 Giuditta Rurale1Emery Di Cicco2
Giuditta Rurale1Emery Di Cicco2 Monica Dentice2Domenico Salvatore3
Monica Dentice2Domenico Salvatore3 Luca Persani1,4
Luca Persani1,4 Federica Marelli1,4*
Federica Marelli1,4* Cristina Luongo3
Cristina Luongo3- 1Division of Endocrine and Metabolic Diseases, IRCCS Istituto Auxologico Italiano, Milan, Italy
- 2Department of Clinical Medicine & Surgery, University of Naples Federico II, Naples, Italy
- 3Department of Public Health, University of Naples Federico II, Naples, Italy
- 4Department of Clinical Sciences and Community Health, University of Milan, Milan, Italy
Thyroid hormone action defects (THADs) have been classically considered conditions of impaired sensitivity to thyroid hormone (TH). They were originally referring to alterations in TH receptor genes (THRA and THRB), but the discovery of genetic mutations and polymorphisms causing alterations in cell membrane transport (e.g., MCT8) and metabolism (e.g., SECISBP2, DIO2) led recently to a new and broader definition of TH hyposensitivity (THH), including not only THADs but all defects that could interfere with the activity of TH. Due to the different functions and tissue-specific expression of these genes, affected patients exhibit highly variable phenotypes. Some of them are characterized by a tissue hypothyroidism or well-recognizable alterations in the thyroid function tests (TFTs), whereas others display a combination of hypo- and hyperthyroid manifestations with normal or only subtle biochemical defects. The huge effort of basic research has greatly aided the comprehension of the molecular mechanisms underlying THADs, dissecting the morphological and functional alterations on target tissues, and defining the related-changes in the biochemical profile. In this review, we describe different pictures in which a specific alteration in the TFTs (TSH, T4, and T3 levels) is caused by defects in a specific gene. Altogether these findings can help clinicians to early recognize and diagnose THH and to perform a more precise genetic screening and therapeutic intervention. On the other hand, the identification of new genetic variants will allow the generation of cell-based and animal models to give novel insight into thyroid physiology and establish new therapeutic interventions.
Background
Thyroid hormones (TH, tetraiodothyronine T4 and triiodothyronine T3) are essential for proper embryonic development and postnatal regulation of growth and function of several organs. TH deficiency or excess during embryonic development results in profound and permanent defects, especially on the nervous system (1).
Circulating levels of TH are maintained within the reference range by feedback mechanisms operated by the Hypothalamic-Pituitary-Thyroid (HPT) axis. At targeted tissues, an adequate TH activity depends on the capability and availability of TH transporters (including but not limited to MCT8, MCT10, OATP1C1), deiodinases (DIO1, 2, and 3), and nuclear TH receptors (TRα1, TRβ1, and TRβ2) (2).
In humans, genetic defects in TH signaling lead to the onset of TH cell membrane transport defects (THCMTD), TH metabolism defects (THMD), or TH action defects (THAD), and are globally referred as TH hyposensitivity (THH) (3, 4).
Mutations of each gene are associated with characteristic alterations of thyroid function tests (TFTs). Nevertheless, some polymorphisms affecting TH transporter and deiodinase functions do not frankly alter TH profile, but a deeper investigation demonstrated that even slight modifications of deiodinase activity could have an impact on the treatment strategies for hypothyroidism (5).
In clinical practice, THH conditions are often difficult to recognize and treat. In fact, the TH profile in THH is not typical and readily recognizable as it happens in classic hyper- or hypothyroidism by routine TFTs. In addition, the clinical manifestations are not straightforward and represent the peculiar associations of peripheral tissue hyper- or hypothyroidism.
During the last two decades, the close collaboration between clinicians and basic researchers has been crucial to better understand the mechanism controlling intracellular TH activity, identify new genetic mutations and polymorphisms, and define the associated phenotypes. Importantly, the generation of specific animal models that mimic human THH conditions helped to unravel the pathophysiological mechanisms underlying these complex and variable phenotypes (6–9).
In this review, we summarize different clinical pictures in which a particular alteration of TH profile correlates with mutations or polymorphisms in a specific gene involved in TH transport (MCT8), TH metabolism (SBP2, DIO2), and TH activity (THRA and THRB).
Normal/High TSH, Low FT4, High FT3: MCT8 Mutations
The transmembrane passage of TH in cells is mediated by specific TH transporters (10). Among them, MCT8 facilitates the uptake and efflux of TH across the plasma membrane. In humans, inactivating mutations of MCT8 cause the Allan-Herndon-Dudley syndrome, characterized by high serum T3, low T4, and slightly elevated TSH and neuromuscular defects (11). The phenotype can be more or less severe due to residual MCT8 activity. In fact, different kinds of mutations (e.g., substitution, deletion, insertion) variably affect the expression and activity of MCT8 (12). Moreover, the compensation operated by alternative TH transporters results in different alterations at tissue levels when MCT8 is absent. In fact, some tissues appear normal or even hyperthyroid, whereas tissues that exclusively express MCT8 present intracellular hypothyroidism (13, 14). Mct8 knockout mice (KO), Mct8/Oatpc1 and Mct8/Dio2 double KO mice provide useful information to understand the mechanism determining these alterations (15–17). The absence of MCT8 determines an increase in D1 and D2 activity that is responsible for the high levels of T3 and low T4 in serum. Moreover, the altered secretion of T3/T4 ratio by thyroid follicles described in Mct8 KO mice may also contribute to explain the low T4 serum content (16). Muscles isolated from Mct8 KO mice are hyperthyroid and showed impaired muscle regeneration, while Mct8/Oatpc1-depleted brains are hypothyroid (18). Recently, pluripotent stem cells (iPSCs) induced from MCT8-deficient patient can be efficiently differentiated into neural cells, and although TH transport is reduced, the TH transcription signature was normal (19). The authors demonstrated that the neurological phenotype is more related to the absence of TH transport throughout the blood-brain barrier than to an intrinsic deficit of MCT8 of the differentiated neurons. In light of these observations, the systemic thyroid status of MCT8-deficient patients cannot be classified as a generalized classical hypothyroidism or hyperthyroidism. This represents an important therapeutic challenge. Treatment with LT4 increased brain TH content but exacerbated the hypermetabolic state due to the increased D1 activity and then T3 production. Concurrently L-T4 and propylthiouracil (D1-inhibitor) administration normalized T4 without affecting T3 but failed to improve the neuromuscular phenotype (20). Lately, thyromimetic drugs as DITPA, Triac, and Tetrac have been proposed as treatment (21–23). In particular, Triac has been shown very effective in promoting neuronal differentiation when administered to Mct8 KO mice during the first postnatal week (24).
Normal/High TSH, High FT4, Normal/Low FT3: SBP2 Mutations
The characteristic thyroid signature of patients with biallelic inactivation of the SBP2 gene is high T4, low/normal T3, and slightly elevated/normal levels of TSH. The SBP2 gene codifies for a SECIS-binding protein involved in the incorporation of selenocysteine (Sec) into a family of selenoproteins (SPs) with diverse, essential, biological roles (25). The defective activity of SPs beside being involved in antioxidant defense and protein folding, affects TH metabolism since the deiodinases are selenocysteine-containing enzymes, thus resulting in a complex phenotype characterized by growth retardation, muscular dystrophy, intellectual disabilities, skin photosensitivity, hearing loss, insulin resistance, azoospermia, and aorthopathy (26). The observed phenotype is complex, but probably reflects three major pathogenic processes: (1) tissue-specific effects mediated by lack of a particular SP (e.g., the musculoskeletal phenotype caused by SEPN1 deficiency) (27); (2) consequences of more generalized tissue oxidative damage due to loss of antioxidant selenoenzymes with excess of cellular reactive oxygen species (e.g., aorthopathy) (26); (3) hypothyroid-related defects due to decreased activity of DIO2 and thus reduced peripheral T4-to-T3 conversion (e.g., growth delay, intellectual disability, and hearing loss) (28). However, whether and how these three mechanisms interplay in different tissues contributing to the SBP2 phenotype remain unknown. Several treatments (Se supplementation and T3 replacement) have been attempted to improve height and normalize TH levels, but only T3 treatment provided some beneficial effects (normal T3 levels, improved linear growth and neurodevelopment) (26, 29). Inducible, hepatocyte and neuron-specific Sbp2-deficient mouse models have not been reported to fully recapitulate SBP2 conditions, and constitutive Sbp2 KO mice die during embryonic life (30–32). Thus, additional model organisms are required to fully assess the pathophysiology responsible for thyroid phenotype and associated manifestations.
TSH, FT3, and FT4 Within the Reference Range: DIO2 Polymorphism
Deiodinases are essential to determine the intracellular concentration of THs. The expression of these three enzymes (D1, D2, and D3) is tissue and time dependent (33). Mice models demonstrated that life without deiodinases is allowed, but at the expense of alteration of sensory organs, metabolism, skeletal development, tissue regeneration, and HPT-axis regulation (34). Until today, mutations in deiodinases have never been reported in human conditions; we cannot discern whether this is due to relatively small effects of these mutations or to an incompatibility with life.
In patients, even slight alterations in TH levels have critical consequences on heart rate and bone mineral density; for this reason, the identification of different deiodinase polymorphisms affecting TH homeostasis is considered a topic of potential interest (5, 35, 36). Among them, the DIO2 Thr92Ala polymorphism has been associated with insulin resistance, obesity, hypertension, and alteration of hypothalamic-pituitary-thyroid axis (37). Notably, while individuals with the Thr92Ala polymorphism do not show an altered thyroid profile, athyreotic subjects carrying the Ala allele were associated with lower plasma fT3 level compared to wild type subjects, suggesting a decreased capability of peripheral T4 to T3 conversion due to a reduced DIO2 activity (5). In this study, the authors confirmed the partial D2 enzymatic deficiency by analyzing T4 sensitivity in pituitary and muscle cells. T4 was less effective in reducing TSH mRNA levels and in inducing apoptosis, respectively in pituitary primary cell culture and muscle cells transfected with the DIO2 Thr92Ala than in those transfected with the WT allele. The generation of a transgenic mouse carrying the DIO2 Thr92Ala polymorphism corroborated these findings. This mouse model showed hypothyroidism in distinct brain areas associated with memory, and behavior issues that improved after T3 administration were aggravated by primary hypothyroidism and responded only partially to LT4 treatment (38).
Normal TSH, Low/Normal FT4, High/Normal FT3: THRA Mutations
Borderline variations in TFTs are frequently observed in patients with Resistance to Thyroid Hormone alpha (RTH α), a rare multisystem disorder caused by heterozygous mutations on the THRA gene (9). Two major receptor isoforms are produced from the THRA loci, TRα1 and TRα2, which differ in their carboxy terminus. Although TRα2 is incapable of binding TH, it is considered a modulator of TH action, antagonizing TRα1 activity (1). TRα1 is mainly expressed in the cerebral cortex, thyroid gland, heart, lung, gastrointestinal tract, liver, kidney, and ovary (39). RTHα patients present a complex and multisystem phenotype characterized by tissue-selective features of hypothyroidism. Based on the reports of nearly 30 patients with RTHα, the clinical phenotype includes, at varying degrees, growth restriction, impaired bone ossification and malformations, intellectual disability, bradycardia, and chronic constipation. A distinct facial dysmorphic phenotype consists of macrocephaly, broad facies, hypertelorism, flattened nose, prominent tongue, and thick lips. Some patients suffer from severe intellectual disability, epilepsy or autism-spectrum disorders, while others manifest only dyspraxia, ataxic gait, and dysarthria. Decreased basal metabolic rate, normochromic normocytic anemia, and elevated levels of creatine kinase (CK) and SHBG are also frequently observed in affected cases (9, 40–42).
RTHα patients typically present low FT4/FT3 ratio with low/normal FT4, high/normal T3, low serum rT3, and normal serum TSH levels (9). Mouse and zebrafish models of RTHα revealed that increased expression of hepatic DIO1 or hypothalamic DIO2 as well as reduced levels of DIO3 may be involved in generating this serum TH profile (43–46).
Because TH levels usually remain within the reference range, RTHα patients are not necessarily referred to endocrinologist, and it is likely that the disease frequently remains unrecognized by clinicians. In fact, only 21 heterozygous variants in THRA have been described in RTHα patients so far, but the latest release of the ExAC database (http://exac.broadinstitute.org) contains 68 THRA missense or frameshift mutations carried by anonymous patients, which are predicted to alter TRα1 function. Moreover, depending on the mutation, the clinical phenotype may vary among patients, ranging from severe to mild or minimal symptoms (41). All of known TRα mutations alter the ligand-binding domain (LBD) and include missense substitutions, deletions, and frameshift/premature stop codon, with dominant-negative (DN) properties. Functional in vitro and in vivo evaluations demonstrated that the missense substitution maintained a partial T3-binding activity, which can be normalized at high T3 doses, whereas the premature truncated variants showed an irreversible inability to recruit the coactivators acting as a constitutive transcriptional repressor (41, 45, 47, 48).
Thus, reduced FT4/FT3 ratio and increased SHBG and CK levels represent helpful serum parameters for diagnosis of RTHα. Moreover, a genetic screening of THRA should be considered in children with decreased growth rate, facial dysmorphic features, and delayed psychomotor development or in adults with unexplained constipation, megacolon, and bradycardia (49, 50). Very recently, characteristic metabolomic fingerprints in biofluids have been described in mice carrying human-like mutations (51). In particular, decreased levels of N-acetylglucosamine and choline/phosphocholine in urine or reduced levels of unsaturated lipids in serum might be used as additional biomarkers for the diagnosis of RTHα, likely discriminating patients with similar TH profiles (e.g., congenital hypothyroidism).
Unsuppressed TSH, High FT4 and FT3: THRB Mutations
Elevated serum TH concentrations accompanied by non-suppressed TSH levels (inappropriate secretion of TSH: IST) are the hallmark of RTHβ, a rare disorder due to heterozygous mutations in the THRB gene causing a decreased response to TH action in the peripheral tissue (52). Two different TRβ (TRβ1 and TRβ2) isoforms are generated by alternate promoters on the THRB gene. TRβ1 is predominantly expressed in the brain, liver, and kidney, whereas TRβ2 is found in the pituitary, retina, and cochlea (39).
RTHβ is often diagnosed at childbirth during the screening for congenital thyroid dysfunction with abnormal level of T4 and TSH due to the impairment of the TRβ2-dependent regulation of the HPT-axis. As a consequence of the hyperstimulation of the thyroid gland, goiter is frequently present in patients with RTHβ (53, 54).
Since TRβ isoforms are expressed in few tissues, the RTHβ patients exhibited tissue-specific hypothyroid-related defects such as hepatic steatosis, dyslipidemia, and impaired hearing and color vision, together with thyrotoxic manifestations including attention-deficit hyperactivity disorder (ADHD), anxiety, tachycardia, and low bone density (53–55). RTHβ can appear in a sporadic form, but most commonly it is a familial syndrome with autosomal dominant inheritance. The clinical manifestations are variable between families with the same mutation and also between members of the same family with identical mutations. Differences in the functional properties of TRβ variants or other genetic and epigenetic modifications may influence the expression and the penetrance of the mutant receptor thus explaining the phenotypic variability of RTHβ. Additionally, high circulating TH levels can compensate the tissue resistance, and patients may appear euthyroid (55–60).
However, in a growing number of individuals, RTHβ occurs in the absence of mutations in the TR genes, likely due to defects in one of cofactors involved in TH signaling (61–63). Polymorphisms in TR-target genes have been associated with alterations in TFTs and may also account for the non-RTH patient's phenotype (64).
Over 160 different mutations in THRB have been identified so far, most of them localized to three hotspots within the LBD of the β receptors, which inhibit in a DN manner the transcriptional activity of TRs (2). As described for the RTHα, the type of the mutation correlates with the severity of the clinical phenotype and with the potential reversion of the DN activity by high T3 administration in vitro and in vivo (44, 65–67). The genetic screening of THRB should be considered in patients with normal/reduced serum SHBG levels in conjunction with high TH and unsuppressed TSH. Moreover, TH effects on neuromuscular and hepatic systems can also be addressed by the measurement of additional metabolic markers (e.g., SGOT, SGPT, cholesterol, triglycerides, ferritin, and CPK). A careful neurological examination looking for signs of hypothyroidism should be done to confirm the diagnosis of RTHβ.
Conclusion
The identification of the physiological functions of genes involved in transport, metabolism, and action of TH in different tissues has been essential for a proper evaluation of the clinical manifestation of TH dysfunction. In particular, the advent of genetic manipulation techniques endorsed the generation of vertebrate knock-out and knock-in models that reproduce human mutations, providing a novel approach to study their functions in normal and pathological settings. Studies in vertebrate models with targeted gene manipulations allow uncovering the molecular mechanisms underlying THH defects and sometimes preceded the identification of the correspondent human genetic mutations (summarized in Table 1). For instance, murine TRα gene deletions were produced 15 years before the identification of the first THRA mutation (78, 94). Differently, the characterization of the phenotypes of DIOs-deficient mouse models was not accompanied by the identification of human genetic mutations so far (72, 73, 76).

Table 1. Animal models of THH.
The impact of neonatal screening programs for congenital hypothyroidism facilitates early diagnosis of such a devastating disorder, resulting in immediate treatment. The possibility to measure TSH and TH in dried spots has huge potential since it facilitates the identification of the different thyroid signatures characterizing THH in the first week of life. What complicates this only apparent simple picture is the high variability of biochemical and clinical phenotypes among patients with similar or even identical genetic mutations. In fact, while some patients present only subtle TH alterations and mild clinical features, others exhibit abnormal TFTs and severe clinical abnormalities. So, changes of metabolic markers together with the observation of certain clinical manifestations are required to recognize the different THH conditions. Moreover, since similar alterations in TFT can be found in patients with different thyroid diseases (e.g., congenital hypothyroidism), these additional indicators should be taken into account during the differential diagnosis. The biochemical signatures associated with each gene mutations are summarized in Figure 1.
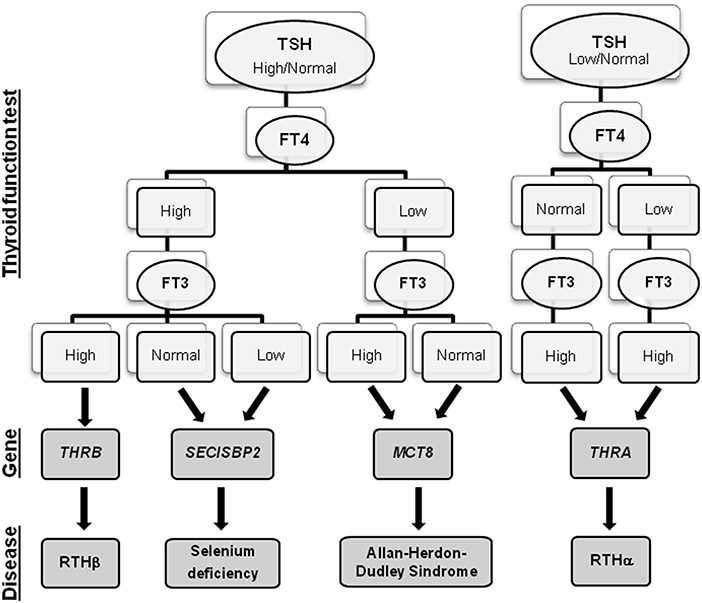
Figure 1. Flowchart of THH diagnosis. For each THH disease, the associated gene and the alterations in the thyroid function tests are reported.
In conclusion, the combined effort of clinical and basic research is the unmet need to improve the diagnosis of the different forms of THH. The identification of new patients and mutations can deeply improve our understanding of the physiologic role of the genes involved in THH but will also provide further insights into the molecular mechanisms underlying each phenotype.
Such information may also facilitate the design of new drugs that can be tested in animal models, as a prelude to subsequently undertaking proof-of-concept clinical trials in patients.
Author Contributions
GR, EC, FM, and CL wrote the manuscript. MD, DS, and LP supervised and finalized the manuscript.
Funding
This work was partially supported by the grant 05C102_2011 of the Ricerca Corrente of the Istituto Auxologico Italiano (Milan, Italy).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. (2010) 31:139–70. doi: 10.1210/er.2009-0007
2. Dumitrescu AM, Refetoff S. Impaired sensitivity to thyroid hormone: defects of transport, metabolism and action. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, editors. Endotext. South Dartmouth, MA: MDText.com, Inc. p. 845–73.
3. Schwartz CE, Stevenson RE. The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract Res Clin Endocrinol Metab. (2007) 21:307–21. doi: 10.1016/j.beem.2007.03.009
4. Singh BK, Yen PM. A clinician's guide to understanding resistance to thyroid hormone due to receptor mutations in the TRα and TRβ isoforms. Clin Diabetes Endocrinol. (2017) 3:8. doi: 10.1186/s40842-017-0046-z
5. Castagna MG, Dentice M, Cantara S, Ambrosio R, Maino F, Porcelli T, et al. DIO2 Thr92Ala reduces deiodinase-2 activity and serum-T3 levels in thyroid-deficient patients. J Clin Endocrinol Metab. (2017) 102:1623–30. doi: 10.1210/jc.2016-2587
6. Bernal J, Guadano-Ferraz A, Morte B. Thyroid hormone transporters–functions and clinical implications. Nat Rev Endocrinol. (2015) 11:506. doi: 10.1038/nrendo.2015.113
7. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. (2002) 23:38–89. doi: 10.1210/edrv.23.1.0455
8. Flamant F. Futures Challenges in Thyroid Hormone Signaling Research. Front Endocrinol. (2016) 7:58. doi: 10.3389/fendo.2016.00058
9. Moran C, Chatterjee K. Resistance to thyroid hormone α-emerging definition of a disorder of thyroid hormone action. J Clin Endocrinol Metab. (2016) 101:2636–9. doi: 10.1210/jc.2016-2317
10. Visser WE, Friesema EC, Visser TJ. Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol. (2011) 25:1–14. doi: 10.1210/me.2010-0095
11. Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, et al. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. (2005) 77:41–53. doi: 10.1086/431313
12. Friesema EC, Visser WE, Visser TJ. Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol. (2010) 322:107–13. doi: 10.1016/j.mce.2010.01.016
13. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. (2004) 74:168–75. doi: 10.1086/380999
14. Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. (2004) 364:1435–7. doi: 10.1016/S0140-6736(04)17226-7
15. Barez-Lopez S, Grijota-Martinez C, Auso E, Fernandez-de Frutos M, Montero-Pedrazuela A, Guadano-Ferraz A. Adult Mice Lacking Mct8 and Dio2 Proteins Present Alterations in Peripheral Thyroid Hormone Levels and Severe Brain and Motor Skill Impairments. Thyroid. (2019) 29:1669–82. doi: 10.1089/thy.2019.0068
16. Di Cosmo C, Liao XH, Dumitrescu AM, Philp NJ, Weiss RE, Refetoff S. Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J Clin Invest. (2010) 120:3377–88. doi: 10.1172/JCI42113
17. Mayerl S, Muller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest. (2014) 124:1987–99. doi: 10.1172/JCI70324
18. Mayerl S, Schmidt M, Doycheva D, Darras VM, Huttner SS, Boelen A, et al. Thyroid hormone transporters MCT8 and OATP1C1 control skeletal muscle regeneration. Stem Cell Reports. (2018) 10:1959–74. doi: 10.1016/j.stemcr.2018.03.021
19. Vatine GD, Al-Ahmad A, Barriga BK, Svendsen S, Salim A, Garcia L, et al. Modeling psychomotor retardation using iPSCs from MCT8-deficient patients indicates a prominent role for the blood-brain barrier. Cell Stem Cell. (2017) 20:831–843 e835. doi: 10.1016/j.stem.2017.04.002
20. Wemeau JL, Pigeyre M, Proust-Lemoine E, d'Herbomez M, Gottrand F, Jansen J, et al. Beneficial effects of propylthiouracil plus L-thyroxine treatment in a patient with a mutation in MCT8. J Clin Endocrinol Metab. (2008) 93:2084–8. doi: 10.1210/jc.2007-2719
21. Groeneweg S, Peeters RP, Moran C, Stoupa A, Auriol F, Tonduti D, et al. Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: an international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. (2019) 7:695–706. doi: 10.1016/S2213-8587(19)30155-X
22. Horn S, Kersseboom S, Mayerl S, Muller J, Groba C, Trajkovic-Arsic M, et al. Tetrac can replace thyroid hormone during brain development in mouse mutants deficient in the thyroid hormone transporter mct8. Endocrinology. (2013) 154:968–79. doi: 10.1210/en.2012-1628
23. Lee JY, Kim MJ, Deliyanti D, Azari MF, Rossello F, Costin A, et al. Overcoming Monocarboxylate Transporter 8 (MCT8)-deficiency to promote human oligodendrocyte differentiation and myelination. EBioMedicine. (2017) 25:122–35. doi: 10.1016/j.ebiom.2017.10.016
24. Barez-Lopez S, Obregon MJ, Martinez-de-Mena R, Bernal J, Guadano-Ferraz A, Morte B. Effect of triiodothyroacetic acid treatment in Mct8 deficiency: a word of caution. Thyroid. (2016) 26:618–26. doi: 10.1089/thy.2015.0388
25. Dumitrescu AM, Liao XH, Abdullah MS, Lado-Abeal J, Majed FA, Moeller LC, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. (2005) 37:1247–52. doi: 10.1038/ng1654
26. Schoenmakers E, Agostini M, Mitchell C, Schoenmakers N, Papp L, Rajanayagam O, et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J Clin Invest. (2010) 120:4220–35. doi: 10.1172/JCI43653
27. Saini AG, Padmanabha H, Kumar S, Sankhyan N, Singhi P. SEPN1-related rigid spine muscular dystrophy. Indian J Pediatr. (2018) 85:1033–4. doi: 10.1007/s12098-018-2713-1
28. Hamajima T, Mushimoto Y, Kobayashi H, Saito Y, Onigata K. Novel compound heterozygous mutations in the SBP2 gene: characteristic clinical manifestations and the implications of GH and triiodothyronine in longitudinal bone growth and maturation. Eur J Endocrinol. (2012) 166:757–64. doi: 10.1530/EJE-11-0812
29. Di Cosmo C, McLellan N, Liao XH, Khanna KK, Weiss RE, Papp L, et al. Clinical and molecular characterization of a novel selenocysteine insertion sequence-binding protein 2 (SBP2) gene mutation (R128X). J Clin Endocrinol Metab. (2009) 94:4003–9. doi: 10.1210/jc.2009-0686
30. Seeher S, Atassi T, Mahdi Y, Carlson BA, Braun D, Wirth EK, et al. Secisbp2 is essential for embryonic development and enhances selenoprotein expression. Antioxid Redox Signal. (2014) 21:835–49. doi: 10.1089/ars.2013.5358
31. Seeher S, Carlson BA, Miniard AC, Wirth EK, Mahdi Y, Hatfield DL, et al. Impaired selenoprotein expression in brain triggers striatal neuronal loss leading to co-ordination defects in mice. Biochem J. (2014) 462:67–75. doi: 10.1042/BJ20140423
32. Seeher S, Schweizer U. Targeted deletion of Secisbp2 reduces, but does not abrogate, selenoprotein expression and leads to striatal interneuron loss. Free Radic Biol Med. (2014) 75:(Suppl. 1):S9. doi: 10.1016/j.freeradbiomed.2014.10.849
33. Luongo C, Dentice M, Salvatore D. Deiodinases and their intricate role in thyroid hormone homeostasis. Nat Rev Endocrinol. (2019) 15:479–88. doi: 10.1038/s41574-019-0218-2
34. Gereben B, McAninch EA, Ribeiro MO, Bianco AC. Scope and limitations of iodothyronine deiodinases in hypothyroidism. Nat Rev Endocrinol. (2015) 11:642–52. doi: 10.1038/nrendo.2015.155
35. Nguyen TT, Chapa F, DiStefano JJ III. Direct measurement of the contributions of type I and type II 5'-deiodinases to whole body steady state 3,5,3'-triiodothyronine production from thyroxine in the rat. Endocrinology. (1998) 139:4626–33. doi: 10.1210/endo.139.11.6323
36. Peeters RP, van Toor H, Klootwijk W, de Rijke YB, Kuiper GG, Uitterlinden AG, et al. Polymorphisms in thyroid hormone pathway genes are associated with plasma TSH and iodothyronine levels in healthy subjects. J Clin Endocrinol Metab. (2003) 88:2880–8. doi: 10.1210/jc.2002-021592
37. Mentuccia D, Proietti-Pannunzi L, Tanner K, Bacci V, Pollin TI, Poehlman ET, et al. Association between a novel variant of the human type 2 deiodinase gene Thr92Ala and insulin resistance: evidence of interaction with the Trp64Arg variant of the β-3-adrenergic receptor. Diabetes. (2002) 51:880–3. doi: 10.2337/diabetes.51.3.880
38. Jo S, Fonseca TL, Bocco B, Fernandes GW, McAninch EA, Bolin AP, et al. Type 2 deiodinase polymorphism causes ER stress and hypothyroidism in the brain. J Clin Invest. (2019) 129:230–45. doi: 10.1172/JCI123176
39. Shahrara S, Drvota V, Sylven C. Organ specific expression of thyroid hormone receptor mRNA and protein in different human tissues. Biol Pharm Bull. (1999) 22:1027–33. doi: 10.1248/bpb.22.1027
40. Demir K, van Gucht AL, Buyukinan M, Catli G, Ayhan Y, Bas VN, et al. Diverse Genotypes and Phenotypes of three novel thyroid hormone receptor-α mutations. J Clin Endocrinol Metab. (2016) 101:2945–54. doi: 10.1210/jc.2016-1404
41. Moran C, Agostini M, McGowan A, Schoenmakers E, Fairall L, Lyons G, et al. Contrasting phenotypes in resistance to thyroid hormone α correlate with divergent properties of thyroid hormone receptor α1 mutant proteins. Thyroid. (2017) 27:973–82. doi: 10.1089/thy.2017.0157
42. Tang Y, Yu M, Lian X. Resistance to thyroid hormone α, revelation of basic study to clinical consequences. J Pediatr Endocrinol Metab. (2016) 29:511–22. doi: 10.1515/jpem-2015-0286
43. Cheng SY. Isoform-dependent actions of thyroid hormone nuclear receptors: lessons from knockin mutant mice. Steroids. (2005) 70:450–4. doi: 10.1016/j.steroids.2005.02.003
44. Marelli F, Carra S, Agostini M, Cotelli F, Peeters R, Chatterjee K, et al. Patterns of thyroid hormone receptor expression in zebrafish and generation of a novel model of resistance to thyroid hormone action. Mol Cell Endocrinol. (2016) 424:102–17. doi: 10.1016/j.mce.2016.01.020
45. Marelli F, Carra S, Rurale G, Cotelli F, Persani L. In vivo functional consequences of human THRA Variants Expressed in the Zebrafish. Thyroid. (2017) 27:279–91. doi: 10.1089/thy.2016.0373
46. Zavacki AM, Ying H, Christoffolete MA, Aerts G, So E, Harney JW, et al. Type 1 iodothyronine deiodinase is a sensitive marker of peripheral thyroid status in the mouse. Endocrinology. (2005) 146:1568–75. doi: 10.1210/en.2004-1392
47. Briet C, Bouhours-Nouet N, Illouz F, Prunier-Mirebeau D, Rodien P. TRα mutations in human. Methods Mol Biol. (2018) 1801:241–5. doi: 10.1007/978-1-4939-7902-8_19
48. van Gucht ALM, Moran C, Meima ME, Visser WE, Chatterjee K, Visser TJ, et al. Resistance to thyroid hormone due to heterozygous mutations in thyroid hormone receptor alpha. Curr Top Dev Biol. (2017) 125:337–55. doi: 10.1016/bs.ctdb.2017.02.001
49. Moran C, Chatterjee K. Resistance to thyroid hormone due to defective thyroid receptor alpha. Best Pract Res Clin Endocrinol Metab. (2015) 29:647–57. doi: 10.1016/j.beem.2015.07.007
50. van Mullem AA, Visser TJ, Peeters RP. Clinical consequences of mutations in thyroid hormone receptor-α1. Eur Thyroid J. (2014) 3:17–24. doi: 10.1159/000360637
51. Boumaza H, Markossian S, Busi B, Rautureau GJP, Gauthier K, Elena-Herrmann B, et al. Metabolomic profiling of body fluids in mouse models demonstrates that nuclear magnetic resonance is a putative diagnostic tool for the presence of thyroid hormone receptor α1 mutations. Thyroid. (2019) 29:1327–35. doi: 10.1089/thy.2018.0730
52. Beck-Peccoz P, Chatterjee VK, Chin WW, DeGroot LJ, Jameson JL, Nakamura H, et al. Nomenclature of thyroid hormone receptor β gene mutations in resistance to thyroid hormone. First workshop on thyroid hormone resistance, July 10-11, 1993, Cambridge, U.K. J Endocrinol Invest. (1994) 17:283–7. doi: 10.1007/BF03348977
53. Beck-Peccoz P, Chatterjee VK. The variable clinical phenotype in thyroid hormone resistance syndrome. Thyroid. (1994) 4:225–32. doi: 10.1089/thy.1994.4.225
54. Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev. (1993) 14:348–99. doi: 10.1210/edrv-14-3-348
55. Refetoff S. Resistance to thyroid hormone. Clin Lab Med. (1993) 13:563–81. doi: 10.1016/S0272-2712(18)30426-8
56. Ribeiro RC, Apriletti JW, Wagner RL, West BL, Feng W, Huber R, et al. Mechanisms of thyroid hormone action: insights from X-ray crystallographic and functional studies. Recent Prog Horm Res. (1998) 53:351–92.
57. Takeda K, Sakurai A, DeGroot LJ, Refetoff S. Recessive inheritance of thyroid hormone resistance caused by complete deletion of the protein-coding region of the thyroid hormone receptor- β gene. J Clin Endocrinol Metab. (1992) 74:49–55. doi: 10.1210/jcem.74.1.1727829
58. Weiss RE, Marcocci C, Bruno-Bossio G, Refetoff S. Multiple genetic factors in the heterogeneity of thyroid hormone resistance. J Clin Endocrinol Metab. (1993) 76:257–9. doi: 10.1210/jcem.76.1.8421095
59. Weiss RE, Weinberg M, Refetoff S. Identical mutations in unrelated families with generalized resistance to thyroid hormone occur in cytosine-guanine-rich areas of the thyroid hormone receptor β gene. Analysis of 15 families. J Clin Invest. (1993) 91:2408–15. doi: 10.1172/JCI116474
60. Zhang XY, Kaneshige M, Kamiya Y, Kaneshige K, McPhie P, Cheng SY. Differential expression of thyroid hormone receptor isoforms dictates the dominant negative activity of mutant β receptor. Mol Endocrinol. (2002) 16:2077–92. doi: 10.1210/me.2002-0080
61. Parikh S, Ando S, Schneider A, Skarulis MC, Sarlis NJ, Yen PM. Resistance to thyroid hormone in a patient without thyroid hormone receptor mutations. Thyroid. (2002) 12:81–6. doi: 10.1089/105072502753452011
62. Reutrakul S, Sadow PM, Pannain S, Pohlenz J, Carvalho GA, Macchia PE, et al. Search for abnormalities of nuclear corepressors, coactivators, and a coregulator in families with resistance to thyroid hormone without mutations in thyroid hormone receptor β or α genes. J Clin Endocrinol Metab. (2000) 85:3609–17. doi: 10.1210/jcem.85.10.6873
63. Weiss RE, Hayashi Y, Nagaya T, Petty KJ, Murata Y, Tunca H, et al. Dominant inheritance of resistance to thyroid hormone not linked to defects in the thyroid hormone receptor α or β genes may be due to a defective cofactor. J Clin Endocrinol Metab. (1996) 81:4196–203. doi: 10.1210/jcem.81.12.8954015
64. Peeters RP, van der Deure WM, Visser TJ. Genetic variation in thyroid hormone pathway genes; polymorphisms in the TSH receptor and the iodothyronine deiodinases. Eur J Endocrinol. (2006) 155:655–62. doi: 10.1530/eje.1.02279
65. Yen PM, Chin WW. Molecular mechanisms of dominant negative activity by nuclear hormone receptors. Mol Endocrinol. (1994) 8:1450–4. doi: 10.1210/me.8.11.1450
66. Yen PM, Chin WW. New advances in understanding the molecular mechanisms of thyroid hormone action. Trends Endocrinol Metab. (1994) 5:65–72. doi: 10.1016/1043-2760(94)90004-3
67. Yen PM, Sugawara A, Refetoff S, Chin WW. New insights on the mechanism(s) of the dominant negative effect of mutant thyroid hormone receptor in generalized resistance to thyroid hormone. J Clin Invest. (1992) 90:1825–31. doi: 10.1172/JCI116058
68. Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. (2007) 117:627–35. doi: 10.1172/JCI28253
69. Trajkovic-Arsic M, Visser TJ, Darras VM, Friesema EC, Schlott B, Mittag J, et al. Consequences of monocarboxylate transporter 8 deficiency for renal transport and metabolism of thyroid hormones in mice. Endocrinology. (2010) 151:802–9. doi: 10.1210/en.2009-1053
70. Vatine GD, Zada D, Lerer-Goldshtein T, Tovin A, Malkinson G, Yaniv K, et al. Zebrafish as a model for monocarboxyl transporter 8-deficiency. J Biol Chem. (2013) 288:169–80. doi: 10.1074/jbc.M112.413831
71. Zada D, Tovin A, Lerer-Goldshtein T, Vatine GD, Appelbaum L. Altered behavioral performance and live imaging of circuit-specific neural deficiencies in a zebrafish model for psychomotor retardation. PLoS Genet. (2014) 10:e1004615. doi: 10.1371/journal.pgen.1004615
72. Christoffolete MA, Linardi CC, de Jesus L, Ebina KN, Carvalho SD, Ribeiro MO, et al. Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes. (2004) 53:577–84. doi: 10.2337/diabetes.53.3.577
73. Dentice M, Marsili A, Ambrosio R, Guardiola O, Sibilio A, Paik JH, et al. The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J Clin Invest. (2010) 120:4021–30. doi: 10.1172/JCI43670
74. Ng L, Goodyear RJ, Woods CA, Schneider MJ, Diamond E, Richardson GP, et al. Hearing loss and retarded cochlear development in mice lacking type 2 iodothyronine deiodinase. Proc Natl Acad Sci USA. (2004) 101:3474–9. doi: 10.1073/pnas.0307402101
75. Schneider MJ, Fiering SN, Pallud SE, Parlow AF, St Germain DL, Galton VA. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol. (2001) 15:2137–48. doi: 10.1210/mend.15.12.0740
76. Houbrechts AM, Delarue J, Gabriels IJ, Sourbron J, Darras VM. Permanent Deiodinase Type 2 Deficiency Strongly Perturbs Zebrafish Development, Growth, and Fertility. Endocrinology. (2016) 157:3668–81. doi: 10.1210/en.2016-1077
77. Houbrechts AM, Van Houcke J, Darras VM. Disruption of deiodinase type 2 in zebrafish disturbs male and female reproduction. J Endocrinol. (2019) 241:111–23. doi: 10.1530/JOE-18-0549
78. Fraichard A, Chassande O, Plateroti M, Roux JP, Trouillas J, Dehay C, et al. The T3R α gene encoding a thyroid hormone receptor is essential for post-natal development and thyroid hormone production. EMBO J. (1997) 16:4412–20. doi: 10.1093/emboj/16.14.4412
79. Gauthier K, Plateroti M, Harvey CB, Williams GR, Weiss RE, Refetoff S, et al. Genetic analysis reveals different functions for the products of the thyroid hormone receptor α locus. Mol Cell Biol. (2001) 21:4748–60. doi: 10.1128/MCB.21.14.4748-4760.2001
80. Kaneshige M, Suzuki H, Kaneshige K, Cheng J, Wimbrow H, Barlow C, et al. A targeted dominant negative mutation of the thyroid hormone α1 receptor causes increased mortality, infertility, and dwarfism in mice. Proc Natl Acad Sci USA. (2001) 98:15095–100. doi: 10.1073/pnas.261565798
81. Markossian S, Guyot R, Richard S, Teixeira M, Aguilera N, Bouchet M, et al. CRISPR/Cas9 editing of the mouse thra gene produces models with variable resistance to thyroid hormone. Thyroid. (2018) 28:139–50. doi: 10.1089/thy.2017.0389
82. Plateroti M, Gauthier K, Domon-Dell C, Freund JN, Samarut J, Chassande O. Functional interference between thyroid hormone receptor α(TRα) and natural truncated TRDeltaα isoforms in the control of intestine development. Mol Cell Biol. (2001) 21:4761–72. doi: 10.1128/MCB.21.14.4761-4772.2001
83. Tinnikov A, Nordstrom K, Thoren P, Kindblom JM, Malin S, Rozell B, et al. Retardation of post-natal development caused by a negatively acting thyroid hormone receptor α1. EMBO J. (2002) 21:5079–87. doi: 10.1093/emboj/cdf523
84. Wikstrom L, Johansson C, Salto C, Barlow C, Campos Barros A, Baas F, et al. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor α1. EMBO J. (1998) 17:455–61. doi: 10.1093/emboj/17.2.455
85. Abel ED, Boers ME, Pazos-Moura C, Moura E, Kaulbach H, Zakaria M, et al. Divergent roles for thyroid hormone receptor β isoforms in the endocrine axis and auditory system. J Clin Invest. (1999) 104:291–300. doi: 10.1172/JCI6397
86. Forrest D, Hanebuth E, Smeyne RJ, Everds N, Stewart CL, Wehner JM, et al. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor β: evidence for tissue-specific modulation of receptor function. EMBO J. (1996) 15:3006–15. doi: 10.1002/j.1460-2075.1996.tb00664.x
87. Gauthier K, Chassande O, Plateroti M, Roux JP, Legrand C, Pain B, et al. Different functions for the thyroid hormone receptors TRα and TRβ in the control of thyroid hormone production and post-natal development. EMBO J. (1999) 18:623–31. doi: 10.1093/emboj/18.3.623
88. Hashimoto K, Curty FH, Borges PP, Lee CE, Abel ED, Elmquist JK, et al. An unliganded thyroid hormone receptor causes severe neurological dysfunction. Proc Natl Acad Sci USA. (2001) 98:3998–4003. doi: 10.1073/pnas.051454698
89. Hayashi Y, Janssen OE, Weiss RE, Murata Y, Seo H, Refetoff S. The relative expression of mutant and normal thyroid hormone receptor genes in patients with generalized resistance to thyroid hormone determined by estimation of their specific messenger ribonucleic acid products. J Clin Endocrinol Metab. (1993) 76:64–9. doi: 10.1210/jcem.76.1.8421105
90. Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, et al. Mice with a targeted mutation in the thyroid hormone β receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA. (2000) 97:13209–14. doi: 10.1073/pnas.230285997
91. Machado DS, Sabet A, Santiago LA, Sidhaye AR, Chiamolera MI, Ortiga-Carvalho TM, et al. A thyroid hormone receptor mutation that dissociates thyroid hormone regulation of gene expression in vivo. Proc Natl Acad Sci USA. (2009) 106:9441–6. doi: 10.1073/pnas.0903227106
92. Ng L, Hurley JB, Dierks B, Srinivas M, Salto C, Vennstrom B, et al. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet. (2001) 27:94–8. doi: 10.1038/83829
93. Suzuki SC, Bleckert A, Williams PR, Takechi M, Kawamura S, Wong RO. Cone photoreceptor types in zebrafish are generated by symmetric terminal divisions of dedicated precursors. Proc Natl Acad Sci USA. (2013) 110:15109–14. doi: 10.1073/pnas.1303551110
Keywords: thyroid hormones, thyroid hormone hyposensitivity, thyroid hormone metabolism defects, thyroid hormone cell membrane transport defects, thyroid hormones action defects
Citation: Rurale G, Cicco ED, Dentice M, Salvatore D, Persani L, Marelli F and Luongo C (2020) Thyroid Hormone Hyposensitivity: From Genotype to Phenotype and Back. Front. Endocrinol. 10:912. doi: 10.3389/fendo.2019.00912
Received: 28 October 2019; Accepted: 16 December 2019;
Published: 24 January 2020.
Edited by:
Noriyuki Koibuchi, Gunma University, JapanReviewed by:
Carmen Grijota-Martinez, Center for Biomedical Research in the Network of Rare Diseases (CIBERER), SpainPieter de Lange, University of Campania Luigi Vanvitelli, Italy
Copyright © 2020 Rurale, Cicco, Dentice, Salvatore, Persani, Marelli and Luongo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federica Marelli, ZmVkZXJpY2EubWFyZWxsaSYjeDAwMDQwO2hvdG1haWwuaXQ=