 Munehiro Kitada
Munehiro Kitada Yoshio Ogura
Yoshio Ogura Itaru Monno
Itaru Monno Daisuke Koya
Daisuke Koya- 1Department of Diabetology and Endocrinology, Kanazawa Medical University, Uchinada, Japan
- 2Division of Anticipatory Molecular Food Science and Technology, Medical Research Institute, Kanazawa Medical University, Uchinada, Japan
The rising incidence of type 2 diabetes mellitus (T2DM) is a major public health concern, and novel therapeutic strategies to prevent T2DM are urgently needed worldwide. Aging is recognized as one of the risk factors for metabolic impairments, including insulin resistance and T2DM. Inflammation, oxidative stress, and mitochondrial dysfunction are closely related to both aging and metabolic disease. Calorie restriction (CR) can retard the aging process in organisms ranging from yeast to rodents and delay the onset of numerous age-related disorders, such as insulin resistance and diabetes. Therefore, metabolic CR mimetics may represent new therapeutic targets for insulin resistance and T2DM. Sirtuin 1 (SIRT1), the mammalian homolog of Sir2, was originally identified as a nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase. The activation of SIRT1 is closely associated with longevity under CR, and it is recognized as a CR mimetic. Currently, seven sirtuins have been identified in mammals. Among these sirtuins, SIRT1 and SIRT2 are located in the nucleus and cytoplasm, SIRT3 exists predominantly in mitochondria, and SIRT6 is located in the nucleus. These sirtuins regulate metabolism through their regulation of inflammation, oxidative stress and mitochondrial function via multiple mechanisms, resulting in the improvement of insulin resistance and T2DM. In this review, we describe the current understanding of the biological functions of sirtuins, especially SIRT1, SIRT2, SIRT3, and SIRT6, focusing on oxidative stress, inflammation, and mitochondrial function, which are closely associated with aging.
Introduction
The rising incidence of type 2 diabetes mellitus (T2DM) has significantly increased worldwide in recent decades, and the development of better treatments for T2DM is urgently needed. Aging is a universal process that affects all organs. Age-related disruptions in cellular homeostasis result in the decline in the responsiveness to physiological stress, including oxidative stress and inflammation, which are implicated in the pathogenesis of metabolic diseases, including insulin resistance and T2DM. Additionally, mitochondria play a central role in energy production and responsiveness to nutrient availability, and they are one of the sources of reactive oxygen species (ROS) (1). Therefore, mitochondrial function decline is also closely related to the impairment of metabolic homeostasis (2) and oxidative stress (3, 4), contributing to the progression of insulin resistance and T2DM, which are associated with aging. Additionally, oxidative stress is closely linked to inflammation (5, 6); therefore, the suppression of oxidative stress/inflammation and preservation of mitochondrial function should be therapeutic targets for insulin resistance and T2DM, as well as for anti-aging treatments.
Calorie restriction (CR) retards aging or extends the life spans of yeast, worms, flies, and rodents (7). The benefits of CR for the suppression of age-related diseases, including glucose intolerance, cardiovascular disease and cancer, have also been observed in rhesus monkeys or humans (8–10), by improving insulin sensitivity and reducing inflammation and oxidative stress. Sirtuins have received attention for their role in modifying lifespan, especially in relation to the benefits of CR. From the initial studies on aging in yeast, silent information regulator 2 (Sir2), a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase, was identified as one of the possible molecules through which CR improves lifespan extension (11). Homologs of Sir2 in higher eukaryotic organisms are known as SIRT1, which may contribute to CR-induced longevity (12–14), and, currently, seven sirtuins, including SIRT1, have been identified in mammals (15, 16) (Table 1). Numerous previous reports have shown the multiple physiological roles of sirtuins, including SIRT1, SIRT2, SIRT3 and SIRT6, in cellular function, such as glucose metabolism, mitochondrial function and resistance against cellular stresses, including oxidative stress and inflammation (15–20). Thus, the modulation of sirtuin activity, as a CR mimetic, may be a novel drug target for insulin resistance and T2DM.
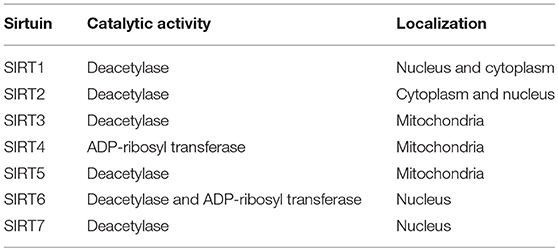
Table 1. Seven sirtuins in mammals.
In this review, we describe the current understanding of the biological functions of sirtuins, especially SIRT1, SIRT2, SIRT3, and SIRT6, focusing on oxidative stress, inflammation and mitochondrial function, which are closely associated with aging. We also discuss their potential as pharmacological targets to prevent the development of metabolic diseases, such as insulin resistance and T2DM.
Inflammation, Oxidative Stress, and Mitochondrial Dysfunction, Which Are Related to the Pathogenesis of Insulin Resistance and Type 2 Diabetes
Chronic inflammation, oxidative stress and impaired mitochondrial function in skeletal muscle, adipose tissue or monocytes/macrophages (21, 22) are closely related to the pathogenesis of insulin resistance and T2DM. Additionally, inflammation and oxidative stress contribute to pancreatic β-cell dysfunction (23, 24), contributing to the progression of T2DM.
The activation of monocytes in the circulation, adipocytes and macrophages residing in adipose tissue leads to the release of various inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and chemoattractant protein-1 (MCP-1), in insulin-resistant and diabetic states. These cytokines activate inflammatory signaling pathways, such as the inhibitor of IκB kinase (IKK) and c-Jun NH2-terminal kinase (JNK) pathways, which impair the insulin signaling pathway by modulating phosphoinositide 3-kinase (PI3K) and Akt (25–27), and they play a crucial role in the pathogenesis of insulin resistance in adipose tissue and skeletal muscle. Oxidative stress also impairs insulin signaling, which contributes to insulin resistance in T2DM. In insulin-resistant or diabetic states, in addition to hyperglycemia, other metabolites, including free fatty acids (FFAs) and certain cytokines, such as TNF-α, induce the overproduction of ROS from mitochondria. ROS trigger the activation of serine/threonine kinases, such as p38 mitogen-activated protein kinase (p38 MAPK), JNK, and IKK, which induce the serine phosphorylation of insulin receptor substrate-1 (IRS-1), and then degrade IRS-1 and reduce IRS-1 tyrosine phosphorylation, leading to the suppression of insulin signaling (28–31), as well as inflammation. Inflammatory mediators and oxidative stress are also related to pancreatic β-cell dysfunction, resulting in the impairment of insulin production or excretion from β cells (23, 24).
The impairment of mitochondrial function in skeletal muscle is involved in the pathogenesis of insulin resistance and progression of T2DM, which may be associated with aging. Mitochondria play a central role in energy production and responsiveness to nutrient availability by regulating mitochondrial oxidative phosphorylation (OXPHOS) and fatty acid oxidation. However, previous studies have shown that the rate of mitochondrial OXPHOS is reduced and that intramyocellular lipid accumulation is increased in the skeletal muscle of patients with insulin resistance and T2DM and in elderly individuals (32–35). Aging is closely linked to the impairment of metabolic homeostasis, such as insulin resistance and T2DM, which are closely related to the decline in mitochondria function. Mitochondrial function decline generates excess ROS from mitochondria, and oxidative stress is linked to inflammation. Thus, there is a vicious cycle among oxidative stress, inflammation and mitochondrial dysfunction, and breaking this cycle may be a therapeutic target for the treatment of age-related insulin resistance and T2DM, focusing on SIRT1, SIRT2, SIRT3, and SIRT6 (Figure 1).
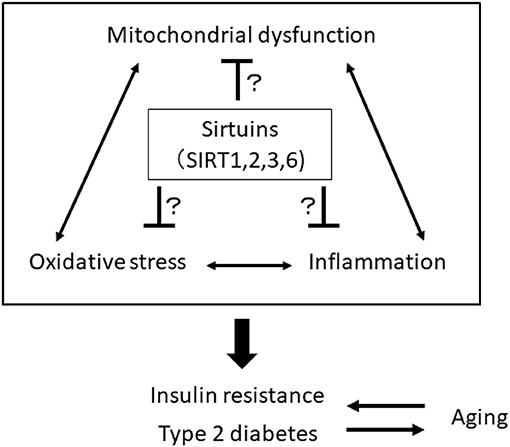
Figure 1. A vicious cycle among oxidative stress, inflammation, and mitochondrial dysfunction is involved in the pathogenesis of insulin resistance, type 2 diabetes, and aging. Sirtuins, including SIRT1, 2, 3, and 6, may be a therapeutic target for the treatment of age-related insulin resistance and T2DM by breaking this vicious cycle.
SIRT1
SIRT1 exists in the nucleus and cytoplasm, and it has NAD+-dependent deacetylase activity (16). Numerous non-histone proteins, including transcription factors, transcriptional coregulatory proteins and histones, serve as substrates for SIRT1, which is associated with a wide variety of cellular processes (16). SIRT1 may play a crucial role in reducing inflammation and oxidative stress and improving mitochondrial function, resulting in both the protection of pancreatic β cells and amelioration of insulin resistance in insulin-sensitive tissues such as skeletal muscle and adipose tissue. Therefore, SIRT1 should be a pharmacological therapeutic target to treat insulin resistance and T2DM (17, 36).
Regulation of Inflammation
Accumulated evidence has demonstrated that SIRT1 suppresses inflammatory processes likely through interfering with nuclear factor kappa-B (NF-κB) signaling. Yeung et al. found that SIRT1 deacetylates the p65 subunit of NF-κB at lysine 310 and inhibits its transcriptional activity (37).
In adipocytes and macrophages, SIRT1 reduces inflammatory process through the deacetylation of NF-κB (p65), leading to improved glucose metabolism (38, 39) (Figure 2A). Additionally, myeloid cell-specific SIRT1 knockout (KO) mice that were fed a high-fat diet (HFD) exhibited macrophage activation and elevated expression of inflammatory mediators in the liver and adipose tissues, which was associated with the development of systemic insulin resistance (40). Furthermore, we previously reported another mechanism regarding SIRT1 inactivation-induced inflammation (Figure 2A): SIRT1 inactivation may enhance the NF-κB signaling pathway by the phosphorylation of NF-κB (p65) via the cellular accumulation of p62/Sqstm1 due to autophagy dysfunction, in THP-1 cells, cultured human monocytes (41). Moreover, SIRT1 inactivation resulted in increased activation of the mammalian target of rapamycin complex 1 (mTORC1) pathway and reduced 5′ AMP-activated kinase (AMPK) activation, possibly contributing to impairment in autophagy (41). In humans, reduction of SIRT1 expression levels in circulating monocytes is correlated with glucose intolerance, insulin resistance and metabolic syndrome in humans (42). Gillum et al. also demonstrated that SIRT1 expression is decreased in adipose tissues from obese males and that the mRNA expression of CD14, a macrophage marker, in adipose tissue is negatively correlated with SIRT1 expression (43).
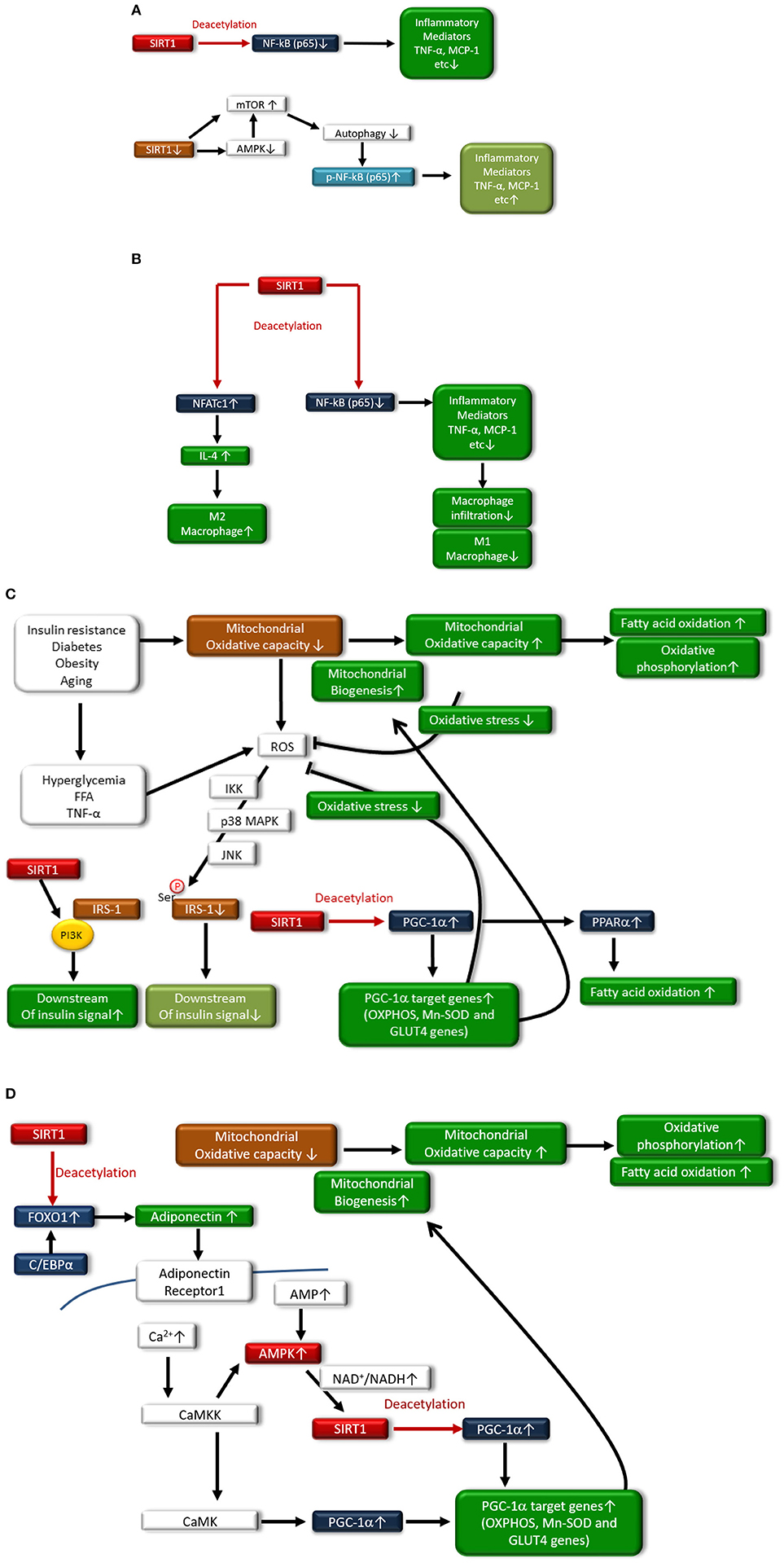
Figure 2. (A) In monocytes/macrophages and adipocytes, SIRT1 deacetylates NF-κB(p65), resulting in reduced expression of inflammatory mediators such as TNF-α and MCP-1. SIRT1 inactivation also induces inflammation through the phosphorylation of the NF-κB pathway via impaired autophagy, which is associated with activation of mammalian target of rapamycin (mTOR) and reduced activation of AMP-activated kinase (AMPK). (B) In adipocytes, SIRT1 deacetylates nuclear factor-κB p65 subunit [NF-κB(p65)], resulting in reduced expression of inflammatory mediators such as tumor necrosis factor-α (TNF-α) and chemoattractant protein-1 (MCP-1), and decreased polarization to M1 macrophages and infiltration to adipose tissue. SIRT also induces polarization to M2 macrophages through increased expression of interleukin-4 (IL-4) expression via deacetylation of nuclear factor of activated T-cells 1 (NFATc1). (C) In skeletal muscle, SIRT1 increases mitochondrial biogenesis and fatty acid oxidation through acetylation and activation of the peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α). Under conditions of insulin resistance, diabetes, obesity, or aging, mitochondrial oxidative capacity is decreased, contributing to the generation of reactive oxygen species (ROS) in mitochondria. Hyperglycemia, free fatty acids (FFAs) and TNF-α stimulate ROS production from the mitochondria, and increased levels of ROS lead to the serine-phosphorylation of insulin receptor substrate-1 (IRS-1), resulting in reduced insulin signaling. However, SIRT1 interacts with phosphoinositide 3-kinase (PI3K), leading to activation of insulin signaling. Additionally, SIRT1 activates PGC-1α transcriptional activity to induce mitochondrial biogenesis and the induction of antioxidative enzymes, which can inhibit the generation of ROS by mitochondria. Expression of glucose transporter 4 (GLUT4) is enhanced through deacetylation of PGC-1α by SIRT1. Moreover, SIRT1 activates peroxisome proliferator-activated receptor-α (PPAR-α), which induces fatty acid oxidation. (D) SIRT1 deacetylates Forkhead box protein O1 (FOXO1) and enhances its interaction with CCAAT/enhancer binding protein α (C/EBPα), resulting in the enhanced transcription of adiponectin in adipocytes. In skeletal muscle, adiponectin is involved in the regulation of Ca2+ signaling and PGC-1α expression through calcium/calmodulin-dependent protein kinase kinase (CaMKK) and calcium/calmodulin-dependent protein kinase (CaMK) activation. Adiponectin activates SIRT1 through AMPK activation, thereby deacetylating PGC-1α and resulting in mitochondrial biogenesis, increased fatty acid oxidation, and oxidative phosphorylation.
Interestingly, adipocyte SIRT1 controls systemic glucose homeostasis and insulin sensitivity through cross talk with adipose-resident macrophages (44) (Figure 2B). Hui et al. recently reported that adipose-specific SIRT1 KO mice showed a higher susceptibility to HFD-induced insulin resistance, which is associated with an increased number of adipose-resident macrophages and their polarization toward the proinflammatory M1 subtype, overexpressing inflammatory mediators (44). SIRT1 in adipocytes modulates the expression and secretion of several adipokines, including adiponectin, MCP-1, TNF-α, and IL-4, which, in turn, alters the recruitment and polarization of macrophages in adipose tissues. In adipocytes, SIRT1 enhances IL-4 expression through deacetylating the transcription factor nuclear factor of activated T cells, cytoplasmic 1 (NFATc1), leading to polarization of the M2 subtype (44). Thus, SIRT1 may diminish inflammation in adipose tissues and monocytes/macrophages and may improve insulin resistance and T2DM.
In addition, SIRT1 protein expression is reduced in skeletal muscle and primary myotubes derived from T2DM patients, suggesting that diminished SIRT1 contributes to insulin resistance induced by TNF-α in skeletal muscle (45). SIRT1 interacts with PI3K adaptor subunit p85 in an insulin-independent manner and activates insulin signaling at physiological insulin concentrations in skeletal muscle cells (45) (Figure 2C). Furthermore, SIRT1 protects pancreatic β cells against various toxic stresses, including oxidative stress and inflammatory cytokines, through suppressing NF-κB signaling (46).
Regulation of Mitochondrial Function
SIRT1 can be involved in the regulation of metabolism and insulin resistance through the modulation of mitochondrial function. Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) maintains mitochondrial biogenesis and OXPHOS proteins, leading to efficient β oxidation of fatty acids in skeletal muscle. However, the levels of PGC-1α in skeletal muscle are decreased in T2DM. SIRT1 regulates mitochondrial function and metabolic homoeostasis, increases the oxygen consumption in skeletal muscle and leads to the expression of OXPHOS genes and mitochondrial biogenesis through the deacetylation of PGC-1α. SIRT1 knockdown largely prevents the upregulation of PGC-1α-induced genes that are involved in mitochondrial fatty acid utilization (47). Furthermore, SIRT1 can regulate peroxisome proliferator-activated receptor-α (PPAR-α) activation by PGC-1α deacetylation, resulting in increased fatty acid oxidation. Thus, the activation of SIRT1 may improve insulin resistance through promoting fatty acid oxidation and mitochondrial biogenesis via deacetylation of PGC-1α and PPAR-α activation in skeletal muscle (Figure 2C). Additionally, PGC-1α remarkably increases the expression of glucose transporter 4 (GLUT4) and activation of glucose transport in murine C2C12 myotubes (48). The effects of PGC-1α on GLUT4 gene expression leads to increased transport of glucose in myocytes, suggesting that the activation of PGC-1α by SIRT1 is involved in insulin sensitization (Figure 2C). Adiponectin has antidiabetic power (49), and the levels of plasma adiponectin are reduced in insulin resistance and T2DM (50, 51). Treatment with adiponectin can decrease glucose levels and ameliorate insulin resistance in mice (52). Mechanistically, adiponectin enhances insulin sensitivity through increasing fatty acid oxidation via AMPK and PPAR-α activation (49). Additionally, SIRT1 deacetylates Forkhead box protein O1 (FOXO1) and enhances its interaction with CCAAT/enhancer binding protein α (C/EBPα), resulting in enhanced transcription of the adiponectin gene in adipocytes (53) (Figure 2D). Iwabu et al. demonstrated that adiponectin signaling plays a crucial role in skeletal muscle cells and is implicated in the regulation of Ca2+ signaling and expression/activation of PGC-1α in muscle adiponectin receptor (adipoR) 1 KO mice (54). Adiponectin activates AMPK by binding to adipoR1, thereby activating SIRT1, and deacetylating PGC-1α improves oxidative stress, mitochondrial function, glucose/lipid metabolism, and exercise endurance (54) (Figure 2D), resulting in improved insulin resistance and T2DM. Thus, SIRT1-induced PGC-1α deacetylation leads to the improvement of mitochondrial function via mitochondrial biogenesis and induction of GLUT4 and adiponectin, demonstrating the beneficial effects against insulin resistance and T2DM.
Regulation of Oxidative Stress
In addition to mitochondrial function modulation, PGC-1α deacetylation by SIRT1 reduces oxidative stress through the overexpression of antioxidative enzymes, including manganese-superoxide dismutase (Mn-SOD) (55). Additionally, Forkhead box protein O3a (FOXO3a) is deacetylated by SIRT1 and translocates to the nucleus, resulting in the upregulation of other antioxidative enzymes and catalases and protection against oxidative stress (56). Thus, SIRT1 may improve insulin resistance and T2DM possibly through reducing oxidative stress (Figure 2C), inducing mitochondrial biogenesis, and increasing mitochondrial function.
SIRT2
SIRT2 is localized in both the cytoplasm and nucleus, and it is widely expressed in various tissues, including the brain, muscle, pancreas, liver, kidney, and adipose tissues. SIRT2 interacts with many histone and non-histone protein substrates, including tubulin and histone H4 (18). SIRT2 is involved in multiple cellular functions, including genomic integrity, cell growth, differentiation, and energy metabolism, and reduced SIRT2 activity has been implicated in cancer, neurodegeneration and metabolic diseases (18). Previous studies have demonstrated that SIRT2 plays an important role in various physiological processes in maintaining metabolic homeostasis, including inflammation, oxidative stress and mitochondrial function, as well as adipocyte differentiation, fatty acid oxidation, gluconeogenesis, and insulin sensitivity (18). A few reports have shown that SIRT2 exerts anti-inflammation and antioxidative stress effects and improves mitochondrial function in metabolic-related tissues, such as skeletal muscle.
Regulation of Inflammation
SIRT2 regulates inflammation by deacetylating the NF-κB p65 subunit (57), similar to SIRT1. Pais et al. demonstrated that SIRT2 plays a crucial role as a major inhibitor of microglia-mediated inflammation and neurotoxicity through the deacetylation of NF-κB (p65) (58) (Figure 2A). In other experimental inflammatory disease models, the anti-inflammatory effect of SITR2 has been demonstrated through the suppression of the NF-κB signaling pathway (59, 60). However, further studies are necessary to elucidate whether this anti-inflammatory effect of SIRT2 may be exerted in metabolic diseases, including insulin resistance and T2DM.
Regulation of Oxidative Stress
SIRT2 regulates redox homeostasis in cells. SIRT2-dependent deacetylation of FOXO3a leads to increased expression of Mn-SOD to improve oxidative stress (61) (Figure 3A). In addition, glucose-6-phosphate dehydrogenase (G6PD) is a key enzyme in the pentose phosphate pathway (PPP) and plays a crucial role in the oxidative stress response by producing nicotinamide adenine dinucleotide phosphate (NADPH) and reduced form glutathione (GSH), the main intracellular reductant (Figure 3B). Wang et al. reported that SIRT2 activates G6PD through deacetylation on lysine 403 in G6PD, which plays an important role in maintaining the cellular redox status and protecting cells from oxidative damage (62).
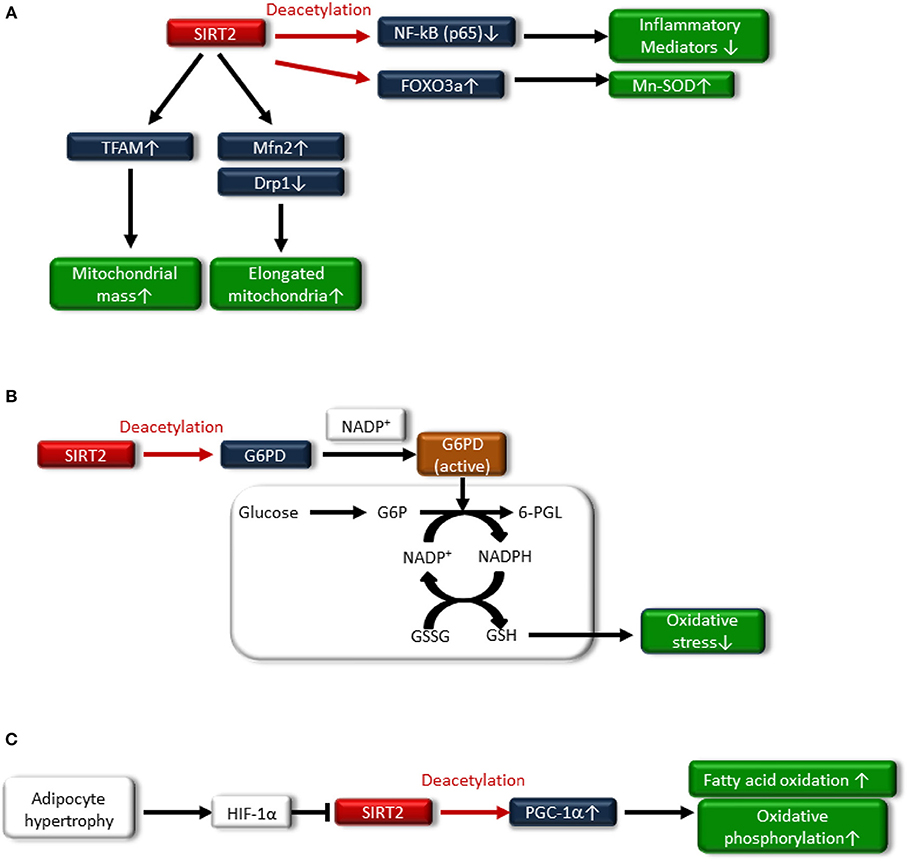
Figure 3. (A) SIRT2 deacetylates nuclear factor-κB p65 subunit [NF-κB (p65)], resulting in decreased expression of inflammatory mediators. Sirt2 also induces Mn-SOD expression by deacetylating Forkhead box protein O3a (FOXO3a). Additionally, SIRT2 increases fusion-related protein mitofusion2 (Mfn2) and decreases mitochondrial-associated dynamin-related protein 1 (Drp1), resulting in an increased number of elongated mitochondria and improved mitochondrial function. SIRT2 also attenuates the downregulation of transcription factor A, mitochondrial (TFAM), a key mitochondrial deoxyribonucleic acid (mtDNA)-associated protein, leading to an increase in mitochondrial mass. (B) Glucose-6-phosphate dehydrogenase (G6PD) plays an important role in the oxidative stress response by producing nicotinamide adenine dinucleotide phosphate (NADPH) and the reduced form glutathione (GSH), which is associated with deacetylating G6PD and binding to nicotinamide adenine dinucleotide phosphate (NADP+). (C) Hypoxia-inducible factor1α (HIF1α), which is accumulated in the adipocytes of hypertrophy, represses SIRT2 expression, resulting in decreased deacetylation of PGC-1α and the expression of β-oxidation and mitochondrial genes.
Regulation of Mitochondrial Function
SIRT2 may be related to the regulation of mitochondrial function. Lemos et al. showed that SIRT2 is downregulated in insulin-resistant hepatocytes and the liver, and is accompanied by increased ROS production, activation of extracellular signal-regulated kinase (ERK1/2), and mitochondrial dysfunction in ob/ob mice (63). SIRT2 overexpression in insulin-resistant hepatocytes improved insulin sensitivity and reduced ROS production. SIRT2 might increase fusion-related protein mitofusion 2 (Mfn2) and decrease mitochondrial-associated dynamin-related protein 1 (Drp1), resulting in an increased number of elongated mitochondria and improving mitochondrial function (Figure 3A). SIRT2 also attenuated the downregulation of transcription factor A mitochondrial (TFAM), a key mitochondrial deoxyribonucleic acid (mtDNA)-associated protein, leading to an increase in the mitochondrial mass (63) (Figure 3A). Furthermore, SIRT2 expression in peripheral blood mononuclear cells (PBMCs) from human subjects was negatively correlated with obesity, insulin resistance and oxidative stress (63).
SIRT2 is most markedly expressed in adipocytes (61). Nutrient overload-induced adipose expansion enhances intra-adipose hypoxia, promoting the accumulation of adipocyte hypoxia-inducible factor 1α (HIF1α). HIF1α suppresses SIRT2 transcription through interaction at a cross-species conserved hypoxic response element (HRE) on the SIRT2 promoter. HIF1α accumulation in the adipocytes of human obese subjects correlates with low levels of SIRT2 in visceral adipose tissue, and reduced SIRT2 activity directly translates into decreased deacetylation of PGC-1α and expression of β-oxidation and mitochondrial genes. HIF-1α suppresses fatty acid catabolism in mitochondria by negatively regulating the SIRT2-PGC-1α axis (64) (Figure 3C).
SIRT3
Preservation of mitochondrial health is crucial to prevent insulin resistance and T2DM during aging. SIRT3 is localized primarily in the mitochondria. SIRT3 is a major mitochondrial deacetylase and plays a major role in deacetylating and modifying the enzymatic activities of several mitochondrial proteins (16). In humans, a polymorphism in the SIRT3 gene has been correlated with reduced enzymatic efficiency and the development of metabolic syndrome (65). SIRT3 is also recognized an anti-aging molecule, and high SIRT3 expression levels are associated with longevity in humans (66, 67). Previous studies have demonstrated that SIRT3 protects organisms against metabolic stress, cancer, the development of cardiac hypertrophy, and oxidative stress (16).
Regulation of Mitochondrial Function and Oxidative Stress
Numerous reports have exhibited that SIRT3 regulates mitochondrial function and maintains redox homeostasis; therefore, the impairment of SIRT3 function is implicated in the pathogenesis of insulin resistance and T2DM. Jing et al. demonstrated that decreased levels of SIRT3 in the skeletal muscle of streptozotocin (STZ)-induced diabetic mice and high-fat diet-induced obese mice were an important component of the pathogenesis of T2DM (68). SIRT3 KO mice exhibited decreased oxygen consumption and increased oxidative stress due to mitochondrial dysfunction via the hyperacetylation of complex I and III in OXPHOS, and these factors led to JNK activation and impaired insulin signaling. In addition, SIRT3 can directly deacetylate succinate dehydrogenase (SDH), a subunit of complex II and that succinate dehydrogenase activity is reduced in SIRT3 KO cells and brown adipose tissue (BAT) from SIRT3 KO mice (69). Thus, SIRT3 may induce mitochondrial oxidative phosphorylation through the deacetylation of complex I, III and SDHA of complex II (Figure 4).
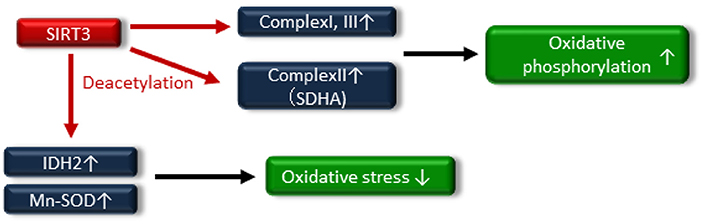
Figure 4. SIRT3 deacetylates the electron transport chain complexes I, II [deacetylate succinate dehydrogenase (SDH) A] and III, leading to increased oxidative phosphorylation. SIRT3 also attenuates oxidative stress by enhancing the glutathione antioxidant defense system via deacetylation and activation of isocitrate dehydrogenase 2 (IDH2) and manganese superoxide dismutase (Mn-SOD).
A reduction in SIRT3 activity contributes to mitochondrial oxidative stress through decreasing the activation of antioxidative enzymes, such as isocitrate dehydrogenase 2 (IDH2) and Mn-SOD (70–72), by increasing the acetylation of antioxidative enzymes (Figure 4). SIRT3 protects pancreatic β cells against lipotoxicity by antagonizing oxidative stress-induced cell damage. Zhou et al. demonstrated that HFD feeding caused elevated oxidative stress accompanied by reduced SIRT3 expression in the pancreatic β cells of wild-type mice (73). Primary pancreatic islets of SIRT3 KO mice and murine pancreatic β-cell line MIN6 cells with downregulated SIRT3 expression showed increased Mn-SOD acetylation and reduced glucose-stimulated insulin secretion and glucose-stimulated ATP generation (73). On the other hand, SIRT3 overexpression, using an adenoviral system, ameliorated palmitate-induced β-cell dysfunction including endoplasmic reticulum (ER) stress in pancreatic β-cell line NIT1 cells (74). In human, reduced expression of SIRT3 in pancreatic β cells from T2DM patients has been linked to impaired β-cell function (75). Thus, novel therapeutic approaches targeting SIRT3 activity may be important in providing new opportunities to treat insulin resistance and T2DM through maintaining mitochondrial health.
SIRT6
SIRT6 is located in the nucleus, and it acts as an adenosine diphosphate (ADP)-ribosyl transferase and NAD+-dependent deacetylase (16). SIRT6 has been associated with longevity regulation. Kanfi et al. reported that the overexpression of SIRT6 extended the lifespan of male mice and was involved in decreased serum levels of insulin-like growth factor (IGF)-1 and increased levels of IGF-binding protein 1 (76). SIRT6 is implicated in DNA repair, telomere maintenance, genomic stability and cell senescence. SIRT6 also attenuates NF-κB signaling via histone H3K9 deacetylation at the chromatin level (77–79) (Figure 5).
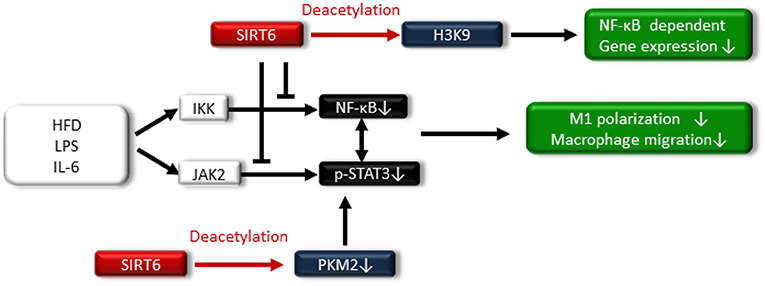
Figure 5. SIRT6 also attenuates NF-κB signaling via histone H3K9 deacetylation at the chromatin level. SIRT6 suppresses the high-fat diet (HFD)-, LPS-, and IL-6-induced I-κ kinase (IKK)-nuclear factor-κB (NF-κB) pathway and Janus activating kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3) pathway, resulting in reduced M1 macrophage polarization and macrophage migration. Additionally, SIRT6 deacetylates pyruvate kinase M2 (PKM2), preventing STAT3 from phosphorylation.
Regulation of Inflammation, Oxidative Stress, and Mitochondrial Function
SIRT6 is involved in vascular inflammation and oxidative stress. Knockdown of SIRT6 in human umbilical vein endothelial cells (HUVECs) increases the expression of proinflammatory cytokines (80). Balestrieri et al. demonstrated that SIRT6 protein expression in atherosclerotic lesions of T2DM patients was downregulated, which was compared with SIRT6 protein expression in atherosclerotic lesions of non-diabetic patients, and the reduced SIRT6 expression was associated with increased oxidative stress and inflammation (81). Additionally, apolipoprotein E-deficient with SIRT6 knockdown using small hairpin RNA (shRNA) lentivirus-injected mice fed a high-cholesterol diet showed the promotion of atherosclerosis that was associated with increased inflammation in endothelial cells (82).
Regarding the relationship between SIRT6 and metabolic disease, SIRT6 levels increase in rats under the CR condition, and transgenic mice that overexpress SIRT6 are protected against HFD-induced several metabolic impairments, including glucose intolerance (83). By contrast, ablation of neural SIRT6 leads to obesity (84). Lee et al. demonstrated that myeloid-specific SIRT6 KO mice exhibit tissue inflammation and insulin resistance when fed an HFD (85). Myeloid SIRT6 deletion promoted proinflammatory M1 polarization of bone marrow macrophages and enhanced the migration potential of macrophages toward adipose-derived inflammatory mediators. SIRT6 deletion in macrophages facilitated activation of the IKK-NF-κB pathway and endogenous production of IL-6, leading to activation of the Janus activating kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3) pathway and positive feedback circuits for NF-κB stimulation; this cross talk expedited M1 polarization (85) (Figure 5). Furthermore, acetylated pyruvate kinase M2 (PKM2) phosphorylates STAT3 in the nucleus (86), PKM2 is deacetylated by SITR6 (87), and the study demonstrated that SIRT6 deacetylates PKM2, preventing STAT3 from being phosphorylated and leading to the suppression of M1 polarization in SIRT6-overexpressed intraperitoneal macrophages treated with LPS (Figure 5).
SIRT6 is involved in the regulation of mitochondrial function in skeletal muscle. Cui et al. demonstrated that muscle-specific SIRT6 KO mice impairs glucose homeostasis and insulin sensitivity, attenuates whole-body energy expenditure, and weakens exercise performance. Mechanistically, the deletion of SIRT6 in muscle decreased the expression of genes associated with glucose and lipid uptake, fatty acid oxidation, and mitochondrial OXPHOS in muscle cells caused by the reduced AMPK activity (88).
In pancreatic β cells, SIRT6 regulates insulin secretion in response to glucose stimulation. Xiong et al. demonstrated that the deletion of SIRT6 in pancreatic β-cells in mice leads to the impairment of glucose-stimulated insulin secretion (89, 90) and they further found that SIRT6 regulates insulin secretion by maintaining mitochondrial function and modulating intracellular Ca2+ dynamics (89, 90). Additionally, SIRT6 plays an important role in the protection of pancreatic β cells from lipotoxicity (palmitic acid, PA)-induced cellular dysfunction or even cell death (90). Oxidative stress generated by fatty acid oxidation is involved in the pathogenesis of PA-induced β-cell dysfunction and apoptosis (91). SIRT6 may exert the effect of antioxidative stress by coactivating NF-E2-related factor 2 (NRF2) (92). However, it is unclear whether SIRT6 plays a role in antioxidative stress in pancreatic β cells.
Thus, SIRT6 may have beneficial effects on glucose metabolism, including insulin resistance and T2DM, by reducing inflammation and improving mitochondrial function. Additionally, SIRT6 protects pancreatic β cells from lipotoxicity-induced cellular damage, through maintaining mitochondrial function and possibly antioxidative stress.
Concluding Remarks
The knowledge of sirtuins has expanded from the original description of a NAD+-dependent deacetylase responsible for longevity in yeast, which is associated with CR. As described above, sirtuin family members, such as SIRT1, 2, 3, and 6, may induce beneficial effects in glucose metabolism, partially through improving inflammation, oxidative stress, and maintaining mitochondrial function. Therefore, pharmacological modulation of sirtuins may represent a novel therapeutic tool to improve insulin resistance and T2DM. Among the sirtuins, several SIRT1 activators, such as resveratrol and synthesized activators, have been evaluated for their antidiabetic effects in animal models (93). In humans, several small trials have shown that SIRT1 activators exert beneficial effects on glucose metabolism and insulin resistance, which resemble the effect of CR (94). However, there is still insufficient clinical data regarding the effect of SIRT1 activators on insulin resistance and T2DM. In addition, SIRT2, SIRT3, and SIRT6, which are induced by CR, play crucial roles in regulating cellular processes, including metabolism, inflammation, oxidative stress and mitochondrial function. However, further investigation into the targets and functions of SIRT1, SIRT2, SIRT3, and SIRT6 will aid in the development of new strategies to treat insulin resistance and T2DM. In addition to SIRT1, SIRT2, SIRT3, and SIRT6, other sirtuins, such as SIRT4, SIRT5, and SIRT7, play crucial roles in cellular homeostasis and functions, including redox homeostasis, anti-inflammation, cell survival, and mitochondrial quality control (95–100), which may be involved in the pathogenesis of insulin resistance and T2DM. However, further basic studies are necessary to elucidate the detailed molecular mechanisms.
Author Contributions
MK and DK designed the manuscript, wrote and edited the manuscript. YO, IM, and DK contributed to the discussion. DK is the guarantor of this work.
Funding
This work was supported by a Grant-in-Aid for Scientific Research (C) (17K09718) to MK, a Grant-in-Aid for Young Scientists (B) (17K16104) to YO, and a Grant-in-Aid for Young Scientists (B) (18K16013) to IM. Boehringer Ingelheim, Mitsubishi Tanabe Pharma, Kyowa Hakko Kirin, Taisho Toyama Pharmaceutical Co., and Ono Pharmaceutical Co. contributed to establishing the Division of Anticipatory Molecular Food Science and Technology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
2. McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. (2006) 16:R551–560. doi: 10.1016/j.cub.2006.06.054
3. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. (2005) 120:483–95. doi: 10.1016/j.cell.2005.02.001
4. Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. (2000) 25:502–8. doi: 10.1016/S0968-0004(00)01674-1
5. Garg AK, Aggarwal BB. Reactive oxygen intermediates in TNF signaling. Mol Immunol. (2002) 39:509–17. doi: 10.1016/S0161-5890(02)00207-9
6. Janssen-Heininger YM, Poynter ME, Baeuerle PA. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radic Biol Med. (2000) 28:1317–27. doi: 10.1016/S0891-5849(00)00218-5
7. Fontana L, Partridge L, Longo VD. Extending healthy life span–from yeast to humans. Science. (2010) 328:321–6. doi: 10.1126/science.1172539
8. Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. (2009) 325:201–4. doi: 10.1126/science.1173635
9. Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci USA. (2004) 101:6659–63. doi: 10.1073/pnas.0308291101
10. Meyer TE, Kovács SJ, Ehsani AA, Klein S, Holloszy JO, Fontana L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J Am Coll Cardiol. (2006) 47:398–402. doi: 10.1016/j.jacc.2005.08.069
11. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. (2000) 403:795–800. doi: 10.1038/35001622
12. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. (2004) 305:390–2. doi: 10.1126/science.1099196
13. Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. (2007) 6:759–67. doi: 10.1111/j.1474-9726.2007.00335.x
14. Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS ONE. (2008) 3:e1759. doi: 10.1371/journal.pone.0001759
15. Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. (2007) 404:1–13. doi: 10.1042/BJ20070140
16. Guarente L. Franklin H. Epstein lecture: sirtuins, aging, and medicine. N Engl J Med. (2011) 364:2235–44. doi: 10.1056/NEJMra1100831
17. Kitada M, Kume S, Kanasaki K, Takeda-Watanabe A, Koya D. Sirtuins as possible drug targets in type 2 diabetes. Curr Drug Targets. (2013) 14:622–36. doi: 10.2174/1389450111314060002
18. Gomes P, Fleming Outeiro T, Cavadas C. Emerging role of Sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol Sci. (2015) 36:756–68. doi: 10.1016/j.tips.2015.08.001
19. McDonnell E, Peterson BS, Bomze HM, Hirschey MD. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol Metab. (2015) 26:486–92. doi: 10.1016/j.tem.2015.06.001
20. Kuang J, Chen L, Tang Q, Zhang J, Li Y, He J. The role of Sirt6 in obesity and diabetes. Front Physiol. (2018) 9:135. doi: 10.3389/fphys.2018.00135
21. Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. (2008) 118:2992–3002. doi: 10.1172/JCI34260
22. de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. (2008) 582:97–105. doi: 10.1016/j.febslet.2007.11.057
23. Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal. (2017) 26:501–18. doi: 10.1089/ars.2016.6755
24. Morgan D, Oliveira-Emilio HR, Keane D, Hirata AE, Santos da Rocha M, Bordin S, et al. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia. (2007) 50:359–69. doi: 10.1007/s00125-006-0462-6
25. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. (2005) 11:191–8. doi: 10.1038/nm1185
26. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. (2010) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
27. Solinas G, Karin M. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J. (2010) 24:2596–611. doi: 10.1096/fj.09-151340
28. Archuleta TL, Lemieux AM, Saengsirisuwan V, Teachey MK, Lindborg KA, Kim JS, et al. Oxidant stress-induced loss of IRS-1 and IRS-2 proteins in rat skeletal muscle: role of p38 MAPK. Free Radic Biol Med. (2009) 47:1486–93. doi: 10.1016/j.freeradbiomed.2009.08.014
29. Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. (2005) 7:1040–52. doi: 10.1089/ars.2005.7.1040
30. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. (2002) 23:599–622. doi: 10.1210/er.2001-0039
31. Bloch-Damti A, Bashan N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid Redox Signal. (2005) 7:1553–67. doi: 10.1089/ars.2005.7.1553
32. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. (2004) 350:664–71. doi: 10.1056/NEJMoa031314
33. Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. (2005) 115:3587–93. doi: 10.1172/JCI25151
34. Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. (2006) 55(Suppl. 2):S9–15. doi: 10.2337/db06-S002
35. Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. (2003) 300:1140–2. doi: 10.1126/science.1082889
36. Kitada M, Koya D. SIRT1 in type 2 diabetes: mechanisms and therapeutic potential. Diabetes Metab J. (2013) 37:315–25. doi: 10.4093/dmj.2013.37.5.315
37. Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. (2004) 23:2369–80. doi: 10.1038/sj.emboj.7600244
38. Yoshizaki T, Milne JC, Imamura T, Schenk S, Sonoda N, Babendure JL, et al. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol Cell Biol. (2009) 29:1363–74. doi: 10.1128/MCB.00705-08
39. Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. (2010) 298:E419–428. doi: 10.1152/ajpendo.00417.2009
40. Schug TT, Xu Q, Gao H, Peres-da-Silva A, Draper DW, Fessler MB, et al. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol. (2010) 30:4712–21. doi: 10.1128/MCB.00657-10
41. Takeda-Watanabe A, Kitada M, Kanasaki K, Koya D. SIRT1 inactivation induces inflammation through the dysregulation of autophagy in human THP-1 cells. Biochem Biophys Res Commun. (2012) 427:191–6. doi: 10.1016/j.bbrc.2012.09.042
42. de Kreutzenberg SV, Ceolotto G, Papparella I, Bortoluzzi A, Semplicini A, Dalla Man C, et al. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes. (2010) 59:1006–15. doi: 10.2337/db09-1187
43. Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, et al. SirT1 regulates adipose tissue inflammation. Diabetes. (2011) 60:3235–45. doi: 10.2337/db11-0616
44. Hui X, Zhang M, Gu P, Li K, Gao Y, Wu D, et al. Adipocyte SIRT1 controls systemic insulin sensitivity by modulating macrophages in adipose tissue. EMBO Rep. (2017) 18:645–57. doi: 10.15252/embr.201643184
45. Fröjdö S, Durand C, Molin L, Carey AL, El-Osta A, Kingwell BA, et al. Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by SIRT1. Mol Cell Endocrinol. (2011) 335:166–76. doi: 10.1016/j.mce.2011.01.008
46. Lee JH, Song MY, Song EK, Kim EK, Moon WS, Han MK, et al. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes. (2009) 58:344–51. doi: 10.2337/db07-1795
47. Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. (2007) 26:1913–23. doi: 10.1038/sj.emboj.7601633
48. Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, et al. Restoration of insulin-sensitive glucose transporter. (GLUT4). gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci USA. (2001) 98:3820–5. doi: 10.1073/pnas.061035098
49. Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. (2006) 116:1784–92. doi: 10.1172/JCI29126
50. Daimon M, Oizumi T, Saitoh T, Kameda W, Hirata A, Yamaguchi H, et al. Decreased serum levels of adiponectin are a risk factor for the progression to type 2 diabetes in the Japanese Population: the Funagata study. Diabetes Care. (2003) 26:2015–20. doi: 10.2337/diacare.26.7.2015
51. Spranger J, Kroke A, Möhlig M, Bergmann MM, Ristow M, Boeing H, et al. Adiponectin and protection against type 2 diabetes mellitus. Lancet. (2003) 361:226–8. doi: 10.1016/S0140-6736(03)12255-6
52. Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. (2001) 7:941–6. doi: 10.1038/90984
53. Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein alpha transcriptional complex. J Biol Chem. (2006) 281:39915–24. doi: 10.1074/jbc.M607215200
54. Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca (2+) and AMPK/SIRT1. Nature . (2010) 464:1313–9. doi: 10.1038/nature08991
55. St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. (2006) 127:397–408. doi: 10.1016/j.cell.2006.09.024
56. Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, et al. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun. (2008) 372:51–6. doi: 10.1016/j.bbrc.2008.04.176
57. Rothgiesser KM, Erener S, Waibel S, Lüscher B, Hottiger MO. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci. (2010) 123:4251–8. doi: 10.1242/jcs.073783
58. Pais TF, Szego EM, Marques O, Miller-Fleming L, Antas P, Guerreiro P, et al. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J. (2013) 32:2603–16. doi: 10.1038/emboj.2013.200
59. Lo Sasso G, Menzies KJ, Mottis A, Piersigilli A, Perino A, Yamamoto H, et al. SIRT2 deficiency modulates macrophage polarization and susceptibility to experimental colitis. PLoS ONE. (2014) 9:e103573. doi: 10.1371/journal.pone.0103573
60. Lin J, Sun B, Jiang C, Hong H, Zheng Y. Sirt2 suppresses inflammatory responses in collagen-induced arthritis. Biochem Biophys Res Commun. (2013) 441:897–903. doi: 10.1016/j.bbrc.2013.10.153
61. Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. (2007) 6:505–14. doi: 10.1111/j.1474-9726.2007.00304.x
62. Wang YP, Zhou LS, Zhao YZ, Wang SW, Chen LL, Liu LX, et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. (2014) 33:1304–20. doi: 10.1002/embj.201387224
63. Lemos V, de Oliveira RM, Naia L, Szegö É, Ramos E, Pinho S, et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum Mol Genet. (2017) 26:4105–17. doi: 10.1093/hmg/ddx298
64. Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, et al. Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. (2012) 26:259–70. doi: 10.1101/gad.180406.111
65. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. (2010) 464:121–5. doi: 10.1038/nature08778
66. Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. (2005) 85:258–63. doi: 10.1016/j.ygeno.2004.11.003
67. Rose G, Dato S, Altomare K, Bellizzi D, Garasto S, Greco V, et al. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp Gerontol. (2003) 38:1065–70. doi: 10.1016/S0531-5565(03)00209-2
68. Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci USA. (2011) 108:14608–13. doi: 10.1073/pnas.1111308108
69. Finley LW, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, et al. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE. (2011) 6:e23295. doi: 10.1371/journal.pone.0023295
70. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. (2010) 143:802–12. doi: 10.1016/j.cell.2010.10.002
71. Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. (2010) 12:662–7. doi: 10.1016/j.cmet.2010.11.015
72. Ogura Y, Kitada M, Monno I, Kanasaki K, Watanabe A, Koya D. Renal mitochondrial oxidative stress is enhanced by the reduction of Sirt3 activity, in Zucker diabetic fatty rats. Redox Rep. (2018) 23:153–9. doi: 10.1080/13510002.2018.1487174
73. Zhou Y, Chung ACK, Fan R, Lee HM, Xu G, Tomlinson B, et al. Sirt3 deficiency increased the vulnerability of pancreatic beta cells to oxidative stress-induced dysfunction. Antioxid Redox Signal. (2017) 27:962–76. doi: 10.1089/ars.2016.6859
74. Kim M, Lee JS, Oh JE, Nan J, Lee H, Jung HS, et al. SIRT3 overexpression attenuates palmitate-induced pancreatic beta-cell dysfunction. PLoS ONE. (2015) 10:e0124744. doi: 10.1371/journal.pone.0124744
75. Caton PW, Richardson SJ, Kieswich J, Bugliani M, Holland ML, Marchetti P, et al. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia. (2013) 56:1068–77. doi: 10.1007/s00125-013-2851-y
76. Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. (2012) 483:218–21. doi: 10.1038/nature10815
77. Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. (2008) 452:492–6. doi: 10.1038/nature06736
78. Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, et al. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle. (2009) 8:2664–6. doi: 10.4161/cc.8.16.9367
79. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. (2009) 136:62–74. doi: 10.1016/j.cell.2008.10.052
80. Lappas M. Anti-inflammatory properties of sirtuin 6 in human umbilical vein endothelial cells. Med Inflamm. (2012) 2012:597514. doi: 10.1155/2012/597514
81. Balestrieri ML, Rizzo MR, Barbieri M, Paolisso P, D'Onofrio N, et al. Sirtuin 6 expression and inflammatory activity in diabetic atherosclerotic plaques: effects of incretin treatment. Diabetes. (2015) 64:1395–406. doi: 10.2337/db14-1149
82. Liu Z, Wang J, Huang X, Li Z, Liu P. Deletion of sirtuin 6 accelerates endothelial dysfunction and atherosclerosis in apolipoprotein E-deficient mice. Transl Res. (2016) 172:18–29. doi: 10.1016/j.trsl.2016.02.005
83. Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, et al. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell. (2010) 9:162–73. doi: 10.1111/j.1474-9726.2009.00544.x
84. Schwer B, Schumacher B, Lombard DB, Xiao C, Kurtev MV, Gao J, et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc Natl Acad Sci USA. (2010) 107:21790–4. doi: 10.1073/pnas.1016306107
85. Lee Y, Ka SO, Cha HN, Chae YN, Kim MK, Park SY, et al. Myeloid Sirtuin 6 deficiency causes insulin resistance in high-fat diet-fed mice by eliciting macrophage polarization toward an M1 phenotype. Diabetes. (2017) 66:2659–68. doi: 10.2337/db16-1446
86. Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki N, et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell. (2013) 52:340–52. doi: 10.1016/j.molcel.2013.09.004
87. Bhardwaj A, Das S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc Natl Acad Sci USA. (2016) 113:E538–547. doi: 10.1073/pnas.1520045113
88. Cui X, Yao L, Yang X, Gao Y, Fang F, Zhang J, et al. SIRT6 regulates metabolic homeostasis in skeletal muscle through activation of AMPK. Am J Physiol Endocrinol Metab. (2017) 313:E493–E505. doi: 10.1152/ajpendo.00122.2017
89. Xiong X, Wang G, Tao R, Wu P, Kono T, Li K, et al. Sirtuin 6 regulates glucose-stimulated insulin secretion in mouse pancreatic beta cells. Diabetologia. (2016) 59:151–60. doi: 10.1007/s00125-015-3778-2
90. Xiong X, Sun X, Wang Q, Qian X, Zhang Y, Pan X, et al. SIRT6 protects against palmitate-induced pancreatic beta-cell dysfunction and apoptosis. J Endocrinol. (2016) 231:159–65. doi: 10.1530/JOE-16-0317
91. Janikiewicz J, Hanzelka K, Kozinski K, Kolczynska K, Dobrzyn A. Islet beta-cell failure in type 2 diabetes–Within the network of toxic lipids. Biochem Biophys Res Commun. (2015) 460:491–6. doi: 10.1016/j.bbrc.2015.03.153
92. Pan H, Guan D, Liu X, Li J, Wang L, Wu J, et al. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. (2016) 26:190–205. doi: 10.1038/cr.2016.4
93. Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. (2007) 450:712–6. doi: 10.1038/nature06261
94. Smoliga JM, Baur JA, Hausenblas HA. Resveratrol and health–a comprehensive review of human clinical trials. Mol Nutr Food Res. (2011) 55:1129–41. doi: 10.1002/mnfr.201100143
95. Tao Y, Huang C, Huang Y, Hong L, Wang H, Zhou Z, et al. SIRT4 suppresses inflammatory responses in human umbilical vein endothelial cells. Cardiovasc Toxicol. (2015) 15:217–23. doi: 10.1007/s12012-014-9287-6
96. Lang A, Anand R, Altinoluk-Hambüchen S, Ezzahoini H, Stefanski A, Iram A, et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging. (2017) 9:2163–89. doi: 10.18632/aging.101307
97. Singh CK, Chhabra G, Ndiaye MA, Garcia-Peterson LM, Mack NJ, Ahmad N. The role of sirtuins in antioxidant and redox signaling. Antioxid Redox Signal. (2018) 28:643–61. doi: 10.1089/ars.2017.7290
98. Zhou L, Wang F, Sun R, Chen X, Zhang M, Xu Q, et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. (2016) 17:811–22. doi: 10.15252/embr.201541643
99. Wang Y, Liu Q, Huan Y, Li R, Li C, Sun S, et al. Sirtuin 5 overexpression attenuates glucolipotoxicity-induced pancreatic β cells apoptosis and dysfunction. Exp Cell Res. (2018) 371:205–13. doi: 10.1016/j.yexcr.2018.08.011
Keywords: SIRT1, SIRT2, SIRT3, SIRT6, Type 2 diabetes
Citation: Kitada M, Ogura Y, Monno I and Koya D (2019) Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function. Front. Endocrinol. 10:187. doi: 10.3389/fendo.2019.00187
Received: 22 July 2018; Accepted: 06 March 2019;
Published: 27 March 2019.
Edited by:
Ralf Jockers, Université Paris-Sorbonne, FranceReviewed by:
Abdu Adem, United Arab Emirates University, United Arab EmiratesHouzao Chen, Chinese Academy of Medical Sciences, China
Zhiping Liu, Augusta University, United States
Sudhanshu Shukla, Amity University, India
Copyright © 2019 Kitada, Ogura, Monno and Koya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Munehiro Kitada, a2l0dGFAa2FuYXphd2EtbWVkLmFjLmpw