 Giusi Taormina
Giusi Taormina Antonio Russo
Antonio Russo Mario A. Latteri
Mario A. Latteri Mario G. Mirisola
Mario G. Mirisola- Surgical, Oncological and Stomatological Disciplines Department, Medical School, University of Palermo, Palermo, Italy
Epigenetic profile is the link between the regulation of nuclear gene expression and the environment. The most important factors capable of significantly affecting the cellular environment are the amount and quality of nutrients available. Mitochondria are both involved in the production of some of the molecules capable of directly affecting the epigenome and have a critical role in the conversion of nutrients into usable energy. Carbohydrate and fats are converted into ATP, acetyl-CoA, SAM, and NADH. These high-energy substrates are, in turn, capable of driving the epigenetic profile. We describe substances capable of affecting this mechanism. On the other hand, nutritional interventions capable of reducing calories or significantly impairing the normal Acetyl-CoA production or the SAM-SAH ratio also impact chromatin methylation and histone modification, suggesting a critical role of mitochondria on nutrient-dependent epigenetic profile.
Introduction
The analysis of the non-genetic factors network, capable of affecting the lifespan of living organisms, revealed nutrition as a major determinant of longevity. On the other hand, many studies found the environment to be an epigenome driver. The predicted existence of a link between nutrition-dependent modulation of longevity and environment-dependent reprogramming of epigenomes has also been demonstrated (1–5). Recent advances suggest that nutrition can affect the epigenome through 2 general mechanisms: either directly, thanks to substances, which interact with the enzymes responsible for “writing” or “erasing” the epigenetic profiles; or indirectly through metabolic rewiring. The latter can be induced by calorie restriction, fasting, fasting mimicking diet, time restricted feeding, or ketogenic diet. These extreme diets are capable of producing massive cellular reprogramming via partially unclear molecular cascades. Interestingly, both mechanisms converge on mitochondria, which produce many factors and substrates essential for epigenetic modifications and are at a crossroads of cellular energy metabolism (Figure 1).
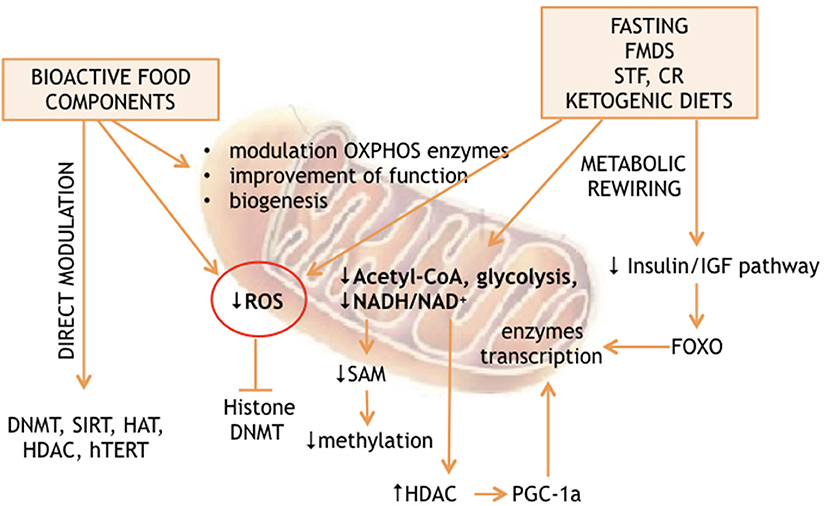
Figure 1. Effect of nutrients and diets on mitochondria and epigenetic.
Nutrient Capable to Directly Affect the Epigenome
Choline, methionine, and folate (see Table 1) deficiencies are responsible for DNA hypomethylation (6) in several species including humans. During embryogenesis, folate has a critical role in the establishment of the epigenome, while in adults, folic acid deficiency is linked to the development of several cancers, such as brain, lung, colorectal, cervix, ovary, and breast cancer (7, 8). On the contrary, a diet rich in folate, vitamins B12, B2, and B6, increases methylation of L1s sequences (long interspersed nucleotide elements) in peripheral blood mononuclear cells, resulting in a decreased risk of developing cervical intraepithelial neoplasia in humans (9). Folate, choline and methionine metabolism are part of the universal metabolism known as one carbon metabolism (1-C). It has an essential role in nucleic acid, methionine and serine biosynthesis. Enzymes of the 1-C metabolism are present in both cytosol and mitochondria with specific roles. While the enzymes and cofactors responsible for homocysteine re-methylation to methionine are localized into the cytosol, the mitochondrial 5,10-methylene-THF dehydrogenase links the 1-C metabolism to NADH availability (10). The observed crosstalk between cytosol and mitochondria is strengthened by the observation that respiratory chain deficiency triggers the expression of serine synthesis genes, thus feeding the 1-C metabolism (11, 12). It must also be noted methionine is an essential amino acid, but it is also the major methyl donor molecule through its adenosylated product S-adenosylmethionine (SAM). SAM, the reactive methyl carrier, is second only to ATP as an enzymatic cofactor for cellular abundance and has a critical role in epigenome establishment and maintenance (13, 14). Interestingly, the effect of folate deficiencies may be reverted by its supplementation. For example, the hypermethylation of the PPARα promoter, induced by folate deficiency during pregnancy, can be reversed by folate supplementation in neonate rats and sheep (15).
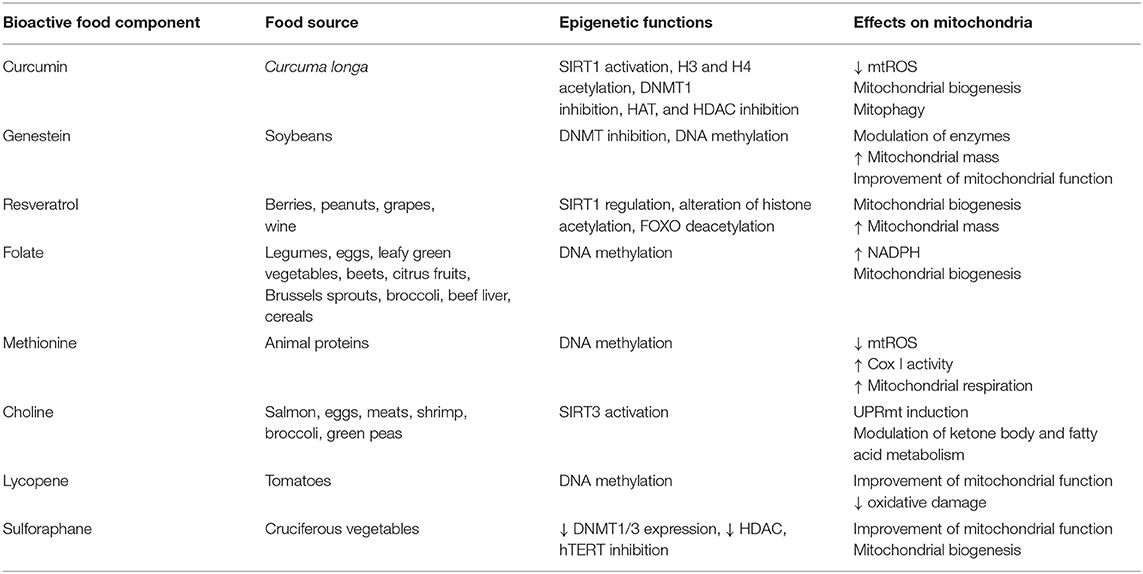
Table 1. Bioactive foods and their role on epigenenome and mitochondria.
Finally, diet methionine concentration can affect the ratio between s-adenosyl methionine and s-adenosyl homocysteine (SAM/SAH), impacting methylation reaction both on DNA and on histone proteins of rodents and humans (16).
Resveratrol (3,5,4′-trihydroxy-trans-stilbene, see Table 1) is the most studied anti-aging compound and is involved in histone modification and DNA methylation. Resveratrol supplementation increases longevity in simple model organisms and in mammals (17, 18). At the molecular level, it regulates SIRT1 (19, 20) and FOXO deacetylation affecting cells survival (21). The major in vivo effects are increased insulin sensitivity (22), anti-inflammatory effects, inhibition of NF-κB, thus inhibiting the development of cancer in cellular models of breast and prostate cancer (23, 24). Resveratrol has many interconnections with mitochondrial metabolism. Its supplementation increases mitochondrial mass (25, 26). SIRT1-dependent deacetylation of PGC-1α results in biogenesis of mitochondria (27, 28), which in turn co-activates the nuclear respiratory factors (NRF-1 and NRF-2), resulting in mitochondrial biogenesis (29). There isn't a general agreement on the mechanism by which resveratrol acts but some hints suggest the involvement of mitochondrial complex III (30–33). Alternatively, AMPK could be the link between resveratrol and SIRT1 through NAD+ concentration modulation (34, 35). SIRT1 is also activated by quercetins, and catechins in different model systems (36, 37).
Curcumin is a polyphenol extracted from the spice Curcuma longa, known as a natural anti-inflammatory agent. Dietary curcumin reduces mitochondrial ROS production (see Table 1) and improves markers of aging in wild-type mice. It is involved in different epigenetic modifications: it regulates H3 and H4 acetylation, DNMT1 and, with a mechanism that involves miRNA, SP1, and PTEN. Its most relevant effect is the inhibition of NF-κB (38). In mice it prevents heart-failure through HATs inhibition, HDACs and p300 degradation induction (39, 40). Interestingly, a homozygous deletion of the mitochondrial uncoupling protein 2 UCP2–/– reverses this effect in mice (41), thus suggesting the active role of mitochondria in this mechanism. In a rat model and in endothelial cell cultures, nitric oxide synthase (eNOS), AMPK phosphorylation, upregulated UCP2, and reduced ROS production are observed after curcumin supplementation, while inhibitors of either AMPK or UCP2 abolish the curcumin effect (41).
Tea polyphenols and catechins are scavengers of free radicals and singlet oxygen. Their efficacy in the prevention and treatment of many diseases has been demonstrated. They affect apoptosis and provoke cell-cycle arrest in human cancer cell lines (42). Epigallocatechin prevents UV-induced carcinogenesis of the mouse skin (43), while epicatechins and catechins have shown anti-aging effects in C. elegans (44).These substances have many epigenetic targets: H3 and H4, NF-κB, IL-6, SUZ12/HAT, HDAC, HMT, P16INK4a, RNRβ, RECK1, hTERT, WIF-1, RXRα, RXRβ, CDX2/DNMTI, and Bcl-2. Epigallocatechin binds to the catalytic region of DNMT1 and inhibits its activity (45). Furthermore, it leads to a decrease in DNMT1, DNMT3a, DNMT3b, and HDAC levels, whereas, it increases the acetylation of histones H3 and H4 at specific sites (46). It has recently been demonstrated that isoflavones stimulates mitochondrial biogenesis and improves mitochondrial function in diabetes, chronic heart failure and renal injury, as well as in aging of rodents (47–49).
Suphorafene induces cell-cycle arrest and apoptosis in mice cancer cells by epigenetic mechanisms (2, 17, 50, 51). A similar effect has been detected for lycopene (52), quercetin (53), and ellagic acid (contained in pomegranate, walnuts, and almonds) (54). Low doses of suphorafene are related to hTERT inhibition and to the reduction in DNMT1 and DNMT3a expression levels (16, see Table 1). Furthermore, suphorafene determines deacetylase down-regulation in melanoma cells in vitro, inhibiting their growth and proliferation (55). Interestingly, sulphoraphane has been demonstrated to enhance ROS and mitochondrial membrane depolarization in human ovarian cancer cell lines (56).
Genistein, contained in soybeans, take part in modulation of chromatin structure and DNA methylation; histones SIRT1, p21, p16, PTEN, p53, FOXO3A, and hTERT are its epigenetic targets (57, 58), which, in turn, are key regulators of cell-cycle regulation and cell survival. A recent study shows that supplementing Laying broiler breed hens with genistein can alter lipid metabolism in the offspring chicks through epigenetic modifications that upregulate PPARδ expression, improving antioxidative capability and growth performance (59). In addition, genistein induces in mitochondria, and modulation of enzymatic activity of components of the oxidative phosphorylation system (see Table 1).
Nutrients Indirectly Affecting the Epigenome
Calorie restriction and the ketogenic diet, as well as fasting and fasting-mimicking diets are nutritional interventions capable of significantly affecting longevity in a wide range of living organisms. The ability of CR to modify the epigenome is suggested by many observations. CR protects against age-related DNA methylation changes as described in different mammalian tissues: Kidney (60), blood (61), liver (62), hippocampus (63), and cerebellum (64) being the most affected in mice and rats. Expression studies of genes coding for proteins involved in mitochondrial function revealed increased expression of PGC1α, TFAM, eNOS, SIRT1, and PARL genes after 6 months of calorie restriction in humans (65). In addition, PGC-1α activation by NAD+-dependent Sirt1 deacetylation connects OXPHOS and calorie restriction (66, 67). PGC-1α and the other two members of this gene family are transcriptional activators (PGC-1α, PERC, and PGC-1β), which affect the transcription of genes involved in mitochondrial energetics modulating thermogenesis, fat oxidation and mitochondrial biogenesis (68, 69). Assumption of very low amount of carbohydrate with high or adequate protein content (ketogenic diets) will force the body to produce, mainly in the liver, ketone bodies which can be subsequently used by many extra-hepatic tissues (e.g., nervous system) as energy sources in place of sugar. This condition can also be transiently triggered by fasting or the fasting-mimicking diet and possibly by calorie restriction. The ketogenic diet has been proposed as an adjuvant therapy for cancer patients based on the idea that most of the cancer cell metabolism is based on high rate glycolysis (known as the Warburg effect). Normal cells are also capable of using glucose as an energy source but can also rely their metabolism on the ketone bodies produced in the liver tissue when glucose concentration is scarce. On the contrary, cancer cells have lost this ability and base their biosynthetic pathways on glucose only. The reduced glucose availability will also provoke a reduction in circulating insulin. Therefore, the insulin dependent (IRS)/RAS/RAF/MEK/MAPK and the RAS/PI3K/AKT/mTOR transduction cascades are turned off (70). This causes downstream reduction of phosphatidylinositol triphosphate (PIP3) production, PI3K and AKT, as well as a drop in MAP kinase activity, and unphosphorylated forkhead box O transcription factor (FOXO) is forced to remain in the nucleus. Nuclear FOXO acts as transcription factor promoting the transcription of hundreds of genes, including mitochondrial glutathione peroxidase, superoxide dismutase 2, and catalase.
Even though chronic calorie restriction delays the growth of many cancers in mice, it imposes weight loss and possibly also immunosuppression, making it infeasible for cancer treatment. In addition, the avoidance of malnutrition is not an easy task for most of the subjects under calorie restriction. On the contrary, complete fasting or a low calorie diet capable of mimicking the effects of fasting (fasting mimicking diets, FMDs) consecutively for 2–7 days have a profound effect on cellular metabolism and cause only a transient loss of weight. Re-feeding is in fact normally accompanied with weight regain unless it is otherwise desired. Fasting for 2–4 days, known as Short Term Fasting (STF), is capable of inducing multiple metabolic changes, blood sugar reduction, drop of circulating IGF1, increased IGF1BP/IGF1 ratio and transient ketone bodies increase. From a metabolic point of view, these nutritional interventions all have the induction of ketosis in common. Many conditions are capable of forcing Ketone bodies oxidation to become a significant part of the mammalian energy metabolism, including diets with very low carbohydrate, strong endurance exercise without carbohydrate supply, fasting, starvation, the neonatal period, pregnancy, and likely calorie restriction. The concentration of ketone bodies normally ranges from 100 to 250 μM in healthy adults and rises up to ~1 mM after prolonged exercise or 24 h of fasting. A higher concentration of ketone bodies is observed in some pathological conditions like diabetic ketoacidosis, where ketone bodies can rise up to a harmful 20 mM (71–75). The mammalian liver uses acetyl-CoA to produce most of the circulating ketone bodies, which are then used by extrahepatic tissues for terminal oxidation (71, 74, 76). Ketone bodie's metabolism is linked to cytoplasmic and mitochondrial pathways. β-oxidation (FAO), gluconeogenesis, the tricarboxylic acid cycle (TCA), de novo lipogenesis (DNL), and sterols biosynthesis are just examples of pathways interconnected with the availability of ketone bodies. Excessive production of acetyl-CoA by β-oxidation and/or depletion of oxalacetate are both capable of triggering liver ketogenesis. The rate of ketogenesis is not linearly dependent on hepatic acetyl-CoA concentration (77, 78). However, hepatocytes aren't capable of metabolizing the ketone bodies that they produce. Heart, brain and skeletal muscles can instead rely on ketone bodie's metabolism, converting them to Acetyl-CoA, which in turn fuels TCA cycle for terminal oxidation. Alternatively, ketone bodies can feed sterol synthetic pathways as well as lipogenesis or can be excreted in the urine (79–81). The amount of acetyl-CoA available can modulate histone acetylation, since this molecule is also the cellular acetyl donor used during histone acetylation. Increased circulating acetyl-CoA, which is directly dependent on the rate of its production, is in fact related to H3K9 acetylation on a genome-wide analysis. It appears that acetyl-CoA availability can force cells to enter growth through histone acetylation and consequential transcription of pro-growth genes (82). In yeast, it has been clearly demonstrated that acetyl-CoA availability is metabolically sufficient to trigger histone acetylation (82). Thus, what fasting, fasting mimicking diet and possibly calorie restriction, as well as ketogenic diet, all have in common is a reduced availability of acetyl-CoA, which results in epigenetic anti-growth modification. However, the energy reduction consequential to fasting or the reduced glycolysis, due to both fasting or ketogenic diet, also affects the available reducing equivalents. NADH will, in fact, be oxidized to NAD+. The latter is a substrate for the class III histone deacetylases, which leads to DNA-binding protein deacetylation with a consequential increase in chromatin positive charge leading to transcription repression and growth arrest.
Energy availability and specific nutrient supply modification, such as those obtained by calorie restriction or by fasting and fasting mimicking diet (see Figure 1), can also modulate nuclear gene expression through DNA or histone methylation by S-adenosyl-L-methionine (SAM). In the cytosol, L-methionine + ATP can give SAM + Pi + PPi. Thus, energy production is linked to methylation of lysine and arginine, as well as cytosine DNA by SAM as already described above. On the other hand, methionine can be obtained by homocysteine methylation in a process regulated by mitochondrial metabolism. The mitochondrial and cytosolic one-carbon metabolisms are interconnected through the exchange of serine and glycine, which are modified in both compartments by the activity of methylene-tetrahydrofolate, thanks to the cytosolic and mitochondrial serine hydroxymethyltransferases. However, methylene-tetrahydrofolate is converted to formyl-tetrahydrofolate in mitochondria, forcing the 1 carbon metabolism to produce formate instead of serine into the mitochondria, which is in turn used for purine biosynthesis. Alternatively, serine is exported to the cytosol. The cytosolic one-carbon metabolism uses serine to convert homocysteine to methionine through methylene-tetrahydrofolate and methyl-tetrahydrofolate, which can then be used for SAM production. Within mitochondria, the direction of the synthesis toward serine or formate production depends on NAD+ availability. Inhibition of OXPHOS increases the NADH/NAD+ ratio, inhibiting the production of methylene-tetrahydrofolate and thus serine, methionine and SAM production (83), while reintroduction of mtDNA into cancer cells is capable of reestablishing the wild type methylation status (83, 84), thus confirming the strict relationship between mitochondrial function and the epigenome profile (Figure 1).
Conclusion
We have briefly reviewed the interconnections between mitochondria and epigenetic profile from the nutrients point of view. It is now becoming clear that epigenome has a critical role in cellular metabolism and that its drift may contribute to the aging process. Since the efficiency and number of mitochondria may be addressed by pharmacological and dietary approaches (85–87), we believe that mitochondria may be a potential target for preventive medicine by dietary, as well as supplemental treatment, and for next-generation anti-aging drugs.
Author Contributions
GT and MM wrote the manuscript. AR, ML, and MM revised the manuscript.
Funding
This work was supported by Laurus project foundation; Create cures foundation to MM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Park LK, Friso S, Choi SW. Nutritional influences on epigenetics and age-related disease. Proc Nutr Soc. (2012) 71:75tr. doi: 10.1017/S0029665111003302
2. Li Y, Daniel M, Tollefsbol TO. Epigenetic regulation of caloric restriction in aging. BMC Med. (2011) 9:98. doi: 10.1186/1741-7015-9-98
3. Ford D, Ions LJ, Alatawi F, Wakeling LA. The potential role of epigenetic responses to diet in ageing. Proc Nutr Soc. (2011) 70:374r S. doi: 10.1017/S0029665111000851
4. McKay JA, Mathers JC. Diet induced epigenetic changes and their implications for health. Acta Physiol. (2011) 202:103iol. doi: 10.1111/j.1748-1716.2011.02278.x
5. Ly A, Hoyt L, Crowell J, Kim YI. Folate and DNA methylation. Antioxid Redox Signal. (2012) 17:302 Re. doi: 10.1089/ars.2012.4554
6. Waterland RA, Lin JR, Smith CA, Jirtle RL. Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum Mol Genet. (2006) 15:705Gen. doi: 10.1093/hmg/ddi484
7. Kim YI. Folate and colorectal cancer: an evidence-based critical review. Mol Nutr Food Res. (2007) 51:267 Fo. doi: 10.1002/mnfr.200600191
8. Yang Q, Bostick RM, Friedman JM, Flanders WD. Serum folate and cancer mortality among U.S. adults: findings from the Third National Health and Nutritional Examination Survey linked mortality file. Cancer Epidemiol Biomarkers Prev. (2009) 18:1439–47. doi: 10.1158/1055-9965.EPI-08-0908
9. Piyathilake CJ, Badiga S, Kabagambe EK, Azuero A, Alvarez RD, Johanning GL, et al. A dietary pattern associated with LINE-1 methylation alters the risk of developing cervical intraepithelial neoplasia. Cancer Prev Res. (2012) 5:385)re. doi: 10.1158/1940-6207.CAPR-11-0387
10. Garcia-Martinez LF, Appling DR. Characterization of the folate-dependent mitochondrial oxidation of carbon 3 of serine. Biochemistry. (1993) 32:4671–6. doi: 10.1021/bi00068a027
11. Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife. (2016) 5:10575. doi: 10.7554/eLife.10575
12. Nikkanen J, Forsstrom S, Euro L, Paetau I, Kohnz RA, Wang L, et al. Mitochondrial DNA replication defects disturb cellular dNTP Pools and remodel one-carbon metabolism. Cell Metab. (2016) 23:635ab. doi: 10.1016/j.cmet.2016.01.019
13. Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. (1990) 1:228Bio. doi: 10.1016/0955-2863(90)90070-2
14. Mudd SH, Brosnan JT, Brosnan ME, Jacobs RL, Stabler SP, Allen RH, et al. Methyl balance and transmethylation fluxes in humans. Am J Clin Nutr. (2007) 85:19in. doi: 10.1093/ajcn/85.1.19
15. Sinclair KD, Allegrucci C, Singh R, Gardner DS, Sebastian S, Bispham J, et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc Natl Acad Sci USA. (2007) 104:19351–9. doi: 10.1073/pnas.0707258104
16. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. (2015) 22:861ab. doi: 10.1016/j.cmet.2015.08.024
17. Barger JL, Kayo T, Vann JM, Arias EB, Wang J, Hacker TA, et al. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE. (2008) 3:e2264. doi: 10.1371/annotation/c54ef754-1962-4125-bf19-76d3ec6f19e5
18. Agarwal B, Baur JA. Resveratrol and life extension. Ann NY Acad Sci. (2011) 1215:138ad. doi: 10.1111/j.1749-6632.2010.05850.x
19. Patel KR, Scott E, Brown VA, Gescher AJ, Steward WP, Brown K. Clinical trials of resveratrol. Ann NY Acad Sci. (2011) 1215:1611d. doi: 10.1111/j.1749-6632.2010.05853.x
20. Subramanian L, Youssef S, Bhattacharya S, Kenealey J, Polans AS, van Ginkel PR. Resveratrol: challenges in translation to the clinic–a critical discussion. Clin Cancer Res. (2010) 16:5942–9. doi: 10.1158/1078-0432.CCR-10-1486
21. Chen Q, Ganapathy S, Singh KP, Shankar S, Srivastava RK. Resveratrol induces growth arrest and apoptosis through activation of FOXO transcription factors in prostate cancer cells. PLoS ONE. (2010) 5:e15288. doi: 10.1371/journal.pone.0015288
22. Boccardi V, Esposito A, Rizzo MR, Marfella R, Barbieri M, Paolisso G. Mediterranean diet, telomere maintenance and health status among elderly. PLoS ONE. (2013) 8:e62781. doi: 10.1371/journal.pone.0062781
23. Tili E, Michaille JJ, Alder H, Volinia S, Delmas D, Latruffe N, et al. Resveratrol modulates the levels of microRNAs targeting genes encoding tumor-suppressors and effectors of TGFbeta signaling pathway in SW480 cells. Biochem Pharmacol. (2010) 80:2057–05. doi: 10.1016/j.bcp.2010.07.003
24. Zhang R, Chen HZ, Liu JJ, Jia YY, Zhang ZQ, Yang RF, et al. SIRT1 suppresses activator protein-1 transcriptional activity and cyclooxygenase-2 expression in macrophages. J Biol Chem. (2010) 285:7097–097. doi: 10.1074/jbc.M109.038604
25. Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. (2006) 444:337rol. doi: 10.1038/nature05354
26. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. (2006) 127:1109–10. doi: 10.1016/j.cell.2006.11.013
27. Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. (2007) 26:1913–91. doi: 10.1038/sj.emboj.7601633
28. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. (2005) 434:113SI. doi: 10.1038/nature03354
29. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. (2011) 1813:1269–26. doi: 10.1016/j.bbamcr.2010.09.019
30. Gledhill JR, Montgomery MG, Leslie AG, Walker JE. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc Natl Acad Sci USA. (2007) 104:13632–3. doi: 10.1073/pnas.0706290104
31. Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. (2010) 11:554abl. doi: 10.1016/j.cmet.2010.04.001
32. Zini R, Morin C, Bertelli A, Bertelli AA, Tillement JP. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp Clin Res. (1999) 25:87xp .
33. Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. (2012) 148:421rol. doi: 10.1016/j.cell.2012.01.017
34. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. (2009) 458:1056–05. doi: 10.1038/nature07813
35. Fulco M, Sartorelli V. Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle. (2008) 7:3669–66. doi: 10.4161/cc.7.23.7164
36. Chung S, Yao H, Caito S, Hwang JW, Arunachalam G, Rahman I. Regulation of SIRT1 in cellular functions: role of polyphenols. Arch Biochem Biophys. (2010) 501:79che. doi: 10.1016/j.abb.2010.05.003
37. Ayissi VB, Ebrahimi A, Schluesenner H. Epigenetic effects of natural polyphenols: a focus on SIRT1-mediated mechanisms. Mol Nutr Food Res. (2014) 58:22r F. doi: 10.1002/mnfr.201300195
38. Mudduluru G, George-William JN, Muppala S, Asangani IA, Kumarswamy R, Nelson LD, et al. Curcumin regulates miR-21 expression and inhibits invasion and metastasis in colorectal cancer. Biosci Rep. (2011) 31:185ep. doi: 10.1042/BSR20100065
39. Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. (2008) 118:868ves. doi: 10.1172/JCI33160
40. Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, et al. Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem. (2006) 2:169m.e. doi: 10.2174/157340606776056133
41. Pu Y, Zhang H, Wang P, Zhao Y, Li Q, Wei X, et al. Dietary curcumin ameliorates aging-related cerebrovascular dysfunction through the AMPK/uncoupling protein 2 pathway. Cell Physiol Biochem. (2013) 32:1167–16. doi: 10.1159/000354516
42. Tsang WP, Kwok TT. Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. J Nutr Biochem. (2010) 21:140io. doi: 10.1016/j.jnutbio.2008.12.003
43. Katiyar S, Elmets CA, Katiyar SK. Green tea and skin cancer: photoimmunology, angiogenesis and DNA repair. J Nutr Biochem. (2007) 18:287ioc. doi: 10.1016/j.jnutbio.2006.08.004
44. Sunagawa T, Shimizu T, Kanda T, Tagashira M, Sami M, Shirasawa T. Procyanidins from apples (Malus pumila Mill.) extend the lifespan of Caenorhabditis elegans. Planta Med. (2011) 77:122–7. doi: 10.1055/s-0030-1250204
45. Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. (2003) 63:7563–56.
46. Nandakumar V, Vaid M, Katiyar SK. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis. (2011) 32:537ene. doi: 10.1093/carcin/bgq285
47. Rasbach KA, Schnellmann RG. Isoflavones promote mitochondrial biogenesis. J Pharmacol Exp Ther. (2008) 325:536ol. doi: 10.1124/jpet.107.134882
48. Nieman DC, Henson DA, Maxwell KR, Williams AS, McAnulty SR, Jin F, et al. Effects of quercetin and EGCG on mitochondrial biogenesis and immunity. Med Sci Sports Exerc. (2009) 41:1467–46. doi: 10.1249/MSS.0b013e318199491f
49. Taub PR, Ramirez-Sanchez I, Ciaraldi TP, Perkins G, Murphy AN, Naviaux R, et al. Alterations in skeletal muscle indicators of mitochondrial structure and biogenesis in patients with type 2 diabetes and heart failure: effects of epicatechin rich cocoa. Clin Transl Sci. (2012) 5:43ra. doi: 10.1111/j.1752-8062.2011.00357.x
50. Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol Carcinog. (2007) 46:24cin. doi: 10.1002/mc.20258
51. Izzotti A, Larghero P, Cartiglia C, Longobardi M, Pfeffer U, Steele VE, et al. Modulation of microRNA expression by budesonide, phenethyl isothiocyanate and cigarette smoke in mouse liver and lung. Carcinogenesis. (2010) 31:894enes. doi: 10.1093/carcin/bgq037
52. King-Batoon A, Leszczynska JM, Klein CB. Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ Mol Mutagen. (2008) 49:36 Mo. doi: 10.1002/em.20363
53. Priyadarsini RV, Vinothini G, Murugan RS, Manikandan P, Nagini S. The flavonoid quercetin modulates the hallmark capabilities of hamster buccal pouch tumors. Nutr Cancer. (2011) 63:218cer. doi: 10.1080/01635581.2011.523503
54. Wen XY, Wu SY, Li ZQ, Liu ZQ, Zhang JJ, Wang GF, et al. Ellagitannin (BJA3121), an anti-proliferative natural polyphenol compound, can regulate the expression of MiRNAs in HepG2 cancer cells. Phytother Res. (2009) 23:778r R. doi: 10.1002/ptr.2616
55. Do DP, Pai SB, Rizvi SA, D'Souza MJ. Development of suphorafene-encapsulated microspheres for cancer epigenetic therapy. Int J Pharm. (2010) 386:114rmt. doi: 10.1016/j.ijpharm.2009.11.009
56. Biswas R, Ahn JC, Kim JS. Sulforaphene synergistically sensitizes cisplatin via enhanced mitochondrial dysfunction and PI3K/PTEN modulation in ovarian cancer cells. Anticancer Res. (2015) 35:3901–8.
57. Fang MZ, Chen D, Sun Y, Jin Z, Christman JK, Yang CS. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res. (2005) 11:7033–03. doi: 10.1158/1078-0432.CCR-05-0406
58. Majid S, Dar AA, Ahmad AE, Hirata H, Kawakami K, Shahryari V, et al. BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis. (2009) 30:662ene. doi: 10.1093/carcin/bgp042
59. Lv Z, Fan H, Song B, Li G, Liu D, Guo Y. Supplementing genistein for breeder hens alters the fatty acid metabolism and growth performance of offsprings by epigenetic modification. Oxid Med Cell Longev. (2019) 2019:9214209. doi: 10.1155/2019/9214209
60. Kim CH, Lee EK, Choi YJ, An HJ, Jeong HO, Park D, et al. Short-term calorie restriction ameliorates genomewide, age-related alterations in DNA methylation. Aging Cell. (2016) 15:1074–07. doi: 10.1111/acel.12513
61. Sziraki A, Tyshkovskiy A, Gladyshev VN. Global remodeling of the mouse DNA methylome during aging and in response to calorie restriction. Aging Cell. (2018) 17:e12738. doi: 10.1111/acel.12738
62. Hahn O, Gronke S, Stubbs TM, Ficz G, Hendrich O, Krueger F, et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. (2017) 18:56. doi: 10.1186/s13059-017-1187-1
63. Hadad N, Unnikrishnan A, Jackson JA, Masser DR, Otalora L, Stanford DR, et al. Caloric restriction mitigates age-associated hippocampal differential CG and non-CG methylation. Neurobiol Aging. (2018) 67:53ol. doi: 10.1016/j.neurobiolaging.2018.03.009
64. Lardenoije R, van den Hove DLA, Vaessen TSJ, Iatrou A, Meuwissen KPV, van Hagen BTJ, et al. Epigenetic modifications in mouse cerebellar Purkinje cells: effects of aging, caloric restriction, and overexpression of superoxide dismutase 1 on 5-methylcytosine and 5-hydroxymethylcytosine. Neurobiol Aging. (2015) 36:3079–89. doi: 10.1016/j.neurobiolaging.2015.08.001
65. Civitarese AE, Carling S, Heilbronn LK, Hulver MH, Ukropcova B, Deutsch WA, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. (2007) 4:e76. doi: 10.1371/journal.pmed.0040076
66. Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA. (2003) 100:10794–9. doi: 10.1073/pnas.1934713100
67. McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, et al. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. (2003) 23:38l B. doi: 10.1128/MCB.23.1.38-54.2003
68. Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. (2004) 18:357–68. doi: 10.1101/gad.1177604
69. Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. (2002) 1576:1762. doi: 10.1016/S0167-4781(02)00343-3
70. Fontana L, Partridge L, Longo VD. Extending healthy life span–from yeast to humans. Science. (2010) 328:321–6. doi: 10.1126/science.1172539
71. Cahill GF Jr. Fuel metabolism in starvation. Annu Rev Nutr. (2006) 26:1ev. doi: 10.1146/annurev.nutr.26.061505.111258
72. Johnson RH, Walton JL, Krebs HA, Williamson DH. Metabolic fuels during and after severe exercise in athletes and non-athletes. Lancet. (1969) 2:452es. doi: 10.1016/S0140-6736(69)90164-0
73. Koeslag JH, Noakes TD, Sloan AW. Post-exercise ketosis. J Physiol. (1980) 301:79lke. doi: 10.1113/jphysiol.1980.sp013190
74. Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. (1980) 60:143Rev. doi: 10.1152/physrev.1980.60.1.143
75. Wildenhoff KE, Johansen JP, Karstoft H, Yde H, Sorensen NS. Diurnal variations in the concentrations of blood acetoacetate and 3-hydroxybutyrate. The ketone body peak around midnight and its relationship to free fatty acids, glycerol, insulin, growth hormone and glucose in serum and plasma. Acta Med Scand. (1974) 195:25d. doi: 10.1111/j.0954-6820.1974.tb08090.x
76. McGarry JD, Foster DW. Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem. (1980) 49:395 Bio. doi: 10.1146/annurev.bi.49.070180.002143
77. Foster DW. Studies in the ketosis of fasting. J Clin Invest. (1967) 46:1283–28. doi: 10.1172/JCI105621
78. Williamson DH, Veloso D, Ellington EV, Krebs HA. Changes in the concentrations of hepatic metabolites on administration of dihydroxyacetone or glycerol to starved rats and their relationship to the control of ketogenesis. Biochem J. (1969) 114:575.n. doi: 10.1042/bj1140575
79. Balasse EO, Fery F. Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab Rev. (1989) 5:247s M. doi: 10.1002/dmr.5610050304
80. Bentourkia M, Tremblay S, Pifferi F, Rousseau J, Lecomte R, Cunnane S. PET study of 11C-acetoacetate kinetics in rat brain during dietary treatments affecting ketosis. Am J Physiol Endocrinol Metab. (2009) 296:E796296:. doi: 10.1152/ajpendo.90644.2008
81. Reichard GA Jr, Owen OE, Haff AC, Paul P, Bortz WM. Ketone-body production and oxidation in fasting obese humans. J Clin Invest. (1974) 53:508nve. doi: 10.1172/JCI107584
82. Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. (2011) 42:426oA. doi: 10.1016/j.molcel.2011.05.004
83. Naviaux RK. Mitochondrial control of epigenetics. Cancer Biol Ther. (2008) 7:1191–1. doi: 10.4161/cbt.7.8.6741
84. Smiraglia DJ, Kulawiec M, Bistulfi GL, Gupta SG, Singh KK. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol Ther. (2008) 7:1182–18. doi: 10.4161/cbt.7.8.6215
85. Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics Effect of exercise and energy depletion off mitochondria fission and metabolism. J Cell Biol. (2016) 212:379olr. doi: 10.1083/jcb.201511036
86. Fujisawa K, Hara K, Takami T, Okada S, Matsumoto T, Yamamoto N, et al. Evaluation of the effects of ascorbic acid on metabolism of human mesenchymal stem cells. Stem Cell Res Ther. (2018) 9:93. doi: 10.1186/s13287-018-0825-1
Keywords: mitochondria and metabolism, nutrition and epigenome, calorie restriction and epigenome, FMD and epigenome, nutrients and epigenome
Citation: Taormina G, Russo A, Latteri MA and Mirisola MG (2019) Mitochondrion at the Crossroad Between Nutrients and Epigenome. Front. Endocrinol. 10:673. doi: 10.3389/fendo.2019.00673
Received: 20 May 2019; Accepted: 17 September 2019;
Published: 04 October 2019.
Edited by:
Patrizia D'Aquila, University of Calabria, ItalyReviewed by:
Marian Beekman, Leiden University Medical Center, NetherlandsMarianna Sadagurski, Wayne State University, United States
Copyright © 2019 Taormina, Russo, Latteri and Mirisola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario G. Mirisola, bWFyaW8ubWlyaXNvbGEmI3gwMDA0MDt1bmlwYS5pdA==