 Kazutaka Ueda
Kazutaka Ueda Yusuke Adachi
Yusuke Adachi Pangyen Liu1
Pangyen Liu1 Nobuaki Fukuma
Nobuaki Fukuma Eiki Takimoto
Eiki Takimoto- 1Department of Cardiovascular Medicine, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
- 2Division of Cardiology, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Premenopausal females have a lower incidence of death from cardiovascular disease (CVD) than male counterparts, supporting the notion that estrogen is protective against the development and progression of CVD. Although large-scale randomized trials of postmenopausal hormone replacement therapy failed to show cardiovascular benefits, recent ELITE study demonstrated anti-atherosclerotic benefits of exogenous estrogen depending on the initiation timing of the therapy. These results have urged us to better understand the mechanisms for actions of estrogens on CVD. Here, we review experimental and human studies, highlighting the emerging role of estrogen's non-nuclear actions linking to NO-cGMP signaling pathways.
Introduction
In developed countries, cardiovascular diseases (CVD) are the leading cause of death in males and females. As females are less likely to develop CVD before menopause, the endogenous female hormone estrogen appears to provide protection against CVD (1). Consistently, numerous experimental studies of CVD models affirmed the beneficial effects of estrogen treatment, including the inhibition of the development of atherosclerosis and endothelial dysfunction, and the reduction in myocardial ischemic injuries (2–4). While earlier large-scale randomized clinical studies of postmenopausal hormone replacement therapy showed rather adverse events without significant cardiovascular benefits (4, 5), a recent clinical study revealed that the timing for therapy initiation might be a critical determination factor (6).
Estrogen binds to multiple receptors, including classical nuclear estrogen receptors (ERs), ERα, and ERβ, and a new class of membrane G protein-coupled receptor, GPR30, also referred to as GPER (7). The ERs signal not only via “classical” regulation of gene transcription in the nucleus, but also via regulation of “non-nuclear” signaling pathways on ligand binding to ERs (1, 8). Accumulating evidence has shown a critical role for the non-nuclear ER signaling in maintaining the homeostasis of the cardiovascular system (9).
The Effects of Estrogen Hormone Therapy on the Cardiovascular System
Multiple cohort studies consistently revealed the lower risk of CVD with hormone therapy (HT), indicating that estrogen loss enhances the risk of CVD in postmenopausal females (2–4). On the other hand, randomized studies including the Women's Health Initiative (WHI) showed no cardiovascular benefits, but rather found an increased risk of stroke and deep vein thrombosis (4, 5). These discordant findings could reflect variations in the time between menopause and HT initiation: earlier cohort studies involved younger women who received HT early after menopause, and patients in randomized studies receive HT 10 years after menopause, where vascular endothelium might have lost its responsiveness to estrogen.
Indeed, several recent studies provided supporting evidence for this “timing hypothesis.” Estrogen alone reduced total mortality in early (<10 years) postmenopausal females (10). After 10 years of randomized treatment, women who received HT early after menopause showed a significantly reduced risk of mortality, heart failure, and myocardial infarction, without increases in the risk of cancer, venous thromboembolism, or stroke (11). The Early vs. Late Intervention Trial with Estradiol (ELITE) study enrolled postmenopausal females with and without histories of prior hysterectomy surgery (6). Approximately half of the participants were considered early menopause (<6 years; mean age, 55 years), and the other half were later menopause (at least 10 years; mean age, 65 years). Participants were randomized to receive either oral 17 β-estradiol or a placebo, and the carotid intima-medial thickness (CIMT) by ultrasound, an atherosclerosis marker predicting cardiovascular events, was assessed as the primary clinical outcome. Over 6 years, the early menopause participants who were randomized to 17 β-estradiol showed a slower progression of CIMT than those randomized to placebo (6). These results strongly indicate that HT exerts cardioprotective effects when initiated at an ideal time point after menopause.
These recent clinical findings encourage researchers to further investigate molecular and physiological function of ER-mediated signaling in the cardiovascular system (12, 13), which provides meaningful steps forward to the development of a next generation of HT for women that capture the beneficial cardiovascular effects of the hormone while minimizing its potential for harm.
Estrogen Receptors and Cardiovascular Cells
ERα and ERβ exhibit high homology and, as with all steroid hormone receptors, function as transcription factors that alter gene expression when activated (3). Functional ERs are expressed in vascular endothelial cells (EC), vascular smooth muscle cells (VSMC), and cardiomyocytes in humans and animals (3) (Figure 1). Some cardiovascular effects might be mediated by a transmembrane G-protein-coupled receptor GPR30 which is extensively reviewed elsewhere (7, 14), and thus this mini review will focus on ERα and ERβ.
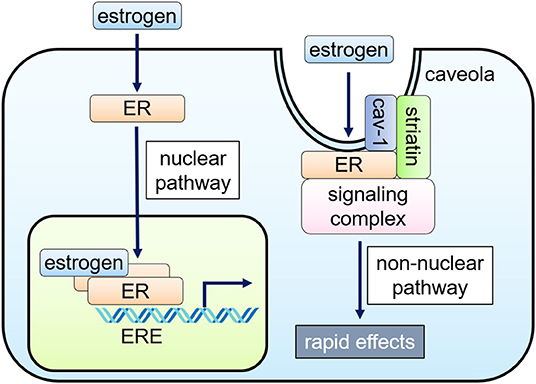
Figure 1. Nuclear and non-nuclear ER signaling pathways. Classically, the hormone-bound estrogen receptor (ER) dimerizes and binds to specific DNA sequences called estrogen response elements (ERE) and activates gene expression (nuclear pathway). Alternatively, ERs localized to caveolae, cell membrane microdomains, can signal without nuclear translocation through inducing a subpopulation of cell membrane–associated ERs to form a signaling complex that results in rapid activation of specific kinases and eNOS in endothelial cells (non-nuclear pathway).
Nuclear ER Signaling
In the nucleus, ligand-bound ERs interact with estrogen response elements (ERE) and function as transcription factors, regulating gene expression (1). Nuclear ER-estrogen complexes also modulate the function of other classes of transcription factors through protein-protein interactions; thus, controlling gene expression without direct binding to DNA (1). Cell type-specific recruitment of co-activators and displacement of co-repressors to DNA binding sites determine cellular response to estrogen (3, 15).
Non-nuclear ER Signaling
Cellular responses to estrogen occur within minutes, which are mediated via enzymatic pathways through the activation of membrane-associated ER, referred to as “rapid” or “non-nuclear” ER signaling (9, 16). Non-nuclear ER signaling has been documented in a variety of cell types in vitro, including oocytes, osteoblasts, osteoclasts, breast cancer cells, adipocytes, VSMC, EC, and cardiac myocytes (17–19). The rapid actions originate at the ERs located at caveolae, small invaginations of the cell membrane, activating kinases, or phosphatases to impact cell physiology (16, 20–22).
The non-nuclear ER signaling in the cardiovascular system has been most studied in EC where rapid eNOS activation by estrogen within 15–30 min was initially identified (23, 24). ERα, co-localized with caveolin-1, binds to a scaffold protein striatin, also associated with caveolin-1 (20, 25, 26). Endogenous ERβ was also detected in the EC membrane, particularly at the caveolae (27), but its associated proteins remain undetermined.
ERs-localized to caveolae activate PI3K, Akt kinase, and ERK1/2, enhancing phosphorylation of Ser-1177 of eNOS (22, 28–30). The activation of PI3K involves a complex process. ERα directly binds to the p85α regulatory subunit of PI3K in a ligand-dependent fashion (30), while PI3K activation requires c-Src kinase, whose SH2 domain interacts with phosphorylated Tyr-537 of ERα (31, 32). Gαi is also involved in this ERα complex at the caveolae, and the physical association of ERα with Gαi is required for eNOS activation (33, 34). On the other hand, striatin serves as a scaffold protein of the ERα complex at the caveolae. Since interruption of ERα-striatin binding, either with a peptide representing ERα amino acids 176-253 or the ERα triple point mutation (lysine 231, arginine 233, and arginine 234 to alanine: KRR), leads to defective non-nuclear signaling without affecting nuclear signaling, striatin also plays a key role to the non-nuclear signaling of ERα (25, 35, 36).
Estrogen-Independent ER Signaling
Interestingly, unliganded ERs or growth factor signaling-mediated ERs play a role in cellular physiology in the absence of estrogen. The presence of unliganded ERα alone decreases EC migration and EC proliferation, and increases SMC proliferation (37). Unliganded ERα also regulates gene expression of ECs. These effects either on physiology or gene expression are reversed by ERα activation in the presence of E2 (37). ERs can be activated in the absence of estrogen by growth factor receptor signaling such as epidermal growth factor and insulin-like growth factor receptors (38), and such estrogen-independent ER activation has been reportedly induced by different intercellular pathways in vascular as well as non-vascular cells (3, 16). Thus, cardioprotective effects of estrogen might be in part attributable to preventing the adverse cardiovascular effects of unliganded ERα by its binding to the receptor and other growth factor signal-mediated ERα activation.
Actions of Estrogen in Cardiovascular Cells
Functional ERs are expressed in multiple cell types which compose the heart. Estrogen regulates diverse cellular functions via nuclear and non-nuclear signaling pathways or ligand-independent signaling pathways; estrogen exerts inhibition of cellular hypertrophy and apoptosis in cardiomyocytes, proliferation of cardiac fibroblasts, proliferation and eNOS signaling activation in vascular endothelium, and anti-proliferative effect on vascular smooth muscle cells. In this section, we review cell-type specific in vitro studies, including cardiomyocytes, cardiac fibroblasts, vascular endothelial, and smooth muscle cells.
Cardiomyocytes
Cardiomyocytes express functional ERs, and estrogen regulates expression of cardiac genes, such as connexin 43, β-myosin heavy chain and ion channels (39, 40). Estrogen also regulates calcineurin abundance, cGMP-PKG activation, Akt activation, and miRNAs in cardiomyocytes to inhibit cellular hypertrophy and confer protection against apoptosis, where both nuclear and non-nuclear pathways might be involved (Figure 2). Sasaki et al. demonstrated rapid activation of cGMP-PKG in response to estrogen in cardiac myocytes, providing the link between estrogen and cGMP-PKG signaling, both of which are known to play protective roles against diverse cardiac pathologies, including cardiac hypertrophy, failure, and ischemic injuries (41). While cGMP-PKG inhibits activation of calcineurin, a central molecule for pathological hypertrophy, estrogen also enhances the degradation of calcineurin. Estrogen activates Akt and inhibits apoptosis via rapid signaling, while it down-regulates miR22 to activate Sp1 for anti-oxidant induction (42–44). Furthermore, Lin et al. reported that estrogen and ERβ-specific agonist diarylpropionitrile increased S-nitrosylation of heat shock proteins and decreased infarct area after ischemia-reperfusion in isolated mice hearts, while these effects were abolished in ERβ knockout mice or with a NOS inhibitor (45).
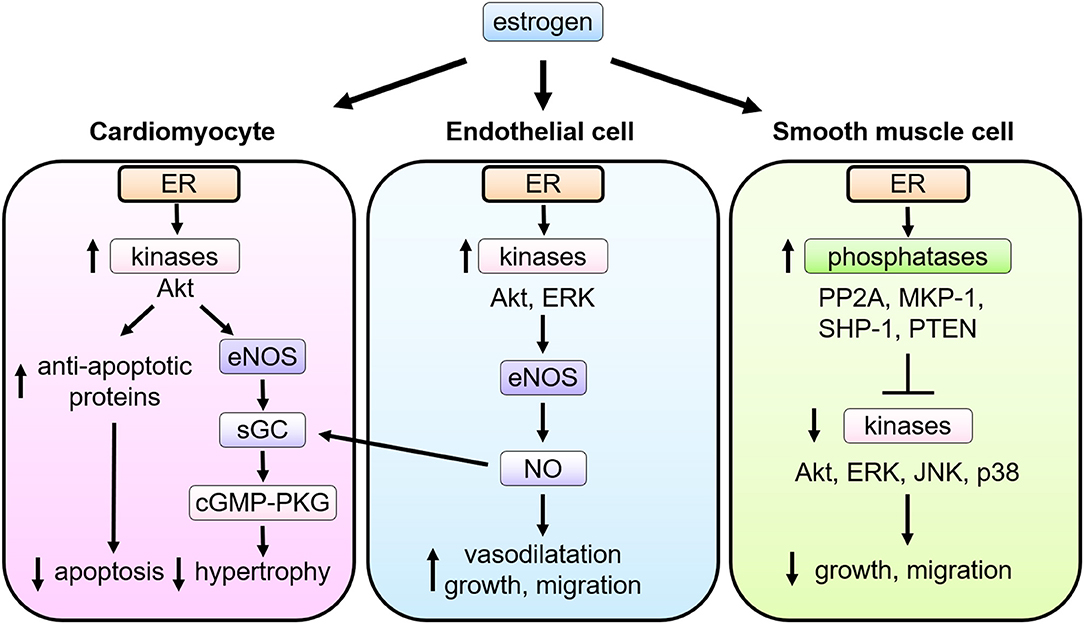
Figure 2. Multiple actions of estrogen-estrogen receptor signaling in cardiovascular cells. In cardiomyocytes, ER signaling leads to the phospho-activation of Akt and eNOS, the latter stimulating soluble guanylate cyclase (sGC) to activate cyclic guanosine monophosphate (cGMP)—PKG (cGMP-dependent protein kinase) signaling pathways and thereby attenuate pathological cardiac hypertrophy. In endothelial cells (EC), upon estrogen binding, ER signaling complex activate the tyrosine kinase Src, the serine/threonine kinase PI3K, and the kinase Akt. Akt then directly phosphorylates eNOS on serine 1177, leading to its enzymatic activation and production of NO, which promotes EC proliferation and migration as well as vasodilatation. NO produced in EC also functions as a stimulator of sGC in cardiomyocytes. In smooth muscle cells (VSMC), upon stimulation of estrogen, ER interacts with and activates several phosphatases including protein phosphatase 2A (PP2A), mitogen-activated protein kinase phosphatase 1 (MKP-1), Src homology region 2 domain-containing phosphatase-1 (SHP-1), and phosphatase and tensin homolog deleted from chromosome 10 (PTEN), and modulates activation of kinases induced by growth stimulation, leading to inhibition of VSMC proliferation and migration.
Cardiac Fibroblasts
Cardiac fibroblasts are the primary source of myocardial extracellular matrix proteins, matrix metalloproteinases (MMPs), growth factors, and cytokines, all of which contribute to cardiac remodeling on physiological and pathological levels (46). Lee and Eghbali-Webb reported that both ERα and β are expressed in fibroblasts isolated from female rats, where ERβ is predominant with both cytosolic and nuclear localization (47). They also reported that 17 β-estradiol enhances cardiac fibroblast proliferation through mitogen-activated protein kinase-dependent mechanisms (47).
Vascular Endothelial Cells
NO derived from eNOS activation exerts potent cardiovascular effects, including vascular relaxation, growth and migration of ECs and VSMCs, and the antagonism of platelet activation, thrombus formation, and leukocyte-EC adhesion (27, 29, 48–50). Estradiol conjugated to bovine serum albumin (E2-BSA), which does not pass through the cell membrane, was originally used to evaluate the role of cell membrane ER (51), and shown to promote phosphorylation of eNOS in cultured EC. Consistently, an estrogen-dendrimer conjugate (EDC) that activates membrane-associated ER, but is excluded from the nucleus (52), was shown to stimulate cultured EC proliferation and migration via ERα, heterotrimeric G protein Gi, and the activation of eNOS (53) (Figure 2).
Recent transcriptome analysis by Lu et al. using EC expressing KRR mutant ER revealed that estrogen regulates 60 genes in ECs expressing wild-type ERα, many of which were associated with cell migration and proliferation identified by pathway analysis. Meanwhile, only 10 genes were regulated by E2 in ECs expressing KRR ERα and thus lacking non-nuclear signaling pathway. These data provide the evidence that non-nuclear signaling also plays a pivotal role for the transcriptional responses of EC to estrogen (35).
Vascular Smooth Muscle Cells
Estrogen exerts anti-proliferative effects on VSMC (54–56). Cell proliferation is considerably regulated by kinase-mediated signal transduction. Treatment with estrogen inhibits phosphorylation of growth-related kinases, such as ERK1/2, JNK, p38, and Akt, which are phosphorylated and activated by growth factor stimulation (54, 55). Estrogen controls the expression and activity of several phosphatases in VSMC (57, 58), including MKP-1, SHP-1, PTEN, and PP2A, balancing the kinases (56, 59, 60). In cultured VSMCs, ERα and PP2A form a complex, and estrogen treatment increases PP2A activity resulting in the inhibition of growth-promoting signal activation (61). The role of non-nuclear ER signaling in VSMC proliferation was evaluated with using a transgenic mouse model (Disrupting Peptide Mouse; DPM), in which non-nuclear ER-mediated signaling was abolished by overexpression of the peptide representing amino acids 176–253 of ERα, preventing ER from forming a signaling complex with striatin (62). Estrogen inhibition on VSMC proliferation was lost in VSMC derived from the DPM (61), supporting the theory that the non-nuclear ER signaling pathway is required for the estrogen-mediated anti-proliferative effects in VSMC (Figure 2).
Estrogen Actions in Animal Models of Cardiovascular Diseases
To date, many studies using experimental models of CVD have been reported to elucidate the role of estrogen in the cardiovascular system. Most studies utilized pharmacological interventions or global gene deletion, whereas studies employing tissue-specific mutant models would provide more insights into our understanding the role of estrogen in CVD.
Ischemic Heart Diseases
ERα and ERβ are both reportedly involved in the cardioprotective effects of estrogen. Global ERβ KO mice revealed increased mortality and exacerbated heart failure after myocardial infarction (63). Consistently, cardiomyocyte-specific ERβ overexpression improved cardiac function and survival after myocardial infarction induced by left anterior descending coronary artery ligation. On the other hand, cardiac fibrosis post-myocardial infarction was attenuated with increased angiogenesis in female mice overexpressing ERα (64, 65).
In an ischemia-reperfusion model of ovariectomized global ERα KO mice, coronary endothelial dysfunction was not reversed by estrogen, while it was normalized in wild types (66). Other studies further revealed ERα KO demonstrated exacerbated ischemia reperfusion phenotype, including markedly impaired cardiac contractility, increased cardiomyocyte death, and mitochondrial damage (67, 68). On the other hand, ERβ KO female hearts exhibited poor functional recovery compared to wild-type or ERα KO mice in an ex vivo model of global ischemia-reperfusion (69). Mechanistically, estrogen mitigates reperfusion injuries after ischemia primarily by activating PI3K-Akt, increasing expression of the anti-apoptotic protein BCL-2 and decreasing the expression of the proapoptotic caspase proteins (43). Importantly, neither ischemic recovery nor PI3K-Akt activation was observed in hearts isolated from female ERβ KO mice (43, 70, 71). Together, ERβ appears to play substantial cardioprotective roles against ischemia reperfusion injury, while the role of ERα seems to differ depending on methodological conditions.
Estrogen might contribute to regenerative process in the heart. Cardiac stem cells treated with estrogen produce protective factors and improve cardiac function and cardiomyocyte survival when injected into hearts after ischemia-reperfusion injury (72). Both ERα and β contribute to estrogen-mediated endothelial progenitor cell activation and mobilization in tissues, helping preserve cardiac function after myocardial infarction (73). ERα selective agonist propylpyrazoletriol reduced apoptosis and increased survival of cardiac cells expressing c-Kit, a potential marker for adult cardiac stem cells, in infarcted hearts; whereas the ERβ selective agonist diarylpropionitrile had no effect (74).
Cardiac Hypertrophy and Failure
The heart develops pathological hypertrophy in response to various pathological stressors, such as genetic, mechanical, or excessive neurohormonal stress. If the stress is sustained, hypertrophy transits to failure. Sex difference has been known to be a modifier of human cardiomyopathy where estrogen also plays a role (75). Genetically-modified mouse models of hypertrophic cardiomyopathy presented sex differences of cardiac phenotype. Mice with a missense mutation (R403Q) in the α-myosin heavy chain exhibited severe biventricular hypertrophy. At 8 months of age, only male mice displayed overt heart failure phenotype (76, 77). Also, transgenic mice with a missense mutation (R92Q) in the cardiac troponin T exhibited more severe fibrosis with marked hypertrophic marker gene induction in males, compared to females (78). Importantly, ovariectomized mutants showed worse phenotype of further impaired contractile function and myocardial energy metabolism, and estrogen supplementation restored these parameters (79), suggesting estrogen's protective effects against cardiac hypertrophy and heart failure in this model.
Studies using global deletion of ERα or ERβ exposed to chronic angiotensin II or pressure-overload have suggested the role for ERβ in the estrogen's protection against hypertrophy and failure, where calcineurin and cGMP-PKG signaling regulation might be significantly involved (80). In particular, the link of estrogen to the latter signaling pathway might be important and deserves further investigation. Myocardial cGMP-PKG signaling pathway is de-activated in human heart failure with preserved ejection fraction (HFpEF), while cGMP-PKG activation is regarded as a new therapeutic strategy to treating heart failure. Considering HFpEF is associated with female sex and increased age independent of obesity and diabetes, it is reasonable to speculate estrogen loss and subsequent cGMP deactivation might contribute to the pathophysiology of HFpEF. Importantly, Sasaki et al. reported that estrogen signal is required in order to activate cGMP-PKG using a PDE5 inhibitor in female cardiac myocytes to ameliorate heart failure in female mice (41, 81, 82). There are several other reported molecular mechanisms including a mammalian target of rapamycin signaling, regulation of phosphorylation of p38 mitogen-activated protein kinase pathway, and regulation of cardiomyocyte histone deacetylases (83–85).
Vascular Injury and Atherosclerosis
Estrogen inhibits excessive responses to vascular injury in a mouse carotid artery injury model. Here, in vivo estrogen treatment inhibits the proliferation of VSMC and promotes re-endothelialization (53, 86–92). In ERβ KO mice, estrogen is still protective against vascular injury (86, 93), whereas, in ERα KO mice, estrogen treatment shows no protective effect on the vascular injury response (86, 90). This supports the concept that ERα is responsible for estrogen's protective effect on vasculature.
The role of the non-nuclear ER signaling pathway in estrogen-induced vascular protection was evaluated in a “gain of function” and “loss of function” study. EDC promoted carotid artery re-endothelialization in an ERα-dependent manner and diminished the development of neointimal hyperplasia following vascular injury in a manner equivalent to estrogen treatment (53). Of note, endometrial carcinoma cell growth in vitro and breast cancer xenograft growth in vivo were stimulated by estrogen, but not EDC (53). This suggests that selective activation of the non-nuclear signaling pathway did not promote cancer growth. Moreover, estrogen significantly decreases injury-induced neointimal hyperplasia while inhibiting VSMC proliferation in wild-type mice. Strikingly, estrogen had no significant effect in DPM mice (62). Taken together, non-nuclear signaling plays a major role in estrogen-induced protection against vascular injury, and compounds that activate non-nuclear signaling might provide a vascular benefit as non-nuclear selective ER modulators without increasing the risk of uterine or breast cancer.
Ligand-bound ERα mediates target gene transcription through the activation function 2 (AF2) domain, located on the C-terminal, and ligand binding domain of ERα. A recent report that used knock-in mice that lacked a functional AF2 domain showed that AF2 is needed to inhibit atherosclerosis (94). In contrast, the effect of estrogen on re-endothelialization after vascular injury was preserved in these mice (94), suggesting an essential role of the non-nuclear ER signaling on endothelial healing after vascular injury.
Conclusion
Large-scale, randomized clinical studies from the early 2000s failed to demonstrate cardiovascular benefits associated with post-menopausal estrogen treatment, and rather showed an increased risk of breast cancer and thrombosis. However, recent clinical trials and meta-analyses have suggested that estrogen treatment has beneficial effects for preventing CVD and does not affect mortality if HT is initiated early after menopause (6, 11). In addition to the timing of HT initiation, various factors including treatment duration, dose, formulation, regimen (estrogen or estrogen plus progesterone), and route of administration may affect cardiovascular outcomes. Greater clarity of the molecular mechanisms by which estrogen regulates specific cardiovascular processes is needed to optimize next-generation HT.
Various ERs are expressed in most cardiovascular cell types, including cardiomyocytes, fibroblasts, vascular EC, and VSMC. Each ER is involved in protective effects of estrogen in multiple animal disease models, including ischemic heart disease, cardiac hypertrophy, heart failure, vascular injury, and atherosclerosis. Emerging evidence has indicated the potential importance of the non-nuclear ER signaling on diverse aspects of the cardiovascular systems, supporting the potential opportunity to design pathway-specific selective ER modulators capable of regulating non-nuclear and nuclear effects, assisting with the development of personalized therapies for preventing and treating CVD. Further research will provide more insight into therapeutic approaches that translate basic science findings into clinical practice innovations.
Author Contributions
KU and ET wrote the manuscript. YA, PL, and NF critically revised the manuscript and contributed to designing the figures.
Funding
This work was supported by Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (KAKENHI) (Grant No. 18K08096), Yamaguchi Endocrine Research Foundation, The Ichiro Kanehara Foundation (KU), Japan Foundation for Applied Enzymology, Japan Heart Foundation Research Grant, SENSHIN Medical Research Foundation (ET), and Takeda Science Foundation (KU and ET).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. (2005) 308:1583–7. doi: 10.1126/science.1112062
2. Gorodeski GI. Update on cardiovascular disease in post-menopausal women. Best Pract Res Clin Obstet Gynaecol. (2002) 16:329–55. doi: 10.1053/beog.2002.0282
3. Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. (1999) 340:1801–11. doi: 10.1056/NEJM199906103402306
4. Turgeon JL, McDonnell DP, Martin KA, Wise PM. Hormone therapy: physiological complexity belies therapeutic simplicity. Science. (2004) 304:1269–73. doi: 10.1126/science.1096725
5. Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, et al. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med. (2003) 349:523–34. doi: 10.1056/NEJMoa030808
6. Hodis HN, Mack WJ, Henderson VW, Shoupe D, Budoff MJ, Hwang-Levine J, et al. Vascular effects of early versus late postmenopausal treatment with estradiol. N Engl J Med. (2016) 374:1221–31. doi: 10.1056/NEJMoa1505241
7. Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. (2009) 104:288–91. doi: 10.1161/CIRCRESAHA.108.190892
8. Sader MA, Celermajer DS. Endothelial function, vascular reactivity and gender differences in the cardiovascular system. Cardiovasc Res. (2002) 53:597–604. doi: 10.1016/S0008-6363(01)00473-4
9. Ueda K, Karas RH. Emerging evidence of the importance of rapid, non-nuclear estrogen receptor signaling in the cardiovascular system. Steroids. (2013) 78:589–96. doi: 10.1016/j.steroids.2012.12.006
10. Rossouw JE, Prentice RL, Manson JE, Wu L, Barad D, Barnabei VM, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. (2007) 297:1465–77. doi: 10.1001/jama.297.13.1465
11. Schierbeck LL, Rejnmark L, Tofteng CL, Stilgren L, Eiken P, Mosekilde L, et al. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ. (2012) 345:e6409. doi: 10.1136/bmj.e6409
12. Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ. (2017) 8:33. doi: 10.1186/s13293-017-0152-8
13. Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res. (2016) 118:994–1007. doi: 10.1161/CIRCRESAHA.115.305376
14. Feldman RD, Limbird LE. GPER (GPR30): a nongenomic receptor (GPCR) for steroid hormones with implications for cardiovascular disease and cancer. Annu Rev Pharmacol Toxicol. (2017) 57:567–84. doi: 10.1146/annurev-pharmtox-010716-104651
15. McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. (2002) 108:465–74. doi: 10.1016/S0092-8674(02)00641-4
16. Mendelsohn ME, Karas RH. Rapid progress for non-nuclear estrogen receptor signaling. J Clin Invest. (2010) 120:2277–9. doi: 10.1172/JCI43756
17. Cheskis BJ. Regulation of cell signalling cascades by steroid hormones. J Cell Biochem. (2004) 93:20–7. doi: 10.1002/jcb.20180
18. Norman AW, Mizwicki MT, Norman DPG. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Disc. (2004) 3:27–41. doi: 10.1038/nrd1283
19. Osborne CK, Schiff R. Estrogen-receptor biology: continuing progress and therapeutic implications. J Clin Oncol. (2005) 23:1616–22. doi: 10.1200/JCO.2005.10.036
20. Chambliss KL, Yuhanna IS, Mineo C, Liu P, German Z, Sherman TS, et al. Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ Res. (2000) 87:E44–52. doi: 10.1161/01.RES.87.11.e44
21. Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol. (2005) 19:1951–9. doi: 10.1210/me.2004-0390
22. Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. (2006) 20:1996–2009. doi: 10.1210/me.2005-0525
23. Caulin-Glaser T, García-Cardeña G, Sarrel P, Sessa WC, Bender JR. 17 beta-estradiol regulation of human endothelial cell basal nitric oxide release, independent of cytosolic Ca2+ mobilization. Circ Res. (1997) 81:885–92. doi: 10.1161/01.RES.81.5.885
24. Lantin-Hermoso RL, Rosenfeld CR, Yuhanna IS, German Z, Chen Z, Shaul PW. Estrogen acutely stimulates nitric oxide synthase activity in fetal pulmonary artery endothelium. Am J Physiol. (1997) 273(1 Pt 1):L119–26. doi: 10.1152/ajplung.1997.273.1.L119
25. Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc Natl Acad Sci USA. (2004) 101:17126–31. doi: 10.1073/pnas.0407492101
26. Razandi M, Alton G, Pedram A, Ghonshani S, Webb P, Levin ER. Identification of a structural determinant necessary for the localization and function of estrogen receptor alpha at the plasma membrane. Mol Cell Biol. (2003) 23:1633–46. doi: 10.1128/MCB.23.5.1633-1646.2003
27. Chambliss KL, Yuhanna IS, Anderson RGW, Mendelsohn ME, Shaul PW. ERbeta has nongenomic action in caveolae. Mol Endocrinol. (2002) 16:938–46. doi: 10.1210/mend.16.5.0827
28. Florian M, Lu Y, Angle M, Magder S. Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids. (2004) 69:637–45. doi: 10.1016/j.steroids.2004.05.016
29. Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors alpha and beta. J Biol Chem. (2005) 280:19704–10. doi: 10.1074/jbc.M501244200
30. Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. (2000) 407:538–41. doi: 10.1038/35035131
31. Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, et al. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem. (2003) 278:2118–23. doi: 10.1074/jbc.M210828200
32. Li L, Hisamoto K, Kim KH, Haynes MP, Bauer PM, Sanjay A, et al. Variant estrogen receptor-c-Src molecular interdependence and c-Src structural requirements for endothelial NO synthase activation. Proc Natl Acad Sci USA. (2007) 104:16468–73. doi: 10.1073/pnas.0704315104
33. Kumar P, Wu Q, Chambliss KL, Yuhanna IS, Mumby SM, Mineo C, et al. Direct interactions with G α i and G βγ mediate nongenomic signaling by estrogen receptor α. Mol Endocrinol. (2007) 21:1370–80. doi: 10.1210/me.2006-0360
34. Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, et al. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha (i). J Biol Chem. (2001) 276:27071–6. doi: 10.1074/jbc.M100312200
35. Lu Q, Schnitzler GR, Ueda K, Iyer LK, Diomede OI, Andrade T, et al. ER Alpha rapid signaling is required for estrogen induced proliferation and migration of vascular endothelial cells. PLoS ONE. (2016) 11:e0152807. doi: 10.1371/journal.pone.0152807
36. Ueda K, Takimoto E, Lu Q, Liu P, Fukuma N, Adachi Y, et al. Membrane-initiated estrogen receptor signaling mediates metabolic homeostasis via central activation of protein phosphatase 2A. Diabetes. (2018) 67:1524–37. doi: 10.2337/db17-1342
37. Lu Q, Schnitzler GR, Vallaster CS, Ueda K, Erdkamp S, Briggs CE, et al. Unliganded estrogen receptor alpha regulates vascular cell function and gene expression. Mol Cell Endocrinol. (2017) 442:12–23. doi: 10.1016/j.mce.2016.11.019
38. Hewitt SC, Winuthayanon W, Korach KS. What's new in estrogen receptor action in the female reproductive tract. J Mol Endocrinol. (2016) 56:R55–71. doi: 10.1530/JME-15-0254
39. Grohé C, Kahlert S, Löbbert K, Stimpel M, Karas RH, Vetter H, et al. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. (1997) 416:107–12. doi: 10.1016/S0014-5793(97)01179-4
40. Stice JP, Chen L, Kim S-C, Jung JS, Tran AL, Liu TT, et al. 17β-estradiol, aging, inflammation, and the stress response in the female heart. Endocrinology. (2011) 152:1589–98. doi: 10.1210/en.2010-0627
41. Sasaki H, Nagayama T, Blanton RM, Seo K, Zhang M, Zhu G, et al. PDE5 inhibitor efficacy is estrogen dependent in female heart disease. J Clin Invest. (2014) 124:2464–71. doi: 10.1172/JCI70731
42. Donaldson C, Eder S, Baker C, Aronovitz MJ, Weiss AD, Hall-Porter M, et al. Estrogen attenuates left ventricular and cardiomyocyte hypertrophy by an estrogen receptor–dependent pathway that increases calcineurin degradation. Circ Res. (2009) 104:265–75. doi: 10.1161/CIRCRESAHA.108.190397
43. Patten RD, Pourati I, Aronovitz MJ, Baur J, Celestin F, Chen X, et al. 17β-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ Res. (2004) 95:692–9. doi: 10.1161/01.RES.0000144126.57786.89
44. Wang L, Tang ZP, Zhao W, Cong BH, Lu JQ, Tang XL, et al. MiR-22/Sp-1 links estrogens with the up-regulation of cystathionine gamma-lyase in myocardium, which contributes to estrogenic cardioprotection against oxidative stress. Endocrinology. (2015) 156:2124–37. doi: 10.1210/en.2014-1362
45. Lin J, Steenbergen C, Murphy E, Sun J. Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation. (2009) 120:245–54. doi: 10.1161/CIRCULATIONAHA.109.868729
46. Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. (2002) 91:1103–13. doi: 10.1161/01.RES.0000046452.67724.B8
47. Lee HW, Eghbali-Webb M. Estrogen enhances proliferative capacity of cardiac fibroblasts by estrogen receptor- and mitogen-activated protein kinase-dependent pathways. J Mol Cell Cardiol. (1998) 30:1359–68. doi: 10.1006/jmcc.1998.0699
48. Jaba IM, Zhuang ZW, Li N, Jiang Y, Martin KA, Sinusas AJ, et al. NO triggers RGS4 degradation to coordinate angiogenesis and cardiomyocyte growth. J Clin Invest. (2013) 123:1718–31. doi: 10.1172/JCI65112
49. Mendelsohn ME. Nongenomic, ER-mediated activation of endothelial nitric oxide synthase: how does it work? What does it mean? Circ Res. (2000) 87:956–60. doi: 10.1161/01.RES.87.11.956
50. Shaul PW. Regulation of endothelial nitric oxide synthase: location, location, location. Annu Rev Physiol. (2002) 64:749–74. doi: 10.1146/annurev.physiol.64.081501.155952
51. Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, et al. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res. (2000) 87:677–82. doi: 10.1161/01.RES.87.8.677
52. Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, et al. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. (2006) 20:491–502. doi: 10.1210/me.2005-0186
53. Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Investig. (2010) 120:2319–30. doi: 10.1172/JCI38291
54. Li Q-y, Chen L, Zhu Y-h, Zhang M, Wang Y-p, Wang M-w. Involvement of estrogen receptor-β in farrerol inhibition of rat thoracic aorta vascular smooth muscle cell proliferation. Acta Pharmacol Sin. (2011) 32:433–40. doi: 10.1038/aps.2011.1
55. Ortmann J, Veit M, Zingg S, Santo SD, Traupe T, Yang Z, et al. Estrogen receptor-α but not -β or GPER inhibits high glucose-induced human VSMC proliferation: potential role of ROS and ERK. J Clin Endocrinol Metab. (2011) 96:220–8. doi: 10.1210/jc.2010-0943
56. Takeda-Matsubara Y, Nakagami H, Iwai M, Cui T-X, Shiuchi T, Akishita M, et al. Estrogen activates phosphatases and antagonizes growth-promoting effect of angiotensin II. Hypertension. (2002) 39:41–5. doi: 10.1161/hy1201.097197
57. Junttila MR, Li S-P, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. (2008) 22:954–65. doi: 10.1096/fj.06-7859rev
58. Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. (1999) 24:186–91. doi: 10.1016/S0968-0004(99)01375-4
59. Lu Q, Surks HK, Ebling H, Baur WE, Brown D, Pallas DC, et al. Regulation of estrogen receptor alpha-mediated transcription by a direct interaction with protein phosphatase 2A. J Biol Chem. (2003) 278:4639–45. doi: 10.1074/jbc.M210949200
60. Yang L, Wang Y, Chen P, Hu J, Xiong Y, Feng D, et al. Na(+)/H(+) exchanger regulatory factor 1 (NHERF1) is required for the estradiol-dependent increase of phosphatase and tensin homolog (PTEN) protein expression. Endocrinology. (2011) 152:4537–49. doi: 10.1210/en.2011-1207
61. Ueda K, Lu Q, Baur W, Aronovitz MJ, Karas RH. Rapid estrogen receptor signaling mediates estrogen-induced inhibition of vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol. (2013) 33:1837–43. doi: 10.1161/ATVBAHA.112.300752
62. Bernelot Moens SJ, Schnitzler GR, Nickerson M, Guo H, Ueda K, Lu Q, et al. Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation. (2012) 126:1993–2004. doi: 10.1161/CIRCULATIONAHA.112.124529
63. Pelzer T, Loza P-AA, Hu K, Bayer B, Dienesch C, Calvillo L, et al. Increased mortality and aggravation of heart failure in estrogen receptor-beta knockout mice after myocardial infarction. Circulation. (2005) 111:1492–8. doi: 10.1161/01.CIR.0000159262.18512.46
64. Mahmoodzadeh S, Leber J, Zhang X, Jaisser F, Messaoudi S, Morano I, et al. Cardiomyocyte-specific estrogen receptor alpha increases angiogenesis, lymphangiogenesis and reduces fibrosis in the female mouse heart post-myocardial infarction. J Cell Sci Ther. (2014) 5:153. doi: 10.4172/2157-7013.1000153
65. Schuster I, Mahmoodzadeh S, Dworatzek E, Jaisser F, Messaoudi S, Morano I, et al. Cardiomyocyte-specific overexpression of oestrogen receptor beta improves survival and cardiac function after myocardial infarction in female and male mice. Clin Sci. (2016) 130:365–76. doi: 10.1042/CS20150609
66. Favre J, Gao J, Henry J-P, Remy-Jouet I, Fourquaux I, Billon-Gales A, et al. Endothelial estrogen receptor α plays an essential role in the coronary and myocardial protective effects of estradiol in ischemia/reperfusion. Arterioscler Thromb Vasc Biol. (2010) 30:2562–7. doi: 10.1161/ATVBAHA.110.213637
67. Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. Estrogen receptor-α mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol. (2006) 290:H2204–9. doi: 10.1152/ajpheart.01219.2005
68. Zhai P, Eurell TE, Cooke PS, Lubahn DB, Gross DR. Myocardial ischemia-reperfusion injury in estrogen receptor-alpha knockout and wild-type mice. Am J Physiol Heart Circ Physiol. (2000) 278:H1640–7. doi: 10.1152/ajpheart.2000.278.5.H1640
69. Gabel SA, Walker VR, London RE, Steenbergen C, Korach KS, Murphy E. Estrogen receptor beta mediates gender differences in ischemia/reperfusion injury. J Mol Cell Cardiol. (2005) 38:289–97. doi: 10.1016/j.yjmcc.2004.11.013
70. Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Kararigas G, et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integr Comp Physiol. (2010) 298:R1597–606. doi: 10.1152/ajpregu.00825.2009
71. Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, et al. Estrogen receptor beta mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol. (2009) 296:R972–8. doi: 10.1152/ajpregu.00045.2009
72. Wang L, Gu H, Turrentine M, Wang M. Estradiol treatment promotes cardiac stem cell (CSC)-derived growth factors, thus improving CSC-mediated cardioprotection after acute ischemia/reperfusion. Surgery. (2014) 156:243–52. doi: 10.1016/j.surg.2014.04.002
73. Hamada H, Kim MK, Iwakura A, Ii M, Thorne T, Qin G, et al. Estrogen receptors alpha and beta mediate contribution of bone marrow-derived endothelial progenitor cells to functional recovery after myocardial infarction. Circulation. (2006) 114:2261–70. doi: 10.1161/CIRCULATIONAHA.106.631465
74. Brinckmann M, Kaschina E, Altarche-Xifro W, Curato C, Timm M, Grzesiak A, et al. Estrogen receptor alpha supports cardiomyocytes indirectly through post-infarct cardiac c-kit+ cells. J Mol Cell Cardiol. (2009) 47:66–75. doi: 10.1016/j.yjmcc.2009.03.014
75. van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. (2013) 123:37–45. doi: 10.1172/JCI62839
76. McKee LA, Chen H, Regan JA, Behunin SM, Walker JW, Walker JS, et al. Sexually dimorphic myofilament function and cardiac troponin I phosphospecies distribution in hypertrophic cardiomyopathy mice. Arch Biochem Biophys. (2013) 535:39–48. doi: 10.1016/j.abb.2012.12.023
77. Olsson MC, Palmer BM, Leinwand LA, Moore RL. Gender and aging in a transgenic mouse model of hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2001) 280:H1136–44. doi: 10.1152/ajpheart.2001.280.3.H1136
78. Maass AH, Ikeda K, Oberdorf-Maass S, Maier SK, Leinwand LA. Hypertrophy, fibrosis, and sudden cardiac death in response to pathological stimuli in mice with mutations in cardiac troponin T. Circulation. (2004) 110:2102–9. doi: 10.1161/01.CIR.0000144460.84795.E3
79. Chen Y, Zhang Z, Hu F, Yang W, Yuan J, Cui J, et al. 17beta-estradiol prevents cardiac diastolic dysfunction by stimulating mitochondrial function: a preclinical study in a mouse model of a human hypertrophic cardiomyopathy mutation. J Steroid Biochem Mol Biol. (2015) 147:92–102. doi: 10.1016/j.jsbmb.2014.12.011
80. Kim JK, Levin ER. Estrogen signaling in the cardiovascular system. Nuclear Recept Signal. (2006) 4:e013. doi: 10.1621/nrs.04013
81. Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. (2005) 111:1601–10. doi: 10.1161/01.CIR.0000160359.49478.C2
82. Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. (2005) 11:214–22. doi: 10.1038/nm1175
83. Gurgen D, Kusch A, Klewitz R, Hoff U, Catar R, Hegner B, et al. Sex-specific mTOR signaling determines sexual dimorphism in myocardial adaptation in normotensive DOCA-salt model. Hypertension. (2013) 61:730–6. doi: 10.1161/HYPERTENSIONAHA.111.00276
84. Pedram A, Razandi M, Narayanan R, Dalton JT, McKinsey TA, Levin ER. Estrogen regulates histone deacetylases to prevent cardiac hypertrophy. Mol Biol Cell. (2013) 24:3805–18. doi: 10.1091/mbc.e13-08-0444
85. van Eickels M, Grohe C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA. 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation. (2001) 104:1419–23. doi: 10.1161/hc3601.095577
86. Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen receptor-beta. Circulation. (2001) 103:423–8. doi: 10.1161/01.CIR.103.3.423
87. Hayashi T, Jayachandran M, Sumi D, Thakur NK, Esaki T, Muto E, et al. Physiological concentration of 17β-estradiol retards the progression of severe atherosclerosis induced by a high-cholesterol diet plus balloon catheter injury role of NO. Arterioscler Thromb Vasc Biol. (2000) 20:1613–21. doi: 10.1161/01.ATV.20.6.1613
88. Iafrati MD, Karas RH, Aronovitz M, Kim S, Sullivan TR Jr, Lubahn DB, et al. Estrogen inhibits the vascular injury response in estrogen receptor alpha-deficient mice. Nat Med. (1997) 3:545–8. doi: 10.1038/nm0597-545
89. Nofer J-R. Estrogens and atherosclerosis: insights from animal models and cell systems. J Mol Endocrinol. (2012) 48:R13–29. doi: 10.1530/JME-11-0145
90. Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, et al. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circ Res. (2002) 90:1087–92. doi: 10.1161/01.RES.0000021114.92282.FA
91. Sullivan TR, Karas RH, Aronovitz M, Faller GT, Ziar JP, Smith JJ, et al. Estrogen inhibits the response-to-injury in a mouse carotid artery model. J Clin Invest. (1995) 96:2482–8. doi: 10.1172/JCI118307
92. Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, et al. 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat Med. (2007) 13:1185–92. doi: 10.1038/nm1641
93. Karas RH, Hodgin JB, Kwoun M, Krege JH, Aronovitz M, Mackey W, et al. Estrogen inhibits the vascular injury response in estrogen receptor beta-deficient female mice. Proc Natl Acad Sci USA. (1999) 96:15133–6. doi: 10.1073/pnas.96.26.15133
94. Billon-Galés A, Krust A, Fontaine C, Abot A, Flouriot G, Toutain C, et al. Activation function 2 (AF2) of estrogen receptor-α is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci USA. (2011) 108:13311–6. doi: 10.1073/pnas.1105632108
Keywords: estrogen, cardiovascular diseases, receptor signaling, non-nuclear pathway, animal study
Citation: Ueda K, Adachi Y, Liu P, Fukuma N and Takimoto E (2020) Regulatory Actions of Estrogen Receptor Signaling in the Cardiovascular System. Front. Endocrinol. 10:909. doi: 10.3389/fendo.2019.00909
Received: 14 May 2019; Accepted: 12 December 2019;
Published: 10 January 2020.
Edited by:
Pierre De Meyts, Université Catholique de Louvain, BelgiumReviewed by:
Richard T. Premont, Harrington Discovery Institute, United StatesDawn A. Lowe, University of Minnesota Twin Cities, United States
Copyright © 2020 Ueda, Adachi, Liu, Fukuma and Takimoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eiki Takimoto, ZXRha2ltb3RvLXRreSYjeDAwMDQwO3VtaW4uYWMuanA=