
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Endocrinol. , 04 July 2019
Sec. Translational and Clinical Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00451
This article is part of the Research Topic Hormones, Inflammation and Immunity: Converging in Immunometabolism View all 5 articles
 Paola de Candia1*
Paola de Candia1* Francesco Prattichizzo1
Francesco Prattichizzo1 Silvia Garavelli2
Silvia Garavelli2 Veronica De Rosa2,3
Veronica De Rosa2,3 Mario Galgani2
Mario Galgani2 Francesca Di Rella4Maria Immacolata Spagnuolo5Alessandra Colamatteo6Clorinda Fusco6Teresa Micillo7Sara Bruzzaniti2Antonio Ceriello1,8,9Annibale A. Puca1,10
Francesca Di Rella4Maria Immacolata Spagnuolo5Alessandra Colamatteo6Clorinda Fusco6Teresa Micillo7Sara Bruzzaniti2Antonio Ceriello1,8,9Annibale A. Puca1,10 Giuseppe Matarese2,6*
Giuseppe Matarese2,6*Type 2 diabetes (T2D) is characterized by a progressive status of chronic, low-grade inflammation (LGI) that accompanies the whole trajectory of the disease, from its inception to complication development. Accumulating evidence is disclosing a long list of possible “triggers” of inflammatory responses, many of which are promoted by unhealthy lifestyle choices and advanced age. Diabetic patients show an altered number and function of immune cells, of both innate and acquired immunity. Reactive autoantibodies against islet antigens can be detected in a subpopulation of patients, while emerging data are also suggesting an altered function of specific T lymphocyte populations, including T regulatory (Treg) cells. These observations led to the hypothesis that part of the inflammatory response mounting in T2D is attributable to an autoimmune phenomenon. Here, we review recent data supporting this framework, with a specific focus on both tissue resident and circulating Treg populations. We also propose that selective interception (or expansion) of T cell subsets could be an alternative avenue to dampen inappropriate inflammatory responses without compromising immune responses.
Type 2 diabetes (T2D) is a multifactorial and multilayer disease, characterized by an altered metabolism of glucose, fat and proteins. Hyperglycemia is the main common feature defining T2D, and clusters of patients are identifiable according to the specific combination of insulin resistance (IR) and absolute or relative insulin deficiency (1), a combination that leads to complex clinical trajectories underlying the development of early metabolic imbalances and later cardiovascular complications (2, 3). T2D and its complications remain major causes of morbidity and mortality in the western world (4).
While it is well established that type 1 diabetes (T1D) results from cell-mediated autoimmune pancreatic β cell destruction, T2D has been historically considered a metabolic disease, and metabolic determinants are traditionally identified as major pathogenetic factors. A more recent line of research has started to focus on low-grade inflammation (LGI) as a pervasive feature of T2D, accompanying the development and the progression of the disease, as well as the genesis of complications (5). Two of the main risk factors to develop T2D are aging and obesity, both known to promote tissue and systemic chronic inflammation, often referred to as inflammaging and metaflammation, respectively (5–8). A number of publications demonstrate that inflammation is not a mere bystander but it plays a key role in the progression of all the main features of T2D disease, e.g., IR, β cell failure or inability to cope with increased insulin demand, and atherosclerotic plaque development and destabilization (5–7). A plethora of putative inflammatory sources and mechanisms have been proposed to explain this evidence (5–9). Seminal discoveries and the majority of studies have mainly focused on cells of the innate immune system (10–12) and more recent data also suggest the direct involvement of acquired immunity. In particular, autoimmunity, a multifactorial process defined by loss of self-tolerance and chronic excess reactivity of B and T cells, has started to be recognized as an overlapping mark of both T1D and T2D. Furthermore, metabolic dysregulation and autoimmune components are able to generate a vicious cycle: the increased production of cytokines characterizing the chronic inflammatory state in T2D concur to destroy pancreatic β cells, and this inflammation-induced tissue damage leads to the release of “self” antigens that promote autoimmune activation. In turn autoimmunity further impairs insulin secretion in β cells and promotes hyperglycaemia (13–15).
A better recognition of the autoimmune components of T2D is relevant because it may lead to a deeper understanding of the mechanisms involved in the insurgence of hyperglycemia; it has also considerable therapeutic consequences, translating into a better classification and treatment of the disease. Indeed, T2D patients with a significant autoimmune component: (i) need insulin earlier during disease progression, (ii) are likely to poorly respond to classical anti-diabetic medications, (iii) may be highly responsive to immunomodulator therapy (16).
Our review focuses on existing evidence on autoimmune aspects of T2D from a molecular, cellular and clinical perspective. We describe how the knowledge regarding the intimate link between metabolism and inflammatory responses in specific immune cell populations is rapidly expanding, and report latest findings regarding the alteration of B and T (with a specific focus on T regulatory subsets) cells, the metabolic mechanisms driving their expansion or dysfunction in T2D (17–20). Finally, we briefly summarize promising clinical data regarding the potential of anti-inflammatory therapies in T2D and hypothesize a framework where inflammatory responses are modulated, rather than suppressed, to intercept and blunt the development and progression of the disease.
The presence of circulating autoantibodies in non-insulin-dependent diabetes mellitus was first identified more than 40 years ago (21). Nowadays, the presence of these autoantibodies characterizes a condition referred to as latent autoimmune diabetes of the adults (LADA). Autoantibodies against glutamic acid decarboxylase (GADA), islet cytoplasm (ICA), insulinoma-associated protein (IA-2A), and zinc transporter (ZnT8A) are commonly found in these patients.
Analyzing a large cohort of T2D patients (n = 3.672) from the UK, aged between 25 and 65 years, the percentage of subjects with ICA and/or GADA autoantibodies was found to be 12% overall and to be significantly higher in younger patients (22). Another study from the Pittsburgh cohort of the Cardiovascular Health Study found that also among diabetic patients aged over 65 years 12% had autoantibodies against GAD65 and/or IA-2, associated with an abnormal glucose control and a pronounced activation of the acute-phase response (increased fibrinogen and C-reactive protein levels), that may in part explain the observed defect in insulin secretion (23). A similar prevalence of diabetes autoimmunity was described in Argentinian elderly T2D patients (24). The largest European study (Action LADA) to date has later analyzed 6,156 T2D patients (age range, 30–70 years) for GADA, IA-2A and ZnT8A and found that 9.7% were positive, with the majority (8.8%) being GADA positive, and that, at diagnosis, these patients are usually non-insulin requiring and do not show categorically distinct clinical features from autoantibody-negative T2D patients (25). Ethnicity may count, as GADA positivity in T2D patients range from 3.8% in Japan (Eihme Study, n = 4,980) (26) to 10% in Norway (HUNT Study, n = 1,134) (27).
At diagnosis, LADA patients do not usually need exogenous insulin and they appear to be clinically affected by T2D, but a large percentage will need it within a few years, showing a much faster decline of β cell function compared to T2D patients, possibly caused by the ongoing immune-mediated β cell destruction. Notably, Turner et al. showed that 94% of patients with ICA and 84% of those with GADA required insulin therapy by 6 years, compared with 14% of those without the antibodies (22). A small study has directly correlated the presence of islet autoantibodies with significantly lower acute insulin response when compared to that of the autoantibody-negative group, but observed similar peripheral IR, providing compelling evidence that the profound impairment of insulin secretion is plausibly determined by the immune-mediated injury of pancreatic β cells (28).
Although formally classified as T1D for the typical presence of autoantibodies, LADA patients present several clinical features that are mixed between T1D and T2D pathologies. Low birthweight results to be a risk factor for LADA of the same strength as for T2D, suggesting LADA etiology includes factors related to T2D (29). Furthermore, LADA is associated with factors well known to promote T2D, such as overweight, physical inactivity, smoking, and sweetened beverage intake, suggesting LADA may in part be preventable through the same lifestyle modifications as T2D (30). In particular, the risk of LADA in relation to overweight/obesity was studied in two large population-based reports from a Swedish case-control study and the Norwegian HUNT Study, whose findings support the hypothesis that, even in the presence of autoimmunity, factors linked to IR, such as excessive weight, could promote LADA onset (31). Metabolomics of LADA, T1D and T2D patients failed to identify a unique metabolite profile for any of the diabetes types. Instead, the metabolome varied along a C-peptide-driven continuum from T1D to T2D, with LADA being an intermediate and patients metabolically closer to T1D showing a faster progression to insulin therapy than those closer to T2D (32). On the other hand, a Danish study analyzing a cohort of 4,374 adults with newly diagnosed diabetes demonstrated that fasting C-peptide and GADA status, but not age at onset, are able to define groups of diabetic patients with clinically relevant differences in glycaemic control and cardiometabolic risk, suggesting that the borders between T1D and LADA may be less discrete than believed (33). Parallel studies also demonstrated that LADA is associated with lower prevalence of microvascular complications, lower mortality, and lower risk of cardiovascular events, compared with T2D (34, 35). The risk of LADA is substantially increased with family history of T1D disease but also, albeit significantly less so, of T2D disease (36). A meta-analysis from 16 independent case-control studies (8,869 cases and 20,829 controls total), aimed at determining the association of T1D and T2D gene variants, demonstrated that some of these polymorphisms are associated with the risk of LADA, further supporting the idea of LADA as a combination of both T1D and T2D and emphasizing its heterogeneity (37). The first genome-wide association study of LADA revealed how the leading genetic signals were principally shared with T1D, although positive genetic correlations genome-wide were registered also with T2D. Authors identified a novel independent signal at the known T1D locus harboring the 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 (PFKFB3) gene (38). This gene encodes a regulator of glycolysis and insulin signaling and thus it had been previously reported as a plausible biological candidate in T2D diabetes (39). PFKFB3 also causes a reduction in T cell glucose consumption and survival, which in turn impairs the immune response in autoimmune conditions (40), calling for further studies to determine whether this genetic factor is truly a distinguishing feature between adult and childhood-onset autoimmune diabetes (38).
To further complicate the picture, while subjects classified as T2D patients are by definition autoantibody negative, autoantibodies may however also fluctuate. The Norwegian HUNT study unveiled that about 3% of subjects classified as T2D show a transient autoantibody positivity associated with earlier disease onset, a pre-diagnostic evidence of autoimmune activity in a sub-group of T2D patients (41).
LADA patients tend to share some clinical and phenotypic characteristics, compared to autoantibody-negative T2D patients: they are usually younger and leaner, suffer from acute symptoms and have a personal and/or familial history of autoimmune diseases (25). Nevertheless, LADA heterogeneity (from patients with clear signs of insulin deficiency associated with strong markers of autoimmunity, to patients showing weak markers of autoimmunity and closely resembling T2D) is possibly the mirror of a progressive and continuous clinical spectrum that blends, instead of discriminating, T1D and T2D (Figure 1). The crucial question arises: how important is the autoimmune component in autoantibody negative T2D patients? To try and answer to this question, we cannot abstain from describing the connection that exists between systemic metabolism and the immune system.
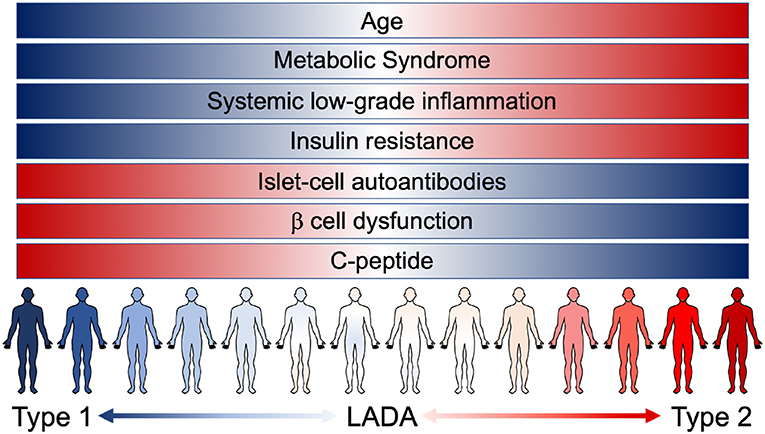
Figure 1. The continuous range of diabetes. Notwithstanding the different risk factors, instead of being clearly confined, T1D, LADA, and T2D patients are now known to present overlaying/overlapping clinical characteristics. LADA patients range from showing clear signs of β cell dysfunction (insulin deficiency and low levels of C-peptide) associated with strong markers of autoimmunity (presence of islet-cell autoantibodies) to patients showing a higher grade of insulin resistance and other pathological components resembling T2D condition [metabolic syndrome and systemic low-grade inflammation (LGI)]. The pathological features of the different forms of diabetes manifest as an uninterrupted spectrum that fails to clearly discriminate T1D and T2D.
Obesity, together with age, is the major risk factor for T2D. A large proportion of T2D patients are obese and the risk of T2D increases with increasing body mass index (BMI) (42). In both human and mice, obesity is associated with chronic LGI, thought to play a pivotal role in the development of IR and T2D pathological process (14). Although obesity is not yet a fully established risk factor for autoimmunity, the altered glucose tolerance and the development of IR due to an abnormal accumulation of adipose tissue (AT) in obese subjects coincide with an elevated incidence of autoimmune diseases (43). Epidemiological data analyzing T1D, multiple sclerosis, and psoriasis patients have recognized a causal risk factor for all these autoimmune conditions in an elevated BMI (44–46), although the mechanistic players of this association remain mostly undetermined. The coexistence of overweight and autoimmunity may be the result of concurrent dysregulation of immune tolerance, involving different organs (47). In the case of T2D, one of the most implicated districts is the AT (48).
The presence of enlarged fat depots that characterizes overweight/obese subjects, may be considered as an autoimmune pathology in which autoreactive T cells attack the adipocytes and fuel adipose inflammation (49, 50). On the other hand, AT is actually contiguous with main immune cell centers, such as lymph nodes, thymus, and bone marrow, in a close embrace that we have recently defined an “anatomical tango” (51). AT surrounds the thymus, hence may influence T cell differentiation in response to metabolic cues (52); furthermore, a high number of adipocytes resides in the bone marrow, with possible involvement in haematopoiesis, lymphopoiesis, and memory B and T cell responses (53). This physical contiguity has been noted by anatomists of the past, but only recently, though, it has been explored from a new and mechanistic point of view, and researchers have hypothesized and tested how it may allow a continuous communication (51).
The AT produces and releases several bioactive molecules, referred to as adipokines with a variety of pro-inflammatory and anti-inflammatory roles, which regulate both metabolism and immune cell function (20). Together with immune-cell derived cytokines, these immune metabolic mediators actually allow a reciprocal regulation between the AT and the immune system at both a paracrine and endocrine level (54, 55). One of the most biologically relevant of these molecules, leptin, is produced by the AT in proportion to the body fat mass and, at a systemic level, regulates appetite and food intake, promoting the entry of glucose into the tissues and reducing hyperglycemia (56). Leptin links lifestyle with inflammatory and/or autoimmune molecular mechanisms (57–59). It is not only known to significantly regulate the innate immune cells, but to also bridge obesity and dysregulated adaptive T cell activity. It seems that metabolic dysregulation both induces and depends on autoimmune alterations (60, 61). As a double edge sword, lymphocyte activation and function are intimately intertwined between metabolic disorders, including T2D, and inflammatory diseases (62, 63).
The research at the interface between the historically distinct disciplines of immunology and metabolism (immunometabolism) focuses on the regulatory connection between the metabolic state of an organism and immune cell physiology/activity. Unveiling this connection is endowed with enormous potential for T2D understanding (64).
While the involvement of AT-resident macrophages and innate immunity has been thoroughly analyzed (65, 66), fewer studies have focused on the role played by the adaptive immune B and T lymphocytes and their released factors in obesity-associated inflammation and T2D triggering events. Next, we critically review these studies.
B lymphocytes are responsible for the humoral immunity component of the adaptive immune system. Beyond antibody secretion, they can also present antigens and secrete cytokines. A plethora of B cell subsets have been identified according to the stage of differentiation, tissue localization and developmental lineage (67). Despite the controversies regarding the correspondence between human and murine B cells (68, 69), different alterations affecting both circulating and tissue-resident B cells have been described in T2D (70). Similarly, both antibody-mediated and contact-dependent mechanisms have been proposed in relation to B cell imbalance and loss of insulin sensitivity.
A clear demonstration supporting the role of B cells also in the development of IR has been provided in murine models. B cell–null New Zealand obese mice do not develop IR in response to obesity (71). Similarly, another study showed that either pharmacological or genetic depletion of B cells is sufficient to significantly attenuate immune cell infiltration and inflammation in the AT, finally promoting insulin sensitivity (72). Mechanistically, high fat diet (HFD) has been proposed to foster the early recruitment of B cells which promote T cell activation in the AT. In turn, this would boost M1 macrophage polarization, and thus induce IR. B cells can also exert detrimental effects systemically through the production of pathogenic IgG antibodies (73). Treatment of B cell-null mice with IgGs derived from WT mice exposed to HFD is sufficient to phenocopy the metabolic alterations induced by HFD (72). In addition, recent findings revealed that hyposialylated IgGs activate endothelial IgG receptor FcγRIIB to promote obesity-induced IR. Of note, the results were replicated with IgGs derived from patients with T2D and transferred to IgG-deficient mice, suggesting the relevance of this mechanism also for human diabetes (74). Consistently, human subjects with IR are characterized by 122 potential IgG targets (72). However, the wide range of possible antigens, their relatively low prevalence, and the absence of an evident tissue lesion (in any diabetes-relevant tissue) argue against a prototypical autoimmune phenomenon for T2D. On the other hand, a similar setting is compatible with low grade inflammation, a subclinical, chronic process without major manifestations of the acute inflammatory response (5). Similar findings expanded these results showing that B-T cell contact is mandatory to develop a pathogenic pro-inflammatory response. Indeed, the addition of a cytokine-permeable trans-well membrane blunted the B cell-induced Th17 response (75). Moreover, B cells from diabetic patients showed reduced secretion of the anti-inflammatory interleukin (IL)-10 upon stimulation (75), a phenomenon observed in multiple cell types exposed to the diabetic environment (9). Regarding specific B cell subpopulations, less is known about their deregulation in T2D, especially considering human data. Naïve B cells are divided into two subsets, the most abundant B-2 follicular and marginal zone B cells, responsible for generating the majority of high-affinity antibodies during an infection and the scarcer B-1 cells, arising from a different developmental pathway and able to generate natural antibodies in a T cell-independent manner (76). AT-resident, B-2 cells were identified to promote the inflammatory response to HFD and IR, possibly through a leukotriene B4 (LTB4) - LTB4 receptor 1 axis (77). Interestingly, two studies reported a protective role for both CD5+ B-1a and CD5− B-1b cells [different at both phenotypic and development level (78)] in HFD-induced metabolic alterations in mouse models. Adoptive transfer of both B-1a and B-1b cells into HFD-fed, B cell–deficient mice ameliorated IR and glucose intolerance through IL-10 and polyclonal IgM-dependent mechanisms, while the transfer of B-2 cells worsened the metabolic imbalances (79, 80). Consistently, diabetic leptin receptor-mutant db/db mice had lower levels of peritoneal B-1a cells, which were also hypo-responsive in terms of differentiation to effector B cells and IgM production (81). On the other hand, one study reported conflicting results, since it showed that the suppression of B cell activation in AT of obese mice fail to discernibly affect systemic inflammation and glucose homeostasis (82). Findings in obese subjects demonstrate that B-1 cells and IgM antibodies in AT inversely correlate to inflammation and IR (80). Circulating populations of B cells in T2D patients further support a B-1 protective, B-2 detrimental paradigm since B-1a cell frequency are inversely correlated with HbA1c, LDL, and triglycerides, while B-2 cells show the opposite trend (83).
The resulting corollary to these observations is that patients with T2D may be characterized by an altered response to vaccination and a higher susceptibility to infections. B cell number and capacity to produce Ig are known to decline with age (84), while few data are available for T2D patients. Elderly patients are characterized by an altered response to vaccinations and infections, despite a higher basal pro-inflammatory status (84, 85). Similarly, B cells derived from T2D patients are characterized by a reduced ability to produce de novo antibody responses, despite a higher basal secretion of pro-inflammatory cytokines (86) but B cell repertoire appears to be affected by obesity, rather than by the diabetic status (87). Nonetheless, the incidence of a wide range of infective diseases is markedly increased in the diabetic population (88, 89). On the other hand, data regarding immune response to vaccination in T2D are not straightforward. In particular, unaltered or even optimal response to influenza vaccination has been reported in young and elderly patients with T2D (90–92). Guidelines for medical care strongly recommend annual influenza vaccination for diabetic patients (93) and a recent systematic review emphasizes and reinforces the need and value of seasonal vaccination to decrease severe complications, hospitalization and/or death in diabetic patients (94). Results regarding other vaccinations are sparser (95, 96).
Overall, more observational and mechanistic data are needed to identify the involvement of B cells in the development of IR and T2D. In particular, few prospective studies have been conducted in humans. Reported alterations in T2D patients could represent either a cause or a consequence of the disease. Nonetheless, the observations that (i) specific antibodies are associated with prevalent T2D (97); (ii) serum IgG2 levels are associated with whole-body insulin-mediated glucose disposal (98); and (iii) general abundance of circulating gamma globulins predicts incident T2D in a large cohort (99), support the postulate that B cell alteration plays a key role in T2D pathogenesis.
CD4+ T helper cells (in particular Th1 and Th17) are among the principal mediators of a pro-inflammatory environment, through the release of inflammatory cytokines (TNF-α, INF-γ, IL-17, IL-22, and IL-26 among others). CD4+CD25highForkhead Box Protein P3 (FoxP3)+ T regulatory (Treg) cells, on the other hand, are a functionally distinct cell lineage committed to exert an anti-inflammatory/immune suppressive control on innate and adaptive immune responses and they represent the most relevant cells in the body to sustain immunological homeostasis (100, 101). Treg cells function by inhibiting the activity of the pro-inflammatory counterpart CD4+ Th1 and Th17 (also referred to as T conventional or Tconv) cell subsets (102, 103). Treg-mediated suppression is based on inhibitory molecules, anti-inflammatory cytokines such as IL-10, IL-35, and Transforming Growth Factor (TGF)β and Cytotoxic T-Lymphocyte Associated protein 4 (CTLA-4), metabolic modulation and direct cytolysis of target cells (101). The transcriptional factor FoxP3 is the master regulator of Treg cell phenotype and function. Treg cells can be subdivided according to their origin into two main groups: one directly originating from the thymus, and the other arising from the peripheral conversion of naive CD4+CD25− Tconv cells (104). Peripheral Treg cell frequency ranges from 5 to 15% of CD4+ T cells, but the ratio between Treg and Tconv cells can diminish upon infection, in order to improve specific immune response activation (105). Notwithstanding the pivotal role of Treg cells in halting unwanted immune responses is certain (106), the contribution of their numerical and/or functional dysregulation to the very development and progression of human autoimmunity, and in particular autoimmune diabetes, is still to be completely clarified. The involvement and causal role of Treg cells in T2D pathogenesis is a fortiori still poorly defined. Relevant knowledge, though, has come from discovering that Treg cells are major sensors of the systemic metabolic state.
In 2009, the group of Diane Mathis showed that about 10% of stromovascular fraction from the visceral adipose tissue (VAT) of C57Bl/6 lean mice fall within the lymphocyte gate, close to half of which are of the CD3+ T lineage. Among them, they revealed the presence of a unique population of Treg cells, as a much higher fraction of the CD4+ T cell compartment than usually observed in lymphoid or other non-lymphoid tissues and, importantly, their numbers were strikingly and specifically reduced at this site in insulin-resistant models of obesity (107). VAT Treg cells show a phenotype clearly distinguishable from that of their counterparts in the spleen and lymph nodes, including a distinct gene-expression profile, T cell receptor repertoire, and pattern of chemokine and chemokine receptor expression. Cytokines differentially synthesized by VAT-resident Treg and Tconv cells directly affect the synthesis of inflammatory mediators and glucose uptake by cultured adipocytes. In particular, Treg cell elevated production of the anti-inflammatory cytokine IL-10 may be essential to curb AT inflammation (107). Moreover, the residence of Treg cells into AT expose them to high concentrations of the adipocytokines, in primis leptin, discovered to halt the generation and proliferation of these cells (60, 62, 108–110). Notably, Treg cells not only express the leptin receptor, but they also secrete leptin, directly contributing to metabolic homeostasis and glucose tolerance (60). Diane Mathis's group has also identified peroxisome proliferator-activated receptor (PPAR)-γ, the “master regulator” of adipocyte differentiation, as a crucial molecular orchestrator of VAT Treg cell accumulation, phenotype and function. By treating obese mice with pioglitazone, an antidiabetic agent whose mechanism of action resides in PPAR-γ activation, it was demonstrated that PPAR-γ expression in VAT Treg cells is necessary for drug-dependent restoration of insulin sensitivity (111). A more recent investigation showed that IL-33 signaling through its receptor ST2 and myeloid differentiation factor MyD88 are also essential for the development and maintenance of VAT-Treg cells and sustains their transcriptional signature. IL-33 administration actually induces vigorous population expansion of VAT Treg cells, which tightly correlates with improvements of metabolic parameters in obese mice (112). Gain-of-function experiments have provided more evidence on how VAT Treg cells are able to directly influence the inflammatory state of AT and, thus, IR in studies analyzing diet-induced obese, leptin-deficient ob/ob and db/db mice. Treg cells are able to decrease not only macrophage number, TNF-α and inflammation in VAT, but also pancreatic islet cell hyperplasia, liver fat accumulation, blood glucose, liver enzymes, IR and kidney damage (113, 114). Notably, restoration of Th1/Foxp3+ balance is able to reverse IR for months, despite continuing high-fat diet (115).
While the biological role of VAT-resident Treg cells seems fully established in mice, published data on human Treg cells have instead been conflicting.
FoxP3 mRNA is readily detectable in both omental and subcutaneous human fat depots and can be titrated by PCR. Despite missing access to non-obese controls due to the rarity of bariatric surgery on normal subjects, Feuerer et al. found a correlation between BMI and the drop in Treg cells in omental vs. subcutaneous fat, that suggested findings on mice may be translatable to humans (107). Another study further confirmed significant lower levels of FoxP3 gene expression in human omental samples in obese compared to lean controls (116). On the other hand, Zeyda et al. analyzed expression of marker genes specific for T cell subsets in visceral and subcutaneous AT from highly obese subjects and lean-to-overweight control subjects. Unexpectedly, proportions of cytotoxic T cells and Th1 cells were unchanged, whereas those of Treg cells were increased in VAT from obese compared to control subjects, and positively correlated with systemic and AT inflammation, thus failing to support a role of protective Treg cell loss in AT inflammation in obese subjects, as clearly indicated by mice studies (117). When FoxP3 mRNA level was quantified in obese subjects differing for insulin sensitivity, it was found decreased in obese insulin-sensitive, but not in insulin resistant patients as compared with lean control subjects. Actually, peripherally induced adaptive FoxP3 Treg cells seem to accumulate in the inflammatory adipose microenvironment in obese subjects with overt IR. Differently, Helios, mainly associated to thymus-derived Treg cells, was significantly lowered in human VAT of all obese subjects irrespective of their IR. Authors themselves commented on the incapability to exclude that FoxP3 mRNA expression may reflect other CD4+ T cells infiltrating VAT of obese subjects with IR (114). A very recent comprehensive investigation into how omental AT immunity changes with obesity and T2D in humans, still revealed important similarities but also differences to paradigms in mice. Patients with T2D had increased proportions of inflammatory cells, including M1 macrophages, with positive correlations to BMI. Treg frequencies did negatively correlate with BMI but were similar in T2D and non-T2D subjects (118). Compared to thymic population, omental AT Treg cells expressed higher levels of PPAR-γ, but failed to show any detectable expression of IL-33 receptor, ST2 (118), thus contradicting a previous report (112).
In 2014, Benoist and co-workers analyzed blood circulating Treg and Tconv cells from 168 donors, either healthy or with established T1D or T2D by performing genome-wide expression profiling. They described a Treg specific transcriptional signature, composed of some very variable transcripts and some almost invariant, with more extensive variability for genes that control effector function than for lineage-specification factors. This Treg signature became sharper with age and with increasing BMI, suggesting a tuning of Treg function with repertoire selection and/or chronic inflammation. Notably, none of the transcripts showed significant association to diabetes, and overall expression of the Treg signature was subtly perturbed in T1D, but not T2D, patients (119).
In Figure 2, we have schematized a possible involvement of adipose resident Treg cells in human T2D pathogenesis but studies on human AT-resident Treg cells are still too scant to provide with conclusive results. In particular, in the next future, it will be necessary to untangle whether in T2D patients Treg cells result further dysregulated, and how, compared to merely obese subjects.
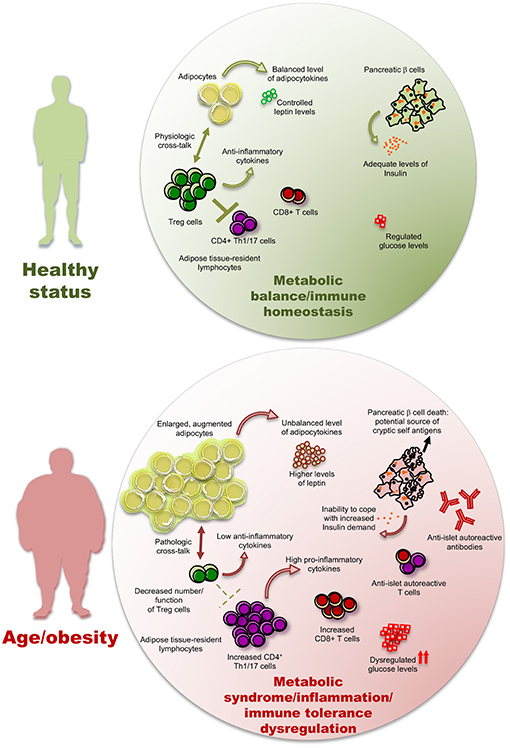
Figure 2. The inflammatory/autoimmune components of type 2 diabetes. Low-grade inflammation (LGI) induced by a high adipocyte mass, together with an elevated abundance of pro-inflammatory cytokines and adipocytokines produced by fat tissue, may hamper the number and function of anti-inflammatory Treg cells and diminish the level of anti-inflammatory cytokines, hence promoting immune responses by the CD4+ and CD8+ T cell subsets, including those with an autoreactive potential. The pancreatic β cell death leads to two pathological consequences in a vicious cycle of autoimmunity and metabolic dysregulation: (i) the potential increase of pancreatic cryptic self-antigens and (ii) a diminished ability to respond to insulin demand, with consequent further dysregulation of glucose levels. The presence of these phenomena is possibly significantly variable from patient to patient.
Treg cells are central in orchestrating the suppression of autoreactive responses by inhibiting the action of Th1 and Th17 cells counterpart. At the central core of autoimmune disease development, there is an imbalance between pro-inflammatory Th1/Th17 and anti-inflammatory Treg (120). Is this imbalance actually observed also in T2D?
In 2006, Seyfert-Margolis et al. published the development of a T cellular immunoblotting assay, with excellent sensitivity and specificity for measuring islet-specific T cell responses in autoimmune diabetes (121). In pivotal works of subsequent years, the same group used this approach to also screen adult phenotypic T2D patients, in parallel with detection of islet autoantibodies. Authors succeeded in identifying a group of adult autoimmune phenotypic T2D patients who are autoantibody-negative but T cell reactivity-positive, and showed that T cell responses to islet proteins fluctuate less than autoantibody responses and are associated with a more severe β cell damage and lower residual insulin secretion. It was concluded that these autoimmune patients would have not been recognized using autoantibody testing alone and thus islet autoimmunity may be more prevalent in adult phenotypic T2D patients than previously estimated (122–124). T cells from both T1D and T2D patients are capable to recognize islet proteins, but these proteins are not the same (123). The development of islet autoimmunity may perhaps follow different pathways in T1D compared to T2D, that is not unexpected since autoimmunity in T2D patients possibly arises as a sequela of obesity-dependent chronic inflammatory responses (125).
Beside self-reactive T cells, obesity and T2D are associated with an imbalance in T cell sub-population ratio. In mice, diet-induced obesity selectively promotes an expansion of the Th17 T cell lineage, a subset with a prominent pro-inflammatory role, and progressively higher production of IL-17 than lean littermates. A pronounced autoimmunity, meaning more severe early disease and histopathology, in two disease murine models of multiple sclerosis and inflammatory bowel disease is known to associate with an increased IL-17 producing Th17 cell bias in target tissues in target tissues (central nervous system and colon, respectively) (126). Jagannathan-Bogdan et al. found that ex vivo T cells from T2D patients appear to be poised for IL-17 production and have increased production of IFN-γ. In contrast, T2D patients had decreased percentages of Treg cells, suggesting that T cells in T2D patients are naturally skewed toward pro-inflammatory subsets that likely promote chronic inflammation in T2D through elevated cytokine production (127). Another study demonstrated that the ratios of Th17/Treg cells and Th1/Treg cells were significantly increased in T2D patients. In particular, the peripheral induced Treg cell number was reduced and Treg cells seemed more prone to cell death, since they showed a decreased Bcl-2/Bax ratio (128).
When TNF receptor superfamily member 5 (CD40)/CD40 ligand (CD40L) pathway, central player in the onset and maintenance of the inflammatory reaction, was studied in human AT, a direct cross-talk between adipocytes (CD40-expressing) and lymphocytes (CD40L expressing) was described, based on actual cell-cell contact (129). CD40 mRNA levels were significantly higher in subcutaneous than in visceral AT of obese subjects and were positively correlated with BMI, and with IL-6 and leptin mRNA levels. T lymphocytes were thus hypothesized to regulate adipocytokine production through both the release of soluble factor(s) and heterotypic contact with adipocytes involving CD40, that may play a direct role in obesity-associated human AT inflammation (129).
What about the pancreas? It is conceivable that a systemic inflammatory state may expose the pancreatic insulin producing cells to immune-mediated damage by random and not specific pathways. LGI induced by a high adipocyte mass, together with an elevated abundance of pro-inflammatory cytokines and adipocytokines produced by fat tissue, promote immune responses by the Th1 and Th17 cell subsets, that favors generic bystander activation of pro-inflammatory immune cells, including those with an autoreactive potential (130). In other words: (i) stressed adipocytes secrete pro-inflammatory adipokines and cytokines, able to recruit macrophages but also B and T cells (131); (ii) pro-inflammatory macrophages and activated lymphocytes secrete cytokines and chemokines, that turn the inflammatory reactions persistent (132). These phenomena may favor the conditions for incidental development of β cell autoreactivity and promote the engagement and activation of self-reactive T cells with direct cytotoxic effects.
In vitro treatment of human pancreatic islets from nondiabetic organ donors with high glucose levels is able to induce Fas expression, caspase-8 and−3 activation, and β cell apoptosis (133). Importantly, an up-regulation of Fas in β cells of T2D patients relative to nondiabetic control subjects has been observed, suggesting elevated glucose levels may contribute to islet destruction by a mechanism based on the interception of β cell expressed Fas Receptor by Fas Ligand (constitutively expressed by many immune cells, including activated T cells), independently of a classic antigen-based autoimmune reaction (133). A subsequent work helped to substantiate that in this detrimental regulatory loop, Fas upregulation, DNA fragmentation, and impaired β cell function were preceded by increased production and release of IL-1β, followed by NF-κB activation in β cells, implicating an inflammatory process in the pathogenesis of T2D glucotoxicity (134). Using high-density microarray analysis of the β cell transcriptome, it was more recently discovered that of those potentially able to activate STAT-1 or NF-κB pathways, TNFR-5 is the most sensitive to high glucose and fatty acid environment, confirming the role of inflammatory pathways in triggering glucolipotoxic islet cell death (135). Experiments performed at the beginning of the 1990s revealed that high glucose is able to also increase β cell immunogenicity (136, 137). Exposing Osborne-Mendel rats to a high-sucrose and high-fat diet with insulin releasing effects was shown to increase both β cell function and islet antigen expression (136). In isolated Sprague-Dawley rat islets cultured at different glucose concentrations, GAD expression was found to be glucose dependent and to correlate with increased functional state of the β cell (137).
It is also important to consider that β cell death and destruction lead to a relevant consequence on the perspective of “self” recognition: the release of a potentially notable amount of “cryptic” β cell antigens with immunogenic potential (Figure 2). These cell-free antigens may now encounter self-reactive T cells, leading to further immune-mediated β cell death and islet destruction (138).
Since accumulating evidence show an etiopathological role of LGI in the development of T2D, clinical trials and observational studies have been designed to explore the effect of anti-inflammatory drugs on glucose parameters (139, 140). On the other side, also glucose-lowering drugs can have anti-inflammatory effects. The description of the pleiotropic, beneficial, anti-inflammatory effects of selected anti-diabetic medications is beyond the scope of this manuscript and it has been largely reviewed elsewhere (5, 6, 139, 141–143).
Given the role of IL-1β in both IR development and β cells deterioration, a number of trials tested different biological drugs blocking IL-1β pathway, e.g., anakinra and canakinumab. These agents have shown a significant beneficial effect on HbA1c, IR, and β cell secretory function (144–146). Some of the outcomes were also long-lasting, since the effect on proinsulin-to-insulin (PI/I) was visible 39 weeks after cessation of the intervention (146). However, the effect on HbA1c was neither enduring nor clinically relevant, if compared to those obtained with conventional glucose-lowering drugs. In addition, a long-term treatment with an immune suppressive agent could be accompanied by relevant adverse effects, especially considering that a continuous presence of the drug would be needed. On the other side, an LGI-targeting intervention could be beneficial in T2D even beyond glycemic control (6, 147). Indeed, a recent trial demonstrated a significant reduction of cardiovascular events in patients treated with canakinumab, forty percent of which constituted by T2D patients, already on optimal pharmaceutical polytherapy (148). Secondary analysis did not reveal a different trend in this specific population. On the contrary, the beneficial effect was particularly evident in patients with high C reactive protein at the beginning of the treatment and diminished levels at the end of the trial, suggesting the existence of a residual (despite polytherapy) inflammatory risk also for T2D (149).
The first discovery linking LGI and T2D found an increased expression of TNFα in the AT of obese subjects (10). A number of observational studies and small-pilot trials have tested the effect of anti-TNFα biologicals on glucose parameters, showing improved insulin sensitivity in subjects without diabetes and with chronic inflammatory diseases, such as rheumatoid arthritis (150–153). While the use of TNF-α antagonism is associated with an improved endothelial function in a wide range of patients (154, 155), however, it has not demonstrated a tangible benefit in patients with T2D (156, 157).
Salicylates are another class of anti-inflammatory drugs tested in settings of T2D. Low-dose aspirin is known to inhibit cyclooxygenase (COX) 1 and 2, while higher doses can also inhibit NF-kB (140). The first clinical trial showed that high-dose aspirin improved IR and both fasting and post-prandial hyperglycemia in patients with T2D (158). Later, the TINSAL-T2D study tested salsalate, a salicylate prodrug marginally affecting the platelet aggregation pathways, in a multicenter randomized trial, demonstrating a significant reduction of HbA1c (159). However, both aspirine and salsalate are associated with a plethora of adverse events, e.g., risk of bleeding, gastrointestinal and renal toxicity, which hampered their full translation in clinical settings (140). Of note, differently from the general population, aspirin use in T2D patients does not appear to provide CV protection in settings of primary prevention (160).
Other conventional immune suppressing drugs such as diacerein, chloroquine, and TLR inhibitors have been shown to affect some markers of glucose metabolism (139, 140). However, immune suppression is always accompanied by side effects, mainly associated with an increased risk of infective diseases (161, 162). In addition, the chronic nature of LGI in T2D implies that such treatments should be applied continuously to obtain enduring effects. Also, a putative beneficial, transient effect of selected inflammatory molecules on glucose homeostasis has been proposed (163). Two alternative approaches to target LGI without suppressing immune function may be represented by: (i) the removal of the pro-inflammatory triggers; (ii) the selective modulation of immune cells function. The first approach is already being exploited with specific interventions known to target LGI and to improve diabetes outcomes, e.g., diet, exercise, and bariatric surgery (5, 164). Also, innovative molecules aimed at attenuating fundamental triggers of LGI are going to be tested in T2D, e.g., microbiota modulators, epigenetic-modifying, and senescence-targeting drugs (5, 143). On the other hand, increasing evidence suggests that immune response can be selectively modulated, rather than suppressed. Indeed, systemic delivery of nanoparticles coated with autoimmune-disease-relevant peptides bound to major histocompatibility complex class II (pMHCII) molecules triggers the generation and expansion of antigen-specific regulatory CD4+ T cell type 1 (TR1)-like cells, restoring normoglycemia in spontaneously diabetic mice. Of note, the effect was evident independently of the prevalence of the disease-relevant antigen and without compromising systemic immunity (18). These promising results may be considered within the panorama of the continuously emerging role of extracellular vesicles (EV)s for the regulation of immune cell behavior. Indeed, EVs exert complex immunomodulatory effects on target cells, acting as both antigen-presenting modules and as shuttles for regulatory biological information (19). EVs can deliver molecules that exert opposite functions, serving as antigens of innate immune receptors or promoting pathogen immune evasion (165). Qualitative and quantitative alterations regarding EVs in settings of T2D are constantly emerging (166–168), as well as their central role in the regulation of immune function (20, 169, 170). Finally, a deeper understanding of this newly described extracellular vesicle-based cell-cell communication machinery will probably enable the design of specific nanodrugs able to produce a long-lasting beneficial effect on the pathological inflammatory component of T2D.
Autoreactivity ranges from a low and physiological level that is actually necessary for proper lymphocyte selection and immune system homeostasis, to an intermediate level that displays circulating antibody with no major tissue infiltrates, to frank pathogenic autoimmunity with manifested immune-mediated tissue damage (47).
The current classification of T1D, T2D, and LADA presents challenges to the diagnosis and treatment of diabetic patients, due to conflicting and confounding definitions, that still fail to incorporate advances in our understanding of these diseases. In 2001, TJ Wilkin formulated a hypothesis to respond to the increasingly fuzzy distinctions between T1 and T2 diabetes mellitus both clinically and aetiologically. His “accelerator hypothesis” identifies three processes which variably accelerate β cell loss: constitution, IR, and autoimmunity. Rather than overlap between two types of diabetes, the accelerator hypothesis envisages overlay: body mass is central to the development and rising incidence of all diabetes. Only tempo distinguishes the “types” (171). A clinical study, led by TJ Wilkin and aimed at testing this hypothesis was launched in Scotland in 2016 by the name of Accelerator Prevention Trial (adAPT). The main objective of the trial is to evaluate the impact of metformin on β cell stress, immune response and incidence of T1D in otherwise healthy children at high risk of T1D. The clinical outcome (incidence of diabetes) is expected in 2023 (172).
Accumulating evidence suggests that B and T cell alterations precede the loss of insulin sensitivity in adipose tissue and contribute to the general pro-inflammatory drift observed in T2D (73). Over-nutrition and advancing age are held to foster an intricate immune trafficking in different diabetes-relevant organs, including liver, muscle, and the adipose tissue (5, 73, 173, 174). Lacking wide-spread identification of specific autoreactive T cell populations and the wide array of antibodies associating to IR do not support the view of a prototypical autoimmune phenomenon for T2D. However, some features of the inflammatory trajectory accompanying all the stages of T2D are likely ascribable to low-grade, subclinical, autoimmune phenomena (68, 69, 72, 115).
Despite the increasing knowledge regarding the inflammatory component of T2D, the existing relevant gap of information has hampered the development of effective anti-inflammatory treatment to manage, prevent, or even revert T2D. Indeed, all the clinical approaches tested so far have tried to suppress the inflammatory response by antagonizing a candidate cytokine or pathway with a putative prominent role, rather than modulating specific cell types and responses. Recent literature is now adding new pieces to the complex crosstalk between cells of innate and acquired immunity, such as extracellular vesicles. Coupled with a continuously progressing technology, these studies may eventually lead to the design of innovative nanoscale drugs able to intercept or expand specific population with pro- or anti-inflammatory characteristics, respectively.
The relevance of the autoimmune phenomena possibly underpinning T2D could be masked by the heterogeneity of the disease. Indeed, many clinicians invoke the case for more personalized data analysis with the aim to move toward precision medicine, that would allow targeting the specific pathological mechanisms of hyperglycemia of the single patient (175). A recently proposed new sub-stratification of diabetes identified five replicable clusters of patients based on six variables (glutamate decarboxylase antibodies, age at diagnosis, BMI, HbA1c, β-cell function, and IR). A similar approach has been proposed to select candidate patients for anti-inflammatory treatments in T2D (140). These efforts are aimed at individualizing treatment regimens and identifying subjects with increased risk of complications at diagnosis, and thus represents a first step toward precision medicine (176).
It remains worth keeping to pursue a better definition of autoimmunity in T2D because a deeper knowledge of its immunogenic basis is not only necessary to better discriminate patient characteristics and select proper treatment approach, but, also importantly, to fully understand the very pathological mechanism of disease, thus possibly developing new strategies to counteract T2D development and progression.
No datasets were generated or analyzed for this study.
PdC, FP, and GM conceived the work and supervised the writing. SG, VD, MG, FD, MS, AlC, AnC, and AP wrote the different paragraphs. CF, TM, and SB conceived the art work.
This paper was supported by grants from the Juvenile Diabetes Research Foundation (JDRF n. 1-SRA-2018-477-S-B to PdC and n. 2-SRA-2018-479-S-B to MG), the National Multiple Sclerosis Society (NMSS PP-1804-30725 to MG and PP-1606-24687 to PdC), Fondazione Italiana Sclerosi Multipla (FISM n. 2016/R/10 to PdC; n. 2016/R/18 to GM; n. 2018/R/4 to VD), Università degli Studi di Napoli Federico II (STAR Program Linea 1 – 2018) and Ministero della Salute (n. GR-2016-02363725) to VD, European Research Council Grant (ERC menTORingTregs n. 310496 to GM), Telethon (n. GGP 17086 to GM). The work was also supported by the Italian Ministry of Health (Ricerca Corrente).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Sladek R. The many faces of diabetes: addressing heterogeneity of a complex disease. Lancet Diabetes Endocrinol. (2018) 6:348–9. doi: 10.1016/S2213-8587(18)30070-6
2. Rawshani A, Rawshani A, Franzen S, Eliasson B, Svensson AM, Miftaraj M, et al. Mortality and cardiovascular disease in type 1 and type 2 diabetes. N Engl J Med. (2017) 376:1407–18. doi: 10.1056/NEJMoa1608664
3. Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. (2014) 383:1068–83. doi: 10.1016/S0140-6736(13)62154-6
4. Twito O, Frankel M, Nabriski D. Impact of glucose level on morbidity and mortality in elderly with diabetes and pre-diabetes. World J Diabetes. (2015) 6:345–51. doi: 10.4239/wjd.v6.i2.345
5. Prattichizzo F, De Nigris V, Spiga R, Mancuso E, La Sala L, Antonicelli R, et al. Inflammageing and metaflammation: the yin and yang of type 2 diabetes. Ageing Res Rev. (2018) 41:1–17. doi: 10.1016/j.arr.2017.10.003
6. Prattichizzo F, De Nigris V, Micheloni S, La Sala L, Ceriello A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: is low-grade inflammation the neglected component? Diabetes Obes Metab. (2018) 20:2515–22. doi: 10.1111/dom.13488
7. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. doi: 10.1038/nature21363
8. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. (2018) 14:576–90. doi: 10.1038/s41574-018-0059-4
9. Prattichizzo F, De Nigris V, Mancuso E, Spiga R, Giuliani A, Matacchione G, et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. (2018) 15:170–81. doi: 10.1016/j.redox.2017.12.001
10. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
11. Bonfigli AR, Spazzafumo L, Prattichizzo F, Bonafe M, Mensa E, Micolucci L, et al. Leukocyte telomere length and mortality risk in patients with type 2 diabetes. Oncotarget. (2016) 7:50835–44. doi: 10.18632/oncotarget.10615
12. Gu Y, Hu K, Huang Y, Zhang Q, Liu L, Meng G, et al. White blood cells count as an indicator to identify whether obesity leads to increased risk of type 2 diabetes. Diabetes Res Clin Pract. (2018) 141:140–7. doi: 10.1016/j.diabres.2018.04.041
13. Wentworth JM, Fourlanos S, Harrison LC. Reappraising the stereotypes of diabetes in the modern diabetogenic environment. Nat Rev Endocrinol. (2009) 5:483–9. doi: 10.1038/nrendo.2009.149
14. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. (2011) 11:98–107. doi: 10.1038/nri2925
15. Kamradt T, Mitchison NA. Tolerance and autoimmunity. N Engl J Med. (2001) 344:655–64. doi: 10.1056/NEJM200103013440907
16. Tuomi T, Carlsson A, Li H, Isomaa B, Miettinen A, Nilsson A, et al. Clinical and genetic characteristics of type 2 diabetes with and without GAD antibodies. Diabetes. (1999) 48:150–7. doi: 10.2337/diabetes.48.1.150
17. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
18. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. (2016) 530:434–40. doi: 10.1038/nature16962
19. Prattichizzo F, Micolucci L, Cricca M, De Carolis S, Mensa E, Ceriello A, et al. Exosome-based immunomodulation during aging: a nano-perspective on inflamm-aging. Mech Ageing Dev. (2017) 168:44–53. doi: 10.1016/j.mad.2017.02.008
20. de Candia P, De Rosa V, Gigantino V, Botti G, Ceriello A, Matarese G. Immunometabolism of human autoimmune diseases: from metabolites to extracellular vesicles. FEBS Lett. (2017) 591:3119–34. doi: 10.1002/1873-3468.12733
21. Irvine WJ, McCallum CJ, Gray RS, Duncan LJ. Clinical and pathogenic significance of pancreatic-islet-cell antibodies in diabetics treated with oral hypoglycaemic agents. Lancet. (1977) 1:1025–7. doi: 10.1016/S0140-6736(77)91258-2
22. Turner R, Stratton I, Horton V, Manley S, Zimmet P, Mackay IR, et al. UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. UK Prospective Diabetes Study Group. Lancet. (1997) 350:1288–93. doi: 10.1016/S0140-6736(97)03062-6
23. Pietropaolo M, Barinas-Mitchell E, Pietropaolo SL, Kuller LH, Trucco M. Evidence of islet cell autoimmunity in elderly patients with type 2 diabetes. Diabetes. (2000) 49:32–8. doi: 10.2337/diabetes.49.1.32
24. Yohena S, Penas-Steinhardt A, Muller C, Faccinetti NI, Cerrone GE, Lovecchio S, et al. Immunological and clinical characteristics of latent autoimmune diabetes in the elderly. Diabetes Metab Res Rev. (2019) 11:e3137. doi: 10.1002/dmrr.3137
25. Hawa MI, Kolb H, Schloot N, Beyan H, Paschou SA, Buzzetti R, et al. Action, adult-onset autoimmune diabetes in Europe is prevalent with a broad clinical phenotype: action LADA 7. Diabetes Care. (2013) 36:908–13. doi: 10.2337/dc12-0931
26. Takeda H, Kawasaki E, Shimizu I, Konoue E, Fujiyama M, Murao S, et al. Clinical, autoimmune, and genetic characteristics of adult-onset diabetic patients with GAD autoantibodies in Japan (Ehime Study). Diabetes Care. (2002) 25:995–1001. doi: 10.2337/diacare.25.6.995
27. Radtke MA, Midthjell K, Nilsen TI, Grill V. Heterogeneity of patients with latent autoimmune diabetes in adults: linkage to autoimmunity is apparent only in those with perceived need for insulin treatment: results from the Nord-Trondelag Health (HUNT) study. Diabetes Care. (2009) 32:245–50. doi: 10.2337/dc08-1468
28. Subauste A, Gianani R, Chang AM, Plunkett C, Pietropaolo SL, Zhang YJ, et al. Islet autoimmunity identifies a unique pattern of impaired pancreatic beta-cell function, markedly reduced pancreatic beta cell mass and insulin resistance in clinically diagnosed type 2 diabetes. PLoS ONE. (2014) 9:e106537. doi: 10.1371/journal.pone.0106537
29. Hjort R, Alfredsson L, Carlsson PO, Groop L, Martinell M, Storm P, et al. Low birthweight is associated with an increased risk of LADA and type 2 diabetes: results from a Swedish case-control study. Diabetologia. (2015) 58:2525–32. doi: 10.1007/s00125-015-3711-8
30. Carlsson S. Environmental (lifestyle) risk factors for Lada. Curr Diabetes Rev. (2018) 15:178–87. doi: 10.2174/1573399814666180716150253
31. Hjort R, Ahlqvist E, Carlsson PO, Grill V, Groop L, Martinell M, et al. Overweight, obesity and the risk of LADA: results from a Swedish case-control study and the Norwegian HUNT Study. Diabetologia. (2018) 61:1333–43. doi: 10.1007/s00125-018-4596-0
32. Al-Majdoub M, Ali A, Storm P, Rosengren AH, Groop L, Spegel P. Metabolite profiling of LADA challenges the view of a metabolically distinct subtype. Diabetes. (2017) 66:806–14. doi: 10.2337/db16-0779
33. Wod M, Yderstraede KB, Halekoh U, Beck-Nielsen H, Hojlund K. Metabolic risk profiles in diabetes stratified according to age at onset, islet autoimmunity and fasting C-peptide. Diabetes Res Clin Pract. (2017) 134:62–71. doi: 10.1016/j.diabres.2017.09.014
34. Wod M, Thomsen RW, Pedersen L, Yderstraede KB, Beck-Nielsen H, Hojlund K. Lower mortality and cardiovascular event rates in patients with Latent Autoimmune Diabetes In Adults (LADA) as compared with type 2 diabetes and insulin deficient diabetes: a cohort study of 4368 patients. Diabetes Res Clin Pract. (2018) 139:107–13. doi: 10.1016/j.diabres.2018.02.042
35. Lu J, Hou X, Zhang L, Hu C, Zhou J, Pang C, et al. Associations between clinical characteristics and chronic complications in latent autoimmune diabetes in adults and type 2 diabetes. Diabetes Metab Res Rev. (2015) 31:411–20. doi: 10.1002/dmrr.2626
36. Hjort R, Alfredsson L, Andersson T, Carlsson PO, Grill V, Groop L, et al. Family history of type 1 and type 2 diabetes and risk of latent autoimmune diabetes in adults (LADA). Diabetes Metab. (2017) 43:536–42. doi: 10.1016/j.diabet.2017.05.010
37. Ramu D, Perumal V, Paul SFD. Association of common type 1 and type 2 diabetes gene variants with latent autoimmune diabetes in adults: a meta-analysis. J Diabetes. (2018) 11:484–96. doi: 10.1111/1753-0407.12879
38. Cousminer DL, Ahlqvist E, Mishra R, Andersen MK, Chesi A, Hawa MI, et al. First genome-wide association study of latent autoimmune diabetes in adults reveals novel insights linking immune and metabolic diabetes. Diabetes Care. (2018) 41:2396–403. doi: 10.2337/dc18-1032
39. Duran J, Obach M, Navarro-Sabate A, Manzano A, Gomez M, Rosa JL, et al. Pfkfb3 is transcriptionally upregulated in diabetic mouse liver through proliferative signals. FEBS J. (2009) 276:4555–68. doi: 10.1111/j.1742-4658.2009.07161.x
40. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. (2013) 210:2119–34. doi: 10.1084/jem.20130252
41. Sorgjerd EP, Asvold BO, Thorsby PM, Grill V. Individuals fulfilling criteria for type 2 diabetes rather than LADA display transient signs of autoimmunity preceding diagnosis with possible clinical implications: the HUNT study. Diabetes Care. (2018) 41:e161–3. doi: 10.2337/dc18-1069
42. Kolb H, Martin S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. (2017) 15:131. doi: 10.1186/s12916-017-0901-x
43. Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev. (2014) 13:981–1000. doi: 10.1016/j.autrev.2014.07.001
44. Ferrara CT, Geyer SM, Liu YF, Evans-Molina C, Libman IM, Besser R, et al. Excess BMI in childhood: a modifiable risk factor for type 1 diabetes development? Diabetes Care. (2017) 40:698–701. doi: 10.2337/dc16-2331
45. Mokry LE, Ross S, Timpson NJ, Sawcer S, Davey Smith G, Richards JB. Obesity and multiple sclerosis: a mendelian randomization study. PLoS Med. (2016) 13:e1002053. doi: 10.1371/journal.pmed.1002053
46. Sterry W, Strober BE, Menter A, International Psoriasis C. Obesity in psoriasis: the metabolic, clinical and therapeutic implications. Report of an interdisciplinary conference and review. Br J Dermatol. (2007) 157:649–55. doi: 10.1111/j.1365-2133.2007.08068.x
47. Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol. (2017) 18:716–24. doi: 10.1038/ni.3731
48. Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov. (2016) 15:639–60. doi: 10.1038/nrd.2016.75
49. Procaccini C, Carbone F, Galgani M, La Rocca C, De Rosa V, Cassano S, et al. Obesity and susceptibility to autoimmune diseases. Expert Rev Clin Immunol. (2011) 7:287–94. doi: 10.1586/eci.11.18
50. Mzimela NC, Ngubane PS, Khathi A. The changes in immune cell concentration during the progression of pre-diabetes to type 2 diabetes in a high-fat high-carbohydrate diet-induced pre-diabetic rat model. Autoimmunity. (2019) 52:27–36. doi: 10.1080/08916934.2019.1575820
51. De Rosa V, La Cava A, Matarese G. Metabolic pressure and the breach of immunological self-tolerance. Nat Immunol. (2017) 18:1190–6. doi: 10.1038/ni.3851
52. Matarese G, La Cava A. The intricate interface between immune system and metabolism. Trends Immunol. (2004) 25:193–200. doi: 10.1016/j.it.2004.02.009
53. Di Rosa F, Gebhardt T. Bone marrow T cells and the integrated functions of recirculating and tissue-resident memory T cells. Front Immunol. (2016) 7:51. doi: 10.3389/fimmu.2016.00051
54. Huh JY, Park YJ, Ham M, Kim JB. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells. (2014) 37:365–71. doi: 10.14348/molcells.2014.0074
55. Keiran N, Ceperuelo-Mallafre V, Calvo E, Hernandez-Alvarez MI, Ejarque M, Nunez-Roa C, et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat Immunol. (2019) 20:581–92. doi: 10.1038/s41590-019-0372-7
56. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. (1995) 269:540–3. doi: 10.1126/science.7624776
57. Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. (1998) 394:897–901. doi: 10.1038/29795
59. Hill JO. Understanding and addressing the epidemic of obesity: an energy balance perspective. Endocr Rev. (2006) 27:750–61. doi: 10.1210/er.2006-0032
60. De Rosa V, Procaccini C, Cali G, Pirozzi G, Fontana S, Zappacosta S, et al. A key role of leptin in the control of regulatory T cell proliferation. Immunity. (2007) 26:241–55. doi: 10.1016/j.immuni.2007.01.011
61. Hasenkrug KJ. The leptin connection: regulatory T cells and autoimmunity. Immunity. (2007) 26:143–5. doi: 10.1016/j.immuni.2007.02.002
62. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
63. Nikolajczyk BS, Jagannathan-Bogdan M, Shin H, Gyurko R. State of the union between metabolism and the immune system in type 2 diabetes. Genes Immun. (2011) 12:239–50. doi: 10.1038/gene.2011.14
64. Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nat Rev Immunol. (2011) 11:81. doi: 10.1038/nri2922
65. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112:1821–30. doi: 10.1172/JCI19451
66. Wensveen FM, Valentic S, Sestan M, Turk Wensveen T, Polic B. The “big bang” in obese fat: events initiating obesity-induced adipose tissue inflammation. Eur J Immunol. (2015) 45:2446–56. doi: 10.1002/eji.201545502
67. Sagaert X, De Wolf-Peeters C. Classification of B-cells according to their differentiation status, their micro-anatomical localisation and their developmental lineage. Immunol Lett. (2003) 90:179–86. doi: 10.1016/j.imlet.2003.09.007
68. Itariu BK, Stulnig TM. Autoimmune aspects of type 2 diabetes mellitus - a mini-review. Gerontology. (2014) 60:189–96. doi: 10.1159/000356747
69. Velloso LA, Eizirik DL, Cnop M. Type 2 diabetes mellitus–an autoimmune disease? Nat Rev Endocrinol. (2013) 9:750–5. doi: 10.1038/nrendo.2013.131
70. Tsai S, Clemente-Casares X, Revelo XS, Winer S, Winer DA. Are obesity-related insulin resistance and type 2 diabetes autoimmune diseases? Diabetes. (2015) 64:1886–97. doi: 10.2337/db14-1488
71. Haskell BD, Flurkey K, Duffy TM, Sargent EE, Leiter EH. The diabetes-prone NZO/HlLt strain. I. Immunophenotypic comparison to the related NZB/BlNJ and NZW/LacJ strains. Lab Invest. (2002) 82:833–42. doi: 10.1097/01.LAB.0000018915.53257.00
72. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. (2011) 17:610–7. doi: 10.1038/nm.2353
73. Duffaut C, Galitzky J, Lafontan M, Bouloumie A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun. (2009) 384:482–5. doi: 10.1016/j.bbrc.2009.05.002
74. Tanigaki K, Sacharidou A, Peng J, Chambliss KL, Yuhanna IS, Ghosh D, et al. Hyposialylated IgG activates endothelial IgG receptor FcgammaRIIB to promote obesity-induced insulin resistance. J Clin Invest. (2018) 128:309–22. doi: 10.1172/JCI89333
75. DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci USA. (2013) 110:5133–8. doi: 10.1073/pnas.1215840110
76. Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol. (2008) 20:149–57. doi: 10.1016/j.coi.2008.03.014
77. Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS, et al. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J Clin Invest. (2017) 127:1019–30. doi: 10.1172/JCI90350
78. Tung JW, Mrazek MD, Yang Y, Herzenberg LA, Herzenberg LA. Phenotypically distinct B cell development pathways map to the three B cell lineages in the mouse. Proc Natl Acad Sci USA. (2006) 103:6293–8. doi: 10.1073/pnas.0511305103
79. Shen L, Chng MH, Alonso MN, Yuan R, Winer DA, Engleman EG. B-1a lymphocytes attenuate insulin resistance. Diabetes. (2015) 64:593–603. doi: 10.2337/db14-0554
80. Harmon DB, Srikakulapu P, Kaplan JL, Oldham SN, McSkimming C, Garmey JC, et al. Protective role for B-1b B cells and IgM in obesity-associated inflammation, glucose intolerance, and insulin resistance. Arterioscler Thromb Vasc Biol. (2016) 36:682–91. doi: 10.1161/ATVBAHA.116.307166
81. Jennbacken K, Stahlman S, Grahnemo L, Wiklund O, Fogelstrand L. Glucose impairs B-1 cell function in diabetes. Clin Exp Immunol. (2013) 174:129–38. doi: 10.1111/cei.12148
82. Subramanian M, Ozcan L, Ghorpade DS, Ferrante AW Jr., Tabas I. Suppression of adaptive immune cell activation does not alter innate immune adipose inflammation or insulin resistance in obesity. PLoS ONE. (2015) 10:e0135842. doi: 10.1371/journal.pone.0135842
83. Hammad RH, El-Madbouly AA, Kotb HG, Zarad MS. Frequency of circulating B1a and B2 B-cell subsets in egyptian patients with type 2 diabetes mellitus. Egypt J Immunol. (2018) 25:71–80.
84. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. (2017) 8:1960. doi: 10.3389/fimmu.2017.01960
85. Fulop T, Witkowski JM, Olivieri F, Larbi A. The integration of inflammaging in age-related diseases. Semin Immunol. (2018) 40:17–35. doi: 10.1016/j.smim.2018.09.003
86. Zhai X, Qian G, Wang Y, Chen X, Lu J, Zhang Y, et al. Elevated B cell activation is associated with type 2 diabetes development in obese subjects. Cell Physiol Biochem. (2016) 38:1257–66. doi: 10.1159/000443073
87. van Beek L, Lips MA, Visser A, Pijl H, Ioan-Facsinay A, Toes R, et al. Increased systemic and adipose tissue inflammation differentiates obese women with T2DM from obese women with normal glucose tolerance. Metabolism. (2014) 63:492–501. doi: 10.1016/j.metabol.2013.12.002
88. Carey IM, Critchley JA, DeWilde S, Harris T, Hosking FJ, Cook DG. Risk of infection in type 1 and type 2 diabetes compared with the general population: a matched cohort study. Diabetes Care. (2018) 41:513–21. doi: 10.2337/dc17-2131
89. Abu-Ashour W, Twells LK, Valcour JE, Gamble JM. Diabetes and the occurrence of infection in primary care: a matched cohort study. BMC Infect Dis. (2018) 18:67. doi: 10.1186/s12879-018-2975-2
90. Frasca D, Diaz A, Romero M, Mendez NV, Landin AM, Ryan JG, et al. Young and elderly patients with type 2 diabetes have optimal B cell responses to the seasonal influenza vaccine. Vaccine. (2013) 31:3603–10. doi: 10.1016/j.vaccine.2013.05.003
91. Sheridan PA, Paich HA, Handy J, Karlsson EA, Schultz-Cherry S, Hudgens M, et al. The antibody response to influenza vaccination is not impaired in type 2 diabetics. Vaccine. (2015) 33:3306–13. doi: 10.1016/j.vaccine.2015.05.043
92. el-Madhun AS, Cox RJ, Seime A, Sovik O, Haaheim LR. Systemic and local immune responses after parenteral influenza vaccination in juvenile diabetic patients and healthy controls: results from a pilot study. Vaccine. (1998) 16:156–60. doi: 10.1016/S0264-410X(97)88328-4
93. American Diabetes Association. Standards of medical care in diabetes-2019 abridged for primary care providers. Clin Diabetes. (2019) 37:11–34. doi: 10.2337/cd18-0105
94. Dos Santos G, Tahrat H, Bekkat-Berkani R. Immunogenicity, safety, and effectiveness of seasonal influenza vaccination in patients with diabetes mellitus: a systematic review. Hum Vaccin Immunother. (2018) 14:1853–66. doi: 10.1080/21645515.2018.1446719
95. Papagianni M, Metallidis S, Tziomalos K. Herpes zoster and diabetes mellitus: a review. Diabetes Ther. (2018) 9:545–50. doi: 10.1007/s13300-018-0394-4
96. Mohan V, Unnikrishnan R, Thomas N, Bhansali A, Wangnoo SK, Thomas K. Pneumococcal infections and immunization in diabetic patients. J Postgrad Med. (2011) 57:78–81. doi: 10.4103/0022-3859.74299
97. Chang DC, Piaggi P, Hanson RL, Knowler WC, Bogardus C, Krakoff J. Autoantibodies against PFDN2 are associated with an increased risk of type 2 diabetes: a case-control study. Diabetes Metab Res Rev. (2017) 33:e2922. doi: 10.1002/dmrr.2922
98. Fiorentino TV, Succurro E, Arturi F, Giancotti A, Peronace C, Quirino A, et al. Serum IgG2 levels are specifically associated with whole-body insulin-mediated glucose disposal in non-diabetic offspring of type 2 diabetic individuals: a cross-sectional study. Sci Rep. (2018) 8:13616. doi: 10.1038/s41598-018-32108-8
99. Lindsay RS, Krakoff J, Hanson RL, Bennett PH, Knowler WC. Gamma globulin levels predict type 2 diabetes in the Pima Indian population. Diabetes. (2001) 50:1598–603. doi: 10.2337/diabetes.50.7.1598
100. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. (2013) 38:414–23. doi: 10.1016/j.immuni.2013.03.002
101. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
102. Yun WJ, Lee DW, Chang SE, Yoon GS, Huh JR, Won CH, et al. Role of CD4CD25FOXP3 regulatory T cells in psoriasis. Ann Dermatol. (2010) 22:397–403. doi: 10.5021/ad.2010.22.4.397
103. Ghoreschi K, Weigert C, Rocken M. Immunopathogenesis and role of T cells in psoriasis. Clin Dermatol. (2007) 25:574–80. doi: 10.1016/j.clindermatol.2007.08.012
104. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. (2014) 259:88–102. doi: 10.1111/imr.12160
105. Su LF, Del Alcazar D, Stelekati E, Wherry EJ, Davis MM. Antigen exposure shapes the ratio between antigen-specific Tregs and conventional T cells in human peripheral blood. Proc Natl Acad Sci USA. (2016) 113:E6192–8. doi: 10.1073/pnas.1611723113
106. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
107. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. (2009) 15:930–9. doi: 10.1038/nm.2002
108. Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev. (2012) 249:14–26. doi: 10.1111/j.1600-065X.2012.01155.x
109. Barbi J, Pardoll D, Pan F. Metabolic control of the Treg/Th17 axis. Immunol Rev. (2013) 252:52–77. doi: 10.1111/imr.12029
110. Matarese G, Colamatteo A, De Rosa V. Metabolic fuelling of proper T cell functions. Immunol Lett. (2014) 161:174–8. doi: 10.1016/j.imlet.2013.12.012
111. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. (2012) 486:549–53. doi: 10.1038/nature11132
112. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. (2015) 16:276–85. doi: 10.1038/ni.3085
113. Ilan Y, Maron R, Tukpah AM, Maioli TU, Murugaiyan G, Yang K, et al. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci USA. (2010) 107:9765–70. doi: 10.1073/pnas.0908771107
114. Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes. (2011) 60:2954–62. doi: 10.2337/db11-0358
115. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. (2009) 15:921–9. doi: 10.1038/nm.2001
116. Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS ONE. (2011) 6:e16376. doi: 10.1371/journal.pone.0016376
117. Zeyda M, Huber J, Prager G, Stulnig TM. Inflammation correlates with markers of T-cell subsets including regulatory T cells in adipose tissue from obese patients. Obesity. (2011) 19:743–8. doi: 10.1038/oby.2010.123
118. Wu D, Han JM, Yu X, Lam AJ, Hoeppli RE, Pesenacker AM, et al. Characterization of regulatory T cells in obese omental adipose tissue in humans. Eur J Immunol. (2019) 49:336–47. doi: 10.1002/eji.201847570
119. Ferraro A, D'Alise AM, Raj T, Asinovski N, Phillips R, Ergun A, et al. Interindividual variation in human T regulatory cells. Proc Natl Acad Sci USA. (2014) 111:E1111–20. doi: 10.1073/pnas.1401343111
120. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev. (2014) 13:668–77. doi: 10.1016/j.autrev.2013.12.004
121. Seyfert-Margolis V, Gisler TD, Asare AL, Wang RS, Dosch HM, Brooks-Worrell B, et al. Analysis of T-cell assays to measure autoimmune responses in subjects with type 1 diabetes: results of a blinded controlled study. Diabetes. (2006) 55:2588–94. doi: 10.2337/db05-1378
122. Brooks-Worrell BM, Reichow JL, Goel A, Ismail H, Palmer JP. Identification of autoantibody-negative autoimmune type 2 diabetic patients. Diabetes Care. (2011) 34:168–73. doi: 10.2337/dc10-0579
123. Brooks-Worrell B, Narla R, Palmer JP. Islet autoimmunity in phenotypic type 2 diabetes patients. Diabetes Obes Metab. (2013) 15(suppl. 3):137–40. doi: 10.1111/dom.12167
124. Goel A, Chiu H, Felton J, Palmer JP, Brooks-Worrell B. T-cell responses to islet antigens improves detection of autoimmune diabetes and identifies patients with more severe beta-cell lesions in phenotypic type 2 diabetes. Diabetes. (2007) 56:2110–5. doi: 10.2337/db06-0552
125. Palmer JP, Hampe CS, Chiu H, Goel A, Brooks-Worrell BM. Is latent autoimmune diabetes in adults distinct from type 1 diabetes or just type 1 diabetes at an older age? Diabetes. (2005) 54(suppl. 2):S62–7. doi: 10.2337/diabetes.54.suppl_2.S62
126. Winer S, Paltser G, Chan Y, Tsui H, Engleman E, Winer D, et al. Obesity predisposes to Th17 bias. Eur J Immunol. (2009) 39:2629–35. doi: 10.1002/eji.200838893
127. Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. (2011) 186:1162–72. doi: 10.4049/jimmunol.1002615
128. Zeng C, Shi X, Zhang B, Liu H, Zhang L, Ding W, et al. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: relationship with metabolic factors and complications. J Mol Med. (2012) 90:175–86. doi: 10.1007/s00109-011-0816-5
129. Poggi M, Jager J, Paulmyer-Lacroix O, Peiretti F, Gremeaux T, Verdier M, et al. The inflammatory receptor CD40 is expressed on human adipocytes: contribution to crosstalk between lymphocytes and adipocytes. Diabetologia. (2009) 52:1152–63. doi: 10.1007/s00125-009-1267-1
130. Caer C, Rouault C, Le Roy T, Poitou C, Aron-Wisnewsky J, Torcivia A, et al. Immune cell-derived cytokines contribute to obesity-related inflammation, fibrogenesis and metabolic deregulation in human adipose tissue. Sci Rep. (2017) 7:3000. doi: 10.1038/s41598-017-02660-w
131. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. (2011) 11:85–97. doi: 10.1038/nri2921
132. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta. (2014) 1843:2563–82. doi: 10.1016/j.bbamcr.2014.05.014
133. Maedler K, Spinas GA, Lehmann R, Sergeev P, Weber M, Fontana A, et al. Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes. (2001) 50:1683–90. doi: 10.2337/diabetes.50.8.1683
134. Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. (2002) 110:851–60. doi: 10.1172/JCI15318
135. Bagnati M, Ogunkolade BW, Marshall C, Tucci C, Hanna K, Jones TA, et al. Glucolipotoxicity initiates pancreatic beta-cell death through TNFR5/CD40-mediated STAT1 and NF-kappaB activation. Cell Death Dis. (2016) 7:e2329. doi: 10.1038/cddis.2016.203
136. McCulloch DK, Barmeier H, Neifing JL, Palmer JP. Metabolic state of the pancreas affects end-point titre in the islet cell antibody assay. Diabetologia. (1991) 34:622–5. doi: 10.1007/BF00400990
137. Hao W, Li L, Mehta V, Lernmark A, Palmer JP. Functional state of the beta cell affects expression of both forms of glutamic acid decarboxylase. Pancreas. (1994) 9:558–62. doi: 10.1097/00006676-199409000-00003
138. Brooks-Worrell B, Palmer JP. Immunology in the clinic review series; focus on metabolic diseases: development of islet autoimmune disease in type 2 diabetes patients: potential sequelae of chronic inflammation. Clin Exp Immunol. (2012) 167:40–6. doi: 10.1111/j.1365-2249.2011.04501.x
139. Pollack RM, Donath MY, LeRoith D, Leibowitz G. Anti-inflammatory agents in the treatment of diabetes and its vascular complications. Diabetes Care. (2016) 39(suppl. 2):S244–52. doi: 10.2337/dcS15-3015
140. Brooks-Worrell B, Narla R, Palmer JP. Biomarkers and immune-modulating therapies for type 2 diabetes. Trends Immunol. (2012) 33:546–53. doi: 10.1016/j.it.2012.07.002
141. Kothari V, Galdo JA, Mathews ST. Hypoglycemic agents and potential anti-inflammatory activity. J Inflamm Res. (2016) 9:27–38. doi: 10.2147/JIR.S86917
142. Bellanne-Chantelot C, Coste J, Ciangura C, Fonfrede M, Saint-Martin C, Bouche C, et al. Monogenic diabetes study group of the societe francophone du, high-sensitivity C-reactive protein does not improve the differential diagnosis of HNF1A-MODY and familial young-onset type 2 diabetes: A grey zone analysis. Diabetes Metab. (2016) 42:33–7. doi: 10.1016/j.diabet.2015.02.001
143. Prattichizzo F, Giuliani A, Mensa E, Sabbatinelli J, De Nigris V, Rippo MR, et al. Pleiotropic effects of metformin: shaping the microbiome to manage type 2 diabetes and postpone ageing. Ageing Res Rev. (2018) 48:87–98. doi: 10.1016/j.arr.2018.10.003
144. Hensen J, Howard CP, Walter V, Thuren T. Impact of interleukin-1beta antibody (canakinumab) on glycaemic indicators in patients with type 2 diabetes mellitus: results of secondary endpoints from a randomized, placebo-controlled trial. Diabetes Metab. (2013) 39:524–31. doi: 10.1016/j.diabet.2013.07.003
145. van Asseldonk EJ, Stienstra R, Koenen TB, Joosten LA, Netea MG, Tack CJ. Treatment with Anakinra improves disposition index but not insulin sensitivity in nondiabetic subjects with the metabolic syndrome: a randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab. (2011) 96:2119–26. doi: 10.1210/jc.2010-2992
146. Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care. (2009) 32:1663–8. doi: 10.2337/dc09-0533
147. Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, et al. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. (2012) 126:2739–48. doi: 10.1161/CIRCULATIONAHA.112.122556
148. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
149. Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. (2018) 391:319–28. doi: 10.1016/S0140-6736(17)32814-3
150. Burska AN, Sakthiswary R, Sattar N. Effects of tumour necrosis factor antagonists on insulin sensitivity/resistance in rheumatoid arthritis: a systematic review and meta-analysis. PLoS ONE. (2015) 10:e0128889. doi: 10.1371/journal.pone.0128889
151. Stagakis I, Bertsias G, Karvounaris S, Kavousanaki M, Virla D, Raptopoulou A, et al. Anti-tumor necrosis factor therapy improves insulin resistance, beta cell function and insulin signaling in active rheumatoid arthritis patients with high insulin resistance. Arthritis Res Ther. (2012) 14:R141. doi: 10.1186/ar3874
152. Otsuka Y, Kiyohara C, Kashiwado Y, Sawabe T, Nagano S, Kimoto Y, et al. Effects of tumor necrosis factor inhibitors and tocilizumab on the glycosylated hemoglobin levels in patients with rheumatoid arthritis; an observational study. PLoS ONE. (2018) 13:e0196368. doi: 10.1371/journal.pone.0196368
153. Wasko MC, Kay J, Hsia EC, Rahman MU. Diabetes mellitus and insulin resistance in patients with rheumatoid arthritis: risk reduction in a chronic inflammatory disease. Arthritis Care Res. (2011) 63:512–21. doi: 10.1002/acr.20414
154. Brezinski EA, Follansbee MR, Armstrong EJ, Armstrong AW. Endothelial dysfunction and the effects of TNF inhibitors on the endothelium in psoriasis and psoriatic arthritis: a systematic review. Curr Pharm Des. (2014) 20:513–28. doi: 10.2174/138161282004140213123852
155. Prattichizzo F, Giuliani A, Recchioni R, Bonafe M, Marcheselli F, De Carolis S, et al. Anti-TNF-alpha treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget. (2016) 7:11945–58. doi: 10.18632/oncotarget.7858
156. Bernstein LE, Berry J, Kim S, Canavan B, Grinspoon SK. Effects of etanercept in patients with the metabolic syndrome. Arch Intern Med. (2006) 166:902–8. doi: 10.1001/archinte.166.8.902
157. Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab. (2000) 85:1316–9. doi: 10.1210/jc.85.3.1316
158. Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, et al. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci. (2008) 1:36–43. doi: 10.1111/j.1752-8062.2008.00026.x
159. Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE, et al. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med. (2010) 152:346–57. doi: 10.7326/0003-4819-152-6-201003160-00004
160. Simpson SH, Gamble JM, Mereu L, Chambers T. Effect of aspirin dose on mortality and cardiovascular events in people with diabetes: a meta-analysis. J Gen Intern Med. (2011) 26:1336–44. doi: 10.1007/s11606-011-1757-y
161. Orlicka K, Barnes E, Culver EL. Prevention of infection caused by immunosuppressive drugs in gastroenterology. Ther Adv Chronic Dis. (2013) 4:167–85. doi: 10.1177/2040622313485275
162. Fishman JA. Opportunistic infections–coming to the limits of immunosuppression? Cold Spring Harb Perspect Med. (2013) 3:a015669. doi: 10.1101/cshperspect.a015669
163. Dror E, Dalmas E, Meier DT, Wueest S, Thevenet J, Thienel C, et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol. (2017) 18:283–92. doi: 10.1038/ni.3659
164. Boido A, Ceriani V, Cetta F, Lombardi F, Pontiroli AE. Bariatric surgery and prevention of cardiovascular events and mortality in morbid obesity: mechanisms of action and choice of surgery. Nutr Metab Cardiovasc Dis. (2015) 25:437–43. doi: 10.1016/j.numecd.2015.01.011
165. Schorey JS, Harding CV. Extracellular vesicles and infectious diseases: new complexity to an old story. J Clin Invest. (2016) 126:1181–9. doi: 10.1172/JCI81132
166. Castano C, Kalko S, Novials A, Parrizas M. Obesity-associated exosomal miRNAs modulate glucose and lipid metabolism in mice. Proc Natl Acad Sci USA. (2018) 115:12158–63. doi: 10.1073/pnas.1808855115
167. Prattichizzo F, Giuliani A, De Nigris V, Pujadas G, Ceka A, La Sala L, et al. Extracellular microRNAs and endothelial hyperglycaemic memory: a therapeutic opportunity? Diabetes Obes Metab. (2016) 18:855–67. doi: 10.1111/dom.12688
168. Freeman DW, Noren Hooten N, Eitan E, Green J, Mode NA, Bodogai M, et al. Altered extracellular vesicle concentration, cargo, and function in diabetes. Diabetes. (2018) 67:2377–88. doi: 10.2337/db17-1308
169. Torri A, Carpi D, Bulgheroni E, Crosti MC, Moro M, Gruarin P, et al. Extracellular MicroRNA signature of human helper T cell subsets in health and autoimmunity. J Biol Chem. (2017) 292:2903–15. doi: 10.1074/jbc.M116.769893
170. de Candia P, De Rosa V, Casiraghi M, Matarese G. Extracellular RNAs: a secret arm of immune system regulation. J Biol Chem. (2016) 291:7221–8. doi: 10.1074/jbc.R115.708842
171. Wilkin TJ. The accelerator hypothesis: weight gain as the missing link between Type I and Type II diabetes. Diabetologia. (2001) 44:914–22. doi: 10.1007/s001250100548
172. Wilkin T, Greene S, McCrimmon R. Testing the accelerator hypothesis: a new approach to type 1 diabetes prevention (adAPT 1). Diabetes Obes Metab. (2016) 18:3–5. doi: 10.1111/dom.12599
173. Yi HS, Kim SY, Kim JT, Lee YS, Moon JS, Kim M, et al. T-cell senescence contributes to abnormal glucose homeostasis in humans and mice. Cell Death Dis. (2019) 10:249. doi: 10.1038/s41419-019-1494-4
174. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest. (2017) 127:43–54. doi: 10.1172/JCI88880
175. Schwartz SS, Epstein S, Corkey BE, Grant SF, Gavin JR III, Aguilar RB. The time is right for a new classification system for diabetes: rationale and implications of the beta-cell-centric classification schema. Diabetes Care. (2016) 39:179–86. doi: 10.2337/dc15-1585
Keywords: diabetes, autoimmunity, immunometabolism, inflammation, T cells
Citation: de Candia P, Prattichizzo F, Garavelli S, De Rosa V, Galgani M, Di Rella F, Spagnuolo MI, Colamatteo A, Fusco C, Micillo T, Bruzzaniti S, Ceriello A, Puca AA and Matarese G (2019) Type 2 Diabetes: How Much of an Autoimmune Disease? Front. Endocrinol. 10:451. doi: 10.3389/fendo.2019.00451
Received: 12 April 2019; Accepted: 21 June 2019;
Published: 04 July 2019.
Edited by:
Oreste Gualillo, Servicio Gallego de Salud, SpainReviewed by:
Morena Scotece, Instituto de Investigación Biomédica de A Coruña (INIBIC), SpainCopyright © 2019 de Candia, Prattichizzo, Garavelli, De Rosa, Galgani, Di Rella, Spagnuolo, Colamatteo, Fusco, Micillo, Bruzzaniti, Ceriello, Puca and Matarese. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola de Candia, cGFvbGEuZGVjYW5kaWFAbXVsdGltZWRpY2EuaXQ=; Giuseppe Matarese, Z2l1c2VwcGUubWF0YXJlc2VAdW5pbmEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.