
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 12 June 2019
Sec. Cancer Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00365
This article is part of the Research Topic Testicular Cancer: New Insights on the Origin, Genetics, Treatment, Fertility, General Health, Quality of Life and Sexual Function View all 18 articles
 Sabrina Chiloiro1†
Sabrina Chiloiro1† Ettore Domenico Capoluongo1†
Ettore Domenico Capoluongo1† Giovanni Schinzari2Paola Concolino3Ernesto Rossi2Maurizio Martini4Alessandra Cocomazzi4
Giovanni Schinzari2Paola Concolino3Ernesto Rossi2Maurizio Martini4Alessandra Cocomazzi4 Giuseppe Grande1
Giuseppe Grande1 Domenico Milardi1
Domenico Milardi1 Brigida Anna Maiorano2Antonella Giampietro1Guido Rindi4
Brigida Anna Maiorano2Antonella Giampietro1Guido Rindi4 Alfredo Pontecorvi1
Alfredo Pontecorvi1 Laura De Marinis1*Antonio Bianchi1
Laura De Marinis1*Antonio Bianchi1Introduction: Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominantly inherited endocrine tumor syndrome characterized by the development of cancer in various endocrine organs, particularly in the pituitary, parathyroid and pancreas. Moreover, in some cases, also non-endocrine tumors can be diagnosed, developing atypical phenotypes.
Case report: We report herein the clinical history of a patient affected by MEN-1 syndrome who developed atypical features for this disease. The patient's clinical history started in August 2015 when he was referred, at the age of 23 years, to the Emergency Department of our Hospital for the occurrence of progressive asthenia, weakness, tremors and syncope. The biochemical test documented hyper-calcemia and severe hypoglycemia. The patient was referred to our Neuroendocrine Tumor and Pituitary Unit and he was diagnosed with pancreatic insulinoma, hypercalcemic hyperparathyroidism, and a prolactin secreting pituitary adenoma. The MEN-1 syndrome was suspected and genetic tests for mutation of menin resulted positive for the pathogenic variant c1548dupG. In January 2016, the patient was diagnosed with intratubular germ cell neoplasia, consisting of a mature teratoma and yolk sac tumor and he underwent a right orchiectomy.
Conclusion: This is the first case report showing the clear association of MEN-1 syndrome with yolk sac tumors and teratomas, as in our case, the c1548dupG represents a pathogenic variant rather than a SNP. This case suggests the opportunity of an accurate evaluation of the testis particularly in young MEN-1 affected patients and that a prompt screening for neoplastic disease should involve all the endocrine glands.
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominantly inherited endocrine tumor syndrome characterized by tumor development in various endocrine organs (1, 2). MEN syndromes are infrequent inherited disorders in which more than one endocrine gland develops noncancerous (benign) or cancerous (malignant) tumors or grows excessively without forming tumors. MEN1 disease is a consequence of the MEN1 gene mutation (3–5). The MEN1 gene synthetizes the protein menin, that acts as a tumor suppressor, as confirmed by microsatellite analysis conducted on cancerous tissues of MEN1 patients (6, 7). The protein menin inhibits the cell proliferation through the interaction with histone-modifying enzymes, with transforming growth factor β1 (TGF-β) signaling and Wnt/β-catenin pathways and with several transcription factors (such as nuclear factor κB (NF-κB), peroxisome proliferator-activated receptors (PPARγ), and vitamin D receptor (VDR) (8). Moreover, menin can act by destroying pro-proliferative factors such as insulin-like growth factors I and II (IGF-I and IGF-II) and parathyroid hormone-related protein (PTHrP) (8). MEN-1 syndrome can present as a familial form (more common) or sporadic form. Specific gene mutations can be identified in 70–95% of cases (3–9). The most commonly diagnosed tumors in MEN-1 syndrome involve the parathyroid glands in around 95% of cases, endocrine pancreatic-gastroenteric tract in around 40% of cases and the anterior pituitary gland, in around 30% of cases (10, 11). The first presentation of MEN1, in up to 85% of patients, is a parathyroid tumor; in other cases, the first manifestation may be prolactinoma or an insulinoma (12). Other tumors can occur in MEN-1 syndrome such as adrenocortical and thyroid tumors, meningiomas, angiofibromas, collagenomas, lipomas and gastric, thymic, and bronchial carcinoids (13–19). Notably, MEN-1 syndrome can show a very variable phenotype (9). We report herein the clinical history of a patient affected by MEN-1 syndrome who developed atypical features for this disease. This feature is peculiar as it has never been described in literature. A written informed consent was obtained from the patient for the publication of this case report and any potentially-identifying images/information.
The patient's clinical history started at the age of 15 years, when he was diagnosed for minor epilepsy. The patient's actual clinical history started in August 2015 when he was referred, at the age of 23 years, to the Emergency Department of our Hospital for the occurrence of progressive asthenia, weakness, tremors and syncope. The biochemical test documented hypercalcemia and severe hypoglycemia. The glycemic value was 27 mg/dL. The patient was treated with a glucose infusion with symptoms reduction. In September 2015, the patient was admitted to our Neuroendocrine Tumor and Pituitary Unit, to perform a 72 h fasting test for a possible insulinoma. After 7 h fasting, the patient was symptomatic for hypoglycemia. The glycemic plasma value resulted as 20 mg/dL, insulin as 18.6 microIU/mL, C-peptide as 1.7 ng/mL. Again symptoms diminished following the glucose infusion. Additionally, blood tests documented a primary hyperparathyroidism with hypercalcemia (Calcium: 11.7 mg/dL, PTH: 134.5 pg/mL) and hyperprolactinemia (PRL: 220 ng/mL).
The abdominal contrast computerized tomography (CT) documented the presence of four hyper-vascular focal lesions, of <1 centimeter and localized at the pancreatic body and tail, which were suggestive for neuroendocrine tumors (NET) (Figure 1). A Gallium-68 labeled somatostatin receptor PET-CT an showed uptake in 3 nodules in the pancreas (Figure 2). Cytological findings of the endoscopic ultrasound-guided fine needle aspiration of the larger pancreatic tumor was consistent with a G2 neuroendocrine tumor, with positive immunohistochemistry for chromogranin A, synaptophysin, CDX2 and a Ki67 proliferation index of 4%. Based on the patient's clinical history, immunohistochemistry was performed for insulin and resulted positive in tumor cells. The patient underwent a thyroid and parathyroid ultrasound which resulted negative for both thyroid nodules and hyperplastic parathyroid. The parathyroid scintigraphy however showed two hyper-functioning parathyroid glands. A pituitary contrasted magnetic resonance evidenced the presence of a small pituitary adenoma with a maximum size of 8 millimeters. Consequently, the patient initiated a prophylactic treatment with diazoxide (at the starting dosage of 25 mg/daily with a subsequent dose titration up to 75 mg/daily) to prevent a potential hypoglycemia crisis, with long acting somatostatin analogs (SSA: Lanreotide Autogel 120 mg/monthly) for the pancreatic NET and with a dopamine agonist (cabergoline 0.5 mg half table twice a week) for the micro-prolactinoma.
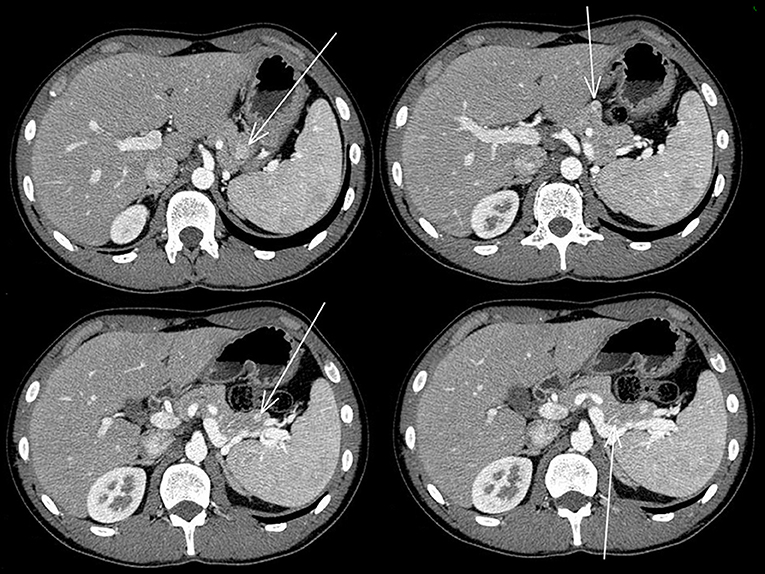
Figure 1. Abdominal contrasted TC scan showed the four pancreatic neuroendocrine tumors that are indicated with the arrows.
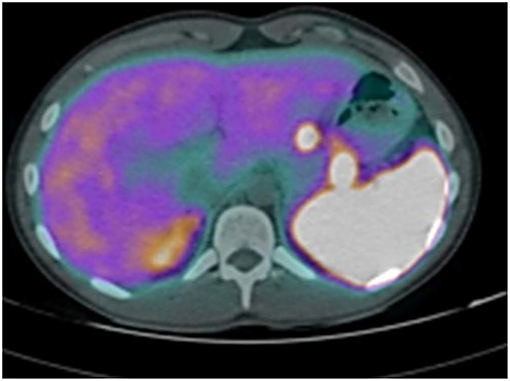
Figure 2. Gallium-68 labeled somatostatin receptor PET-CT showed uptake in 3 Gallium-68 labeled somatostatin receptor PET-CT showed an uptake in 3 nodules in the pancreas.
In the family history, the patient's sister underwent successful neurosurgery to remove a pituitary prolactinoma at the age of 18 years.
According to the patient's clinical assessment and family history, a MEN-1 syndrome was suspected. Genetic testing for the mutation of menin resulted positive for the pathogenic variant c1548dupG, in heterozygosis.
All of the patients' first-degree relatives were tested for MEN-1 syndrome, after signing informed consents. Figure 3 shows the index pedigree.
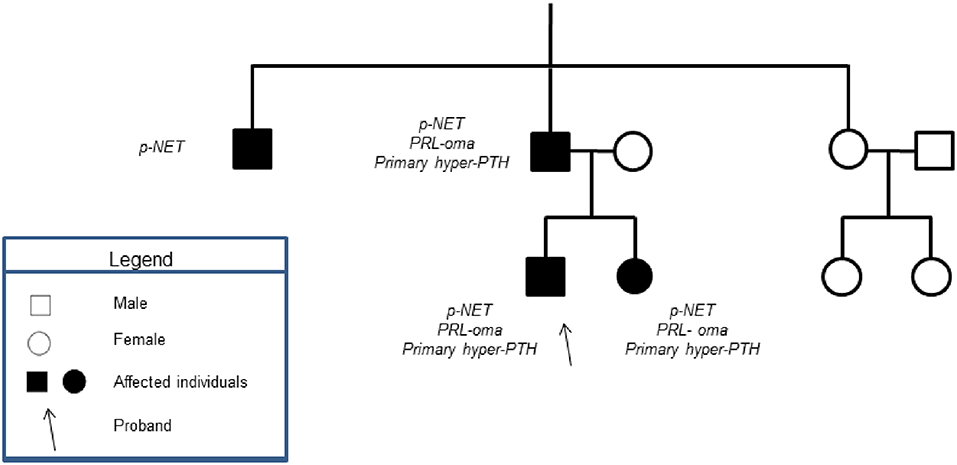
Figure 3. Showed the patient's family tree. None of the patient's male relatives had history of testicular mass.
According to the multidisciplinary decision of the Neuroendocrine tumor (NET) board, the patient first received a total parathyroidectomy before the scheduled sub-total pancreatectomy treatment.
In November 2015, a total parathyroidectomy and a thymectomy were performed. The dosage of intra-operatory serum PTH showed a progressive reduction, from the initial value of 191.3 pg/mL to the final value of 20 pg/mL. The patient was treated with calcitriol without any occurrence of hypocalcemia. The histological examination documented a diffuse hyperplasia of all four removed parathyroid glands in the absence of thymic neoplasia/hyperplasia and only initial adipose thymic involution. One month after surgery, serum PTH concentration was of 5 pg/mL (range 14–72).
In January 2016, the patient was referred with right testicular swelling. The alpha-fetoprotein serum level was 43 ng/mL (<9). A testis ultrasound documented a hypoechoic and hyper-vascularized nodule. The patient underwent a testicular nodule resection. The histological examination showed an intratubular germ cell neoplasia (IGCNU), consisting of a mature teratoma and yolk sac tumor, with signs of pre-invasive lesion, such as presence of peri-neoplastic, placental alkaline phosphatase (PLAP) and CD117 positive seminiferous tubules, with basal nuclei and abundant clear cytoplasm (Figure 4). According to the presence of germ cell neoplasia, in March 2016, a right orchiectomy was conducted, following germ cell cryopreservation. No neoplastic cells were detected at the histological examination of the resected testis. The post-surgery total body CT and 18F-FDG PET/CT were negative for metastasis 6 months later.
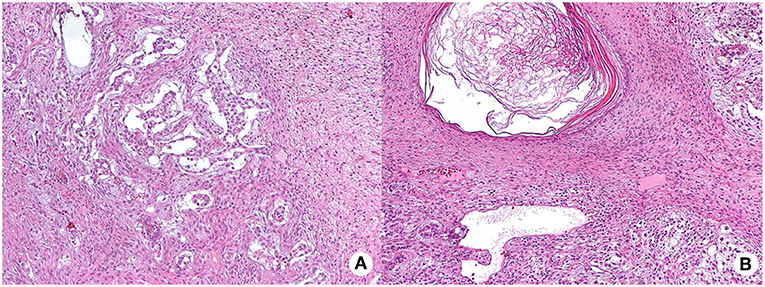
Figure 4. Hematoxylin and eosin (HE) staining of testis intra-tubular germ cell neoplasia (200X magnification) composed by the yolk sac tumor, with mainly anastomosing channels that focally expand to form variably sized cysts lined by primitive tumor cells with varying amounts of clear, glycogenated cytoplasm (mycrocistic or reticular pattern, panel A) and of the mature teratoma, with different type of mature tissue such as squamous epithelium with keratinization (panel B shows a dermoid cyst).
In February 2017, the patient underwent a sub-total pancreatectomy. Treatment with diazoxide was withdrawn.
The histological examination proved the presence of four pancreatic NETs: two of the pancreas body and the other two at the pancreas tail. All the lesions showed a positive immunohistochemistry for chromogranin A and synaptophysin and one only was positive for insulin. The higher mitotic index was 7 per 10 high-power fields with the Ki67 proliferation index ranging from 4 to 8%. These lesions were diagnosed as three non-functioning G2 NET and one insulinoma G2 NET. No other histological alterations were identified in the endocrine and in the exocrine residual pancreas.
At present the patient is in good clinical condition, in the absence of disease recurrence or adverse event such as diabetes mellitus or episodes of hypocalcemia or hypoglycemia. He is still on treatment with SSA and DA, with normal prolactin values. Hormonal replacement therapy with testosterone analogs was not prescribed, given the absence of referred symptoms and according to the laboratory assessment.
Blood test, abdominal CT, Gallium-68 labeled somatostatin receptor PET-CT and pituitary MR are periodically scheduled at our Neuroendocrine Tumor and Pituitary Unit.
To our knowledge, this is the first MEN1 patient who also developed an intra-tubular germ cell neoplasia of the testis. It is well-known that endocrine glands are very sensitive to the development of noncancerous or cancerous lesions. In a recent study by Wautot et al. (20) the menin expression (detected as a 68 KDalton protein) was demonstrated in the brain cortex, the kidney, the pituitary, the testes, the thymus and in the thyroid, providing a rationale for the high risk of neoplasia development at these sites in MEN1 patients.
We can speculate that the germline c1548dupG pathogenic variant could also play a role in the onset of this peculiar tumor of the testis. In this regard, we also tried to evaluate the status of MEN1 copy number (CNV), however and unfortunately, we could not assess CNV since our method is set for germline blood-derived fresh DNA rather than on somatic formalin-fixed paraffin-embedded (FFPE) DNA (data not shown). As reported, MEN1 gene encodes for menin that act as tumor suppressor, as confirmed by microsatellite analysis conducted on cancerous tissues of MEN1 patients. Therefore, although in the absence of a direct evidence, we cannot exclude the relationship between MEN1-mutations and testis tumor development. In addition, the c.1548dupG (p.Lys517Glufs; rs761695866) is very well-established as a pathogenic variant and reported as very rare within the population (Varsome Database). The absence of MEN1 mutations reported for yolk sac and mature teratoma testis tumor (the tumor described in this report), within the ATLAS genome and COSMIC databases may well be due to the rarity of this tumor histotype. The testis tumor affecting our patient was classified as a non-germinomatous germ cell tumor (NGGCTs) (21) or as a type II testicular germ cell tumor (22). This group of testis neoplasia typically occurs in the third and fourth decade of life and includes seminoma, embryonal carcinoma, teratoma, yolk sac tumor, choriocarcinoma, and mixed germ cell tumors (22). All type II testicular germ cell tumors develop from a pre-invasive lesion called intratubular germ cell neoplasia unclassified (IGCNU), defined as malignant germ cells confined to the seminiferous tubules, which usually lack normal spermatogenesis (22).
Similarly, teratomas derive from pluripotent cells (23) and can be differentiated in mature and immature, according to the differentiation grade of tissue within the tumors. Fully differentiated neuroectodermal, mesodermal and endodermal elements are detected in mature teratomas. Instead, embryonic elements deriving from any or all of the three germinal cell layers are typically detected in immature teratomas (23). Teratomas are commonly located in gonads, anterior mediastinum, retroperitoneum, and sacrococcygeal region but can also involve atypical organs, such as the pituitary gland (24).
Similar to other neoplasia in the testis, several factors were suggested as being involved into the onco-genesis, such as genetic disorders and a history of cryptorchidism or testis dysgenesis. Genetic studies have suggested an association between testis oncogenesis and mutations of several genes. In particular, since 2009 there are new genetic insights starting from two testicular germ cell tumors (TGCT)- genome wide association studies (GWAS), followed by several additional TGCT-GWAS (25). In these studies some SNPs with significant associations were identified in or near the genes KITLG (KIT ligand), SPRY4 (sprouty 4: sprout-related, EVH1 domain containing 2), BAK1 (BCL2-antagonist/killer 1), DMRT1 (doublesex and mab-3-related transcription factor 1), TERT (telomerase reverse transcriptase), ATF7IP (activating transcription factor 7 interacting protein), HPGDS (hematopoietic prostaglandin D synthase), MAD1L1 (mitotic arrest deficient-like 1), RFWD3 (ring finger WD domain 3), TEX14 (testis expressed 14), and PPM1E (protein phosphatase, Mg2+/Mn2+ dependent, 1E) (25).
We underline the fact that the Elzinga-Tinke et al. review paper does not associate MEN1 gene pathogenic variants to the etiopathogenesis of TGCT, so this is the first case report showing a clear association with yolk sac tumors and with teratomas. Furthermore, all the above mentioned GWAS studies identified only SNPs within the called genes. In our case, however, the c1548dupG represents a pathogenic variant rather than a SNP variant. This data can further support the association between our peculiar phenotype with the genotype.
This case report confirms that the early diagnosis of MEN-1 syndrome, along with appropriate screening and prompt therapeutic management of MEN-1 related neoplasia, can improve prognosis, particularly in cases of pancreatic NET, as shown in our previous experience (26, 27). In fact, in most cases, MEN-1 related tumors are diagnosed for local mass effects or for symptoms due to the overproduction of hormones (12). Although MEN-1 related tumors are usually benign, an aggressive behavior, with high risk of malignancy, as for carcinoid tumors and gastrinomas can occur (28). Consequently, in individuals with two or more MEN1-related tumors and in first-degree family members, a genetic test for MEN-1 syndrome should be offered (29). In addition, patients with the genetic diagnosis of MEN-1 syndrome should also be offered an appropriate screening and follow-up for all MEN-1 related tumors.
According to our experience and clinical practice, the integration of diagnostic modalities can improve the sensibility of each diagnostic test allowing an earlier and effective diagnosis. In particular, in our case the integration of neck ultrasound and parathyroid scintigraphy allowed the diagnosis of primary hyperparathyroidism. The sensitivity of ultrasonography is 76–87% with a positive predictive value of 93–97% and a diagnostic accuracy of 88% (30). By converse, 99 mTc-sestamibi scintigraphy has a higher sensitivity (90%) and accuracy (97.2%) then ultrasound (30). However, the concordance between scintigraphy and ultrasound is nor reached in all cases (30). On the same line CDX2 immunohistochemistry was conducted on the diagnostic cytological specimens to confirm the digestive source (and namely pancreatic) origin of tumor cells. CDX2 protein expression was reported positive in a percentage of pancreatic neuroendocrine tumors (31, 32). Similarly, PDX1, a transcription factor, was identified in metastatic NET of gastro-intestinal and pancreatic origin (31, 32).
This case suggests the opportunity of an accurate evaluation of testis particularly in affected young MEN-1 patients. Others neoplasia such as thyroid and breast tumors can be detected in patients affected by MEN1 syndrome and required an appropriate screening (33). However, as these neoplasms are common also in the general population and since the role of MEN1 gene in the thyroid and breast cancers is uncertain, the association of thyroid and breast tumors and MEN1 is considered incidental (33).
In conclusion, this unique case report suggests that a prompt screening for neoplastic disease should involve all the endocrine glands (not only pituitary, parathyroids, and pancreas), in patients diagnosed for MEN-1 syndrome, in order to have the opportunity and the benefit of an early diagnosis of neoplasia.
No datasets were generated or analyzed for this study.
This study represents a case report. All the procedures in this case were conducted according to guidelines and according to clinical practice. All procedures performed in studies involving human participants were in accordance with the ethical standards of the Ethics committee of Fondazione Policlinico Gemelli, Rome and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. A written informed consent was obtained from the patient for the publication of this case report and any potentially-identifying images/information.
SC and EC wrote the manuscript. EC, PC, and AC conducted the genetic analysis. GS, ER, and BM conducted the oncological management and follow-up. MM conducted the pathological examination of testis tumor. GR conducted the pathological examination of the neuroendocrine tumors. GG and DM conducted the clinical diagnosis of testis tumor. LD, AB, SC, AG, and AP conducted the endocrinological diagnosis and follow-up. All the authors reviewed and approved the manuscript version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med. (1954) 16:363–71. doi: 10.1016/0002-9343(54)90353-8
2. Wermer P. Endocrine adenomatosis, peptic ulcer disease in a large kindred: inherited multiple tumors, mosaic pleiotropism in man. Am J Med. (1963) 35:205–8. doi: 10.1016/0002-9343(63)90212-2
3. Concolino P, Costella A, Capoluongo E. Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet. (2016) 209:6–41. doi: 10.1016/j.cancergen.2015.12.002
4. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. (1997) 276:404–7. doi: 10.1126/science.276.5311.404
5. Byström C, Larsson C, Blomberg C, Sandelin K, Falkmer U, Skogseid B, et al. Localization of the MEN1 gene to a small region within chromosome 11q13 by deletion mapping in tumors. Proc Natl Acad Sci USA. (1990) 87:1968–72. doi: 10.1073/pnas.87.5.1968
6. Farnebo F, Teh BT, Kytölä S, Svensson A, Phelan C, Sandelin K., et al. Alterations of the MEN1 gene in sporadic parathyroid tumors. J Clin Endocrinol Metab. (1998) 83:2627–30. doi: 10.1210/jc.83.8.2627
7. Larsson C, Skogseid B, Öberg K, Nakamura Y, Nordenskjöld M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature. (1988) 332:85–7. doi: 10.1038/332085a0
8. Khatami F, Tavangar SM. Multiple endocrine neoplasia syndromes from genetic and epigenetic perspectives. Biomark Insights. (2018) 13:1–9. doi: 10.1177/1177271918785129
9. Marini F, Falchetti A, Luzi E, Tonelli F, Maria Luisa B. Multiple endocrine neoplasia type 1 (MEN1) syndrome. In: Riegert-Johnson DL, Boardman LA, Hefferon T, Roberts M, editors. Cancer Syndromes. Bethesda, MD: National Center for Biotechnology Information (US) (2008).
10. Grajo JR, Paspulati RM, Sahani DV, Kambadakone A. Multiple endocrine neoplasia syndromes: a comprehensive imaging review. Radiol Clin North Am. (2016) 54:441–51. doi: 10.1016/j.rcl.2015.12.001
11. Walls GV. Multiple endocrine neoplasia (MEN) syndromes. Semin Pediatric Surg. (2014) 23:96–10. doi: 10.1053/j.sempedsurg.2014.03.008
12. Syro LV, Scheithauer BW, Kovacs K, Toledo RA, London FJ, Ortiz LD, et al. Uribe Pituitary tumors in patients with MEN1 syndrome. Clinics. (2012) 67:43–8. doi: 10.6061/clinics/2012(Sup01)09
13. Burgess JR, Harle RA, Tucker P, Parameswaran V, Davies P, Greenaway TM, et al. Adrenal lesions in a large kindred with multiple endocrine neoplasia type 1. Arch Surg. (1996) 131:699–702. doi: 10.1001/archsurg.1996.01430190021006
14. Skogseid B, Larsson C, Lindgren PG, Kvanta E, Rastad J, Eodorsson E, et al. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. (1992) 75:76–81. doi: 10.1210/jcem.75.1.1352309
15. Debelenko LV, Brambilla E, Agarwal SK, Swalwell JI, Kester MB, Lubensky IA, et al. Identification of MEN1 gene mutations in sporadic carcinoid tumors of the lung. Hum Mol Genet. (1997) 6:2285–90.
16. Debelenko LV, Emmert-Buck MR, Zhuang Z, Epshteyn E, Moskaluk CA, Jensen RT, et al. The multiple endocrine neoplasia type I gene locus is involved in the pathogenesis of type II gastric carcinoids. Gastroenterology. (1997) 113:773–81. doi: 10.1016/S0016-5085(97)70171-9
17. Teh BT. Thymic carcinoids in multiple endocrine neoplasia type 1. J Intern Med. (1998) 243:501–4. doi: 10.1046/j.1365-2796.1998.00329.x
18. Darling TN, Skarulis MC, Steinberg SM, Marx SJ, Spiegel AM, Turner M. Multiple facial angiobromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. (1997) 133:853–7. doi: 10.1001/archderm.133.7.853
19. Pack S, Turner ML, Zhuang Z, Vortmeyer AO, Boni R, Skarulis M, et al. Cutaneous tumors in patients with multiple endocrine neoplasia type 1 show allelic deletion of the MEN1 gene. J Invest Dermatol. (1998) 110:438–40. doi: 10.1046/j.1523-1747.1998.00140.x
20. Wautot V, Khodaei S, Frappart L, Buisson N, Baro E, Lenoir GM, et al. Expression analysis of endogenous menin, the product of the multiple endocrine neoplasia type 1 gene, in cell lines and human tissues. Int J Cancer. (2000) 85:877. doi: 10.1002/(SICI)1097-0215(20000315)85:6<877::AID-IJC23>3.0.CO;2-F
21. Yagi K, Kageji T, Nagahiro S, Horiguchi H. Growing teratoma syndrome in a patient with a non-germ germinomatous germ cell tumor in the neurohypophysis. Neurol Med. Chir. (2004) 44:33–7. doi: 10.2176/nmc.44.33
22. Al-Hussain T, Bakshi N, Akhtar M. Intratubular germ cell neoplasia of the testis: a brief review. Adv Anat Pathol. (2015) 22:3. doi: 10.1097/PAP.0000000000000066
23. Mazumdar D, Goel A, Desai K, Shenoy A. Mature teratoma arising from the sella. Neurol MedChir. (2001) 41:356–9. doi: 10.2176/nmc.41.356
24. Chiloiro S, Giampietro A, Bianchi A, De Marinis L. Clinical management of teratoma, a rare hypothalamic-pituitary neoplasia. Endocrine. (2016) 53:636–42. doi: 10.1007/s12020-015-0814-4
25. Elzinga-Tinke JE, Dohle GR, Looijenga LH. Etiology and early pathogenesis of malignant testicular germ cell tumors: towards possibilities for preinvasive diagnosis. Asian J Androl. (2015) 17:381–93. doi: 10.4103/1008-682X.148079
26. Palermo A, Capoluongo E, Del Toro R, Manfrini S, Pozzilli P, Maggi D, et al. A novel germline mutation at exon 10 of MEN1 gene: a clinical survey and positive genotype-phenotype analysis of a MEN1 Italian family, including monozygotic twins. Hormones. (2018) 17:427–35. doi: 10.1007/s42000-018-0044-2
27. Chiloiro S, Lanza F, Bianchi A, Schinzari G, Brizi MG, Giampietro A, et al. Pancreatic neuroendocrine tumors in MEN1 disease: a mono-centric longitudinal and prognostic study. Endocrine. (2018) 60:362–7. doi: 10.1007/s12020-017-1327-0
28. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab. (2010) 24:355–70. doi: 10.1016/j.beem.2010.07.003
29. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PE, Melmed S, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. (2012) 97:2990−3011. doi: 10.1210/jc.2012-1230
30. Kunstman JW, Kirsch JD, Mahajan A, Udelsman R. Clinical review: parathyroid localization and implications for clinical management. J Clin Endocrinol Metab. (2013) 98:902–12. doi: 10.1210/jc.2012-3168
31. Hermann G, Konukiewitz B, Schmitt A, Perren A, Klöppel G. Hormonally defined pancreatic and duodenal neuroendocrine tumors differ in their transcription factor signatures: expression of ISL1, PDX1, NGN3, and CDX2. Virchows Arch. (2011) 459:147–54. doi: 10.1007/s00428-011-1118-6
32. Yang Z, Klimstra DS, Hruban RH, Tang LH. Immunohistochemical characterization of the origins of metastatic well-differentiated neuroendocrine tumors to the liver. Am J Surg Pathol. (2017) 41:915–22. doi: 10.1097/PAS.0000000000000876
Keywords: menin, SNP, neuroendocrine tumor, insulinoma, hyperparathyroidism
Citation: Chiloiro S, Capoluongo ED, Schinzari G, Concolino P, Rossi E, Martini M, Cocomazzi A, Grande G, Milardi D, Maiorano BA, Giampietro A, Rindi G, Pontecorvi A, De Marinis L and Bianchi A (2019) First Case of Mature Teratoma and Yolk Sac Testis Tumor Associated to Inherited MEN-1 Syndrome. Front. Endocrinol. 10:365. doi: 10.3389/fendo.2019.00365
Received: 17 February 2019; Accepted: 22 May 2019;
Published: 12 June 2019.
Edited by:
Wen Zhou, Case Western Reserve University, United StatesReviewed by:
Jean-Yves Scoazec, Institut Gustave Roussy, FranceCopyright © 2019 Chiloiro, Capoluongo, Schinzari, Concolino, Rossi, Martini, Cocomazzi, Grande, Milardi, Maiorano, Giampietro, Rindi, Pontecorvi, De Marinis and Bianchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura De Marinis, bGF1cmFkZW1hcmluaXNAeWFob28uaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.