
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 22 November 2019
Sec. Endocrinology of Aging
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00811
This article is part of the Research TopicOvarian Ageing: Pathophysiology and Recent Development of Maintaining Ovarian ReserveView all 7 articles
 Hiroyuki Sasaki1
Hiroyuki Sasaki1 Toshio Hamatani1*Shintaro Kamijo1Maki Iwai1Masato Kobanawa1Seiji Ogawa1
Toshio Hamatani1*Shintaro Kamijo1Maki Iwai1Masato Kobanawa1Seiji Ogawa1 Kenji Miyado2Mamoru Tanaka1
Kenji Miyado2Mamoru Tanaka1Reproductive capacity in women starts to decline beyond their mid-30s and pregnancies in older women result in higher rates of miscarriage with aneuploidy. Age-related decline in fertility is strongly attributed to ovarian aging, diminished ovarian reserves, and decreased developmental competence of oocytes. In this review, we discuss the underlying mechanisms of age-related decline in oocyte quality, focusing on oxidative stress (OS) in oocytes. The primary cause is the accumulation of spontaneous damage to the mitochondria arising from increased reactive oxygen species (ROS) in oocytes, generated by the mitochondria themselves during daily biological metabolism. Mitochondrial dysfunction reduces ATP synthesis and influences the meiotic spindle assembly responsible for chromosomal segregation. Moreover, reproductively aged oocytes produce a decline in the fidelity of the protective mechanisms against ROS, namely the ROS-scavenging metabolism, repair of ROS-damaged DNA, and the proteasome and autophagy system for ROS-damaged proteins. Accordingly, increased ROS and increased vulnerability of oocytes to ROS lead to spindle instability, chromosomal abnormalities, telomere shortening, and reduced developmental competence of aged oocytes.
In reproductive health, a decrease in pregnancy rate and an increase in miscarriage rate are observed in women aged 35 and older. While the rate of live baby born per oocyte is 26% for women under age 35, it deteriorates at an accelerating pace and finally drops to 1% for those over age 42 (1). There has been extensive discussion about reduced quality of oocytes during aging, which is considered to cause chromosomal aberration in embryos and reduced embryonic developmental competence. The aging phenomena can be attributed to accumulated oxidative damage in somatic cells (2, 3), as mammalian oocytes also show increased ROS levels with age (4–6). The relationship between oxidative stress and oocyte deterioration has been well-investigated (7–10). Moreover, antioxidants such as melatonin and coenzyme Q10 have anti-aging effects on mouse oocytes by regulating mitochondrial functions and ROS levels in oocytes during reproductive aging (11–13). In this review, using the latest literature, we discuss the pathophysiology in which oocytes are more exposed and vulnerable to OS as they age, and the mechanisms by which oocyte quality is degraded.
Mitochondria are important organelles that act as sites of energy production in aerobic respiration. Mitochondria produce ATP during energy metabolism utilizing the redox reaction in the respiratory chain complex located on the inner membrane. At the same time, most of the superoxides produced in vivo are generated in mitochondria (14). ROS are constantly generated in the mitochondria of aerobic organisms, but are also eliminated by antioxidant enzymes in the mitochondria thus maintaining redox balance and homeostasis. However, when ROS are excessively generated or the antioxidant ability is reduced because of aging or disease, the redox balance is lost and ROS are accumulated. The increase in OS is closely related to mitochondrial dysfunction (15–17).
Mitochondrial dysfunction is indeed correlated with aging in somatic cells. For example, mammalian aging has been correlated with the accumulation of mtDNA deletions/mutations and reduced mitochondrial respiratory chain function (18, 19). mtDNA is vulnerable and easily mutated owing to its proximity to the respiratory chain producing ROS, its lack of protective histones, and deficiency of efficient repair mechanisms (20). Mitochondrial gene mutations in turn lead to reduced mitochondrial function, i.e., disturbance of the redox balance and OS. A mouse model called “mtDNA mutator mouse,” which harbors a D257A mutation in the exonuclease “proofreading” domain of DNA polymerase-γ (Polg) gene, exhibits a progeroid phenotype. Accelerated accumulation of mtDNA mutations and mitochondrial dysfunction lead to a systemic premature aging phenotype including reduced fertility (17, 21). Likewise, in oocytes, mitochondria are closely related to the decline of oocyte quality with age (22). Point mutations and deletions in the mtDNA in oocytes are also found with maternal aging (23–25). Moreover, embryos obtained from elderly women have an abnormally high copy number of mtDNA and such embryos do not implant (26–28). It can be presumed that a vicarious increase in mtDNA copy number via mitochondrial biogenesis may effectively compensate for heteroplasmic mtDNA mutations and mitochondrial dysfunction (15). However, considering the qualitative aspect, the relationship between mtDNA integrity in oocytes and ovarian aging is controversial. The accumulation of the 4977-bp deletion is related to age due to OS in somatic cells (29). Although the 4977-bp deletion also tends to increase with age in oocytes, the frequency of the mitochondrial mutation and IVF failure are not significantly correlated (30).
Morphological and ultrastructural changes of oocytes with age are also observed. They include increase of irregular mitochondria with changed matrix density, ooplasmic fraction of vacuoles and dilated smooth endoplasmic reticulum (ER) and Golgi complex (31, 32). In vitro matured eggs from aged mice did not have a cortical distribution of active mitochondria shown in those from young mice (33). Mitochondrial fusion and fission, adapting the size and shape, also play a critical role in maintaining the function in response to changes in the metabolic milieu. Mitofusin 1 and 2 mediate mitochondrial fusion. Oocyte-specific targeted deletion of either gene results in severely reduced oocyte quality with elevated ROS levels (34, 35). Moreover, mitochondrial fission maintains the competency of oocytes via multi-organelle rearrangement. A study on Dynamin-related protein 1 (Drp1) known as an oocyte-specific mitochondrial fission factor showed that mitochondria are highly aggregated with other organelles (e.g., ER and secretory vesicles) in oocytes from Drp1-deficient mice, resulting in impaired Ca2+ signaling and meiotic resumption. Oocytes from aged mice also showed a decrease in Drp1-dependent mitochondrial fission and defective organelle morphogenesis, which is similarly observed in Drp1 KO oocytes (36). Since Drp1 is also recruited by OS and plays a protective role (37), the decline in oocyte quality with age may be associated with a decrease in Drp1 responsiveness to OS (38).
Mitochondrial dysfunction, which is caused by or causes OS, induces chromosomal non-disjunction, fertilization failure, and decrease in embryo competence (16, 39, 40). Since a large amount of ATP is consumed in the process of meiosis completion, fertilization, and embryonic development, it is possible that the decrease in ATP production due to deterioration of mitochondrial function results in a decrease in oocyte quality (41, 42). A study has shown that ATP contents ≧ 2 pmol/oocyte are necessary for oocytes to support normal embryo development (43). The reduced ATP production in oocyte may lead to a dysfunction of the spindle assembly checkpoint (SAC). A predominant mechanism of SAC silencing is dynein-mediated transport of certain kinetochore proteins along microtubules. ATP reduction prevents the release of dynein and its cargoes from the spindle poles and the redistribution of the core SAC proteins from attached kinetochores to spindle poles in metaphase-arrested cells, at a time when the SAC should be satisfied and silenced (44). The fidelity of SAC is compromised in aged oocytes, suggesting that SAC failure is a likely contributor to the increased incidence of chromosome abnormalities documented in oocytes and embryos of older women (45).
The efficacy of DNA double-strand break (DSB) repair mechanisms is attenuated in aged oocytes. Moreover, oocytes are acutely susceptible to accumulated DNA damage by reason of their extended prophase arrest. Increased oxidative damage brought about by mutations in mtDNA and the oxidative DNA repair enzyme OGG1 leads to accelerated aging phenotypes including spindle and chromosomal abnormalities in senescence-accelerated mice (46). An increase in the expression of the DNA DSB damage marker γH2AX in primordial follicles and germinal vesicle oocytes from aged mice and humans correlates with a decline in the expression of several DNA DSB repair genes including Brca1, Mre11, Atm, and Rad51 (47). RNAi-mediated reduction of Brca1 in oocytes results in abnormal spindle formation, chromosome misalignment, and a significant increase in hyperploid oocytes (48).
A global gene expression analysis of aged oocytes in mice revealed the decrease in mRNA expression of mitochondrial antioxidant genes, peroxiredoxin 3 (Prdx3) and thioredoxin 2 (Txn2), as well as cytosolic antioxidant genes, glutaredoxin 1 (Glrx1), glutathione S-transferase mu 2 (Gstm2), and superoxide dismutase 1 (Sod1) (49, 50). Prdx3, abundantly distributed in mitochondria, plays a key role as a regulator of mitochondrial H2O2 concentration and apoptosis (51). Another analysis has also reported reduced expression of Sod1 and Txn family members in MII oocytes from aged mice (48). SOD1 is highly expressed in human oocytes (52) and the addition of Sod1 protein improves preimplantation development in mice (53). Embryos from SOD1-deficient mouse oocytes have significantly higher levels of superoxide than wild-type embryos and their preimplantation development is halted at the 2-cell stage under atmospheric oxygen. Instead of any treatments with antioxidants, only hypoxic culture with 1% O2 negated the 2-cell arrest (54). Interestingly, knockdown of either the cytoplasmic or mitochondrial SOD in Drosophila significantly increases the percentage of oocytes showing arm cohesion defects and provokes segregation errors (55). Chemical inhibition of SOD activity in porcine oocytes elicits a reduction in meiotic progression, decreased GSH levels, and diminished rates of cleavage and blastocyst formation (56). The depletion of GSH is also associated with altered spindle morphology, disturbed microtubule function, and chromosome clumping in hamster and bovine MII oocytes (57, 58). Furthermore, thioredoxins are also involved in the reduction and protection against oxidative stress-induced apoptosis, and the targeted mutation of Txn1 causes embryonic lethality shortly after implantation (59). The decline in the fidelity of these protective mechanisms against OS collectively renders aged oocytes vulnerable to OS.
OS is involved in the effect of aging on reproductive function by causing ER stress (60, 61). Acting as a major site for the biosynthesis of proteins, lipids and secretory proteins, the ER plays a key role in meeting the oocyte's increased demand for new proteins during oocyte maturation and embryo development. Therefore, ER stress and homeostasis play an important role in determining oocyte quality. OS can induce ER stress and an adaptive signaling cascade known as the unfolded protein response (UPR) by impeding correct protein folding and calcium homeostasis (62, 63). If the UPR-mediated response fails in correcting the protein-folding defect, apoptosis is activated. Interestingly, lycium barbarum polysaccharide (LBP), extracted from the traditional Chinese herbal medicine goji berry, has antioxidant and cryoprotective properties, and improves the developmental competence of mouse oocytes that were vitrified/warned at the germinal vesicle stage with cumulus cells (64). LBP may reduce ER stress, activate both PI3K/AKT and MAPK3/1, and prevent cell death (64).
There is also a theory that proteasome dysfunction due to accumulation of oxidatively induced damage of functional proteins is also involved in the deterioration of oocyte quality with age. An age-related decline in proteasome activity has been demonstrated in a multitude of mammalian tissues and cells (60–68). In fact, a comprehensive analysis revealed that in oocytes, many proteasome-related genes are expressed less with increasing age (50). Decreased proteasome activity in naturally aged mouse oocytes are positively correlated with increased protein modification by 4-hydroxynonenal (4-HNE), which is elevated by the lipid peroxidation chain reaction in conditions involving oxidative stress (65). An exposure of germinal vesicle oocytes to either H2O2 or 4-HNE contributes to decreased meiotic completion, increased spindle abnormalities, chromosome misalignments and aneuploidy (66).
Autophagy is an evolutionarily conserved phenomenon by which unwanted intracellular proteins and organelles are sequestered within autophagosomes and delivered to lysosomes for degradation. Autophagy was found as a cell survival mechanism in starving cells, and also has a role in cell death. It is generally accepted that autophagy induces ROS, but also reduces oxidative damage (69). Autophagy has an important role in removing damaged mitochondria by mitophagy and in reducing ER stress. Autophagy has been observed in mouse, rat, and porcine oocytes. In rat ovaries, all phases of the estrous cycle contain oocytes that simultaneously express proteins involved both in apoptosis and autophagy (67). Autophagy-defective oocytes, obtained from oocyte-specific Atg5 knockout mice, could not develop beyond the four- and eight-cell stages after fertilization with Atg5-null sperm, suggesting a critical role of autophagy in pre-implantation mammalian development (68). In bovine embryos, a transient increase in autophagy also leads to decreased ER stress, which has a positive influence on in vitro preimplantation development (70). The consequences of autophagy modulation, including those that are mediated by OS during aging, may either promote cell survival or be associated with cell death (71). Decreased autophagy may provide a cellular environment allowing for the accumulation of dysfunctional mitochondria (72).
The sirtuin family is involved in regulating the energy metabolism and stress resistance of mammalian cells (73). Recently, SIRT1 and SIRT3 have been revealed to have an important role as sensors and protectors of the redox balance in oocytes, granulosa cells, and early embryos (74). SIRT1 and SIRT2 have been found in both the nucleus and cytosol, while SIRT3, SIRT4, and SIRT5 have been detected exclusively in mitochondria; SIRT6 and SIRT7 have been localized only in the nuclear compartment (75–77). The antioxidant response, the “FoxO3a-MnSod axis” orchestrated by SIRT1, is attenuated with age in oocytes (78). Several studies have investigated anti-aging treatment with resveratrol or calorie restriction focusing on SIRT1 (69, 79). SIRT3 also acts in a protective role against stress conditions in preimplantation embryos (80). Decreased expression of SIRT3 correlates with lower embryonic developmental competence (81). Melatonin, which enhances SIRT1 and SIRT3 activity, is considered effective as a treatment for aging oocytes (81, 82).
OS and telomere shortening are correlated exponentially with aging of somatic cells (83). The ROS generated by compromised mitochondria could potentially oxidize proteins necessary for telomere maintenance (84). Telomeres lack protective proteins and sit in the nuclear membrane, where they are susceptible to lipid peroxidation (85). Intrinsically, their sequences are rich in guanine, which is quite susceptible to oxidation. Telomere shortening is also involved in failure of spindle formation, arrest of embryonic development, and fragmentation in oocytes (86). In an experiment administering a cigarette smoke condensate (CSC) to mouse 1-cell zygotes, OS induced chromosomal aberrations in mouse embryos via telomere shortening and loss, but the antioxidant N-acetyl-L-cysteine (NAC) prevented the defects induced by CSC (8). Fertilized mouse eggs treated with FCCP, which uncouples the mitochondrial electron transport pathway, also show significantly increased ROS and decreased developmental competence with telomere shortening and chromosomal fusion compared to control embryos (84). In reproductively aged mouse oocytes, Q-PCR and quantitative fluorescence in situ hybridization analyses show significant increase of OS and shortening of telomere lengths (6, 87). Human oocytes with shorter telomeres develop into more fragmented and more aneuploid preimplantation embryos with lower implantation rates (88, 89), whereas relative telomere length was comparable in aneuploid and euploid first polar bodies and blastomeres (90). Further, SIRT6, associated with oxidative homeostasis, has been identified as an important modulator of telomeres in age-related deterioration of mouse oocytes (91, 92). Overexpression of SIRT6 in oocytes from aged mice promotes telomere elongation in 2-cell embryos and lowers the incidence of apoptotic blastomeres (91). Further studies on the age-related alteration of telomere length in mammalian oocytes seem necessary.
In recent years, social progress of women has resulted in delayed reproduction. Reproductive medicine faces serious challenges because aging causes oocyte quality to deteriorate. This deterioration of oocyte quality is influenced by OS. OS damages many cellular components, including mitochondria, lipids, proteins, enzymes, and DNA, leading to ATP shortage, DNA break, chromosomal segregation error, dysregulation of autophagy and proteasome system (Figure 1). In particular, mitochondria are the most significant targets of OS as they are pivotal in controlling cell survival and death. Moreover, a theory showing DNA methylation and epigenetic errors as influencing OS on germ cells has also been proposed (50, 93, 94). However, direct evidence for a participation of OS in the aging process of human oocytes and mechanisms of protection against OS is still to be gathered.
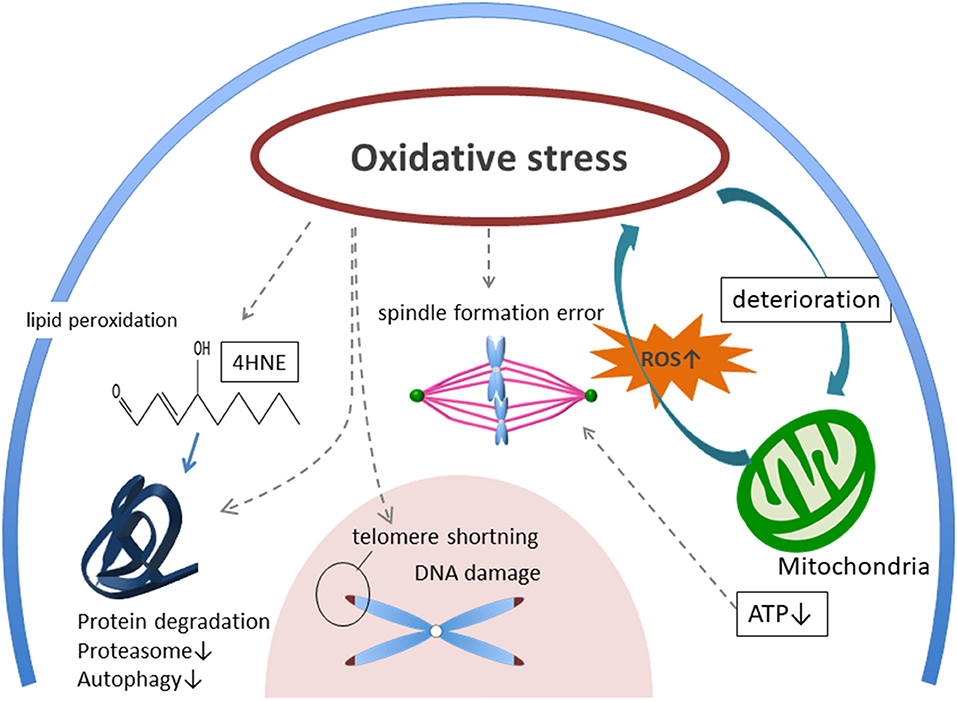
Figure 1. Possible mechanism of aged oocyte deterioration with accumulating oxidative stress. External ROS, AGEs, and accumulation of internal ROS from mitochondria are burdening oocytes as oxidative stress (OS). Then, OS induces deterioration of mitochondria, telomere shortening, spindle formation error, DNA damage, and protein degradation. ROS, reactive oxygen species. AGEs, glycation end-products. RAGE, receptor for advanced glycation end-products. 4-HNE, 4-hydroxynonenal.
HS and TH contributed to the drafting. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported in part, by Grants-in-Aid from the Japan Society for the Promotion of Science (Kiban-B-16H05475 to TH and Kiban-A-19H01067 to TH and KM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Silber SJ, Kato K, Aoyama N, Yabuuchi A, Skaletsky H, Fan Y, et al. Intrinsic fertility of human oocytes. Fertil Steril. (2017) 107:1232–7. doi: 10.1016/j.fertnstert.2017.03.014
2. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. (2000) 408:239–47. doi: 10.1038/35041687
3. Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. (1996) 273:59–63. doi: 10.1126/science.273.5271.59
4. Igarashi H, Takahashi T, Nagase S. Oocyte aging underlies female reproductive aging: biological mechanisms and therapeutic strategies. Reprod Med Biol. (2015) 14:159–69. doi: 10.1007/s12522-015-0209-5
5. Li YJ, Han Z, Ge L, Zhou CJ, Zhao YF, Wang DH, et al. C-phycocyanin protects against low fertility by inhibiting reactive oxygen species in aging mice. Oncotarget. (2016) 7:17393–409. doi: 10.18632/oncotarget.8165
6. Yamada-Fukunaga T, Yamada M, Hamatani T, Chikazawa N, Ogawa S, Akutsu H, et al. Age-associated telomere shortening in mouse oocytes. Rep Biol Endocrinol. (2013) 11:108. doi: 10.1186/1477-7827-11-108
7. Wiener-Megnazi Z, Vardi L, Lissak A, Shnizer S, Reznick AZ, Ishai D, et al. Oxidative stress indices in follicular fluid as measured by the thermochemiluminescence assay correlate with outcome parameters in in vitro fertilization. Fertil Steril. (2004) 82 (Suppl. 3):1171–6. doi: 10.1016/j.fertnstert.2004.06.013
8. Huang J, Okuka M, McLean M, Keefe DL, Liu L. Telomere susceptibility to cigarette smoke-induced oxidative damage and chromosomal instability of mouse embryos in vitro. Free Rad Biol Med. (2010) 48:1663–76. doi: 10.1016/j.freeradbiomed.2010.03.026
9. Appasamy M, Jauniaux E, Serhal P, Al-Qahtani A, Groome NP, Muttukrishna S. Evaluation of the relationship between follicular fluid oxidative stress, ovarian hormones, and response to gonadotropin stimulation. Fertil Steril. (2008) 89:912–21. doi: 10.1016/j.fertnstert.2007.04.034
10. Becatti M, Fucci R, Mannucci A, Barygina V, Mugnaini M, Criscuoli L, et al. A biochemical approach to detect oxidative stress in infertile women undergoing assisted reproductive technology procedures. Int J Mol Sci. (2018) 19:E592. doi: 10.3390/ijms19020592
11. Zhang L, Zhang Z, Wang J, Lv D, Zhu T, Wang F, et al. Melatonin regulates the activities of ovary and delays the fertility decline in female animals via MT1/AMPK pathway. J Pineal Res. (2019) 66:e12550. doi: 10.1111/jpi.12550
12. Ben-Meir A, Burstein E, Borrego-Alvarez A, Chong J, Wong E, Yavorska T, et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell. (2015) 14:887–95. doi: 10.1111/acel.12368
13. Tamura H, Kawamoto M, Sato S, Tamura I, Maekawa R, Taketani T, et al. Long-term melatonin treatment delays ovarian aging. J Pineal Res. (2017) 62:e12381. doi: 10.1111/jpi.12381
14. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
15. Kauppila TES, Kauppila JHK, Larsson NG. Mammalian mitochondria and aging: an update. Cell Metab. (2017) 25:57–71. doi: 10.1016/j.cmet.2016.09.017
16. Van Blerkom J. Mitochondrial function in the human oocyte and embryo and their role in developmental competence. Mitochondrion. (2011) 11:797–813. doi: 10.1016/j.mito.2010.09.012
17. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. (2005) 309:481–4. doi: 10.1126/science.1112125
18. Lakshmanan LN, Yee Z, Ng LF, Gunawan R, Halliwell B, Gruber J. Clonal expansion of mitochondrial DNA deletions is a private mechanism of aging in long-lived animals. Aging Cell. (2018) 17:e12814. doi: 10.1111/acel.12814
19. Damas J, Samuels DC, Carneiro J, Amorim A, Pereira F. Mitochondrial DNA rearrangements in health and disease–a comprehensive study. Hum Mutation. (2014) 35:1–14. doi: 10.1002/humu.22452
20. Lynch M, Koskella B, Schaack S. Mutation pressure and the evolution of organelle genomic architecture. Science. (2006) 311:1727–30. doi: 10.1126/science.1118884
21. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. (2004) 429:417–23. doi: 10.1038/nature02517
22. May-Panloup P, Boucret L, Chao de la Barca JM, Desquiret-Dumas V, Ferre-L'Hotellier V, Moriniere C, et al. Ovarian ageing: the role of mitochondria in oocytes and follicles. Hum Reprod Update. (2016) 22:725–43. doi: 10.1093/humupd/dmw028
23. Chan CC, Liu VW, Lau EY, Yeung WS, Ng EH, Ho PC. Mitochondrial DNA content and 4977 bp deletion in unfertilized oocytes. Mol Hum Reprod. (2005) 11:843–6. doi: 10.1093/molehr/gah243
24. Keefe DL, Niven-Fairchild T, Powell S, Buradagunta S. Mitochondrial deoxyribonucleic acid deletions in oocytes and reproductive aging in women. Fertil Steril. (1995) 64:577–83. doi: 10.1016/S0015-0282(16)57796-6
25. Barritt JA, Cohen J, Brenner CA. Mitochondrial DNA point mutation in human oocytes is associated with maternal age. Reprod Biomed Online. (2000) 1:96–100. doi: 10.1016/S1472-6483(10)61946-3
26. Fragouli E, Spath K, Alfarawati S, Kaper F, Craig A, Michel CE, et al. Altered levels of mitochondrial DNA are associated with female age, aneuploidy, and provide an independent measure of embryonic implantation potential. PLoS Genet. (2015) 11:e1005241. doi: 10.1371/journal.pgen.1005241
27. Fragouli E, McCaffrey C, Ravichandran K, Spath K, Grifo JA, Munne S, et al. Clinical implications of mitochondrial DNA quantification on pregnancy outcomes: a blinded prospective non-selection study. Hum Reprod. (2017) 32:2340–7. doi: 10.1093/humrep/dex292
28. Lledo B, Ortiz JA, Morales R, Garcia-Hernandez E, Ten J, Bernabeu A, et al. Comprehensive mitochondrial DNA analysis and IVF outcome. Hum Reprod Open. (2018) 2018:hoy023. doi: 10.1093/hropen/hoy023
29. Zheng Y, Luo X, Zhu J, Zhang X, Zhu Y, Cheng H, et al. Mitochondrial DNA 4977 bp deletion is a common phenomenon in hair and increases with age. Bosn J Basic Med Sci. (2012) 12:187–92. doi: 10.17305/bjbms.2012.2480
30. Mirabutalebi SH, Karami N, Ashrafzadeh HR, Akhvansales Z, Tavakoli M, Ghasemi N. Detection of 4977-bp deletion of mitochondrial DNA in in vitro fertilization failure women: a case-control study. Int J Reprod Biomed. (2018) 16:571–6. doi: 10.29252/ijrm.16.9.571
31. de Bruin JP, Dorland M, Spek ER, Posthuma G, van Haaften M, Looman CW, et al. Ultrastructure of the resting ovarian follicle pool in healthy young women. Biol Reprod. (2002) 66:1151–60. doi: 10.1095/biolreprod66.4.1151
32. de Bruin JP, Dorland M, Spek ER, Posthuma G, van Haaften M, Looman CW, et al. Age-related changes in the ultrastructure of the resting follicle pool in human ovaries. Biol Reprod. (2004) 70:419–24. doi: 10.1095/biolreprod.103.015784
33. Pasquariello R, Ermisch AF, Silva E, McCormick S, Logsdon D, Barfield JP, et al. Alterations in oocyte mitochondrial number and function are related to spindle defects and occur with maternal aging in mice and humansdagger. Biol Reprod. (2019) 100:971–81. doi: 10.1093/biolre/ioy248
34. Zhang M, Bener MB, Jiang Z, Wang T, Esencan E, Scott Iii R, et al. Mitofusin 1 is required for female fertility and to maintain ovarian follicular reserve. Cell Death Dis. (2019) 10:560. doi: 10.1038/s41419-019-1799-3
35. Zhang M, Bener MB, Jiang Z, Wang T, Esencan E, Scott R, et al. Mitofusin 2 plays a role in oocyte and follicle development, and is required to maintain ovarian follicular reserve during reproductive aging. Aging. (2019) 11:3919–38. doi: 10.18632/aging.102024
36. Udagawa O, Ishihara T, Maeda M, Matsunaga Y, Tsukamoto S, Kawano N, et al. Mitochondrial fission factor Drp1 maintains oocyte quality via dynamic rearrangement of multiple organelles. Curr Biol. (2014) 24:2451–8. doi: 10.1016/j.cub.2014.08.060
37. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. (2012) 337:1062–5. doi: 10.1126/science.1219855
38. Zhou L, Zhang Q, Zhang P, Sun L, Peng C, Yuan Z, et al. c-Abl-mediated Drp1 phosphorylation promotes oxidative stress-induced mitochondrial fragmentation and neuronal cell death. Cell Death Dis. (2017) 8:e3117. doi: 10.1038/cddis.2017.524
39. Schon EA, Kim SH, Ferreira JC, Magalhaes P, Grace M, Warburton D, et al. Chromosomal non-disjunction in human oocytes: is there a mitochondrial connection? Hum Reprod. (2000) 15 (Suppl. 2):160–72. doi: 10.1093/humrep/15.suppl_2.160
40. Reynier P, May-Panloup P, Chretien MF, Morgan CJ, Jean M, Savagner F, et al. Mitochondrial DNA content affects the fertilizability of human oocytes. Mol Hum Reprod. (2001) 7:425–9. doi: 10.1093/molehr/7.5.425
41. Zhang X, Wu XQ, Lu S, Guo YL, Ma X. Deficit of mitochondria-derived ATP during oxidative stress impairs mouse MII oocyte spindles. Cell Res. (2006) 16:841–50. doi: 10.1038/sj.cr.7310095
42. Zhou CX, Shi LY, Li RC, Liu YH, Xu BQ, Liu JW, et al. GTPase-activating protein Elmod2 is essential for meiotic progression in mouse oocytes. Cell Cycle. (2017) 16:852–60. doi: 10.1080/15384101.2017.1304329
43. Van Blerkom J, Davis PW, Lee J. ATP content of human oocytes and developmental potential and outcome after in-vitro fertilization and embryo transfer. Hum Reprod. (1995) 10:415–24. doi: 10.1093/oxfordjournals.humrep.a135954
44. Silva PM, Reis RM, Bolanos-Garcia VM, Florindo C, Tavares AA, Bousbaa H. Dynein-dependent transport of spindle assembly checkpoint proteins off kinetochores toward spindle poles. FEBS Lett. (2014) 588:3265–73. doi: 10.1016/j.febslet.2014.07.011
45. Marangos P, Stevense M, Niaka K, Lagoudaki M, Nabti I, Jessberger R, et al. DNA damage-induced metaphase I arrest is mediated by the spindle assembly checkpoint and maternal age. Nat Commun. (2015) 6:8706. doi: 10.1038/ncomms9706
46. Tanisawa K, Mikami E, Fuku N, Honda Y, Honda S, Ohsawa I, et al. Exome sequencing of senescence-accelerated mice (SAM) reveals deleterious mutations in degenerative disease-causing genes. BMC Genomics. (2013) 14:248. doi: 10.1186/1471-2164-14-248
47. Titus S, Li F, Stobezki R, Akula K, Unsal E, Jeong K, et al. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci Transl Med. (2013) 5:172ra21. doi: 10.1126/scitranslmed.3004925
48. Pan H, Ma P, Zhu W, Schultz RM. Age-associated increase in aneuploidy and changes in gene expression in mouse eggs. Dev Biol. (2008) 316:397–407. doi: 10.1016/j.ydbio.2008.01.048
49. Lim J, Luderer U. Oxidative damage increases and antioxidant gene expression decreases with aging in the mouse ovary. Biol Reprod. (2011) 84:775–82. doi: 10.1095/biolreprod.110.088583
50. Hamatani T, Falco G, Carter MG, Akutsu H, Stagg CA, Sharov AA, et al. Age-associated alteration of gene expression patterns in mouse oocytes. Hum Mol Genet. (2004) 13:2263–78. doi: 10.1093/hmg/ddh241
51. Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. (2004) 279:41975–84. doi: 10.1074/jbc.M407707200
52. El Mouatassim S, Guerin P, Menezo Y. Expression of genes encoding antioxidant enzymes in human and mouse oocytes during the final stages of maturation. Mol Hum Reprod. (1999) 5:720–5. doi: 10.1093/molehr/5.8.720
53. Noda Y, Matsumoto H, Umaoka Y, Tatsumi K, Kishi J, Mori T. Involvement of superoxide radicals in the mouse two-cell block. Mol Reprod Dev. (1991) 28:356–60. doi: 10.1002/mrd.1080280408
54. Kimura N, Tsunoda S, Iuchi Y, Abe H, Totsukawa K, Fujii J. Intrinsic oxidative stress causes either 2-cell arrest or cell death depending on developmental stage of the embryos from SOD1-deficient mice. Mol Hum Reprod. (2010) 16:441–51. doi: 10.1093/molehr/gaq007
55. Perkins AT, Das TM, Panzera LC, Bickel SE. Oxidative stress in oocytes during midprophase induces premature loss of cohesion and chromosome segregation errors. Proc Natl Acad Sci USA. (2016) 113:E6823–30. doi: 10.1073/pnas.1612047113
56. Tatemoto H, Muto N, Sunagawa I, Shinjo A, Nakada T. Protection of porcine oocytes against cell damage caused by oxidative stress during in vitro maturation: role of superoxide dismutase activity in porcine follicular fluid. Biol Reprod. (2004) 71:1150–7. doi: 10.1095/biolreprod.104.029264
57. Curnow EC, Ryan J, Saunders D, Hayes ES. Bovine in vitro oocyte maturation as a model for manipulation of the gamma-glutamyl cycle and intraoocyte glutathione. Reprod Fertil Dev. (2008) 20:579–88. doi: 10.1071/RD08041
58. Zuelke KA, Jones DP, Perreault SD. Glutathione oxidation is associated with altered microtubule function and disrupted fertilization in mature hamster oocytes. Biol Reprod. (1997) 57:1413–9. doi: 10.1095/biolreprod57.6.1413
59. Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, et al. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. (1996) 178:179–85. doi: 10.1006/dbio.1996.0208
60. Cao B, Camden AJ, Parnell LA, Mysorekar IU. Autophagy regulation of physiological and pathological processes in the female reproductive tract. Am J Reprod Immunol. (2017) 77:e12650. doi: 10.1111/aji.12650
61. Liu H, Zhang X, Zhang S, Huang H, Wu J, Wang Y, et al. Oxidative stress mediates Microcystin-LR-Induced Endoplasmic Reticulum Stress and Autophagy in KK-1 Cells and C57BL/6 mice ovaries. Front Physiol. (2018) 9:1058. doi: 10.3389/fphys.2018.01058
62. Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants Redox Signal. (2007) 9:2277–93. doi: 10.1089/ars.2007.1782
63. Latham KE. Endoplasmic reticulum stress signaling in mammalian oocytes and embryos: life in balance. Int Rev Cell Mol Biol. (2015) 316:227–65. doi: 10.1016/bs.ircmb.2015.01.005
64. Yang L, Lei L, Zhao Q, Gao Z, Xu X. Lycium barbarum polysaccharide improves the development of mouse oocytes vitrified at the germinal vesicle stage. Cryobiology. (2018) 85:7–11. doi: 10.1016/j.cryobiol.2018.10.265
65. Mihalas BP, Bromfield EG, Sutherland JM, De Iuliis GN, McLaughlin EA, Aitken RJ, et al. Oxidative damage in naturally aged mouse oocytes is exacerbated by dysregulation of proteasomal activity. J Biol Chem. (2018) 293:18944–64. doi: 10.1074/jbc.RA118.005751
66. Mihalas BP, De Iuliis GN, Redgrove KA, McLaughlin EA, Nixon B. The lipid peroxidation product 4-hydroxynonenal contributes to oxidative stress-mediated deterioration of the ageing oocyte. Sci Rep. (2017) 7:6247. doi: 10.1038/s41598-017-06372-z
67. Escobar Sanchez ML, Echeverria Martinez OM, Vazquez-Nin GH. Immunohistochemical and ultrastructural visualization of different routes of oocyte elimination in adult rats. Eur J Histochem. (2012) 56:e17. doi: 10.4081/ejh.2012.17
68. Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science. (2008) 321:117–20. doi: 10.1126/science.1154822
69. Sugiyama M, Kawahara-Miki R, Kawana H, Shirasuna K, Kuwayama T, Iwata H. Resveratrol-induced mitochondrial synthesis and autophagy in oocytes derived from early antral follicles of aged cows. J Reprod Dev. (2015) 61:251–9. doi: 10.1262/jrd.2015-001
70. Song BS, Yoon SB, Kim JS, Sim BW, Kim YH, Cha JJ, et al. Induction of autophagy promotes preattachment development of bovine embryos by reducing endoplasmic reticulum stress. Biol Reprod. (2012) 87:1–11. doi: 10.1095/biolreprod.111.097949
71. Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. (2011) 36:30–8. doi: 10.1016/j.tibs.2010.07.007
72. Alexeyev MF, Ledoux SP, Wilson GL. Mitochondrial DNA and aging. Clin Sci. (2004) 107:355–64. doi: 10.1042/CS20040148
73. Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family's role in aging and age-associated pathologies. J Clin Invest. (2013) 123:973–9. doi: 10.1172/JCI64094
74. Tatone C, Di Emidio G, Vitti M, Di Carlo M, Santini S Jr, D'Alessandro AM, et al. Sirtuin functions in female fertility: possible role in oxidative stress and aging. Oxid Med Cell Longev. (2015) 2015:659687. doi: 10.1155/2015/659687
75. Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Rad Biol Med. (2013) 56:133–71. doi: 10.1016/j.freeradbiomed.2012.10.525
76. Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. (2010) 31:212–20. doi: 10.1016/j.tips.2010.02.003
77. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. (2009) 460:587–91. doi: 10.1038/nature08197
78. Di Emidio G, Falone S, Vitti M, D'Alessandro AM, Vento M, Di Pietro C, et al. SIRT1 signalling protects mouse oocytes against oxidative stress and is deregulated during aging. Hum Reprod. (2014) 29:2006–17. doi: 10.1093/humrep/deu160
79. Liu WJ, Zhang XM, Wang N, Zhou XL, Fu YC, Luo LL. Calorie restriction inhibits ovarian follicle development and follicle loss through activating SIRT1 signaling in mice. Eur J Med Res. (2015) 20:22. doi: 10.1186/s40001-015-0114-8
80. Kawamura Y, Uchijima Y, Horike N, Tonami K, Nishiyama K, Amano T, et al. Sirt3 protects in vitro-fertilized mouse preimplantation embryos against oxidative stress-induced p53-mediated developmental arrest. J Clin Invest. (2010) 120:2817–28. doi: 10.1172/JCI42020
81. Zhao HC, Ding T, Ren Y, Li TJ, Li R, Fan Y, et al. Role of Sirt3 in mitochondrial biogenesis and developmental competence of human in vitro matured oocytes. Hum Reprod. (2016) 31:607–22. doi: 10.1093/humrep/dev345
82. Song C, Peng W, Yin S, Zhao J, Fu B, Zhang J, et al. Melatonin improves age-induced fertility decline and attenuates ovarian mitochondrial oxidative stress in mice. Sci Rep. (2016) 6:35165. doi: 10.1038/srep35165
83. Richter T, von Zglinicki T. A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp Gerontol. (2007) 42:1039–42. doi: 10.1016/j.exger.2007.08.005
84. Liu L, Trimarchi JR, Smith PJ, Keefe DL. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell. (2002) 1:40–6. doi: 10.1046/j.1474-9728.2002.00004.x
85. Passos JF, von Zglinicki T. Mitochondria, telomeres and cell senescence. Exp Gerontol. (2005) 40:466–72. doi: 10.1016/j.exger.2005.04.006
86. Keefe DL. Telomeres, reproductive aging, and genomic instability during early development. Reprod Sci. (2016) 23:1612–5. doi: 10.1177/1933719116676397
87. Ramos-Ibeas P, Pericuesta E, Peral-Sanchez I, Heras S, Laguna-Barraza R, Perez-Cerezales S, et al. Longitudinal analysis of somatic and germ-cell telomere dynamics in outbred mice. Mol Reprod Dev. (2019) 86:1033–43. doi: 10.1002/mrd.23218
88. Keefe DL, Franco S, Liu L, Trimarchi J, Cao B, Weitzen S, et al. Telomere length predicts embryo fragmentation after in vitro fertilization in women–toward a telomere theory of reproductive aging in women. Am J Obstet Gynecol. (2005) 192:1256–60. doi: 10.1016/j.ajog.2005.01.036
89. Keefe DL, Liu L, Marquard K. Telomeres and aging-related meiotic dysfunction in women. Cell Mol Life Sci. (2007) 64:139–43. doi: 10.1007/s00018-006-6466-z
90. Turner K, Lynch C, Rouse H, Vasu V, Griffin DK. Direct single-cell analysis of human polar bodies and cleavage-stage embryos reveals no evidence of the telomere theory of reproductive ageing in relation to aneuploidy generation. Cells. (2019) 8:E163. doi: 10.3390/cells8020163
91. Ge J, Li C, Li C, Huang Z, Zeng J, Han L, et al. SIRT6 participates in the quality control of aged oocytes via modulating telomere function. Aging. (2019). 11:1965–76. doi: 10.18632/aging.101885
92. Liao CY, Kennedy BK. SIRT6, oxidative stress, and aging. Cell Res. (2016) 26:143–4. doi: 10.1038/cr.2016.8
93. Eini F, Novin MG, Joharchi K, Hosseini A, Nazarian H, Piryaei A, et al. Intracytoplasmic oxidative stress reverses epigenetic modifications in polycystic ovary syndrome. Reprod Fertil Dev. (2017) 29:2313–23. doi: 10.1071/RD16428
Keywords: oxidative stree, mitochondrial dysfunction, antioxidants, oocyte aging, ER stress
Citation: Sasaki H, Hamatani T, Kamijo S, Iwai M, Kobanawa M, Ogawa S, Miyado K and Tanaka M (2019) Impact of Oxidative Stress on Age-Associated Decline in Oocyte Developmental Competence. Front. Endocrinol. 10:811. doi: 10.3389/fendo.2019.00811
Received: 16 June 2019; Accepted: 06 November 2019;
Published: 22 November 2019.
Edited by:
Osamu Hiraike, Tokyo University of Science, JapanReviewed by:
Claudio Acuña-Castillo, Universidad de Santiago de Chile, ChileCopyright © 2019 Sasaki, Hamatani, Kamijo, Iwai, Kobanawa, Ogawa, Miyado and Tanaka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshio Hamatani, dG9zaGlvaGFtYXRhbmlAa2Vpby5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.