 Denise Kreuch1
Denise Kreuch1 Damien J. Keating2,3
Damien J. Keating2,3 Tongzhi Wu1
Tongzhi Wu1 Michael Horowitz1
Michael Horowitz1 Christopher K. Rayner1
Christopher K. Rayner1 Richard L. Young1,3*
Richard L. Young1,3*- 1Faculty of Health and Medical Sciences & Centre of Research Excellence in Translating Nutritional Science to Good Health, Adelaide Medical School, The University of Adelaide, Adelaide, SA, Australia
- 2College of Medicine and Public Health, Flinders University, Bedford Park, SA, Australia
- 3Nutrition and Metabolism, South Australian Health and Medical Research Institute, Adelaide, SA, Australia
Sensing nutrients within the gastrointestinal tract engages the enteroendocrine cell system to signal within the mucosa, to intrinsic and extrinsic nerve pathways, and the circulation. This signaling provides powerful feedback from the intestine to slow the rate of gastric emptying, limit postprandial glycemic excursions, and induce satiation. This review focuses on the intestinal sensing of sweet stimuli (including low-calorie sweeteners), which engage similar G-protein-coupled receptors (GPCRs) to the sweet taste receptors (STRs) of the tongue. It explores the enteroendocrine cell signals deployed upon STR activation that act within and outside the gastrointestinal tract, with a focus on the role of this distinctive pathway in regulating glucose transport function via absorptive enterocytes, and the associated impact on postprandial glycemic responses in animals and humans. The emerging role of diet, including low-calorie sweeteners, in modulating the composition of the gut microbiome and how this may impact glycemic responses of the host, is also discussed, as is recent evidence of a causal role of diet-induced dysbiosis in influencing the gut-brain axis to alter gastric emptying and insulin release. Full knowledge of intestinal STR signaling in humans, and its capacity to engage host and/or microbiome mechanisms that modify glycemic control, holds the potential for improved prevention and management of type 2 diabetes.
Introduction
It is now widely recognized that the gastrointestinal tract is a major determinant of metabolic homeostasis, and the largest endocrine organ of the body. This is due to the diversity and wide signaling repertoire of the gastrointestinal enteroendocrine cells (EECs) which can, collectively, release over 30 different peptide hormones and neurotransmitters (1). To subserve this signaling function gastrointestinal EECs are configured either as “open” cells—possessing long, slim, finger-like extensions on their apical side to sense the luminal milieu and, in turn, release signaling molecules, or as “closed” cells which do not access the lumen, but can respond indirectly to luminal content (2). EEC have classically been sub-divided according to their hormone or transmitter content, and regional location within the gastrointestinal tract. However, the substantial overlap in transcriptional expression and subcellular stores that has recently been identified now supports a more heterogeneous EEC population (3, 4).
EECs Respond to Intestinal Carbohydrates
Exposure to luminal glucose generates signals that have a profound influence on intestinal motor and absorptive function. These signals include release of the gut peptides glucose-dependent insulinotropic polypeptide (GIP) from K-cells located in the proximal intestine and glucagon-like peptide-1 and 2 (GLP-1, GLP-2) from L-cells located in more distal regions of the intestine (5, 6), and release of the bioamine serotonin (5-HT) from enterochromaffin (EC) cells located throughout the gastrointestinal tract (7–9). GLP-1 and GIP, the “íncretin” peptide hormones, are degraded rapidly upon release by the ubiquitous enzyme dipeptidyl peptidase-IV and neutral endopeptidase, with <50% of secreted hormone entering the circulation. However, they powerfully augment glucose-dependent insulin release in response to an enteral glucose load (in comparison to an intravenous isoglycemic glucose load) (10, 11). GLP-1 and 5-HT also activate GLP-1 and 5-HT3 receptors on intestinal vagus nerve endings as key signals in the “gut-brain axis,” which, in turn, triggers vagal reflexes to slow the subsequent emptying of carbohydrate from the stomach, and induce satiation (12, 13). Accordingly, the release of GLP-1, GIP, and 5-HT is crucial to the regulation of postprandial glycemia. In contrast, GLP-2, which is co-released with GLP-1, is intestinotrophic and a potent signal to upregulate the expression and function of the primary intestinal glucose transporter, sodium-glucose cotransporter-1 (SGLT-1) (14).
Sweet Taste Machinery
Lingual Sweet Taste
All known sweet tastants, including hexose sugars, D-amino acids, sweet proteins (such as monellin and thaumatin), and low-calorie sweeteners (LCS) are sensed by a single broadly-tuned sweet taste receptor (STR), comprised of a heterodimer of class C, G-protein coupled receptors, T1R2, and T1R3 (15). In lingual sweet taste cells, where sweet taste transduction has been most fully characterized, the interaction of sweet tastants with STRs initiates dissociation of the G-protein, gustducin, into Gα and Gβγ subunits and activation of phospholipase C β2 (PLCβ2); intracellular Ca2+ is then released from inositol 1,4,5-trisphosphate-sensitive (IP3) stores, leading to opening of the melastatin type-5 transient receptor potential cation channel (TRPM5) to sodium influx [for review, see (16)]. Increases in intracellular Na+ and Ca2+ then depolarize the basolateral membrane and, via 5-HT and ATP-dependent pathways, activate intermediary taste cells and chorda tympani and glossopharyngeal nerves that convey taste information centrally to the insular cortex [for review, see (17)] (18–20).
Intestinal Sweet Taste
STRs are well-described on subsets of EEC in the proximal small intestine, with evidence of STR-equipped K-cells, L-cells, and EC cells in humans (21, 22). STRs are also documented widely in metabolic tissues that sense and respond to carbohydrates, such as pancreatic β-cells, hepatocytes, adipocytes, and hypothalamic neurons [for review, see (23, 24)]. Expression of intestinal STR, like many GPCR, is of low magnitude, and optimally detected with high sensitivity SYBR-based PCR approaches rather than Taq-based PCR. Evidence in rodents, and in human cells and tissues, provides strong support that intestinal STRs function as upstream sensors linked to the release of GLP-1 from L-cells, and 5-HT from EC cells, and genetic deletion of T1R3, or pharmacological blockade of STRs with lactisole, decreases glucose and LCS-evoked GLP-1 and 5-HT release (21, 25–27). This is also true for GLP-2 release, which is STR-dependent in rodents (28, 29) and inhibited by the murine STR inhibitor, gurmarin (30).
Clinical studies have also reported acute effects of LCS to augment GLP-1 release in the presence of glucose and have shown a dose-dependent effect of lactisole to attenuate glucose-induced GLP-1 release in healthy subjects (31–34). Despite this, the balance of clinical evidence indicates that, at least in acute settings, LCS do not contribute substantially to the circulating pool of GLP-1 in humans (35–37).
Interplay Between STRs and SGLT-1 Can Regulate Glycemic Responses
Enterocytes account for around 90% of all intestinal epithelial cells and are polarized cells consisting of apical and basolateral membrane domains (38). These cells transport nutrients from the gut lumen to the circulation, and for glucose, apical SGLT-1 is the primary intestinal glucose transporter in both humans and animals. SGLT-1 is expressed primarily in the small intestine with highest density in the jejunum followed by the duodenum and then ileum (39, 40). SGLT-1 enables glucose absorption by co-transporting sodium along the electrochemical gradient established by the basolateral sodium-potassium ATPase (38, 41). Glucose then enters the systemic circulation via the facilitative monosaccharide transporter, GLUT2, located on the basolateral membrane of enterocytes; GLUT2 is bidirectional and capable of moving glucose in or out of enterocytes depending on glucose concentration gradients (38).
Importantly, transport of the monosaccharide substrates of SGLT-1 (e.g., glucose and galactose) triggers incretin hormone secretion (20), an action attenuated when SGLT-1 is pharmacologically inhibited with the competitive antagonist phlorizin, or absent through genetic deletion in rodents (42, 43). Our group provided the first evidence that SGLT-1 substrates, even if not metabolized (such as the glucose analog 3-O-methyl-glucose, 3-OMG), have the capacity to stimulate GLP-1 and GIP secretion in humans (44). We have also established that SGLT-1-based transport is critical for ex vivo release of GLP-1 in human ileum, while blocking SGLT-1 with phlorizin or replacing extracellular Na+ with N-methyl-D-glucamine abolishes this response (26).
In animals, a wide range of sweet stimuli are capable of upregulating SGLT-1 expression and function, including LCS (45–48), indicating that SGLT-1 activity is modulated by an upstream and broadly tuned sweet taste sensor. Accordingly, STRs may have the capacity to stimulate gut hormone release both directly, and indirectly by augmenting SGLT-1 function. The latter is evidenced in mice lacking T1R3 or α-gustducin, where SGLT-1 expression and function are not increased in response to dietary glucose or LCS supplementation as occurs in control mice (42). Moreover, the 3-fold increase in jejunal SGLT-1 expression following 4 days of sucralose gavage (100 mg, twice-daily) in control mice was absent in our mice lacking both T1R2 and T1R3 subunits of the STR (Marino Z, Young RL; Figure 1). Together, these experiments attest to the importance of intestinal STRs in regulating SGLT-1 function in mice, and support the notion that LCS can potentiate postprandial glycemic excursions via STR-dependent gains in SGLT-1 function and glucose absorption, in response to habitual consumption of sugars or LCS (Figure 2).
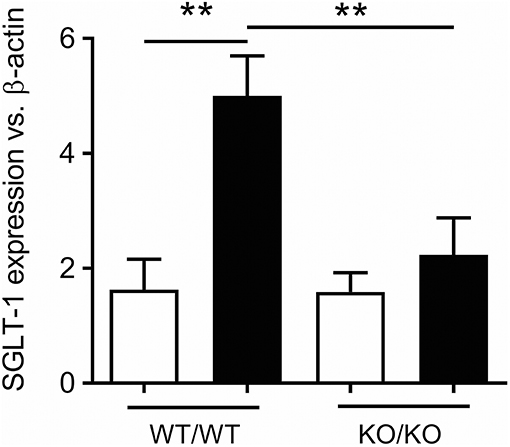
Figure 1. STR-dependence of SGLT-1 expression in mice. Increased jejunal expression of SGLT-1 mRNA in 10 week-old control (WT/WT) mice gavaged for 4 days with sucralose (black bars) compared to water (white bars), and to mice homozygous for both Tas1r2 and Tas1r3 genes (KO/KO). Breeding pairs of mice homozygous for the Tas1r2 or Tas1r3 gene (129X1/SvJ mice backcrossed for at least 3 generations with C57BL/6 mice) were provided by Prof Charles Zuker (University of California, San Diego, USA). Mice homogenous for each gene were then paired to produce mice heterozygous for Tas1r2 and Tas1r3. These mice, in turn, were paired to generate mice heterozygous, homozygous, and wild-type for both genes. From these mice, double homozygous (KO/KO) and wild-type littermate controls (WT/WT) were the subject of gavage experiments. Ten-week old male mice (N = 5 per group) maintained under standard housing and diet conditions in the SA Pathology Animal Care Facility were gavaged twice daily with 100 mg sucralose (Redox Chemicals, Minto, NSW Australia) in 200 μL water, or 200 μL water, at 0800 and 1800 over 4 days. These mice were fasted overnight then humanely killed at 0800, total RNA extracted from the jejunal mucosa, and real-time RT-PCR performed using primer assays for SGLT-1 (QT00112679) and β-actin (QT01136772, Qiagen, Sydney, NSW Australia) relative to expression of β-actin, as described (49); SGLT-1 expression was compared between groups and gavage regime by analysis of variance (ANOVA), adjusted for multiple comparisons by Holm-Sidak's correction (GraphPad Prism 7.02, San Diego, CA, USA). This experiment was approved and performed in accordance with guidelines of the Animal Ethics Committees of The University of Adelaide and SA Pathology (Adelaide, Australia). Data is shown as Mean ± SEM; **P < 0.01. We thank Prof Charles Zuker for generously supplying the homozygous Tas1r2 and Tas1r3 mice.
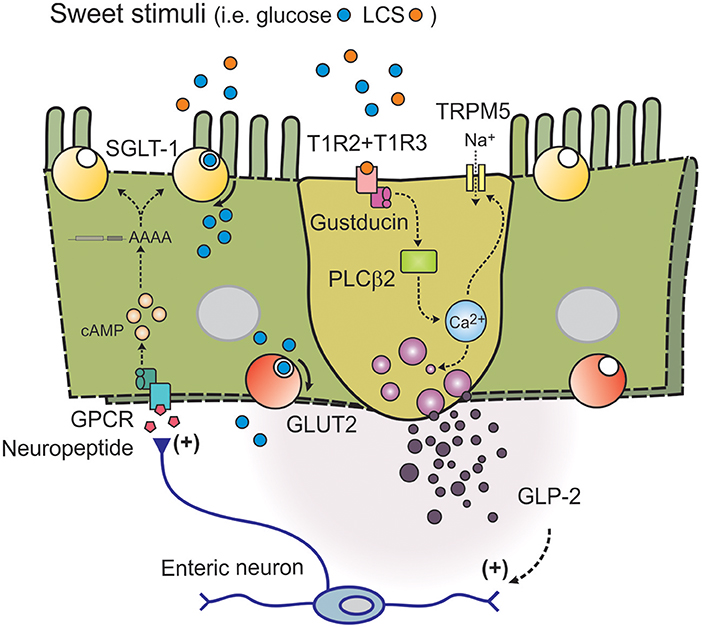
Figure 2. Model of intestinal sweet taste sensing and signaling effectors. Sweet stimuli, including LCS, bind to STR comprised of a heterodimer of G-protein coupled receptors T1R2 and T1R3. Upon receptor binding an intracellular signaling cascade is activated, initiated by dissociation of G-protein gustducin into Gα and Gβγ subunits and activation of phospholipase C β2 (PLCβ2); intracellular Ca2+ is then released from inositol 1,4,5-trisphosphate-sensitive stores, leading to opening of the melastatin type-5 transient receptor potential cation channel (TRPM5) to sodium influx. Increases in intracellular Na+ and Ca2+ then depolarize the basolateral membrane, to facilitate release of peptide hormones such as GLP-2. GLP-2 may then trigger an enteric neuron pathway to release an unknown neuropeptide at nearby absorptive enterocytes leading to adenylate cyclase-dependent stablilization of the 3' end of SGLT-1 mRNA (to increase half-life), and SGLT-1 translation and insertion into the apical brush border membrane.
There is evidence that enteric neurons link glucose sensing in EEC to glucose transport function in enterocytes (50). Studies in rodents have shown that intestinal areas adjacent to regions exposed to LCS have increased SGLT-1 expression (46). This communication between STR-equipped L cells and SGLT-1-bearing absorptive enterocytes is likely to involve gut hormone intermediaries, such as GLP-1 and/or GLP-2. Indeed, GLP-2 receptors are present on enteric neurons in guinea pig ileum, mouse jejunum, mouse and pig intestine (20, 51, 52) and absorptive enterocyte progenitors in mouse jejunum respond to GLP-2 in an enteric neuron-dependent manner (52). GLP-2 is also capable of upregulating SGLT-1 expression in vivo (28), and STR-dependent release of both GLP-1 and GLP-2 is detected at higher concentrations in the portal and lymphatic circulation than the systemic circulation in rodents (28, 53). This indicates that local release of either mediator in response to sweet stimuli, including LCS, may be sufficient to increase SGLT-1 function. It may also, in part, explain the equivocal nature of human data on LCS-evoked gut hormone release, as paracrine signaling in the mucosa could occur in the absence of a substantial contribution to circulating hormone levels. To this end, we provided the first evidence that LCS evoke ex vivo GLP-1 release from human ileal tissue (26). However, the precise signal transduction pathways utilized by LCS to trigger gut hormone release in human mucosa remain to be identified.
An increase in SGLT-1 protein in the apical brush border of enterocytes occurs in a cyclic AMP (cAMP)-dependent manner in response to transduction of basolateral signals (54, 55), and secondary to an increase in SGLT-1 transcription (56) and stabilization (increased half-life) of the 3′-untranslated region of the SGLT-1 transcript (57, 58). This facilitates an increase in apical SGLT-1 protein translation and insertion in response to gut hormone signaling. Jugular vein infusion of GLP-2 increases the abundance of SGLT-1 protein and rate of SGLT-1-dependent glucose transport in the apical membrane of jejunal enterocytes in rats, a response abolished when protein translocation is inhibited with brefeldin (29, 59). This highlights the importance of GLP-2 in the regulation of SGLT-1 function at the apical brush border membrane.
While enteric neurons express receptors for other gut hormones, including GIP, GIP is unlikely to be responsible for glucose or LCS effects on SGLT-1 (20, 60). GIP receptor knockout and wild type mice show similar increases in jejunal SGLT-1 expression on a high carbohydrate diet, compared to mice on a low carbohydrate diet (20). Irrespective of which mucosal mediator is a trigger upon intestinal STR activation, the interplay between these broadly-tuned receptors and SGLT-1 is critical for glucose absorption and represents a major mechanism regulating overall glycemic control.
Type 2 Diabetes is Associated With STR Dysregulation
Globally, over 400 million people are living with diabetes, projected to rise to over 600 million by 2040 (61). Effective control of glycemia, as assessed by glycated hemoglobin (HbA1c) <6.5–7.0% (48–53 mmol/mol), is important to minimize the risk of the development and progression of microvascular complications (i.e., eye, kidney, and nerve damage), and to a lesser extent, macrovascular complications. In the majority of patients with type 2 diabetes, who are relatively well-controlled, postprandial glycaemic excursions predominate over fasting blood glucose levels in contributing to HbA1c (62), and are determined by meal composition, the rate of gastric emptying, hepatic and peripheral glucose metabolism, intestinal glucose absorption, and insulin secretion and resistance (63). Meal-related secretion of insulin is augmented through the insulinotropic actions of the incretin hormones GIP and GLP-1 to reduce postprandial glycemic excursions in health (64, 65); in type 2 diabetes, a markedly attenuated insulinotropic action of GIP (66) and, in some cases, attenuated secretion of GLP-1 (67), contribute to an impairment of postprandial insulin secretion, so that the latter is insufficient to maintain euglycaemia. Furthermore, gut-derived 5-HT can also modulate glucose and energy homeostasis (68–70), and is augmented in patients with type 2 diabetes (71) and the obese (9).
The recognition that the gut, and EEC signals, are major determinants of glycemic control is attested to by the successful deployment of incretin-based therapies for type 2 diabetes. These include mimetics of GLP-1, GLP-1/GIP dual receptor agonists, and inhibitors of dipeptidyl peptidase-IV, which inactivates endogenous GLP-1 (72). These pharmaceutical compounds have improved clinical management of type 2 diabetes substantially, but their use is compromised by cost, compliance with administration, adverse gastrointestinal effects, or suboptimal efficacy in some patients.
While experiments in animal models and patients with type 2 diabetes have shown a gain in function of SGLT-1 and corresponding increase in the rate of intestinal glucose absorption (73, 74), the targeting of intestinal glucose absorption has received comparatively little attention. Indeed, it is likely that a proportion of the clinical benefits of the anti-diabetic gliflozin-class agents (SGLT-2 inhibitors) are due to actions at intestinal SGLT-1. This is particularly true for first-in-class examples, such as the dual SGLT-1/SGLT-2 inhibitor sotagliflozin, which has lower selectivity for SGLT-2 and acts beyond inhibition of renal glucose reabsorption by SGLT-2 to induce partial inhibition of intestinal SGLT-1, leading to augmented GLP-1 and insulin secretion, and a reduction in postprandial glucose excursions (75).
To assess whether regulation of intestinal STR was disrupted in patients with type 2 diabetes, and had an unfavorable impact on glucose absorption and postprandial hyperglycemia, we compared intestinal STR expression in individuals with and without type 2 diabetes. We first established that STRs were expressed at similar levels in the duodenum in both groups when sampled at euglycemia (49). However, we found that T1R2 expression was decreased following enteral glucose exposure under hyperglycemic conditions in non-diabetic subjects, but remained elevated in patients with type 2 diabetes, where it was linked to an increase in glucose absorption (assessed by serum levels of 3-OMG which had been co-administered with the glucose load) (22). These findings support the notion that intestinal STR dysregulation in type 2 diabetes can exacerbate postprandial glycemic excursions. Furthermore, given that patients with type 2 diabetes are 3-fold more likely to consume beverages sweetened with LCS than healthy individuals (76), it is possible that high dietary LCS consumption contributes to, rather than alleviates, postprandial glycemic dysregulation.
Low-Calorie Sweeteners and Glycemic Control
Sugar-sweetened beverages contain high levels of sucrose or high fructose corn syrup (77) and represent a major source of added sugars in western diets. They account for around 16% of daily caloric intake of adults in the United States (78) and 11% in Canada and Australia (79), a level that exceeds the World Health Organization recommendation that added sugar consumption should be limited to 10% of daily caloric intake (80). These sugars are rapidly absorbed by the small intestine to increase glycemic load, which, when associated with increased peripheral insulin resistance, increases the risk of developing type 2 diabetes (81).
The outcomes of epidemiological studies indicate that high and habitual consumption of sugar-sweetened beverages is associated with an increased risk of developing type 2 diabetes, independent of total energy intake or body mass (77, 82). While these findings do not establish causality (83, 84), the adverse health outcomes linked to high sugar consumption have led to changes in global health policy to limit such intake, with several countries now implementing a sugar tax (80, 85). Not surprisingly, beverages sweetened with LCS have become a popular alternative.
Diet beverages contain a single LCS, or more frequently, LCS combinations, in place of sugars (86), with specific LCS commonly identified by their European Food Safety Authority E-number, i.e., aspartame (E951), sucralose (E955), and acesulfame-K (E950). LCS differ substantially in their oral bioavailability and, therein, exposure to intestinal regions and their microbiota. For example, aspartame is completely hydrolyzed in the proximal intestine to methanol and constituent amino acids, aspartate and phenylalanine, and has no effective oral bioavailability or exposure to the distal intestine and its microbiota. Sucralose has low oral bioavailability (around 15%), but full exposure to the intestine and microbiota due to excretion in largely unchanged form in feces; minor absorbed sucralose and glucoronidation end-products undergo renal excretion. Finally, acesulfame-K has high oral bioavailability (90–100%) due to rapid absorption in the proximal intestine and has limited exposure to the distal intestine and its microbiota; acesulfame-K is cleared via renal excretion in largely unchanged form [for reviews, see (87–89)]. These distinct properties should be considered in interpreting effects of LCS both within, and outside, the gastrointestinal tract.
LCS are 200 to 13,000 times sweeter than sucrose by weight, and were expected to be beneficial in the setting of obesity and type 2 diabetes due to their low calorie content. There is, however, only equivocal evidence of this benefit, with several epidemiological studies indicating little or no benefit, or even an increased risk of weight gain (90–92). Moreover, some epidemiological studies suggest that a high habitual intake of beverages sweetened with LCS is associated with an increased risk of developing type 2 diabetes (93–97). Reverse causality (e.g., people opting for LCS-sweetened beverages in response to weight gain and/or obesity, or subclinical disease including pre-diabetes) is unlikely to fully account for the increased risk, which is evident even after adjusting for differences in body mass and energy intake. Furthermore, two studies have reported an elevated risk of developing type 2 diabetes in normal weight individuals (93, 97).
The outcomes of studies that have prospectively investigated the effects of LCS intake on long-term glycaemic control (assessed by HbA1c) or insulin resistance have been equivocal, and several failed to adjust for differences in sugar intake (76, 98–102). Despite this, high habitual patterns of LCS consumption have been reported to increase HbA1c levels in healthy adults, independent of body mass (101), while daily LCS consumption has been dose-dependently associated with HbA1c increases in type 2 diabetes (76). A negative impact of LCS on acute glycemic control has also been shown in obese individuals, where a sucralose preload consumed in advance of an oral glucose tolerance test augmented blood glucose levels over the following 5 h substantially, when compared to water or no preload (103).
Collectively, the potential for LCS to impair glycemic control remains uncertain, in large part due to the small number of prospective clinical studies (104, 105). Proposed mechanisms linking LCS to an increased risk of developing type 2 diabetes in humans include a reduced fidelity of central responses to nutritive stimuli, effects on gut microbiota, and an effect of LCS to augment glucose absorption.
We recently reported early findings of a randomized placebo-controlled clinical study investigating the effect of diet supplementation with combined LCS (sucralose 276 mg, acesulfame-K 156 mg in capsules; equivalent to 1.5 L diet beverage/day) over 2 weeks on glycemic responses to enteral glucose. We observed a clinically significant effect of LCS to increase the rate of glucose absorption and augment blood glucose responses to enteral glucose in healthy subjects consuming LCS, relative to placebo. Moreover, glucose-evoked GLP-1 and GLP-2 release was decreased in LCS-consuming participants, which may relate to the more rapid proximal absorption of glucose limiting the exposure of more distally located L-cells (106). These findings indicate a negative impact of habitual high LCS intake on glucose absorption and acute glycaemic control in health, and add support for the concept that high habitual intake of LCS may increase the magnitude of postprandial glycemic excursions.
LCS and the Gut Microbiome
The gut microbiome comprises the diverse range of bacteria, yeasts, and other microorganisms which exist in a largely symbiotic relationship with the host (107). These prevent potentially harmful microorganisms from colonizing the gut by competing for energy resources (108). Use of these resources liberates nutrients which would be otherwise inaccessible to the host, i.e., microbial conversion of indigestible polysaccharides to short chain fatty acids (SCFAs) such as acetate, propionate, and butyrate, which act as substrates for cellular metabolism, gluconeogenesis and lipogenesis. Moreover, SCFAs play a crucial role in satiety signaling, and modulate appetite directly and indirectly via leptin synthesis in adipose tissue (109). SCFAs also have a beneficial impact on glycemia, with propionate shown to improve insulin sensitivity, and butyrate to prevent or improve insulin resistance in mice fed a high fat diet (110–112). Microbial-derived signals from the gut, therefore, have the potential to influence glycemic control substantially.
The composition of the gut microbiome of individuals with type 2 diabetes differs from that of non-diabetic individuals, in its relative and BMI-independent decrease in abundance of species from the Clostridium phylum (113, 114). These species are negatively correlated with markers of poor glycemic control such as fasting glucose, HbA1c and insulin, but positively correlated with the insulin sensitizing hormone adiponectin (113). Alterations in the gut microbiome of individuals with type 2 diabetes are also associated with changes in functional microbial genes, with a specific enrichment of pathways for starch, glucose, fructose, and mannose metabolism, which increases the potential for energy harvest and metabolism (113). These changes are causally related to the development of insulin insensitivity and resistance, as allogenic transplantation of intestinal microbiota from lean donors to recipients with the metabolic syndrome improved insulin sensitivity (115). This highlights the importance of the gut microbiome composition with respect to the development of metabolic disorders, including type 2 diabetes.
Exposure to LCS has been shown to drive glucose intolerance in mice via a LCS-dependent shift in composition of the gut microbiome (“dysbiosis”). Transplantation of fecal microbiota from donor mice supplemented chronically with LCS (saccharin) to germ-free recipient mice resulted in glucose intolerance after 6 days. Changes in abundance of more than 40 operational taxonomic units were demonstrated in the recipient mice, along with an upregulation of microbial carbohydrate-related metabolic pathways, and an increase in fecal SCFA levels (101). This increase in SCFAs was speculated to represent increased microbial energy harvest, but may equally represent the outcome of differences in intestinal transit time or absorption (116, 117). It is also been unclear whether fecal bacterial samples accurately represent the microbiome of the proximal gut (118). Indeed, Daly et al. showed that supplementation with SUCRAM (neohesperidin dihydrochalcone and saccharin) over 2 weeks in weaned piglets increased the abundance of Lactobacillaceae in cecal, but not fecal, samples, while cecal SCFA levels were comparable in the LCS and control diet groups (117). These findings underscore the importance of testing regional (or mucosa-associated) bacteria in the gut, and of establishing causal mechanisms as opposed to microbial followers of changes in host metabolism.
Causal mechanisms linking dysbiosis to impaired GLP-1 signaling in the gut-brain axis were recently investigated in mouse models of diet-induced type 2 diabetes. Grasset et al., identified a subset of ileal bacteria in these mice that disrupted GLP-1-dependent nitric oxide production in ileal enteric neurons via an attenuation of GLP-1 receptor expression, and showed that this drove GLP-1 resistance in the regulation of gastric emptying and insulin release (119). A GLP-1 resistant phenotype in germ-free mice was rescued through conventionalization with ileal bacteria from control-fed mice, but not from mice fed the diabetogenic diet, while antibiotic treatment led to GLP-1 resistance in control-fed mice, but improved GLP-1 resistance in diabetogenic diet-fed mice. This study demonstrated that diabetogenic diet-induced gut dysbiosis was causally related to dysglycemia via disruption of GLP-1 signaling in the gut-brain axis, but did not extend to an assessment of specific bacterial populations or products that mediated this effect.
Accordingly, clinical studies are now required to determine whether LCS induce intestinal dysbiosis in humans, whether this is causally related to disruption of the gut-brain axis that controls glycemia, and which microbiome-derived signals effect this change. Such investigation holds the potential to usher in new classes of anti-diabetic therapy which would correct defects in microbiome composition and/or associated signaling pathways that impact glycemic control adversely.
Tasting Sweet Via Non-STR Pathways
Several studies have reported the existence of a lingual and STR-independent sensor tuned to detect the nutritive value of complex carbohydrates. Behavioral studies in rodents have shown that rats prefer consumption of polycose (glucose oligomer) solutions above that of water or solutions of the disaccharides sucrose and maltose, particularly at low concentrations (120, 121). This was further supported by electrophysiology studies of lingual nerve activity, which indicated that rats could distinguish the tastes of polycose and sucrose (122, 123). Importantly, mice lacking one or both STR subunits had limited, or no behavioral or lingual nerve responses to simple sugars, while responses to polycose remained normal (124–127). More recently, behavioral research on human taste detection have added support for a human polycose taste receptor, showing that humans can detect glucose oligomer solutions on an equimolar basis to simple sugars, even when lingual STR were blocked with lactisole and amylase activity inhibited by an α-glucosidase inhibitor (to prevent oral breakdown of glucose oligomers to STR-detectable mono- and disaccharides) (128–130). The latter study also indicated that oligosaccharides of 4 or higher degrees of polymerization (i.e., maltotetraose) were detected by a STR-independent lingual taste pathway in humans. While a polycose receptor is yet to be cloned, future characterization may also reveal its potential as an intestinal nutrient sensor, and whether there are associated consequences for glycemic control in humans.
Conclusion
Although foods and beverages sweetened with LCS have become a popular alternative to their sugar-sweetened counterparts, research relating to their impact on acute and chronic human health has been inappropriately limited, and the outcomes equivocal. However, the outcomes of the hitherto small number of well-conducted studies raises concerns regarding their health impact. Further research is now required to better characterize the EEC biology of intestinal sweet taste signaling in humans, characterize the mechanisms utilized by LCS to impact glycemic control, and identify potential targets capable of modifying STR signaling for clinical benefits (Figure 3). In addition, studies are needed to determine whether patterns of LCS consumption can trigger gut dysbiosis, with consequences for human health as are subsequent metagenomic, metabolomic, and functional investigations of causal mechanisms. These hold the high potential for improved prevention and novel management of type 2 diabetes.
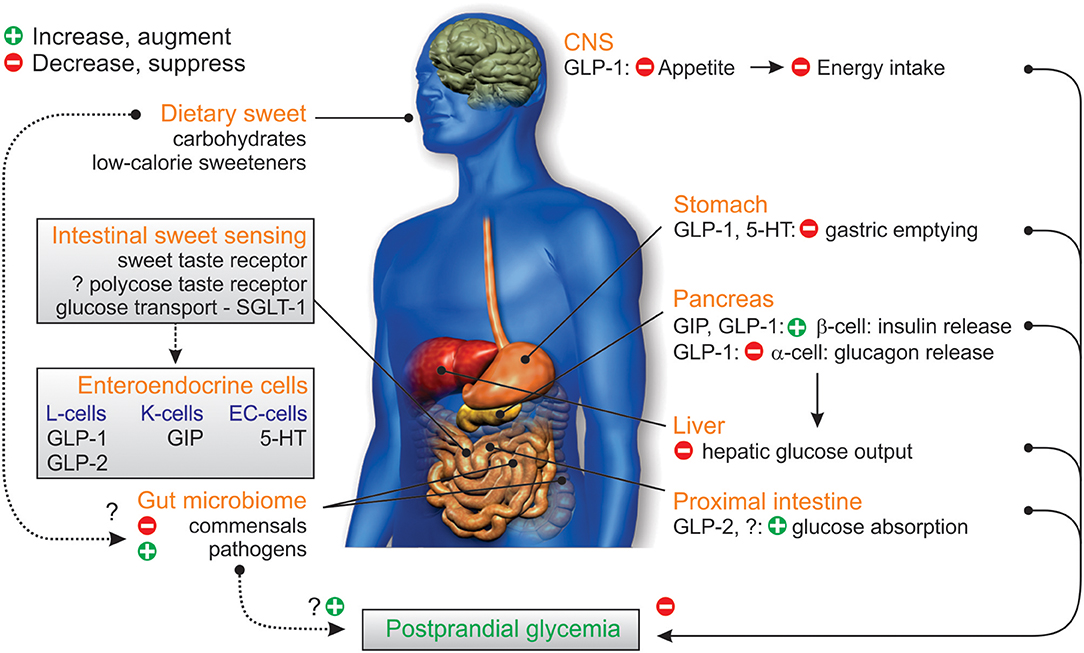
Figure 3. Gastrointestinal factors influencing glycemic control. Dietary sweet stimuli can activate STR in the proximal intestine facilitating the enteroendocrine cell release of the incretin peptides GIP from K-cells and GLP-1 from L-cells, as well as 5-HT from EC-cells; substrates of SGLT-1 (glucose, galactose) also trigger GIP and GLP-2 release. GIP and GLP-1 stimulate glucose-dependent insulin release, to increase glucose disposal; GLP-1 and 5-HT also slow the rate of gastric emptying via vagus nerve signals (not shown) while GLP-1 inhibits pancreatic glucagon release, leading to reduced hepatic glucose output. GLP-2 co-released from L-cells acts to increase intestinal glucose absorption via an increase in the capacity for SGLT-1-based glucose transport. Dietary sweet stimuli can also alter the composition of the gut microbiome in favor of colonization of gut pathogens over fermentative gut commensals, which can affect energy harvest, and disrupt microbiome signaling to the host and glycemic control. Together these influences can disrupt the homeostatic balance between glucose-evoked gut hormone release, glucose absorption, and microbiome composition, leading to dysglycemia which would potentially be harmful in the setting of type 2 diabetes. In addition, complex carbohydrates (oligosaccharides) may contribute to these processes via a yet to be identified polycose taste receptor.
Author Contributions
DK, DJK, TW, MH, CR, and RY were all involved in conception, design, and writing of the manuscript. All authors have approved the publication of this final version of the manuscript.
Funding
The authors' work in this area has been supported by the National Health and Medical Research Council (NHMRC) of Australia (ID: 627127, 1081182), Diabetes Australia Research Trust, The Hospital Research Foundation and the Royal Adelaide Hospital Research Foundation. DJK is supported by an NHMRC Career Development Fellowship, and TW by a Royal Adelaide Hospital Research Foundation Florey Fellowship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sternini C, Anselmi L, Rozengurt E. Enteroendocrine cells: a site of 'taste' in gastrointestinal chemosensing. Curr Opin Endocrinol Diabetes Obes. (2008) 15:73–8. doi: 10.1097/MED.0b013e3282f43a73
2. Rehfeld JF. A centenary of gastrointestinal endocrinology. Horm Metab Res. (2004) 36:735–41. doi: 10.1055/s-2004-826154
3. Gribble FM, Reimann F. Signalling in the gut endocrine axis. Physiol Behav. (2017) 176:183–8. doi: 10.1016/j.physbeh.2017.02.039
4. Fothergill LJ, Callaghan B, Hunne B, Bravo DM, Furness JB. Costorage of enteroendocrine hormones evaluated at the cell and subcellular levels in male mice. Endocrinology (2017) 158:2113–23. doi: 10.1210/en.2017-00243
5. Sjolund K, Sanden G, Hakanson R, Sundler F. Endocrine cells in human intestine: an immunocytochemical study. Gastroenterology (1983) 85:1120–30.
6. Holst JJ. On the physiology of GIP and GLP-1. Horm Metab Res. (2004) 36:747–54. doi: 10.1055/s-2004-826158
7. Zelkas L, Raghupathi R, Lumsden AL, Martin AM, Sun E, Spencer NJ, et al. Serotonin-secreting enteroendocrine cells respond via diverse mechanisms to acute and chronic changes in glucose availability. Nutr Metab. (2015) 12:55. doi: 10.1186/s12986-015-0051-0
8. Martin AM, Lumsden AL, Young RL, Jessup CF, Spencer NJ, Keating DJ. Regional differences in nutrient-induced secretion of gut serotonin. Physiol Rep. (2017) 5:e13199. doi: 10.14814/phy2.13199
9. Young RL, Lumsden AL, Martin AM, Schober G, Pezos N, Thazhath SS, et al. Augmented capacity for peripheral serotonin release in human obesity. Int J Obes. (2018) 42:1880–9. doi: 10.1038/s41366-018-0047-8
10. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
11. Lund A, Bagger JI, Christensen M, Grondahl M, van Hall G, Holst JJ, et al. Higher endogenous glucose production during OGTT vs isoglycemic intravenous glucose infusion. J Clin Endocrinol Metab. (2016) 101:4377–84. doi: 10.1210/jc.2016-1948
12. Imeryuz N, Yegen BC, Bozkurt A, Coskun T, Villanueva-Penacarrillo ML, Ulusoy NB. Glucagon-like peptide-1 inhibits gastric emptying via vagal afferent-mediated central mechanisms. Am J Physiol Gastrointest Liver Physiol. (1997) 273 (4 Pt 1):G920–7. doi: 10.1152/ajpgi.1997.273.4.G920
13. Raybould HE, Glatzle J, Robin C, Meyer JH, Phan T, Wong H, et al. Expression of 5-HT3 receptors by extrinsic duodenal afferents contribute to intestinal inhibition of gastric emptying. Am J Physiol Gastrointest Liver Physiol. (2003) 284:G367–72. doi: 10.1152/ajpgi.00292.2001
14. Marathe CS, Rayner CK, Jones KL, Horowitz M. Glucagon-like peptides 1 and 2 in health and disease: a review. Peptides (2013) 44:75–86. doi: 10.1016/j.peptides.2013.01.014
15. Nelson G, Hoon MA, Chandrashekar J, Zhang Y, Ryba NJ, Zuker CS. Mammalian sweet taste receptors. Cell (2001) 106:381–90. doi: 10.1016/S0092-8674(01)00451-2
16. Thompson MD, Cole DE, Jose PA, Chidiac P. G protein-coupled receptor accessory proteins and signaling: pharmacogenomic insights. Methods Mol Biol. (2014) 1175:121–52. doi: 10.1007/978-1-4939-0956-8_7
17. Yarmolinsky DA, Zuker CS, Ryba NJ. Common sense about taste: from mammals to insects. Cell (2009) 139:234–44. doi: 10.1016/j.cell.2009.10.001
18. Dyer J, Salmon KS, Zibrik L, Shirazi-Beechey SP. Expression of sweet taste receptors of the T1R family in the intestinal tract and enteroendocrine cells. Biochem Soc Trans. (2005) 33 (Pt 1):302–5. doi: 10.1042/BST0330302
19. Liu D, Liman ER. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc Natl Acad Sci USA. (2003) 100:15160–5. doi: 10.1073/pnas.2334159100
20. Shirazi-Beechey SP, Moran AW, Batchelor DJ, Daly K, Al-Rammahi M. Glucose sensing and signalling; regulation of intestinal glucose transport. Proc Nutr Soc. (2011) 70:185–93. doi: 10.1017/S0029665111000103
21. Jang HJ, Kokrashvili Z, Theodorakis MJ, Carlson OD, Kim BJ, Zhou J, et al. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc Natl Acad Sci USA. (2007) 104:15069–74. doi: 10.1073/pnas.0706890104
22. Young RL, Chia B, Isaacs NJ, Ma J, Khoo J, Wu T, et al. Disordered control of intestinal sweet taste receptor expression and glucose absorption in type 2 diabetes. Diabetes (2013) 62:3532–41. doi: 10.2337/db13-0581
23. Laffitte A, Neiers F, Briand L. Functional roles of the sweet taste receptor in oral and extraoral tissues. Curr Opin Clin Nutr Metab Care (2014) 17:379–85. doi: 10.1097/MCO.0000000000000058
24. Rother KI, Conway EM, Sylvetsky AC. How non-nutritive sweeteners influence hormones and health. Trends Endocrinol Metab. (2018) 29:455–67. doi: 10.1016/j.tem.2018.04.010
25. Kidd M, Modlin IM, Gustafsson BI, Drozdov I, Hauso O, Pfragner R. Luminal regulation of normal and neoplastic human EC cell serotonin release is mediated by bile salts, amines, tastants, and olfactants. Am J Physiol Gastrointest Liver Physiol. (2008) 295:G260–72. doi: 10.1152/ajpgi.00056.2008
26. Sun EW, de Fontgalland D, Rabbitt P, Hollington P, Sposato L, Due SL, et al. Mechanisms controlling glucose-induced GLP-1 secretion in human small intestine. Diabetes (2017) 66:2144–9. doi: 10.2337/db17-0058
27. Zopun M, Lieder B, Holik AK, Ley JP, Hans J, Somoza V. Noncaloric sweeteners induce peripheral serotonin secretion via the T1R3-dependent pathway in human gastric parietal tumor cells (HGT-1). J Agric Food Chem. (2018) 66:7044–53. doi: 10.1021/acs.jafc.8b02071
28. Sato S, Hokari R, Kurihara C, Sato H, Narimatsu K, Hozumi H, et al. Dietary lipids and sweeteners regulate glucagon-like peptide-2 secretion. Am J Physiol Gastrointest Liver Physiol. (2013) 304:G708–14. doi: 10.1152/ajpgi.00282.2012
29. Cheeseman CI. Upregulation of SGLT-1 transport activity in rat jejunum induced by GLP-2 infusion in vivo. Am J Physiol Regul Integr Comp Physiol. (1997) 273 (6 Pt 2):R1965–71. doi: 10.1152/ajpregu.1997.273.6.R1965
30. Daly K, Al-Rammahi M, Arora DK, Moran AW, Proudman CJ, Ninomiya Y, et al. Expression of sweet receptor components in equine small intestine: relevance to intestinal glucose transport. Am J Physiol Regul Integr Comp Physiol. (2012) 303:R199–208. doi: 10.1152/ajpregu.00031.2012
31. Brown RJ, Walter M, Rother KI. Ingestion of diet soda before a glucose load augments glucagon-like peptide-1 secretion. Diabetes Care (2009) 32:2184–6. doi: 10.2337/dc09-1185
32. Steinert RE, Gerspach AC, Gutmann H, Asarian L, Drewe J, Beglinger C. The functional involvement of gut-expressed sweet taste receptors in glucose-stimulated secretion of glucagon-like peptide-1 (GLP-1) and peptide YY (PYY). Clin Nutr. (2011) 30:524–32. doi: 10.1016/j.clnu.2011.01.007
33. Gerspach AC, Steinert RE, Schonenberger L, Graber-Maier A, Beglinger C. The role of the gut sweet taste receptor in regulating GLP-1, PYY, and CCK release in humans. Am J Physiol Endocrinol Metab. (2011) 301:E317–25. doi: 10.1152/ajpendo.00077.2011
34. Temizkan S, Deyneli O, Yasar M, Arpa M, Gunes M, Yazici D, et al. Sucralose enhances GLP-1 release and lowers blood glucose in the presence of carbohydrate in healthy subjects but not in patients with type 2 diabetes. Eur J Clin Nutr. (2015) 69:162–6. doi: 10.1038/ejcn.2014.208
35. Ma J, Bellon M, Wishart JM, Young R, Blackshaw LA, Jones KL, et al. Effect of the artificial sweetener, sucralose, on gastric emptying and incretin hormone release in healthy subjects. Am J Physiol Gastrointest Liver Physiol. (2009) 296:G735–9. doi: 10.1152/ajpgi.90708.2008
36. Ma J, Chang J, Checklin HL, Young RL, Jones KL, Horowitz M, et al. Effect of the artificial sweetener, sucralose, on small intestinal glucose absorption in healthy human subjects. Br J Nutr. (2010) 104:803–6. doi: 10.1017/S0007114510001327
37. Wu T, Bound MJ, Standfield SD, Bellon M, Young RL, Jones KL, et al. Artificial sweeteners have no effect on gastric emptying, glucagon-like peptide-1, or glycemia after oral glucose in healthy humans. Diabetes Care (2013) 36:e202–3. doi: 10.2337/dc13-0958
38. Shirazi-Beechey SP. Molecular biology of intestinal glucose transport. Nutr Res Rev. (1995) 8:27–41. doi: 10.1079/NRR19950005
39. Balen D, Ljubojevic M, Breljak D, Brzica H, Zlender V, Koepsell H, et al. Revised immunolocalization of the Na+-D-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol. (2008) 295:C475–89. doi: 10.1152/ajpcell.00180.2008
40. Binder HJ. Role of colonic short-chain fatty acid transport in diarrhea. Annu Rev Physiol. (2010) 72:297–313. doi: 10.1146/annurev-physiol-021909-135817
41. Wright EM, Hirayama BA, Loo DF. Active sugar transport in health and disease. J Intern Med. (2007) 261:32–43. doi: 10.1111/j.1365-2796.2006.01746.x
42. Margolskee RF, Dyer J, Kokrashvili Z, Salmon KS, Ilegems E, Daly K, et al. T1R3 and gustducin in gut sense sugars to regulate expression of Na+-glucose cotransporter 1. Proc Natl Acad Sci USA. (2007) 104:15075–80. doi: 10.1073/pnas.0706678104
43. Moriya R, Shirakura T, Ito J, Mashiko S, Seo T. Activation of sodium-glucose cotransporter 1 ameliorates hyperglycemia by mediating incretin secretion in mice. Am J Physiol Endocrinol Metab. (2009) 297:E1358–65. doi: 10.1152/ajpendo.00412.2009
44. Wu T, Zhao BR, Bound MJ, Checklin HL, Bellon M, Little TJ, et al. Effects of different sweet preloads on incretin hormone secretion, gastric emptying, and postprandial glycemia in healthy humans. Am J Clin Nutr. (2012) 95:78–83. doi: 10.3945/ajcn.111.021543
45. Dyer J, Vayro S, King TP, Shirazi-Beechey SP. Glucose sensing in the intestinal epithelium. Eur J Biochem. (2003) 270:3377–88. doi: 10.1046/j.1432-1033.2003.03721.x
46. Stearns AT, Balakrishnan A, Rhoads DB, Tavakkolizadeh A. Rapid upregulation of sodium-glucose transporter SGLT1 in response to intestinal sweet taste stimulation. Ann Surg. (2010) 251:865–71. doi: 10.1097/SLA.0b013e3181d96e1f
47. Moran AW, Al-Rammahi MA, Arora DK, Batchelor DJ, Coulter EA, Daly K, et al. Expression of Na+/glucose co-transporter 1 (SGLT1) is enhanced by supplementation of the diet of weaning piglets with artificial sweeteners. Br J Nutr. (2010) 104:637–46. doi: 10.1017/S0007114510000917
48. Gorboulev V, Schurmann A, Vallon V, Kipp H, Jaschke A, Klessen D, et al. Na+-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes (2012) 61:187–96. doi: 10.2337/db11-1029
49. Young RL, Sutherland K, Pezos N, Brierley SM, Horowitz M, Rayner CK, et al. Expression of taste molecules in the upper gastrointestinal tract in humans with and without type 2 diabetes. Gut (2009) 58:337–46. doi: 10.1136/gut.2008.148932
50. Cummings DE, Overduin J. Gastrointestinal regulation of food intake. J Clin Invest. (2007) 117:13–23. doi: 10.1172/JCI30227
51. Baldassano S, Liu S, Qu MH, Mule F, Wood JD. Glucagon-like peptide-2 modulates neurally evoked mucosal chloride secretion in guinea pig small intestine in vitro. Am J Physiol Gastrointest Liver Physiol. (2009) 297:G800–5. doi: 10.1152/ajpgi.00170.2009
52. Bjerknes M, Cheng H. Modulation of specific intestinal epithelial progenitors by enteric neurons. Proc Natl Acad Sci USA. (2001) 98:12497–502. doi: 10.1073/pnas.211278098
53. Pal A, Rhoads DB, Tavakkoli A. Foregut exclusion disrupts intestinal glucose sensing and alters portal nutrient and hormonal milieu. Diabetes (2015) 64:1941–50. doi: 10.2337/db14-1578
54. Sharp PA, Debnam ES. The role of cyclic AMP in the control of sugar transport across the brush-border and basolateral membranes of rat jejunal enterocytes. Exp Physiol. (1994) 79:203–14. doi: 10.1113/expphysiol.1994.sp003753
55. Williams M, Sharp P. Regulation of jejunal glucose transporter expression by forskolin. Biochim Biophys Acta (2002) 1559:179–85. doi: 10.1016/S0005-2736(01)00449-7
56. Loflin P, Lever JE. HuR binds a cyclic nucleotide-dependent, stabilizing domain in the 3' untranslated region of Na+/glucose cotransporter (SGLT1) mRNA. FEBS Lett. (2001) 509:267–71. doi: 10.1016/S0014-5793(01)03176-3
57. Lee WY, Loflin P, Clancey CJ, Peng H, Lever JE. Cyclic nucleotide regulation of Na+/glucose cotransporter (SGLT1) mRNA stability. Interaction of a nucleocytoplasmic protein with a regulatory domain in the 3'-untranslated region critical for stabilization. J Biol Chem. (2000) 275:33998–4008. doi: 10.1074/jbc.M005040200
58. Martin MG, Wang J, Solorzano-Vargas RS, Lam JT, Turk E, Wright EM. Regulation of the human Na+-glucose cotransporter gene, SGLT1, by HNF-1 and Sp1. Am J Physiol Gastrointest Liver Physiol. (2000) 278:G591–603. doi: 10.1152/ajpgi.2000.278.4.G591
59. Helms JB, Rothman JE. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature (1992) 360:352–4. doi: 10.1038/360352a0
60. Singh SK, Bartoo AC, Krishnan S, Boylan MO, Schwartz JH, Michael Wolfe M. Glucose-dependent insulinotropic polypeptide (GIP) stimulates transepithelial glucose transport. Obesity (2008) 16:2412–6. doi: 10.1038/oby.2008.393
61. Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al. IDF diabetes atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract. (2017) 128:40–50. doi: 10.1016/j.diabres.2017.03.024
62. Standl E, Schnell O, Ceriello A. Postprandial hyperglycemia and glycemic variability: should we care? Diabetes Care (2011) 34 (Suppl. 2):S120–7. doi: 10.2337/dc11-s206
63. Marathe CS, Rayner CK, Jones KL, Horowitz M. Relationships between gastric emptying, postprandial glycemia, and incretin hormones. Diabetes Care (2013) 36:1396–405. doi: 10.2337/dc12-1609
64. Holst JJ, Vilsboll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol. (2009) 297:127–36. doi: 10.1016/j.mce.2008.08.012
65. Vilsboll T, Krarup T, Madsbad S, Holst JJ. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept. (2003) 114:115–21. doi: 10.1016/S0167-0115(03)00111-3
66. Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. (1993) 91:301–7. doi: 10.1172/JCI116186
67. Toft-Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab. (2001) 86:3717–23. doi: 10.1210/jcem.86.8.7750
68. Martin AM, Lumsden AL, Young RL, Jessup CF, Spencer NJ, Keating DJ. The nutrient-sensing repertoires of mouse enterochromaffin cells differ between duodenum and colon. Neurogastroenterol Motil. (2017) 29. doi: 10.1111/nmo.13046
69. Martin AM, Young RL, Leong L, Rogers GB, Spencer NJ, Jessup CF, et al. The diverse metabolic roles of peripheral serotonin. Endocrinology (2017) 158:1049–63. doi: 10.1210/en.2016-1839
70. Young RL, Lumsden AL, Keating DJ. Gut serotonin is a regulator of obesity and metabolism. Gastroenterology (2015) 149:253–5. doi: 10.1053/j.gastro.2015.05.020
71. Takahashi T, Yano M, Minami J, Haraguchi T, Koga N, Higashi K, et al. Sarpogrelate hydrochloride, a serotonin2A receptor antagonist, reduces albuminuria in diabetic patients with early-stage diabetic nephropathy. Diabetes Res Clin Pract. (2002) 58:123–9. doi: 10.1016/S0168-8227(02)00105-5
72. Sneha P, Doss CG. Gliptins in managing diabetes—Reviewing computational strategy. Life Sci. (2016) 166:108–20. doi: 10.1016/j.lfs.2016.10.009
73. Dyer J, Garner A, Wood IS, Sharma AK, Chandranath I, Shirazi-Beechey SP. Changes in the levels of intestinal Na+/glucose co-transporter (SGLT1) in experimental diabetes. Biochem Soc Trans. (1997) 25:479S. doi: 10.1042/bst025479s
74. Dyer J, Wood IS, Palejwala A, Ellis A, Shirazi-Beechey SP. Expression of monosaccharide transporters in intestine of diabetic humans. Am J Physiol Gastrointest Liver Physiol. (2002) 282:G241–8. doi: 10.1152/ajpgi.00310.2001
75. Cariou B, Charbonnel B. Sotagliflozin as a potential treatment for type 2 diabetes mellitus. Expert Opin Investig Drugs (2015) 24:1647–56. doi: 10.1517/13543784.2015.1100361
76. Mackenzie T, Brooks B, O'Connor G. Beverage intake, diabetes, and glucose control of adults in America. Ann Epidemiol. (2006) 16:688–91. doi: 10.1016/j.annepidem.2005.11.009
77. Malik VS, Hu FB. Fructose and cardiometabolic health: what the evidence from sugar-sweetened beverages tells us. J Am Coll Cardiol. (2015) 66:1615–24. doi: 10.1016/j.jacc.2015.08.025
78. Ervin RB, Ogden CL. Consumption of added sugars among U.S. adults, 2005–2010. NCHS Data Brief (2013) 122:1–8.
79. Brisbois TD, Marsden SL, Anderson GH, Sievenpiper JL. Estimated intakes and sources of total and added sugars in the Canadian diet. Nutrients (2014) 6:1899–912. doi: 10.3390/nu6051899
80. World Health Organisation. Guideline: Sugars Intake for Adults and Children. Geneva: WHO Guidelines Approved by the Guidelines Review Committee (2015).
81. Schulze MB, Liu S, Rimm EB, Manson JE, Willett WC, Hu FB. Glycemic index, glycemic load, and dietary fiber intake and incidence of type 2 diabetes in younger and middle-aged women. Am J Clin Nutr. (2004) 80:348–56. doi: 10.1093/ajcn/80.2.348
82. Malik VS, Popkin BM, Bray GA, Despres JP, Willett WC, Hu FB. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care (2010) 33:2477–83. doi: 10.2337/dc10-1079
83. Kahn R, Sievenpiper JL. Dietary sugar and body weight: have we reached a crisis in the epidemic of obesity and diabetes?: we have, but the pox on sugar is overwrought and overworked. Diabetes Care (2014) 37:957–62. doi: 10.2337/dc13-2506
84. Rippe JM, Angelopoulos TJ. Added sugars and risk factors for obesity, diabetes and heart disease. Int J Obes. (2016) 40 (Suppl. 1):S22–7. doi: 10.1038/ijo.2016.10
85. Johnson RK, Appel LJ, Brands M, Howard BV, Lefevre M, Lustig RH, et al. Dietary sugars intake and cardiovascular health: a scientific statement from the American Heart Association. Circulation (2009) 120:1011–20. doi: 10.1161/CIRCULATIONAHA.109.192627
86. Sylvetsky AC, Rother KI. Trends in the consumption of low-calorie sweeteners. Physiol Behav. (2016) 164 (Pt B):446–50. doi: 10.1016/j.physbeh.2016.03.030
87. Stanley L. External Study Report. Review of Data on the Food Additive Aspartame (2013). Available online at: https://efsa.onlinelibrary.wiley.com/doi/pdf/10.2903/sp.efsa.2013.EN-399
88. Schiffman SS, Rother KI. Sucralose, a synthetic organochlorine sweetener: overview of biological issues. J Toxicol Environ Health B Crit Rev. (2013) 16:399–451. doi: 10.1080/10937404.2013.842523
89. Daly K, Darby AC, Shirazi-Beechey SP. Low calorie sweeteners and gut microbiota. Physiol Behav. (2016) 164 (Pt B):494–500. doi: 10.1016/j.physbeh.2016.03.014
90. Raben A, Vasilaras TH, Moller AC, Astrup A. Sucrose compared with artificial sweeteners: different effects on ad libitum food intake and body weight after 10 wk of supplementation in overweight subjects. Am J Clin Nutr. (2002) 76:721–9. doi: 10.1093/ajcn/76.4.721
91. Ebbeling CB, Feldman HA, Chomitz VR, Antonelli TA, Gortmaker SL, Osganian SK, et al. A randomized trial of sugar-sweetened beverages and adolescent body weight. N Engl J Med. (2012) 367:1407–16. doi: 10.1056/NEJMoa1203388
92. Miller PE, Perez V. Low-calorie sweeteners and body weight and composition: a meta-analysis of randomized controlled trials and prospective cohort studies. Am J Clin Nutr. (2014) 100:765–77. doi: 10.3945/ajcn.113.082826
93. Fowler SP, Williams K, Resendez RG, Hunt KJ, Hazuda HP, Stern MP. Fueling the obesity epidemic? Artificially sweetened beverage use and long-term weight gain. Obesity (2008) 16:1894–900. doi: 10.1038/oby.2008.284
94. Nettleton JA, Polak JF, Tracy R, Burke GL, Jacobs DR Jr. Dietary patterns and incident cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis. Am J Clin Nutr. (2009) 90:647–54. doi: 10.3945/ajcn.2009.27597
95. Brown RJ, de Banate MA, Rother KI. Artificial sweeteners: a systematic review of metabolic effects in youth. Int J Pediatr Obes. (2010) 5:305–12. doi: 10.3109/17477160903497027
96. Duffey KJ, Steffen LM, Van Horn L, Jacobs DR Jr, Popkin BM. Dietary patterns matter: diet beverages and cardiometabolic risks in the longitudinal Coronary Artery Risk Development in Young Adults (CARDIA) Study. Am J Clin Nutr. (2012) 95:909–15. doi: 10.3945/ajcn.111.026682
97. Fagherazzi G, Vilier A, Saes Sartorelli D, Lajous M, Balkau B, Clavel-Chapelon F. Consumption of artificially and sugar-sweetened beverages and incident type 2 diabetes in the Etude Epidemiologique aupres des femmes de la Mutuelle Generale de l'Education Nationale-European Prospective Investigation into Cancer and Nutrition cohort. Am J Clin Nutr. (2013) 97:517–23. doi: 10.3945/ajcn.112.050997
98. Grotz VL, Henry RR, McGill JB, Prince MJ, Shamoon H, Trout JR, et al. Lack of effect of sucralose on glucose homeostasis in subjects with type 2 diabetes. J Am Diet Assoc. (2003) 103:1607–12. doi: 10.1016/j.jada.2003.09.021
99. Ferri LA, Alves-Do-Prado W, Yamada SS, Gazola S, Batista MR, Bazotte RB. Investigation of the antihypertensive effect of oral crude stevioside in patients with mild essential hypertension. Phytother Res. (2006) 20:732–6. doi: 10.1002/ptr.1944
100. Maersk M, Belza A, Stodkilde-Jorgensen H, Ringgaard S, Chabanova E, Thomsen H, et al. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: a 6-mo randomized intervention study. Am J Clin Nutr. (2012) 95:283–9. doi: 10.3945/ajcn.111.022533
101. Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O, et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature (2014) 514:181–6. doi: 10.1038/nature13793
102. Madjd A, Taylor MA, Delavari A, Malekzadeh R, Macdonald IA, Farshchi HR. Effects on weight loss in adults of replacing diet beverages with water during a hypoenergetic diet: a randomized, 24-wk clinical trial. Am J Clin Nutr. (2015) 102:1305–12. doi: 10.3945/ajcn.115.109397
103. Pepino MY, Tiemann CD, Patterson BW, Wice BM, Klein S. Sucralose affects glycemic and hormonal responses to an oral glucose load. Diabetes Care (2013) 36:2530–5. doi: 10.2337/dc12-2221
104. Azad MB, Abou-Setta AM, Chauhan BF, Rabbani R, Lys J, Copstein L, et al. Nonnutritive sweeteners and cardiometabolic health: a systematic review and meta-analysis of randomized controlled trials and prospective cohort studies. CMAJ (2017) 189:E929–39. doi: 10.1503/cmaj.161390
105. Swithers SE. Artificial sweeteners produce the counterintuitive effect of inducing metabolic derangements. Trends Endocrinol Metab. (2013) 24:431–41. doi: 10.1016/j.tem.2013.05.005
106. Young RL, Isaacs NJ, Schober G, Wu T, Cvijanovic N, Pezos N, et al. Impact of artificial sweeteners on glycaemic control in healthy humans. Diabetologia (2017) 60 (Suppl. 1):S91.
107. Rogers GB, Keating DJ, Young RL, Wong ML, Licinio J, Wesselingh S. From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol Psychiatr. (2016) 21:738–48. doi: 10.1038/mp.2016.50
109. Shen J, Obin MS, Zhao L. The gut microbiota, obesity and insulin resistance. Mol Aspects Med. (2013) 34:39–58. doi: 10.1016/j.mam.2012.11.001
110. Neyrinck AM, Delzenne NM. Potential interest of gut microbial changes induced by non-digestible carbohydrates of wheat in the management of obesity and related disorders. Curr Opin Clin Nutr Metab Care (2010) 13:722–8. doi: 10.1097/MCO.0b013e32833ec3fb
111. Al-Lahham SH, Peppelenbosch MP, Roelofsen H, Vonk RJ, Venema K. Biological effects of propionic acid in humans; metabolism, potential applications and underlying mechanisms. Biochim Biophys Acta (2010) 1801:1175–83. doi: 10.1016/j.bbalip.2010.07.007
112. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes (2009) 58:1509–17. doi: 10.2337/db08-1637
113. Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature (2013) 498:99–103. doi: 10.1038/nature12198
114. Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE (2010) 5:e9085. doi: 10.1371/journal.pone.0009085
115. Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology (2012) 143:913–6.e7. doi: 10.1053/j.gastro.2012.06.031
116. Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (2010) 18:190–5. doi: 10.1038/oby.2009.167
117. Daly K, Darby AC, Hall N, Wilkinson MC, Pongchaikul P, Bravo D, et al. Bacterial sensing underlies artificial sweetener-induced growth of gut Lactobacillus. Environ Microbiol. (2016) 18:2159–71. doi: 10.1111/1462-2920.12942
118. Hollister EB, Gao C, Versalovic J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology (2014) 146:1449–58. doi: 10.1053/j.gastro.2014.01.052
119. Grasset E, Puel A, Charpentier J, Collet X, Christensen JE, Terce F, et al. A specific gut microbiota dysbiosis of type 2 diabetic mice induces GLP-1 resistance through an enteric NO-dependent and gut-brain axis mechanism. Cell Metab. (2017) 25:1075–90.e5. doi: 10.1016/j.cmet.2017.04.013
120. Feigin MB, Sclafani A, Sunday SR. Species differences in polysaccharide and sugar taste preferences. Neurosci Biobehav Rev. (1987) 11:231–40. doi: 10.1016/S0149-7634(87)80031-3
121. Sclafani A, Hertwig H, Vigorito M, Sloan H, Kerzner B. Influence of saccharide length on polysaccharide appetite in the rat. Neurosci Biobehav Rev. (1987) 11:197–200. doi: 10.1016/S0149-7634(87)80026-X
122. Giza BK, Scott TR, Sclafani A, Antonucci RF. Polysaccharides as taste stimuli: their effect in the nucleus tractus solitarius of the rat. Brain Res. (1991) 555:1–9. doi: 10.1016/0006-8993(91)90852-M
123. Sako N, Shimura T, Komure M, Mochizuki R, Matsuo R, Yamamoto T. Differences in taste responses to polycose and common sugars in the rat as revealed by behavioral and electrophysiological studies. Physiol Behav. (1994) 56:741–5. doi: 10.1016/0031-9384(94)90236-4
124. Treesukosol Y, Blonde GD, Spector AC. T1R2 and T1R3 subunits are individually unnecessary for normal affective licking responses to polycose: implications for saccharide taste receptors in mice. Am J Physiol Regul Integr Comp Physiol. (2009) 296:R855–65. doi: 10.1152/ajpregu.90869.2008
125. Treesukosol Y, Smith KR, Spector AC. Behavioral evidence for a glucose polymer taste receptor that is independent of the T1R2+3 heterodimer in a mouse model. J Neurosci. (2011) 31:13527–34. doi: 10.1523/JNEUROSCI.2179-11.2011
126. Treesukosol Y, Spector AC. Orosensory detection of sucrose, maltose, and glucose is severely impaired in mice lacking T1R2 or T1R3, but polycose sensitivity remains relatively normal. Am J Physiol Regul Integr Comp Physiol. (2012) 303:R218–35. doi: 10.1152/ajpregu.00089.2012
127. Zukerman S, Glendinning JI, Margolskee RF, Sclafani A. T1R3 taste receptor is critical for sucrose but not polycose taste. Am J Physiol Regul Integr Comp Physiol. (2009) 296:R866–76. doi: 10.1152/ajpregu.90870.2008
128. Lapis TJ, Penner MH, Lim J. Humans can taste glucose oligomers independent of the hT1R2/hT1R3 sweet taste receptor. Chem Senses (2016):41:755–762. doi: 10.1093/chemse/bjw088
129. Low JYQ, Lacy KE, McBride RL, Keast RSJ. Evidence supporting oral sensitivity to complex carbohydrates independent of sweet taste sensitivity in humans. PLoS ONE (2017) 12:e0188784. doi: 10.1371/journal.pone.0188784
Keywords: intestinal sweet taste receptors, L-cells, glucose transport, SGLT-1, glycemic control, type 2 diabetes mellitus
Citation: Kreuch D, Keating DJ, Wu T, Horowitz M, Rayner CK and Young RL (2018) Gut Mechanisms Linking Intestinal Sweet Sensing to Glycemic Control. Front. Endocrinol. 9:741. doi: 10.3389/fendo.2018.00741
Received: 05 July 2018; Accepted: 22 November 2018;
Published: 04 December 2018.
Edited by:
Sandhya Srikant Visweswariah, Indian Institute of Science (IISc), IndiaReviewed by:
Miles Douglas Thompson, Rady Children's Hospital-San Diego, University of California, San Diego, United StatesGuillermo Romero, University of Pittsburgh, United States
Copyright © 2018 Kreuch, Keating, Wu, Horowitz, Rayner and Young. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard L. Young, richard.young@adelaide.edu.au