
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 27 September 2019
Sec. Pediatric Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00648
 Sigrid Aslaksen1,2*
Sigrid Aslaksen1,2* Paal Methlie1,2,3Magnus D. Vigeland4,5Dag E. Jøssang6
Paal Methlie1,2,3Magnus D. Vigeland4,5Dag E. Jøssang6 Anette B. Wolff1,2
Anette B. Wolff1,2 Ying Sheng5
Ying Sheng5 Bergithe E. Oftedal1,2
Bergithe E. Oftedal1,2 Beate Skinningsrud5Dag E. Undlien4,5Kaja K. Selmer7,8Eystein S. Husebye1,2,3
Beate Skinningsrud5Dag E. Undlien4,5Kaja K. Selmer7,8Eystein S. Husebye1,2,3 Eirik Bratland1,2
Eirik Bratland1,2Background: Underlying causes of adrenal insufficiency include congenital adrenal hyperplasia (CAH) and autoimmune adrenocortical destruction leading to autoimmune Addison's disease (AAD). Here, we report a patient with a homozygous stop-gain mutation in 3β-hydroxysteroid dehydrogenase type 2 (3βHSD2), in addition to impaired steroidogenesis due to AAD.
Case Report: Whole exome sequencing revealed an extremely rare homozygous nonsense mutation in exon 2 of the HSD3B2 gene, leading to a premature stop codon (NM_000198.3: c.15C>A, p.Cys5Ter) in a patient with AAD and premature ovarian insufficiency. Scrutiny of old medical records revealed that the patient was initially diagnosed with CAH with hyperandrogenism and severe salt-wasting shortly after birth. However, the current steroid profile show complete adrenal insufficiency including low production of pregnenolone, dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEA-S), without signs of overtreatment with steroids.
Conclusion: To the best of our knowledge, this is the first description of autoimmune adrenalitis in a patient with 3βHSD2 deficiency and suggests a possible association between AAD and inborn errors of the steroidogenesis.
3β-hydroxysteroid dehydrogenase type 2 (3βHSD2) catalyzes the conversion of Δ5-steroids into Δ4-steroids (Figure 1). Deficiency of 3βHSD2 causes a rare autosomal recessive form of congenital adrenal hyperplasia (CAH) characterized by high levels of pregnenolone, 17-hydroxypregnenolone, dehydroepiandrosterone (DHEA), DHEA sulfate (DHEA-S), and androstenediol, and lack of cortisol, aldosterone, and androstenedione (1). There are two isozymes of 3βHSD encoded by HSD3B1 and HSD3B2. 3βHSD2 is expressed in the gonads and the adrenal cortex, whereas 3βHSD1 is expressed in peripheral tissues, converting circulating DHEA to testosterone (1). 3βHSD2 deficiency can therefore cause relatively high levels of testosterone in females, whereas it cannot compensate for the absence of adrenal and gonadal synthesis of testosterone in males. This causes ambiguous genitalia in males, whereas female newborns exhibit mild virilization or normal sexual differentiation, and may remain undiagnosed until a salt-wasting crisis occurs (1–3).
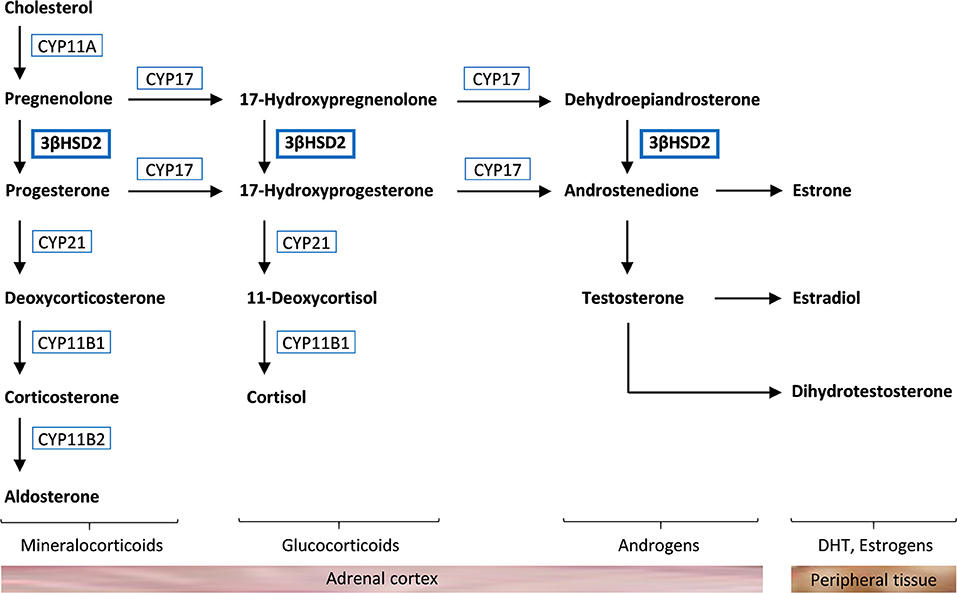
Figure 1. Steroidogenesis in the adrenal cortex. Cholesterol is converted to aldosterone, cortisol, and androgens through different pathways that require specific enzymes [cholesterol side-chain cleavage enzyme (CYP11A), 17α-hydroxylase (CYP17), 3β-hydroxysteroid dehydrogenase type 2 (3βHSD2), 21-hydroxylase (CYP21), 11β-hydroxylase (CYP11B1), and aldosterone synthase (CYP11B2)]. Androstenedione and testosterone are further converted to dihydrotestosterone (DHT) and estrogens in peripheral tissue.
Adrenal insufficiency can also be due to autoimmune adrenalitis, or autoimmune Addison's disease (AAD), characterized by an immunological attack of the adrenal cortex leading to decreased production of cortisol and aldosterone (4). The self-antigen 21-hydroxylase (21OH) is the dominant target of adrenal autoantibodies and autoreactive T cells (4). Therefore, autoantibodies against 21OH and low serum cortisol levels are important diagnostic markers for AAD.
To the best of our knowledge, we here report the first patient affected by both inborn 3βHSD2 deficiency and acquired AAD.
A whole-exome sequencing study involving 142 AAD patients (5) revealed a patient with a rare homozygous mutation in exon 2 of HSD3B2 [frequency ~0.00003 in the Genome Aggregation Database (gnomAD)] at nucleotide position 15 (NM_000198.3:c.15C>A), resulting in the exchange of the cysteine codon to a premature stop codon (p.Cys5Ter). Subsequent screening of the Norwegian Addison Registry identified the patient harboring the mutation, a 55-year-old female with AAD, premature ovarian insufficiency and vitamin B12 deficiency, accompanied by 21OH- and parietal cell autoantibodies (Figure 2A). Notably, she did not carry any of the major histocompatibility complex (MHC) alleles conferring high risk to develop AAD (4). She tested negative for other autoantibodies such as anti-thyroid peroxidase, and had normal levels of thyroid stimulating hormone (0.40–4.50 mIU/L). Scrutiny of early medical records showed that she exhibited hyperpigmentation of genitalia and clitoris hypertrophy already at birth. One week of age, she started to vomit and developed hyponatremia (127 mmol/L) and hyperkalemia (6.1 mmol/L). Elevated levels of 17-ketosteroids were detected. She was therefore diagnosed with CAH and supplemented with cortisone acetate and eventually fludrocortisone. Her sister had also been diagnosed with CAH, but unfortunately died in an adrenal crisis at 2 years of age.
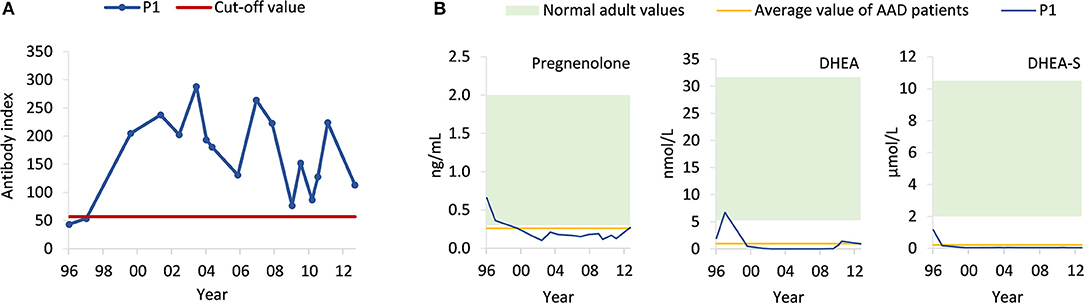
Figure 2. Levels of circulating 21OH autoantibodies in the patient (P1) and steroid profiling. (A) Using radioimmunoprecipitation assay, levels of 21OH autoantibodies (blue) were measured in serum samples taken at different time points from 1996 to 2013. The cut-off value (red) was set to obtain the maximal accuracy as calculated by an interlaboratory study (6). (B) Using ELISA, levels of pregnenolone, DHEA, and DHEA-S were measured in serum samples from the patient (P1, blue) taken from different time points from 1996 to 2013. Normal adult values (green) are based on established reference values from the textbook “Gynecologic Endocrinology” (pregnenolone) (7), hormone laboratory at Oslo University Hospital (DHEA) (https://ehandboken.ous-hf.no/api/File/GetFile?entityId=105475) and hormone laboratory at Haukeland University Hospital (DHEA-S) (https://analyseoversikten.no/analyse/14). The average value of AAD patients (yellow) is measured from serum samples of patients included in our biobank.
Given these conflicting findings, computer tomography (CT) and magnetic resonance (MR) scans, taken from age 43 to 51, were re-evaluated and the adrenals were found to be in the lower range of normal thickness 2–3 mm, indicating adrenocortical atrophy rather than hyperplasia as would be expected in case of isolated CAH. We then obtained a steroid profile by liquid chromatography tandem mass spectroscopy (LC-MS/MS) of a serum sample taken after an overnight medication fast (Table 1). Levels of all mineralocorticoids, and most glucocorticoids and androgens were below the detection limit, except for tetrahydrocortisol (0.308 nmol/L), 5α-tetrahydrocortisol (0.187 nmol/L), and testosterone (0.066 nmol/L). Total pregnenolone, DHEA and DHEA-S levels were measured by enzyme-linked immunosorbent assay (ELISA) from previously collected serum samples spanning the years 1996–2013 (Figure 2B). Normal levels of pregnenolone and DHEA were found in samples from the first 2 years, but then levels decreased toward the subnormal levels generally seen in AAD patients. DHEA-S was also below the normal range, typical of AAD patients, at all time points. ACTH was measured at several time points, revealing elevated levels on multiple occasions. The highest levels were observed in 2011 at 130 pmol/L (normal range 2.0–11.6 pmol/L). Importantly, we detected no subsequent increase in pregnenolone, DHEA, and DHEA-S in spite of elevated ACTH levels (Figure 2B).
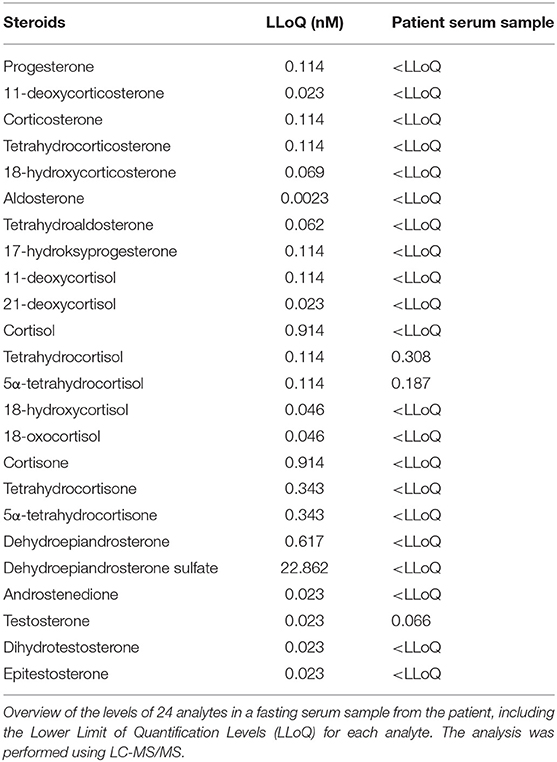
Table 1. Steroid profile of the patient.
She is currently, at age 57, treated with 20 mg hydrocortisone (Plenadren™) and 100 μg fludrocortisone, and does not have suppressed ACTH-values.
This is the first report of a patient with primary adrenal insufficiency due to both 3βHSD2 deficiency and AAD. At inclusion in the national Addison registry, she was classified as having AAD with vitamin B12 deficiency, and 21OH- and parietal cell autoantibodies. However, genetic screening and scrutiny of old records revealed a rare form of CAH due to a stop-gain mutation in HSD3B2. The presence of small sized adrenal glands instead of hyperplastic glands, normally seen in CAH, and no overproduction of Δ5-steroids following elevated ACTH levels, suggest that the adrenal cortex is not functioning. Presence of tetrahydrocortisol and 5α-tetrahydrocortisol is consistent with her ongoing replacement therapy which includes hydrocortisone. The low, but detectable, level of testosterone may be due to conversion of DHEA by 3βHSD1 in the periphery. Previously measureable pregnenolone levels suggest that the autoimmune destruction of the adrenal cortex commenced sometimes in the 1990-ties.
According to literature, we could only find one reported case of CAH occurring together with complete adrenal cortex insufficiency suspected to be autoimmune adrenalitis. This patient, however, had neither 21OH autoantibodies, nor MHC risk alleles, but was positive for autoantibodies against 17α-hydroxylase (8). Therefore, we speculate there might be other rare unreported cases of autoimmune adrenalitis due to early diagnosis of CAH, masking the clinical symptoms of AAD. Interestingly, several other AAD patients included in our exome sequencing analysis carry rare heterozygous non-synonymous variants in HSD3B2 (Table 2). Although it appears that family members of CAH patients, carrying heterozygous HSD3B2 mutations, maintain normal 3βHSD2 activity in vivo (14), genetic variations in HSD3B2 are associated with other conditions such as idiopathic hypospadias and prostate cancer (15, 16). Therefore, both subtle molecular abnormalities and deleterious mutations in HSD3B2 could have biological consequences, and may play a role in the pathogenesis of the immune-mediated adrenocortical destruction in AAD.

Table 2. Overview of rare variants of HSD3B2 (NM_000198.3) found in AAD patients.
This manuscript contains previously unpublished data. The name of the repository and accession number(s) are not available.
The studies involving human participants were reviewed and approved by Regional Committee for Medical and Health Ethics. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the Novo Nordisk Foundation (grant number NNF14OC0011005); and the Research Council of Norway (grant numbers 250030, 262677).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patients participating in our research, and Nina Henne, Elisabeth Halvorsen, Elin Theodorsen, and Hajirah Muneer for great technical assistance.
1. Al Alawi AM, Nordenstrom A, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase type 2 deficiency. Endocrine. (2019) 63:407–21. doi: 10.1007/s12020-018-01835-3
2. Alos N, Moisan A-M, Ward L, Desrochers M, Legault L, Leboeuf G, et al. A novel A10E homozygous mutation in the HSD3B2 gene causing severe salt-wasting 3β-hydroxysteroid dehydrogenase deficiency in 46,XX and 46,XY French-Canadians: evaluation of gonadal function after puberty*. J Clin Endocrinol Metab. (2000) 85:1968–74. doi: 10.1210/jc.85.5.1968
3. Burckhardt MA, Udhane SS, Marti N, Schnyder I, Tapia C, Nielsen JE, et al. Human 3β-hydroxysteroid dehydrogenase deficiency seems to affect fertility but may not harbor a tumor risk: lesson from an experiment of nature. Eur J Endocrinol. (2015) 173:K1–2. doi: 10.1530/EJE-15-0599
4. Bratland E, Husebye ES. Cellular immunity and immunopathology in autoimmune Addison's disease. Mol Cell Endocrinol. (2011) 336:180–90. doi: 10.1016/j.mce.2010.12.015
5. Aslaksen S, Wolff AB, Vigeland MD, Breivik L, Sheng Y, Oftedal BE, et al. Identification and characterization of rare Toll-like receptor 3 variants in patients with autoimmune Addison's disease. J Transl Autoimmun. (2019) 2019:100005. doi: 10.1016/j.jtauto.2019.100005
6. Falorni A, Bini V, Betterle C, Brozzetti A, Castaño L, Fichna M, et al. Determination of 21-hydroxylase autoantibodies: inter-laboratory concordance in the Euradrenal International Serum Exchange Program. Clin Chem Lab Med. (2015) 53:1761–70. doi: 10.1515/cclm-2014-1106
8. Reinehr T, Rothermel J, Wegener-Panzer A, Hartmann MF, Wudy SA, Holterhus PM. Vanishing 17-hydroxyprogesterone concentrations in 21-hydroxylase deficiency. Horm Res Paediatr. (2018) 90:138–44. doi: 10.1159/000487927
9. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
10. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. (2013) Chapter7:Unit7.20. doi: 10.1002/0471142905.hg0720s76
11. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890
12. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. (2012) 7:e46688. doi: 10.1371/journal.pone.0046688
13. Kircher M, Witten DM, Jain P, O'roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
14. Pang S, Carbunaru G, Haider A, Copeland KC, Chang YT, Lutfallah C, et al. Carriers for type II 3β-hydroxysteroid dehydrogenase (HSD3B2) deficiency can only be identified by HSD3B2 genotype study and not by hormone test. Clin Endocrinol. (2003) 58:323–31. doi: 10.1046/j.1365-2265.2003.01716.x
15. Codner E, Okuma C, Iñiguez Gn, Boric MAl, Avila A, Johnson MC, et al. Molecular study of the 3β-hydroxysteroid dehydrogenase gene type II in patients with hypospadias. J Clin Endocrinol Metab. (2004) 89:957–64. doi: 10.1210/jc.2002-020873
Keywords: adrenal insufficiency, congenital adrenal hyperplasia, 3β-hydroxysteroid dehydrogenase type 2 deficiency, autoimmune adrenalitis, autoimmune Addison's disease
Citation: Aslaksen S, Methlie P, Vigeland MD, Jøssang DE, Wolff AB, Sheng Y, Oftedal BE, Skinningsrud B, Undlien DE, Selmer KK, Husebye ES and Bratland E (2019) Coexistence of Congenital Adrenal Hyperplasia and Autoimmune Addison's Disease. Front. Endocrinol. 10:648. doi: 10.3389/fendo.2019.00648
Received: 10 July 2019; Accepted: 06 September 2019;
Published: 27 September 2019.
Edited by:
Sandro Loche, Ospedale Microcitemico, ItalyReviewed by:
Alberto Falorni, University of Perugia, ItalyCopyright © 2019 Aslaksen, Methlie, Vigeland, Jøssang, Wolff, Sheng, Oftedal, Skinningsrud, Undlien, Selmer, Husebye and Bratland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sigrid Aslaksen, c2lncmlkLmFzbGFrc2VuQHVpYi5ubw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.