 Sonya Galcheva
Sonya Galcheva Hüseyin Demirbilek
Hüseyin Demirbilek Sara Al-Khawaga
Sara Al-Khawaga Khalid Hussain
Khalid Hussain- 1Department of Paediatrics, University Hospital St. Marina, Varna Medical University, Varna, Bulgaria
- 2Department of Paediatric Endocrinology, Hacettepe University Faculty of Medicine, Ankara, Turkey
- 3Division of Endocrinology, Department of Paediatric Medicine, Sidra Medicine, Doha, Qatar
Congenital hyperinsulinism (CHI) is a heterogenous and complex disorder in which the unregulated insulin secretion from pancreatic beta-cells leads to hyperinsulinaemic hypoglycaemia. The severity of hypoglycaemia varies depending on the underlying molecular mechanism and genetic defects. The genetic and molecular causes of CHI include defects in pivotal pathways regulating the secretion of insulin from the beta-cell. Broadly these genetic defects leading to unregulated insulin secretion can be grouped into four main categories. The first group consists of defects in the pancreatic KATP channel genes (ABCC8 and KCNJ11). The second and third categories of conditions are enzymatic defects (such as GDH, GCK, HADH) and defects in transcription factors (for example HNF1α, HNF4α) leading to changes in nutrient flux into metabolic pathways which converge on insulin secretion. Lastly, a large number of genetic syndromes are now linked to hyperinsulinaemic hypoglycaemia. As the molecular and genetic basis of CHI has expanded over the last few years, this review aims to provide an up-to-date knowledge on the genetic causes of CHI.
Introduction
Congenital hyperinsulinism (CHI) is a heterogeneous and complex biochemical disorder which is characterized by the dysregulated release of insulin from pancreatic β-cell (1). In normal physiological state, the secretion of insulin is tightly coupled to glucose metabolism within the β-cell so that the insulin release is regulated to keep the plasma glucose concentration around 3.5–5.5 mmol/L. However, in CHI the secretion of insulin becomes unrelated to glucose metabolism, so that there is inappropriate insulin release for the plasma glucose level (2).
The genetic and molecular cause of CHI includes defects in key genes regulating insulin secretion from the pancreatic β-cell. Molecular defects in previously described genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, UCP2, HNF4A, HNF1A, HK1, PGM1, and PMM2) have been reported (3). However, recent studies have linked the role of other genes (CACNA1D, FOXA2) to hyperinsulinaemic hypoglycaemia (HH) but in some of these cases the underlying molecular mechanisms are still not fully elucidated (Table 1). Understanding the molecular mechanisms of CHI due to these genetic abnormalities has provided unique insight into the normal physiological mechanisms which regulate the insulin release.
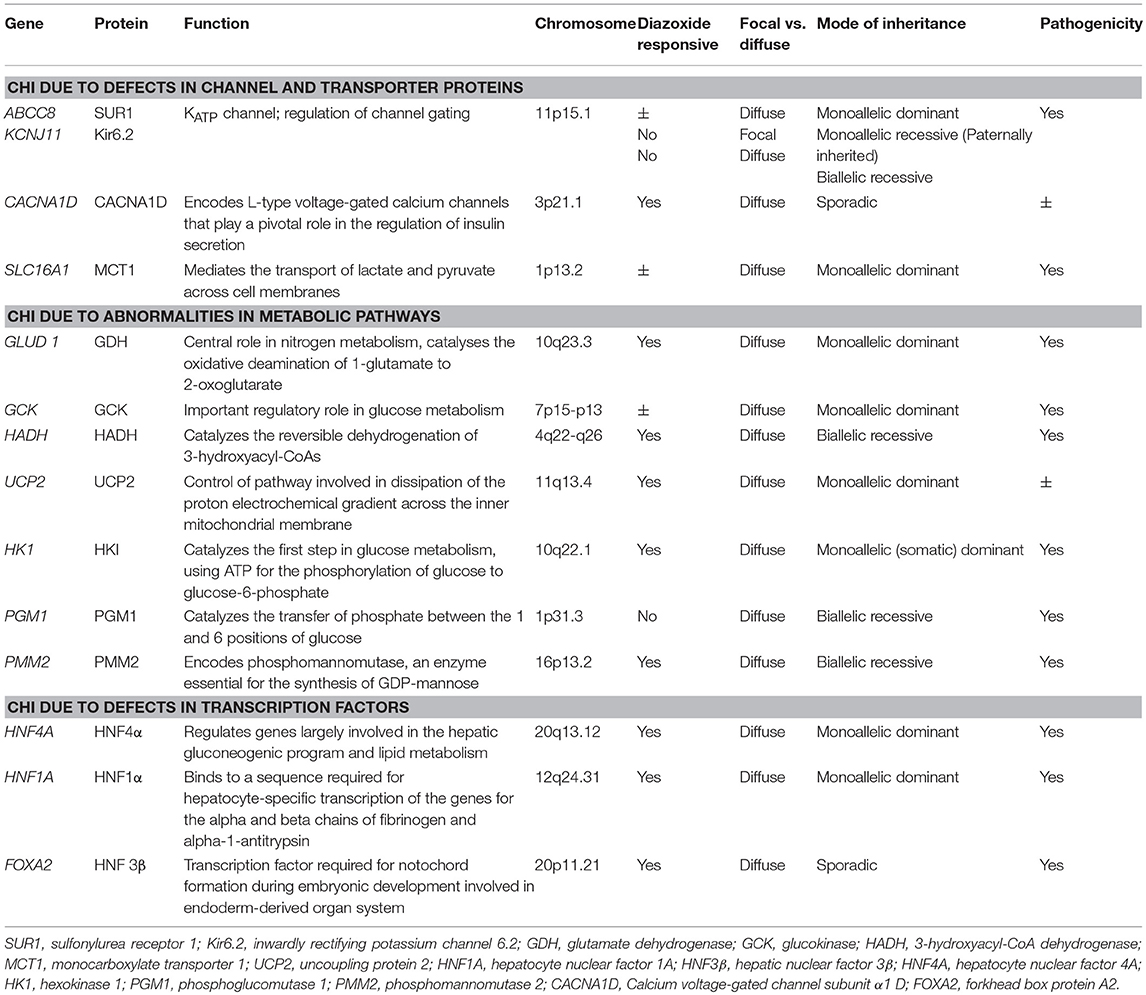
Table 1. Monogenic causes of CHI.
Broadly, these genetic defects leading to unregulated insulin secretion can be grouped into four main categories. The first group consists of defects in genes encoding the pancreatic KATP channels (ABCC8 and KCNJ11) and other channel/transporter proteins (KCNQ1, CACNA1D, SLC16A1) (Figure 1). Pancreatic KATP channels have a critical role in the regulation of insulin release and defects in their encoding genes cause the most prevalent and severe forms of CHI (4). The second and third categories of conditions are enzymatic gene defects (GLUD1, GCK, HADH, UCP2, HK1, PMM2, PGM1) and defects in genes encoding the transcription factors (HNF1A, HNF4A, FOXA2) leading to changes in nutrient flux into metabolic pathways which converge on insulin secretion (5) (Figure 2). The β-cell insulin release is coupled to the metabolic signals and so any perturbation in these pathways will ultimately result in inappropriate insulin secretion. Lastly, there are a large number of syndromic conditions (like Beckwith-Weidemann syndrome) which feature HH as a part of the syndrome while the underlying molecular mechanism leading to unregulated insulin secretion has yet to be clarified in most of the syndromes (6).
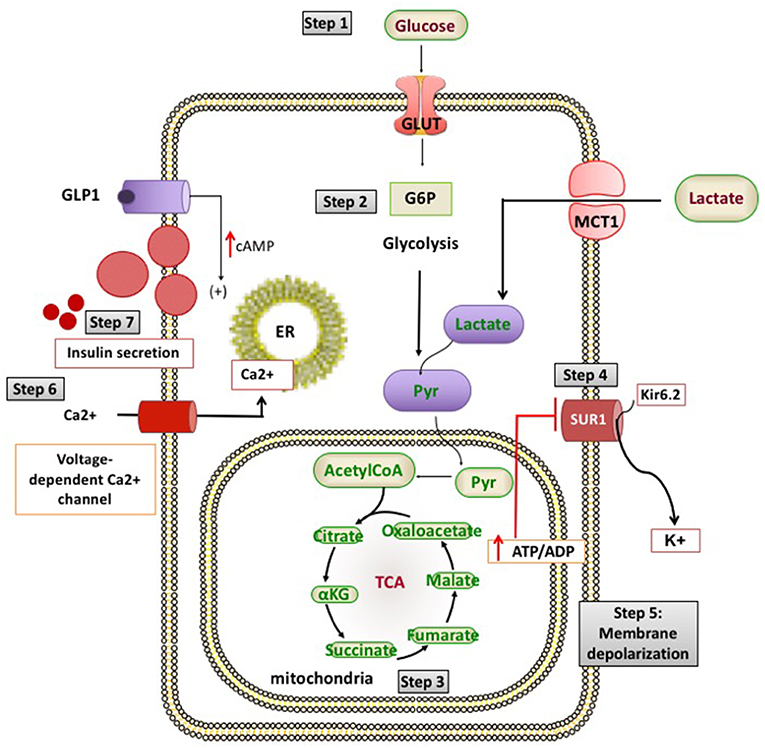
Figure 1. Channel and transporter proteins involved in the process of insulin secretion from pancreatic β-cells. ADP, adenosine diphosphate; ATP, adenosine triphosphate; Ca2+, calcium ions; cAMP, cyclic adenosine monophosphate; G6P, glucose 6-phosphate; GLP1, glucagon like peptide 1; GLUT 2, glucose transporter 2; K+, potassium; Kir6.2, inward rectifier potassium channel 6.2; MCT1, monocarboxylate transporter 1; Pyr, pyruvate; SUR1, sulfonylurea receptor 1; TCA, tricarboxylic acid.
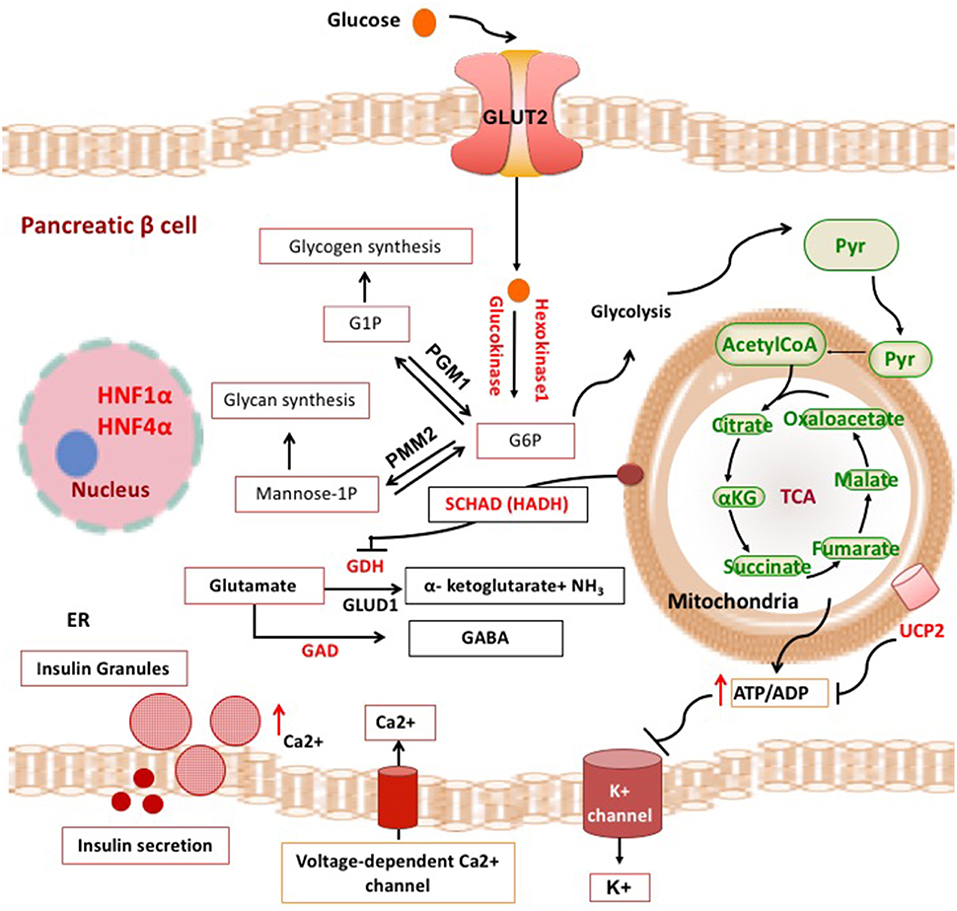
Figure 2. Metabolic pathways and transcription factors related to the development of CHI. ADP, adenosine diphosphate; ATP, adenosine triphosphate; αKG, alpha ketoglutarate; Ca2+, calcium ions; ER, endoplasmic reticulum; G1P, gluscose-1-phosphate; G6P, glucose 6-phosphate; GABA, γ-aminobutyric acid; GAD, glutamate decarboxylase enzyme; GDH, glutamate dehydrogenase; GLUT, glucose transporter; HADH, hydroxyacyl-CoA dehydrogenase; HNF1α, hepatocyte nuclear factor 1α; HNF4α, hepatocyte nuclear factor 4α; K+, potassium; NH3, ammonia; PGM1, phosphoglucomutase 1; PMM2, phosphomannomutase 2; Pyr, pyruvate; TCA, tricarboxylic acid; UCP2, mitochondrial uncoupling protein 2.
In this article, we review the state of the art knowledge on the genetic and molecular basis of all the different types of CHI and discuss a possible new classification of the molecular basis of CHI.
β-Cell Glucose Sensing and the Role of KATP Channels in Insulin Release
The rise of plasma glucose levels after feeding triggers insulin release from the β-cells. The glucose enters the β-cells via the facilitated diffusion driven by the glucose transporter 2 (GLUT2), where it undergoes glycolytic phosphorylation mediated by the enzyme glucokinase (GCK, a hexokinase) and converts to glucose-6-phosphate (G6P). Recently, it has been shown that other hexokinases, particularly HK1, are also expressed in the neonatal β-cells but the gene expression is downregulated after birth during the maturation of the β-cells (7, 8).
GCK is a glucose sensor linking the extracellular concentration of glucose and its metabolism in the β-cells (9, 10). The rise of the plasma glucose leads to an enhanced GCK activity producing energy rich compounds (ATP) and results in an increase in the cytoplasmic ATP:ADP ratio. The β-cell insulin release is mostly regulated by the ATP-sensitive potassium (KATP) channel located in the membrane of the pancreatic β-cells (11). These channels have a crucial role in the glucose homeostasis as they link its metabolism to the membrane electrical excitability with a consequent β-cells secretion of insulin (12, 13).
The β-cell KATP channel is a large hetero-octamer with a 4:4 stoichiometry, composed of 4 pore-forming inwardly-rectifying K+ channel subunits (Kir6.2) and 4 high-affinity regulatory SUR1 subunits. The Kir6.2 subunit, a “weak” inward rectifier (14), contains two putative transmembrane domains (TMD1 and TMD2), bound by an extracellular loop (H5), and cytosolic -NH2 and -COOH terminal domains containing ~70 and ~220 amino acid residues, respectively.
The Kir6.2 subunit of KATP channel affects the biophysical characteristics of the channel complex such as K+ selectivity, rectification, activation by acyl-CoAs and inhibition by ATP (11, 15). The SUR1 subunit provides the channels with a susceptibility to the stimulatory effects of MgADP and confers on the channels a responsiveness to pharmacological activators (diazoxide) and inhibitiors (sulfonylureas) (16). It promotes ATP hydrolysis without directly transporting the substrates and regulates the Kir6.2 activity in the KATP channel complex.
Only fully-assembled channel complexes which are properly transported to the surface of the cell membrane (trafficking) can operate correctly (17). Hence, only octameric KATP channels can be expressed on the plasma membrane surface due to the masking of the endoplasmic reticulum (ER) retention signals (RKR) exposed in partially-assembled channels (18).
The key regulators of KATP channels are the changing concentrations of intracellular nucleotides. Adenine nucleotides are capable of stimulatory interacting with Mg2+. However, the non-hydrolytic ATP binding takes advantage over KATP channels in the lack of Mg2+. The increased cytosolic ATP:ADP ratio inhibits the KATP channel activity and reduces the potassium ions efflux across the plasma membrane adjusting its potential. Thus, the basal activity of the channels produce a slow depolarization until a threshold membrane potential is reached that increases the open probability of calcium channels. This process changes the physiological conformation of the potassium channels, which results in their closure, followed by chronic plasma membrane depolarization and Ca2+ influx due to the voltage-gated calcium channels activation (19). The increased intracellular Ca2+ activates specific pathways, such as protein kinases A and C, leading to impaired glucose sensing, insulin release from the storage granules and its subsequent secretion into the bloodstream (20, 21). In a similar way, when glucose concentrations are low, the KATP channels open, causing membrane hyperpolarization and subsequently inhibited release of insulin (22).
CHI Due to Defects in Channel and Transporter Proteins
Pancreatic β-Cell KATP Channel Defects (ABCC8 and KCNJ11 Gene Mutations) and CHI
Mutations in ABCC8 and KCNJ11 genes account for the main genetic causes of CHI, characterized by defective pancreatic β-cell KATP channel subunits (SUR1 and Kir6.2, respectively). Both genes are mapped on the same chromosome (11p15.1), being divided by a small part of DNA (4.5 kb) (16, 20, 23). The ABCC8 gene is a large gene spanning more than 100 kb of DNA, divided in 39 exons (20). The KCNJ11 gene consists only 1 exon encoding a protein (Kir6.2) with a molecular weight of about 43kDa (17).
ABCC8 or KCNJ11 gene mutations have been found in ~50% of CHI patients (24). Based on the mutation data from the Human Gene Mutation Database (HGMD) (www.hgmd.org, accessed February 2018), 448 homozygous, compound heterozygous and heterozygous inactivating ABCC8 mutations (25–28) and around 66 KCNJ11 (26, 29) mutations have been reported, associated with differentially expressed clinical symptoms.
Recessive and dominant ABCC8/KCNJ11 mutations may influence the proper function of the KATP channels either by disrupting their cell surface expression or by affecting the MgADP stimulation of their activity. As a result, in both cases, the pancreatic β-cell membrane will be continuously depolarized with uncontrolled release of insulin despite severe hypoglycaemia (15, 30). The first type of ABCC8/KCNJ11 mutations affecting the channel membrane expression impairs the SUR 1 synthesis or maturation, leading to proteins which do not reach the plasma membrane. These mutations can also cause impaired SUR1 trafficking. Since the Kir6.2 expression depends on the surface co-expression of SUR1, in these cases the Kir6.2 surface expression is also disturbed and it is missing from the membrane (4). The second type of mutations produce non-functional KATP channel complexes which are insensitive to the changing MgADP concentrations and stay closed even when the glucose level drops too low (4).
Recessive Inactivating Mutations (Homozygous or Compound Heterozygous)
In ABCC8 and KCNJ11 genes generally cause the most severe cases of CHI, which usually do not respond to diazoxide therapy and often require a resection of the pancreas (31). However, some compound heterozygous mutations may cause milder hyperinsulinism which is responsive to diazoxide (32). The molecular basis of these recessive mutations comprises of defects in the biogenesis and turnover of KATP channels (33), their abnormal exit from the ER and trafficking to the cytoplasmic membrane (34), and channel changes as a result of nucleotide regulation and frequency of open-state (35).
Recessive ABCC8/KCNJ11 Mutations With Defects in Channel Biogenesis and Turnover
A pulse labeling study has shown that both channel subunits have long-lived species with a half-life of about 24 h, which is in accordance with the idea for a slow assembling of the KATP channel (33). The Kir6.2 turnover is biphasic when the subunit is expressed individually, as about 60% having a half-life of 36 min, while the rest changes to species with a half-life of 26 h. Expressed alone SUR1 is long-lived in the ER with a half-life of more than 25 h. Being co-expressed, both channel subunits associate rapidly with an elimination of the rapid degradation of Kir6.2 (33). After the termination of their assembly, the channels pass to the Golgi apparatus and SURs change into the mature, completely glycosylated form.
For example, the F1388del and W91R mutations in ABCC8 and KCNJ11 genes, respectively, which are related to the development of a critical form of CHI in patients, change the subunit turnover rate (33). The mutant subunits connect with their relevant subunits but then dissociate and undergo a rapid degradation, affecting the KATP channel biogenesis and turnover. Homozygous patients have no functional KATP channels on the pancreatic β-cell surface, while heterozygous carriers possess a reduced complement of normal channels.
Recessive ABCC8/KCNJ11 Mutations With Defects in Channel Trafficking
Channel trafficking requires the RKR signals, present in both channel subunits, to be masked during their assembly. ABCC8 mutations (F1388del, L1544P) result in a defective trafficking as they influence the exit of the subunits from the ER (36, 37). Several KCNJ11 mutations (Y12X, H259R, E282K) have also been described as causing a trafficking defect and truncated non-functional proteins (33, 38, 39).
Recessive ABCC8/KCNJ11 Mutations With Defects in Channel Regulation
Since the SUR1 protein operates as a conductance regulator of Kir6.2, both channel subunits are susceptible to alterations in adenosine and guanosine nucleotides. The KATP channel regulation involves interactions of nucleotides at the subunits with an ATP-provoked Kir6.2 inhibition, which is opposed by the ADP-activation at SUR1. Some mutations in ABCC8 (T1139M, R1215Q) have been reported to affect the channel conductance and lead to a loss of ADP-dependent gating, causing the ATP-induced channel inhibition (40). The R1420C mutation in ABCC8 gene, located in NBD2 of SUR1 subunit, also reduces the channel affinity for ATP and ADP (41).
Dominant Inactivating ABCC8/KCNJ11 Mutations
Show normal KATP channel assembly and trafficking to the plasma membrane (42). They are uncommon and typically lead to a milder form of CHI presented at a later age, although the phenotype may vary from asymptomatic to persistent HH (43).
Interestingly, in adult carriers dominant mutations can cause dominantly inherited diabetes (44, 45). The possible mechanisms for that have been imputed to the apoptosis of the β-cell (46) mediated by the increased cell depolarization and subsequent Ca2+ flow into the β-cell. However, the predisposition to diabetes later in life related to dominant mutations is controversial. Huopio et al. (45) have reported a development of impaired glucose tolerance in 75% of the mothers during pregnancy while Pinney et al. (42) have not seen such a positive trend toward diabetes in mutation carriers. More recently, we reported a family carrying a novel missense c.511C>T (p.L171F) variant in exon 4 of ABCC8 which lead to adult onset diabetes in heterozygous carriers, while a homozygous carrier developed HH at neonatal period and diabetes later in life (47).
An important feature of the dominant inactivating ABCC8/KCNJ11 mutations is the responsiveness of the patients to diazoxide treatment compared to those with a recessive condition (42, 44, 45, 48). However, dominant forms unresponsive to medications have also been described (49). For example, MacMullen et al. have reported patients with medically unresponsive diffuse CHI caused by dominant ABCC8 mutations, where 15 of all 17 patients have need a near-total pancreatectomy (50). These patients are born large for their gestational age having a similar age at clinical presentation to that of individuals with recessive mutations. The functional analyses have shown severely impaired channel responses to diazoxide and MgADP-activation; however, the channels have normal trafficking to the membrane like mutations which result in a dominant disease responsive to diazoxide therapy (50). It has been hypothesized that the mutations change the nucleotide hydrolysis rates affecting the diazoxide activity or they alter the coupling of MgATP-hydrolysis to the activation of the channel. It is believed that the MgATP-hydrolysis lead to desensitization to the ATP inhibition, so diazoxide and MgADP stabilize the desensitized condition of the KATP channel (51).
Most of the reported dominant medically-unresponsive variants have been located in the NBDs of the SUR1 subunit, including the same gene regions as many of the dominant medically-responsive SUR1 mutations (50) as well as the regions of many of the known recessive mutations (49). It has been shown that the involvement of Walker A and B motifs of NBD1 seems to be restricted to dominant mutations unresponsive to diazoxide treatment, whereas, the mutations affecting NBD2 are associated with both diazoxide-responsive and unresponsive hyperinsulinism (51).
A dominant Kir6.2 missense mutation (F55L) causing CHI has been reported to decrease KATP channels open probability without having an influence on the channel expression (35). The reduced channel activity is related to its low response to membrane phosphoinositides/long-chain acyl-CoAs (41). Recently, a novel phenomenon has been described in a transient CHI patient, reporting the co-existance of heterozygous ABCC8 and KCNJ11 gene mutations (52).
The detection of a paternally inherited monoallelic KATP channel mutation with a post-zygotic loss of the corresponding maternal region on chromosome 11 usually cause focal adenomatous hyperplasia accounting for 30–40% of the cases with CHI (15, 25, 53, 54). The loss of heterozygosity leading to paternal isodisomy makes the β-cells channel defects biallelic in the abnormal foci, changing the channels and causing abnormal secretion of insulin into the lesion (53). The subsequent disproportion in the expression of several imprinted genes involved in cell proliferation within the 11p15 region (decreased expression of the maternal tumor suppressor genes CDKN1C and H19 and expression of the paternal IGF2) results in focal hyperplasia of the islet cells (55, 56).
Focal CHI due to paternally inherited monoallelic ABCC8/KCNJ11 mutations is usually unresponsive to medical treatment (57), although a diazoxide-responsive focal form has recently been reported (58). In the medically unresponsive children, genetic testing should be followed by 18F-DOPA PET/CT scanning in order to determine the focal or diffuse subtypes of hyperinsulinism prior to initiating the appropriate treatment to cure the patient—targeted lesionectomy in focal forms or near-total pancreatectomy in diffuse CHI (59). Figure 3 outlines the genetic causes and treatment approaches to different histological subtypes of CHI.
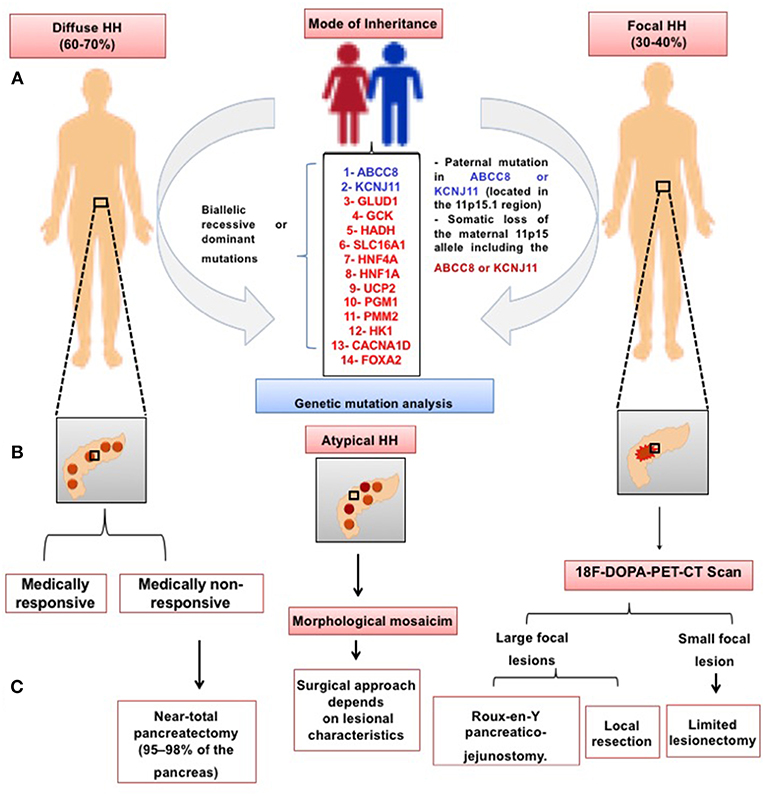
Figure 3. Schematic presentation of the histological subtypes of CHI. In the diffuse CHI, there is a global hyperchromatic β-cell enlargement and hyperplasia. In the focal subtype, β-cell hyperplasia is limited to a certain region of the pancreas with a superficial or deep localization. (A) Mode of inheritance and the genetic causes of CHI in the diffuse and focal subtype, respectively; (B) Schematic illustration of diffuse, atypical and focal subtypes; (C) Management approaches to different forms of CHI.
There are studies showing that some individuals with paternally inherited KATP mutations may respond to diazoxide or resolve spontaneously (57, 60). They are likely to have dominant acting CHI, exerting the effect of the heterozygous ABCC8/KCNJ11 mutations by a dominant-negative mechanism leading to diffuse CHI (27, 36, 60, 61). Possible mechanisms of how heterozygous mutations can cause a diffuse disease could be a post-zygotic mutation affecting the other allele (“second hit” in the pancreas) or inability to identify a channel mutation inherited from the mother due to localization in a non-coding region of the genes (62).
A novel genetic mechanism of some atypical diffuse forms of CHI has been described in an individual with a nonsense heterozygous ABCC8 mutation (Q54X) due to mosaic segmental paternal isodisomy (63). The absence of focal disease in this case has been explicated by the absence of uniparental disomy at the 11p15.5 region.
Mutations in KCNQ1 and HH
It has been reported that the pore-forming region of the α-subunit of a voltage-gated potassium channel (Kv7.1) is encoded by the KCNQ1 gene. The channel, located in the heart, inner ear, stomach, colon and pancreatic β-cells, is crucial for ion homeostasis in the tissues. The gene mutations are associated with cardiac arrhythmias (i.e., the hereditary long QT syndrome (LQTS), deafness and defects of the gastrointestinal system (64). Recently, it has been reported the presence of HH in individuals with LQTS caused by KCNQ1 mutations (65). These patients experience increased release of insulin during an oral glucose tolerance test with hypoglycaemic episodes following the prolonged test. Although the Kv7.1 role in the glucose metabolism is not fully ellucidated, data suggest that this channel may regulate insulin secretion by controlling the process of repolarization of plasma membrane.
Mutations in CACNA1D and HH
Calcium Voltage-Gated Channel Subunit Alpha1 D (CACNA1D) encodes an L-type voltage-sensitive Ca2+ channel that affects the regulation of insulin release from the β-cells (66, 67). Activating germline mutations in CACNA1D gene have previously been found in patients with primary hyperaldosteronism, neuromuscular abnormalities, and transient diazoxide-responsive hypoglycaemia (68). In a recent study the same CACNA1D gene mutation has been shown in a patient with HH, cardiac defects and severe hypotonia (69). This mutation causes an activation of the L-type Ca2+ channel and leading the channel remain open at a lower membrane potential, thereby results in dysregulated insulin secretion (67, 68). Taking into consideration the role of these channels in the process of β-cell insulin release, the heterozygous c.1208G>A (p.G403D) mutation detected in present and previously published cases, suggested CACNA1D as one of the candidate genes for the underlying molecular genetic etiology of CHI. In addition, although the reported patient was diazoxide-responsive, considering the insulin secretion physiology from the β-cells and the role of Ca2+ influx from the voltage gated Ca2+ channels, it was suggested that calcium channel blockers would be a more effective option for the medical management of these patients (69). However, the underlying molecular mechanism leading to CHI is still not clear and requires further investigations and more experiences on HH patients with mutations of this gene.
Mutations in SLC16A1 Gene and CHI
The monocarboxylate transporter 1 (MCT1) catalyses the rapid transport of monocarboxylates (pyruvate and lactate) into the cells. It is encoded by the SLC16A1 gene mapped on chromosome 1p13.2-p12. This gene consists of 5 exons and 4 introns, spanning approximately 44 kb (70). Under normal physiological conditions, there is a low MCT1 expression in the pancreatic β-cells, with minimized intracellular concentrations of pyruvate and lactate which do not increase the insulin release (67). However, dominant activating mutations in the SLC16A1 gene promoter cause an enhanced MCT1 expression in the pancreatic islets, followed by an increased uptake of pyruvate and its metabolism in the Krebs cycle, with a subsequently increased production of ATP and insulin exocytosis (71).
The activating SLC16A1 mutations cause CHI known as exercise-induced hyperinsulinism. The condition is characterized by hypoglycaemic episodes which usually occur within 30–45 min. after strenuous activity, in response to the accumulation of pyruvate and lactate, acting as insulin secretagogues. Sometimes diazoxide therapy may not prevent hypoglycaemia, so avoiding intense physical activity and carbohydrate intake during or after the exercise may require as a part of the treatment (72).
Although previously reported mutations have been limited to the SLC16A1 promoter region, recently the first intragenic heterozygous mutation (c.556C>G, p.L186V) has been described in a child with CHI (73). This mutation has been found to be “probably damaging” leading to gene overexpression or baseline low expression of a mutant transport protein, followed by its enhanced transport into the pancreatic cells with subsequent increase in insulin release (73).
CHI Due to Abnormalities in Metabolic Pathways
Other known genetic causes of CHI involved in β-cell dysregulation and abnormal insulin secretion have been identified. These involve defects in metabolic pathways converging on insulin secretory mechanisms.
Mutations in GLUD1 Gene and CHI (Hyperinsulinism/Hyperammonaemia Syndrome)
The GLUD1 gene is mapped on chromosome 10q23.3. The gene consists of 13 exons and encodes the homohexameric enzyme glutamate dehydrogenase (GDH) (74), present in the mitochondrial matrix and mainly expressed in the liver, kidneys, brain and pancreas (75). GDH catalyses the reversible oxidation of the amino acid L-glutamate to α-ketoglutarate (αKG) and ammonia in the liver and kidney requiring NAD+ or NADP+ as a co-enzyme. In the pancreatic β-cell GDH catalyses the synthesis of αKG, a substrate for the citric acid cycle and causes an increased intracellular ATP:ADP ratio, activating the KATP channel with subsequent cell depolarization and insulin exocytosis (76, 77). GDH activity can be influenced by allosteric effectors working as activators (ADP and leucine) or inhibitors (GTP and ATP) (78).
Activating “de novo” or dominant missense GLUD1 mutations decrease the enzyme sensitivity to the GTP allosteric inhibition and upregulate its activity in the presence of amino acid leucine, followed by increased insulin secretion (76). Interestingly, in a mutant GDH mouse model carrying the H454Y mutation, in addition to the loss of GTP inhibition on GDH activity, there is also an inhibition of glucagon secretion which contributes to the hypoglycaemic phenotype (79).
Activating GLUD1 mutations are the second most common cause of CHI. They are identified mainly in the GTP allosteric binding site (exons 11 and 12) (80). Mutations have also been found in the catalytic site (exons 6 and 7), leading to a regular enzyme activity but reduced sensitivity to GTP (81). The exon 10 in the antenna-like region is the third domain where GLUD1 mutations have been reported (82). The mutations cause a hyperinsulinism/hyperammonaemia syndrome (HI/HA), which is a protein/leucine-sensitive form of CHI (83, 84). It is characterized by dysregulated insulin release and persistent elevated production of ammonia (mainly in the kidney) (85).
The HI/HA syndrome, a milder form of CHI, usually presents in late infancy and early childhood. It is characterized by a normal birth-size, fasting or postprandial protein/leucine-stimulated hypoglycaemia, with persistent but asymptomatic hyperammonaemia (86, 87). The latter biochemical characteristic (hyperammonaemia) is typical for the syndrome. The ammonia levels are usually 3–5 times above the normal values but the hyperammonaemia does not appear to cause symptoms of ammonia toxicity such as lethargy, headache, vomiting, coma, etc. However, some patients (mainly those with mutations in exons 6 and 7) have episodes of epilepsy, suggesting that the condition may have a direct effect on the brain (86, 88). The suspected mechanisms for the pathogenesis of generalized epilepsy, learning disorders and developmental delay in HI/HA syndrome involve the influence of the persistent hyperammonaemia, hypoglycaemic injury of the brain and shift in the glutamate metabolism leading to decreased synthesis of inhibitory neurotransmitter γ-amino-butyric acid (GABA) in the brain due to GDH hyperactivity (88). However, there is no confirmative data for the association between ammonia levels and the risk for epilepsy or other neurological sequelae in these patients. Furthermore, there are individuals with GLUD1 mutations who have normal ammonia concentrations, probably as a result of the mosaicism for the GDH activity(86).
Recently, a study has reported the first homozygous activating mutation of GLUD1 in a neonate who has presented with severe hypoglycaemia, hyperammonaemia, and seizures soon after birth (89). The mutation analysis has shown a novel frameshift mutation (c.37delC) inherited from the asymptomatic mother and a de novo activating mutation (p.S445L) near the GTP binding site, leading to a more severe loss of the GTP inhibiting effects on GDH (89).
Mutations in Glucokinase (GCK) Gene and CHI
The glucokinase (GCK) gene is mapped on chromosome 7 and contains 12 exons encoding for a monomeric protein with a molecular weight of about 52 kDa. The gene has tissue specific promoters, responsible for the transcription initiation in a mutually exclusive manner in distinct tissues such as the pancreas, liver, and the brain (90).
The GCK is a hexokinase isoenzyme, that has a substantial role in the carbohydrate metabolism by acting as a glucosensor for the β-cells, regulating glucose-induced insulin release. It mediates the phosphorylation of glucose molecules on carbon 6 to produce G6P, which is the first step in glycolysis (91). GCK activity has a crucial role in coupling plasma glucose levels to insulin release due to its biochemical characteristics such as a low affinity for glucose, lacking inhibition by G6P and a cooperativity with respect to glucose (92).
Heterozygous GCK activating mutations cause an increased glucose affinity of the enzyme leading to hyperinsulinism. So far, 15 activating mutations associated with HH have been reported, the majority of which group at the allosteric activator domain of the enzyme (91, 93, 94). The latter is the site where small-molecule activators bind, converting the GCK enzyme from its closed (active) to an open inactive state, including three intermediate stages (95). Natural ligands and activating mutations delay the changes in the shape of the GCK macromolecule when passing through the intermediate stages, suggesting the pivotal role of the allosteric domain in regulating the enzyme activity (96, 97).
Patients presenting with GCK-CHI have a wide spectrum of clinical presentations with a varying severity of the symptoms. Children usually have a family history of hypoglycaemia and age at clinical diagnosis ranging from birth to adulthood (98–101). Although most of the reported GCK mutations lead to CHI responsive to diazoxide therapy (98, 99), some patients may need more aggressive treatment to control hypoglycaemia such as octreotide administration or surgery (100, 102). For example, the Y214C mutation in the putative allosteric activator site has been found in a patient with medically-unresponsive CHI (103).
Mutations in HADH Gene and CHI
The oxidation of fatty acids is an essential process that generates ketone bodies serving as alternative metabolic substrates, used in the state of prolonged fasting. The enzyme L-3-hydroxyacyl-CoA-dehydrogenase (HADH) is a mitochondrial oxidoreductase that catalyses the third step of fatty acid β-oxidation, converting the 3-hydroxyacyl–CoA to 3-ketoacyl-CoA. The coding HADH gene is mapped on chromosome 4q22-26 and comprises 8 exons spanning approximately 49 kb (104). Its expression in the β-cells is controlled by different transcription factors, pivotal for the correct cellular differentiation and function (105).
Recessive inactivating HADH gene mutations have been described in CHI patients leading to a reduction in the enzyme levels (106–108). The HADH deficiency results in a loss of its inhibitory effect on GDH, causing a GDH overstimulation, followed by an increased intracellular ATP and enhanced secretion of insulin (109). This GDH activation is restricted mainly to the pancreatic islets and does not lead to elevated ammonia levels compared to the cases with HI/HA syndrome. Therefore, HADH mutations cause a protein/leucine-sensitive CHI due to the close link between the oxidation of fatty acids, amino acid metabolism and insulin release (110).
Patients may have heterogeneous clinical presentations which vary between mild late-onset fasting or protein/leucine-sensitive hypoglycaemia and severe hypoglycaemia soon after birth (106, 108, 111). Some affected individuals may also have abnormally raised metabolites of acylcarnitine such as plasma 3-hydroxybutyrylcarnitine and urinary 3-hydroxyglutaric acid, with no signs of liver or cardiac dysfunction (107, 110). Most children respond to diazoxide therapy, and genetic testing is suggested when patients are from consanguineous families and negative for KATP channel mutations (112).
Mutations in the Mitochondrial UCP2 Gene and CHI
The uncoupling protein 2 (UCP2) belongs to an inner mitochondrial anion-carrier family and is encoded by the UCP2 gene. It is widely expressed in many tissues, including the pancreas (113, 114). UCP2 facilitates the transfer of protons across the inner membrane of mitochondria, thereby separates the mitochondrial oxidative phosphorylation from the synthesis of ATP. Since the increased intracellular ATP synthesis is the key stimulator for sensing glucose and insulin release from the pancreatic islets, the expression of UCP2 decreases the ATP synthesis and negatively regulates the glucose-induced release of insulin (114–116). Furthermore, it has been reported that the UCP2 overexpression in isolated rat β-cells downregulates the ATP synthesis and inhibits the glucose-mediatied secretion of insulin, while in UCP2 knockdown models glucose mediated insulin secretion has shown to be enhanced (117–119). Therefore, inactivating heterozygous mutations of this gene would enhance the glucose oxidation and increase intracellular ATP synthesis leading to inappropriate insulin secretion (114, 118).
CHI due to UCP2 mutations can present with a variable clinical phenotype ranging from transient CHI to prolonged diazoxide-responsive HH (114, 120, 121). In one study UCP2 variants were found in 2.4% of 211 diazoxide-responsive CHI patients (121). However, in a more recent study, no protein truncated variants have been detected in the UCP2 gene among 206 diazoxide-responsive CHI patients (122). The only variant detected was considered to be a common polymorphism suggesting the necessity of further investigations of the UCP2 role in CHI.
Somatic Overexpression of Hexokinase 1 (HK1) and CHI
Hexokinases are a group of enzymes that catalyse the initial step in the glucose utilization, phosphorylating the hexoses (including glucose) due to the enzyme ability to transfer a phosphate group from ATP to the substrates. The phosphorylated glucose (G6P) is the most important product which serves as an intermediate substrate for glycolysis, glycogen synthesis and pentose phosphate pathway. There are four hexokinase isoenzymes (HKI-IV) encoded by different genes, which are expressed at a different rate in various tissues (123). Of those, the HK-IV or GCK, has a much lower affinity for glucose and it is expressed only when an excessive amount of glucose is present. However, HK-I has about 50-fold higher affinity for glucose and at the low glucose levels, HK-I is the dominant isoenzyme catalyzing the glucose phosphorylation. The HK1 gene, mapped on chromosome 10q22.1, encodes the enzyme HK-I. This enzyme predominatly serves G6P to the glycolytic pathway for energy production. Its enzymatic activity is allosterically inhibited by the end-product, G6P. The elevated G6P plays a signal transducing role indicating the lack of requirement of G6P for energy production, thus suppressing the expression of HK1. On the other hand, the HK1 expression at low glucose levels is silenced in the pancreatic β-cells preventing from inappropriate insulin release (8).
Recently, in a large family with dominant CHI, the responsible region has been mapped on chromosome 10, containing 48 genes (124). The first cases from this family had been reported by McQuarrie in 1954 as “idiopathic infantile hypoglycaemia” (120). Pinney et al. have investigated this large four-generation Northern European family with multiplex members with diazoxide-responsive hypoglycaemia and detected three novel variants in the HK1 non-coding regions, suggesting the possibility of an inhibition of the HK1 suppression in the β-cells as a result of a mutation (124). In an in-vitro study evaluating the pancreatic specimens of five CHI patients the role of the HK1 overexpression has been shown in a group of β-cells (125). In these patients, although the KATP channels were functional, there was an inappropriate insulin release in the presence of hypoglycaemia (125).
Mutations in Phosphoglucomutase 1 (PGM1) Gene and CHI
Phosphoglucomutases (PGM) are a group of enzymes that catalyse the interconversion of G6P and G1P (126). There are five different isoenzymes of PGM encoded by different genes. The enzyme phosphoglucomutase-1 is encoded by the PGM1 gene, mapped on chromosome 1p31. It is a phosphotransferase that catalyses the reversible phosphate transfer between positions 1- and 6- of the glucose molecule, thus being involved in the glycogen synthesis, glycogen degradation and glycoprotein synthesis (126). Mutations in PGM1 typically show a broad range of clinical manifestations resembling glycogenosis (Type XIV) as well as mixed-type congenital disorders of glycosylation (CDG type1t) (127–130). Recently, Tegtmeyer et al. have shown 19 patients from 16 families with a variety of clinical features suggesting CDG and atypical isoelectric focusing findings (128). Interestingly, the first clinical finding at birth (in 16 of 19 patients) was a bifid uvvula, but later the patients developed various manifestations including malignant hyperthermia, hepatopathy, secondary hypogonadism, short stature, hypoglycaemia, dilated cardiomyopathy and cardiac arrest. These patients were found to have recessive inactivating PGM1 mutations, which has previously demonstrated to be related to the development of hypoglycaemia, similar to glycogenoses (127, 128). The PGM1 mutations normally cause fasting hyperketotic hypoglycaemia, but in some cases a postprandial hypoketotic HH has been reported (131). It is proposed that the PGM1 mutations cause HH due to the decreased threshold for glucose-mediated insulin release from the pancreatic cells (131). A later report (132) on two of the patients from the publication by Tegtmeyer (128) has shown more pronounced hypoglycosylation in childhood than in adults with a total PGM1-mRNA level reduced to 0.25% of that of normal controls. The study has also reported that hypoglycaemia might occur in PGM1 deficient patients by starving and would be exaggerated by strong exercise (132).
Mutations in Phosphomannomutase 2 (PMM2) Gene and CHI
The enzyme phosphomannomutase 2 (PMM2) is involved in the N-glycosylation of proteins leading to glycopreotein synthesis. In the process of N-glycosylation large carbohydrate molecules (glycans) are covalently-attached to the N-terminal of the proteins (133). Although, the N-glycosylation does not change the conformation of protein structures, it reduces their dynamic fluctuation, leading to an enhanced protein stability (133). The PMM 2 enzyme is encoded by the PMM2 gene, mapped on chromosome 16p13.2. The enzyme is a phosphotransferase catalyzing the interconversion of mannose-6-phosphate to mannose-1-phosphate which constitutes one of the early steps of the glycosylation process. Homozygous recessive mutations of PMM2 are responsible for a variety of symptoms related to many organ systems, which include CDG type 1a (134, 135). The severity of the disease is closely associated with the remaining enzymatic activity, therefore, it is extremely variable ranging from a lethal embryopathy to a mild subclinical disease diagnosed in adulthood (136, 137). HH has previously been reported as a part of the wide-range manifestations of CDG type Ia (138). Recently, in 17 children from 11 unrelated families, a recessively inherited mutation (c.-167G>T) found in the PMM2 gene promoter has been shown to result in a medically-responsive form of hyperinsulinism, associated with congenital polycystic kidney disease (139). This mutation changes the formation of the tissue-specific chromatin loop with an organ-specific gene expression which could explain the selective organ involvement in this condition. The functional analysis of the mutation has revealed dysregulated insulin secretion. Therefore, the PMM2 gene might take a part in regulating insulin release from the pancreatic islets.
CHI Due to Defects in Transcription Factors
Muations in HNF1A&4A and CHI
Transcription factors for nuclear hormone receptors (hepatocyte nuclear factors 1α and 4α (HNF1α and HNF4α) are expressed in the β-cells regulating the glucose-induced secretion of insulin (140–142). These factors are encoded by the HNF1A and HNF4A genes, respectively. Heterozygous inactivating variants of the genes cause two distinct conditions—CHI in newborns and maturity onset-diabetes (MODY type 1 and 3) in later life (143–146). This type of CHI due to HNF1A/HNF4A gene mutations is characterized with macrosomia and a clinical severity varying form mild transient hypoglycaemia to severe HH responsive to diazoxide therapy (25, 143, 147–149). In addition, a glycogenosis-like phenotype, characterized with glycogen accumulation in red blood cells, elevated liver enzymes and increased hepatic echogenicity on ultrasound examination, has been reported in CHI due to HNF4A gene mutations (150, 151). Although CHI due to HNF1A and HNF4A mutations is rare, in some series of diazoxide-responsive HH, these mutations have been shown as one of the most common genetic etiology (149, 152). The exact mechanisms by which the mutations in HNF4A and HNF1A could cause CHI are yet to be elucidated.
Mutations in FOXA2 and CHI
The forkhead box A2 transcription factor (FOXA2), also known as hepatocyte nuclear factor 3β (HNF3β), is encoded by FOXA2 gene, localized on chromosome 20p11.21. It is required for the embryogenesis and organogenesis of endoderm-derived tissues including pancreas and forebrain structures including pitutary gland (153–157). FOXA2 is a major upstream regulator of Pdx1 which is a transcription factor implicated in the pancreas development (155, 158). In mouse models it has been demonstrated that foxa2 is necessary for the pancreatic α-cells differentiation and tissue-specific foxa2 ablation leads to an imbalance in pancreatic β/α-cell ratio, profound hypoglucagonemia, inappropriate hyperinsulinaemia and hypoglycaemia (105, 155, 158, 159). FOXA2 also regulates the expression of KCNJ11 and ABCC8 genes and has a binding site on the intronic region of HADH gene playing a role in its transactivation (154, 157, 159).
The effects of FOXA2 mutations on the pituitary function have previously been shown in cases with deletions of the proximal 20p11 region (160, 161). The first mutation of FOXA2 causing both pituitary dysfunction and CHI was recently reported in a case with congenital hypopituitarism, HH and endoderm-derived organ abnormalities (154). In this report a “de novo” heterozygous c.505T>C (p.S169P) variant was detected in the FOXA2 DNA-binding domain. The patient had a specific clinical presentation including hypoplasia of the anterior pituitary, absent neurohypophysis with interrupted pitutary stalk and hypoplasia of corpus collosum, associated with multiple pituitary hormone deficiency, persistent HH, craniofacial dysmorphism and abnormalities of organs of endodermal origin, e.g., liver, lungs and gastrointestinal tract (154). The authors have shown increased pancreatic hFOXA2 expression in the immunohistochemical analysis of human embryos and lower transcriptional activity of hFOXA2-p.S169P variant in the functional analysis. Soon after the first report, another research group has reported a “de novo” heterozygous p.R257L mutation in an infant with congenital hypopituitarism, HH and atypical features including coarse face, increased distance between the orbits, low-set ears, and widely-spaced nipples (157). The functional analysis of this mutation demonstrated decreased SHH, Gli2 and NKX2.2 gene expression suggesting the importance of FOXA2 mutation for the abnormal findings of the pituitary gland. Besides, the partially reduced expression of ABCC8, KCNJ11, and HADH has been suggested as an evidence for the mutation effects on the developement of HH in this case. Although, the molecular basis of the HH observed in the patients has not been fully elucidated, results from previously published mouse models and latest case reports suggest that mutation in FOXA2 could potentially be a monogenic cause of CHI.
Hyperinsulinaemic Hypoglycaemia due to Syndromes
HH has been described in several different syndromes (6) (Table 2). In most cases the underlying biochemical and molecular basis of the HH is unknown. Beckwith-Wiedemann syndrome (BWS), the most frequent syndromic condition associated with HH, is an overgrowth disorder characterized with macrosomia, macroglossia, neonatal hypoglycaemia, hemi-hypertrophy, omphalocele. BWS patients are also prone to the development of embryonal tumors (Wilm's tumor, hepatoblastoma, neuroblastoma, rhabdomyosarcoma). BWS is linked to genetic and/or epigenetic abnormalities that alter the expression of imprinted genes on chromosome 11p15.5 (178). HH develops in approximately 50% of patients with BWS and in most cases it is transient. However, in some cases treatment with diazoxide will be required or a near total pancreatectomy (179). Although the molecular basis of HH due to BWS is not known, patients with BWS due to mosaic paternal isodisomy for chromosome 11 have the most severe hypoglycaemia (180). In one patient with BWS due to paternal uniparental disomy for this chromosome, electrophysiological studies of β-cells received at the time of surgery showed defects in pancreatic KATP channels (162). HH also occurs in other genetic forms of BWS but tends to be milder and shows marked clinical heterogeneity even with the same genetic cause (181). In some BWS patients, HH may be the main clinical presentation (182).
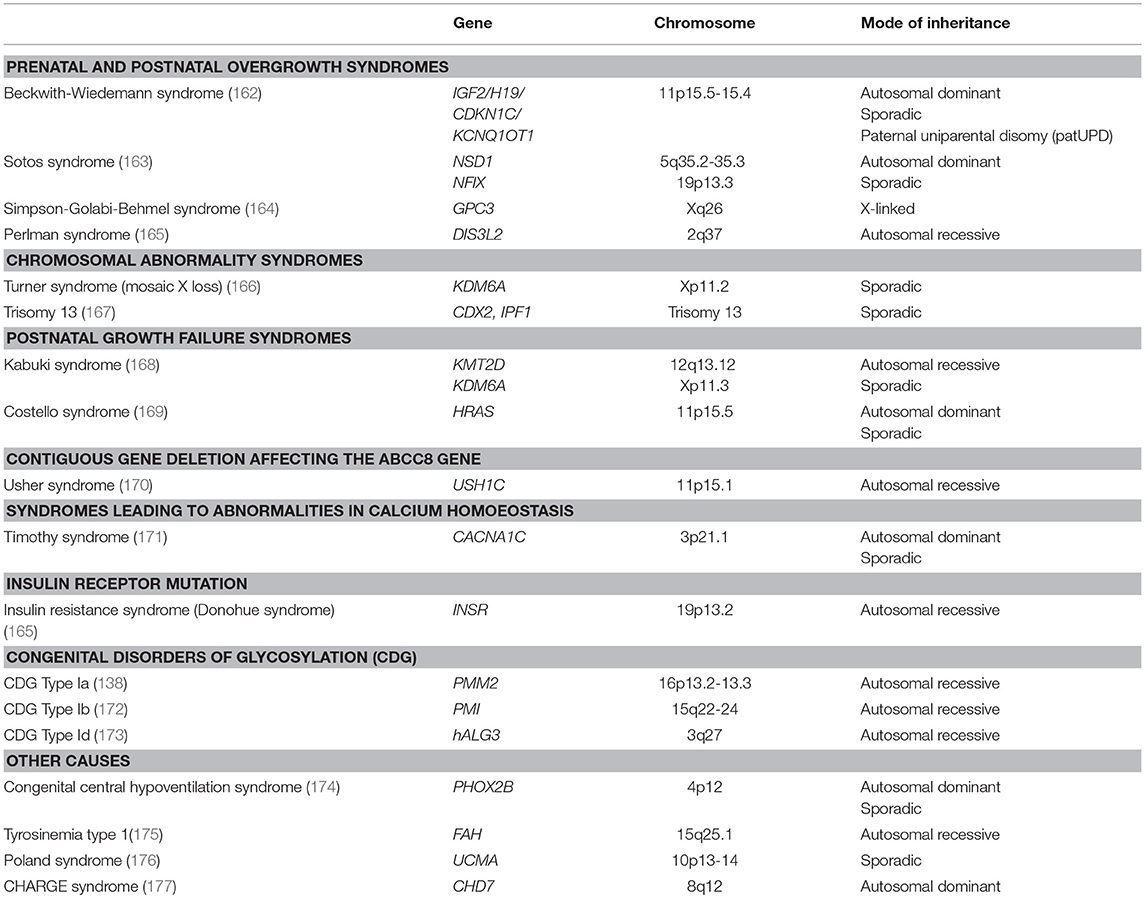
Table 2. Syndromic and metabolic causes of hyperinsulinaemic hypoglycaemia.
Kabuki syndrome is a rare genetic multisystem disorder characterized by developmental delay, “peculiar” face with elongated palpebral fissures, eversion of the lateral part of the lower eyelids, dermatoglyphic abnormalities with persistent fingertip pads, multiple congenital skeletal and visceral anomalies. Most of the cases with Kabuki syndrome have been associated with mutations in MLL2 and KDM6A genes. HH has been reported in a few patients with Kabuki syndrome (168, 183, 184). However, a group has recently identified 10 patients with the syndrome (5 with MLL2 and 5 with KDM6A mutations) associated with HH. Following description of these 10 patients the authors have analyzed 100 HH patients with unknown etiology and detected Kabuki syndrome in one of those patients (1%) (185). They, therefore, have suggested that Kabuki syndrome might account for the etiology of HH in more patients. Nevertheless, the role/s of MLL2 and KDM6A in insulin release and glucose physiology is/are not ellucidated.
Sotos syndrome is also an overgrowth disorder. More than 75% of the cases with Sotos syndrome are due to intragenic mutations and deletions of the nuclear receptor binding SET-Domain 1 (NSD1) which is located at chromosome 5q35. Transient HH has been identified in several patients with Sotos syndrome (163, 186). NSD1 is not known to be directly involved in regulating insulin secretion but patients with Sotos syndrome have alterations in the IGF-1 axis which could play a role in β-cell hyperplasia (187).
Turner syndrome (partial or total monosomy X) leads to short stature in females and is associated with impaired glucose tolerance later in life. Until recently, only three patients had been described with mild diazoxide-responsive HH (166, 188, 189), all being mosaic and having the ring X chromosome. Therefore, the abnormal mosaic expression of unknown X-chromosomal gene(s) in the β-cells was suggested leading to HH. However, recently, Gibson et al. reported that patients with Turner syndrome account for 10 out of 1,050 patients with HH suggesting a frequency of 48 times more than expected (190). The authors showed that a half of the girls with Turner syndrome and HH were medically unresponsive, with 3 patients requiring a partial or near-total pancreatectomy. Furthermore, all patients with HH had at least one monosomic cell line for the X–chromosome. In the same study the authors also demonstrated increased cytosolic calcium and enhanced amino acid-induced secretion of insulin in isolated pancreatic islets from 1 case and mouse islets exposed to a KDM6A inhibitor. They suggested that the KDM6A haploinsufficiency caused by mosaic X chromosome monosomy may explain the HH in Turner syndrome.
Timothy syndrome is a rare multi-system disorder characterized by lethal arrhythmias, congenital heart defects, syndactyly, immune deficiency, intermittent HH, intellectual disability, autism and autistic spectrum disorders. It is due to novel Ca(V)1.2 missense mutations (171). This is a calcium channel protein and theoretically could be connected with the regulation of the calcium homeostasis and secretion of insulin. One patient with another calcium channel gene (CACNA1D) mutation has been described with HH, hypotonia and congenital heart defects (69).
Other syndromes associated with HH include Costello (169), Trisomy 13 (167), central hypoventilation syndrome (174) and recently in CHARGE syndrome (177) (Table 2).
Conclusion
CHI is a complex condition characterized by upregulated β-cell insulin secretion leading to HH. Mutations in the ABCC8/KCNJ11 genes of the KATP channel result in the most severe forms of CHI. Abnormal variants in several other genes with a particular importance in the regulation of insulin release and several syndromic conditions may cause milder forms of HH. The molecular cause of CHI is determined in only half of the patients. The understanding of novel molecular mechanisms of the dysregulated release of insulin will provide novel insights into the pancreatic β-cells function.
Author Contributions
SG: literature review, wrote the section on non KATP causes of hyperinsulinism; SA-K: literature review, created all the figures and tables; HD: literature review, wrote the section on introduction and Katp channel causes of hyperinsulinism; KH: conception, planning, writing, organizing, checking and communicating.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The publication of this article was funded by the Qatar National Library.
References
1. Ferrara C, Patel P, Becker S, Stanley CA, Kelly A. Biomarkers of insulin for the diagnosis of hyperinsulinemic hypoglycemia in infants and children. J Pediatr. (2016) 168:212–19. doi: 10.1016/j.jpeds.2015.09.045
2. Aynsley-Green A, Hussain K, Hall J, Saudubray JM, Nihoul-Fékété C, De Lonlay-Debeney P, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed. (2000) 82:F98–107. doi: 10.1136/fn.82.2.F98
3. Demirbilek H, Hussain K. Congenital hyperinsulinism: diagnosis and treatment update. J Clin Res Pediatr Endocrinol. (2017) 9(Suppl. 2):69–87. doi: 10.4274/jcrpe.2017.S007
4. Saint-Martin C, Arnoux JB, de Lonlay P, Bellanné-Chantelot C. KATP channel mutations in congenital hyperinsulinism. Semin Pediatr Surg. (2011) 20:18–22. doi: 10.1053/j.sempedsurg.2010.10.012
5. Lu M, Li C. Nutrient sensing in pancreatic islets: lessons from congenital hyperinsulinism and monogenic diabetes. Ann N Y Acad Sci. (2018) 1411:65–82. doi: 10.1111/nyas.13448
6. Kapoor RR, James C, Hussain K. Hyperinsulinism in developmental syndromes. Endocr Dev. (2009) 14:95–113. doi: 10.1159/000207480
7. Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, et al. DNA methylation directs functional maturation of pancreatic β-cells. J Clin Invest. (2015) 125:2851–60. doi: 10.1172/JCI79956
8. Quintens R, Hendrickx N, Lemaire K, Schuit F. Why expression of some genes is disallowed in beta cells. Biochem Soc Trans. (2008) 36:300–5. doi: 10.1042/BST0360300
9. Matschinsky FM. Banting lecture. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes (1996) 45:223–41. doi: 10.2337/diab.45.2.223
10. Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. (2013) 18:162–85. doi: 10.1016/j.cmet.2013.05.018
11. Nessa A, Rahman SA, Hussain K. Hyperinsulinemic hypoglycemia - the molecular mechanisms. Front Endocrinol. (2016) 7:29. doi: 10.3389/fendo.2016.00029
12. Cook DL, Hales CN. Intracellular ATP directly blocks K+channels in pancreatic B-cells. Nature (1984) 311:271-3.
13. Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature (1984) 312:446-48.
14. Doupnik CA, Davidson N, Lester HA. The inward rectifier potassium channel family. Curr Opin Neurobiol. (1995) 5:268–77. doi: 10.1016/0959-4388(95)80038-7
15. Rahman SA, Nessa A, Hussain K. Molecular mechanisms of congenital hyperinsulinism. J Mol Endocrinol. (2015) 54:R119–29. doi: 10.1530/JME-15-0016
16. Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP IV, Boyd AE III, González G, et al. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science (1995) 268:423-26.
17. Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa J, Gonzalez G, et al. Reconstitution of IKATP: An inward rectifier subunit plus the sulfonylurea receptor. Science (1995) 270:1166–70. doi: 10.1126/science.270.5239.1166
18. Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K (ATP) channels. Neuron (1999) 22:537-48.
19. Li L, Geng X, Drain P. Open state destabilization by atp occupancy is mechanism speeding burst exit underlying KATP channel inhibition by ATP. J Gen Physiol. (2002) 119:105–16. doi: 10.1085/jgp.119.1.105
20. Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev. (1999) 20:101–35. doi: 10.1210/er.20.2.101
21. Shimono D, Fujimoto S, Mukai E, Takehiro M, Nabe K, Radu RG, et al. ATP enhances exocytosis of insulin secretory granules in pancreatic islets under Ca2+ depleted condition. Diabetes Res Clin Pract. (2005) 69:216–23. doi: 10.1016/j.diabres.2005.01.010
22. Cryer PE. Glucose counterregulation: prevention and correction of hypoglycemia in humans. Am J Phys. (1993) 264:E149–55.
23. Stanley CA. Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. J Clin Endocrinol Metab. (2016) 101:815–26. doi: 10.1210/jc.2015-3651
24. Flanagan SE, Clauin S, Bellanne-Chantelot C, de Lonlay P, Harries LW, Gloyn AL, et al. Update of mutations in the genes encoding the pancreatic beta-cell K (ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat. (2009) 30:170-80. doi: 10.1002/humu.20838
25. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. (2013) 98:E355–63. doi: 10.1210/jc.2012-2169
26. Mohnike K, Wieland I, Barthlen W, Vogelgesang S, Empting S, Mohnike W, et al. Clinical and genetic evaluation of patients with KATP channel mutations from the German registry for congenital hyperinsulinism. Horm Res Paediatr. (2014) 81:156–68. doi: 10.1159/000356905
27. Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol. (2013) 168:557–64. doi: 10.1530/EJE-12-0673
28. Bellanné-Chantelot C, Saint-Martin C, Ribeiro MJ, Vaury C, Verkarre V, Arnoux JB, et al. ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism. J Med Genet. (2010) 47:752–59. doi: 10.1136/jmg.2009.075416
29. Park SE, Flanagan SE, Hussain K, Ellard S, Shin CH, Yang SW. Characterization of ABCC8 and KCNJ11 gene mutations and phenotypes in Korean patients with congenital hyperinsulinism. Eur J Endocrinol. (2011) 164:919–26. doi: 10.1530/EJE-11-0160
30. Kane C, Shepherd RM, Squires PE, Johnson PR, James RF, Milla PJ, et al. Loss of functional KATP channels in pancreatic beta-cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nat Med (1996) 2:1344-47.
31. Flanagan SE, Kapoor RR, Hussain K. Genetics of congenital hyperinsulinemic hypoglycemia. Semin Pediatr Surg. (2011) 20:13–17. doi: 10.1053/j.sempedsurg.2010.10.004
32. Dekel B, Lubin D, Modan-Moses D, Quint J, Glaser B, Meyerovitch J. Compound heterozygosity for the common sulfonylurea receptor mutations can cause mild diazoxide-sensitive hyperinsulinism. Clin Pediatr. (2002) 41:183–6. doi: 10.1177/000992280204100310
33. Crane A, Aguilar-Bryan L. Assembly, maturation, and turnover of K(ATP) channel subunits. J Biol Chem. (2004) 279:9080–90. doi: 10.1074/jbc.M311079200
34. Yan FF, Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes (2007) 56:2339–48. doi: 10.2337/db07-0150
35. Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. A novel KCNJ11 mutation associated with congenital hyperinsulinism reduces the intrinsic open probability of beta-cell ATP-sensitive potassium channels. J Biol Chem. (2006) 281:3006–12. doi: 10.1074/jbc.M511875200
36. Cartier EA, Conti LR, Vandenberg CA, Shyng SL. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci USA. (2001) 98:2882–7. doi: 10.1073/pnas.051499698
37. Taschenberger G, Mougey A, Shen S, Lester LB, LaFranchi S, Shyng SL. Identification of a familial hyperinsulinism-causing mutation in the sulfonylurea receptor 1 that prevents normal trafficking and function of KATP channels. J Biol Chem. (2002) 277:7139–46. doi: 10.1074/jbc.M200363200
38. Taneja TK, Mankouri J, Karnik R, Kannan S, Smith AJ, Munsey T, et al. Sar1-GTPase-dependent ER exit of KATP channels revealed by a mutation causing congenital hyperinsulinism. Hum Mol Genet. (2009) 18:2400–13. doi: 10.1093/hmg/ddp179
39. Marthinet E, Bloc A, Oka Y, Tanizawa Y, Wehrle-Haller B, Bancila V, et al. Severe congenital hyperinsulinism caused by a mutation in the Kir6.2 subunit of the adenosine triphosphate-sensitive potassium channel impairing trafficking and function. J Clin Endocrinol Metab. (2005) 90:5401–6. doi: 10.1210/jc.2005-0202
40. Shyng SL, Ferrigni T, Shepard JB, Nestorowicz A, Glaser B, Permutt MA, et al. Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes (1998) 47:1145–51. doi: 10.2337/diabetes.47.7.1145
41. Matsuo M, Trapp S, Tanizawa Y, Kioka N, Amachi T, Oka Y, et al. Functional analysis of a mutant sulfonylurea receptor, SUR1-R1420C, that is responsible for persistent hyperinsulinemic hypoglycemia of infancy. J Biol Chem. (2000) 275:41184–91. doi: 10.1074/jbc.M006503200
42. Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest. (2008) 118:2877–86. doi: 10.1172/JCI35414
43. Kapoor RR, Flanagan SE, James CT, McKiernan J, Thomas AM, Harmer SC, et al. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/ KCNJ11 mutations. Diabetologia (2011) 54:2575–83. doi: 10.1007/s00125-011-2207-4
44. Thornton PS, MacMullen C, Ganguly A, Ruchelli E, Steinkrauss L, Crane A, et al. Clinical and molecular characterization of a dominant form of congenital hyperinsulinism caused by a mutation in the high-affinity sulfonylurea receptor. Diabetes (2003) 52:2403–10. doi: 10.2337/diabetes.52.9.2403
45. Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, et al. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest. (2000) 106:897-906. doi: 10.1172/JCI9804
46. Glaser B, Ryan F, Donath M, Landau H, Stanley CA, Baker L, et al. Hyperinsulinism caused by paternal-specific inheritance of a recessive mutation in the sulfonylurea-receptor gene. Diabetes (1999) 48:1652–7. doi: 10.2337/diabetes.48.8.1652
47. Işik E, Demirbilek H, Houghton JAL, Ellard S, Flanagan SE, Hussain K. Congenital hyperinsulinism and evolution to sulfonylurea-responsive diabetes later in life due to a novel homozygous p.L171F ABCC8 mutation. J Clin Res Pediatr Endocrinol. (2018). doi: 10.4274/jcrpe.0077 [Epub ahead of print].
48. Nessa A, Aziz QH, Thomas AM, Harmer SC, Tinker A, Hussain K. Molecular mechanisms of congenital hyperinsulinism due to autosomal dominant mutations in ABCC8. Hum Mol Genet. (2015) 24:5142–53. doi: 10.1093/hmg/ddv233
49. Flanagan SE, Kapoor RR, Banerjee I, Hall C, Smith VV, Hussain K, et al. Dominantly acting ABCC8 mutations in patients with medically unresponsive hyperinsulinaemic hypoglycaemia. Clin Genet. (2011) 79:582–87. doi: 10.1111/j.1399-0004.2010.01476.x
50. MacMullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, et al. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the β-cell sulfonylurea receptor SUR1. Diabetes (2011) 60:1797–804. doi: 10.2337/db10-1631
51. Shyng SL, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. J Gen Physiol. (1997) 110:643–54. doi: 10.1085/jgp.110.6.643
52. Rozenkova K, Nessa A, Obermannova B, Elblova L, Dusatkova P, Sumnik Z, et al. Could a combination of heterozygous ABCC8 and KCNJ11 mutations cause congenital hyperinsulinism? J Pediatr Endocrinol Metab. (2017) 30:1311–15. doi: 10.1515/jpem-2017-0163
53. Damaj L, le Lorch M, Verkarre V, Werl C, Hubert L, Nihoul-Fékété C, et al. Chromosome 11p15 paternal isodisomy in focal forms of neonatal hyperinsulinism. J Clin Endocrinol Metab. (2008) 93:4941–47. doi: 10.1210/jc.2008-0673
54. De Lonlay P, Fournet JC, Rahier J, Gross-Morand MS, Poggi-Travert F, Foussier V, et al. Somatic deletion of the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J Clin Invest. (1997) 100:802–07. doi: 10.1172/JCI119594
55. Shuman C, Smith AC, Steele L, Ray PN, Clericuzio C, Zackai E, et al. Constitutional UPD for chromosome 11p15 in individuals with isolated hemihyperplasia is associated with high tumor risk and occurs following assisted reproductive technologies. Am J Med Genet. Part A (2006) 140:1497–503. doi: 10.1002/ajmg.a.31323
56. Sempoux C, Capito C, Bellanné-Chantelot C, Verkarre V, de Lonlay P, Aigrain Y, et al. Morphological mosaicism of the pancreatic islets: a novel anatomopathological form of persistent hyperinsulinemic hypoglycemia of infancy. J Clin Endocrinol Metab. (2011) 96:3785–93. doi: 10.1210/jc.2010-3032
57. Arya VB, Guemes M, Nessa A, Alam S, Shah P, Gilbert C, et al. Clinical and histological heterogeneity of congenital hyperinsulinism due to paternally inherited heterozygous ABCC8/KCNJ11 mutations. Eur J Endocrinol. (2014) 171:685–95. doi: 10.1530/EJE-14-0353
58. Maiorana A, Barbetti F, Boiani A, Rufini V, Pizzoferro M, Francalanci P, et al. Focal congenital hyperinsulinism managed by medical treatment: a diagnostic algorithm based on molecular genetic screening. Clin Endocrinol. (2014) 81:679–88. doi: 10.1111/cen.12400
59. Banerjee I, Avatapalle B, Padidela R, Stevens A, Cosgrove KE, Clayton PE, et al. Integrating genetic and imaging investigations into the clinical management of congenital hyperinsulinism. Clin Endocrinol. (2013) 78:803–13. doi: 10.1111/cen.12153
60. Salomon-Estebanez M, Flanagan SE, Ellard S, Rigby L, Bowden L, Mohamed Z, et al. Conservatively treated Congenital Hyperinsulinism (CHI) due to K-ATP channel gene mutations: reducing severity over time. Orphanet J Rare Dis. (2016) 11:163. doi: 10.1186/s13023-016-0547-3
61. Chandran S, Peng FY, Rajadurai VS, Lu YT, Chang KT, Flanagan SE, et al. Paternally inherited ABCC8 mutation causing diffuse congenital hyperinsulinism. Endocrinol Diabetes Metab Case Rep. (2013) 2013:130041. doi: 10.1530/EDM-13-0041
62. Flanagan SE, Xie W, Caswell R, Damhuis A, Vianey-Saban C, Akcay T, et al. Next-generation sequencing reveals deep intronic cryptic ABCC8 and HADH splicing founder mutations causing hyperinsulinism by pseudoexon activation. Am J Hum Genet. (2013) 92:131–36. doi: 10.1016/j.ajhg.2012.11.017
63. Hussain K, Flanagan SE, Smith VV, Ashworth M, Day M, Pierro A, et al. An ABCC8 gene mutation and mosaic uniparental isodisomy resulting in atypical diffuse congenital hyperinsulinism. Diabetes (2008) 57:259–63. doi: 10.2337/db07-0998
64. Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med. (1997) 336:1562–7. doi: 10.1056/NEJM199705293362204
65. Torekov SS, Iepsen E, Christiansen M, Linneberg A, Pedersen O, Holst JJ, et al. KCNQ1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes (2014) 63:1315–25. doi: 10.2337/db13-1454
66. Iwashima Y, Pugh W, Depaoli AM, Takeda J, Seino S, Bell GI, et al. Expression of calcium channel mRNAs in rat pancreatic islets and downregulation after glucose infusion. Diabetes (1993) 42:948–55. doi: 10.2337/diab.42.7.948
67. Shah P, Rahman SA, Demirbilek H, Guemes M, Hussain K. Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol. (2017) 5:729–42. doi: 10.1016/S2213-8587(16)30323-0
68. Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet. (2013) 45:1050–54. doi: 10.1038/ng.2695
69. Flanagan SE, Vairo F, Johnson MB, Caswell R, Laver TW, Lango Allen H et al. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes (2017) 18:320–23. doi: 10.1111/pedi.12512
70. Cuff MA, Shirazi-Beechey SP. The human monocarboxylate transporter, MCT1: genomic organization and promoter analysis. Biochem Biophys Res Commun. (2002) 292:1048–56. doi: 10.1006/bbrc.2002.6763
71. Pullen TJ, Sylow L, Sun G, Halestrap AP, Richter EA, Rutter GA. Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic beta-cells leads to relative hyperinsulinism during exercise. Diabetes (2012) 61:1719–25. doi: 10.2337/db11-1531
72. Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, et al. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes (2003) 52:199–204. doi: 10.2337/diabetes.52.1.199
73. Tosur M, Jeha GS. A novel intragenic SLC16A1 mutation associated with congenital hyperinsulinism. Glob Pediatr Health (2017) 4:2333794X17703462. doi: 10.1177/2333794X17703462
74. Deloukas P, Dauwerse JG, Moschonas NK, van Ommen GJ, van Loon AP. Three human glutamate dehydrogenase genes (GLUD1, GLUDP2, and GLUDP3) are located on chromosome 10q, but are not closely physically linked. Genomics (1993) 17:676–81. doi: 10.1006/geno.1993.1389
75. Hudson RC, Daniel RM. L-glutamate dehydrogenases: distribution, properties and mechanism. Comp Biochem Physiol B (1993) 106:767–92. doi: 10.1016/0305-0491(93)90031-Y
76. Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. (1998) 338:1352–57. doi: 10.1056/NEJM199805073381904
77. Li C, Najafi H, Daikhin Y, Nissim IB, Collins HW, Yudkoff M, et al. Regulation of leucine-stimulated insulin secretion and glutamine metabolism in isolated rat islets. J Biol Chem. (2003) 278:2853–58. doi: 10.1074/jbc.M210577200
78. Li M, Li C, Allen A, Stanley CA, Smith TJ. The structure and allosteric regulation of glutamate dehydrogenase. Neurochem Int. (2011) 59:445–55. doi: 10.1016/j.neuint.2010.10.017
79. Kibbey RG, Choi CS, Lee HY, Cabrera O, Pongratz RL, Zhao X, et al. Mitochondrial GTP insensitivity contributes to hypoglycemia in hyperinsulinemia hyperammonemia by inhibiting glucagon release. Diabetes (2014) 63:4218–29. doi: 10.2337/db14-0783
80. Stanley CA, Fang J, Kutyna K, Hsu BY, Ming JE, Glaser B, et al. Molecular basis and characterization of the hyperinsulinism/hyperammonemia syndrome: predominance of mutations in exons 11 and 12 of the glutamate dehydrogenase gene. HI/HA Contributing Investigators. Diabetes (2000) 49:667–73. doi: 10.2337/diabetes.49.4.667
81. Santer R, Kinner M, Passarge M, Superti-Furga A, Mayatepek E, Meissner T, et al. Novel missense mutations outside the allosteric domain of glutamate dehydrogenase are prevalent in European patients with the congenital hyperinsulinism–hyperammonemia syndrome. Hum Genet. (2001) 108:66–71. doi: 10.1007/s004390000432
82. Fujioka H, Okano Y, Inada H, Asada M, Kawamura T, Hase Y, et al. Molecular characterisation of glutamate dehydrogenase gene defects in Japanese patients with congenital hyperinsulinism/hyperammonaemia. Eur J Hum Genet. (2001) 9:931–7. doi: 10.1038/sj.ejhg.5200749
83. Stanley CA. Hyperinsulinism/hyperammonemia syndrome: insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Mol Genet Metab. (2004) 81:S45–51. doi: 10.1016/j.ymgme.2003.10.013
84. Senniappan S, Arya VB, Hussain K. The molecular mechanisms, diagnosis and management of congenital hyperinsulinism. Ind J Endocrinol Metab. (2013) 17:19–30. doi: 10.4103/2230-8210.107822
85. Treberg JR, Clow KA, Greene KA, Brosnan ME, Brosnan JT. Systemic activation of glutamate dehydrogenase increases renal ammoniagenesis: implications for the hyperinsulinism/hyperammonemia syndrome. Am J Phys Endocrinol Metab. (2010) 298:E1219–25. doi: 10.1152/ajpendo.00028.2010
86. Kapoor RR, Flanagan SE, Fulton P, Chakrapani A, Chadefaux B, Ben-Omran T, et al. Hyperinsulinism-hyperammonaemia syndrome: novel mutations in the GLUD1 gene and genotype-phenotype correlations. Eur J Endocrinol. (2009) 161:731–35. doi: 10.1530/EJE-09-0615
87. Hsu BY, Kelly A, Thornton PS, Greenberg CR, Dilling LA, Stanley CA. Proteinsensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. J Pediatr. (2001) 138:383–89. doi: 10.1067/mpd.2001.111818
88. Bahi-Buisson N, Roze E, Dionisi C, Escande F, Valayannopoulos V, Feillet F, et al. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev Med Child Neurol. (2008) 50:945-9. doi: 10.1111/j.1469-8749.2008.03114.x
89. Barrosse-Antle M, Su C, Chen P, Boodhansingh KE, Smith TJ, Stanley CA, et al. A severe case of hyperinsulinism due to hemizygous activating mutation of glutamate dehydrogenase. Pediatr Diabetes (2017) 18:911–16. doi: 10.1111/pedi.12507
90. Iynedjian PB, Mobius G, Seitz HJ, Wollheim CB, Renold AE. Tissue-specific expression of glucokinase: identification of the gene product in liver and pancreatic islets. Proc Natl Acad Sci USA. (1986) 83:1998–2001. doi: 10.1073/pnas.83.7.1998
91. Matschinsky FM. Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes (2002) 51:S394–404. doi: 10.2337/diabetes.51.2007.S394
92. Zelent D, Najafi H, Odili S, Buettger C, Weik-Collins H, Li C, et al. Glucokinase and glucose homeostasis: proven concepts and new ideas. Biochem Soc Trans. (2005) 33:306–10. doi: 10.1042/BST0330306
93. Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. (2009) 30:1512–26. doi: 10.1002/humu.21110
94. Barbetti F, Cobo-Vuilleumier N, Dionisi-Vici C, Toni S, Ciampalini P, Massa O, et al. Opposite clinical phenotypes of glucokinase disease:description of a novel activating mutation and contiguous inactivating mutations in human Glucokinase (GCK). Gene Mol Endocrinol. (2009) 23:1983–89. doi: 10.1210/me.2009-0094
95. Huang M, Lu S, Shi T, Zhao Y, Chen Y, Li X, et al. Conformational transition pathway in the activation process of allosteric glucokinase. PLoS ONE (2013) 8:e55857. doi: 10.1371/journal.pone.0055857
96. Ralph EC, Thomson J, Almaden J, Sun S. Glucose modulation of glucokinase activation by small molecules. Biochemistry (2008) 47:5028–36. doi: 10.1021/bi702516y
97. Christesen HB, Tribble ND, Molven A, Siddiqui J, Sandal T, Brusgaard K, et al. Activating Glucokinase (GCK) mutations as a cause of medically responsive congenital hyperinsulinism: prevalence in children and characterisation of a novel GCK mutation. Eur J Endocrinol. (2008) 159:27–34. doi: 10.1530/EJE-08-0203
98. Martinez R, Gutierrez-Nogues A, Fernandez-Ramos C, Velayos T, Vela A; Spanish Congenital Hyperinsulinism Group, et al. Heterogeneity in phenotype of hyperinsulinism caused by activating glucokinase mutations: a novel mutation and its functional characterization. Clin Endocrinol. (2017) 86:778–83. doi: 10.1111/cen.13318
99. Morishita K, Kyo C, Yonemoto T, Kosugi R, Ogawa T, Inoue T. Asymptomatic congenital hyperinsulinism due to a Glucokinase-activating mutation, treated as adrenal insufficiency for twelve years. Case Rep Endocrinol. (2017) 2017:4709262. doi: 10.1155/2017/4709262
100. Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, et al. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes (2009) 58:1419–27. doi: 10.2337/db08-1792
101. Ajala ON, Huffman DM, Ghobrial II. Glucokinase mutation-a rare cause of recurrent hypoglycemia in adults: a case report and literature review. J Community Hosp Intern Med Perspect. (2016) 6:32983. doi: 10.3402/jchimp.v6.32983
102. Kassem S, Bhandari S, Rodriguez-Bada P, Motaghedi R, Heyman M, García-Gimeno MA, et al. Large islets, beta cell proliferation, and a glucokinase mutation. N Engl J Med. (2010) 362:1348–50. doi: 10.1056/NEJMc0909845
103. Cuesta-Munoz AL, Huopio H, Otonkoski T, Gomez-Zumaquero JM, Näntö-Salonen K, Rahier J, et al. Severe persistent hyperinsulinemic hypoglycemia due to a de novo glucokinase mutation. Diabetes (2004) 53:2164–8. doi: 10.2337/diabetes.53.8.2164
104. Vredendaal PJ, van den Berg IE, Malingre HE, Stroobants AK, Olde Weghuis DE, Berger R. Human short-chain L-3-hydroxyacyl-CoA dehydrogenase: cloning and characterization of the coding sequence. Biochem Biophys Res Commun. (1996) 223:718–23. doi: 10.1006/bbrc.1996.0961
105. Lantz KA, Vatamaniuk MZ, Brestelli JE, Friedman JR, Matschinsky FM, Kaestner KH. Foxa2 regulates multiple pathways of insulin secretion. J Clin Invest. (2004) 114:512–20. doi: 10.1172/JCI21149
106. Kapoor RR, James C, Flanagan SE, Ellard S, Eaton S, Hussain K. 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency and hyperinsulinemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensitivity. J Clin Endocrinol Metab. (2009) 94:2221–25. doi: 10.1210/jc.2009-0423
107. Clayton PT, Eaton S, Aynsley Green A, Edginton M, Hussain K, Krywawych S, et al. Hyperinsulinism in short chain L 3 hydroxyacyl CoA dehydrogenase deficiency reveals the importance of beta oxidation in insulin secretion. J Clin Invest. (2001) 108:457–65. doi: 10.1172/JCI200111294
108. Martins E, Cardoso ML, Rodrigues E, Barbot C, Ramos A, Bennett MJ, et al. Short chain 3 hydroxyacyl CoA dehydrogenase deficiency: the clinical relevance of an early diagnosis and report of four new cases. J Inherit Metab Dis. (2011) 34:835–42. doi: 10.1007/s10545-011-9287-7
109. Li C, Chen P, Palladino A, Narayan S, Russell LK, Sayed S, et al. Mechanism of hyperinsulinism in short chain 3 hydroxyacyl CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem. (2010) 285:31806–18. doi: 10.1074/jbc.M110.123638
110. Heslegrave AJ, Hussain K. Novel insights into fatty acid oxidation, amino acid metabolism, and insulin secretion from studying patients with loss of function mutations in 3-hydroxyacyl-CoA dehydrogenase. J Clin Endocrinol Metab. (2013) 98:496–501. doi: 10.1210/jc.2012-3134
111. Babiker O, Flanagan SE, Ellard S, Al Girim H, Hussain K, Senniappan S. Protein-induced hyperinsulinaemic hypoglycaemia due to a homozygous HADH mutation in three siblings of a Saudi family. J Pediatr Endocrinol Metab. (2015) 28:1073–77. doi: 10.1515/jpem-2015-0033
112. Flanagan SE, Patch AM, Locke JM, Akcay T, Simsek E, Alaei M, et al. Genome wide homozygosity analysis reveals HADH mutations as a common cause of diazoxide responsive hyperinsulinemic hypoglycemia in consanguineous pedigrees. J Clin Endocrinol Metab. (2011) 96:E498–502. doi: 10.1210/jc.2010-1906
113. Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, et al. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet. (1997) 15:269–72. doi: 10.1038/ng0397-269
114. González-Barroso MM, Giurgea I, Bouillaud F, Anedda A, Bellanné-Chantelot C, Hubert L, et al. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS ONE (2008) 3:e3850. doi: 10.1371/journal.pone.0003850
115. Krauss S, Zhang CY, Lowell BB. A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc Natl Acad Sci USA. (2002) 99:118–22. doi: 10.1073/pnas.012410699
116. Nicholls DG. The pancreatic beta-cell: a bioenergetic perspective. Physiol Rev. (2016) 96:1385–447. doi: 10.1152/physrev.00009.2016
117. Chan CB, Harper ME. Uncoupling proteins: role in insulin resistance and insulin insufficiency. Curr Diabetes Rev. (2006) 2:271–83. doi: 10.2174/157339906777950660
118. Chan CB, De Leo D, Joseph JW, McQuaid TS, Ha XF, Xu F, et al. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes (2001) 50:1302–10. doi: 10.2337/diabetes.50.6.1302
119. Affourtit C, Brand MD. Uncoupling protein-2 contributes significantly to high mitochondrial proton leak in INS-1E insulinoma cells and attenuates glucose-stimulated insulin secretion. Biochem J. (2008) 409:199–204. doi: 10.1042/BJ20070954
120. McQuarrie I. Idiopathic spontaneously occurring hypoglycemia in infants; clinical significance of problem and treatment. AMA Am J Dis Child (1954) 87:399–428. doi: 10.1001/archpedi.1954.02050090387001
121. Ferrara CT, Boodhansingh KE, Paradies E, Giuseppe F, Steinkrauss LJ, Topor LS, et al. Novel hypoglycemia phenotype in congenital hyperinsulinism due to dominant mutations of Uncoupling Protein 2 (UCP2). J Clin Endocrinol Metab. (2017) 102:942–49. doi: 10.1210/jc.2016-3164
122. Laver TW, Weedon MN, Caswell R, Hussain K, Ellard S, Flanagan SE. Analysis of large-scale sequencing cohorts does not support the role of variants in UCP2 as a cause of hyperinsulinaemic hypoglycaemia. Hum Mutat. (2017) 38:1442–44. doi: 10.1002/humu.23289
123. Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. (2003) 206:2049–57. doi: 10.1242/jeb.00241
124. Pinney SE, Ganapathy K, Bradfield J, Stokes D, Sasson A, Mackiewicz K, et al. Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm Res Paediatr. (2013) 80:18–27. doi: 10.1159/000351943
125. Henquin JC, Sempoux C, Marchandise J, Godecharles S, Guiot Y, Nenquin M, et al. Congenital hyperinsulinism caused by hexokinase I expression or glucokinase-activating mutation in a subset of beta-cells. Diabetes (2013) 62:1689–96. doi: 10.2337/db12-1414
126. Putt W, Ives JH, Hollyoake M, Hopkinson DA, Whitehouse DB, Edwards YH. Phosphoglucomutase 1: a gene with two promoters and a duplicated first exon. Biochem J. (1993) 296(Pt 2):417–22. doi: 10.1042/bj2960417
127. Stojkovic T, Vissing J, Petit F, Piraud M, Orngreen MC, Andersen G, et al. Muscle glycogenosis due to phosphoglucomutase 1 deficiency. N Engl J Med. (2009) 361:425–27. doi: 10.1056/NEJMc0901158
128. Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld ME, Timal S, et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med. (2014) 370:533–42. doi: 10.1056/NEJMoa1206605
129. Timal S, Hoischen A, Lehle L, Adamowicz M, Huijben K, Sykut-Cegielska J, et al. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum Mol Genet. (2012) 21:4151–61. doi: 10.1093/hmg/dds123
130. Perez B, Medrano C, Ecay MJ, Ruiz-Sala P, Martínez-Pardo M, Ugarte M, et al. A novel congenital disorder of glycosylation type without central nervous system involvement caused by mutations in the phosphoglucomutase 1 gene. J Inherit Metab Dis. (2013) 36:535–42. doi: 10.1007/s10545-012-9525-7
131. Ackermann AM LC, Freeze HH, Ficicioglu C, Kaestner KH, Stanley CA. Hypoglycemia due to lower threshold of glucose-stimulated insulin secretion in phosphoglucomutase 1 deficiency. In: Platform Presentation at: Annual Meeting of the Pediatric Academic Societies, April 25–28. San Diego, CA (2015).
132. Schrapers E, Tegtmeyer LC, Simic-Schleicher G, Debus V, Reunert J, Balbach S, et al. News on Clinical details and treatment in PGM1-CDG. JIMD Rep. (2016) 26:77–84. doi: 10.1007/8904_2015_471
133. Lee HS, Qi Y, Im W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci Rep. (2015) 5:8926. doi: 10.1038/srep08926
134. Jaeken J, Carchon H, Stibler H. The carbohydrate-deficient glycoprotein syndromes: pre-Golgi and Golgi disorders? Glycobiology (1993) 3:423–28. doi: 10.1093/glycob/3.5.423
135. Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman JJ, et al. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat Genet. (1997) 16:88–92. doi: 10.1038/ng0597-88
136. Grunewald S, Schollen E, Van Schaftingen E, Jaeken J, Matthijs G. High residual activity of PMM2 in patients' fibroblasts: possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am J Hum Genet. (2001) 68:347–54. doi: 10.1086/318199
137. Matthijs G, Schollen E, Heykants L, Grunewald S. Phosphomannomutase deficiency: the molecular basis of the classical Jaeken syndrome (CDGS type Ia). Mol Gen Metab. (1999) 68:220–26. doi: 10.1006/mgme.1999.2914
138. Shanti B, Silink M, Bhattacharya K, Howard NJ, Carpenter K, Fietz M, et al. Congenital disorder of glycosylation type Ia: heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inh Metab Dis. (2009) 32:S241–51. doi: 10.1007/s10545-009-1180-2
139. Cabezas OR, Flanagan SE, Stanescu H, García-Martínez E, Caswell R, Lango-Allen H, et al. Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. J Am Soc Nephrol. (2017) 28:2529–39. doi: 10.1681/ASN.2016121312
140. Gupta RK, Vatamaniuk MZ, Lee CS, Flaschen RC, Fulmer JT, Matschinsky FM, et al. The MODY1 gene HNF-4alpha regulates selected genes involved in insulin secretion. J Clin Invest. (2005) 115:1006–15. doi: 10.1172/JCI200522365
141. Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science (2004) 303:1378–81. doi: 10.1126/science.1089769
142. Miura A, Yamagata K, Kakei M, Hatakeyama H, Takahashi N, Fukui K, et al. Hepatocyte nuclear factor-4alpha is essential for glucose-stimulated insulin secretion by pancreatic beta-cells. J Biol Chem. (2006) 281:5246–57. doi: 10.1074/jbc.M507496200
143. Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. (2007) 4:e118. doi: 10.1371/journal.pmed.0040118
144. Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature (1996) 384:458–60. doi: 10.1038/384458a0
145. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature (1996) 384:455–58. doi: 10.1038/384455a0
146. Arya VB, Rahman S, Senniappan S, Flanagan SE, Ellard S, Hussain K. HNF4A mutation: switch from hyperinsulinaemic hypoglycaemia to maturity-onset diabetes of the young, and incretin response. Diab Med. (2014) 31:e11–5. doi: 10.1111/dme.12369
147. Fajans SS, Bell GI. Macrosomia and neonatal hypoglycaemia in RW pedigree subjects with a mutation (Q268X) in the gene encoding hepatocyte nuclear factor 4alpha (HNF4A). Diabetologia (2007) 50:2600–01. doi: 10.1007/s00125-007-0833-7
148. Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol. (2010) 162:987–92. doi: 10.1530/EJE-09-0861
149. Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, et al. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes (2008) 57:1659–63. doi: 10.2337/db07-1657
150. Stanik J, Skopkova M, Brennerova K, Danis D, Rosolankova M, Salingova A, et al. Congenital hyperinsulinism and glycogenosis-like phenotype due to a novel HNF4A mutation. Diabetes Res Clin Pract. (2017) 126:144–50. doi: 10.1016/j.diabres.2017.02.014
151. Stanescu DE, Hughes N, Kaplan B, Stanley CA, De Leon DD. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J Clin Endocrinol Metab. (2012) 97:E2026–30. doi: 10.1210/jc.2012-1356
152. Rozenkova K, Malikova J, Nessa A, Dusatkova L, Bjørkhaug L, Obermannova B, et al. High incidence of heterozygous ABCC8 and HNF1A mutations in Czech patients with congenital hyperinsulinism. J Clin Endocrinol Metab. (2015) 100:E1540–49. doi: 10.1210/jc.2015-2763
153. Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. (2008) 22:3435–48. doi: 10.1101/gad.1752608
154. Giri D, Vignola ML, Gualtieri A, Scagliotti V, McNamara P, Peak M, et al. Novel FOXA2 mutation causes hyperinsulinism, hypopituitarism with craniofacial and endoderm-derived organ abnormalities. Hum Mol Genet. (2017) 26:4315–26. doi: 10.1093/hmg/ddx318
155. Lee CS, Sund NJ, Behr R, Herrera PL, Kaestner KH. Foxa2 is required for the differentiation of pancreatic alpha-cells. Dev Biol. (2005) 278:484–95. doi: 10.1016/j.ydbio.2004.10.012
156. Lee CS, Sund NJ, Vatamaniuk MZ, Matschinsky FM, Stoffers DA, Kaestner KH. Foxa2 controls Pdx1 gene expression in pancreatic beta-cells in vivo. Diabetes (2002) 51:2546–51. doi: 10.2337/diabetes.51.8.2546
157. Vajravelu ME, Chai J, Krock B, Baker S, Langdon D, Alter C, et al. Congenital hyperinsulinism and hypopituitarism attributable to a novel mutation in FOXA2. J Clin Endocrinol Metab. (2018) 103:1042–47. doi: 10.1210/jc.2017-02157
158. Wu Z, Fei A, Liu Y, Pan S. Conditional tissue-specific Foxa2 ablation in mouse pancreas causes hyperinsulinemic hypoglycemia. Am J Ther. (2016) 23:e1442–8. doi: 10.1097/MJT.0000000000000399
159. Sund NJ, Vatamaniuk MZ, Casey M, Ang SL, Magnuson MA, Stoffers DA, et al. Tissue-specific deletion of Foxa2 in pancreatic beta cells results in hyperinsulinemic hypoglycemia. Genes Dev. (2001) 15:1706–15. doi: 10.1101/gad.901601
160. Dayem-Quere M, Giuliano F, Wagner-Mahler K, Massol C, Crouzet-Ozenda L, Lambert JC, et al. Delineation of a region responsible for panhypopituitarism in 20p11.2. Am J Med Genet Part A (2013) 161a:1547–54. doi: 10.1002/ajmg.a.35921
161. Williams PG, Wetherbee JJ, Rosenfeld JA, Hersh JH. 20p11 deletion in a female child with panhypopituitarism, cleft lip and palate, dysmorphic facial features, global developmental delay and seizure disorder. Am J Med Genet Part A (2011) 155a:186–91. doi: 10.1002/ajmg.a.33763
162. Hussain K, Cosgrove KE, Shepherd RM, Luharia A, Smith VV, Kassem S, et al. Hyperinsulinemic hypoglycemia in Beckwith-Wiedemann syndrome due to defects in the function of pancreatic beta-cell adenosine triphosphate-sensitive potassium channels. J Clin Endocrinol Metab. (2005) 90:4376–82. doi: 10.1210/jc.2005-0158
163. Matsuo T, Ihara K, Ochiai M, Kinjo T, Yoshikawa Y, Kojima-Ishii K, et al. Hyperinsulinemic hypoglycemia of infancy in Sotos syndrome. Am J Med Genet A (2013) 161A:34–7. doi: 10.1002/ajmg.a.35657
164. Terespolsky D, Farrell SA, Siegel-Bartelt J, Weksberg R. Infantile lethal variant of Simpson-Golabi-Behmel syndrome associated with hydrops fetalis. Am J Med Genet. (1995) 59:329–33. doi: 10.1002/ajmg.1320590310
165. Güemes M, Hussain K. Hyperinsulinemic hypoglycemia. Pediatr Clin North Am. (2015) 62:1017–36. doi: 10.1016/j.pcl.2015.04.010
166. Alkhayyat H, Christesen HB, Steer J, Stewart H, Brusgaard K, Hussain K. Mosaic Turner syndrome and hyperinsulinaemic hypoglycaemia. J Pediatr Endocrinol Metab. (2006) 19:1451–7. doi: 10.1515/JPEM.2006.19.12.1451
167. Tamame T, Hori N, Homma H, Yoshida R, Inokuchi M, Kosaki K, et al. Hyperinsulinemic hypoglycemia in a newborn infant with trisomy 13. Am J Med Genet A (2004) 129A:321–2. doi: 10.1002/ajmg.a.30147
168. Subbarayan A, Hussain K. Hypoglycemia in Kabuki syndrome. Am J Med Genet A (2014) 164A:467–71. doi: 10.1002/ajmg.a.36256
169. Alexander S, Ramadan D, Alkhayyat H, Al-Sharkawi I, Backer KC, El-Sabban F, et al. Costello syndrome and hyperinsulinemic hypoglycemia. Am J Med Genet A (2005) 139:227–30. doi: 10.1002/ajmg.a.31011
170. Al Mutair AN, Brusgaard K, Bin-Abbas B, Hussain K, Felimban N, Al Shaikh A, et al. Heterogeneity in phenotype of usher-congenital hyperinsulinism syndrome: hearing loss, retinitis pigmentosa, and hyperinsulinemic hypoglycemia ranging from severe to mild with conversion to diabetes. Diabetes Care (2013) 36:557–61. doi: 10.2337/dc12-1174
171. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell (2004) 119:19–31. doi: 10.1016/j.cell.2004.09.011
172. de Lonlay P, Cuer M, Vuillaumier-Barrot S, Beaune G, Castelnau P, Kretz M, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: a new manifestation of carbohydrate-deficient glycoprotein syndrome treatable with mannose. J Pediatr. (1999) 135:379–83. doi: 10.1016/S0022-3476(99)70139-3
173. Sun L, Eklund EA, Chung WK, Wang C, Cohen J, Freeze HH. Congenital disorder of glycosylation Id presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia. J Clin Endocrinol Metab. (2005) 90:4371–75. doi: 10.1210/jc.2005-0250
174. Farina MI, Scarani R, Po C, Agosto C, Ottonello G, Benini F. Congenital central hypoventilation syndrome and hypoglycaemia. Acta Paediatr. (2012) 101:e92–6. doi: 10.1111/j.1651-2227.2011.02533.x
175. Baumann U, Preece MA, Green A, Kelly DA, McKiernan PJ. Hyperinsulinism in tyrosinaemia type I. J Inherit Metab Dis. (2005) 28:131–35. doi: 10.1007/s10545-005-5517-1
176. Giri D, Patil P, Hart R, Didi M, Senniappan S. Congenital hyperinsulinism and Poland syndrome in association with 10p13–14 duplication. Endocrinol Diabetes Metab Case Rep. (2017). doi: 10.1530/EDM-16-0125 [Epub ahead of print].
177. Sekiguchi K, Itonaga T, Maeda T, Fukami M, Yorifuji T, Ihara K. A case of CHARGE syndrome associated with hyperinsulinemic hypoglycemia in infancy. Eur J Med Genet. (2018) 61:312–14. doi: 10.1016/j.ejmg.2018.01.008
178. Shuman C, Beckwith JB, Weksberg R. Beckwith-Wiedemann Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. Source GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993-2018 (2000) (Accessed August 11, 2016).
179. DeBaun MR, King AA, White N. Hypoglycemia in Beckwith-Wiedemann syndrome. Semin Perinatol. (2000) 24:164–71. doi: 10.1053/sp.2000.6366
180. Smith AC, Shuman C, Chitayat D, Steele L, Ray PN, Bourgeois J, et al. Severe presentation of Beckwith-Wiedemann syndrome associated with high levels of constitutional paternal uniparental disomy for chromosome 11p15. Am J Med Genet A. (2007) 143A:3010–5. doi: 10.1002/ajmg.a.32030
181. Senniappan S, Ismail D, Shipster C, Beesley C, Hussain K. The heterogeneity of hyperinsulinaemic hypoglycaemia in 19 patients with Beckwith-Wiedemann syndrome due to KvDMR1 hypomethylation. J Pediatr Endocrinol Metab. (2015) 28:83–6. doi: 10.1515/jpem-2013-0390
182. Adachi H, Takahashi I, Higashimoto K, Tsuchida S, Noguchi A, Tamura H, et al. Congenital hyperinsulinism in an infant with paternal uniparental disomy on chromosome 11p15: few clinical features suggestive of Beckwith-Wiedemann syndrome. Endocr J. (2013) 60:403–8. doi: 10.1507/endocrj.EJ12-0242
183. Gohda Y, Oka S, Matsunaga T, Watanabe S, Yoshiura K, Kondoh T, et al. Neonatal case of novel KMT2D mutation in Kabuki syndrome with severe hypoglycemia. Pediatr Int. (2015) 57:726–8. doi: 10.1111/ped.12574
184. Toda N, Ihara K, Kojima-Ishii K, Ochiai M, Ohkubo K, Kawamoto Y, et al. Hyperinsulinemic hypoglycemia in Beckwith-Wiedemann, Sotos, and Kabuki syndromes: a nationwide survey in Japan. Am J Med Genet A (2017) 173:360–67. doi: 10.1002/ajmg.a.38011
185. Yap KL, Johnson AEK, Fischer D, Kandikatla P, Deml J, Nelakuditi V, et al. Congenital hyperinsulinism as the presenting feature of Kabuki syndrome: clinical and molecular characterization of 10 affected individuals. GenetMed. (2019) 21:233–42 doi: 10.1038/s41436-018-0013-9
186. Nakamura Y, Takagi M, Yoshihashi H, Miura M, Narumi S, Hasegawa T, et al. A case with neonatal hyperinsulinemic hypoglycemia: It is a characteristic complication of Sotos syndrome. Am J Med Genet A (2015) 167A:1171–4. doi: 10.1002/ajmg.a.36996
187. De Boer L, Van Duyvenvoorde HA, Willemstein-Van Hove EC, Hoogerbrugge CM, Van Doorn J, Maassen JA, et al. Mutations in the NSD1 gene in patients with Sotos syndrome associate with endocrine and paracrine alterations in the IGF system. Eur J Endocrinol. (2004) 151:333–41. doi: 10.1530/eje.0.1510333
188. Cappella M, Graziani V, Pragliola A, Sensi A, Hussain K, Muratori C, et al. Hyperinsulinemic hypoglycaemia in a Turner syndrome with ring (X). Case Rep Pediatr. (2015) 2015:561974. doi: 10.1155/2015/561974
189. Pietzner V, Weigel JF, Wand D, Merkenschlager A, Bernhard MK. Low-level hyperinsulinism with hypoglycemic spells in an infant with mosaic Turner syndrome and mild Kabuki-like phenotype: a case report and review of the literature. J Pediatr Endocrinol Metab. (2014) 27:165–70. doi: 10.1515/jpem-2013-0090
Keywords: hyperinsulinism, hypoglycaemia, molecular mechanisms, genetics, mutation
Citation: Galcheva S, Demirbilek H, Al-Khawaga S and Hussain K (2019) The Genetic and Molecular Mechanisms of Congenital Hyperinsulinism. Front. Endocrinol. 10:111. doi: 10.3389/fendo.2019.00111
Received: 02 November 2018; Accepted: 06 February 2019;
Published: 26 February 2019.
Edited by:
Tsutomu Ogata, Hamamatsu University School of Medicine, JapanReviewed by:
Tohru Yorifuji, Osaka City General Hospital, JapanTatsuhiko Urakami, Nihon University, Japan
Copyright © 2019 Galcheva, Demirbilek, Al-Khawaga and Hussain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khalid Hussain, a2h1c3NhaW5Ac2lkcmEub3Jn