 Fan Zhong
Fan Zhong Yan Jiang
Yan Jiang- 1Department of Gastroenterology, Songjiang Hospital Affiliated First People's Hospital, Shanghai Jiao Tong University, Shanghai, China
- 2Institutes of Biomedical Sciences of Shanghai Medical College, Fudan University, Shanghai, China
Endogenous pancreatic β cell regeneration is a potential strategy for β cell expansion or neogenesis to treat diabetes. Regeneration can occur through stimulation of existing β cell replication or conversion of other pancreatic cells into β cells. Recently, various strategies and approaches for stimulation of endogenous β cell regeneration have been evaluated, but they were not suitable for clinical application. In this paper, we comprehensively review these strategies, and further discuss various factors involved in regulation of β cell regeneration under physiological or pathological conditions, such as mediators, transcription factors, signaling pathways, and potential pharmaceutical drugs. Furthermore, we discuss possible reasons for the failure of regenerative medicines in clinical trials, and possible strategies for improving β cell regeneration. As β cell heterogeneity and plasticity determines their function and environmental adaptability, we focus on β cell subtype markers and discuss the importance of research evaluating the characteristics of new β cells. In addition, based on the autoimmunologic features of type 1 diabetes, NOD/Lt-SCID-IL2rgnull (NSG) mice grafted with human immune cells and β cells are recommended for use in evaluation of antidiabetic regenerative medicines. This review will further understand current advances in endogenous β cell regeneration, and provide potential new strategies for the treatment of diabetes focused on cell therapy.
Introduction
The pancreas plays an essential role in energy consumption and metabolism. It consists of two functionally and morphologically distinct components: the exocrine and endocrine. The exocrine pancreas is composed of acinar and ductal cells that secrete digestive enzymes. The endocrine pancreas is composed of five different hormone-secreting cell types that include glucagon-secreting α cells, insulin-producing β cells, somatostatin-releasing δ cells, ghrelin-releasing ε cells, and pancreatic polypeptide (PP)-secreting cells. These cells aggregate to form the islets of Langerhans, which are intermingled with the intra-islet microvascular network and play an essential role in regulation of blood glucose levels by directly secreting insulin and glucagon into the bloodstream. Type 1 diabetes (T1D) and type 2 diabetes (T2D) are defined as blood hyperglycemia caused by an absolute or relative deficiency of pancreatic β cells. Autopsy studies have shown deficits in β cell mass in approximately 70~100 and 0~65% in patients with T1D and T2D, respectively (1, 2). Therefore, β cell mass regeneration is a potential therapeutic strategy for recovery of β cell loss in patients with diabetes. Regeneration of β cells occurs through endogenous regeneration or exogenous supplementation, such as transplantation of cadaveric islets or grafting of new β cells generated from in vitro cell engineering. Recently, numerous strategies and technologies for producing human insulin-secreting cells have emerged, including in vivo stimulation of existing β cell replication, reprogramming of other pancreatic cells to differentiate into β cells, in vitro differentiation of induced pluripotential stem (iPS) cells into new β cells, and generation of human islets from genetically engineered pigs (3, 4). However, clinical application has remained a challenge. For example, strategies for enhancing replication of residual β cells have been successful in rodent but not in humans. In addition, drugs that stimulated conversion of α cells into β cells in animal experiments did not do so in clinical trials. As such, it is critical to determine the causes for limited success of clinical trials, and to determine possible strategies for improving cell therapy for T1D. In this review, we summarize advanced strategies and approaches for endogenous β cell regeneration, discuss regenerative mechanisms under physiological and pathological conditions, focus on various factors involved in stimulation of regeneration, and discuss promising potential pharmaceutical drugs. Moreover, as T1D is characterized by autoimmune-mediated β cells death, and heterogeneity and plasticity of β cells determine their function and environmental adaptability, we believe that thorough understanding associations between neogenetic β cells and diabetogenic autoimmune cells can lead to strategies to enhance the immunologic tolerance of neogenetic β cells, thus improving T1D cell therapy. In this review we introduce β cell subtyping markers that correspond with their functional features, and highlight the importance of using the humanized diabetic mice grafted with autoimmune cells and β cells in future studies.
Replication of Existing Pancreatic β Cells
Pancreatic β cells replicate readily in the fetal and neonatal stages. However, this ability to replicate rapidly declines after these stages. Furthermore, this ability to replicate is different in rodents and humans. Proliferation of β cells is precisely controlled by cell cycle regulators and circulating soluble factors. Studies have shown that many mitogenic agents could stimulate β cell replication in young rodents, but not in humans. However, using high-throughput chemical screening, a series of inhibitors of DYRK1A-NFAT, GSK3, and NF-κB signaling pathways were shown to increase human pancreatic β cell replication, suggesting that these inhibitors have unique potential for treatment of diabetes.
Replicative Ability of β Cells Over the Lifetime
During embryonic development, insulin-positive β cells appear at approximately embryonic day 13.5 in mice or during weeks 8–9 in humans. During the fetal period, β cells are mainly generated by differentiation of endocrine progenitor cells (5). During the late gestational and neonatal stages, β cells are generated by replication of existing β cells (6, 7). The rate of β cell replication reduces after weaning, and the renewal capacity of β cells becomes limited during adulthood or late adolescence. Nevertheless, β cell mass, which is determined on the basis of cell numbers and individual cell volumes, correlates in a linear fashion with body weight throughout the lifespan of an organism (5, 8). For example, in rats, the number and size of β cells expands with body weight during the first few months of life. The rate of β cell replication then progressively declines, to 1% in young rats (1 month of age), and <0.2% in adults (3~7 months) (8). In aging rats (15~20 months), β cell mass primarily increases through increased cell size (9). In healthy rodents, individual β cells have long lifespans, and replication of mature β cells is limited during adulthood (5, 10). Under some physiological or pathological conditions, rates of β cell proliferation are elevated. For example, β cells proliferate adaptively in response to pregnancy or obesity via self-replication (11–14). Moreover, in young rodents, β cell proliferation can be induced by increased metabolic demands or β cell deficiency resulting from tissue injury (8, 15).
Different β Cell Replicative Ability Between Rodent and Human
Human and rodent islets have distinct structural and molecular characteristics (16). Replicative ability of human and rodent β cells have common and different features. For example, β cell mass increases during the earlier stages of life and declines with aging in both species. Adaptive β cell proliferation during pregnancy and obesity occurs extensively in rodents, but is limited in humans (17). Pregnancy-associated insulin resistance induces amplified insulin production to maintain glucose homeostasis. In rodents, elevated insulin production is accompanied by increased β cell numbers mediated by lactotrophic hormones (13, 14, 18). Humans also exhibit a compensatory increase in insulin secretion. New β cells originate from other pancreatic cell lineages and existing β-cells. Moreover, β cell proliferation mediated by lactotrophic hormones or other mitogenic stimuli is limited in humans (19). In addition, obesity-induced insulin resistance is associated with dramatic expansion of β cell mass in several rodent models (20), but not in human islets (20). Various mitogenic agents, hormones, and growth factors (GFs) such as Glp-1, Gip-1, exendin-4, prolactin, Hgf, and Igf-1 stimulate β cell proliferation in rodents but not in humans (21–27).
Mediators of β Cell Replication
Cell Cycle Regulators
β cell replication is mediated by multiple mitogenic signaling pathways such as Irs–Pi3k–Akt, Gsk3, mTor, ChREBP/cMyc, Ras/Raf/Erk, and Nfats. These mechanisms also involve upstream activators of mitogenic signaling pathways, including nutrients (glucose, calcium), epidermal and platelet-derived GFs (Glp1, Gip), and hormones (leptin, estrogen, prolactin, and progesterone). Mitogenic signals stimulate quiescent β cells to re-enter the cell cycle by regulating the expression of downstream cell cycle regulators such as cyclins, cyclin-dependent kinases (Cdks), cell-cycle inhibitors, and E2F factors (28–33). For example, exendin-4 and glucagon-like peptide 1 (Glp-1) exert mitogenic effects on β cell proliferation by activating cell cycle activators (cyclin A and Cdk1) and proliferation-activating transcription factors (TFs) through the cAMP-dependent calcineurin/Nfat pathway (24, 25, 34–37). Menin is an endocrine tumor suppressor that suppresses β cell proliferation by epigenetically promoting the expression of the cell-cycle inhibitors p27 and p18 or by inhibiting K-Ras signaling (38–40). Moreover, Ezh2 mediates increased trimethylation of p16INK4a and p19Arf by H3K27, which epigenetically represses Ink4a/Arf production and contributes to proliferation of pancreatic β cells (41).
Circulatory Regulators
Circulating soluble factors derived from other organs act as systemic regulators that control β cell proliferation during puberty, pregnancy, and obesity. Multiple circulatory regulators have been implicated in control of β cell proliferation in response to insulin resistance. Examples of circulating regulators include intestinal peptides such as Glp-1 and Gip-1 (24, 25), adipose-tissue-derived adipokines such as adipsin (42, 43), resistin, and leptin (44, 45), and skeletal-muscle-secreted factors such as Il6 and Il10 (46, 47). In addition, crosstalk between hepatic and pancreatic tissues modulates β cell growth in response to insulin resistance. Many hepatocyte-derived factors have been identified as stimulators of β cell proliferation in mice and humans (48). In an insulin receptor knockout mouse model of insulin resistance, hepatocyte-derived secretory SerpinB1, and its partial mimic GW311616A, enhanced β cell proliferation by inhibiting elastase activity and activating key proteins in GF signaling (49). In addition, in mouse models of diabetes, exogenic expression of hepatic GFs, such as Hgf, Igf1, and Igf2, can regulate β cell mass by increasing β cell replication (50–53).
Strategies for the Stimulation of β Cell Replication
Multiple approaches have been evaluated to rapidly and robustly replenish β cell masses. Numerous stimuli that promote β cell proliferation have been identified (38, 54–56). Studies have shown that administration of exogenous stimuli can stimulate β cell proliferation in young rodents. Whether adult rodent β cells can be induced to proliferate by exogenous stimuli remains unclear. Some studies have suggested that the replication of existing β cells induced by pancreatectomy (Px) or β cell apoptosis is the major source of new insulin-expressing cells in adult mice (6, 57). Other studies have indicated that various diabetogenic injuries, including partial Px, streptozotocin administration, and pancreatic duct ligation (PDL), cannot stimulate β cell proliferation in adult mice (21, 58, 59). Recently, high-throughput chemical screening has identified multiple potential agents for stimulation of β cell replication (60). As shown in Table 1, these agents include DYRK1A inhibitors (harmine, aminopyrazine compounds, and 5-iodotubercidin), which increase β cell proliferation by inhibiting calcineurin/Nfat/Dyrk1a signaling (61, 62, 64, 66). Osteoprotegerin and denosumab stimulate human β cell proliferation through inhibition of the receptor activator of NF-κB ligand pathway (63). Moreover, high-throughput RNAi screening has demonstrated that CDKN2C/p18 or CDKN1A/p21 silencing facilitated cell-cycle re-entry of quiescent adult human β cells (65).
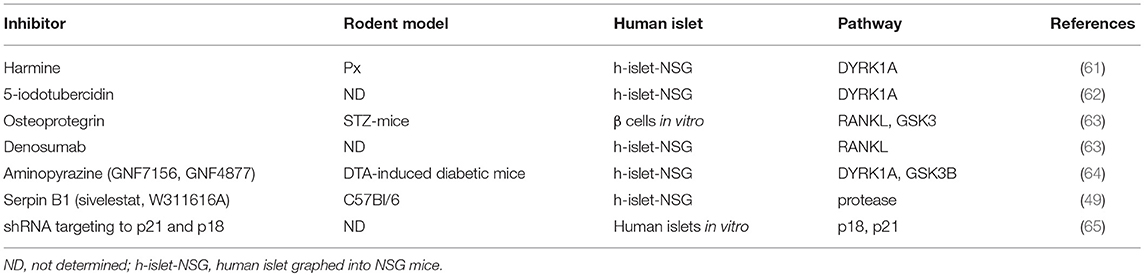
Table 1. Potential drugs for increasing pancreatic β cell replication.
Reprogramming of Other Pancreatic Cells Into β Cells
During embryonic development, pancreatic β cells are generated from multipotent pancreatic progenitors in a sequential and gradual process that is elaborately controlled by defined transcription factors. Pancreatic ductal epithelium cells and Ngn3 positive pancreatic cells are generally considered progenitors of β cells. They differentiated into β cells when islets were destroyed in rodent models. In this section, we introduce advanced strategies for conversion of non-β cells to β cells through ectopic expression of specific TFs or by pharmaceutical stimuli.
TFs Regulate the Differentiation of Endocrine Cells
Development of embryonic pancreatic β cells is elaborately controlled by TFs involved in pancreatic determination (67) (Figure 1). During early pancreatic bud outgrowth, maintenance and specialization of multipotent pancreatic progenitor cells (MPCs) is modulated by the pancreatic TFs Gata4/6, Foxa1/2, Pdx1, Ptf1a, Mnx1, Sox9, Nkx6.1, and Hnf1β. Depletion of any of these TFs impairs pancreatic bud formation (68–72). Although Pdx1 is extensively expressed in pancreatic cells, it is only highly expressed in adult β cells. Lineage tracing studies in mice have demonstrated that Pdx1-positive MPCs possess high proliferative ability and can differentiate into all cell types in each of the three major pancreatic compartments (exocrine, endocrine, and ductal) (73, 74). Furthermore, the ectopic expression of Pdx1 or Ptf1a in the endoderm can induce ectopic pancreatic bud formation (71, 75). Ptf1a+/Gata4+ MPCs differentiate into exocrine progenitors, whereas Sox9+/Ndx6.1+ MPCs differentiate into endocrine/ductal bipotent progenitors (69, 76–79). Early endocrine progenitors originate from bipotent trunk duct-endocrine progenitors, and their differentiation is initiated by the expression of neurogenin 3 (Ngn3). Ngn3-deficient mice fail to generate endocrine cells, but undergo duct enlargement, and ectopic expression of Ngn3 directs pancreatic progenitors toward an endocrine fate. These phenomena suggest that Ngn3 is necessary for differentiation of islet cells (80–82). Endocrine development requires participation of other endocrine progenitor TFs such as Isl1, Neurod1, Pax6, Mafb, Nkx2.2, and Rfx6. These TFs are activated by Ngn3 and are involved in differentiation of endocrine cell lineages (80, 83). Finally, monohormonal islet cell lineages differentiated from Ngn3-positive endocrine progenitors are also regulated by a specific combination of TFs. For example, Pax4 and Arx participate in islet cell specialization via cross-inhibitory interactions. Pax4 and Arx promote the differentiation of islet progenitor cells into β/δ or α/PP cells, respectively (84). The α cell positive TF profile includes Arx, Mafb, Rfx6, Nkx2.2, Neurod1, and Pax6, whereas the β cell positive TF profile includes Nkx2.2, Pax4/6, Pdx1, Nkx6.1, and Mafα.
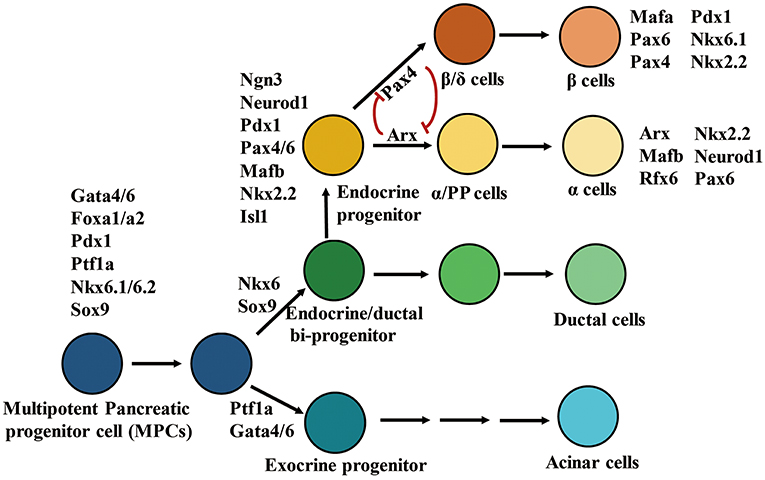
Figure 1. Transcription factors involve in differentiation of pancreatic cell lineages. In the diagrammatic sketch for pancreatic cell differentiation, the process of differentiation from multipotent pancreatic progenitors to α or β cells is presented; and the combination of key TFs determining the specialization of endocrine and exocrine pancreatic cell lineages is displayed. The figure was reproduced with permission from Elsevier and Copyright Clearance Center. This figure was adapted from Hang and Stein (67).
Differentiation of Pancreatic Progenitors Into β Cells
Increasing β cell proliferation is a possible approach for recovery of β cell deficiency in diabetes. Stimulation of β cell neogenesis, however, may be a more feasible approach for treatment of diabetes than elevation of β cell proliferation given the nearly complete loss of β cells in T1D. Neogenesis is defined as formation of insulin-producing β cells through differentiation from stem/progenitor cells or conversion from other pancreatic cells. The existence of adult β cell progenitors remains the most controversial topic in diabetes research despite evidence showing that pancreatic cell lineages including ductal, endocrine, and exocrine, are derived from embryonic multipotent progenitors. Several studies have shown that β cells develop from other progenitor cells. The pancreatic ductal epithelium is a potential progenitor of islet and acinar tissues after birth (85). Foci of regeneration induced by partial Px comprise new ductal cells that express markers of embryonic pancreatic epithelium, including Pdx1, Hnf6, Foxa2, Tcf1/2, and Sox9, resulting in formation of new pancreatic lobes. These behaviors suggest that new ductal cells act as progenitors for the regenerating pancreas (86). In adult rodents, ductal cells differentiate into β cells in response to specific stimuli. For example, in adult mice Sox9 positive ductal cells differentiate into β cells in response to moderate hyperglycemia combined with long-term administration of low dose epidermal GFs (69, 87). In the PDL mouse model, pancreatic duct cells positive for the expression of carbonic anhydrase II, a duct cell-specific marker, act as progenitors of new islets and exocrine cells after injury (88–90). Recently, a selective Cdk5 inhibitor was identified that could promote β cell differentiation from ductal progenitors in zebrafish. This effect was observed in adult mice treated with PDL and human induced pluripotent stem (iPS) cells, which indicated that Cdk5 acts as an endogenous suppressor for β-cell differentiation (91). Moreover, in injured adult mouse pancreases, Ngn3-positive pancreatic cells act as endocrine progenitors and give rise to all islet cell types, including glucose-responsive β cells through the notch signaling pathway (73, 92, 93).
Conversion of Other Pancreatic Cells to β Cells
β cell regeneration through transdifferentiation from other pancreatic cells, including exocrine and endocrine pancreatic cells, has been well-characterized. Results of genetic lineage tracing studies involving mouse models of severe β cell ablation have suggested that new insulin-producing β cells are generated from conversion of pancreatic α or δ cells in adult and adolescent mice (94, 95). Moreover, in diabetic mice transient treatment with epidermal growth and ciliary neurotrophic factors induces reprogramming of acinar cells to β cell masses (96). Differentiation of pancreatic lineages is sequentially and regionally regulated by pancreatic TFs (97). In adult mice, re-expression of the specific combination of Ngn3, Pdx1, and Mafα by adenoviral transduction contributes to reprogramming of pancreatic exocrine cells into insulin-expressing cells that are similar to β cells (98). Similarly, ectopic overexpression of β cell-specific TFs also induces reprogramming of exocrine or endocrine cells into β cells. In adult mice, ectopic expression of β cell-specific single TFs such as Pax1 or Pax4, ectopic expression of a combination of Pax1 and Mafα, or targeted disruption of α cell-specific TFs such as Dnmt1 and Arx, can induce conversion of α cells to β cells (74, 99–102). In addition, forced expression of Pax4 also mediates transdifferentiation of δ cells, Ngn3 positive endocrine progenitors, and duct-lining precursor cells into β cells (74, 103, 104).
Potential Drugs for Stimulation of β Cells Conversion
Although ectopic expression of TFs effectively induces conversion of other pancreatic cell into β cells in mice, viral or transgene-mediated overexpression may be difficult to achieve in humans. As such, drug-stimulated conversion may be a potential alternative approach for T1D treatment. Recently, several small molecules have been identified as activators of β cell neogenesis.
γ-Aminobutyric Acid
γ-Aminobutyric acid (GABA), an inhibitory neurotransmitter in the central nervous system, is synthesized from glutamate by glutamate decarboxylase (GAD) (105). High levels of GABA and GAD are present in pancreatic islet cells, particularly β cells (106, 107). GAD65, an isoform of GAD, acts as a major autoantigen in T1D (108). GABA promotes β cell replication and inhibits β cell apoptosis in mouse models of STZ-induced diabetes and grafted human islets (109–112). GABA released from β cells interacts with and activates the ionotropic receptor GABAA (a Cl− ion channel) and the metabotropic G-protein-coupled receptor GABAB in plasma membranes of islet cells (113–116). Binding of ligands to receptors enhance insulin secretion from β cells and suppress glucagon release from α cells (109, 117). Recently, Ben Othman reported that prolonged GABA exposure induced conversion of α cells into β cell-like cells in a mouse model of STZ-induced diabetes. Moreover, GABA treatment results in loss of α cells in grafted human islets and concomitantly increased islet mass and β cell-like counts. The mechanism of GABA-mediated conversion of α cells into β cells, however, requires further elucidation. The ability of GABA to downregulate Arx expression suggests that it acts on GABAA receptors on α cells (118). In addition, GABA may act as an immunosuppressive regulator in T1D by mediating cytokine secretion from human peripheral blood mononuclear cells and CD4+ T cells (119, 120). In summary, administration of GABA contributes to replication of β cells, enhances conversion of α cells to β cells, and suppresses immune reactions in rodent models of diabetes. Given these actions, GABA has a potential antidiabetic role and clinical value for treatment of T1D.
Artemisinin
Artemisinin may act as a potential activator of conversion of α cells to β cells. One report showed that artemisinin impaired α cell identity and induce insulin expression in α cells through translocation of Arx from the nucleus to the cytoplasm, which then inhibited Arx. Moreover, the mechanism of action of artemisinin on transdifferentiation of α cells into β-like cells involves enhancement of GABA receptor signaling in a gephyrin-dependent manner (121). However, Meulen et al. reported that stimulation of intact islets with high doses of artemether failed to promote transdifferentiation of primary α cells to β cells. Moreover, artemisinin reduces Ins2 gene expression, suppresses glucose uptake, and abrogates calcium responses and insulin secretion in response to glucose (122). These paradoxical effects of artemisinin on β cell regeneration warrant further verification.
Diet Therapy
Interestingly, a novel diet therapy with 4-day fasting-mimicking diet (FMD) cycles can reverse β cell failure and can reverse diabetes in mice. FMD promotes Ngn3-driven β cell regeneration by inducing re-expression of prenatal development genes, such as Sox17 and Pdx1, in the adult pancreas (123).
In addition, the mechanism underlying maintenance of the correct proportion of cellular components in neogenesis of islets derived from conversion of non-β cells has yet to be determined. For example, how the pancreas compensates for loss of α cells during conversion of α cells to β cells requires further study. One potential mechanism is that α cell conversion includes mobilization of duct-lining precursor cells (reawakening of the epithelia-to-mesenchymal transition), regeneration of α cells, and conversion of α cells to β cells (118). This mechanism suggests that neogenetic β cells originate from neogenetic α cells that differentiated from mobilized Ngn3-positive endocrine progenitor cells. Accordingly, formation of new islets is a complex and dynamic process, and new islets may contain different cell types or even intermediate transitional cells. Tracking the fate of these cells through lineage tracing, and identifying them by single cell analysis would help reveal mechanisms of α cell conversion (124–126).
Rodent Models For Study of β Cell Regeneration
Diabetic rodent models, which generally include classical and genetic models, play important roles in the study of the molecular mechanisms of β cell regeneration and the evaluation of the effects of potential pharmaceutical drugs for diabetes treatment. Classical models are defined by damage of β cells by surgery or treatment with chemical compounds, such as Px, PDL, and STZ-mediated β cell ablation. Genetic models are constructed through crossing special transgenic mouse strains, which leads to specific and inducible β cell ablation or allows for tracking of target islet cells (Figure 2). In this section, we comprehensively introduce principles and protocols for construction of these rodent diabetic models, and review the recent advances in β cell regeneration through use of these models. In addition, based on autoimmunologic features of T1D and difficulties in stimulation of β cell regeneration in humans, we introduce a humanized diabetic mouse model, and suggest use of this model for study of β cell regeneration.
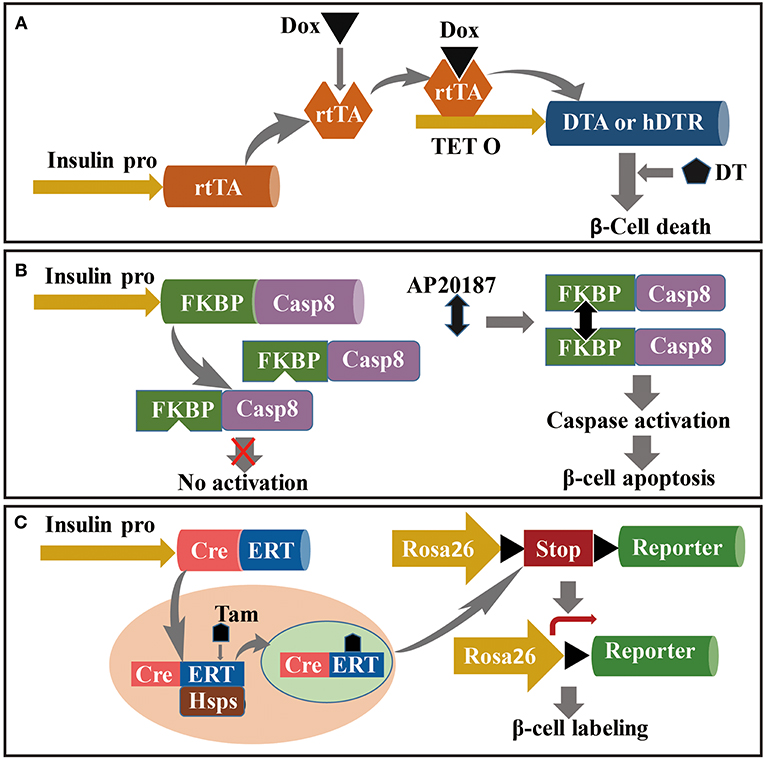
Figure 2. Mouse model for the study of β cell regeneration. The targeting to pancreatic β cells is relayed on the driving of insulin promoter, and conditional ablation (or labeling) is dependent on the induction under Dox, AP20187 or Tam administration in specific transgenic strains. (A) Dox-induced DT-dependent specific ablation of β cells; (B) Caspase-FKBP-induced apoptosis of pancreatic β cells conditionally activated by AP2018; and (C) Tam-induced lineage tracing of pancreatic β cells based on CreERT: Rosa26-LoxP-Reporter transgenic strains.
Classical Diabetic Rodent Models
Classical rodent models of β cell regeneration include partial Px, PDL, and STZ-mediated β cell ablation. Px: Removal of 60~90% of the adult rat pancreas through Px administration induces extensive pancreatic regeneration with formation of new lobes and islets, and proliferation of acinar cells. The Px model has been used to study β cell neogenesis and replication (86, 127, 128). PDL: PDL is defined as surgical ligation of the pancreatic duct at the level of the pylorus. This procedure obstructs drainage of exocrine secretions and results in loss of acinar cells through death and dedifferentiation. During the early years of diabetes research, this model was widely used to study mechanisms of β cell formation (129–131). More recently, the PDL model has been used to demonstrate expansion of β cells generated from Ngn3-positive endogenous progenitors (92). A controversial finding, however, suggested that PDL failed to elevate β cell levels in mice (132, 133). STZ-mediated β cell ablation: STZ, a cytotoxic chemical produced by Streptomycetes achromogenes, can be used to cause extensive damage to endogenous β cells and induce hyperglycemia. It binds to the Glut-2 transporter, which is abundantly distributed on plasma membranes of β cells. STZ causes DNA alkylation and generates high levels of free radicals that cause DNA damage and cell death. STZ can be used alone or in combination with other chemicals to induce diabetes. STZ-induced β cell damage causes spontaneous β cell regeneration in neonatal and adult rodents (134–136). STZ administration, however, has failed to stimulate an adaptive increase in β cells in adult monkeys (137). The STZ model has been used to study β cell regeneration induced by various stimuli, including transgenes and signaling pathway activators (52, 138–142).
Genetic Modification Induces β Cell Ablation
Diphtheria Toxin-Targeted Specific and Inducible β Cell Ablation
Corynebacterium diphtheriae produces diphtheria toxin (DT) as a single secretory polypeptide. In vitro, mature DT generates two components, fragments A (DTA) and B (DTB). DT binds to a DT receptor on cell surfaces and is incorporated into cells through receptor-mediated endocytosis. DT can inactivate elongation factor (EF)-2 in cells by catalyzing transfer of ADP-ribose to EF-2. Inactivation of EF-2 inhibits protein synthesis and causes cell death (143). Thus, specific and inducible killing of pancreatic β cells is based on doxycycline (Dox)-induced expression of DT. The approach for construction of this model is crossing an insulin-reverse tetracycline-dependent transactivator (insulin-rtTA) transgenic mouse strain with the TetO-DTA mouse. RtTA expression in the insulin-rtTA mouse strain is driven by a rat insulin promoter, whereas DTA subunit expression in the TetO-DTA mouse strain is driven by a rtTA-responsive promoter. In the Tet–on system, rtTA interacts with the tet-resistance operon in the presence of Dox and activates transcription (Figure 2A) (144). Accordingly, administration of Dox to double-transgenic mice induces DTA expression in β cells, resulting in widespread β cell apoptosis (15). Moreover, DT binding affinity for human DT receptors is 105-fold higher than that of murine DT receptors (145). Conditional and targeted cell ablation in mice can be achieved through transgenic human DTR expression driven by specific promoters in the presence of DT. Numerous studies have documented that ectopic expression of human DTR driven by insulin or glucagon promoters resulted in targeted ablation of 99% of α or β cells following DT administration (94, 95, 146).
Caspase 8–FKBP Transgene Induces β Cell Apoptosis
In this section, mechanisms of caspase-FKBP-induced specific-cell ablation are summarized in context of conditional caspase activation driven by cell-specific promoters. Using the caspase-FKBP transgenic model, multiple tissue cells have undergone conditional and specific ablation such as cardiac myocytes, adipocytes, hepatocytes, and pancreatic β cells (147–151). The caspase-FKBP fusion protein was designed with human caspase catalytic domains such as p20 and p10 from caspase 8 fused with a series of FKBPv domains such as Phe36Val mutant FKBP. The binding affinity of the caspase–FKBP fusion protein for the FK506 analog AP20187 is 1,000-fold greater than that of endogenous FKBP. The mechanism underlying caspase activation through the forced dimerization of adjacent FKBP molecules by AP20187 is shown in Figure 2B (148–150). Accordingly, a PANIC–ATTAC mouse model was constructed through transgenic expression of FKBPv–caspase 8 fusion protein driven by a rat insulin promoter. This model has been used to study inducible and reversible β cell ablation and other aspects of diabetes (150, 151).
Genetic Cell Lineage Tracing Model
Genetic cell lineage tracing has been used for tracking target cells in the body, and enables visualization of the source and fate of target cells. This approach has been used to analyze formation and regeneration of β cells under physiological or pathological conditions (152). For the past several decades, the Cre/loxP genetic lineage tracing system has been the most widely used method for tracking cell fate (153). The Cre/loxP system functions through expression of Cre recombinase driven by the loxP-stop-loxP cassette, a cell-specific promoter located upstream of the reporter gene. Cre recombinase excises the floxed STOP cassette and subsequently activates reporter gene expression. Conditional or inducible expression of Cre recombinase is based on the TetO-Cre or CreERTM transgenic mouse strains, which are crossed with the ROSA26/reporter mouse strain and result in inducible tracking for target cells. The CreERTM transgenic cassette contains a fusion of Cre recombinase with a mutated ligand-binding domain (ERTM), the latter of which preferentially binds to the antiestrogen tamoxifen instead of endogenous 17β-estradiol (154). Under normal conditions, the CreERT fusion protein is sequestered by HSPs in the cytoplasm. Following treatment with tamoxifen, tamoxifen binds to CreERT, resulting in disruption of the interaction with Hsp90. Released CreERT transfers to the nucleus, initiates recombination, and activates reporter gene expression (Figure 2C) (155, 156). Numerous experiments aimed at identifying the source or fate of pancreatic cells in rodent models have been performed using the conditional Cre-LoxP genetic lineage tracing system (6, 103, 104, 123). For example, Fabrizio crossed RIP-DTR, RIP-CreERTM, and ROSA26-LoxP-YFP transgenic mouse strains to achieve DT-dependent conditional β cell ablation and tamoxifen-dependent β cell lineage tracing. Moreover, hybridization among the transgenic mouse strains Glucagon-rtTA, TetO-Cre, and ROSA26-LoxP-YFP was used to track the source of insulin+ cells. This study found that 65% of insulin-expressing cells following β cell ablation were YFP+, which indicated that neogenetic insulin-expressing cells originated from α cells (94).
Humanized Diabetic Mice Model
T1D is an autoimmune disease caused by immune-mediated destruction of pancreatic β cells. The etiology of T1D involves interactions among genetic, environmental, and immune factors. Multiple approaches for prevention or treatment of T1D have been have been successfully used in the non-obese diabetic (NOD) mouse model, but have not been successfully reproduced in humans. These failures may be attributed to structural and compositional variations between NOD murine and human islet cells, as well as to differences between human and murine immune systems (157, 158). These differences include genetic susceptibility loci, immune responses to environmental factors, leukocyte subsets, and immunological factor compositions (159, 160). To define the interaction between the human immune system and β cells, and to improve treatments for T1D, a humanized diabetes mouse model in which mice are grafted with a functional human immune system and human β cells closely imitates the physiological conditions of human T1D. The immunodeficient NOD/Lt-SCID-IL2rgnull (NSG) genotype constructed on NOD mice is suitable for grafting of human immune cells and human β cells because it contains genetic modifications of severe combined immunodeficiency (SCID) mutation and a complete null mutation of the IL2rg gene (160–163). NSG mice can be grafted with human tissue, hematopoietic stem cells, and peripheral blood mononuclear cells (162, 164). Recently, studies of immunological mechanisms of T1D, treatment strategies for diabetes, and transplantation of islet cells using NSG mice have made significant progress (165–170). Thus, this model will be a powerful platform for finding potential drug targets for T1D therapy and evaluating antidiabetic drugs in preclinical trials.
Characterizing Neogenetic Islet Cells
New β cells are typically described as β cell-like cells, which often possess the basic characteristics of β cells such as glucose-stimulated insulin secretion. However, β cells are heterogeneous and exhibit plasticity during development, under pathological conditions, or following specific treatments. Therefore, it is necessary to identify characteristics and subtypes of new β cells.
Features of Mature β Cells
Embryonic and neonatal β cells are immature and can produce insulin, but lack the ability to respond to glucose stimulation. Within days after birth, β cells develop the ability to secrete insulin in response to glucose (glucose-stimulated insulin secretion; GSIS) and become mature functional β cells (171). Mafa, NeuroD, and Errγ drive β cell maturation, and urocorin3 (Ucn3) acts as a marker of β cell maturation (172–176). Immature β cells that lack Ucn3 are present throughout life. These cells are involved in the intermediate stage of transdifferentiation of α cells into β cells and may be potential sources for β cell regeneration (177). Therefore, given that the maturation state of β cells affects insulin production, secretion, and GSIS, subtyping new β cells will allow for better understanding of β cell characteristics and functions.
New Markers for Subtyping of β Cells
Under pathological conditions or in response to specific treatments, adult β cells exhibit heterogeneous responses (178). During the past few decades, a series of markers of β cell heterogeneity have been identified and characterized (Table 2). For example, insulin and Pdx1 levels reflect different maturation states of β cell subpopulations (179, 185). Expression of Glut2lower characterizes rare subpopulations of β cells with low insulin content, properties of stem/progenitor cells, and lineage plasticity that appears during β cell regeneration induced by β cell ablation (180). The correlation between E-cadherin and insulin levels in adult rodent β cells suggests the importance of tight cell-to-cell junctions to the function of β cells (181, 186). Recently, novel proteins have been identified as phenotypic and functional markers for discrimination of β cell populations (187). Flattop, a Wnt/planar cell polarity effector, can be used to distinguish the proliferative competence of mature β cells. Subpopulations of β cells labeled by Flattop present distinct molecular (gene expression level of Ucn3 and MafA), physiological (expansive ability to respond to stimulation), and ultrastructural features (182, 188, 189). Other subtyping markers, St8sia1 and Cd9, based on their expression levels, human β cells have been divided into four subtypes, which have diverse gene expression profiles and distinct basal and GSIS ability (183). In addition, hubs are the markers for β cell subpopulations with transcriptional immaturity and high metabolism. Hubs were discovered through studies of optogenetics and photopharmacology (184).

Table 2. The markers for subtyping of pancreatic β cells.
Functional cooperation among islet cells is dependent on the three-dimensional architecture and cellular composition of islet cells (190). β cell subpopulations combine with other islet cells to form a three-dimensional islet architecture that contributes to distinct functions and influences development of diabetes mellitus. In addition, the physiological and pathological conditions of the pancreas also affect β cell heterogeneity (191, 192). In this process, β cells adapt physiologically, morphologically, and functionally to specific environmental cues. Therefore, given that heterogeneity and plasticity of β cells determine their functional and environmental adaptability, subtyping new insulin expressing cells would provide new clues for treatment of diabetes.
Discussion
Over the past several decades, studies examining endogenous β cell regeneration have proposed numerous strategies for treatment of β cell-deficient diabetes. Most of these strategies, however, have only been successfully applied to animals. Although some treatment strategies for diabetes have been successful in rodent models, most have failed in humans. It is generally accepted that the autoimmunologic features of T1D are the primary causes of clinical failures. Specifically, neogenetic β cells are always recognized and attacked by diabetogenic T cells, which results in death of these new β cells. Fortunately, the approaches and technologies for protection of new β cells in islet transplantation have improved. For example, encapsulating technologies protect grafted islets from the host immune system (193, 194). Another strategy is to improve immunologic tolerance of new β cells through replenishment of regulatory T cells (195–198). Clinical trial results showed that prolonged immunosuppression in chronic T1D patients slightly increased native pancreatic insulin production, which demonstrated the effect of the immune system on endogenous pancreatic β cell regeneration (199). Moreover, technologies for generation of insulin producing cells derived from iPS cells has allowed for autologous β cell transplantation for T1D treatment (200, 201). However, because T1D is an autoimmune disease, new β cells are attacked by immune cells. Thus, further studies should focus on increasing the autoimmunologic tolerance of new β cells. First, we suggest focusing on the functionality and immunogenicity of new pancreatic β cells to improve adaptability in clinical applications. Subsequently, steps should be taken to improve understanding of the characteristics of pancreatic islets, islet cells, and new insulin-expressing cells. Furthermore, studies aimed at determining molecular mechanisms and potential regenerative medicines should use humanized mouse models of diabetes, which will provide significant information for development of new T1D therapies.
Author Contributions
YJ designed and wrote the paper. FZ collected the data.
Funding
This work was also supported by the National Key Research and Development Program of China [grant numbers 2018YFC0910701, 2016YFC0901903]. We declare all sources of funding received for the research being submitted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
National Natural Science Foundation of China [grant number 91659301].
References
1. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes (2003) 52:102–10. doi: 10.2337/diabetes.52.1.102
2. Matveyenko AV, Butler PC. Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab. (2008) 10(Suppl. 4):23–31. doi: 10.1111/j.1463-1326.2008.00939.x
3. Zhou Q, Melton DA. Pancreas regeneration. Nature (2018) 557:351–8. doi: 10.1038/s41586-018-0088-0
4. Aguayo-Mazzucato C, Bonner-Weir S. Pancreatic beta cell regeneration as a possible therapy for diabetes. Cell Metab. (2018) 27:57–67. doi: 10.1016/j.cmet.2017.08.007
5. Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes (1995) 44:249–56. doi: 10.2337/diab.44.3.249
6. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature (2004) 429:41–6. doi: 10.1038/nature02520
7. Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes (2008) 57:1584–94. doi: 10.2337/db07-1369
8. Montanya E, Nacher V, Biarnes M, Soler J. Linear correlation between beta-cell mass and body weight throughout the lifespan in Lewis rats: role of beta-cell hyperplasia and hypertrophy. Diabetes (2000) 49:1341–6. doi: 10.2337/diabetes.49.8.1341
9. Bock T, Pakkenberg B, Buschard K. Increased islet volume but unchanged islet number in ob/ob mice. Diabetes (2003) 52:1716–22. doi: 10.2337/diabetes.52.7.1716
10. Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes (2005) 54:2557–67. doi: 10.2337/diabetes.54.9.2557
11. Gargani S, Thevenet J, Yuan JE, Lefebvre B, Delalleau N, Gmyr V, et al. Adaptive changes of human islets to an obesogenic environment in the mouse. Diabetologia (2013) 56:350–8. doi: 10.1007/s00125-012-2775-y
12. Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia (2010) 53:2167–76. doi: 10.1007/s00125-010-1809-6
13. Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab. (2010) 21:151–8. doi: 10.1016/j.tem.2009.11.001
14. Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. (2010) 16:804–8. doi: 10.1038/nm.2173
15. Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest. (2007) 117:2553–61. doi: 10.1172/JCI32959
16. Dai C, Brissova M, Hang Y, Thompson C, Poffenberger G, Shostak A, et al. Islet-enriched gene expression and glucose-induced insulin secretion in human and mouse islets. Diabetologia (2012) 55:707–18. doi: 10.1007/s00125-011-2369-0
17. Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. (2004) 114:963–8. doi: 10.1172/JCI22098
18. Xu Y, Wang X, Gao L, Zhu J, Zhang H, Shi H, et al. Prolactin-stimulated survivin induction is required for beta cell mass expansion during pregnancy in mice. Diabetologia (2015) 58:2064–73. doi: 10.1007/s00125-015-3670-0
19. Baeyens L, Hindi S, Sorenson RL, German MS. beta-Cell adaptation in pregnancy. Diabetes Obes Metab. (2016) 18(Suppl. 1):63–70. doi: 10.1111/dom.12716
20. Linnemann AK, Baan M, Davis DB. Pancreatic beta-cell proliferation in obesity. Adv Nutr. (2014) 5:278–88. doi: 10.3945/an.113.005488
21. Menge BA, Tannapfel A, Belyaev O, Drescher R, Muller C, Uhl W, et al. Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes (2008) 57:142–9. doi: 10.2337/db07-1294
22. Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. beta-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care (2013) 36:111–7. doi: 10.2337/dc12-0421
23. Chen H, Kleinberger JW, Takane KK, Salim F, Fiaschi-Taesch N, Pappas K, et al. Augmented Stat5 signaling bypasses multiple impediments to lactogen-mediated proliferation in human beta-Cells. Diabetes (2015) 64:3784–97. doi: 10.2337/db15-0083
24. Fusco J, Xiao X, Prasadan K, Sheng Q, Chen C, Ming YC, et al. GLP-1/Exendin-4 induces beta-cell proliferation via the epidermal growth factor receptor. Sci Rep. (2017) 7:9100. doi: 10.1038/s41598-017-09898-4
25. Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes (2003) 52:124–32. doi: 10.2337/diabetes.52.1.124
26. Tella SH, Rendell MS. Glucagon-like polypeptide agonists in type 2 diabetes mellitus: efficacy and tolerability, a balance. Ther Adv Endocrinol Metab. (2015) 6:109–34. doi: 10.1177/2042018815580257
27. Stamateris RE, Sharma RB, Kong Y, Ebrahimpour P, Panday D, Ranganath P, et al. Glucose induces mouse beta-cell proliferation via IRS2, MTOR, and Cyclin D2 but Not the Insulin Receptor. Diabetes (2016) 65:981–95. doi: 10.2337/db15-0529
28. Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF. Human beta-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes (2012) 61:2205–13. doi: 10.2337/db12-0018
29. Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocana A. Human beta-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes (2014) 63:819–31. doi: 10.2337/db13-1146
30. Stewart AF, Hussain MA, Garcia-Ocana A, Vasavada RC, Bhushan A, Bernal-Mizrachi E, et al. Human beta-cell proliferation and intracellular signaling: part 3. Diabetes (2015) 64:1872–85. doi: 10.2337/db14-1843
31. Rieck S, Zhang J, Li Z, Liu C, Naji A, Takane KK, et al. Overexpression of hepatocyte nuclear factor-4alpha initiates cell cycle entry, but is not sufficient to promote beta-cell expansion in human islets. Mol Endocrinol. (2012) 26:1590–602. doi: 10.1210/me.2012-1019
32. Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocana A, Vasavada R, et al. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev. (2006) 27:356–70. doi: 10.1210/er.2006-0004
33. Huang C, Snider F, Cross JC. Prolactin receptor is required for normal glucose homeostasis and modulation of beta-cell mass during pregnancy. Endocrinology (2009) 150:1618–26. doi: 10.1210/en.2008-1003
34. Song WJ, Schreiber WE, Zhong E, Liu FF, Kornfeld BD, Wondisford FE, et al. Exendin-4 stimulation of cyclin A2 in beta-cell proliferation. Diabetes (2008) 57:2371–81. doi: 10.2337/db07-1541
35. Fujitani Y. How does glucagon-like peptide 1 stimulate human beta-cell proliferation? A lesson from islet graft experiments. J Diabetes Investig. (2018) 9:1255–7. doi: 10.1111/jdi.12861
36. Dai C, Hang Y, Shostak A, Poffenberger G, Hart N, Prasad N, et al. Age-dependent human beta cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J Clin Invest. (2017) 127:3835–44. doi: 10.1172/JCI91761
37. Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature (2006) 443:345–9. doi: 10.1038/nature05097
38. Chamberlain CE, Scheel DW, McGlynn K, Kim H, Miyatsuka T, Wang J, et al. Menin determines K-RAS proliferative outputs in endocrine cells. J Clin Invest. (2014) 124:4093–101. doi: 10.1172/JCI69004
39. Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci USA. (2005) 102:14659–64. doi: 10.1073/pnas.0503484102
40. Karnik SK, Chen H, McLean GW, Heit JJ, Gu X, Zhang AY, et al. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science (2007) 318:806–9. doi: 10.1126/science.1146812
41. Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. (2009) 23:975–85. doi: 10.1101/gad.1742509
42. Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, et al. Adipsin is an adipokine that improves beta cell function in diabetes. Cell (2014) 158:41–53. doi: 10.1016/j.cell.2014.06.005
43. Retnakaran R. Adiponectin and beta-cell adaptation in pregnancy. Diabetes (2017) 66:1121–2. doi: 10.2337/dbi17-0001
44. Pham MN, Kolb H, Mandrup-Poulsen T, Battelino T, Ludvigsson J, Pozzilli P, et al. Serum adipokines as biomarkers of beta-cell function in patients with type 1 diabetes: positive association with leptin and resistin and negative association with adiponectin. Diabetes Metab Res Rev. (2013) 29:166–70. doi: 10.1002/dmrr.2378
45. Park S, Hong SM, Sung SR, Jung HK. Long-term effects of central leptin and resistin on body weight, insulin resistance, and beta-cell function and mass by the modulation of hypothalamic leptin and insulin signaling. Endocrinology (2008) 149:445–54. doi: 10.1210/en.2007-0754
46. Dirice E, Kahraman S, Jiang W, El Ouaamari A, De Jesus DF, Teo AK, et al. Soluble factors secreted by T cells promote beta-cell proliferation. Diabetes (2014) 63:188–202. doi: 10.2337/db13-0204
47. Dauriz M, Trombetta M, Boselli L, Santi L, Brangani C, Pichiri I, et al. Interleukin-6 as a potential positive modulator of human beta-cell function: an exploratory analysis-the Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 6. Acta Diabetol. (2016) 53:393–402. doi: 10.1007/s00592-015-0807-z
48. El Ouaamari A, Kawamori D, Dirice E, Liew CW, Shadrach JL, Hu J, et al. Liver-derived systemic factors drive beta cell hyperplasia in insulin-resistant states. Cell Rep. (2013) 3:401–10. doi: 10.1016/j.celrep.2013.01.007
49. El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou JY, Shirakawa J, et al. SerpinB1 promotes pancreatic beta cell proliferation. Cell Metab. (2016) 23:194–205. doi: 10.1016/j.cmet.2015.12.001
50. Gaddy DF, Riedel MJ, Pejawar-Gaddy S, Kieffer TJ, Robbins PD. In vivo expression of HGF/NK1 and GLP-1 From dsAAV vectors enhances pancreatic ss-cell proliferation and improves pathology in the db/db mouse model of diabetes. Diabetes (2010) 59:3108–16. doi: 10.2337/db09-1886
51. Agudo J, Ayuso E, Jimenez V, Salavert A, Casellas A, Tafuro S, et al. IGF-I mediates regeneration of endocrine pancreas by increasing beta cell replication through cell cycle protein modulation in mice. Diabetologia (2008) 51:1862–72. doi: 10.1007/s00125-008-1087-8
52. Alvarez-Perez JC, Ernst S, Demirci C, Casinelli GP, Mellado-Gil JM, Rausell-Palamos F, et al. Hepatocyte growth factor/c-Met signaling is required for beta-cell regeneration. Diabetes (2014) 63:216–23. doi: 10.2337/db13-0333
53. Modi H, Cornu M, Thorens B. Glutamine stimulates biosynthesis and secretion of insulin-like growth factor 2 (IGF2), an autocrine regulator of beta cell mass and function. J Biol Chem. (2014) 289:31972–82. doi: 10.1074/jbc.M114.587733
54. Wang W, Walker JR, Wang X, Tremblay MS, Lee JW, Wu X, et al. Identification of small-molecule inducers of pancreatic beta-cell expansion. Proc Natl Acad Sci USA. (2009) 106:1427–32. doi: 10.1073/pnas.0811848106
55. Guthalu Kondegowda N, Joshi-Gokhale S, Harb G, Williams K, Zhang XY, Takane KK, et al. Parathyroid hormone-related protein enhances human ss-cell proliferation and function with associated induction of cyclin-dependent kinase 2 and cyclin E expression. Diabetes (2010) 59:3131–8. doi: 10.2337/db09-1796
56. Hakonen E, Ustinov J, Mathijs I, Palgi J, Bouwens L, Miettinen PJ, et al. Epidermal growth factor (EGF)-receptor signalling is needed for murine beta cell mass expansion in response to high-fat diet and pregnancy but not after pancreatic duct ligation. Diabetologia (2011) 54:1735–43. doi: 10.1007/s00125-011-2153-1
57. Cano DA, Rulifson IC, Heiser PW, Swigart LB, Pelengaris S, German M, et al. Regulated beta-cell regeneration in the adult mouse pancreas. Diabetes (2008) 57:958–66. doi: 10.2337/db07-0913
58. Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes (2009) 58:1365–72. doi: 10.2337/db08-1198
59. Rankin MM, Wilbur CJ, Rak K, Shields EJ, Granger A, Kushner JA. beta-Cells are not generated in pancreatic duct ligation-induced injury in adult mice. Diabetes (2013) 62:1634–45. doi: 10.2337/db12-0848
60. Walpita D, Hasaka T, Spoonamore J, Vetere A, Takane KK, Fomina-Yadlin D, et al. A human islet cell culture system for high-throughput screening. J Biomol Screen (2012) 17:509–18. doi: 10.1177/1087057111430253
61. Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med. (2015) 21:383–8. doi: 10.1038/nm.3820
62. Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, et al. Inhibition of DYRK1A stimulates human beta-cell proliferation. Diabetes (2016) 65:1660–71. doi: 10.2337/db15-1127
63. Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocana A, Penninger JM, et al. Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-kappaB ligand pathway. Cell Metab. (2015) 22:77–85. doi: 10.1016/j.cmet.2015.05.021
64. Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang YQ, et al. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nat Commun. (2015) 6:8372. doi: 10.1038/ncomms9372
65. Robitaille K, Rourke JL, McBane JE, Fu A, Baird S, Du Q, et al. High-throughput functional genomics identifies regulators of primary human beta cell proliferation. J Biol Chem. (2016) 291:4614–25. doi: 10.1074/jbc.M115.683912
66. Shirakawa J, Kulkarni RN. Novel factors modulating human beta-cell proliferation. Diabetes Obes Metab. (2016) 18(Suppl. 1):71–7. doi: 10.1111/dom.12731
67. Hang Y, Stein R. MafA and MafB activity in pancreatic β cells. Trends Endocrinol Metab. (2011) 22:364–73. doi: 10.1016/j.tem.2011.05.003
68. Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. (2002) 32:128–34. doi: 10.1038/ng959
69. Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci USA. (2007) 104:1865–70. doi: 10.1073/pnas.0609217104
70. Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature (1997) 386:399–402. doi: 10.1038/386399a0
71. Grapin-Botton A, Majithia AR, Melton DA. Key events of pancreas formation are triggered in gut endoderm by ectopic expression of pancreatic regulatory genes. Genes Dev. (2001) 15:444–54. doi: 10.1101/gad.846001
72. Romer AI, Sussel L. Pancreatic islet cell development and regeneration. Curr Opin Endocrinol Diabetes Obes. (2015) 22:255–64. doi: 10.1097/MED.0000000000000174
73. Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development (2002) 129:2447–57.
74. Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, et al. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell (2009) 138:449–62. doi: 10.1016/j.cell.2009.05.035
75. Afelik S, Chen Y, Pieler T. Combined ectopic expression of Pdx1 and Ptf1a/p48 results in the stable conversion of posterior endoderm into endocrine and exocrine pancreatic tissue. Genes Dev. (2006) 20:1441–6. doi: 10.1101/gad.378706
76. Schaffer AE, Freude KK, Nelson SB, Sander M. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell (2010) 18:1022–9. doi: 10.1016/j.devcel.2010.05.015
77. Rodriguez-Segui S, Akerman I, Ferrer J. GATA believe it: new essential regulators of pancreas development. J Clin Invest. (2012) 122:3469–71. doi: 10.1172/JCI65751
78. Xuan S, Borok MJ, Decker KJ, Battle MA, Duncan SA, Hale MA, et al. Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J Clin Invest. (2012) 122:3516–28. doi: 10.1172/JCI63352
79. Seymour PA, Freude KK, Dubois CL, Shih HP, Patel NA, Sander M. A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev Biol. (2008) 323:19–30. doi: 10.1016/j.ydbio.2008.07.034
80. Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA. (2000) 97:1607–11. doi: 10.1073/pnas.97.4.1607
81. Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G, et al. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell (2007) 12:457–65. doi: 10.1016/j.devcel.2007.02.010
82. Oropeza D, Horb M. Transient expression of Ngn3 in Xenopus endoderm promotes early and ectopic development of pancreatic beta and delta cells. Genesis (2012) 50:271–85. doi: 10.1002/dvg.20828
83. Jeon J, Correa-Medina M, Ricordi C, Edlund H, Diez JA. Endocrine cell clustering during human pancreas development. J Histochem Cytochem. (2009) 57:811–24. doi: 10.1369/jhc.2009.953307
84. Napolitano T, Avolio F, Courtney M, Vieira A, Druelle N, Ben-Othman N, et al. Pax4 acts as a key player in pancreas development and plasticity. Semin Cell Dev Biol. (2015) 44:107–14. doi: 10.1016/j.semcdb.2015.08.013
85. Bonner-Weir S, Toschi E, Inada A, Reitz P, Fonseca SY, Aye T, et al. The pancreatic ductal epithelium serves as a potential pool of progenitor cells. Pediatr Diabetes (2004) 5(Suppl. 2):16–22. doi: 10.1111/j.1399-543X.2004.00075.x
86. Li WC, Rukstalis JM, Nishimura W, Tchipashvili V, Habener JF, Sharma A, et al. Activation of pancreatic-duct-derived progenitor cells during pancreas regeneration in adult rats. J Cell Sci. (2010) 123:2792–802. doi: 10.1242/jcs.065268
87. Zhang M, Lin Q, Qi T, Wang T, Chen CC, Riggs AD, et al. Growth factors and medium hyperglycemia induce Sox9+ ductal cell differentiation into beta cells in mice with reversal of diabetes. Proc Natl Acad Sci USA. (2016) 113:650–5. doi: 10.1073/pnas.1524200113
88. Inada A, Nienaber C, Katsuta H, Fujitani Y, Levine J, Morita R, et al. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci USA. (2008) 105:19915–9. doi: 10.1073/pnas.0805803105
89. Inada A, Nienaber C, Fonseca S, Bonner-Weir S. Timing and expression pattern of carbonic anhydrase II in pancreas. Dev Dyn. (2006) 235:1571–7. doi: 10.1002/dvdy.20754
90. Bonner-Weir S, Inada A, Yatoh S, Li WC, Aye T, Toschi E, et al. Transdifferentiation of pancreatic ductal cells to endocrine beta-cells. Biochem Soc Trans. (2008) 36:353–6. doi: 10.1042/BST0360353
91. Liu KC, Leuckx G, Sakano D, Seymour PA, Mattsson CL, Rautio L, et al. Inhibition of Cdk5 promotes beta-cell differentiation from ductal progenitors. Diabetes (2018) 67:58–70. doi: 10.2337/db16-1587
92. Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell (2008) 132:197–207. doi: 10.1016/j.cell.2007.12.015
93. Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, et al. Notch signalling controls pancreatic cell differentiation. Nature (1999) 400:877–81. doi: 10.1038/23716
94. Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature (2010) 464:1149–54. doi: 10.1038/nature08894
95. Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature (2014) 514:503–7. doi: 10.1038/nature13633
96. Baeyens L, Lemper M, Leuckx G, De Groef S, Bonfanti P, Stange G, et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol. (2014) 32:76–83. doi: 10.1038/nbt.2747
97. Conrad E, Stein R, Hunter CS. Revealing transcription factors during human pancreatic beta cell development. Trends Endocrinol Metab. (2014) 25:407–14. doi: 10.1016/j.tem.2014.03.013
98. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature (2008) 455:627–32. doi: 10.1038/nature07314
99. Matsuoka TA, Kawashima S, Miyatsuka T, Sasaki S, Shimo N, Katakami N, et al. Mafa enables Pdx1 to effectively convert pancreatic islet progenitors and committed islet alpha-cells into beta-cells in vivo. Diabetes (2017) 66:1293–300. doi: 10.2337/db16-0887
100. Zhang Y, Fava GE, Wang H, Mauvais-Jarvis F, Fonseca VA, Wu H. PAX4 gene transfer induces alpha-to-beta cell phenotypic conversion and confers therapeutic benefits for diabetes treatment. Mol Ther. (2016) 24:251–60. doi: 10.1038/mt.2015.181
101. Yang YP, Thorel F, Boyer DF, Herrera PL, Wright CV. Context-specific alpha- to-beta-cell reprogramming by forced Pdx1 expression. Genes Dev. (2011) 25:1680–5. doi: 10.1101/gad.16875711
102. Chakravarthy H, Gu X, Enge M, Dai X, Wang Y, Damond N, et al. Converting adult pancreatic islet alpha cells into beta cells by targeting both Dnmt1 and Arx. Cell Metab. (2017) 25:622–34. doi: 10.1016/j.cmet.2017.01.009
103. Druelle N, Vieira A, Shabro A, Courtney M, Mondin M, Rekima S, et al. Ectopic expression of Pax4 in pancreatic delta cells results in beta-like cell neogenesis. J Cell Biol. (2017) 216:4299–311. doi: 10.1083/jcb.201704044
104. Al-Hasani K, Pfeifer A, Courtney M, Ben-Othman N, Gjernes E, Vieira A, et al. Adult duct-lining cells can reprogram into beta-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell (2013) 26:86–100. doi: 10.1016/j.devcel.2013.05.018
105. Represa A, Ben-Ari Y. Trophic actions of GABA on neuronal development. Trends Neurosci. (2005) 28:278–83. doi: 10.1016/j.tins.2005.03.010
106. Adeghate E, Ponery AS. GABA in the endocrine pancreas: cellular localization and function in normal and diabetic rats. Tissue Cell (2002) 34:1–6. doi: 10.1054/tice.2002.0217
107. Sorenson RL, Garry DG, Brelje TC. Structural and functional considerations of GABA in islets of Langerhans. Beta-cells and nerves. Diabetes (1991) 40:1365–74. doi: 10.2337/diab.40.11.1365
108. Yoon JW, Yoon CS, Lim HW, Huang QQ, Kang Y, Pyun KH, et al. Control of autoimmune diabetes in NOD mice by GAD expression or suppression in beta cells. Science (1999) 284:1183–7. doi: 10.1126/science.284.5417.1183
109. Soltani N, Qiu H, Aleksic M, Glinka Y, Zhao F, Liu R, et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc Natl Acad Sci USA. (2011) 108:11692–7. doi: 10.1073/pnas.1102715108
110. Purwana I, Zheng J, Li X, Deurloo M, Son DO, Zhang Z, et al. GABA promotes human beta-cell proliferation and modulates glucose homeostasis. Diabetes (2014) 63:4197–205. doi: 10.2337/db14-0153
111. Weir GC, Bonner-Weir S. GABA signaling stimulates beta cell regeneration in diabetic mice. Cell (2017) 168:7–9. doi: 10.1016/j.cell.2016.12.006
112. Tian J, Dang H, Chen Z, Guan A, Jin Y, Atkinson MA, et al. gamma-Aminobutyric acid regulates both the survival and replication of human beta-cells. Diabetes (2013) 62:3760-5. doi: 10.2337/db13-0931
113. Braun M, Ramracheya R, Bengtsson M, Clark A, Walker JN, Johnson PR, et al. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes (2010) 59:1694–701. doi: 10.2337/db09-0797
114. Wendt A, Birnir B, Buschard K, Gromada J, Salehi A, Sewing S, et al. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes (2004) 53:1038–45. doi: 10.2337/diabetes.53.4.1038
115. Rorsman P, Berggren PO, Bokvist K, Ericson H, Mohler H, Ostenson CG, et al. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature (1989) 341:233–6. doi: 10.1038/341233a0
116. Braun M, Wendt A, Buschard K, Salehi A, Sewing S, Gromada J, et al. GABAB receptor activation inhibits exocytosis in rat pancreatic beta-cells by G-protein-dependent activation of calcineurin. J Physiol. (2004) 559:397–409. doi: 10.1113/jphysiol.2004.066563
117. Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, et al. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab. (2006) 3:47–58. doi: 10.1016/j.cmet.2005.11.015
118. Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, et al. Long-term GABA administration induces alpha cell-mediated beta-like cell neogenesis. Cell (2017) 168:73–85 e11. doi: 10.1016/j.cell.2016.11.002
119. Bhandage AK, Jin Z, Korol SV, Shen Q, Pei Y, Deng Q, et al. GABA regulates release of inflammatory cytokines from peripheral blood mononuclear cells and CD4(+) T cells and is immunosuppressive in type 1 diabetes. EBioMedicine (2018) 30:283–94. doi: 10.1016/j.ebiom.2018.03.019
120. Jin Z, Mendu SK, Birnir B. GABA is an effective immunomodulatory molecule. Amino Acids (2013) 45:87–94. doi: 10.1007/s00726-011-1193-7
121. Li J, Casteels T, Frogne T, Ingvorsen C, Honore C, Courtney M, et al. Artemisinins target GABAA receptor signaling and impair alpha cell identity. Cell (2017) 168:86–100 e15. doi: 10.1016/j.cell.2016.11.010
122. van der Meulen T, Lee S, Noordeloos E, Donaldson CJ, Adams MW, Noguchi GM, et al. Artemether does not turn alpha cells into beta cells. Cell Metab. (2018) 27:218–25 e4. doi: 10.1016/j.cmet.2017.10.002
123. Cheng CW, Villani V, Buono R, Wei M, Kumar S, Yilmaz OH, et al. Fasting-Mimicking diet promotes Ngn3-driven beta-cell regeneration to reverse diabetes. Cell (2017) 168:775–88 e12. doi: 10.1016/j.cell.2017.01.040
124. Wang YJ, Golson ML, Schug J, Traum D, Liu C, Vivek K, et al. Single-cell mass cytometry analysis of the human endocrine pancreas. Cell Metab. (2016) 24:616–26. doi: 10.1016/j.cmet.2016.09.007
125. Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andersson AC, Sun X, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. (2016) 24:593–607. doi: 10.1016/j.cmet.2016.08.020
126. Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature (2014) 510:363–9. doi: 10.1038/nature13437
127. Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes (2008) 57:3069–77. doi: 10.2337/db08-0878
128. Tellez N, Vilaseca M, Marti Y, Pla A, Montanya E. beta-cell dedifferentiation, reduced duct cell plasticity, and impaired beta-cell mass regeneration in middle-aged rats. Am J Physiol Endocrinol Metab. (2016) 311:E554–63. doi: 10.1152/ajpendo.00502.2015
129. Rooman I, Lardon J, Bouwens L. Gastrin stimulates beta-cell neogenesis and increases islet mass from transdifferentiated but not from normal exocrine pancreas tissue. Diabetes (2002) 51:686–90. doi: 10.2337/diabetes.51.3.686
130. Lardon J, Huyens N, Rooman I, Bouwens L. Exocrine cell transdifferentiation in dexamethasone-treated rat pancreas. Virchows Arch. (2004) 444:61–5. doi: 10.1007/s00428-003-0930-z
131. Wang RN, Kloppel G, Bouwens L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia (1995) 38:1405–11. doi: 10.1007/BF00400600
132. Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, et al. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development (2011) 138:653–65. doi: 10.1242/dev.056499
133. Van de Casteele M, Leuckx G, Cai Y, Yuchi Y, Coppens V, De Groef S, et al. Partial duct ligation: beta-cell proliferation and beyond. Diabetes (2014) 63:2567–77. doi: 10.2337/db13-0831
134. Kataoka M, Kawamuro Y, Shiraki N, Miki R, Sakano D, Yoshida T, et al. Recovery from diabetes in neonatal mice after a low-dose streptozotocin treatment. Biochem Biophys Res Commun. (2013) 430:1103–8. doi: 10.1016/j.bbrc.2012.12.030
135. Movassat J, Portha B. Beta-cell growth in the neonatal Goto-Kakisaki rat and regeneration after treatment with streptozotocin at birth. Diabetologia (1999) 42:1098–106. doi: 10.1007/s001250051277
136. Thyssen S, Arany E, Hill DJ. Ontogeny of regeneration of beta-cells in the neonatal rat after treatment with streptozotocin. Endocrinology (2006) 147:2346–56. doi: 10.1210/en.2005-0396
137. Saisho Y, Manesso E, Butler AE, Galasso R, Kavanagh K, Flynn M, et al. Ongoing beta-cell turnover in adult nonhuman primates is not adaptively increased in streptozotocin-induced diabetes. Diabetes (2011) 60:848–56. doi: 10.2337/db09-1368
138. Kaya-Dagistanli F, Ozturk M. The role of clusterin on pancreatic beta cell regeneration after exendin-4 treatment in neonatal streptozotocin administrated rats. Acta Histochem. (2013) 115:577–86. doi: 10.1016/j.acthis.2012.12.007
139. Golson ML, Maulis MF, Dunn JC, Poffenberger G, Schug J, Kaestner KH, et al. Activated FoxM1 attenuates streptozotocin-mediated beta-cell death. Mol Endocrinol. (2014) 28:1435–47. doi: 10.1210/me.2014-1024
140. Shu L, Zien K, Gutjahr G, Oberholzer J, Pattou F, Kerr-Conte J, et al. TCF7L2 promotes beta cell regeneration in human and mouse pancreas. Diabetologia (2012) 55:3296–307. doi: 10.1007/s00125-012-2693-z
141. Chen S, Bastarrachea RA, Roberts BJ, Voruganti VS, Frost PA, Nava-Gonzalez EJ, et al. Successful beta cells islet regeneration in streptozotocin-induced diabetic baboons using ultrasound-targeted microbubble gene therapy with cyclinD2/CDK4/GLP1. Cell Cycle (2014) 13:1145–51. doi: 10.4161/cc.27997
142. Grossman EJ, Lee DD, Tao J, Wilson RA, Park SY, Bell GI, et al. Glycemic control promotes pancreatic beta-cell regeneration in streptozotocin-induced diabetic mice. PLoS ONE (2010) 5:e8749. doi: 10.1371/journal.pone.0008749
143. Yamaizumi M, Mekada E, Uchida T, Okada Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell (1978) 15:245–50. doi: 10.1016/0092-8674(78)90099-5
144. Das AT, Tenenbaum L, Berkhout B. Tet-on systems for doxycycline-inducible gene expression. Curr Gene Ther. (2016) 16:156–67. doi: 10.2174/1566523216666160524144041
145. Pappenheimer AM Jr, Harper AA, Moynihan M, Brockes JP. Diphtheria toxin and related proteins: effect of route of injection on toxicity and the determination of cytotoxicity for various cultured cells. J Infect Dis. (1982) 145:94–102. doi: 10.1093/infdis/145.1.94
146. Thorel F, Damond N, Chera S, Wiederkehr A, Thorens B, Meda P, et al. Normal glucagon signaling and beta-cell function after near-total alpha-cell ablation in adult mice. Diabetes (2011) 60:2872–82. doi: 10.2337/db11-0876
147. Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. (2003) 111:1497–504. doi: 10.1172/JCI17664
148. Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, et al. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. (2005) 11:797–803. doi: 10.1038/nm1262
149. Mallet VO, Mitchell C, Guidotti JE, Jaffray P, Fabre M, Spencer D, et al. Conditional cell ablation by tight control of caspase-3 dimerization in transgenic mice. Nat Biotechnol. (2002) 20:1234–9. doi: 10.1038/nbt762
150. Wang ZV, Mu J, Schraw TD, Gautron L, Elmquist JK, Zhang BB, et al. PANIC-ATTAC: a mouse model for inducible and reversible beta-cell ablation. Diabetes (2008) 57:2137–48. doi: 10.2337/db07-1631
151. Ye R, Wang M, Wang QA, Spurgin SB, Wang ZV, Sun K, et al. Autonomous interconversion between adult pancreatic alpha-cells and beta-cells after differential metabolic challenges. Mol Metab. (2016) 5:437–48. doi: 10.1016/j.molmet.2016.05.001
152. Herrera PL, Nepote V, Delacour A. Pancreatic cell lineage analyses in mice. Endocrine (2002) 19:267–78. doi: 10.1385/ENDO:19:3:267
153. Kawaguchi Y, Takaori K, Uemoto S. Genetic lineage tracing, a powerful tool to investigate the embryonic organogenesis and adult organ maintenance of the pancreas. J Hepatobiliary Pancreat Sci. (2011) 18:1–5. doi: 10.1007/s00534-010-0307-z
154. Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. (1995) 23:1686–90. doi: 10.1093/nar/23.10.1686
155. Picard D. Regulation of protein function through expression of chimaeric proteins. Curr Opin Biotechnol. (1994) 5:511–5. doi: 10.1016/0958-1669(94)90066-3
156. Kretzschmar K, Watt FM. Lineage tracing. Cell (2012) 148:33–45. doi: 10.1016/j.cell.2012.01.002
157. Brehm MA, Powers AC, Shultz LD, Greiner DL. Advancing animal models of human type 1 diabetes by engraftment of functional human tissues in immunodeficient mice. Cold Spring Harb Perspect Med. (2012) 2:a007757. doi: 10.1101/cshperspect.a007757
158. Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, et al. Unique arrangement of alpha- and beta-cells in human islets of Langerhans. Diabetes (2010) 59:1202–10. doi: 10.2337/db09-1177
159. Abiru N, Maniatis AK, Yu L, Miao D, Moriyama H, Wegmann D, et al. Peptide and major histocompatibility complex-specific breaking of humoral tolerance to native insulin with the B9-23 peptide in diabetes-prone and normal mice. Diabetes (2001) 50:1274–81. doi: 10.2337/diabetes.50.6.1274
160. Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med. (1999) 5:601–4. doi: 10.1038/9442
161. Greiner DL, Hesselton RA, Shultz LD. SCID mouse models of human stem cell engraftment. Stem Cells (1998) 16:166–77. doi: 10.1002/stem.160166
162. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. (2007) 7:118–30. doi: 10.1038/nri2017
163. Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature (1983) 301:527–30. doi: 10.1038/301527a0
164. Brehm MA, Cuthbert A, Yang C, Miller DM, DiIorio P, Laning J, et al. Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rgamma(null) mutation. Clin Immunol. (2010) 135:84–98. doi: 10.1016/j.clim.2009.12.008
165. Viehmann Milam AA, Maher SE, Gibson JA, Lebastchi J, Wen L, Ruddle NH, et al. A humanized mouse model of autoimmune insulitis. Diabetes (2014) 63:1712–24. doi: 10.2337/db13-1141
166. Presa M, Chen YG, Grier AE, Leiter EH, Brehm MA, Greiner DL, et al. The presence and preferential activation of regulatory T cells diminish adoptive transfer of autoimmune diabetes by polyclonal Nonobese Diabetic (NOD) T cell effectors into NSG versus NOD-scid mice. J Immunol. (2015) 195:3011–9. doi: 10.4049/jimmunol.1402446
167. Abdulreda MH, Rodriguez-Diaz R, Caicedo A, Berggren PO. Liraglutide compromises pancreatic beta cell function in a humanized mouse model. Cell Metab. (2016) 23:541–6. doi: 10.1016/j.cmet.2016.01.009
168. Tan S, Li Y, Xia J, Jin CH, Hu Z, Duinkerken G, et al. Type 1 diabetes induction in humanized mice. Proc Natl Acad Sci USA. (2017) 114:10954–9. doi: 10.1073/pnas.1710415114
169. Brehm MA, Bortell R, Diiorio P, Leif J, Laning J, Cuthbert A, et al. Human immune system development and rejection of human islet allografts in spontaneously diabetic NOD-Rag1null IL2rgammanull Ins2Akita mice. Diabetes (2010) 59:2265–70. doi: 10.2337/db10-0323
170. Wu H, Wen D, Mahato RI. Third-party mesenchymal stem cells improved human islet transplantation in a humanized diabetic mouse model. Mol Ther. (2013) 21:1778–86. doi: 10.1038/mt.2013.147
171. Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, et al. Weaning triggers a maturation step of pancreatic beta cells. Dev Cell (2015) 32:535–45. doi: 10.1016/j.devcel.2015.01.002
172. Aguayo-Mazzucato C, Koh A, El Khattabi I, Li WC, Toschi E, Jermendy A, et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia (2011) 54:583–93. doi: 10.1007/s00125-010-2026-z
173. Nishimura W, Takahashi S, Yasuda K. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia (2015) 58:566–74. doi: 10.1007/s00125-014-3464-9
174. Yoshihara E, Wei Z, Lin CS, Fang S, Ahmadian M, Kida Y, et al. ERRgamma is required for the metabolic maturation of therapeutically functional glucose-responsive beta cells. Cell Metab. (2016) 23:622–34. doi: 10.1016/j.cmet.2016.03.005
175. Gu C, Stein GH, Pan N, Goebbels S, Hornberg H, Nave KA, et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. (2010) 11:298–310. doi: 10.1016/j.cmet.2010.03.006
176. Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. (2012) 30:261–4. doi: 10.1038/nbt.2141
177. van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dolleman S, et al. Virgin beta cells persist throughout life at a neogenic niche within pancreatic islets. Cell Metab. (2017) 25:911–26 e6. doi: 10.1016/j.cmet.2017.03.017
178. Roscioni SS, Migliorini A, Gegg M, Lickert H. Impact of islet architecture on beta-cell heterogeneity, plasticity and function. Nat Rev Endocrinol. (2016) 12:695–709. doi: 10.1038/nrendo.2016.147
179. Szabat M, Luciani DS, Piret JM, Johnson JD. Maturation of adult beta-cells revealed using a Pdx1/insulin dual-reporter lentivirus. Endocrinology (2009) 150:1627–35. doi: 10.1210/en.2008-1224
180. Beamish CA, Strutt BJ, Arany EJ, Hill DJ. Insulin-positive, Glut2-low cells present within mouse pancreas exhibit lineage plasticity and are enriched within extra-islet endocrine cell clusters. Islets (2016) 8:65–82. doi: 10.1080/19382014.2016.1162367
181. Bosco D, Rouiller DG, Halban PA. Differential expression of E-cadherin at the surface of rat beta-cells as a marker of functional heterogeneity. J Endocrinol. (2007) 194:21–9. doi: 10.1677/JOE-06-0169
182. Bader E, Migliorini A, Gegg M, Moruzzi N, Gerdes J, Roscioni SS, et al. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature (2016) 535:430–4. doi: 10.1038/nature18624
183. Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, et al. Human islets contain four distinct subtypes of beta cells. Nat Commun. (2016) 7:11756. doi: 10.1038/ncomms11756
184. Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, et al. Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab. (2016) 24:389–401. doi: 10.1016/j.cmet.2016.06.020
185. Katsuta H, Akashi T, Katsuta R, Nagaya M, Kim D, Arinobu Y, et al. Single pancreatic beta cells co-express multiple islet hormone genes in mice. Diabetologia (2010) 53:128–38. doi: 10.1007/s00125-009-1570-x
186. Geron E, Boura-Halfon S, Schejter ED, Shilo BZ. The edges of pancreatic islet beta cells constitute adhesive and signaling microdomains. Cell Rep. (2015). 10:317–25. doi: 10.1016/j.celrep.2014.12.031
187. Liu JS, Hebrok M. All mixed up: defining roles for beta-cell subtypes in mature islets. Genes Dev. (2017) 31:228–40. doi: 10.1101/gad.294389.116
188. Gegg M, Bottcher A, Burtscher I, Hasenoeder S, Van Campenhout C, Aichler M, et al. Flattop regulates basal body docking and positioning in mono- and multiciliated cells. Elife (2014) 3:e0384. doi: 10.7554/eLife.03842
189. Migliorini A, Roscioni SS, Lickert H. Targeting insulin-producing beta cells for regenerative therapy. Diabetologia (2016) 59:1838–42. doi: 10.1007/s00125-016-3949-9
190. Pipeleers D, In't Veld PI, Maes E, Van De Winkel M. Glucose-induced insulin release depends on functional cooperation between islet cells. Proc Natl Acad Sci USA (1982) 79:7322–5.
191. Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology (1992) 130:1459–66.
192. Bonner-Weir S, Li WC, Ouziel-Yahalom L, Guo L, Weir GC, Sharma A. Beta-cell growth and regeneration: replication is only part of the story. Diabetes (2010) 59:2340–8. doi: 10.2337/db10-0084
193. Vegas AJ, Veiseh O, Gurtler M, Millman JR, Pagliuca FW, Bader AR, et al. Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice. Nat Med. (2016) 22:306–11. doi: 10.1038/nm.4030
194. Orlando G, Gianello P, Salvatori M, Stratta RJ, Soker S, Ricordi C, et al. Cell replacement strategies aimed at reconstitution of the beta-cell compartment in type 1 diabetes. Diabetes (2014) 63:1433–44. doi: 10.2337/db13-1742
195. Bluestone JA, Tang Q. Treg cells-the next frontier of cell therapy. Science (2018) 362:154–5. doi: 10.1126/science.aau2688
196. Esensten JH, Muller YD, Bluestone JA, Tang Q. Regulatory T-cell therapy for autoimmune and autoinflammatory diseases: the next frontier. J Allergy Clin Immunol. (2018) 142:1710–18. doi: 10.1016/j.jaci.2018.10.015
197. Szot GL, Yadav M, Lang J, Kroon E, Kerr J, Kadoya K, et al. Tolerance induction and reversal of diabetes in mice transplanted with human embryonic stem cell-derived pancreatic endoderm. Cell Stem Cell (2015) 16:148–57. doi: 10.1016/j.stem.2014.12.001
198. Trotta E, Bessette PH, Silveria SL, Ely LK, Jude KM, Le DT, et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat Med. (2018) 24:1005–14. doi: 10.1038/s41591-018-0070-2
199. Liu EH, Digon BJ III, Hirshberg B, Chang R, Wood BJ, Neeman Z, et al. Pancreatic beta cell function persists in many patients with chronic type 1 diabetes, but is not dramatically improved by prolonged immunosuppression and euglycaemia from a beta cell allograft. Diabetologia (2009) 52:1369–80. doi: 10.1007/s00125-009-1342-7
200. Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic beta cells in vitro. Cell (2014) 159:428–39. doi: 10.1016/j.cell.2014.09.040
Keywords: pancreatic β cells, endogenous regeneration, pharmaceutical stimuli, rodent model, diabetes
Citation: Zhong F and Jiang Y (2019) Endogenous Pancreatic β Cell Regeneration: A Potential Strategy for the Recovery of β Cell Deficiency in Diabetes. Front. Endocrinol. 10:101. doi: 10.3389/fendo.2019.00101
Received: 24 October 2018; Accepted: 04 February 2019;
Published: 20 February 2019.
Edited by:
Stanley Steven Schwartz, Main Line Health, United StatesReviewed by:
Jan Brož, Charles University, CzechiaAngela Lombardi, Albert Einstein College of Medicine, United States
Copyright © 2019 Zhong and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Jiang, amlhbmd5YW41MTVAZnVkYW4uZWR1LmNu
†These authors share co-first authorship