 Rosemary McDonald
Rosemary McDonald Carolyn Sadler1
Carolyn Sadler1 T. Rajendra Kumar
T. Rajendra Kumar- 1Division of Reproductive Sciences, Department of Obstetrics and Gynecology, University of Colorado Anschutz Medical Campus, Aurora, IL, United States
- 2Integrated Physiology Graduate Program, University of Colorado Anschutz Medical Campus, Aurora, IL, United States
- 3Division of Reproductive Endocrinology and Infertility, Department of Obstetrics and Gynecology, University of Colorado Anschutz Medical Campus, Aurora, IL, United States
Follicle–stimulating hormone (FSH) is a pituitary-derived gonadotropin that plays key roles in male and female reproduction. The physiology and biochemistry of FSH have been extensively studied for many years. Beginning in the early 1990s, coincident with advances in the then emerging transgenic animal technology, and continuing till today, several gain-of-function (GOF) models have been developed to understand FSH homeostasis in a physiological context. Our group and others have generated a number of FSH ligand and receptor GOF mouse models. An FSH GOF model when combined with Fshb null mice provides a powerful genetic rescue platform. In this chapter, we discuss different GOF models for FSH synthesis, secretion and action and describe additional novel genetic models that could be developed in the future to further refine the existing models.
Introduction
Follicle–Stimulating Hormone (FSH) is a gonadotropin synthesized in gonadotropes of the anterior pituitary gland. FSH is a heterodimeric glycoprotein, consisting of two distinct α- and β (FSHβ) subunits (1–4). The α-subunit is structurally identical to both gonadotropins–luteinizing hormone (LH), and chorionic gonadotropin (CG) as well as thyroid-stimulating hormone (TSH). FSH subunits are encoded by distinct genes. The FSHβ subunit is unique and confers the biological specificity for FSH functions (1–4). The β-subunits exist in comparatively lower amounts within the pituitary than their corresponding α-subunit. FSH subunits are synthesized and assembled non-covalently in gonadotropes (1–4). Non-covalent linkage of the subunits allows for easy separation and hybridization, yet free α and β-subunits are typically expressed by other tissues under a variety of pathological conditions (1–4).
FSH signaling in the hypothalamic-pituitary-gonadal axis (HPG) regulates critical reproductive functions such as steroidogenesis and gametogenesis. In males, FSH contributes to spermatogenesis and testicular development by binding to Sertoli cells and regulating their development and differentiation (5–7). In females, FSH contributes to ovarian follicular development by upregulating aromatase expression in granulosa cells, which results in increased estrogen production. Increased estrogen synthesis is required for normal follicular growth (8).
FSH and LH are synthesized and released in response to gonadotropin-releasing hormone (GnRH) secreted from the hypothalamus. GnRH binds to GnRH- receptors on pituitary gonadotropes (4, 9). Differing GnRH release frequencies favor either FSH or LH synthesis as gonadotropes are sensitive to patterns of GnRH stimulation ad respond by altering hormone-specific subunit gene transcription. LH is secreted in a regulated, pulsatile fashion in response to increased GnRH pulse frequency, whereas FSH is released is mostly constitutive and responds to decreased GnRH pulse frequency (10, 11).
FSH synthesis and release are also regulated by several other proteins, such as follistatin, inhibin, and activin (12–17). Activin exerts positive effects on FSH by stimulating transcription, biosynthesis, and ultimately secretion as well as stimulating GnRH receptor gene expression. In rodents, after activin binds to gonadotrope membrane activin receptors, transcription factors, such as Smad4 are recruited and directly interact with the FSHβ gene promoter to upregulate its expression (15, 18). Another important transcription factor in gonadotropin regulation is the forkhead box (FOX) protein, FoxL2, which is a transcription factor known for its role in folliculogenesis and female sex determination (19). Foxl2 knockout mice have substantially decreased Fshb mRNA and serum FSH levels as well as reduced activin induction of FSHβ (20). Both inhibin and follistatin prevent the stimulatory effects of activin, causing suppression of FSHβ synthesis by blocking activin binding, thereby inhibiting intracellular pathways and subsequent FSHβ transcription (18, 21, 22).
There are several clinical conditions under which FSH expression or signaling via its receptor is increased, resulting in higher circulating levels and ultimately creating a FSH “gain of function” effect. These include ovarian hyperstimulation syndrome (23–26), certain ovarian cancers (27–29), and the recent discovery of non-gonadal FSH actions on bone in transgenic mouse models with elevated human FSH levels (30). GOF effects of FSH in ovaries were also occasionally noted in patients with FSH hypersecreting pituitary adenomas (31). The existence of such conditions highlights the importance of generating animal models that closely mimic human disease phenotypes, allowing us to expand the medical knowledge of these conditions and ultimately providing opportunities to learn how to treat them. While mouse models do not always accurately mimic human pathology, they provide a quick genetic test to address the function of human proteins in a physiological context that cannot be reliably achieved using in vitro experimental approaches. In this review, we highlight and describe previously generated FSH gain of function animal models and how they can potentially be used to develop new approaches for treating clinical conditions involving FSH.
GOF Mouse Models for FSH
Several GOF genetic models have been generated and used to study the physiological consequences of FSH. These models are described below in detail and summarized in Table 1.
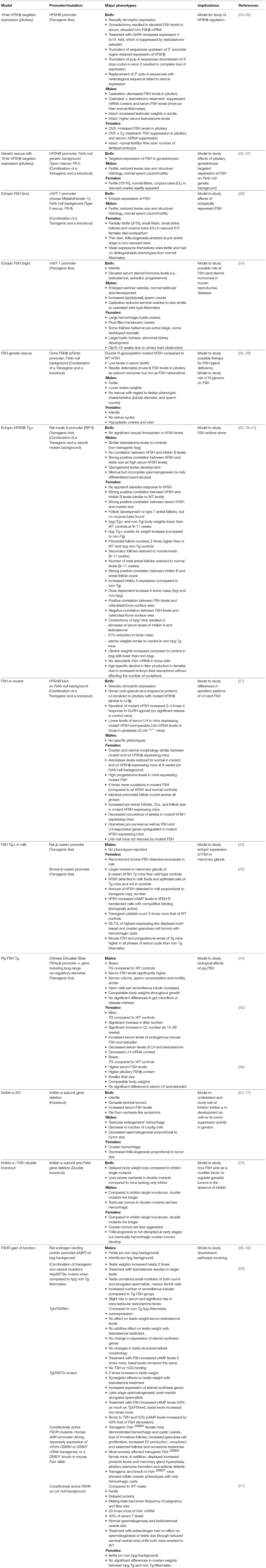
Table 1. Major phenotypes of FSH gain-of-function genetic models.
Expression of Human FSHB in vivo
A transgenic mouse model harboring a 10 kb HFSHB transgene was the first mouse model generated to test cell-specific expression of FSH in gonadotrope cells and to identify that species-specific differences exist in FSH regulation (32). The HFSHB transgene was cloned into an EcoR1-Sph1 genomic fragment and microinjected into fertilized one-cell embryos. The resulting transgenic mice exhibit only pituitary-specific HFSHB transgene expression, with no ectopic expression in non-pituitary tissues (32). Expression of hFSHβ was found to be localized to only gonadotrope cells in the anterior pituitary gland. The FSH heterodimer presumably incorporated the mouse-α subunit, creating an interspecies hybrid heterodimer with hFSHβ, because no free hFSHβ was detected in serum (32). FSH dimer secretion and pituitary HFSHB mRNA expression were higher in both transgenic and normal males than in their female counterparts. The retention of normal gonadotrope-specific expression of FSH and its function in mice expressing hFSHβ demonstrates conservation of regulatory elements and transcription factors for this subunit gene in both mice and humans (32). This mouse model provided a novel approach for studying molecular mechanisms and regulatory elements that are involved in control of the human FSHβ-encoding gene and its expression.
Gonadal Steroid Regulation of HFSHB
The same transgenic mouse model described above was also used for experiments designed to analyze steroid regulation of HFSHB in vivo (32, 33). This study included several experimental groups including castrated male and ovariectomized female mice. Castration resulted in elevated serum FSH levels in both normal controls and transgenic males. Similarly, increased serum FSH levels were observed in ovariectomized normal and transgenic females (33). Testosterone replacement after castration in male transgenic mice resulted in suppressed serum FSH levels. Estradiol (E2) replacement in ovariectomized females similarly resulted in suppressed serum and tissue FSH content. The sexually dimorphic pattern previously observed, in which both normal and transgenic males exhibiting greater tissue and serum FSH levels than the corresponding females, was also observed in these studies (33). These studies highlight the species-specific differences and suggest that the elements responsible for continued synthesis and secretion of hFSHβ in response to androgens are not present in the mouse pituitary environment (32, 33).
GnRH-Independent Androgen Inhibition of HFSHB Transgene
To further elucidate the direct roles that steroid hormones play in regulation of hFSHβ at the pituitary level, HFSHB transgenic mice were used to observe the effect of androgen in the presence or absence of gonadotropin-releasing hormone (GnRH) using both in vitro and in vivo approaches (34). Since there was an apparent species-specific difference in FSH secretion and molecular mechanisms in response to androgens, GnRH was identified as a possible key regulatory site in the androgen response of human FSHβ. For in vitro studies, primary pituitary cultures were obtained from GnRH-deficient hypogonadal (hpg) mice (9, 52) carrying the HFSHB transgene (34). Testosterone treatment in the absence of GnRH resulted in suppression of HFSHB mRNA and confirmed the inhibitory action of androgens directly at the pituitary level independent of GnRH (34). In vivo experiments in the hpg HFSHB mice included daily GnRH injections, which induced HFSHB expression in both males and females. Simultaneous administration of testosterone propionate in males completely blocked the stimulatory effect of GnRH, whereas simultaneous E2 administration in females only partially inhibited GnRH effects (34). These results demonstrated direct effects of testosterone and E2 on hFSHβ subunit expression at the pituitary level as well as an indirect suppression of GnRH as an additional regulatory mechanism (34). Additional hypothalamic site of E2 action cannot also be ruled out based on the above data.
Having established that the 10 kb HFSHB transgene is appropriately targeted to and hormonally regulated in mouse gonadotropes, a series of deletions were made on the 10 kb HFSHB transgene (35). Several independent transgenic lines expressing 5′ and 3′ truncated versions of HFSHB transgene were produced and systematically analyzed. These in vivo models helped to identify that truncation of sequences upstream of 5′ promoter region retained expression of hFSHβ in mouse gonadotropes, truncation of poly-A sequences downstream of 3′ stop codon in exon 3 resulted in complete loss of expression. Replacement of 3′ poly-A sequences with heterologous sequences (for example, lacZ reporter sequences) similarly failed to confer expression (35).
Since FSH is normally released from the pituitary in response to GnRH, it is of great interest to observe the physiological response to targeted expression of FSH in non-pituitary tissues. Accordingly, mouse models have been generated that drive expression from either specific or multiple ectopic tissues.
Use of HFSHB Transgenes to Achieve Genetic Rescue of Fshb Null Mice
An FSH-deficient mouse model was created in 1997 through targeted mutation (Fshbm1) in exon 3 of the FSHβ-encoding gene (53). Mice that were homozygous, i.e., Fshb null (Fshbm1/ Fshbm1), and therefore FSH-deficient, were generated by intercrossing heterozygous mice. Fshb null males displayed decreased testis size, yet were fertile (53). Sperm number was decreased by 75%, however, viability remained unchanged. In contrast, Fshb null females with the Fshbm1/ Fshbm1 genotype were infertile, with small ovaries and thin uteri. Ovaries had arrested follicular development at the secondary stage, and lacked any corpora lutea (53).
Genetic rescue of FSH-deficient mice was achieved using two independent methods (36). The type 1 genetic rescue (FR-I) consisted of targeting the previously described 10-kb HFSHB transgene specifically to pituitary gonadotrope cells. This genetic strategy resulted in complete rescue of both males and females lacking endogenous Fshb (36). Testis size and sperm counts in FR-I males were restored to those observed in wild-type values. Similar results were obtained in FR-1 females, as uterine and ovarian sizes also returned to wild-type values. Normal follicular development, restored estrous cycles, and production of normal litter sizes were observed in FR-I females.
Low- level ectopic expression of HFSHB was achieved in multiple tissues using a mouse metallothionein (mMT-1) gene promoter with the goal of genetically restoring reproductive phenotypes in FSH-deficient mice, designated as type 2 rescue (FR-II) (36). The mMT-1 promoter was used to drive ectopic expression of both a hCGα-encoding minigene and a hFSHβ-encoding gene, thereby resulting in expression of hFSH dimer in multiple tissues (36). Male mice expressing ectopic hFSH (FR-II) showed complete restoration of testis size and sperm counts. However, restoration of normal reproductive phenotypes was incomplete in FR-II females. Only 3 out of 10 FR-II females were able to conceive, and litter sizes were small. Arrested folliculogenesis was frequently observed in FR-II females. The small number of FR-II females that were able to become pregnant produced one litter, had obvious corpora lutea, yet small antral follicles (36). The results with the type 2 genetic rescue suggest that ectopic expression of human FSH can completely rescue Fshb null male mice, yet only partially rescue Fshb null females (36).
Ectopic Overexpression of HFSH Dimer in Transgenic Mice
Overexpression of FSH may lead to high serum levels and clinical conditions that negatively affect fertility. A transgenic mouse model ectopically expressing human FSHβ using an mMT-1 promoter resulted in male and female mice overexpressing hFSH in several tissues (24). Mice expressing either only transgenic MT-α subunit or only MT-FSHβ were crossed to obtain mice expressing hFSH ectopically (24). Founders expressing hFSH dimer at very high levels were chosen for further analysis. Both males and females were infertile; males showed enlarged seminal vesicles and elevated testosterone levels yet normal testis size and spermatogenesis (24). These high level FSH expressing males were infertile, presumably due to male sexual behavioral deficits secondary to excess testosterone. However, this behavioral phenotype was not tested in these studies. Females displayed arrested folliculogenesis, along with increased serum estrogen, progesterone, and testosterone concentrations. Females also developed urinary tract obstruction and hemorrhagic and cystic ovaries, yet exhibited no signs of tumors (24). Their symptoms were comparable, but not identical to human conditions such as polycystic ovarian and ovarian hyperstimulation syndromes. Most females died between 6 and 13 weeks of age. (24). The overexpression of hFSH in multiple tissues gave insight into these clinical conditions and provided a model that may be used in the future for developing treatments.
Ectopic Expression of FSH in hpg Mice
The role of FSH in gonadal physiology was investigated using a mouse model similar to the models described above that carry a HFSHB transgene (39). However, this model expressed transgenic human-FSH (tg-FSH) on a gonadotropin-deficient hypogonadal (hpg) background to observe FSH effects independent of LH. The HFSHB transgene was cloned into a vector containing the rat insulin II promoter (RIP) and injected into mouse oocytes (39). RIP directed ectopic expression of tg-FSH to the pancreas. Hypogonadism was accomplished by breeding tg-FSH mice to an hpg strain containing a truncating mutation that caused GnRH depletion, thereby creating tg-FSH+ hpg mice (39, 52). Varying serum tg-FSH levels were found in different strains of mice, allowing for analysis of a range of circulating FSH concentrations. Tg-FSH seemed to have no effect on androgen levels in the tg-FSH+ hpg mice, which appeared to be due to underdeveloped epididymis and seminal vesicles. In male tg-FSH+ hpg mice, testis size increased as compared to non-tg-FSH hpg controls, however, this was only observed in males exhibiting high serum FSH levels (>1 IU/liter). Tg-FSH+ hpg female mice secreting high levels of FSH exhibited dose-dependent, elevated inhibin B secretion. Ovaries of tg-FSH+ hpg females were also enlarged and exhibited increased follicular development to the antral stage (39).
Additional studies were performed using female tg-FSH+ hpg mice to determine the effect of FSH alone on primordial follicle reserve and the role of FSH in early follicular development (40). Partial disruption of follicular development was observed in non-tg hpg ovaries. Although development past the primary follicle stage occurred, there were small numbers of early antral follicles. In contrast, tg-FSH+ hpg females showed advanced follicular development up to the antral stage, although no corpora lutea were observed in any tg-FSH+ or non-tg FSH hpg ovaries due to the absence of LH (40). Significant increases in total primordial and secondary follicle numbers were seen in tg-FSH+ hpg females as compared with both non-tg hpg and wild-type mice. The total antral follicle count was 15-fold higher in tg-FSH+ hpg ovaries than non-tg FSH hpg levels, which restored values to wild-type levels (40). The findings from this study indicate an important role of FSH in early follicular development, showing an increase in primordial follicle reserve and stimulation of follicle growth.
Interestingly, when tg-FSH+ female mice alone with progressively rising hFSH levels (2.5–10 IU/ml) were monitored across the life span, age-specific phenotypes were observed. Whereas, tg-FSH+ female mice <22 week of age delivered increased litter sizes, those that were older (>23 week of age) produced decreased litter sizes despite increased ovulations and demonstrated premature infertility due to embryo resorptions and parturition failure. Thus, this model provided a novel in vivo scenario in which age-related rise in FSH contributes to female reproductive aging and infertility by a post-implantation defect (embryo-fetal resorption) without directly affecting the ovarian reserve (41).
Contrary to the proposed deleterious and direct effects of FSH on bone osteoclasts in mice (54), ectopic human FSH expression in the above described genetic model caused an increase in bone mass in female mice (30). Similar phenotypes were also observed when the HFSHB transgene was expressed on the hpg genetic background with a total suppression of endogenous gonadotropins and E2. Expression analysis indicated osteoclasts did not express Fshr mRNA and the bone phenotypes manifest only when ovaries were intact. Further studies indicated that bone volume in these transgenic mice positively correlated with ovary-derived inhibin A and androgens. Thus, ectopic human FSH expression in this model suggests FSH acts indirectly to enhance bone function in an ovary-dependent and LH-independent manner (30). The controversy with regard to non-gonadal actions of FSH is ongoing and has been recently described in detail (55).
Rerouting of FSH Into the LH Secretion Pathway
Transcriptional responses of FSHβ and LHβ encoding genes are different and dependent on GnRH pulse frequencies (10, 11, 56). In immortalized gonadotrope cells, Fshb gene transcription is favored by slow GnRH pulses whereas Lhb gene transcription is dependent on fast GnRH pulses (10, 11, 56). Once the heterodimers are assembled, FSH is largely released constitutively, while LH is released as pulses via a regulated secretory pathway (10, 11). LH contains a carboxy terminal (C′) heptapeptide that directs its secretion via this regulated pathway. A novel mouse model took advantage of this heptapeptide to observe the physiological response to a mutant FSH that contained this peptide (37). Human transgenes encoding either a wild type (HFSHBWT) or mutant (HFSHBMut) FSHβ were introduced onto an Fshb-null genetic background.
The presence of interspecies heterodimers of mouse-α-and WT or mutant FSHβ subunits in different mouse lines was confirmed by Western blot analysis (37). LH is stored in dense-core granules (DCG) prior to release (57–59). To determine whether mutant FSH was secreted via the same pathway as LH, co-localization of mutant FSHβ subunit and DCG-specific Rab27 was evaluated in gonadotrope cells. Interestingly, the number of mutant FSHβ subunit and Rab27 co-localized gonadotropes was 6 to 8- fold higher than seen in gonadotropes of control mice, where FSH is secreted via the constitutive pathway (37). Co-localization of mutant FSHβ and a chaperone protein chromogranin-A (Chr-A), which is found in the Golgi network in gonadotropes and important for regulated release of LH, was also examined. Co-localization of mutant FSHβ and Chr-A was higher when compared to both control and Fshb null mice expressing a HFSHBWT transgene and was similar to levels seen with co-localization of LHβ (37). These results suggest that the engineered mutant FSHβ (containing the C′ heptapeptide normally only found in LH)- containing FSH heterodimer successfully entered the regulated secretory pathway.
As described above, mice deficient in FSH are infertile, have small ovaries and thin uteri, along with disrupted folliculogenesis (53). HFSHBMut transgene was able to genetically rescue Fshb-null mice, restoring ovarian and uterine size as well as estrus cycles leading to the presence of antral follicles and corpora lutea in ovaries. Progesterone levels were higher in HFSHBMut–expressing mice than in control and wild-type mice (37). Interestingly, the number of ovulations was increased from 9 to 10 per cycle in control mice to 55 per cycle in mutant FSH–expressing Fshb null mice (37). This was accompanied by the presence of more pre-antral follicles and reduced follicular atresia in ovaries. Mice expressing HFSHBMut also demonstrated increased follicle size and enhanced granulosa cell proliferation, leading to a longer reproductive lifespan and follicle survival (37). This mouse model provided a novel approach to studying differential secretory pathways of FSH and LH, and demonstrated a potential role of rerouted FSH in treating age-associated reproductive conditions (37).
Expression of a Transgene Encoding the Non-glycosylated Human FSHβ Subunit
One of the characteristic features of glycoprotein hormones, including FSH is the presence of N-linked sugar chains on both the α- and β-subunits. FSH possesses four (2 on each subunit) potential N-glycan attachment sites (1–4, 60, 61). The presence or absence of both N-glycans on FSHβ subunit contributes to macroheterogeneity, significantly affects serum half–life and may alter bioactivity (60, 61). To genetically determine the role of N-glycans on the human FSHβ subunit, the nucleotides corresponding to two Thr residues following Asn residues on which N-glycans are added, Asn7 and Asn24, were mutated to Ala, thereby abolishing N-glycosylation events at these two sites (38, 60).
The mutant cDNA transgene encoding the N-glycosylation double mutant FSHβ subunit was targeted to gonadotropes in transgenic mice first and the transgene was subsequently introduced onto Fshb null genetic background using the well-established genetic rescue scheme. An HFSHB WT transgene similarly was introduced onto Fshb null genetic background and the resultant Fshb null mice carrying the HFSHB WT transgene were used as positive controls (38, 60). Biochemical studies confirmed that the mutant FSHβ subunit inefficiently assembled with the endogenous mouse α-subunit and very little FSH heterodimer was present in pituitary extracts (38). Moreover, media collected from short term pituitary organ culture experiments and serum from mutant FSHβ-expressing Fshb null mice showed very low or undetectable levels of FSH by radioimmunoassays. These data indicated that the double N-glycosylation mutant FSHβ subunit was secretion incompetent (38). Moreover, the mutant transgene, unlike the HFSHB WT transgene, did not rescue Fshb null mice, confirming that even if secreted in low levels, the mutant FSHβ-subunit containing FSH dimer was biologically inactive (38). Thus, these studies provided in vivo genetic evidence that N-glycosylation on FSHβ subunit is critical for FSH heterodimer assembly, secretion and action (38).
Expression of FSH in the Mammary Gland
Ectopic expression of bovine FSH has been achieved in a model targeting its expression in milk secreted from mouse mammary glands (42). This mouse model was created using a rat β-casein gene promoter driving expression of bovine α and FSHβ subunits. The β-casein promoter drives targeted expression in only the mammary gland, making it possible to observe the effects of ectopic bovine FSH expression in a single tissue (42). Transgenic (Tg) mice expressing the transgene were created either by microinjection of both subunits, or by breeding of mice that expressed each one of them separately (42). The presence of tg-FSH was confirmed by northern blot and radioimmunoassay. Bioactivity of tg-FSH was also confirmed by measuring granulosa cell counts and the ability of the cells to produce estrogen (42). The amount of tg-FSH positively correlated with granulosa cell number and estrogen production, and therefore suggesting successful bioactivity of the transgene-derived FSH (42). No other overt phenotypes were observed in transgenic mice. This mouse model provided a novel approach for expressing FSH in mammary glands and releasing it into milk.
A similar model was later generated that also ectopically expressed FSH in mammary glands, but in this case, the transgene encoded human FSH. In these studies, the investigators used the bovine β-casein gene promoter to specifically express human FSH in mammary glands of transgenic mice (43). Milk was collected from tg-hFSH mice to determine FSH concentration using an enzyme-linked immunosorbent assay (ELISA). Two mouse lines showed nearly undetectable levels of hFSH, as compared to one line that had high levels (300 mIU/mL). This variation in hFSH concentration in different lines was most likely due to differing transgene copy numbers (43). In vitro biological activity of hFSH was determined by measuring cyclic-AMP (cAMP) production after exposing hFSH receptor-transfected cells to sterilized milk from each cell line. Milk containing hFSH increased cAMP production in the assay, indicating receptor binding and intracellular signaling, and therefore, biological activity of hFSH (43).
Blood cell counts were performed to analyze the effects of any hFSH leakage into the circulation. Several of the transgenic lines showed increased white blood cell and platelet levels as compared to normal mice, red blood cells in the affected animals were also smaller in size (43). Ovarian and breast tumors were observed in one transgenic line, along with collapsed alveoli within the lactating glands. Human FSH also seemed to have a stimulatory effect on endogenous mouse FSH, as mFSH levels were higher at all estrus cycle phases in transgenic mice (43). This unique mouse model displayed distinct physiological responses to ectopic expression of hFSH in mammary glands and secretion into milk. Many of them negatively affected blood circulation and reproductive health. Ectopic hFSH leaking into the bloodstream appeared to lead to overproduction of endogenous mouse FSH, possibly contributing to the observed ovarian and breast tumors by increased estrogen production as a secondary consequence (43).
Expression of Porcine FSH in Mice
Transgenic expression of porcine FSH using bacterial artificial chromosome (BAC) methods has been achieved using a gain-of-function mouse model (45). A BAC containing porcine (p) α and β subunits was constructed and isolated from a porcine BAC library from a male Erhualian pig, a highly prolific pig breed. BAC clones containing pFSH α and β were then digested and microinjected into fertilized mouse zygotes (45). Transgenic (Tg) mice expressing pFSH were identified by PCR and Southern blot. After further breeding of Tg mice to wild-type mice, both pFSHβ and pFSHα BACs were transmitted in identical Mendelian ratios to offspring, indicating proper hybridization of the Tg subunits. Expression of Tg pFSH mRNA was confirmed by RT-PCR and northern blot, with expression localized specifically to the pituitary gland. Circulating pFSH was confirmed by evaluation of serum samples by ELISA, showing levels ranging from 6.36 to 19.83 IU/L, which were within the physiological range (45).
Female fecundity was analyzed in both Tg and WT mice. Interestingly, Tg females had litter sizes that were significantly larger than WT females as well as the total number of pups than WT littermates. This increase in fecundity in Tg females seemed to be due to enhanced ovulation, as histological examination revealed a significant increase in the number of corpora lutea at 14 and 28 weeks of age in Tg mice compared to WT. Serum hormone levels were analyzed to determine if increased ovulation was due to differential hormonal regulation (45).
Endogenous mouse FSH levels were elevated in Tg mice, as was estradiol. Serum levels of LH and testosterone were significantly lower in Tg mice compared to WT. Pituitary expression of LHβ mRNA was also lower in Tg mice than in WT mice (45). The increased estradiol levels could be due to greater aromatization of androgens to estrogens due to elevated levels of endogenous mFSH in addition to transgene-derived pFSH (45). This enhanced conversion of testosterone to estrogen as a result of increased aromatase activity could explain the lower serum levels of testosterone. However, the mechanism for reduced LHβ mRNA and serum LH despite low levels of serum testosterone is unclear. The results from this study provided confirmation of successful expression of porcine FSH in a transgenic mouse model with no reproductive effects in males but enhanced ovulation in female Tg mice (45).
Expression of Transgenic Porcine FSH in Large White Boars
Porcine FSH was further analyzed by the same group that produced BAC pFSH subunits from Chinese Erhualian pigs, and introduced them into Large White Boars (44). As the Large White Boar previously showed poor reproductive performance, the investigators sought to determine if overexpression of pFSH from a highly prolific pig breed could improve fertility. Successful integration of pFSH into transgenic (Tg) boars was confirmed using genomic PCR as well as RT-qPCR analysis to determine mRNA expression of porcine FSHα and FSHβ. Expression of FSHα was observed in multiple tissues including heart, liver, spleen, kidney, brain, testis, and epididymis of both Tg and wild type (WT) boars, with higher expression in Tg than WT.
Expression of FSHβ was localized specifically to the pituitary and was significantly higher in Tg than WT boars (44). Higher serum levels of both FSHα and FSHβ were observed in Tg boars suggesting overexpression of pFSH. Male reproductive performance was measured by evaluating semen volume, sperm motility, sperm concentration, and total sperm number per ejaculation (44). There was no significant difference between semen quality parameters of Tg and WT boars. However, the number of germ cells per seminiferous tubule was significantly higher in Tg boars than WT (44). The elevated germ cell counts in seminiferous tubules suggested increased spermatogenesis capacity, but the lack of significant results from semen evaluation leaves room for further analysis to confirm this possibility (44).
In a third study done using the same BAC system containing pFSH α and β, analysis of reproductive phenotypes in female transgenic (Tg) Large White Boars were analyzed. Methods for pFSH BAC transfer were the same as previously described for this model (44). The specific integration site of pFSH into Large White Boars was determined using whole-genome sequencing, identifying exogenous FSHα/β genes at 140, 646, 456 bp on chromosome 9 (46). The transgene integration site was mapped to perhaps rule out that the integration itself did not result in modification of any endogenous loci that regulate fertility.
Analysis of Tg female boars revealed elevated levels of serum FSH and FSHβ protein in the pituitary, but Tg females produced reduced numbers of total newborn piglets as compared to WT. Reduced expression of Fshr, Lhr, Esr1, and Esr2 was also observed using RT-qPCR in Tg boars as compared to WT at 300 days of age. Reproductive organ weights, blood cell counts, and histological analysis revealed no differences between Tg and WT boars in overall reproductive health (46). The reduced expression of mRNAs encoding receptors for FSH, LH, and estrogen suggest a possible negative effect of pFSH overexpression in female pigs as well as the observation of reduced total newborn piglets (46). Further studies using this model are needed to confirm the effects. However, these studies provided novel information on the physiological role of porcine FSH in vivo in the homologous species.
Inhibin Knock Out Mouse Model
Important regulators of FSH production and secretion are gonad-derived dimeric growth factors, activin and inhibin. Inhibin is a heterodimer, consisting of an α and β subunit, whereas activin is a homodimer, consisting of various combinations of the two homologous β subunits (βA and βB) (21). The inhibin and activin subunits are expressed in multiple tissues throughout the body. Inhibins suppress and whereas activins promote FSH synthesis and secretion. To investigate the role of inhibin in reproduction and general physiology, a knockout mouse model with a targeted deletion of the α-inhibin–encoding gene was achieved using homologous recombination technology in mouse embryonic stem cells (47). Targeted deletion of only the α-subunit of inhibin ensured successful inhibin deficiency without unwanted deletion of the activins.
Male and female heterozygous mice produced normal litter sizes and were fertile. However, homozygous males and females proved to be infertile when crossed with wild-type mice, despite developing normal external genitalia (47). In addition to infertility, all homozygous mice tested showed gonadal tumors when examined histologically, which were evident as early as 4 weeks of age. Testicular enlargement was visible in males, along with gradual regression of spermatogenesis and a decrease in Leydig cell count starting at 5–7 weeks of age (47). In females, ovarian tumors disrupted follicular development and morphology from 7 to 16 weeks of age. FSH levels in both males and female homozygous, inhibin-deficient mice were 2- to 3-fold higher when compared to both heterozygous and WT control mice (47). The results of this study suggested a novel secreted tumor suppressor role for inhibin that was highly specific to the gonads. Development of normal external sexual organs and gametes followed by regression and disruption at a later age indicated normal embryogenesis and early sexual development, therefore suggesting gonadal tumors as the cause for infertility (47).
To further investigate the role of inhibin and FSH in gonadal tumorigenesis, a double-mutant knockout approach was taken. Since the inhibin-deficient mice developed aggressive gonadal tumors accompanied by elevated FSH levels, the contribution of FSH to tumorigenesis was directly assessed using a genetic strategy. To achieve this, double-homozygous mutant mice were created by intercrossing heterozygotes for each knock-out mutation (Inham1 and Fshbm1) to generate mice deficient in both inhibin and FSH (Inham1/Inham1; Fshbm1/Fshbm1) (24). The first parameter examined in the double-knockout mice was weight, as the previous study showed that mice deficient in inhibin only, exhibit a severe cachexia-like syndrome and die by 12 weeks of age (24). Most of double-knockout mutant males survived for 1 year and showed no dramatic weight loss or testis phenotypes. Approximately, 95% of the inhibin-deficient female mice died by 17 weeks. In contrast, double null mutant females, about 70% survived past 17 weeks, but these all eventually died by 39 weeks and all of them exhibited severe weight loss (24).
Gonadal tumor progression was also altered in the double-mutant mice, as development of tumors was slower and less aggressive than in mice deficient in inhibin alone (24). In 12 week old double mutant males, the tumors were small, there were no signs of hemorrhage, and tubule morphology was also unaltered by tumor growth, despite proliferation of tumor cells (24). Beyond 1 year of age, some males showed no tumor development, as compared to inhibin-deficient males which all had tumors as early as 4 weeks (24).
In female double-mutants, ovaries appeared morphologically normal at 12 weeks of age. Histological analysis revealed hemorrhage, cysts, as well as granulosa cell tumors. However, these tumors in the double-knockout females appeared less invasive and developed more slowly than in inhibin-deficient mice. Both male and female double-mutant mice showed reduced serum levels of activin A and estradiol as compared to mice lacking only inhibin (24). In addition to this, aromatase mRNA expression levels were reduced in double-mutant mice compared to those in inhibin-deficient mice. These results confirm the role of inhibin in gonadal tumorigenesis and identify FSH as an important modifier in the progression and aggressive growth of inhibin-deficient gonadal tumors (24).
FSH Receptor Gain of Function
The FSH receptor (FSHR) is a transmembrane, G-protein coupled receptor expressed on testicular Sertoli cells and ovarian granulosa cells in males and females, respectively (2, 4). Signaling via FSHRs results in steroidogenesis (production of estrogen) and is essential for gonadal development in both sexes. A novel approach to studying the gain-of-function effects of FSH receptor was undertaken by generating a mouse model exhibiting constitutively active FSH receptor action on an hpg genetic background (48). The use of the hpg genetic background allowed observation of the effects of active FSHR completely independent of endogenous gonadotropins, FSH and LH.
The gain of function receptor mutation (FSHR+) was a single amino acid substitution (Asp567Gly) that was specifically expressed in Sertoli cells by using the rat androgen binding protein (rABP) promoter (48). Bioactivity of the ligand-independent FSH receptor was confirmed by measuring cAMP production, which was significantly higher in Tg-Sertoli cells than in non-Tg-hpg Sertoli cells in vitro (48). The in vivo effects of the FSHR+ mutation were first examined by measuring testicular weight. Tg-FSHR+ hpg testis weights were increased up to 5-fold, with an average of a 2-fold increase as compared to non-Tg hpg controls. Histological examination of FSHR+ testes showed round and elongated spermatids and signs of Sertoli cell maturation, as compared with non-FSHR+ hpg controls that lacked mature Sertoli cells and exhibited blocked spermatogenesis. Tg-FSHR+ mice also had elevated testosterone levels, and undetectable levels of LH and FSH, suggesting that the observed physiological responses to the constitutively active FSH receptor was indeed gonadotropin-independent (48).
To determine whether the results from the FSHR+ mutation were solely due to the amino-acid substitution or if they were in response to overexpression of mutant FSH receptor, a comparative study was performed using the same mouse model in parallel with a transgenically overexpressed wild-type human FSH receptor (TgFSHRwt) (49). Transgenic male and female mice overexpressing TgFSHRwt were created by microinjection of a human FSHR DNA construct into mouse oocytes. Overexpression of TgFSHRwt was confirmed by measuring radioactive 125I-FSH binding to testicular membranes. Significantly elevated 125I-FSH binding was observed in TgFSHRwt testes as compared to non-Tg controls, similar to levels in FSHR+ mice, thereby confirming overexpression.
TgFSHRwt mice displayed no difference in testis weights or serum testosterone (T) levels, as compared to FSHR+ mice, which had larger average testis weights and elevated T concentrations when compared to non-Tg controls. TgFSHRwt Sertoli cells showed higher cAMP activity in vitro, however basal levels did not exhibit the same increased activity as FSHR+ cells (49). Receptor ligand-specificity was decreased in FSHR+ mice, as exposure to human chorionic gonadotropin and TSH resulted in intracellular signaling. However, the same response was not observed in TgFSHRwt mice. Steroidogenic enzyme-encoding mRNAs such as Cyp11a1 and Star, were elevated in FSHR+ mice, suggesting increased steroidogenic potential (49). However, this increase was also not observed in TgFSHRwt mice. Sertoli and germ cell maturation that was observed in FSHR+ mice was also absent in TgFSHRwt mice, as they exhibited immature development similar to non-Tg hpg mice. Together, all of these results suggest that the physiological responses to constitutively active mutant FSH receptors in FSHR+ mice were due to the mutation itself and not the result of receptor overexpression (49).
Activating mutations in human FSHR are very rare. To identify the phenotypic consequences of such mutations in humans, mouse models harboring mutant versions of Fshr were first developed (26, 50). The rationale was that phenotypic analysis of these mice would provide information to look for similar phenotypes in patients carrying analogous mutations. Two independent Fshr point mutants D580H and D580Y were created and expressed using human AMH promoter to achieve ovary-specific expression in transgenic mice. Additionally, an Fshr BAC clone was engineered to carry the D580Y mutation and knocked-in to the endogenous Fshr locus in ES cells first and subsequently, the knock-in mutant mice were generated (26, 50).
Both D580H and D580Y mutants displayed increased basal receptor activity and they both demonstrated FSH binding. D580H mutant FSHR was neither ligand-dependent nor promiscuous toward LH/CG stimulation (26, 50). Granulosa cell-specific expression of mFshr D580H resulted in multiple ovarian abnormalities in transgenic female mice. Ovaries in the majority of transgenic females displayed hemorrhagic cysts, accelerated loss of immature follicles, increased granulosa cell proliferation and E2 biosynthesis, and luteinized but unruptured follicles, and teratomas (26, 50). A subset of the most severely affected transgenic females were infertile due to disrupted estrus cycles, and decreased gonadotropin, and increased prolactin levels. The increase in E2 and PRL levels led to secondary abnormalities including mammary gland enlargement and hyperplasia, pituitary adenoma formation, and defects in adrenal glands (26, 50). In contrast to phenotypic consequences of mFshr D580H expression, either transgenic or knock-in expression of mFshr D580Y mutant resulted in milder phenotypes, mostly hemorrhagic cysts in ovaries (26, 50). Thus, these GOF Fshr mutant mice resulted in distinct changes in ovarian function and proved valuable in the search of similar mutations in humans.
The most recent FSH receptor gain of function model challenged the traditional dogma that testosterone is essential for spermatogenesis (51). A mouse model was created that possessed a constitutively active mutant FSH receptor on an LH receptor null background (Fshr-CAM/Lhr−/−) (51). As LH regulates testosterone production via binding of LH receptors (LHRs) in Leydig cells, it was hypothesized that an Lhr knockout approach would eliminate testosterone action. However, testosterone production persisted in the initial Fshr-CAM/Lhr−/− male mice, as serum levels recovered to about 40% of wild-type concentrations. Therefore, the rescue of testicular size and spermatogenesis was probably due to normal testosterone actions (51). To eliminate any basal T activity, a treatment using the antiandrogen, flutamide, was employed. In WT control mice that had no Fshr-CAM, reduction in the size of testes and seminal vesicles was observed as well as arrested spermatogenesis. Interestingly, the Fshr-CAM/Lhr−/− males had only reduced seminal vesicle size after anti-androgen treatment, with no change in testicular size, and normal spermatogenesis (51). In addition to this, expression of androgen-dependent genes (Drd4, Rhox 5, Aqp8, and Eppin) were tested in the Fshr-CAM/Lhr−/− males. Anti-androgen treated WT males showed reduced expression profiles of these genes, whereas mutant Fshr-CAM/Lhr−/− showed no reduction (51). These results suggested that even in the absence of testosterone, constitutively active FSH receptor alone is able to maintain androgen-dependent gene expression as well as normal spermatogenesis and testicular development.
Conclusions and Future Directions
The first GOF mouse model for FSH was generated by our group more than 25 years ago (32). Since then investigators have used different promoters to achieve gonadotrope-specific as well as ectopic expression of the FSH ligand. Most commonly, human FSHB gene or individual subunit-encoding cDNAs (CGA or FSHB) were used in these experiments. More recently, BACS, or genes encoding porcine FSHβ subunit were also used to create transgenic pigs. GOF models for FSH action were generated using constitutively active FSH receptor–expressing mice and the reproductive consequences were studied either in these models directly or on an Lhr null genetic background (Figure 1). Combination of Fshb null mice and FSH GOF models resulted in genetic rescue that was used as an efficient in vivo functional assay for testing bioactivities of FSH and FSH analogs (Figure 2). A summary of the major phenotypes observed in each model is listed in Table 1.
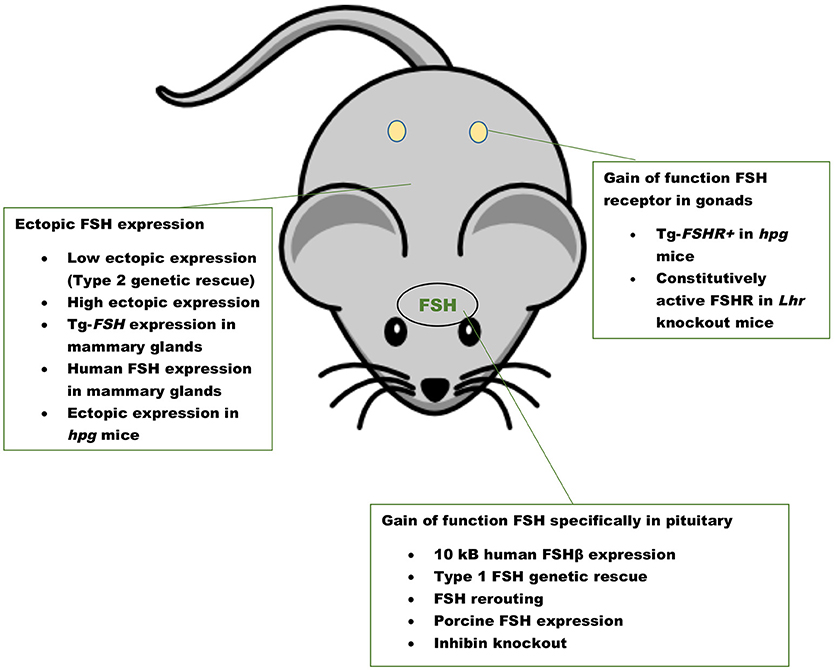
Figure 1. Gain of function mouse models for FSH ligand and FSH receptor. A summary of mouse models with pituitary-targeted and ectopic expression of FSH and gain of function mouse models for FSHR activation. Inhibin knockout mice have high levels of FSH as a result of loss suppression by inhibin.
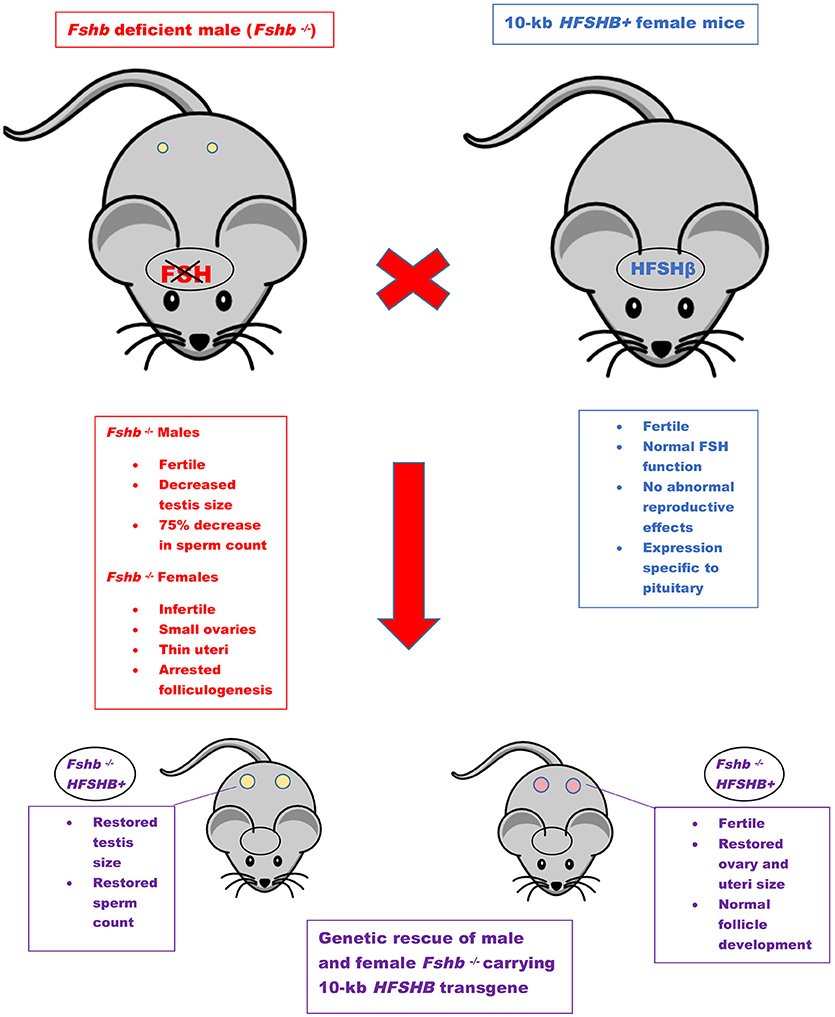
Figure 2. Genetic rescue scheme. Mice lacking Fshb show sexually dimorphic phenotypes (red box). The features of HFSHB+ mice were shown in blue box. HFSHB+ mice by themselves do not exhibit any overt phenotypes. The Fshb null male mice are typically intercrossed with HFSHB+ transgenic female mice to eventually generate Fshb−/− HFSHB+ mice. The HFSHB+ transgene fully rescues both male and female Fshb null mice (purple boxes).
Promoters used to generate FSH GOF models have also proved useful to express different reporters specifically in the gonadotrope cell lineage (62, 63). Recent advances in temporally regulated gene expression (64–66) will allow us in the future to tightly control FSH expression in desired tissues at desirable times across the life span of a mouse. Such refined genetic models will be useful to identify age-dependent gene/protein networks in FSH target tissues. These genetic models to conditionally “turning on” FSH in desired cells will also allow us to test if FSH receptor expression in non-gonadal cells has any physiological significance (55).
Author Contributions
RM wrote the first draft of the manuscript and created the Figures. CS generated the entire Table. TK edited the manuscript, Table, and Figures. He also wrote the abstract, about Glycosylation, and Conclusions, and Future Directions. He created and formatted the entire Bibliography.
Funding
Work reported in this Chapter was supported in part by NIH grants CA166557, AG029531, AG056046, HD081162, and The Makowski Endowment (to TK). RM is a Makowski Summer Scholar in the Kumar lab and is a graduate student supported by the Integrated Physiology program at the University of Colorado Anschutz Medical Campus, Aurora, CO, USA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. George Bousfield for critically reading the manuscript and many helpful suggestions.
Abbreviations
BAC, Bacterial Artificial Chromosome; cAMP, 3′,5′-cyclic adenosine monophosphate; DCG, Dense Core Granules; E2, Estradiol; FSH, Follicle-stimulating hormone; FSHR, FSH-receptor; FoxL2, Forkhead box L2; FR-I, Fshb type - I genetic rescue; FR-II, Fshb type - II genetic rescue; GnRH, Gonadotropin releasing hormone; GOF, Gain-of-function; GPCR, G-protein Coupled Receptor; hCG, Human chorionic gonadotropin; HPG, Hypothalamus-Pituitary-Gonadal; hpg, Hypogonadal; LH, Luteinizing hormone; LHR, Luteinizing hormone receptor; mMT-1, mouse metallothionein- 1; RIP, Rat insulin II promoter; RT-PCR, Reverse transcription- polymerase chain reaction; T, Testosterone; Tg, Transgenic; TSH, Thyroid-stimulating hormone; m, mouse; h, human; p, porcine; o, ovine; WT, Wild-type.
References
1. Bousfield GR, Jia L, Ward DN. Gonadotropins: chemistry and biosynthesis A2. 3rd ed. In: Neill, Jimmy D, editors. Knobil and Neill's Physiology of Reproduction. St Louis, MO: Academic Press (2006). p. 1581–634. doi: 10.1016/B978-012515400-0/50035-X
2. Narayan P, Ulloa-Aguirre A, Dias JA. Gonadotropin hormones and their receptors. In: Strauss J, Barbieri A, Gargiulo A, editors. Yen & Jaffe's Reproductive Endocrinology. Philadelphia, PA: Elsevier (2018). p. 25–57.
3. Pierce JG, Parsons TF. Glycoprotein hormones: structure and function. Annu Rev Biochem. (1981) 50:465–95. doi: 10.1146/annurev.bi.50.070181.002341
4. Ulloa-Aguirre A, Dias JA, Bousfield GR. Gonadotropins. In: M.SImoni, Huhtaniemi I, editors. Endocrinology of the Testis and Male Reproduction. Springer International (2017). p. 1–52.
5. Simoni M, Gromoll J, Nieschlag E. The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocr Rev. (1997) 18:739–73. doi: 10.1210/er.18.6.739
6. Heckert L, Griswold MD. Expression of the FSH receptor in the testis. Recent Prog Horm Res. (1993) 48:61–77. doi: 10.1016/B978-0-12-571148-7.50006-3
7. Heckert LL, Griswold MD. The expression of the follicle-stimulating hormone receptor in spermatogenesis. Recent Prog Horm Res. (2002) 57:129–48. doi: 10.1210/rp.57.1.129
8. Richards JS, Pangas SA. The ovary: basic biology and clinical implications. J Clin Invest. (2010) 120:963–72. doi: 10.1172/JCI41350
9. Mason AJ, Pitts SL, Nikolics K, Szonyi E, Wilcox JN, Seeburg PH, et al. The hypogonadal mouse: reproductive functions restored by gene therapy. Science (1986) 234:1372–8. doi: 10.1126/science.3097822
10. Thompson IR, Kaiser UB. GnRH pulse frequency-dependent differential regulation of LH and FSH gene expression. Mol Cell Endocrinol. (2014) 385:28–35. doi: 10.1016/j.mce.2013.09.012
11. Stamatiades GA, Kaiser UB. Gonadotropin regulation by pulsatile GnRH: signaling and gene expression. Mol Cell Endocrinol. (2018) 463:131–41. doi: 10.1016/j.mce.2017.10.015
12. Bernard DJ, Fortin J, Wang Y, Lamba P. Mechanisms of FSH synthesis: what we know, what we don't, and why you should care. Fertil Steril. (2010) 93:2465–85. doi: 10.1016/j.fertnstert.2010.03.034
13. Ciccone NA, Kaiser UB. The biology of gonadotroph regulation. Curr Opin Endocrinol Diabetes Obes. (2009) 16:321–7. doi: 10.1097/MED.0b013e32832d88fb
14. Coss D. Regulation of reproduction via tight control of gonadotropin hormone levels. Mol Cell Endocrinol. (2018) 463:116–30. doi: 10.1016/j.mce.2017.03.022
15. Coss D, Mellon PL, Thackray VG. A FoxL in the smad house: activin regulation of FSH. Trends Endocrinol Metab. (2010) 21:562–8. doi: 10.1016/j.tem.2010.05.006
16. Das N, Kumar TR. Molecular regulation of follicle-stimulating hormone synthesis, secretion and action. J Mol Endocrinol. (2018) 60:R131–55. doi: 10.1530/JME-17-0308
17. Matzuk MM, Kumar TR, Shou W, Coerver KA, Lau AL, Behringer RR, et al. Transgenic models to study the roles of inhibins and activins in reproduction, oncogenesis, and development. Recent Prog Horm Res. (1996) 51:123–54; discussion 155–27.
18. Fortin J, Ongaro L, Li Y, Tran S, Lamba P, Wang Y, et al. Minireview: activin signaling in gonadotropes: what does the FOX say…to the SMAD? Mol Endocrinol. (2015) 29:963–77. doi: 10.1210/me.2015-1004
19. Uhlenhaut NH, Treier M. Forkhead transcription factors in ovarian function. Reproduction (2011) 142:489–95. doi: 10.1530/REP-11-0092
20. Thackray VG. Fox tales: regulation of gonadotropin gene expression by forkhead transcription factors. Mol Cell Endocrinol. (2014) 385:62–70. doi: 10.1016/j.mce.2013.09.034
21. Gregory SJ, Kaiser UB. Regulation of gonadotropins by inhibin and activin. Semin Reprod Med. (2004) 22:253–67. doi: 10.1055/s-2004-831901
22. Fortin J, Boehm U, Deng CX, Treier M, Bernard DJ. Follicle-stimulating hormone synthesis and fertility depend on SMAD4 and FOXL2. FASEB J. (2014) 28:3396–410. doi: 10.1096/fj.14-249532
23. Desai SS, Roy BS, Mahale SD. Mutations and polymorphisms in FSH receptor: functional implications in human reproduction. Reproduction (2013) 146:R235–48. doi: 10.1530/REP-13-0351
24. Kumar TR, Palapattu G, Wang P, Woodruff TK, Boime I, Byrne MC, et al. Transgenic models to study gonadotropin function: the role of follicle-stimulating hormone in gonadal growth and tumorigenesis. Mol Endocrinol. (1999) 13:851–65. doi: 10.1210/mend.13.6.0297
25. Montanelli L, Delbaere A, Di Carlo C, Nappi C, Smits G, Vassart G, et al. A mutation in the follicle-stimulating hormone receptor as a cause of familial spontaneous ovarian hyperstimulation syndrome. J Clini Endocrinol Metabol. (2004) 89:1255–8. doi: 10.1210/jc.2003-031910
26. Peltoketo H, Rivero-Muller A, Ahtiainen P, Poutanen M, Huhtaniemi I. Consequences of genetic manipulations of gonadotrophins and gonadotrophin receptors in mice. Ann Endocrinol. (2010) 71:170–6. doi: 10.1016/j.ando.2010.02.022
27. Ghinea N. Vascular endothelial FSH receptor, a target of interest for cancer therapy. Endocrinology (2018) 159:3268–74. doi: 10.1210/en.2018-00466
28. Papadimitriou K, Kountourakis P, Kottorou AE, Antonacopoulou AG, Rolfo C, Peeters M, et al. Follicle-stimulating hormone receptor (FSHR): a promising tool in oncology? Mol Diagn Ther. (2016) 20:523–30. doi: 10.1007/s40291-016-0218-z
29. Valdelievre C, Sonigo C, Comtet M, Simon C, Eskenazi S, Grynberg M. [Impact of gonadotropins in women suffering from cancer]. Bull Cancer (2016) 103:282–8. doi: 10.1016/j.bulcan.2016.01.004
30. Allan CM, Kalak R, Dunstan CR, McTavish KJ, Zhou H, Handelsman DJ, et al. Follicle-stimulating hormone increases bone mass in female mice. Proc Natl Acad Sci USA. (2010) 107:22629–34. doi: 10.1073/pnas.1012141108
31. Macchia E, Simoncini T, Raffaelli V, Lombardi M, Iannelli A, Martino E. A functioning FSH-secreting pituitary macroadenoma causing an ovarian hyperstimulation syndrome with multiple cysts resected and relapsed after leuprolide in a reproductive-aged woman. Gynecol Endocrinol. (2012) 28:56–9. doi: 10.3109/09513590.2011.588758
32. Kumar TR, Fairchild-Huntress V, Low MJ. Gonadotrope-specific expression of the human follicle-stimulating hormone beta-subunit gene in pituitaries of transgenic mice. Mol Endocrinol. (1992) 6:81–90.
33. Kumar TR, Low MJ. Gonadal steroid hormone regulation of human and mouse follicle stimulating hormone beta-subunit gene expression in vivo. Mol Endocrinol. (1993) 7:898–906.
34. Kumar TR, Low MJ. Hormonal regulation of human follicle-stimulating hormone-beta subunit gene expression: GnRH stimulation and GnRH-independent androgen inhibition. Neuroendocrinology (1995) 61:628–37. doi: 10.1159/000126889
35. Kumar TR, Schuff KG, Nusser KD, Low MJ. Gonadotroph-specific expression of the human follicle stimulating hormone β gene in transgenic mice. Mol Cell Endocrinol. (2006) 247:103–15. doi: 10.1016/j.mce.2005.12.006
36. Kumar TR, Low MJ, Matzuk MM. Genetic rescue of follicle-stimulating hormone beta-deficient mice. Endocrinology (1998) 139:3289–95. doi: 10.1210/endo.139.7.6111
37. Wang H, Larson M, Jablonka-Shariff A, Pearl CA, Miller WL, Conn PM, et al. Redirecting intracellular trafficking and the secretion pattern of FSH dramatically enhances ovarian function in mice. Proc Natl Acad Sci USA. (2014) 111:5735–40. doi: 10.1073/pnas.1321404111
38. Wang H, Butnev V, Bous GR, Kumar TR. A human FSHB transgene encoding the double N-glycosylation mutant (Asn 7 D Asn 24 D) FSH b subunit fails to rescue Fshb null mice. Mol Cell Endocrinol. (2016) 426:113–24. doi: 10.1016/j.mce.2016.02.015
39. Allan CM, Haywood M, Swaraj S, Spaliviero J, Koch A, Jimenez M, et al. A novel transgenic model to characterize the specific effects of follicle-stimulating hormone on gonadal physiology in the absence of luteinizing hormone actions. Endocrinology (2001) 142:2213–20. doi: 10.1210/endo.142.6.8092
40. Allan CM, Wang Y, Jimenez M, Marshan B, Spaliviero J, Illingworth P, et al. Follicle-stimulating hormone increases primordial follicle reserve in mature female hypogonadal mice. J Endocrinol. (2006) 188:549–57. doi: 10.1677/joe.1.06614
41. McTavish KJ, Jimenez M, Walters KA, Spaliviero J, Groome NP, Themmen AP, et al. Rising follicle-stimulating hormone levels with age accelerate female reproductive failure. Endocrinology (2007) 148:4432–39. doi: 10.1210/en.2007-0046
42. Greenberg NM, Anderson JW, Hsueh AJ, Nishimori K, Reeves JJ, deAvila DM, et al. Expression of biologically active heterodimeric bovine follicle-stimulating hormone in milk of transgenic mice. Proc Natl Acad Sci USA. (1991) 88:8327–31. doi: 10.1073/pnas.88.19.8327
43. Kim MO, Kim SH, Lee SR, Shin MJ, Min KS, Lee DB, et al. Ectopic expression of tethered human follicle-stimulating hormone (hFSH) gene in transgenic mice. Transgenic Res. (2007) 16:65–75. doi: 10.1007/s11248-006-9031-5
44. Xu P, Li Q, Jiang K, Yang Q, Bi M, Jiang C, et al. BAC mediated transgenic large white boars with FSHalpha/beta genes from Chinese erhualian pigs. Transgenic Res. (2016) 25:693–709. doi: 10.1007/s11248-016-9963-3
45. Bi M, Tong J, Chang F, Wang J, Wei H, Dai Y, et al. Pituitary-specific overexpression of porcine follicle-stimulating hormone leads to improvement of female fecundity in BAC transgenic mice. PLoS ONE (2012) 7:e42335. doi: 10.1371/journal.pone.0042335
46. Jiang K, Xu P, Li W, Yang Q, Li L, Qiao C, et al. The increased expression of follicle-stimulating hormone leads to a decrease of fecundity in transgenic large white female pigs. Transgenic Res. (2017) 26:515–27. doi: 10.1007/s11248-017-0026-1
47. Matzuk MM, Finegold MJ, Su JG, Hsueh AJ, Bradley A. Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature (1992) 360:313–9. doi: 10.1038/360313a0
48. Haywood M, Tymchenko N, Spaliviero J, Koch A, Jimenez M, Gromoll J, et al. An activated human follicle-stimulating hormone (FSH) receptor stimulates FSH-like activity in gonadotropin-deficient transgenic mice. Mol Endocrinol. (2002) 16:2582–91. doi: 10.1210/me.2002-0032
49. Allan CM, Lim P, Robson M, Spaliviero J, Handelsman DJ. Transgenic mutant D567G but not wild-type human FSH receptor overexpression provides FSH-independent and promiscuous glycoprotein hormone Sertoli cell signaling. Am J Physiol Endocrinol Metabol. (2009) 296:E1022–8. doi: 10.1152/ajpendo.90941.2008
50. Peltoketo H, Strauss L, Karjalainen R, Zhang M, Stamp GW, Segaloff DL, et al. Female mice expressing constitutively active mutants of FSH receptor present with a phenotype of premature follicle depletion and estrogen excess. Endocrinology (2010) 151:1872–83. doi: 10.1210/en.2009-0966
51. Oduwole OO, Peltoketo H, Poliandri A, Vengadabady L, Chrusciel M, Doroszko M, et al. Constitutively active follicle-stimulating hormone receptor enables androgen-independent spermatogenesis. J Clin Invest. (2018) 128:1787–92. doi: 10.1172/JCI96794
52. Mason AJ, Hayflick JS, Zoeller RT, Young WS, Phillips HS, Nikolics K, et al. A deletion truncating the gonadotropin-releasing hormone gene is responsible for hypogonadism in the hpg mouse. Science (1986) 234:1366–71. doi: 10.1126/science.3024317
53. Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. (1997) 15:201–4. doi: 10.1038/ng0297-201
54. Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, et al. FSH directly regulates bone mass. Cell (2006) 125:247–60. doi: 10.1016/j.cell.2006.01.051
55. Kumar TR. Extragonadal actions of FSH: a critical need for novel genetic models. Endocrinology (2018) 159:2–8. doi: 10.1210/en.2017-03118
56. Stamatiades GA, Carroll RS, Kaiser UB. GnRH-A key regulator of FSH. Endocrinology (2019) 160:57–67. doi: 10.1210/en.2018-00889
57. McNeilly AS, Crawford JL, Taragnat C, Nicol L, McNeilly JR. The differential secretion of FSH and LH: regulation through genes, feedback and packaging. Reprod Suppl. (2003) 61:463–76.
58. Jablonka-Shariff A, Boime I. Secretory trafficking signal encoded in the carboxyl-terminal region of the CGbeta-subunit. Mol Endocrinol. (2009) 23:316–23. doi: 10.1210/me.2008-0351
59. Jablonka-Shariff A, Garcia-Campayo V, Boime I. Evolution of lutropin to chorionic gonadotropin generates a specific routing signal for apical release in vivo. J Biol Chem. (2002) 277:879–82. doi: 10.1074/jbc.C100402200
60. Bousfield GR, May JV, Davis JS, Dias JA, Kumar TR. In Vivo and in vitro impact of carbohydrate variation on human follicle-stimulating hormone function. Front Endocrinol. (2018) 9:216. doi: 10.3389/fendo.2018.00216
61. Kumar TR. Fshb knockout mouse model, two decades later and into the future. Endocrinology (2018) 159:1941–9. doi: 10.1210/en.2018-00072
62. Kumar TR. Mouse Models for the Study of Synthesis, Secretion, and Action of Pituitary Gonadotropins. New York, NY: Elsevier Inc. (2016). doi: 10.1016/bs.pmbts.2016.08.006
63. Wang H, Hastings R, Miller WL, Kumar TR. Fshb -i Cre mice are ef fi cient and speci fi c Cre deleters for the gonadotrope lineage. Mol Cell Endocrinol. (2016) 419:124–38. doi: 10.1016/j.mce.2015.10.006
64. Jeong JH. Inducible mouse models for cancer drug target validation. J Cancer Prev. (2016) 21:243–8. doi: 10.15430/JCP.2016.21.4.243
65. Morozov A. Conditional gene expression and targeting in neuroscience research. Curr Protoc Neurosci. (2008) 4:4.31. doi: 10.1002/0471142301.ns0431s44
Keywords: pituitary, follicle-stimulating hormone, transgenic mice, testis, ovary
Citation: McDonald R, Sadler C and Kumar TR (2019) Gain–of–Function Genetic Models to Study FSH Action. Front. Endocrinol. 10:28. doi: 10.3389/fendo.2019.00028
Received: 07 September 2018; Accepted: 15 January 2019;
Published: 07 February 2019.
Edited by:
Ilpo Huhtaniemi, Imperial College London, United KingdomReviewed by:
Prema Narayan, Southern Illinois University Carbondale, United StatesAlfredo Ulloa-Aguirre, National Autonomous University of Mexico, Mexico
Hellevi Peltoketo, University of Oulu, Finland
Copyright © 2019 McDonald, Sadler and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: T. Rajendra Kumar, cmFqLmt1bWFyQHVjZGVudmVyLmVkdQ==