
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 09 October 2019
Sec. Pituitary Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00690
This article is part of the Research Topic Molecular network study of pituitary adenomas View all 16 articles
 Na Li1,2,3
Na Li1,2,3 Xianquan Zhan1,2,3,4,5*
Xianquan Zhan1,2,3,4,5*Mitochondrion is a multi-functional organelle, which is associated with various signaling pathway networks, including energy metabolism, oxidative stress, cell apoptosis, cell cycles, autophagy, and immunity process. Mitochondrial proteins have been discovered to modulate these signaling pathway networks, and multiple biological behaviors to adapt to various internal environments or signaling events of human pathogenesis. Accordingly, mitochondrial dysfunction that alters the bioenergetic and biosynthetic state might contribute to multiple diseases, including cell transformation and tumor. Multiomics studies have revealed that mitochondrial dysfunction, oxidative stress, and cell cycle dysregulation signaling pathways operate in human pituitary adenomas, which suggest mitochondria play critical roles in pituitary adenomas. Some drugs targeting mitochondria are found as a therapeutic strategy for pituitary adenomas, including melatonin, melatonin inhibitors, temozolomide, pyrimethamine, 18 beta-glycyrrhetinic acid, gossypol acetate, Yougui pill, T-2 toxin, grifolic acid, cyclosporine A, dopamine agonists, and paeoniflorin. This article reviews the latest experimental evidence and potential biological roles of mitochondrial dysfunction and mitochondrial dynamics in pituitary adenoma progression, potential molecular mechanisms between mitochondria and pituitary adenoma progression, and current status and perspectives of mitochondria-based biomarkers and targeted drugs for effective management of pituitary adenomas.
Pituitary adenomas are intracranial tumors that develop in the pituitary gland, and account for 10 to 25% of all intracranial neoplasms. Most pituitary adenomas are benign, nearly 35% sufferers present invasiveness and just 0.1–0.2% are diagnosed as carcinomas (1). Pituitary adenomas are commonly divided into functional pituitary adenomas, and non-functional pituitary adenomas according to the clinical level of hormone secretion (2). Functional pituitary adenomas are hormone-secreting pituitary adenomas that could cause hyperpituitarism, such as Cushing's syndrome, acromegaly, and hyperprolactinaemia; and non-functional pituitary adenomas are non-hormone-secreting pituitary adenomas (3). Pituitary adenomas are also divided into microadenomas (<10 mm) and macroadenomas (≥10 mm) according to tumor size (4). The clinically chief complaints of pituitary adenomas are visual field defects, headache, and increased intracranial pressure, which are usually derived from a compression of the neighboring tissues and structures. Another type of clinical problem is an inappropriate hormone secretion in hormone-secreting pituitary adenomas (5).
Currently, high-throughput omic technologies have been extensively used to study pituitary adenomas (6) from a multi-parameter systematic biology angle to overcome the irrationality that use a single molecule as biomarker for accurate predictive, preventive, and personalized medicine (PPPM) practice, because numerous molecules alter at the different levels of DNAs (genome), RNAs (transcriptome), proteins (proteome), and metabolites (metabolome), and are involved in different pathway network systems for tumorigenesis (7). Among the field of multiomics, transcriptomics, and proteomics are two important ways to systematically study the functions of genes (8, 9). Thousands of differentially expressed genes have been identified in human pituitary adenomas (10–12), which have addressed at a certain degree the functions of genes. However, transcriptiomics study cannot fully reveal the functions of genes, because proteins are the final performer of the corresponding genes, there are lots of regulations and modifications occurred in the process from mRNA to proteins. The number of proteins is much more than the number of transcripts, and the correlation coefficient only reaches 0.4 between proteomics and transcriptomics (13, 14). Therefore, proteomics is a more important way to address the functions of genes, especially subcellular proteomics such as mitochondrial proteomics is an effective method to reveal the specialized functions of a organelle to associate with a given diseases such as cancer (15). Currently mitochondrial proteomics has become a research hotpot because mitochondria are ubiquitously subcellular organelles responsible for providing energy to eukaryotic cells, and are the key links of metabolism, oxidative stress, cell apoptosis, cell cycles, autophagy, and immunity process (16), which are involved in a wide range of diseases including cancers (17). This review article will mainly focus on the mitochondrial dysfunction pathway alterations in pituitary adenomas from a systematic viewpoint.
Pituitary adenoma proteomics-based molecular network study have revealed that mitochondrial dysfunction, oxidative stress, cell cycle dysregulation, and MAPK signaling abnormality are significantly associated with the pathogenesis of pituitary adenomas (18–21). Mitochondria are actually center of oxidative stress, which clearly demonstrate that mitochondrial dysfunction pathway plays important roles in pituitary adenomas. Furthermore, electron microscopy morphology study demonstrates that mitochondria are abundantly filled in cytoplasm of pituitary oncocytoma cells (22–24). Some studies demonstrate that the volume of mitochondria is different among different subtypes in pituitary adenomas; for example, the volume of mitochondria of prolactinoma is larger than acromegaly (25). More important are that some drugs targeting mitochondria have been reported as a therapeutic strategy for pituitary adenomas (Table 1) (26–39), including melatonin, melatonin inhibits, temozolomide and pyrimethamine, 18 beta-glycyrrhetinic acid, gossypol acetate, Yougui pill, T-2 toxin, grifolic acid, and paeoniflorin. Those evidences clearly demonstrate the important roles of mitochondrial biological functions and dynamic shift in pituitary adenoma pathogenesis, however, their molecular mechanisms remain unclear yet (2, 40). Mitochondria-based study might provide new insights into molecular mechanisms of pituitary adenomas, discover new biomarkers and molecular targets for effective management of pituitary adenomas. This review article discusses observations in the context of how mitochondrial dysfunction can influence the biological status in pituitary adenoma, including energy metabolism, oxidative stress, cell apoptosis, autophagy, and immunity (Figure 1; Table 2).
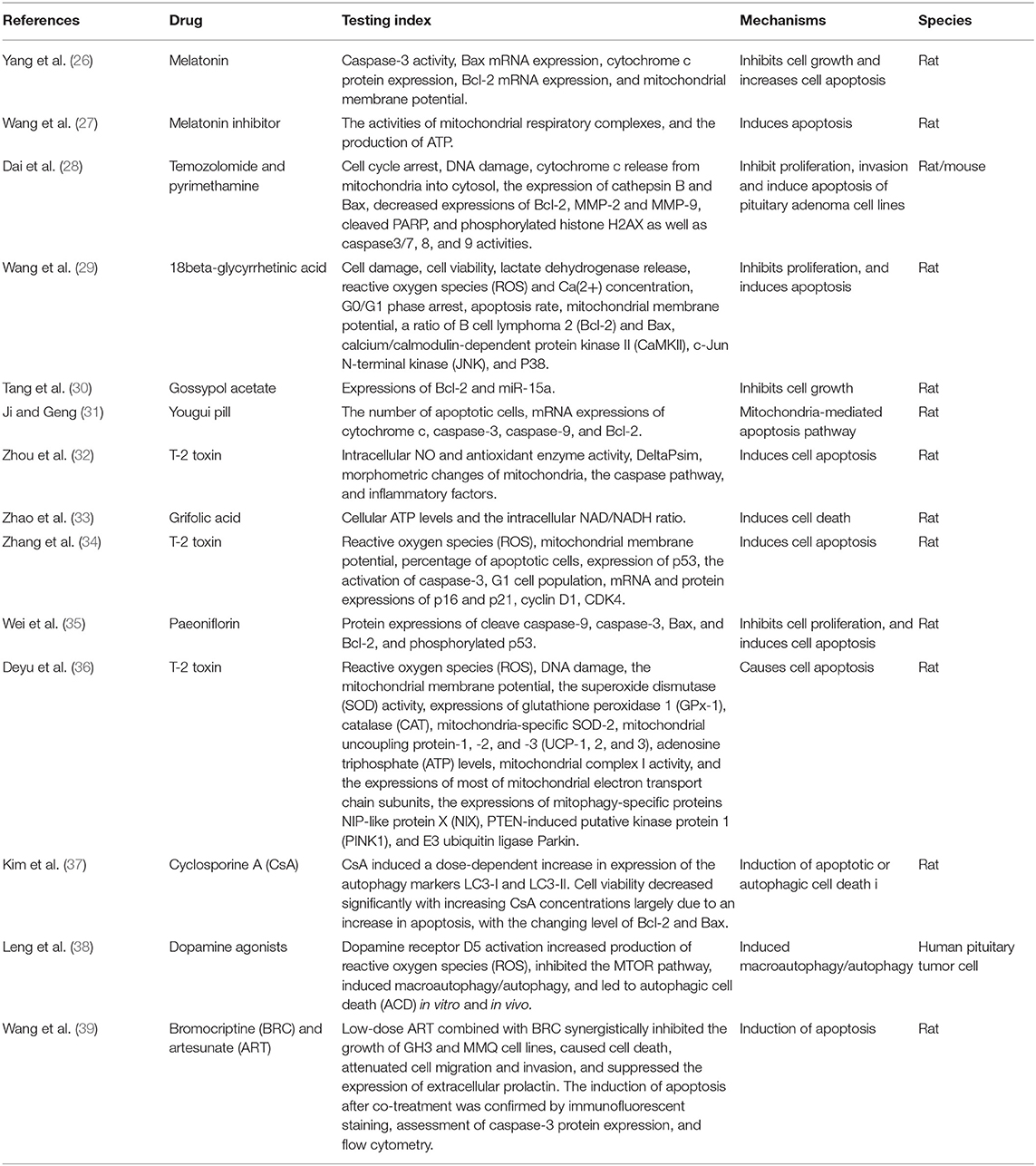
Table 1. Some drugs targeting mitochondria as a therapeutic strategy for pituitary adenomas.
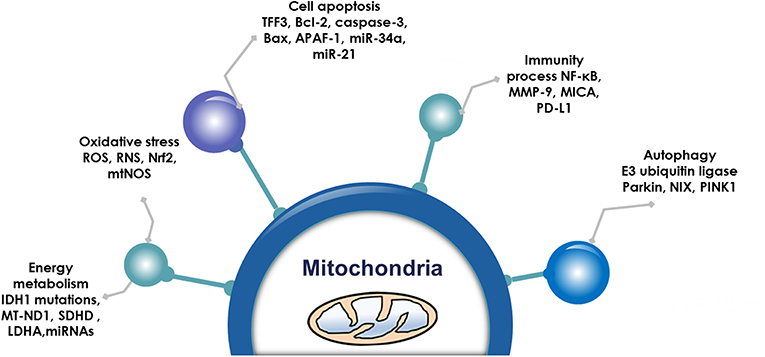
Figure 1. Mitochondrial functions. Emerging data show that mitochondria are associated with energy metabolism, oxidative stress, cell apoptosis autophagy, and immunity process in pituitary adenomas.
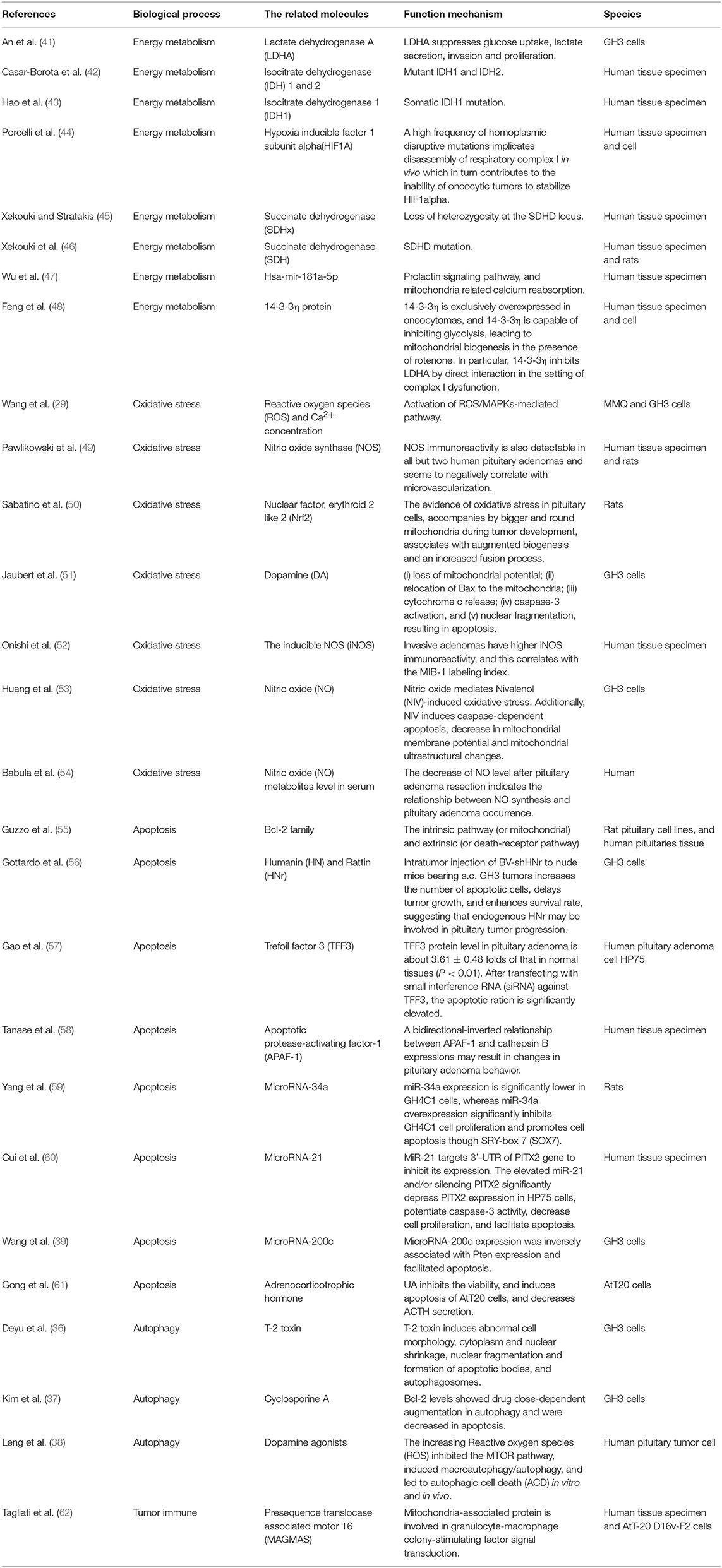
Table 2. Mitochondrial dysfunction pathway in the pathogenesis of pituitary adenomas.
Energy metabolism alterations are an emerging hallmark in tumor, which are still an unresolved issue that how energy metabolism system plays in formation and progression of tumors or metastases. Tumor cell energy metabolism has mainly focused on glucose metabolism and lipid metabolism. The metabolism of glucose to lactic acid in the presence of oxygen have been recognized in cancer cells, commonly called the Warburg effect (63). Further, the reverse Warburg effect put forward in 2009 provides complementary mechanisms for cancer energy metabolism (64). In addition, novel evidence is shedding light on alterations in lipid metabolism-associated pathways that have been discussed for past years. A gene expression study find that the upregulated lipogenesis pathways are associated with poor survival outcomes (65), and the elevated levels of lipid droplets are associated with cancer aggressiveness, which has been proposed to predict prognosis of cancer (66). More interesting is that lipids could be transferred from adipocytes to cancer cells by co-culture condition to promote cancer cell growth (67). It clearly indicates that lipid metabolism disorder is closely associated with tumorigenesis, whose study would be significantly enhanced with the development of lipidomics based on electrospray ionization/mass spectrometry (ESI-MS) (68).
The citric acid cycle, oxidative phosphorylation (OXPHOS), and fatty acid beta-oxidation occur in the matrix of the mitochondrion in eukaryotic cells. Mitochondrial dysfunction is closely associated with energy metabolism reprogramming to associate cancer formation or progression (69). In the Warburg and reverse Warburg effects, cancer cells have metabolic symbiosis with mesenchymal cells, especially cancer-associated fibroblasts (CAFs), namely cancer cells produce reactive oxygen species (ROS) to induce oxidative stress and aerobic glycolysis of CAFs; in turn, CAFs produce lots of nourishments (especially lactate and pyruvate) to feed the adjacent cancer cells to produce more ATPs (64). The reverse Warburg effect shows metabolic interplay between high glycolytic cells and mitochondrial OXPHOS activates cells via lactate shuttle. Important enzymes involved in mitochondrial OXPHOS include the enzymes in the citric acid cycle and electron transport chain. The citric acid cycle is a series of enzymatic reactions to release energy through the oxidation of acetyl-CoA into ATP and carbon dioxide. The citric acid cycle is undergoing 10 steps to complete ATP production with a series of enzymatic reactions, including citrate synthase, aconitase, isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, succinyl-CoA synthetase, succinate dehydrogenase, fumarase, and malate dehydrogenase. OXPHOS is the metabolic pathway in which cells use enzymes to oxidize nutrients for releasing energy to produce ATP. The eukaryotic electron transport chain contains NADH-coenzyme Q oxidoreductase (complex I), succinate-Q oxidoreductase (complex II), Q-cytochrome c oxidoreductase (complex III), cytochrome coxidase (complex IV), and ATP synthase (complex V). Furthermore, fatty acid molecules are broken down in the eukaryotic mitochondria to transform into acetyl-CoA to enter the citric acid cycle, and generate NADH and FADH2 that are co-enzymes used in the electron transport chain (Figure 2).
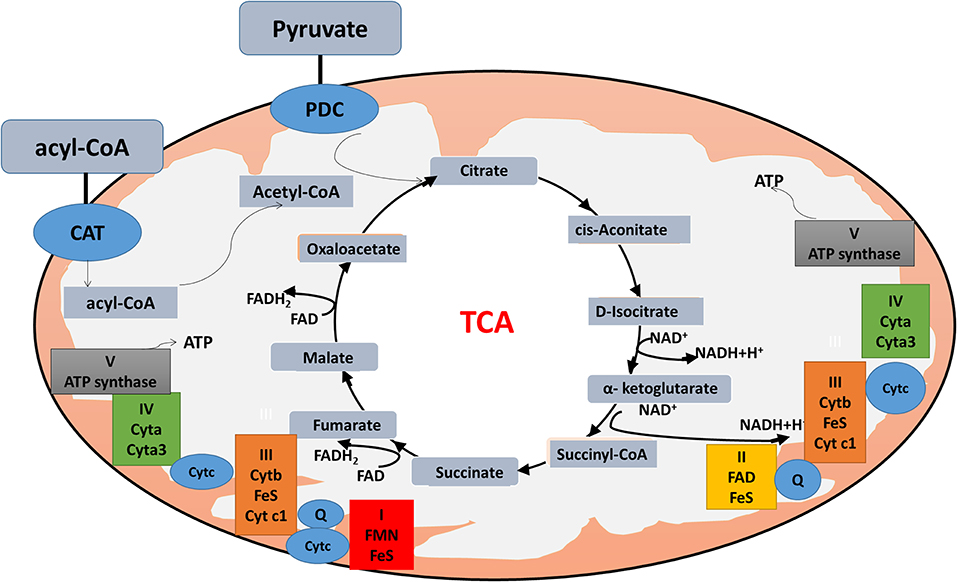
Figure 2. Mitochondrial physiology. Acetyl-CoA enters the mitochondrion via pyruvate or fatty acids. Pyruvate is imported through the mitochondrial inner membrane by the pyruvate dehydrogenase complex (PDC), and is oxidatively decarboxylated to produce acetyl-CoA. Fatty acids form acyl-CoA in the cytosol, and are transported into mitochondrion through carnitine (CAT) for β-oxidation. Acyl-CoA enters the citric acid cycle, and generates NADH and FADH2 (co-enzymes used in the electron transport chain) to produce ATP.
Mitochondria are the main location of energy metabolism pathways (citric acid cycle, OXPHOS, and fatty acid beta-oxidation). Some mitochondria-associated proteins have been reported to play a critical role in pituitary adenomas. For example, overexpression of lactate dehydrogenase A significantly promotes proliferation and invasion of pituitary adenomas, and positively correlates with higher Ki-67 index (41). Mutant succinate dehydrogenase in the citric acid cycle occurs in the pituitary adenomas (42). In addition, DNA sequencing-based genotypic studies demonstrate identical IDH1 mutations (c.394 C > T) in pituitary adenomas tissues (43). The high frequency of respiratory complex I mutations are found in mitochondrial DNA in a large panel of oncocytic pituitaries, which indicates dysfunction of respiratory complex I to cause instability of HIF1alpha in pituitary adenomas. Briefly, mutations in the mitochondria-coded MT-ND1 gene, an important composition of respiratory complex I, is closely associated with energy metabolic impairment to influence balance of succinate and alpha-ketoglutarate, which leads to the abnormal citric acid cycle metabolites (succinate and alpha-ketoglutarate) to be responsible for HIF1alpha stabilization in pituitary adenomas (44). The multifunctional succinate dehydrogenase (SDH) is located in the inner membrane of mitochondria, and serves as a critical step in Krebs cycle and a crucial member of the respiratory chain. SDH subunit D (SDHD) mutation is found in an aggressive GH-secreting pituitary adenoma, indicating SDHD mutation might link to the progression of pituitary adenomas (45). The study on SDH subunit B (SDHB) (+/–) mice finds that SDHx-deficiency is a main initiator to result in the cascade of molecular events for the formation of pituitary adenomas (46). The whole-exome sequencing analysis of pituitary oncocytomas found mitochondrial DNA mutations, respiratory complex I dysfunction, and reductions of lactate and lactate dehydrogenase A (LDHA) (48). Besides glycometabolism change, lipid metabolism is also alerted in pituitary adenomas. The gene microarray analysis of miRNAs expression profile between invasive and non-invasive non-functional pituitary adenomas finds that fatty acid metabolism plays a prominent role in pituitary adenomas (47). Therefore, energy metabolism alteration plays important roles in pituitary adenomas with high metabolic demand, which also influences cell proliferation, growth, and angiogenesis. The development of new drugs targeted mitochondria might be an new approach to block energy metabolism pathways for effective treatment of pituitary adenomas (70).
Oxidative stress reflects an imbalance between free radical/reactive oxygen/nitrogen species (ROS/RNS) productions and endogenous antioxidant defense mechanisms in the cells and body, which results in damage to proteins, DNA, membrane, and cellular organelles so on (71). ROS/RNS can be beneficial because they take part in attacking and killing pathogens by the immune system (72). Short-term oxidative stress might also be meaningful in prevention from aging (73). However, oxidative stress is also involved in the development of various diseases including cancers (74–79). Severely oxidative stresses even cause cell death, apoptosis, necrosis, cell migration, fibrosis, and angiogenesis. The lipid peroxidation of fatty acids as a type of oxidative stress increased ROS/RNS to injury bilayer lipid membranes. The secondary products of lipid peroxides such as malondialdehyde, aldehydes, 4-hydroxynonenal (HNE), hexanal, or acrolein have very long and broad effects (80).
Elevated level of ROS was a key constituent in cancer survival and resistance to treatment. The mitochondrial OXPHOS system is the major sites where produce endogenous ROS, including OH and superoxide radicals () (81). Mitochondrial complexes I, II, and III play a crucial role in the generation of mitochondrial ROS. Electrons tend to be leaky at complexes I and III to cause the incomplete reduction of oxygen to generate a free radical such as superoxide (80). Mitochondrial dysfunction could result in the increased ROS in cancer cells to mediate tumor-related signaling pathways and activate pro-oncogenic signaling, which regulate cancer progression, angiogenesis, metastasis, and survival (Figure 3). Although increased ROS does not benefit the cancer cell survival, however, antioxidant substance system is also activated in cancer cells to help cancer cell death escape (82). Moreover, the presence of mitochondrial nitric oxide synthase (mtNOS) provided the opportunity to review complementary aspects of mitochondrial physiology. The mtNOS could cause the generation of a partial nitric oxide (NO) in mitochondria (83), besides large amounts of NO produced by inducible NOS (iNOS) in pathophysiological status (84). NO can react quickly with the superoxide radicals to generate more toxic peroxynitrite anion (ONOO−) or hydroxyl radical (.OH). Mitochondria, as generators and targets of NO, determine the steady-state of NO though modulating the rates of consumption and production at the subcellular levels. Thus, mtNOS plays a crucial role in this process, which was activated by calcium and transcriptional/translational regulation (85). Furthermore, NO production in mitochondria was still decided by subcellular localization of mtNOS due to post-translational modification or protein-protein interactions. Therefore, mitochondria could produce NO by temporospatial distribution of mtNOS, and receive NO signal to regulate mitochondrial events (86).
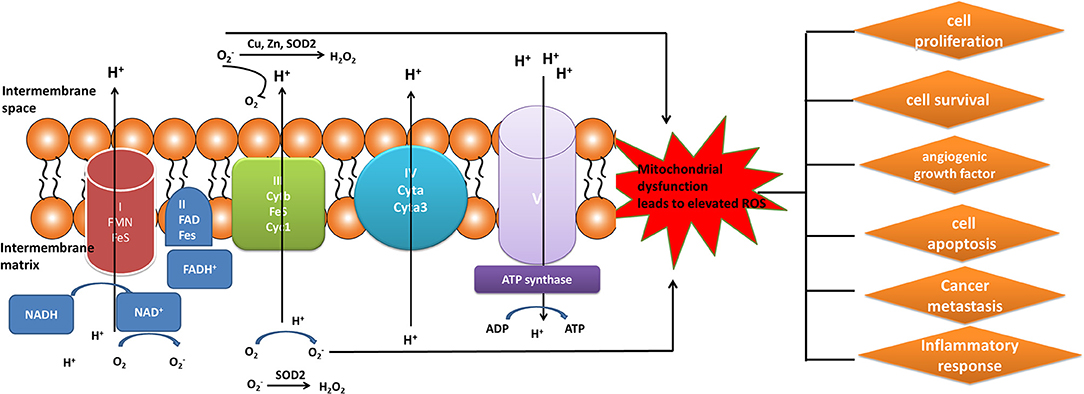
Figure 3. Mitochondrial generation of ROS. Complexes I, II, and III (complexes located on electron transport chain) play a pivotal role in the generation of ROS during the process of oxidative phosphorylation. The increased ROS regulated cancer progression, angiogenesis, metastasis, and survival.
It needs further study for imbalance of ROS and RNS production resulted from mitochondria dysfunction in pituitary adenomas. Many studies found the presence of ROS/RNS in human pituitaries, and the increased activities of ROS/RNS in pituitary adenoma compared to control tissues (41, 49). Also, oxidative stress in pituitary adenoma cells is accompanied by mitochondria swelling during tumor development, and associated with an increased fusion process and augmented biogenesis. An activation of the nuclear factor erythroid 2 like 2 (Nrf2) pathway and the reduction of oxidative damage signals were also observed during tumor development, which might provide survival advantages to pituitary adenoma cells (50). ROS pathway tends to be a medium in human pituitary cells. The pro-apoptotic effects are regulated partly by the dopamine transporter in GH3 pituitary cell lines, and involve oxidative stress as well as ROS formation. The use of only dopamine to treat hypophysis cells found that intracellular ROS was increased rapidly, and antioxidant N-acetyl-L-cysteine effectively inhibited dopamine-induced ROS generation and apoptosis (51). These data clearly demonstrated that ROS formation was closely related to signaling pathways in pituitary adenomas to affect tumor biological behavior. Studies found that 18beta-glycyrrhetinic acid had significantly antitumor effects on pituitary adenomas because this drug activated mitochondria-mediated ROS-mitogen-activated protein kinase (MAPK) pathways to induce cell apoptosis in pituitary adenomas, and that these activating effects were attenuated in pituitary adenomas by pretreatment with N-acetyl-L-cysteine, a ROS inhibitor (29). Moreover, many studies found the presence of NOS in human pituitaries, and NOSs were markedly higher expressed in invasive relative to non-invasive pituitary adenomas (52). NO mediated oxidative stress in pituitary adenoma cell lines to induce caspase-dependent apoptosis (53). Another study found that serum NO concentration was significantly decreased after the surgery of patients with pituitary adenomas (n = 21), thus monitoring serum NO level after pituitary adenoma surgery might benefit the prediction of its occurrence (54).
Thereby, oxidative stress has been considered as one of essential factors to contribute in the pathogenesis of pituitary adenomas. However, its molecular mechanisms remain unclear. The previous studies have been provided clues to the mechanism; for example, “mitochondrial theory of aging” increased production of ROS with altered expression of caveolae (87). It is meaningful to explore the roles of oxidative stress-mediated apoptosis, ER stress, DNA damage, metabolism, autophagy, migration, and anticancer drugs. The in-depth understanding of the relationship between oxidative stress and mitochondrial dysfunction might benefit improvement of chemotherapeutic approaches based on ROS/RNS-modulating drugs in the treatments of pituitary adenomas.
Apoptosis is a gene-controlled form of programmed cell death, and is closely related to cancer. Apoptosis activation mechanisms include extrinsic and intrinsic pathways (88). The extrinsic pathway including FAS path and TNF path is activated by receptor-ligand-mediated model. Extracellular ligands binding to membrane death receptors result in the formation of death-inducing signaling complex (89). The intrinsic pathway including mitochondrial apoptosis and endoplasmic reticulum apoptosis pathways is activated by intracellular signals. Internal mitochondrial apoptosis pathway could be activated by internal apoptosis stimulators, such as persistent DNA damage, cell hypoxia, and cell growth factor deletion (90), or by death ligands and caspase 12. The Bcl-2 family proteins decrease the mitochondrial membrane potential promotion and increase mitochondrial outer membrane permeabilization, which leads to release pro-apoptotic factors such as AIF, cytochrome c, SMAC/DIABL, HTRA2/OMI, and ENDOG from the mitochondria into the cytosol (91). The increased mitochondrial outer membrane permeabilization is generally considered to activate the apoptotic pathway because the formation of apoptosome in the cytosol induces the caspase cascade (Figure 4) (92). Mitochondrial dysfunction occurred in a human pituitary tumor with mitochondrial morphological and functional changes, including large mitochondria, mitochondrial irregular swelling, and partly or fully disintegrated cristaes (16). Mitochondrial dysfunction caused change of mitochondrial membrane potential and internal apoptosis stimulator responses, which leads to mitochondria-mediated apoptosis signaling pathway alteration (93).
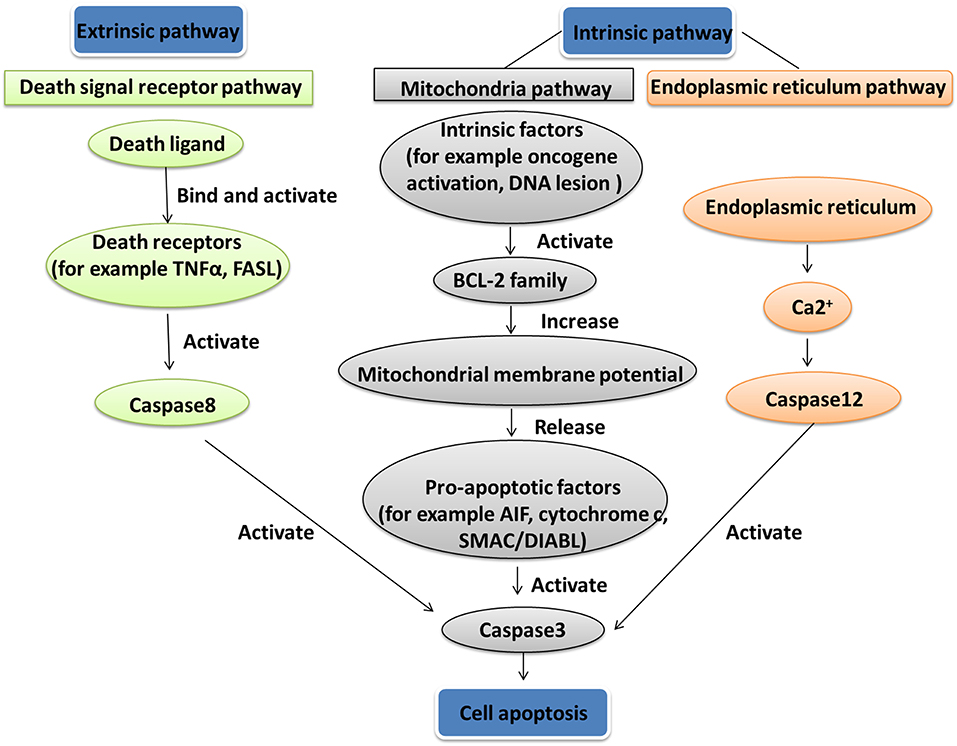
Figure 4. Apoptosis pathway. Two apoptosis activation mechanisms are the extrinsic and intrinsic pathways. The extrinsic pathway, including Fas path and TNF path, is activated by receptor-ligand-mediated model. The intrinsic pathway, including the mitochondrial apoptosis pathway and endoplasmic reticulum apoptosis pathway, is activated by intracellular signals.
From the embryology angle, many apoptotic cells are formed in Rathke's pouch tissues from the roof of oral ectoderm at an early stage of pituitary gland formation. However, when adenohypophysis is formed in the distal part of the gland, the ratio of apoptotic cells was significantly lower than early stage. It means that an imbalance in apoptosis process might be the boundary between embryonic development and tumor progress. It is interested that pituitary adenoma cells undergo the imbalanced expressions of apoptosis-related genes/proteins to cause uncontrolled cell proliferation (55). Study found that targeting mitochondria could have an effective impact on the treatment of pituitary tumors through apoptosis pathway (56). For example, trefoil factor 3 (TFF3) is an apoptosis-related protein, and its knockout in human pituitary adenoma cell line decreased the levels of apoptosis-related proteins Bcl-2 and caspase-3, and increased the levels of Bax and cleaved caspase-3. It clearly demonstrated that TFF3 protein knockout can accelerate the apoptosis in human pituitary adenoma cells via mitochondrial apoptosis pathway (57). Moreover, apoptotic protease-activating factor-1 (APAF-1) is a pivotal functional protein to involve in the intrinsic mitochondrial apoptosis pathway. Low expression of APAF-1 was detected in most invasive pituitary adenomas, and was negatively correlated with the aggressive behavior of invasive pituitary adenoma, which suggested that shifting the balance of apoptosis mediators in cells could lead to changes of pituitary tumor behaviors (58). In addition, some microRNAs-target genes to mediate apoptosis pathway also have been found in pituitary adenomas. For example, tumor suppressor microR-34a overexpression significantly inhibited cell proliferation and promoted cell apoptosis in pituitary adenoma cells (59). miR-21 expression was lower in invasive relative to non-invasive pituitary adenoma tissues, and miR-21 targeted 3'-UTR of PITX2 gene to enhance caspase-3 activity, which inhibits cell proliferation and facilitates apoptosis in pituitary adenoma cells (60). Dysregulation of apoptosis-related proteins might be meaningful indicator of tumor progression because mitochondrial dysfunction pathway might facilitate tumorigenesis and tumor development (94).
The novel mechanisms in mitochondrial dysfunction-mediated cell apoptosis would facilitate the development of effective anti-cancer drugs. For example, the classical antitumor effect of paclitaxel is to target on tubulin in the cytoplasm. However, further study found that paclitaxel induced apoptosis by promoting the release of Cyt C after binding with Bcl-2 (95), which promoted one to accurately deliver paclitaxel though well-designed nanocarrier to improve its treatment performance (94). Furthermore, for adrenocorticotrophic hormone (ACTH)-producing pituitary adenomas (96), ursolic acid was found to be a potential agent targeting ACTH-producing AtT20 cells because ursolic acid inhibited cell proliferation, reduced ACTH secretion, and induced cell apoptosis in AtT20 cells with the decreased ratio of Bcl-2 to Bcl2-associated X protein to cause the release of mitochondrial cytochrome c from mitochondria to the cytosol, and activate subsequently caspase-9, -3/7, and -8. It indicates that ursolic acid may be a promising candidate drug for the treatment of ACTH-producing pituitary adenomas (61). Therefore, insights into mitochondria-mediated apoptosis might benefit the development of novel pro-apoptotic therapeutic drugs and discovery of biomarkers for early detection to treat a pituitary adenoma.
Autophagy or autophagocytosis meaning “self-devouring” is the natural process and common cellular phenomenon, which is involved in the processes of phagocytosis and degradation of dysfunctional or unnecessary cell components, and also reusing of cellular components (97, 98). Briefly, the dysfunctional or unnecessary components are engulfed to form a double membrane called autophagosome, and then autophagosome is fused with the lysosome, followed by degradation of the contents into smaller constituents via acidic lysosomal hydrolase within lysosome (99). Autophagy takes part in various cellular functions, and particular attention has been paid to dual functions of autophagy in cancer—both protection cells against cancer and a potentially factor in cancer cell survival. Autophagy is regulated by many of the proteins, including oncogene and tumor suppressor proteins. Specifically, tumor suppressor proteins that negatively regulate mTOR pathway, such as LKB1, PTEN, TSC1/2, and AMPK, stimulate autophagy, while oncogenes that activate mTOR pathway, such as Ras, class I PI3K, AKT, and Rheb, inhibit autophagy, indicating the contribution of autophagy to cancer growth or tumor suppression. Moreover, the inhibition of autophagy induces genomic instability, oxidative stress, and tumorigenesis. Nevertheless, autophagy also functions as a protective factor under stress conditions, including nutrient starvation, and hypoxia that facilitates tumor cell survival and sensitivity and resistance to chemotherapy (100).
Mitophagy is the complex biological process that cells selectively eliminate mitochondria by autophagy. The engulfment of mitochondria forms a double-membrane-enclosed autophagosome and then fuses with lysosomes. The process emits high-energy substances to recycle cell compartment, including fatty acids and amino acids (Figure 5) (101). Defective mitochondria undergoing damage or stress tend to the induction of mitophagy. However, the occurrence of mitophagy is not only restricted to the defective mitochondria but also involves normal ones (102). Mitophagy promotes turnover and the selective degradation of mitochondria, and prevents accumulation of dysfunctional mitochondria, which can lead to keep steady-state mitochondrial turnover, cellular metabolic needs, and certain cellular developmental stages (102). In this process, mitophagy depends on the general autophagy mechanism, meanwhile, both “mitophagy adaptors” and regulatory molecules are involved, such as p62, FUNDC1, prohibitin2, BNIP3L (NIX), PGAM5, OPA1, TBK1, CK, OPTN, and Bcl2-L13 (103). Those autophagy-related proteins activate downstream mitophagy pathways by post-translational modifications (104). Two distinct principal mitophagy mechanisms had been proved in mammalian cells. Firstly, receptor-mediated mitophagy is activated, and then recruits Atg8-like proteins to mitochondria to increase combination. Secondly, the highly ubiquitylated mitochondrial outer membrane proteins recruit bifunctional adapter proteins, which in turn increases the binding of Atg8-like proteins (105). Atg8-like proteins prompts encapsulation of mitochondria into autophagosomes through the expansion of the phagophore membrane; and fusion with the lysosome results in the formation of autolysosomes facilitating degradation of selected dysfunctional mitochondria (106). Also, mitophagy receptors exist some complex regulation mechanisms. An unmodified receptor is phosphorylated by a kinase, activating or deactivating downstream mitophagy pathway by increasing or decreasing Atg8-like proteins binding. This effect can be reversed by phosphatases. For NIX (BNIP3L), BCL2L13, and BNIP3, only the activated phosphorylation and the modified residue mechanism are understood but the kinases or phosphatases have not been identified yet for FUNDC1, the activated and deactivated phosphorylation intracellular mechanisms, the modified residues and participated kinases or phosphatases are well-known (107).
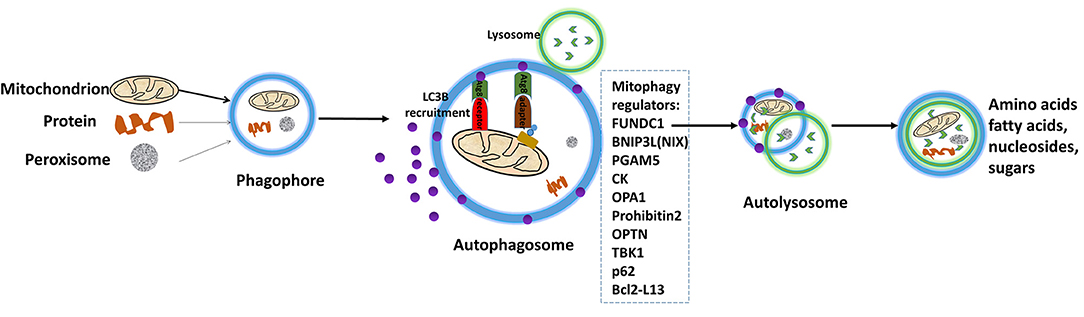
Figure 5. Depiction of the process of mitophagy. The engulfment of mitochondria forms a double-membrane-enclosed autophagosome, and then fuses with lysosomes. The process emits high-energy substances to recycle cell compartment, for example fatty acids and amino acids. Modified from Li et al. (15), with permission from Bioscientifica Limt.
Mitochondrial dysfunction might affect mitophagy that can be related to metabolic reprogramming, inflammatory signaling, cell fate determination and differentiation, DNA damage responses in response to stress, which in turn lead to human disease incidence and etiology, including malignant tumor (103). It is well-known that mitophagy and mitochondrial dysfunction are related to pituitary adenomas. The mitochondrial toxicity and protective mechanisms of T-2 toxin are not fully understood in mammalian cells (108). The investigation of the cellular and mitochondrial toxicity of T-2 toxin shows that T-2 toxin significantly increases mitophagic activity, ROS and DNA damage in rat pituitary GH3 cells. With the increased expression of mitophagy-specific proteins, including E3 ubiquitin ligase Parkin, NIP-like protein X (NIX), and PTEN-induced putative kinase protein 1 (PINK1), T-2 toxin can be increased (109). The regulating mechanism of mitophagy is also mediated by nuclear factor (erythroid-derived 2)-like 2 (Nrf2)/PINK1/Parkin pathway in pituitary GH3 cells. The relevant drug activates the protective protein kinase A signaling pathway, which activates the Nrf2/PINK1/Parkin pathway to mediate mitophagy. Taken together, increasing mitophagy and mitochondrial dysfunction might increase chemo-resistance in pituitary GH3 cells (36). Sometimes apoptosis and autophagy coexist. Dopamine agonists such as bromocriptine and cabergoline have been successfully used in the treatment of pituitary prolactinomas. DRD5 activation increases production of ROS, inhibits the MTOR pathway, induces macroautophagy/autophagy, and leads to autophagic cell death (ACD) in human pituitary tumor cells (38). In addition, when cyclosporine A (CsA) induces apoptotic and ACD in pituitary GH3 cells, Bcl-2 levels show dose-dependent augmentation in autophagy and are decreased in apoptosis (37).
Autophagy can promote survival of tumor cells in starvation mode, or degrade cell apoptotic mediators to maintain the tumor clone. In such cases, treated patients with the late stage of autophagy—blockers (such as chloroquine), on the cells that depend on autophagy to survive, may be one of viable therapeutic measurements in fighting cancer (110). Thus, the qualities of mitophagy can be used as a therapeutic method for cancer prevention. Mitophagy plays a role in tumor suppression and tumor cell survival. One strategy is to induce mitophagy and enhance the function of antitumor. The other strategy is to inhibit mitophagy and thus induce apoptosis (111). The first strategy has been tested by monitoring dose-response anti-tumor effects during autophagy-targeted therapies. These treatment effects have shown that autophagy has some dose-dependence in tumor suppression and tumor cell survival progression. The result supports the development of therapies through autophagy (112). In addition, inhibition of the protein related to autophagy pathways may also serve as an anticancer therapy (113). Autophagy is a protein degradation system to play a role in maintaining homeostasis and inducing apoptosis. Thus, sometimes inhibition of autophagy has on the probability of existential risk as it may lead to tumor development instead of the desired cell death (114).
Tumor immune escape is an important hallmark in cancer (115). Tumor-evading immune destruction is closely correlated with prognosis or survival in various tumors (116). A study found a number of tumor immunity-related inflammatory cells, for example, cytotoxic T cells (CTLs), regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and natural killer (NK) cells (37). It also found a number of tumor immunity-related pathways, such as altered interleukin signaling (117), MHC-I pathway (118), type 1 cytokine-induced T-cell (119), interferon gamma signaling (120), type I interferon-mediated responses (121), and transcription factor nuclear factor-kappa B (122). Along with the advancement in tumor immunology, the immune-checkpoint blockade therapy has been an important aspect in the mode of combined therapy of tumor. One of the most important immune checkpoint pathways has been applied between the PD-1 receptor expressed on activated T cells and its ligands, programmed death-1 ligand (PD-L1) and PD-L2 (123).
Immunity process is interlinked with mitochondrial function. Mitochondria can regulate immunity in different ways (Figure 6): (i) Current literature shows that many changes of cancers occurred in substance metabolism pathways such as TCA cycle, oxidative phosphorylation, fatty acid oxidation, and amino acid metabolism. Mitochondria that induce transcriptional key enzymes or important molecule changes can lead to completely different results in immune cells (116). Thus, mitochondria can regulate differentiation, activation, and survival of immune cells (124). (ii) Mitochondrial DNA (mtDNA) could translocate from the mitochondria to cytoplasm and activate the NLRP3 inflammasome to induce IL-1β and IL-18 release (125). (iii) Mitochondria can transmit signals through mtDNA or mitochondrial ROS (mtROS) to regulate gene expressions of immune cells (126). (iv) Mitophagy is crucial for degradation of the damaged mitochondria, and the decreased mitophagy causes ROS increasing which further makes the susceptibility to infections (127). (v) Immune functions are influenced by fission and fusion of mitochondria, which determines mitochondrial mobility and mass. (vi) When mitochondria are located near endoplasmic reticulum (ER), the mitochondria and ER junction signaling could be activated in immune cells to influence immune cell metabolism (128). (vii) The inflammatory response can be initiated by mitochondrial antiviral signaling (MAVS). Therefore, mitochondrial machinery is crucial for immune functions, such as metabolic pathways, mtDNA, mtROS, mitochondrial dynamics, and mitophagy.

Figure 6. Immunity and mitochondria are interlinked with each other. Mitochondria can regulate immunity in different ways, including metabolic pathways, mitochondrial dynamics, mtDNA, mitophagy, mtROS, MAVS, UCP2, and ER-mitochondria junction.
Tumor immune microenvironment is gradually recognized as a critical contributor in tumor progression, development, and control. The increasing studies show that immune cells infiltrate in pituitary adenomas. Dysregulation of several genes in granulocyte-macrophage colony-stimulating factor signal transduction has been regarded as a possible alteration underlying the occurrence and development of pituitary tumors, such as mitochondria-associated protein Tim 16. High-expression of Tim 16 is identified in mouse and human ACTH-secreting pituitary adenomas compared to normal pituitary to protect pituitary cells from apoptosis (62). Also, more macrophages are identified in larger pituitary adenomas, and more T cells are detected in GH-secreting pituitary adenomas. A positive correlation is found between the numbers of CD68+ macrophages and tumor sizes and grades for pituitary adenoma invasiveness. The density of infiltrated CD4+ and CD8+ T-lymphocytes may be relatively insufficient in these pituitary adenomas, but CD4+ and CD8+ T lymphocytes are significantly more in GH-secreting adenomas than non-GH adenomas. Both densely and sparsely granulated GH-adenomas had significantly more CD4+ cells than ACTH-adenomas, and significantly more CD8+ cells than null cell adenomas. These results suggest an association of the enhanced T-lymphocytes infiltration and invasiveness in pituitary adenomas, and that adjuvant immunotherapy might block the tumor enlargement and invasiveness of pituitary adenomas (129). Furthermore, study shows that low expression levels of immune-related genes induce the occurrence of pituitary adenomas (130). Another study show reveals the association of NF-κB, MMP-9, and MICA in pituitary adenomas, the higher expressions of MICA, MMP-9, and NF-κB in mRNA and protein levels in pituitary adenomas relative to healthy tissues; which found that the upregulation of NF-κB can activate the expression of MICA and increase MMP-9 expression to hydrolyze MICA into sMICA to facilitate tumor immune escape (131). Although most pituitary adenomas are treated successfully, it remains challenging to treat invasive non-functional pituitary adenomas as well as functional pituitary adenomas unresponsive to traditional therapy. Immunotherapy might be a potential alternative therapy for pituitary tumors that are resistant to traditional therapy (132). The positive PD-L1 immunostaining is significantly more frequent in functional relative to non-functional pituitary adenomas (p = 0.000). The expression level of PD-L1 is more related to the increased blood levels of ACTH-, PRL-, GH-, and cortisol-secreting pituitary adenomas. PD-L1 expression is also associated with GH and PRL immunostaining density and higher Ki-67 index (133). Thereby, immunotherapy might be a promising therapy option of functional pituitary adenomas on the basis of in-depth understanding of mitochondria-mediated immunity in pituitary adenomas.
Mitochondrion is a highly dynamic organelle under the coordination between fission and fusion cycles, which is referred as “mitochondrial dynamics.” Fission-fusion cycles affect mitochondria shape, size, and distribution (134). Mitochondria transient and rapid morphological adaptations are crucial for various cellular processes such as apoptosis (135), energy metabolism (136), cell cycle (137), ROS (138), immunity (139), mitophagy (140), and mitochondrial quality control (Figure 7). Mutations of the key machinery components or defects of mitochondrial dynamics are associated with lots of human diseases, including cancer (141). These dynamic transitions are primarily regulated by large GTPases. Mitochondrial fission and fusion cycle is a multi-step process. Mitochondrial fission is controlled by recruitment of dynamin-related protein 1 (Drp1) by adaptors at ER- and actin-mediated mitochondrial constriction sites. Drp1 oligomerization results in mitochondrial constriction, which leads to dynamin 2 recruitment to terminate mitochondrial outer membrane scission. Inner mitochondrial membrane constriction is an independent process, and mediated by calcium influx. Mitochondrial fusion is driven by mitofusins 1 and 2 within the outer mitochondrial membrane, and mediated by optic atrophy 1 with inner membrane (142). Moreover, several members of membrane lipid composition could undergo post-translational modifications to regulate these processes (143). Therefore, it is crucial for one to in-depth understand molecular mechanisms of mitochondrial dynamics for further studying various cellular processes associated with human diseases.
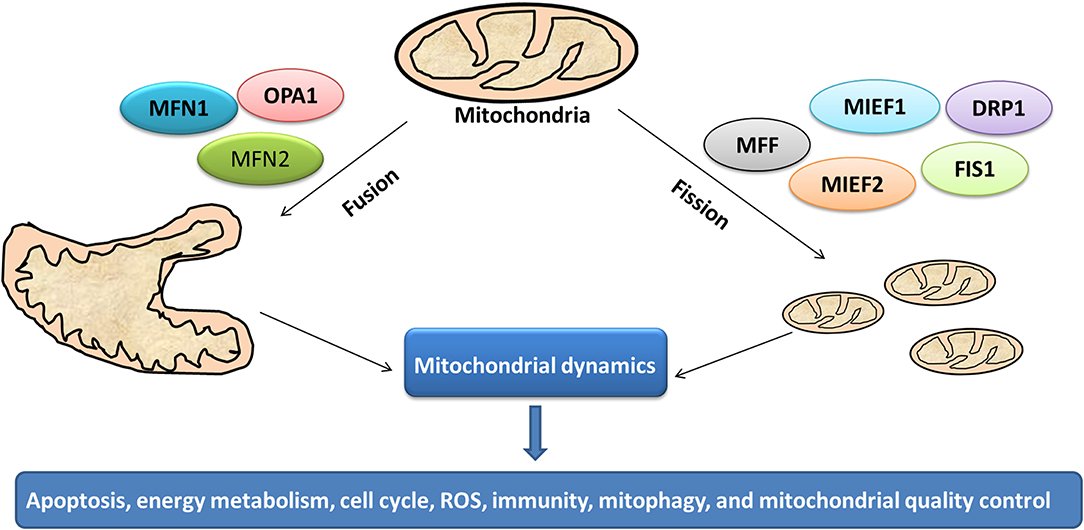
Figure 7. Mitochondrial dynamics in cancer. Cycles of fission and fusion are crucial for various cellular processes such as apoptosis, energy metabolism, cell cycle, ROS, immunity, mitophagy, and mitochondrial quality control.
Various cellular processes, such as apoptosis (135), energy metabolism (136), cell cycle (137), ROS (138), immunity (139), and mitophagy (140), are closely related to mitochondrial dynamics and mitochondrial dysfunction. The relationship study between cell apoptosis and mitochondrial fission found that IR-783 induces Drp1 translocation from cytoplasm to mitochondria, makes the expression of mitochondrial fission proteins (MFF) and mitochondrial fission factor fission-1 (Fis1) increased, and the expression of optic atrophy 1 (OPA1) and mitochondrial fusion proteins mitofusin1 (Mfn1) decreased. The process of mitochondrial translocation of Drp1 mediated mitochondrial fission and markedly induced apoptosis in vivo and xenograft model (144). The signaling pathways involved in mitochondrial dynamics regulation and their roles in maintaining energy metabolism has become an active area of research. Mitochondrial dynamic events, such as fusion, fission, and transport, affect the mitochondrial shape, size, function, and subcellular localization. Mitochondria dynamic changes play a crucial role in assisting metabolite transfer, biogenesis, and degradation to maintain energy homeostasis (136). Mitochondrial electron transport chain-derived ROS and mitochondrial fission/fusion rates influence this delicate balance between mitochondrial dynamics and mitochondria-derived ROS production, which plays main roles in malignant diseases (138). In addition, mitochondrial dynamics role in cancer growth connects with the immune system activity, especially T cells. Although it has not been directly verified whether or not mitochondrial dynamics are associated with lymphocytes memory formation, Drp1-dependent mitochondrial fission has the potential contribution to regulate NK memory phase. It is indicated that mitochondrial dynamics is also possible to play role in the cytotoxic activity of these lymphocytes against cancer. Thereby, mitochondria can control local calcium influx to regulate the inner mitochondrial membrane constriction. It would be then interesting to see how mitochondrial dynamics to regulate the release of cytotoxic granules by T lymphocytes (139). The close interactions between mitochondrial dynamics and mitophagy become main players in the physiological cell processes in cancers. The new metabolic changes that mainly lead to mitochondrial functions and dysfunctions are strongly related to cancers, mitochondrial dynamics, and mitophagy (140). Study demonstrates that the BCL2/BCLXL inhibitor ABT737 mediates intrinsic apoptotic pathways and mitophagy through increasing levels of DRP1 in mitochondria and rates of mitochondrial fission (145).
In spite of considerable progresses in understanding mitochondrial dysfunction pathway networks and mitochondrial dynamics in the pathogenesis of pituitary adenomas, many key issues remain unclear. Several lines of evidence indicate that mitochondrial dysfunction emerge cross-links with various complex biological processes, including energy metabolism, oxidative stress, cell apoptosis, cell cycle, mitophagy, and immunity process. Moreover, mitochondrial dynamics is closely associated with mitochondrial dysfunction, and also plays a critical role in many biological processes. This review breaks new ground in the comprehensive understanding of potential mechanisms underlying between mitochondrial homeostasis and tumorigenesis in pituitary adenomas. The association of mitochondrial dysfunction, mitochondrial dynamics, and the complex biological processes helps to broaden the knowledge of mitochondrial functions in cancer and perspectives regarding tumor treatment. It offers the new promising to develop new candidate targets based on mitochondrial dysfunction pathway and mitochondrial dynamics, for effective therapy in pituitary adenomas.
NL collected and analyzed references, prepared figures and tables, and wrote the manuscript. XZ conceived the concept, designed, coordinated, critically revised/wrote manuscript, and was responsible for its financial supports and the corresponding works. All authors approved the final manuscript.
This work was supported by the grants from Hunan Provincial Hundred Talent Plan (to XZ), the China 863 Plan Project (Grant No. 2014AA020610-1 to XZ), National Natural Science Foundation of China (Grant No. 81572278 and 81272798), the Xiangya Hospital Funds for Talent Introduction (to XZ), and the Hunan Provincial Natural Science Foundation of China (Grant No. 14JJ7008 to XZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACTH, Adrenocorticotrophic hormone; AIF, Apoptosis-inducing factor mitochondria-associated 1; AMPK, Protein kinase AMP-activated catalytic subunit alpha 1; APAF-1, Apoptotic protease-activating factor-1; ATP, Adenosine triphosphate; Bcl-2, Apoptosis regulator; BNIP3L, BCL2 interacting protein 3 like; CAF, Cancer-associated fibroblasts; CAPN2, Calpain 2; CK, Choline kinase alpha; CTLs, Certain cytotoxic T cells; DNA, Deoxyribonucleic acid; Drp1, Dynamin-related protein 1; ER, Endoplasmic reticulum; ESI-MS, Electro-spray ionization-mass spectrometry; FADH2, Flavin adenine dinucleotide reduced; Fis1, Mitochondrial fission factor fission-1; FUNDC1, FUN14 domain containing 1; GH, Growth hormone; HNE, 4-hydroxynonenal; HTRA2/OMI, HtrA serine peptidase 2; IDH1, Isocitrate dehydrogenase 1; IL-18, Interleukin-18; IL-1β, Interleukin 1β; ITPR1, Inositol 1, 4, 5-trisphosphate receptor type 1; KIi-67, Nuclcar-associated antigen; LDHA, Lactate dehydrogenase A; LKB1, Serine/threonine kinase 11; MAVS, Mitochondrial antiviral signaling; MDSCs, Myeloid-derived suppressor cells; MFF, Mitochondrial fission proteins; Mfn1, Mitochondrial fusion proteins mitofusin 1; MHC-I, Major histocompatibility complex, class I; MMP-9, Matrix metallopeptidase 9; MT/ND1, Mitochondrially encoded NADH dehydrogenase 1; mtNOS, Mitochondrial nitric oxide synthase; mtROS, Mitochondrial ROS; NADH, Nicotinamide adenine dinucleotide; NF-κB/IRF, Nuclear factor kappa B subunit 1/tripartite motif containing 63; NK, Natural killer; NLRP3, NLR family pyrin domain containing 3; NO, Nitric oxide; Nrf2, Nuclear factor erythroid 2 like 2; OPA1, Optic atrophy 1; OPTN, Optineurin; OXPHOS, Oxidative phosphorylation; PD-L1, Programmed cell death 1 ligand 1; PD-L2, Programmed cell death 1 ligand 2; PGAM5, PGAM family member 5; PI3K, Phosphatidylinositol-4, 5-bisphosphate 3-kinase catalytic subunit beta; PINK1, PTEN-induced putative kinase protein 1; PITX2, Paired like homeodomain 2; PPP3CA, Protein phosphatase 3 catalytic subunit alpha; PPPM, Predictive, preventive, and personalized Medicine; PRKCA, Protein kinase C alpha; PTEN, Phosphatase and tensin homolog; Ras, RAS proto-oncogene GTPase; Rheb, Ras homolog mTORC1 binding; RIG-I, Retinoic acid-inducible gene I; RNA, Ribonucleic acid; RNS, Reactive nitrogen species; ROS, Reactive oxygen species; SDH, Succinate dehydrogenase; SIRT3, Programmed cell death 1PD-1, sirtuin 3; SMAC/DIABL, Diablo IAP-binding mitochondrial protein; TBK1, TANK binding kinase 1; TCA, Tricarboxylic acid cycle; TFF3, ENDOG trefoil factor 3; TLR2, Toll-like receptor 2; TNF, Tumor necrosis factor; Tregs, Regulatory T cells; TSC1/2, TSC complex subunit .
1. Stalla GK, Ciato D, Dimopoulou C. “The adrenal gland: central relay in health and disease - Current challenges and perspectives 2018” - Cushing's disease. Exp Clin Endocrinol Diabetes. (2018) 127:147–55. doi: 10.1055/a-0664-7632
2. Zhan X, Wang X, Cheng T. Human pituitary adenoma proteomics: new progresses and perspectives. Front Endocrinol. (2016) 7:54. doi: 10.3389/fendo.2016.00054
3. Qian S, Yang Y, Li N, Cheng T, Wang X, Liu J, et al. Prolactin variants in human pituitaries and pituitary adenomas identified with two-dimensional gel electrophoresis and mass spectrometry. Front Endocrinol. (2018) 9:468. doi: 10.3389/fendo.2018.00468
4. Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. (2017) 134:521–35. doi: 10.1007/s00401-017-1769-8
5. Reimondo G, Puglisi S, Pia A, Terzolo M. Autonomous hypercortisolism: definition and clinical implications. Minerva Endocrinol. (2018) 44:33–42. doi: 10.23736/s0391-1977.18.02884-5
6. Zhan X, Huang Y, Long Y. Two-dimensional gel electrophoresis coupled with mass spectrometry methods for an analysis of human pituitary adenoma tissue proteome. J Vis Exp. (2018) 134:e56739. doi: 10.3791/56739
7. Cheng T, Zhan X. Pattern recognition for predictive, preventive, and personalized medicine in cancer. Epma J. (2017) 8:51–60. doi: 10.1007/s13167-017-0083-9
8. Zhu H, Tamura T, Hamachi I. Chemical proteomics for subcellular proteome analysis. Curr Opin Chem Biol. (2018) 48:1–7. doi: 10.1016/j.cbpa.2018.08.001
9. Robin T, Bairoch A, Muller M, Lisacek F. Large-scale reanalysis of publicly available hela cell proteomics data in the context of the human proteome project. (2018) 17:4160–70. doi: 10.1021/acs.jproteome.8b00392
10. Moreno CS, Evans CO, Zhan X, Okor M, Desiderio DM, Oyesiku NM. Novel molecular signaling and classification of human clinically nonfunctional pituitary adenomas identified by gene expression profiling and proteomic analyses. Cancer Res. (2005) 65:10214–22. doi: 10.1158/0008-5472.can-05-0884
11. Evans CO, Moreno CS, Zhan X, McCabe MT, Vertino PM, Desiderio DM, et al. Molecular pathogenesis of human prolactinomas identified by gene expression profiling, RT-qPCR, and proteomic analyses. Pituitary. (2008) 11:231–245. doi: 10.1007/s11102-007-0082-2
12. Tanase C, Codrici E, Popescu ID, Cruceru ML, Enciu AM, Albulescu R, et al. Angiogenic markers: molecular targets for personalized medicine in pituitary adenoma. Per Med. (2013) 10:539–48. doi: 10.2217/pme.13.61
13. Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. (1999) 19:1720–30.
14. Anderson L, Seilhamer J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis. (1997) 18:533–7. doi: 10.1002/elps.1150180333
15. Li N, Li H, Cao L, Zhan X. Quantitative analysis of the mitochondrial proteome in human ovarian carcinomas. Endocr Relat Cancer. (2018) 25:909–31. doi: 10.1530/erc-18-0243
17. Magalhaes J, Venditti P, Adhihetty PJ, Ramsey JJ, Ascensao A. Mitochondria in health and disease. Oxid Med Cell Longev. (2014) 2014:814042. doi: 10.1155/2014/814042
18. Zhan X, Desiderio DM. Signaling pathway networks mined from human pituitary adenoma proteomics data. BMC Med Genomics. (2010) 3:13. doi: 10.1186/1755-8794-3-13
19. Zhan X, Wang X, Long Y, Desiderio DM. Heterogeneity analysis of the proteomes in clinically nonfunctional pituitary adenomas. BMC Med Genomics. (2014) 7:69. doi: 10.1186/s12920-014-0069-6
20. Lu M, Wang Y, Zhan X. The MAPK pathway-based drug therapeutic targets in pituitary adenomas. Front Endocrinol. (2019) 10:330. doi: 10.3389/fendo.2019.00330
21. Tanase CP, Neagu M, Albulescu R. Key signaling molecules in pituitary tumors. Expert Rev Mol Diagn. (2009) 9:859–77. doi: 10.1586/erm.09.60
22. Fujisawa H, Tohma Y, Muramatsu N, Kida S, Kaizaki Y, Tamamura H. Spindle cell oncocytoma of the adenohypophysis with marked hypervascularity. Case report. Neurol Med Chir. (2012) 52:594–8. doi: 10.2176/nmc.52.594
23. Saeger W, Rubenach-Gerz K, Caselitz J, Ludecke DK. Electron microscopical morphometry of GH producing pituitary adenomas in comparison with normal GH cells. Virchows Arch A Pathol Anat Histopathol. (1987) 411:467–72.
24. Yamada S, Asa SL, Kovacs K. Oncocytomas and null cell adenomas of the human pituitary: morphometric and in vitro functional comparison. Virchows Arch A Pathol Anat Histopathol. (1988) 413:333–9.
25. Saeger W, Kant P, Caselitz J, Ludecke DK. Electron microscopical morphometry of pituitary adenomas. Comparison of tumours in acromegaly and hyperprolactinemia. Pathol Res Pract. (1988) 183:17–24.
26. Yang QH, Xu JN, Xu RK, Pang SF. Antiproliferative effects of melatonin on the growth of rat pituitary prolactin-secreting tumor cells in vitro. J Pineal Res. (2007) 42:172–9. doi: 10.1111/j.1600-079X.2006.00403.x
27. Wang BQ, Yang QH, Xu RK, Xu JN. Elevated levels of mitochonrial respiratory complexes activities and ATP production in 17-beta-estradiol-induced prolactin-secretory tumor cells in male rats are inhibited by melatonin in vivo and in vitro. Chin Med J. (2013) 126:4724–30. doi: 10.3760/cma.j.issn.0366-6999.20131965
28. Dai C, Zhang B, Liu X, Guo K, Ma S, Cai F, et al. Pyrimethamine sensitizes pituitary adenomas cells to temozolomide through cathepsin B-dependent and caspase-dependent apoptotic pathways. Int J Cancer. (2013) 133:1982–93. doi: 10.1002/ijc.28199
29. Wang D, Wong HK, Feng YB, Zhang ZJ. 18beta-glycyrrhetinic acid induces apoptosis in pituitary adenoma cells via ROS/MAPKs-mediated pathway. J Neurooncol. (2014) 116:221–30. doi: 10.1007/s11060-013-1292-2
30. Tang J, Wang Z, Chen L, Huang G, Hu X. Gossypol acetate induced apoptosis of pituitary tumor cells by targeting the BCL-2 via the upregulated microRNA miR-15a. Int J Clin Exp Med. (2015) 8:9079–85.
31. Ji YZ, Geng L. Chinese herbal medicine Yougui Pill reduces exogenous glucocorticoid-induced apoptosis in anterior pituitary cells. Neural Regen Res. (2016) 11:1962–8. doi: 10.4103/1673-5374.197138
32. Zhou HB, Wei HC, Chen HD, Liu X, Guo P, Liu A, et al. Nitric oxide (NO)-mediated mitochondrial damage plays a critical role in T-2 toxin-induced apoptosis and growth hormone deficiency in rat anterior pituitary GH3 cells. Food Chem Toxicol. (2017) 102:11–23. doi: 10.1016/j.fct.2017.01.017
33. Zhao Y, Zhang L, Yan A, Chen D, Xie R, Liu Y, et al. Grifolic acid induces GH3 adenoma cell death by inhibiting ATP production through a GPR120-independent mechanism. BMC Pharmacol Toxicol. (2018) 19:26. doi: 10.1186/s40360-018-0215-4
34. Zhang Q, Li C, Zhou Y, Zhao S, Hong L, Song Q, et al. The critical role of p16/Rb pathway in the inhibition of GH3 cell cycle induced by T-2 toxin. Toxicology. (2018) 400–1:28–39. doi: 10.1016/j.tox.2018.03.006.
35. Wei Y, Zhou X, Ren L, Wang C, Li Y. The prolactin-release inhibitor paeoniflorin suppresses proliferation and induces apoptosis in prolactinoma cells via the mitochondria-dependent pathway. J Cell Biochem. (2018) 119:5704–14. doi: 10.1002/jcb.26752
36. Deyu H, Luqing C, Xianglian L, Pu G, Qirong L, Xu W, et al. Protective mechanisms involving enhanced mitochondrial functions and mitophagy against T-2 toxin-induced toxicities in GH3 cells. Toxicol Lett. (2018) 295:41–53. doi: 10.1016/j.toxlet.2018.05.041
37. Kim HS, Choi SI, Jeung EB, Yoo YM. Cyclosporine A induces apoptotic and autophagic cell death in rat pituitary GH3 cells. PLoS ONE. (2014) 9:e108981. doi: 10.1371/journal.pone.0108981
38. Leng ZG, Lin SJ, Wu ZR, Guo YH, Cai L, Shang HB, et al. Activation of DRD5 (dopamine receptor D5) inhibits tumor growth by autophagic cell death. Autophagy. (2017) 13:1404–19. doi: 10.1080/15548627.2017.1328347
39. Wang X, Du Q, Mao Z, Fan X, Hu B, Wang Z, et al. Combined treatment with artesunate and bromocriptine has synergistic anticancer effects in pituitary adenoma cell lines. Oncotarget. (2017) 8:45874–87. doi: 10.18632/oncotarget.17437
40. Wang X, Guo T, Peng F, Long Y, Mu Y, Yang H, et al. Proteomic and functional profiles of a follicle-stimulating hormone positive human nonfunctional pituitary adenoma. Electrophoresis. (2015) 36:1289–304. doi: 10.1002/elps.201500006
41. An J, Zhang Y, He J, Zang Z, Zhou Z, Pei X, et al. Lactate dehydrogenase A promotes the invasion and proliferation of pituitary adenoma. Sci Rep. (2017) 7:4734. doi: 10.1038/s41598-017-04366-5
42. Casar-Borota O, Oystese KA, Sundstrom M, Melchior L, Popovic VA. high-throughput analysis of the IDH1(R132H) protein expression in pituitary adenomas. Pituitary. (2016) 19:407–14. doi: 10.1007/s11102-016-0720-7
43. Hao S, Hong CS, Feng J, Yang C, Chittiboina P, Zhang J, et al. Somatic IDH1 mutation in a pituitary adenoma of a patient with Maffucci syndrome. J Neurosurg. (2016) 124:1562–7. doi: 10.3171/2015.4.jns15191
44. Porcelli AM, Ghelli A, Ceccarelli C, Lang M, Cenacchi G, Capristo M, et al. The genetic and metabolic signature of oncocytic transformation implicates HIF1alpha destabilization. Hum Mol Genet. (2010) 19:1019–32. doi: 10.1093/hmg/ddp566
45. Xekouki P, Stratakis CA. Succinate dehydrogenase (SDHx) mutations in pituitary tumors: could this be a new role for mitochondrial complex II and/or Krebs cycle defects? Endocr Relat Cancer. (2012) 19:C33–40. doi: 10.1530/erc-12-0118
46. Xekouki P, Szarek E, Bullova P, Giubellino A, Quezado M, Mastroyannis SA, et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J Clin Endocrinol Metab. (2015) 100:E710–9. doi: 10.1210/jc.2014-4297
47. Wu S, Gu Y, Huang Y, Wong TC, Ding H, Liu T, et al. Novel biomarkers for non-functioning invasive pituitary adenomas were identified by using analysis of microRNAs expression profile. Biochem Genet. (2017) 55:253–67. doi: 10.1007/s10528-017-9794-9
48. Feng J, Zhang Q, Li C, Zhou Y, Zhao S, Hong L, et al. Enhancement of mitochondrial biogenesis and paradoxical inhibition of lactate dehydrogenase mediated by 14-3-3eta in oncocytomas. J Pathol. (2018) 245:361–72. doi: 10.1002/path.5090
49. Pawlikowski M, Winczyk K, Jaranowska M. Immunohistochemical demonstration of nitric oxide synthase (NOS) in the normal rat pituitary gland, estrogen-induced rat pituitary tumor and human pituitary adenomas. Folia Histochem Cytobiol. (2003) 41:87–90.
50. Sabatino ME, Grondona E, Sosa LDV, Mongi Bragato B, Carreno L, Juarez V, et al. Oxidative stress and mitochondrial adaptive shift during pituitary tumoral growth. Free Radic Biol Med. (2018) 120:41–55. doi: 10.1016/j.freeradbiomed.2018.03.019
51. Jaubert A, Ichas F, Bresson-Bepoldin L. Signaling pathway involved in the pro-apoptotic effect of dopamine in the GH3 pituitary cell line. Neuroendocrinology. (2006) 83:77–88. doi: 10.1159/000094044
52. Onishi K, Kamida T, Momii Y, Abe T, Fujiki M. The clinical and pathological significance of nitric oxide synthase in human pituitary adenomas: a comparison with MIB-1. Endocrine. (2014) 46:154–9. doi: 10.1007/s12020-013-0046-4
53. Huang D, Cui L, Guo P, Xue X, Wu Q, Hussain HI, et al. Nitric oxide mediates apoptosis and mitochondrial dysfunction and plays a role in growth hormone deficiency by nivalenol in GH3 cells. Sci Rep. (2017) 7:17079. doi: 10.1038/s41598-017-16908-y
54. Babula D, Horecka A, Luchowska-Kocot D, Kocot J, Kurzepa J. Decreased nitric oxide serum level after pituitary adenoma resection. J Neurosurg Sci. (2017). doi: 10.23736/s0390-5616.17.04083-8. [Epub ahead of print].
55. Guzzo MF, Carvalho LR, Bronstein MD. Apoptosis: its role in pituitary development and neoplastic pituitary tissue. Pituitary. (2014) 17:157–62. doi: 10.1007/s11102-013-0481-5
56. Gottardo MF, Pidre ML, Zuccato C, Asad AS, Imsen M, Jaita G, et al. Baculovirus-based gene silencing of Humanin for the treatment of pituitary tumors. Apoptosis. (2018) 23:143–51. doi: 10.1007/s10495-018-1444-0
57. Gao F, Pan S, Liu B, Zhang H. TFF3 knockout in human pituitary adenoma cell HP75 facilitates cell apoptosis via mitochondrial pathway. Int J Clin Exp Pathol. (2015) 8:14568–73.
58. Tanase C, Albulescu R, Codrici E, Calenic B, Popescu ID, Mihai S, et al. Decreased expression of APAF-1 and increased expression of cathepsin B in invasive pituitary adenoma. Onco Targets Ther. (2015) 8:81–90. doi: 10.2147/ott.s70886
59. Yang Z, Zhang T, Wang Q, Gao H. Overexpression of microRNA-34a attenuates proliferation and induces apoptosis in pituitary adenoma cells via SOX7. Mol Ther Oncol. (2018) 10:40–7. doi: 10.1016/j.omto.2018.07.001
60. Cui M, Zhang M, Liu HF, Wang JP. Effects of microRNA-21 targeting PITX2 on proliferation and apoptosis of pituitary tumor cells. Eur Rev Med Pharmacol Sci. (2017) 21:2995–3004.
61. Gong YY, Liu YY, Yu S, Zhu XN, Cao XP, Xiao HP. Ursolic acid suppresses growth and adrenocorticotrophic hormone secretion in AtT20 cells as a potential agent targeting adrenocorticotrophic hormone-producing pituitary adenoma. Mol Med Rep. (2014) 9:2533–9. doi: 10.3892/mmr.2014.2078
62. Tagliati F, Gentilin E, Buratto M, Mole D, degli Uberti EC, Zatelli MC. Magmas, a gene newly identified as overexpressed in human and mouse ACTH-secreting pituitary adenomas, protects pituitary cells from apoptotic stimuli. Endocrinology. (2010) 151:4635–42. doi: 10.1210/en.2010-0441
64. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
65. Vargas T, Moreno-Rubio J, Herranz J, Cejas P, Molina S, Gonzalez-Vallinas M, et al. ColoLipidGene: signature of lipid metabolism-related genes to predict prognosis in stage-II colon cancer patients. Oncotarget. (2015) 6:7348–63. doi: 10.18632/oncotarget.3130
66. Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. (2014) 19:393–406. doi: 10.1016/j.cmet.2014.01.019
67. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. (2011) 17:1498–503. doi: 10.1038/nm.2492
68. Griffiths WJ, Wang Y. An update on oxysterol biochemistry: new discoveries in lipidomics. Biochem Biophys Res Commun. (2018) 504:617–22. doi: 10.1016/j.bbrc.2018.02.019
69. Wanet A, Arnould T, Najimi M, Renard P. Connecting mitochondria, metabolism, and stem cell fate. Stem Cells Dev. (2015) 24:1957–71. doi: 10.1089/scd.2015.0117
70. Oldfield EH, Merrill MJ. Apoplexy of pituitary adenomas: the perfect storm. J Neurosurg. (2015) 122: 1444–9. doi: 10.3171/2014.10.jns141720
71. Sinha N, Dabla PK. Oxidative stress and antioxidants in hypertension-a current review. Curr Hypertens Rev. (2015) 11:132–42. doi: 10.2174/1573402111666150529130922
72. Xu H, Wang X, Burchiel SW. Toxicity of environmentally-relevant concentrations of arsenic on developing T lymphocyte. Environ Toxicol Pharmacol. (2018) 62:107–13. doi: 10.1016/j.etap.2018.07.003
73. Kaarniranta K, Kajdanek J, Morawiec J, Pawlowska E, Blasiak J. PGC-1alpha protects RPE cells of the aging retina against oxidative stress-induced degeneration through the regulation of senescence and mitochondrial quality control. The significance for AMD pathogenesis. Int J Mol Sci. (2018) 19:E2317. doi: 10.3390/ijms19082317
74. Hassanzadeh K, Rahimmi A. Oxidative stress and neuroinflammation in the story of Parkinson's disease: could targeting these pathways write a good ending? J Cell Physiol. (2018) 234:23–32. doi: 10.1002/jcp.26865
75. Matsuzaki J, Ochiya T. Extracellular microRNAs and oxidative stress in liver injury: a systematic mini review. J Clin Biochem Nutr. (2018) 63:6–11. doi: 10.3164/jcbn.17-123
76. Siasos G, Tsigkou V, Kosmopoulos M, Theodosiadis D, Simantiris S, Tagkou NM, et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. J Cell Physiol. (2018) 6:256. doi: 10.21037/atm.2018.06.21
77. van der Vliet A, Janssen-Heininger YMW, Anathy V. Oxidative stress in chronic lung disease: from mitochondrial dysfunction to dysregulated redox signaling. Mol Aspects Med. (2018). 63:59–69. doi: 10.1016/j.mam.2018.08.001
78. Tsuchiya T, Kijima A, Ishii Y, Takasu S, Yokoo Y, Nishikawa A, et al. Mechanisms of oxidative stress-induced in vivo mutagenicity by potassium bromate and nitrofurantoin. J Toxicol Pathol. (2018) 31:179–88. doi: 10.1293/tox.2018-0024
79. Rekha VR, Sunil S, Rathy R. Evaluation of oxidative stress markers in oral lichen planus. J Oral Maxillofac Pathol. (2017) 21:387–93. doi: 10.4103/jomfp.JOMFP_19_17
80. Kumari S, Badana AK, G MM GS, Malla R. Reactive oxygen species: a key constituent in cancer survival. Biomark Insights. (2018) 13:1177271918755391. doi: 10.1177/1177271918755391
81. Fischer N, Seo EJ, Efferth T. Prevention from radiation damage by natural products. Phytomedicine. (2018) 47:192–200. doi: 10.1016/j.phymed.2017.11.005
82. Tang JY, Farooqi AA, Ou-Yang F, Hou MF, Huang HW, Wang HR, et al. Oxidative stress-modulating drugs have preferential anticancer effects - involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin Cancer Biol. (2018) 58:109–17. doi: 10.1016/j.semcancer.2018.08.010
83. Kato K, Giulivi C. Critical overview of mitochondrial nitric-oxide synthase. Front Biosci. (2006) 11:2725–38. doi: 10.2741/2002
84. Shvedova M, Anfinogenova Y, Popov SV, Atochin DN. Connexins and nitric oxide inside and outside mitochondria: significance for cardiac protection and adaptation. Front Physiol. (2018) 9:479. doi: 10.3389/fphys.2018.00479
85. Navarro A, Boveris A. Mitochondrial nitric oxide synthase, mitochondrial brain dysfunction in aging, and mitochondria-targeted antioxidants. Adv Drug Deliv Rev. (2008) 60:1534–44. doi: 10.1016/j.addr.2008.05.002
86. Zaobornyj T, Ghafourifar P. Strategic localization of heart mitochondrial NOS: a review of the evidence. Am J Physiol Heart Circ Physiol. (2012) 303:H1283–93. doi: 10.1152/ajpheart.00674.2011
87. Caravia L, Dudau M, Gherghiceanu M, Tanase C, Enciu AM. Could caveolae be acting as warnings of mitochondrial ageing? Mech Ageing Dev. (2015) 146–8:81–7. doi: 10.1016/j.mad.2015.04.003
88. Jin Y, Chen S, Li N, Liu Y, Cheng G, Zhang C, et al. Defect-related luminescent bur-like hydroxyapatite microspheres induced apoptosis of MC3T3-E1 cells by lysosomal and mitochondrial pathways. Sci China Life Sci. (2018) 61:464–75. doi: 10.1007/s11427-017-9258-3
89. Zakeri Z, Lockshin RA. Cell death: history and future. Adv Exp Med Biol. (2008) 615:1–11. doi: 10.1007/978-1-4020-6554-5_1
90. Visalli G, Baluce B, Bertuccio M, Picerno I, Di Pietro A. Mitochondrial-mediated apoptosis pathway in alveolar epithelial cells exposed to the metals in combustion-generated particulate matter. J Toxicol Environ Health A. (2015) 78:697–709. doi: 10.1080/15287394.2015.1024081
91. Hantusch A, Rehm M, Brunner T. Counting on Death - Quantitative aspects of Bcl-2 family regulation. FEBS J. (2018) 285:4124–38. doi: 10.1111/febs.14516
92. Pena-Blanco A, Garcia-Saez AJ. Bax, Bak and beyond - mitochondrial performance in apoptosis. FEBS J. (2018) 285:416–31. doi: 10.1111/febs.14186
93. Xiong S, Mu T, Wang G, Jiang X. Mitochondria-mediated apoptosis in mammals. Protein Cell. (2014) 5:737–49. doi: 10.1007/s13238-014-0089-1
94. Hou XS, Wang HS, Mugaka BP, Yang GJ, Ding Y. Mitochondria: promising organelle targets for cancer diagnosis and treatment. Biomater Sci. (2018) 6:2786–97. doi: 10.1039/c8bm00673c
95. Andre N, Braguer D, Brasseur G, Goncalves A, Lemesle-Meunier D, Guise S, et al. Paclitaxel induces release of cytochrome c from mitochondria isolated from human neuroblastoma cells'. Cancer Res. (2000) 60:5349–53.
96. Ogando-Rivas E, Alalade AF, Boatey J, Schwartz TH. Double pituitary adenomas are most commonly associated with GH- and ACTH-secreting tumors: systematic review of the literature. Pituitary. (2017) 20:702–8. doi: 10.1007/s11102-017-0826-6
97. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
98. Dlugonska H. Autophagy as a universal intracellular process. A comment on the 2016 Nobel Prize in Physiology or Medicine. Ann Parasitol. (2017) 63:153–7. doi: 10.17420/ap6303.100
99. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. (2016) 12:1–222. doi: 10.1080/15548627.2015.1100356
100. Avalos Y, Canales J, Bravo-Sagua R. Tumor suppression and promotion by autophagy. Biomed Res Int. (2014) 2014:603980. doi: 10.1155/2014/603980
101. Martins WK, Santos NF, Rocha CS, Bacellar IOL, Tsubone TM, Viotto AC, et al. Parallel damage in mitochondria and lysosomes is an efficient way to photoinduce cell death. Autophagy. (2018) 15:259–79. doi: 10.1080/15548627.2018.1515609
102. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. (2011) 12:9–14. doi: 10.1038/nrm3028
103. Drake LE, Springer MZ, Poole LP, Kim CJ, Macleod KF. Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol. (2017) 47:110–24. doi: 10.1016/j.semcancer.2017.04.008
104. Wu H, Wei H, Sehgal SA, Liu L, Chen Q. Mitophagy receptors sense stress signals and couple mitochondrial dynamic machinery for mitochondrial quality control. Free Radic Biol Med. (2016) 100:199–209. doi: 10.1016/j.freeradbiomed.2016.03.030
105. Zimmermann M, Reichert A S. How to get rid of mitochondria: crosstalk and regulation of multiple mitophagy pathways. Biol Chem. (2017) 399:29–45. doi: 10.1515/hsz-2017-0206
106. Boyle KB, Randow F. The role of ‘eat-me' signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol. (2013) 16:339–48. doi: 10.1016/j.mib.2013.03.010
107. Zhang L, Qin Y, Chen M. Viral strategies for triggering and manipulating mitophagy. Autophagy. (2018) 14:1665–73. doi: 10.1080/15548627.2018.1466014
108. Yang L, Tu D, Zhao Z, Cui J. Cytotoxicity and apoptosis induced by mixed mycotoxins (T-2 and HT-2 toxin) on primary hepatocytes of broilers in vitro. Toxicon. (2017) 129:1–10. doi: 10.1016/j.toxicon.2017.01.001
109. Zhu CC, Zhang Y, Duan X, Han J, Sun SC. Toxic effects of HT-2 toxin on mouse oocytes and its possible mechanisms. Arch Toxicol. (2016) 90:1495–505. doi: 10.1007/s00204-015-1560-3
110. Vlahopoulos S, Critselis E, Voutsas IF, Perez SA, Moschovi M, Baxevanis CN, et al. New use for old drugs? Prospective targets of chloroquines in cancer therapy. Curr Drug Targets. (2014) 15:843–51. doi: 10.2174/1389450115666140714121514
111. Wu J, Li J, Wang H. Mitochondrial-targeted penetrating peptide delivery for cancer therapy. Expert Opin Drug Deliv. (2018) 15:951–64. doi: 10.1080/17425247.2018.1517750
112. Desantis V, Saltarella I, Lamanuzzi A, Mariggio MA, Racanelli V, Vacca A, et al. Autophagy: a new mechanism of prosurvival and drug resistance in multiple myeloma. Transl Oncol. (2018) 11:1350–7. doi: 10.1016/j.tranon.2018.08.014
113. Rademacher BL, Meske LM, Matkowskyj KA, Hanlon BM, Carchman EH. Genetic inhibition of autophagy in a transgenic mouse model of anal cancer. J Carcinog. (2018) 17:3. doi: 10.4103/jcar.JCar_4_18
114. Bishop E, Bradshaw TD. Autophagy modulation: a prudent approach in cancer treatment? Cancer Chemother Pharmacol. (2018) 82:913–22. doi: 10.1007/s00280-018-3669-6
115. Leone K, Poggiana C, Zamarchi R. The interplay between circulating tumor cells and the immune system: from immune escape to cancer immunotherapy. Diagnostics. (2018) 8:E59. doi: 10.3390/diagnostics8030059
116. Sica A, Massarotti M. Myeloid suppressor cells in cancer and autoimmunity. J Autoimmun. (2017) 85:117–25. doi: 10.1016/j.jaut.2017.07.010
117. Watza D, Lusk CM, Dyson G, Purrington KS, Chen K, Wenzlaff AS, et al. Prognostic modeling of the immune-centric transcriptome reveals interleukin signaling candidates contributing to differential patient outcomes. Carcinogenesis. (2018) 39:1447–54. doi: 10.1093/carcin/bgy119
118. Going CC, Tailor D, Kumar V, Birk AM, Pandrala M, Rice MA, et al. Quantitative proteomic profiling reveals key pathways in the anti-cancer action of methoxychalcone derivatives in triple negative breast cancer. J Proteome Res. (2018). 17:3574–85. doi: 10.1021/acs.jproteome.8b00636
119. Ma W, Concha-Benavente F, Santegoets S, Welters MJP, Ehsan I, Ferris RL, et al. EGFR signaling suppresses type 1 cytokine-induced T-cell attracting chemokine secretion in head and neck cancer. PLoS ONE. (2018) 13:e0203402. doi: 10.1371/journal.pone.0203402
120. Bhat MY, Solanki HS, Advani J, Khan AA, Keshava Prasad TS, Gowda H, et al. Comprehensive network map of interferon gamma signaling. J Cell Commun Signal. (2018) 12:745–51. doi: 10.1007/s12079-018-0486-y
121. Budhwani M, Mazzieri R, Dolcetti R. Plasticity of type I interferon-mediated responses in cancer therapy: from anti-tumor immunity to resistance. Front Oncol. (2018) 8:322. doi: 10.3389/fonc.2018.00322
122. Giuliani C, Bucci I, Napolitano G. The role of the transcription factor nuclear factor-kappa B in thyroid autoimmunity and cancer. Front Endocrinol. (2018) 9:471. doi: 10.3389/fendo.2018.00471
123. Sui H, Ma N. Anti-PD-1/PD-L1 Therapy for non-small-cell lung cancer: toward personalized medicine and combination strategies. J Immunol Res. (2018) 2018:6984948. doi: 10.1155/2018/6984948
124. Angajala A, Lim S, Phillips JB, Kim JH, Yates C, You Z, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol. (2018) 9:1605. doi: 10.3389/fimmu.2018.01605
125. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
126. Agod Z, Fekete T, Budai MM, Varga A, Szabo A, Moon H, et al. Regulation of type I interferon responses by mitochondria-derived reactive oxygen species in plasmacytoid dendritic cells. Redox Biol. (2017) 13:633–45. doi: 10.1016/j.redox.2017.07.016
127. Lawlor KE, Vince JE. Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochim Biophys Acta. (2014) 1840:1433–40. doi: 10.1016/j.bbagen.2013.08.014
128. Misawa T, Takahama M, Saitoh T. Mitochondria-endoplasmic reticulum contact sites mediate innate immune responses. Adv Exp Med Biol. (2017) 997:187–97. doi: 10.1007/978-981-10-4567-7_14
129. Lu JQ, Adam B, Jack AS, Lam A, Broad RW, Chik CL. Immune cell infiltrates in pituitary adenomas: more macrophages in larger adenomas and more T cells in growth hormone adenomas. Endocr Pathol. (2015) 26:263–72. doi: 10.1007/s12022-015-9383-6
130. Wang W, Xu Z, Fu L, Liu W, Li X. Pathogenesis analysis of pituitary adenoma based on gene expression profiling. Oncol Lett. (2014) 8:2423–30. doi: 10.3892/ol.2014.2613
131. Chen Z, Li Z, Chang Y, Ma L, Xu W, Li M, et al. Relationship between NF-kappaB, MMP-9, and MICA expression in pituitary adenomas reveals a new mechanism of pituitary adenomas immune escape. Neurosci Lett. (2015) 597:77–83. doi: 10.1016/j.neulet.2015.04.025
132. Maghathe T, Miller WK, Mugge L, Mansour TR, Schroeder J. Immunotherapy and potential molecular targets for the treatment of pituitary adenomas resistant to standard therapy: a critical review of potential therapeutic targets and current developments. J Neurosurg Sci. (2018). doi: 10.23736/s0390-5616.18.04419-3. [Epub ahead of print].
133. Wang PF, Wang TJ, Yang YK, Yao K, Li Z, Li YM. The expression profile of PD-L1 and CD8(+) lymphocyte in pituitary adenomas indicating for immunotherapy. J Neurooncol. (2018) 139:89–95. doi: 10.1007/s11060-018-2844-2
134. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. (2018) 62:341–60. doi: 10.1042/ebc20170104
135. Cook SJ, Stuart K, Gilley R, Sale MJ. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. (2017) 284:4177–95. doi: 10.1111/febs.14122
136. Yu SB, Pekkurnaz G. Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J Mol Biol. (2018) 430:3922–41. doi: 10.1016/j.jmb.2018.07.027
137. Rasmussen ML, Ortolano NA. Wnt signaling and its impact on mitochondrial and cell cycle dynamics in pluripotent stem cells. Genes. (2018) 9:E109. doi: 10.3390/genes9020109
138. Jezek J, Cooper KF, Strich R. Reactive oxygen species and mitochondrial dynamics: the Yin and Yang of mitochondrial dysfunction and cancer progression. Antioxidants. (2018) 7:E13. doi: 10.3390/antiox7010013
139. Simula L, Nazio F, Campello S. The mitochondrial dynamics in cancer and immune-surveillance. Semin Cancer Biol. (2017) 47:29–42. doi: 10.1016/j.semcancer.2017.06.007
140. Bordi M, Nazio F, Campello S. The close interconnection between mitochondrial dynamics and mitophagy in cancer. Front Oncol. (2017) 7:81. doi: 10.3389/fonc.2017.00081
141. van der Ende M, Grefte S, Plas R, Meijerink J, Witkamp R, Keijer J, et al. Mitochondrial dynamics in cancer-induced cachexia. Biochim Biophys Acta. (2018) 1870:137–50. doi: 10.1016/j.bbcan.2018.07.008
142. Lopez-Lluch G, Hernandez-Camacho JD, Fernandez-Ayala DJM, Navas P. Mitochondrial dysfunction in metabolism and ageing: shared mechanisms and outcomes? Biogerontology. (2018) 19:461–80. doi: 10.1007/s10522-018-9768-2
143. Kameoka S, Adachi Y, Okamoto K, Iijima M, Sesaki H. Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. (2018) 28:67–76. doi: 10.1016/j.tcb.2017.08.011
144. Tang Q, Liu W, Zhang Q, Huang J, Hu C, Liu Y, et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to IR-783-induced apoptosis in human breast cancer cells. J Cell Mol Med. (2018) 22:4474–85. doi: 10.1111/jcmm.13749
Keywords: mitochondrial dysfunction, mitochondrial dynamics, pituitary adenomas, omics, systems biology
Citation: Li N and Zhan X (2019) Mitochondrial Dysfunction Pathway Networks and Mitochondrial Dynamics in the Pathogenesis of Pituitary Adenomas. Front. Endocrinol. 10:690. doi: 10.3389/fendo.2019.00690
Received: 28 September 2018; Accepted: 23 September 2019;
Published: 09 October 2019.
Edited by:
Corin Badiu, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Leila Warszawski, Instituto Estadual de Diabetes e Endocrinologia Luiz Capriglione, BrazilCopyright © 2019 Li and Zhan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianquan Zhan, eWp6aGFuMjAxMUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.