 Letizia Canu1
Letizia Canu1 Massimo Mannelli
Massimo Mannelli
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 04 June 2019
Sec. Molecular and Structural Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00333
This article is part of the Research Topic Endocrine Forms of Hypertension: Clinical and Emerging Molecular Aspects View all 7 articles
Chromaffin tumors are included among the causes of secondary hypertension because of the release of catecholamines. Nevertheless, the clinical, cardiovascular, and hypertensive picture of patients affected by pheochromocytomas/paragangliomas (PPGL) is extremely variable, due to the different quantitative and qualitative releasing activity of these tumors. A consistent percentage of these patients, about 20%, is normotensive and not affected by the characteristic symptomatic crises due to sudden release of catecholamines. The factors causing such wide clinical variability are many and probably not all known. It is well known that many of these tumors are genetically determined and that the genetic profile influences the biochemical characteristics and the biology of the tumors as well as the clinical presentation of the affected patients. The number of asymptomatic or poorly symptomatic patients is increased after the introduction of genetic screening and the early diagnosis in mutation carriers. In this paper we can review the genotype-phenotype correlation of PPGLs with a focus on the cardiovascular picture.
Pheochromocytomas and Paragangliomas (PPGL) are included, together with Primary Aldosteronism and Cushing's Syndrome, among the main causes of endocrine hypertension in view of their catecholamine releasing property (1). They are in fact tumors composed by neural crest derived cells that during their development acquire and maintain a sympatho-adrenergic phenotype characterized by the enzymatic and storage machineries present in the mature chromaffin cells (2). Nevertheless, at variance with the chromaffin cells of the normal adrenal medulla whose secreting activity is finely regulated by cholinergic sympathetic neurons, tumor chromaffin cells are not controlled in their discharging activity. Moreover, they sometimes display an incomplete enzymatic pathway causing a variable biochemical secretory pattern which may also include the co-release of peptides (3) differently active at the cardiovascular level.
These characteristics explain why PPGL show an extremely variable clinical pattern with very different blood pressure profiles (4, 5).
The variability of PPGL clinical presentation is since long well known (6) and underlined by the definition of “the great mimic” assigned to PPGL since the beginning of their discovery.
The increase in blood pressure (BP) is caused by the actions of tumor catecholamines on the adrenergic receptors. Activation of vascular α1 receptors causes peripheral vasoconstriction and an increase in vascular resistance. Activation of cardiac β1 receptors causes a chronotropic and inotropic effect on the myocardium thus leading to an increased output. Finally, high plasma concentrations of norepinephrine may act on the β1 receptors of the juxta-glomerular cells in the kidney, causing an activation of the renin-angiotensin-aldosterone system. As a whole, the tumor catecholamine discharge determines an increase in BP.
This increase may differ in many aspects, depending on many factors. Among these, the pattern of catecholamine release (continuous or sporadic), the amount of catecholamine release (determined mainly by the size of the PPGL), the type of catecholamine released (adrenaline, noradrenaline or dopamine), the possible co-release of peptides with different actions on vascular resistance (Table 1).

Table 1. Peptides possibly secreted by chromaffin tumors influencing blood pressure.
Hypertension may present as continuous or intermittent, with or without sudden spikes, associated or not to other symptoms of adrenergic activation such as palpitations, sweating, trembling, anxiety.
The hypertensive crises can be so severe to be associated to cardiovascular complications such as myocardial ischemia, arrhythmias, heart failure, cerebral hemorrhage, sudden death.
In a retrospective collaborative study conducted on behalf of the Italian Endocrine Society on 284 patients affected by a PPGL, only 60% presented hypertensive crises, about 20% presented a hypertension not different from that of patients with essential hypertension, and 21% resulted normotensive (5) (Figure 1).
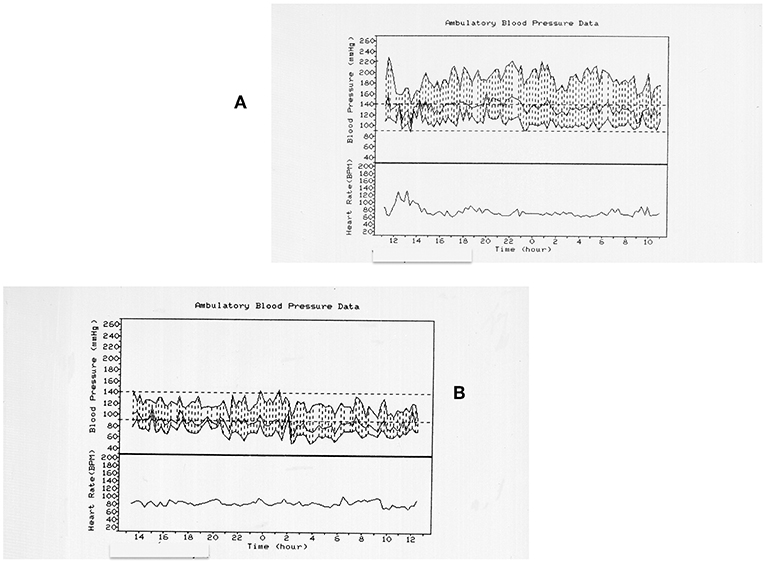
Figure 1. Twenty four hours blood pressure (BP) profiles in two different patients with pheochromocytoma. (A) refers to a 72 years old women with a right pheochromocytoma. The patient is hypertensive with BP spurts. (B) refers to a 79 years old man with a right pheochromocytoma. The patient is normotensive. The only common feature is the non-dipping profile of both the patients.
The secretory pattern, continuous or intermittent, can also influence the arterial vasoconstriction modifying the vascular responsiveness to catecholamines. In fact, a continuous exposure to high concentrations of catecholamines causes a down regulation of adrenergic receptors (7), thus counteracting the increase in vascular resistance.
In the very rare occurrence of a PPGL in pregnancy, the clinical presentation is influenced by the physiological cardiovascular changes occurring in this condition such as the decrease in vascular resistance and the expansion of circulating volume. In pregnancy, the PPGL-induced hypertension if often erroneously diagnosed as pre-eclampsia with deleterious consequences on the mother and the fetus (8).
The surgical removal of the tumor normalizes the catecholamine plasma levels and abolishes the risks of acute cardiovascular complications but not always is able to normalize BP in hypertensive patients (9).
In the last 20 years the spectrum of the PPGL susceptibility genes has progressively enlarged so that at present about 35–40% of PPGL is caused by a germ-line mutation of one of these genes (10, 11) (Table 2). The genetic profiling of PPGL has shown the occurrence of two main clusters characterized by the activation of two different pathogenic pathways (12). Cluster 1 includes mainly tumors linked to mutations of VHL (von Hippel-Lindau) and SDHx (succinate-dehydrogenase) genes and characterized by the induction of a pseudohypoxia mechanism. Cluster 2 includes tumors mainly linked to mutations of NF1 (neurofibromatosis type 1) and RET (responsible for the occurrence of Multiple Endocrine Neoplasia type 2) genes and characterized by the activation of tyrosine-kynase pathway.
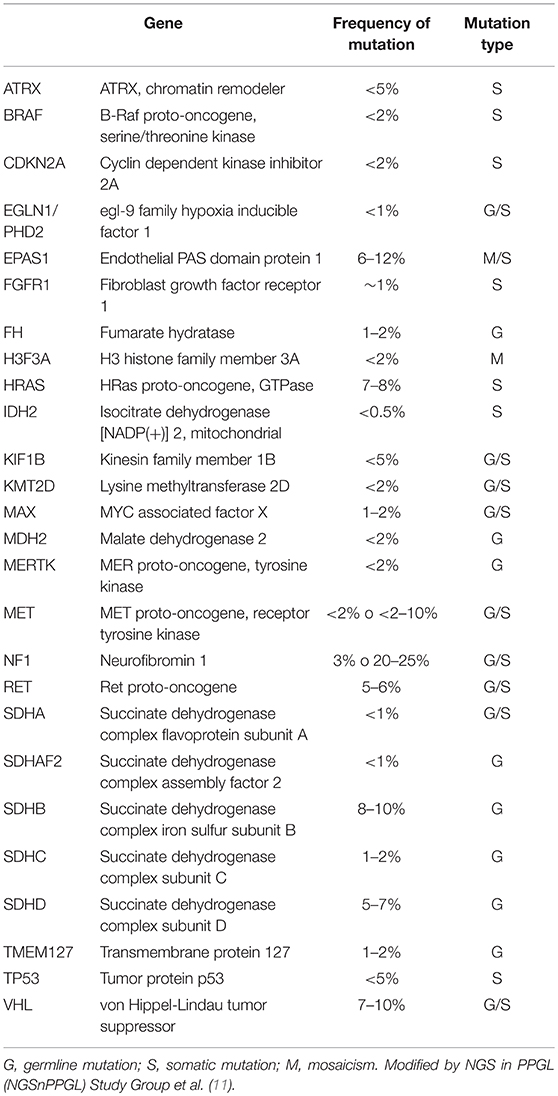
Table 2. Genes implied in the pathogenesis of Pheochromocytoma/paraganglioma.
The two different pathogenic pathways cause different PPGL phenotypes in terms of secretory pattern and biochemistry. It has demonstrated that VHL and MEN2 PPGL are indeed different (13).
In VHL syndrome, PPGL present a lower expression of TH (tyrosine hydroxylase), the rate limiting enzyme in catecholamine biosynthesis and an almost absent activity of the PNMT (phenylethanolamine-N–methyltransferase), responsible for the conversion of norepinephrine to epinephrine. In fact, in VHL tumors, the PNMT gene has been found hypermethylated and therefore downregulated (14). As a consequence, VHL PPGL have a lower total tissue content of catecholamines, represented by norepinephrine while MEN2 PPGL have higher tissue content of catecholamines and release both epinephrine and norepinephrine (15). These biochemical differences explain why MEN2 patients result more symptomatic and have a higher incidence of hypertension, mainly paroxysmal in comparison with VHL patients.
Moreover, cluster 1 is characterized by a more immature phenotype, especially in SDHB related PPGL. These tumors, which display the higher occurrence of metastatic forms, not only do not express PNMT but often also lack dopamine-β-hydroxylase activity (the enzyme responsible for the conversion of dopamine to norepinephrine) (16). Therefore, many SDHB related PPGL release dopamine which, acting at the vascular and renal DA1 receptors causes vasodilation and natriuresis, respectively, thus counteracting the hypertensive effects of norepinephrine.
Many other factors might contribute to a different BP profile. Succinate, which is increased in SDHx mutated PPGL, causes an increase of plasma renin activity in rats (17) but circulating succinate does not differ between hypertensive patients and normotensive controls (18).
Finally, the familial genetic screening permits the discovery of mutation carriers and the early diagnosis of small PPGL whose scanty releasing activity does not cause hypertension.
The therapy of PPGL is surgical and in the sporadic forms, the removal of the tumor leads to the disease cure in a very high percentage of cases. Nevertheless, a pre-surgical medical therapy is generally recommended (19) and the drugs of choice are α-blockers. The two most often used drugs are phenoxybenzamine (PBZ) and doxazosine (DXZ). PBZ is a non-selective (blocks both α1 and α2 receptors), non-competitive drug while DXZ is a selective (blocks only α1 receptors), competitive (can be displaced from the receptors by endogenous norepinephrine) drug. The α2-receptor blocking activity of PBZ exerted at the presynaptic level causes an increased discharge of norepinephrine from the sympathetic terminals so that almost always it causes a painful tachycardia which needs the addition of a β blocker.
It is worth mentioning that, in the absence of α-blocking therapy, β-blockers are contraindicated in patients with PPGL. In fact, inhibiting the vasodilation mediated by vascular β2-receptors, they can worsen a supervening catecholamine-induced hypertensive crisis.
The α blocking action causes a vasodilation, a reversal of the receptor dawn-regulation and a decrease in BP. This latter causes in turn an increase in blood volume and all these actions not only limit the pre-surgical hypertensive spikes but also avoid the dangerous hypotensive post-surgical crises.
According to the Endocrine Society guidelines (20), a target BP of <130/80 mmHg while seated and >90 mm Hg systolic while standing seems reasonable, with a target heart rate of 60–70 bpm seated and 70–80 bpm standing. If this target is not achieved by the use of only α blockers, other hypotensive drugs such as calcium channel blockers, angiotensin converting enzyme inhibitors or angiotensin II receptors antagonists can be added in the pre-surgical treatment.
PPGL are among the causes of endocrine hypertension. Nevertheless, at variance with the other causes, the BP pattern they present is extremely variable, being the hypertension often intermittent or paroxysmal and even absent in about 20% of cases.
MM, LC, GD, and GP contributed conception of the paper. LC wrote the first draft of the manuscript. GD and GP wrote sections of the manuscript. MM revised the manuscript. All authors read and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past collaboration with one of the authors LC.
1. Manger WM, Gifford RW. Pheochromocytoma. New York, NY: Springer-Verlag (1977). doi: 10.1007/978-1-4612-9900-4
2. Unsicker K. The chromaffin cell: paradigm in cell, developmental and growth factor biology. J Anat. (1993) 183:207–21.
3. Baldi E, De Feo ML, Geppetti P, Carlà V, Maggi M, Pupilli C, et al. Measurement of catecholamines, met-enkephalin, somaostatin and substance P-like immunoreactivities in 12 human pheochromocytomas. J Endocrinol Invest. (1988) 11:133–8. doi: 10.1007/BF03350121
4. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. (2005) 366:665–75. doi: 10.1016/S0140-6736(05)67139-5
5. Mannelli M, Ianni L, Cilotti A, Conti A, the National Study Group on Adrenal Tumors of the Italian Society of Endocrinology. Pheochromocytoma in Italy: a multicentric retrospective study. Eur J Endocrinol. (1999) 141:619–24. doi: 10.1530/eje.0.1410619
6. Manger WM. The protean manifestations of pheochromocytoma. Horm Metab Res. (2009) 41:658–63. doi: 10.1055/s-0028-1128139
7. Leeb-Lundberg LM, Corecchia S, De Blasi A, Caron MG, Lefkowitz RJ. Regulation of adrenergic receptor function by phosphorylation.I. Agonist-promoted desensitization and phosphorylation of α1-adrenergic receptors coupled to inositol phospholipid metabolism in DDT, MF-2 smooth muscle cells. J Biol Chem. (1987) 262:3098–105.
8. Mannelli M, Bemporad D. Diagnosis and management of pheochromocytoma during pregnancy. J Endocrinol Invest. (2002) 25:567–71. doi: 10.1007/BF03345503
9. Timmers HJ, Brouwers FM, Hermus AR, Sweep FC, Verhofstad AA, Verbeek AL, et al. Metastases but not cardiovascular mortality reduces life expectancy following surgical resection in apparently benign pheochromocytoma. Endocr Relat Cancer. (2008) 15:1127–33. doi: 10.1677/ERC-08-0049
10. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. (2014) 14:108–19. doi: 10.1038/nrc3648
11. NGS in PPGL (NGSnPPGL) Study Group, Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol. (2017) 13:233–47. doi: 10.1038/nrendo.2016.185
12. Burnichon N, Vescovo L, Amar L, Libé R, de Reynies A, Venisse A, et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet. (2011) 20:3974–85. doi: 10.1093/hmg/ddr324
13. Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. (2001) 86:1999–2008. doi: 10.1210/jc.86.5.1999
14. Castro-Vega LJ, Letouzé E, Burnichoan N, Buffet A, Disderot PH, Khalifa E, et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun. (2015) 6:6044. doi: 10.1038/ncomms7044
15. Eisenhofer G, Pacak K, Huynh TT, Qin N, Bratslavsky G, Linehan WM, et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer. (2010) 18:97–111. doi: 10.1677/ERC-10-0211
16. Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. (2012) 48:1739–49. doi: 10.1016/j.ejca.2011.07.016
17. He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. (2004) 429:188–93. doi: 10.1038/nature02488
18. Sadagopan N, Li W, Roberds SL, Major T, Preston GM, Yu Y, et al. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am J Hypertens. (2017) 20:1209–15. doi: 10.1016/j.amjhyper.2007.05.010
19. Mannelli M. Management and treatment of pheochromocytomas and paragangliomas. Ann N Y Acad Sci. (2006) 1073:405–16. doi: 10.1196/annals.1353.044
Keywords: pheochromocytoma, paraganglioma, endocrine hypertension, chromaffin tumors, genetic-disease susceptibility
Citation: Canu L, Parenti G, De Filpo G and Mannelli M (2019) Pheochromocytomas and Paragangliomas as Causes of Endocrine Hypertension. Front. Endocrinol. 10:333. doi: 10.3389/fendo.2019.00333
Received: 05 March 2019; Accepted: 08 May 2019;
Published: 04 June 2019.
Edited by:
Peter Igaz, Semmelweis University, HungaryReviewed by:
Joakim Crona, Uppsala University, SwedenCopyright © 2019 Canu, Parenti, De Filpo and Mannelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimo Mannelli, bWFzc2ltby5tYW5uZWxsaUB1bmlmaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.