with a lower false-positive rate (6).
Treatment and Monitoring of FD/MAS
Comprehensive guidelines regarding the management of the skeletal and extra-skeletal manifestations of FD/MAS were recently published, and should be considered when caring for patients along this clinical disease spectrum (6). The mainstay of therapy in FD/MAS remains adequate pain control, optimization of phosphate and vitamin D status, treatment of IGF-1 excess if present, and judicious consideration of surgical resection of FD lesions once they have stabilized. Unfortunately, there are no effective medical treatments available for FD/MAS. Bisphosphonate therapy in IV formulation has been reported to provide some benefits for pain control in patients with persistent moderate-to-severe pain from FD lesions, but why this helps in only some patients remains unclear (7–9). In addition, there is no evidence to suggest that bisphosphonates decrease the progression of FD lesions, and may not adequately control pain in some patients (7, 10).
Presently, there is minimal evidence for the use of denosumab and other anti-resorptive agents in FD, although there are case reports suggesting potential clinical benefits (8, 11–16). However, there are major concerns about rebound fractures and FD lesion progression after drug cessation (17–19). Ongoing clinical trials to address the utility of denosumab in FD are underway (NCT03571191). In addition, the TOCIDYS trial is evaluating the efficacy of IL-6 inhibition in patients with FD who did not have improvement in pain with prior bisphosphonate treatment (NCT01791842). These exciting trials hold promise for identifying potential medical strategies for mitigating the complications from FD.
Mouse Models for Understanding FD
One contributor to the dearth of effective treatments for FD/MAS is the complexity of the GNAS locus. This complexity has made it challenging to develop robust mouse and human models to dissect the mechanisms of FD/MAS. During the past several years, novel strategies for uncovering the roles of Gs-GPCR signaling in bone have been developed. These models provide critical insights into the pathogenesis and potential therapeutic approaches for FD.
One of the earliest models utilized the PTH/PTH-related protein (PTH/PTHrP) receptor (PPR), a GPCR, to study Jansen's metaphyseal chondrodysplasia (JMC). JMC is a rare form of short-limbed dwarfism caused by activating mutations of the PPR, leading to constitutive receptor activation and ligand-independent intracellular cAMP accumulation. Calvi et al. generated a mouse (Col1-caPPR) that expressed the human mutant PPR HKrk-H223R (caPPR), one of the causative mutations associated with JMC, in osteoblastic lineage cells in mice using a Col1 (2.3 kb) promoter (20). At 1 week of age, these mice showed increased osteoblast number and function in both the trabecular bone and the endosteal surface of cortical bone in the long bones. However, periosteal osteoblast activity was inhibited. This resulted in an increase in trabecular bone volume and a decrease in cortical bone thickness in the long bones. Calvarial thickness remained unchanged but there was increased porosity and bone remodeling on the endosteal surface of the skull. There was also an increased number of mature osteoclasts in these mice, which led to increased porosity of the cortical bone. At 2 weeks of age, excess bone formed in the bone marrow space (21). The area between the trabeculae was occupied by fibrous cells, blood vessels, and osteoclasts. There was delayed formation of bone marrow cavities, adipocytes, and hematopoietic cells. Surprisingly, these dysplastic bone and fibrous tissue phenotypes gradually resolved over time, and were limited to the metaphyseal area at 4 months. These studies showed that when the constitutively active PPR was expressed in osteoblastic lineage cells, the receptor could mediate both the anabolic and resorptive effects of PTH, and that PPR is involved in the regulation of both bone marrow and stromal tissues.
Our group used a different approach, creating an engineered GPCR RASSL (receptor activated solely by a synthetic ligand) to regulate GPCR signaling (22). The Gs-coupled receptor, Rs1, was created by inserting a D100A mutation into the wild-type human 5HT4 serotonin receptor (23). This engineered receptor has a high basal level of constitutively active Gsα activity and is not responsive to the endogenous serotonin ligand. The Col1(2.3)/Rs1 mouse model limits Rs1 expression spatially to osteoblastic lineage cells by using a Col1a1(2.3 kb) promotor, and temporally using the tet-off system, which allows for controlled expression of Rs1 in the absence of doxycycline. The FD-like phenotype of mice born off doxycycline was first apparent at 6 days (24). These mice showed age-dependent, increased trabecular bone formation with loss of marrow space and thinned cortical bone. The histologic and radiographic features strongly resemble human FD of the bone. There was also a dramatic increase in the number of immature osteoblasts present in the FD lesions, deposition of immature bone tissue, and reduced mineralization seen in analyses by FTIR spectroscopy and synchrotron radiation micro-computed tomography (25). The mice also showed lower numbers of hematopoietic stem cells (26). Col1(2.3)/Rs1 mouse bones showed dramatically reduced mature adipocyte differentiation, and higher osteoblastic glucose utilization than control mice (27). RNA analysis on whole bone samples showed increased Wnt signaling, suggesting that Wnt proteins may be a major driver of this effect (27). Importantly, blocking Gs signaling by using doxycycline at 4 weeks gradually reversed the bone phenotype (24), providing a proof-of-concept that therapies inhibiting Gs signaling may be effective for reversing fibrous dysplastic bone lesions. This model provides a powerful tool for understanding the effects of Gs-GPCR signaling on dynamic bone growth and remodeling.
In contrast to strategies that did not directly manipulate GNAS, Saggio et al. generated a transgenic mouse model of human FD that constitutively expressed GNAS-R201C (28). The transgene expression did not show any appreciable effect on embryonic skeletal formation and unlike human FD, could be vertically transmitted. FD lesions were detected radiographically at 6 months of age. The lesions first appeared in the tail and later progressed to the femurs and the skull. The onset and progression of lesions were defined in three stages: an early phase (<2 months), defined by abnormal trabecular bone formation, endosteal thickening, and ectopic cortical lesions; an intermediate phase (2–6 months), with narrowed marrow cavity, expanded cortical bone and increased osteoclastic activity; and a late phase (>10 months) described as the fibrous dysplastic phase, with abnormal bone trabeculae and matrix fibrous tissue. At 1 year of age, the skeletal features resembled human FD.
When GNAS-R201C expression was limited to maturing osteoblasts using the Col1a1 (2.3 kb) promoter, changes in bone structure were detected on radiograph at 3 weeks in the tail, with excess bone mass (29). At 3 months, increased bone density was noted in all bones. The transgenic mice showed excess bone formation and remodeling of the bone marrow space. However, the expression in Col1a1(2.3 kb) cells failed to reproduce other features of human FD, such as bone marrow space fibrosis, and the loss of adipocytes and hematopoietic cells seen in other models (23, 28).
Palmisano et al. tested the effect of RANKL (receptor activator of nuclear factor kappa-B ligand) inhibition by treating the mice with an anti-RANKL antibody (30). They found that new and highly mineralized bone replaced the FD-like lesions in 2-month old mice, and the bone density increased. The treatment also stopped the growth of pre-existing FD lesions. In addition, there was a higher maximum load and stiffness of the bones in the treated group. However, the FD-like lesions progressed after RANKL inhibitor cessation.
More recently, Zhao et al. developed a mouse model expressing GNAS-R201C in the skeletal stem cell lineage using a tetracycline-inducible Cre-mediated Prrx1 driver (31). The FD bone phenotype was observed in embryos and adult mice <2 weeks after doxycycline administration (to activate the transgene) at E4.5. The FD lesions in the long bones showed reduced endochondral ossification, and in craniofacial bones showed decreased mineralization in the calvarial sutures. Poorly mineralized trabeculae and a dense fibrous matrix were present, and the bone marrow spaces were also decreased. When doxycycline was administered to 3-week old mice for 2 weeks (to induce expression), all limbs developed FD-like lesions with expansile bone deformities, fractures in limb bones, and defects in calvarial sutures. Doxycycline withdrawal resulted in reversal of the bone lesions. Hematopoietic cells and adipocytes appeared in the bone marrow of former FD lesions. The skulls of the reversed GNAS-R201C mice also showed normal morphology with healed sutures.
Finally, Khan et al. generated a new conditional knock-in mouse model [GNASf(R201H)] using a minigene cassette to express GNAS-R201H, a human FD mutation, at the endogenous mouse GNAS locus (32). They found that expression of GNAS-R201H in the germ-line using Sox2-Cre resulted in embryonic lethality. Using Prrx1-Cre, they limited the expression of GNAS-R201H to osteochondral progenitor cells and early limb bud mesenchyme cells. At P0, control mice had well-formed cortical bone, cartilaginous epiphyses, and a normal marrow cavity. In contrast, Prrx1-Cre; GNASf(R201H)/+ showed severely deficient long bone formation. At P6, Prrx1-Cre; GNASf(R201H)/+ mice showed bone formation, but the marrow space was replaced by fibrous tissue and trabecular bone. In mutant long bones, higher levels of Wnt/β-catenin were also noted. At 3 weeks of age, there was more FD-like bone formation in the Prrx1-Cre; GNASf(R201H)/+ mice; expansion of woven trabecular bone; lack of cortical bone; and increased osteoclasts. Furthermore, in this mouse model, Xu et al. demonstrated that Gsα signaling mediated intramembranous ossification of cranial bones by regulating both Hh and Wnt/β-catenin signaling (33).
Together, these mouse models demonstrate that FD bone lesions can develop from activating mutations at the Gs-GPCR level as well as from transgenic expression or knock-in expression of a GNAS allele carrying the R201C or R201H activating mutation. In addition, data from several mouse models suggest that blocking Gs pathway hyperactivity can lead to reversal of the FD bone lesions and may be a viable treatment strategy for human FD.
Human Cellular Models of Gs−Signaling
Human cellular models have contributed significantly to our understanding of FD/MAS pathophysiology, and patient-derived tissue samples have been an invaluable source. In 1998, Bianco et al. performed one of the earliest studies using FD/MAS patient bone marrow samples and showed that bone marrow stromal cell (BMSC) progenitors isolated from fibrous dysplastic lesions were either heterozygous for the Gsα activating mutant allele, or homozygous wildtype (34). This discovery demonstrated that the skeletal progenitor cell population in FD lesions is mosaic, with a resident population of bone marrow stromal cells harboring the mutation and another population that is unaffected (34). When these cells were transplanted into immunocompromised mice, the wildtype colonies developed normal ossicles, but the colonies containing only the GNAS mutant allele did not survive and did not form ossicles. However, when a mixture of wildtype and mutant cell colonies was transplanted, an abnormal ossicle formed, with histopathologic features resembling FD. These experiments provided strong evidence that both wildtype and mutant cells were necessary to form a FD lesion, and that the mosaicism inherent to FD/MAS can be recapitulated within an FD lesion (34).
This novel concept of somatic mosaicism within an FD lesion was explored further by Kuznetsov et al. (35). In their study, they isolated colony forming unit-fibroblasts (CFU-Fs) from FD lesions of patients with FD/MAS and calculated the frequency of mutation-bearing CFU-Fs vs. normal CFU-Fs. They noted an inverse relationship between patient age and the number of mutated skeletal stem cell colonies present within an FD lesion, and concluded that the number of mutated stem cells must undergo apoptosis as patients age. Similarly, the bone histology in older patients (32–52 years of age) with FD/MAS was less severe than that of younger patients, and was more likely to be associated with a lower GNAS mutational burden. They hypothesized that as skeletal stem cells aged, there may be preferential apoptosis of mutated cells, resulting in loss of these cells and self-renewal of non-mutated cell populations. This could account for the clinically observed decreased incidence of new FD lesions and the relative stability of existing FD lesions as patients age. Kuznetsov et al. also transplanted cell colonies containing CFU-Fs into immunocompromised mice and showed that FD ossicles formed, whereas non-mutant CFU-Fs or mutation-positive strains without mutant skeletal stem cell populations did not form FD ossicles. Thus, they concluded that GNAS mutations within the skeletal stem cell population was sufficient to induce formation of FD lesions (35).
Piersanti et al. developed a model in which human skeletal progenitor cells were engineered to stably over-express the GNAS-R201C mutation using a lentiviral vector (36). These cells demonstrated elevated cAMP production, consistent with over-expression of Gsα. When cultured in vitro, they did not exhibit mineralization and had lower osteocalcin levels than controls. Levels of the osteogenic markers alkaline phosphatase and bone sialoprotein were elevated compared to controls, and the cells exhibited robust RANKL expression, consistent with the profound osteoclastogenesis seen in most human FD lesions. Additionally, genes in the phosphodiesterase pathway were upregulated in these cells, suggesting an adaptive response to Gsα over-expression. When these cells were transplanted into immunocompromised mice, the stably-transduced GNAS-R201C cells formed ossicles but were unable to differentiate into adipocytes or hematopoietic components. Finally, silencing of the GNAS-R201C allele with lentiviral vectors containing short hairpin interfering RNA sequences caused these cells to revert to their normal state and no longer exhibit a mutant phenotype.
In 2019, de Castro et al. used human FD-derived BMSCs from a well-characterized cohort of FD patients at the NIH to show that RANKL expression in FD skeletal lesions may directly contribute to osteoclast induction in FD lesions (37). They showed that serum levels of RANKL were 16-fold higher in FD patients compared with healthy controls, and the serum RANKL/OPG ratio was 12-fold higher. The magnitude of increase in RANKL and RANKL/OPG was positively correlated with total body skeletal disease burden score, a well-validated scoring system used to determine the severity of FD.
de Castro et al. also isolated BMSCs from these FD patients and healthy volunteers and showed that RANKL levels were higher in the conditioned media of FD BMSCs compared with BMSCs derived from healthy volunteers when stimulated with prostaglandin E2 (PGE2) and 1,25 vitamin D3 (37). These cells also released OPG, but to a much lower degree than healthy volunteer controls. Additionally, when FD BMSCs were co-cultured in osteogenic media with peripheral monocytes from healthy volunteers, the monocytes differentiated into TRAP+ osteoclasts; however, when cultured in non-osteogenic media, they did not induce osteoclastogenesis. They subsequently showed that osteoclastogenesis could be inhibited when the cell co-cultures were treated with denosumab. This study provided strong evidence that RANKL is expressed in human FD lesions and is correlated with disease burden, thus implicating RANKL over-expression as an important contributor to FD pathogenesis and informing the design of ongoing studies testing denosumab in FD (NCT03571191).
Conclusions
The past 15 years have shown major advances in our understanding of FD/MAS. There are striking consistencies among the mouse models of FD, and a number of features of the human disease are replicated in the mouse genetic models, suggesting that direct targeting of the Gs pathway has strong potential as a therapeutic strategy for FD. In addition, the human cell models of FD are providing new tools for understanding FD pathogenesis and cell-type specific effects of the GNAS activating mutations. Finally, international collaborations among clinicians and researchers with strong experience in FD/MAS are yielding best-practice recommendations and treatment guidelines for optimal management of FD/MAS.
Author Contributions
EH, KW, and HL conceived of this manuscript, wrote the manuscript, and edited the manuscript.
Funding
This work was supported by the University of California Department of Medicine (to EH), a Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine Doctoral Training Fellowship (to HL), and National Institutes of Health Career Development Grant K08 DE028946 (to KW).
Conflict of Interest
EH and KW receive clinical trial research support through their institution from Clementia Pharmaceuticals for clinical trials unrelated to this work. EH is also a volunteer on the editorial board for the Journal of Bone and Mineral Research. EH serves in an unpaid capacity on the International FOP Association Medical Registry Advisory Board, on the International Clinical Council on FOP, and on the Fibrous Dysplasia Foundation Medical Advisory Board. EH has also received clinical trial research support through his institution from Regeneron Pharmaceuticals.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. (2003) 63:1256–72. doi: 10.1124/mol.63.6.1256
2. Karchin R, Karplus K, Haussler D. Classifying G-protein coupled receptors with support vector machines. Bioinformatics. (2002) 18:147–59. doi: 10.1093/bioinformatics/18.1.147
3. Brink CB, Harvey BH, Bodenstein J, Venter DP, Oliver DW. Recent advances in drug action and therapeutics: relevance of novel concepts in G-protein-coupled receptor and signal transduction pharmacology. Br J Clin Pharmacol. (2004) 57:373–87. doi: 10.1111/j.1365-2125.2003.02046.x
4. Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. (2000) 21:90–113. doi: 10.1210/edrv.21.1.0390
5. Chapurlat RD, Orcel P. Fibrous dysplasia of bone and McCune-Albright syndrome. Best Pract Res Clin Rheumatol. (2008) 22:55–69. doi: 10.1016/j.berh.2007.11.004
6. Javaid MK, Boyce A, Appelman-Dijkstra N, Ong J, Defabianis P, Offiah A, et al. Best practice management guidelines for fibrous dysplasia/McCune-Albright syndrome: a consensus statement from the FD/MAS international consortium. Orphanet J Rare Dis. (2019) 14:139. doi: 10.1186/s13023-019-1102-9
7. Majoor BC, Appelman-Dijkstra NM, Fiocco M, van de Sande MA, Dijkstra PS, Hamdy NA. Outcome of long-term bisphosphonate therapy in McCune-albright syndrome and polyostotic fibrous dysplasia. J Bone Miner Res. (2017) 32:264–76. doi: 10.1002/jbmr.2999
8. Boyce AM, Chong WH, Yao J, Gafni RI, Kelly MH, Chamberlain CE, et al. Denosumab treatment for fibrous dysplasia. J Bone Miner Res. (2012) 27:1462–70. doi: 10.1002/jbmr.1603
9. Florenzano P, Pan KS, Brown SM, Paul SM, Kushner H, Guthrie LC, et al. Age-related changes and effects of bisphosphonates on bone turnover and disease progression in fibrous dysplasia of bone. J Bone Miner Res. (2019) 34:653–60. doi: 10.1002/jbmr.3649
10. Boyce AM, Kelly MH, Brillante BA, Kushner H, Wientroub S, Riminucci M, et al. A randomized, double blind, placebo-controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab. (2014) 99:4133–40. doi: 10.1210/jc.2014-1371
11. Eller-Vainicher C, Rossi DS, Guglielmi G, Beltramini GA, Cairoli E, Russillo A, et al. Prompt clinical and biochemical response to denosumab in a young adult patient with craniofacial fibrous dysplasia. Clin Cases Miner Bone Metab. (2016) 13:253–6. doi: 10.11138/ccmbm/2016.13.3.253
12. Majoor BCJ, Papapoulos SE, Dijkstra PDS, Fiocco M, Hamdy NAT, Appelman-Dijkstra NM. Denosumab in patients with fibrous dysplasia previously treated with bisphosphonates. J Clin Endocrinol Metab. (2019) 104:6069–78. doi: 10.1210/jc.2018-02543
13. Benhamou J, Gensburger D, Chapurlat R. Transient improvement of severe pain from fibrous dysplasia of bone with denosumab treatment. Joint Bone Spine. (2014) 81:549–50. doi: 10.1016/j.jbspin.2014.04.013
14. Wang HD, Boyce AM, Tsai JY, Gafni RI, Farley FA, Kasa-Vubu JZ, et al. Effects of denosumab treatment and discontinuation on human growth plates. J Clin Endocrinol Metab. (2014) 99:891–7. doi: 10.1210/jc.2013-3081
15. Chapurlat RD, Gensburger D, Jimenez-Andrade JM, Ghilardi JR, Kelly M, Mantyh P. Pathophysiology and medical treatment of pain in fibrous dysplasia of bone. Orphanet J Rare Dis. (2012) 7(Suppl 1):S3. doi: 10.1186/1750-1172-7-S1-S3
16. Ganda K, Seibel MJ. Rapid biochemical response to denosumab in fibrous dysplasia of bone: report of two cases. Osteoporos Int. (2014) 25:777–82. doi: 10.1007/s00198-013-2585-1
17. Tsourdi E, Langdahl B, Cohen-Solal M, Aubry-Rozier B, Eriksen EF, Guanabens N, et al. Discontinuation of denosumab therapy for osteoporosis: a systematic review and position statement by ECTS. Bone. (2017) 105:11–7. doi: 10.1016/j.bone.2017.08.003
18. Bone HG, Wagman RB, Brandi ML, Brown JP, Chapurlat R, Cummings SR, et al. 10 years of denosumab treatment in postmenopausal women with osteoporosis: results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol. (2017) 5:513–23. doi: 10.1016/S2213-8587(17)30138-9
19. Cummings SR, Ferrari S, Eastell R, Gilchrist N, Jensen JB, McClung M, et al. Vertebral fractures after discontinuation of denosumab: a post-hoc analysis of the randomized placebo-controlled FREEDOM trial and its extension. J Bone Miner Res. (2018) 33:190–8. doi: 10.1002/jbmr.3337
20. Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. (2001) 107:277–86. doi: 10.1172/JCI11296
21. Kuznetsov SA, Riminucci M, Ziran N, Tsutsui TW, Corsi A, Calvi L, et al. The interplay of osteogenesis and hematopoiesis: expression of a constitutively active PTH/PTHrP receptor in osteogenic cells perturbs the establishment of hematopoiesis in bone and of skeletal stem cells in the bone marrow. J Cell Biol. (2004) 167:1113–22. doi: 10.1083/jcb.200408079
22. Chang WC, Ng JK, Nguyen T, Pellissier L, Claeysen S, Hsiao EC, et al. Modifying ligand-induced and constitutive signaling of the human 5-HT4 receptor. PLoS ONE. (2007) 2:e1317. doi: 10.1371/journal.pone.0001317
23. Hsiao EC, Boudignon BM, Chang WC, Bencsik M, Peng J, Nguyen TD, et al. Osteoblast expression of an engineered Gs-coupled receptor dramatically increases bone mass. Proc Natl Acad Sci USA. (2008) 105:1209–14. doi: 10.1073/pnas.0707457105
24. Hsiao EC, Boudignon BM, Halloran BP, Nissenson RA, Conklin BR. Gs G protein-coupled receptor signaling in osteoblasts elicits age-dependent effects on bone formation. J Bone Miner Res. (2010) 25:584–93. doi: 10.1002/jbmr.3
25. Kazakia GJ, Speer D, Shanbhag S, Majumdar S, Conklin BR, Nissenson RA, et al. Mineral composition is altered by osteoblast expression of an engineered G(s)-coupled receptor. Calcif Tissue Int. (2011) 89:10–20. doi: 10.1007/s00223-011-9487-z
26. Schepers K, Hsiao EC, Garg T, Scott MJ, Passegue E. Activated Gs signaling in osteoblastic cells alters the hematopoietic stem cell niche in mice. Blood. (2012) 120:3425–35. doi: 10.1182/blood-2011-11-395418
27. Cain CJ, Valencia JT, Ho S, Jordan K, Mattingly A, Morales BM, et al. Increased Gs signaling in osteoblasts reduces bone marrow and whole-body adiposity in male mice. Endocrinology. (2016) 157:1481–94. doi: 10.1210/en.2015-1867
28. Saggio I, Remoli C, Spica E, Cersosimo S, Sacchetti B, Robey PG, et al. Constitutive expression of Gsalpha(R201C) in mice produces a heritable, direct replica of human fibrous dysplasia bone pathology and demonstrates its natural history. J Bone Miner Res. (2014) 29:2357–68. doi: 10.1002/jbmr.2267
29. Remoli C, Michienzi S, Sacchetti B, Consiglio AD, Cersosimo S, Spica E, et al. Osteoblast-specific expression of the fibrous dysplasia (FD)-causing mutation Gsalpha(R201C) produces a high bone mass phenotype but does not reproduce FD in the mouse. J Bone Miner Res. (2015) 30:1030–43. doi: 10.1002/jbmr.2425
30. Palmisano B, Spica E, Remoli C, Labella R, Di Filippo A, Donsante S, et al. RANKL inhibition in fibrous dysplasia of bone: a preclinical study in a mouse model of the human disease. J Bone Miner Res. (2019) 34:2171–82. doi: 10.1002/jbmr.3828
31. Zhao X, Deng P, Iglesias-Bartolome R, Amornphimoltham P, Steffen DJ, Jin Y, et al. Expression of an active Galphas mutant in skeletal stem cells is sufficient and necessary for fibrous dysplasia initiation and maintenance. Proc Natl Acad Sci USA. (2018) 115:E428–E37. doi: 10.1073/pnas.1713710115
32. Khan SK, Yadav PS, Elliott G, Hu DZ, Xu R, Yang Y. Induced Gnas(R201H) expression from the endogenous Gnas locus causes fibrous dysplasia by up-regulating Wnt/beta-catenin signaling. Proc Natl Acad Sci USA. (2018) 115:E418–E27. doi: 10.1073/pnas.1714313114
33. Xu R, Khan SK, Zhou T, Gao B, Zhou Y, Zhou X, et al. Galphas signaling controls intramembranous ossification during cranial bone development by regulating both Hedgehog and Wnt/beta-catenin signaling. Bone Res. (2018) 6:33. doi: 10.1038/s41413-018-0034-7
34. Bianco P, Kuznetsov SA, Riminucci M, Fisher LW, Spiegel AM, Robey PG. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gsalpha-mutated skeletal progenitor cells. J Clin Invest. (1998) 101:1737–44. doi: 10.1172/JCI2361
35. Kuznetsov SA, Cherman N, Riminucci M, Collins MT, Robey PG, Bianco P. Age-dependent demise of GNAS-mutated skeletal stem cells and “normalization” of fibrous dysplasia of bone. J Bone Miner Res. (2008) 23:1731–40. doi: 10.1359/jbmr.080609
36. Piersanti S, Remoli C, Saggio I, Funari A, Michienzi S, Sacchetti B, et al. Transfer, analysis, and reversion of the fibrous dysplasia cellular phenotype in human skeletal progenitors. J Bone Miner Res. (2010) 25:1103–16. doi: 10.1359/jbmr.091036
Keywords: GPCR (G protein-coupled receptors), McCune-Albright syndrome (MAS), fibrous dysplasia (FD), GNAS (guanine nucleotide-binding protein/[alpha]-subunit, Gsα, cAMP, mouse models, human cell models
Citation: Lung H, Hsiao EC and Wentworth KL (2020) Advances in Models of Fibrous Dysplasia/McCune-Albright Syndrome. Front. Endocrinol. 10:925. doi: 10.3389/fendo.2019.00925
Received: 05 November 2019; Accepted: 18 December 2019;
Published: 24 January 2020.
Edited by:
Wim Van Hul, University of Antwerp, BelgiumReviewed by:
Paula H. Stern, Northwestern University, United StatesDavid M. Findlay, University of Adelaide, Australia
Copyright © 2020 Lung, Hsiao and Wentworth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward C. Hsiao, ZWR3YXJkLmhzaWFvQHVjc2YuZWR1; Kelly L. Wentworth, a2VsbHkud2VudHdvcnRoQHVjc2YuZWR1