 Silvano Paternoster
Silvano Paternoster Marco Falasca
Marco Falasca- Metabolic Signalling Group, School of Pharmacy and Biomedical Sciences, Curtin Health Innovation Research Institute, Curtin University, Perth, WA, Australia
An aging world population exposed to a sedentary life style is currently plagued by chronic metabolic diseases, such as type-2 diabetes, that are spreading worldwide at an unprecedented rate. One of the most promising pharmacological approaches for the management of type 2 diabetes takes advantage of the peptide hormone glucagon-like peptide-1 (GLP-1) under the form of protease resistant mimetics, and DPP-IV inhibitors. Despite the improved quality of life, long-term treatments with these new classes of drugs are riddled with serious and life-threatening side-effects, with no overall cure of the disease. New evidence is shedding more light over the complex physiology of GLP-1 in health and metabolic diseases. Herein, we discuss the most recent advancements in the biology of gut receptors known to induce the secretion of GLP-1, to bridge the multiple gaps into our understanding of its physiology and pathology.
Introduction
The gastrointestinal (GI) tract is a complex organ that monitors the body's energetical state and provides it with water and macro and micronutrients extracted from the ingested food. Along its length, the enteroendocrine cells (EECs) constitute a complex endocrine organ that communicates with the central nervous system (CNS) and the enteric nervous system (ENS) to orchestrate the homeostatic balance of the body in response to the GI luminal content.
This enteroendocrine system has traditionally been divided into 12 different cell types, based entirely on their hormonal content and cellular morphology. This endocrine organ is not organized in a glandular structure; on the contrary, it is dispersed heterogeneously, mainly as single cells, along the epithelium of the GI tract, from the stomach to the rectum with a defined cephalocaudal, crypt-to-villus in the small intestine and crypt-to-surface distribution in the colon (1, 2).
Despite representing just 1% of the adult gut epithelium, in the last decade it has become clear that the EECs constitute the largest endocrine organ in mammalia (3). Recent analysis of the expression of specific hormones at the cellular level, demonstrated that the EECs subdivision introduced above is outdated. Each enteroendocrine cell co-secretes multiple hormones with spatio-temporal, crypt-to-villus, and rostro-caudal variability, leading to the formation of overlapped gradients of individual hormones along the GI tract; the concept of well-defined subclasses of cells committed to express a specific subset of hormones independent of their location is currently untenable, thus detailed description of the topographical location of the cells needs to be implemented for future clarity (4).
Collectively, the EECs are responsible for the production of more than 30 different hormones that help to orchestrate the fate of the intermediary metabolism; acting upon different organs such as the pancreatic islets, the hypothalamus or the stomach, for the release of insulin, to regulate food intake or gastric emptying respectively (5–8).
Surprisingly, this heterogeneous and highly plastic population of cells is known to differentiate from a single staminal progenitor that gives also rise to enterocytes, goblet and paneth cells (1, 9).
It has been known for more than a century that the gut is capable to stimulate the endocrine portion of the pancreas and even improve the hyperglycaemic state of diabetic patients (10, 11). In 1932, the Belgian investigator LaBarre referred to these “factors” extracted from the intestinal mucosa as “incrétine,” deriving it from: INtestinal seCRETion of insulin (12). In the 60s, different authors demonstrated that oral glucose was capable to induce a 2-fold increase in insulin compared to an in-vein isoglycaemic administration (13).
In the last three decades, the incretin-effect has been attributed primarily to two peptide hormones, the gastric-insulinotropic peptide (GIP) and glucagon-like peptide-1 (GLP-1), excreted primarily by duodenal (K) and ileo-colonic (L) enteroendocrine cells respectively (14). Indeed, type 2 diabetes (T2D) is a metabolic disease reported to involve an impaired intestinal release of GLP-1 and its co-secreted peptides oxyntomodulin and glicentin (15–17), together with an insulinotropic resistance to GIP in the pancreas (18) which lead to a deficient incretin system, purportedly causing the disease (19, 20). Despite being still largely unknown how hyper caloric diets are disrupting the incretin signaling, some authors have shown that even circadian rhythms disruption, and the saturated fat palmitate, are significant stressors capable to hamper GLP-1 secretion (21, 22).
Obesity and Type 2 diabetes are chronic diseases for which the most effective treatment is bariatric surgery. These invasive gut surgical procedures, aimed to reduce absorptive surface area of the proximal GI tract, such as Roux-en-Y gastric by-pass (RYGB) or Sleeve Gastrectomy (SLG), are associated with an improved glycaemic control, weight loss, and often with complete remission from T2DM (23).
Despite this, the complete remittance of a great fraction of RYGB patients represents a fascinating new case-series that points at the importance of the EECs and its modulation of the whole-body metabolism (24). As such, the study of this complex endocrine organ, might help us to create new pharmacological tools to amend the specific molecular axis that drive T2DM and the associated co-morbidities known to affect the cardiovascular (25, 26) and renal system (27, 28).
A panoply of contradictory studies have attempted to establish what is the possible role of GLP-1 or other gut peptides in the rapid, and long-lasting remittance from T2D after bariatric surgery, but no consensus about the identity of the molecular players has yet been reached (29–39).
Since 2005, there are on the market only two classes of drugs that attempt to bolster glucagon-like peptide-1 signaling, GLP-1 receptor agonists and DPP-IV inhibitors, for a supra-physiological GLP-1 activity. Unexpected safety-issues and important side-effects (40) prove that the peripheral hijack of this peptide is not sufficient, and does not replicate the remittance seen in bariatric surgery.
This review summarizes the most recent studies that reframe our understanding of the physiology of GLP-1 in health and disease.
Chemosensation in GLP-1-Producing Cells
Intestinal proglucagon expressing cells were historically named L-cells more than 4 decades ago because of their large 500 nm secretory granules seen under electron microscopy (41). Today, we know that these are nutrient-responsive enteroendocrine cells that secrete a variety of peptide hormones, primarily derived from the proglucagon gene (GCG) (42). Once translated, the 180 amino acid long GCG protein is processed by two proteases, Psck1 and Psck3, to give GLP-1, GLP-2 but also the less studied and understood glicentin and oxyntomodulin (43). Other peptide hormones, such as insulin-like peptide 5 (INSL5) (44, 45), PYY (46), GIP and neurotensin (17, 47) can be co-expressed with the GCG products depending on the topographical localization of the cell; surprisingly, it appears that GLP-1 and PYY can be excreted independently possibly due to the existence of compartmentalized secretory vesicles (48).
There appear to be considerable species-specificity in terms of anatomical localization of GLP-1 production as summarized in Figure 1. Independently of other hormones, in mice the distal colon and rectum show the higher levels of GLP-1 per gram of tissue. Conversely, in rats the distal ileum and in pigs the caecum are the anatomical regions with the highest amounts of GLP-1 (49). In humans, the density of GLP-1 and PYY positive cells increase steadily along the small intestine, decreasing in the colon, and then raising again reaching a maximum density in the rectum with the highest values of around 150 GLP-1-expressing cells per square millimeter. Curiously in type 2 diabetes, an equally distributed gradient of GCG and PC1/3 mRNA appears upregulated, but with normal GLP-1+ cell densities, indicating a possible translational resistance (51).
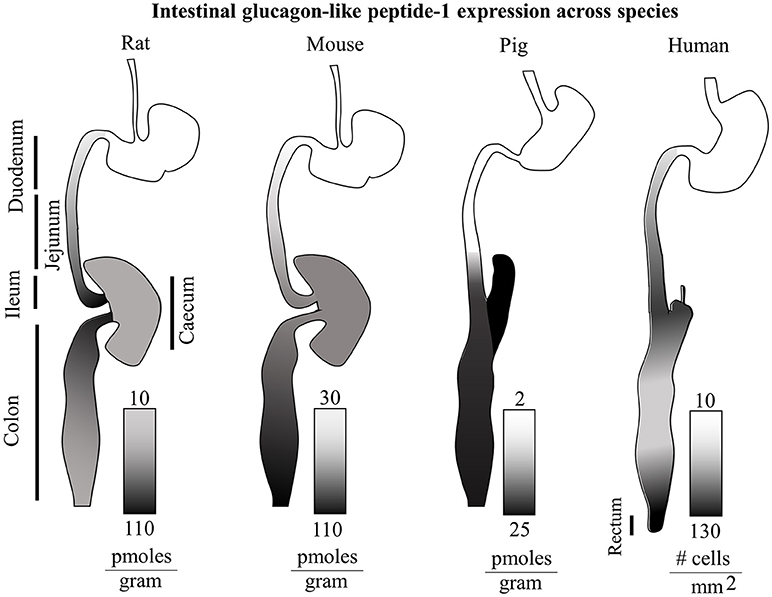
Figure 1. Intestinal glucagon-like peptide-1 expression across species. Total GLP-1 expression along the rat, mouse, pig and human intestinal tracts (relative lengths not to scale) is displayed with gradients as individually indicated in figure. The rat GI tract shows the highest levels of GLP-1 in the ileum and proximal colon. On the other hand the murine gut, displays the highest GLP-1 levels in the distal colon. The porcine intestine shows highest levels in the caecum and distal colon, and virtually none in the proximal small intestine. In humans, a steady increasing gradient along the small intestine is followed by a decrease in expression in the colon, and a second steeper gradient culminating in the rectum with the highest GLP-1 expression (49–51).
The L-cells derived cocktail of hormones is believed to play pivotal roles in digestion, for example slowing down the GI motility (PYY) and suppressing the appetite in vivo (GLP-1, oxyntomodulin, PYY), apparently in response to direct sensing of the gut luminal content via G-protein coupled receptors or through neuronal circuits (43, 52).
Current in vitro technologies are not capable to support for long-term ex vivo the growth of isolated GLP-1 producing-cells. The available knowledge about the biology of GLP-1 is primarily drawn upon studies operated with the murine-derived GLUTag or STC-1, and the human-derived NCI-H716 cell lines. It is important to understand that these in vitro models express a different hormonal cocktail and respond to different chemical stimuli than intestinal L-cells in vivo (53, 54). Primary cultures are another useful short-term system; nonetheless GLP-1-producing cells amount to only 1–2% of the whole cultured mucosal population, with considerable intra and inter-assay variability (53).
The more physiologically relevant studies make use of in vivo transgenic mice, ex vivo perfused intestines or, more recently, crypt organoids derived from human, mouse or porcine guts (55).
In situ immunostaining and FACS studies have demonstrated that the hormonal secretome of GLP-1-secreting-cells is anatomically dependent. In the upper gut where these cells are more sparse and rare, GLP-1 is co-expressed with GIP, a K-cell feature, but also with cholecystokinin (CCK) and Neurotensin (NT). Conversely in the colonic mucosa, GLP-1 co-localizes with PYY, CCK and the orexigenic Insulin-Like peptide 5 (INSL5) (4, 43, 45, 53, 56, 57). Interestingly, colonic L-cells possess twice as much total GLP-1 compared to L-cells from the upper GI tract (53). Furthermore, considering the differential response to glucose, it is clear that the physiology of this population of EECs is distinct, and evolved under a different evolutionary pressure dictated by the exposure to a different luminal content (53, 58).
L-cells are known to modulate the release of their hormonal cargo in response to the activation of a plethora of receptors capable to sense fats, carbohydrates, proteins and many other compounds. Enteroendocrine cells, like other endocrine, muscle and neuronal cells, are electrically excitable. Membrane depolarization, triggered by a ligand-bound receptor, results in a spike of intracellular calcium (Ca2+) which leads to the fusion of the endocrine granules with the lateral and the broader basal side, resulting in the discharge of a hormonal cargo in the capillaries of the mucosa.
Surprisingly, the EECs in the colon have been demonstrated to physically connect through a basal process named Neuropod, with afferent nerve cells residing in the lamina propria, defining a neuroepithelial circuit that expands the physiology of these cells (59). In fact, the idea of a direct neuronal regulation has been demonstrated decades ago in rats, where a bilateral vagotomy massively downregulates circulating PYY and GLP-1 levels after a glucose load (60). Furthermore, intracerebral acute, but not chronic administration of GLP-1 in mice, improves pancreatic glucose stimulated insulin secretion (61).
GPCRs as Molecular Tastants
G-protein coupled receptors (GPCRs) are evolutionary ancient proteins spanning seven times across the plasma membrane of virtually any known cell type. In metazoans, these proteins evolved into thousands different molecular transducers capable to translate the presence of extracellular molecules into intracellular cascades of messages amplified by different G-proteins, which in turn enforce a myriad of different cellular processes via secondary messengers (62). The transmembrane domain of these chemosensors being exposed to a tighter evolutionary pressure lead to a relative evolutionary stability of the same 3-dimensional structure. On the contrary, the extracellular facing portion is what primarily defines the identity of a myriad of different receptors, capable to sense a panoply of molecular entities ranging in size from a single atom to hundreds aminoacids long proteins. The intracellular portion of these nano-sensors, has evolved in humans in a complex hub that triggers multiple molecular cascades that results in short-term and long-term modifications of the target cell and even the whole-body metabolism.
Different receptors, expressed by the same cell type or tissue, can trigger the same molecular cascade. With this notion, the study of these molecular transducers has been approached by some authors in recent years from a top-down point of view, whereby sub-type specific, allosteric positive or negative modulators (PAM, NAMs), as well as direct agonists, are utilized as tools for pathway dissection and analysis (63, 64). In the last decade, technological advancements in techniques such as circular dichroism (65), Cryo-electron microscopy (Cryo-EM) (66) and crystallography (67) have expanded our understanding of the physiology of multiple chemosensors expressed by L-cells, which led to the discovery of new molecular tools with possible future clinical applications in diseases such as type 2 diabetes (64, 68–70).
The expression of different GPCRs to restricted anatomical regions, such as the enteroendocrine cell system, is a finely tuned system that evolved in metazoan. Macronutrients, bile acids (BAs), and microbiota-derived compounds activate many of these GPCRs expressed by GLP-1 expressing cells (71). Nonetheless, not all intestinal stimuli signals through these chemosensors; for example glucose induces the release of GLP-1 from human duodenum and ileum via electrogenic transporters (SGLT1) and voltage-gated Calcium and Sodium channels responsible for the membrane depolarization and hormonal release (53, 72).
The main G protein-coupled receptors which activation appears to cause the release of GLP-1 are: GPRC6A (73), GPR40-41-42-43-93-119-120 (43), GPR142, GHS-R1A (74), Tas1R2-Tas2R3(T1R2-T1R3) (75), GPBAR1 (TGR5), and CasR (6, 76, 77) (Table 1). The functional differences seen between Jejunum-Ileal and colonic GLP-1 producing cells, could be explained by a different pool of GPCRs, or possibly by the presence of heteromers displaying a more complex pharmacology than with each individual receptor.
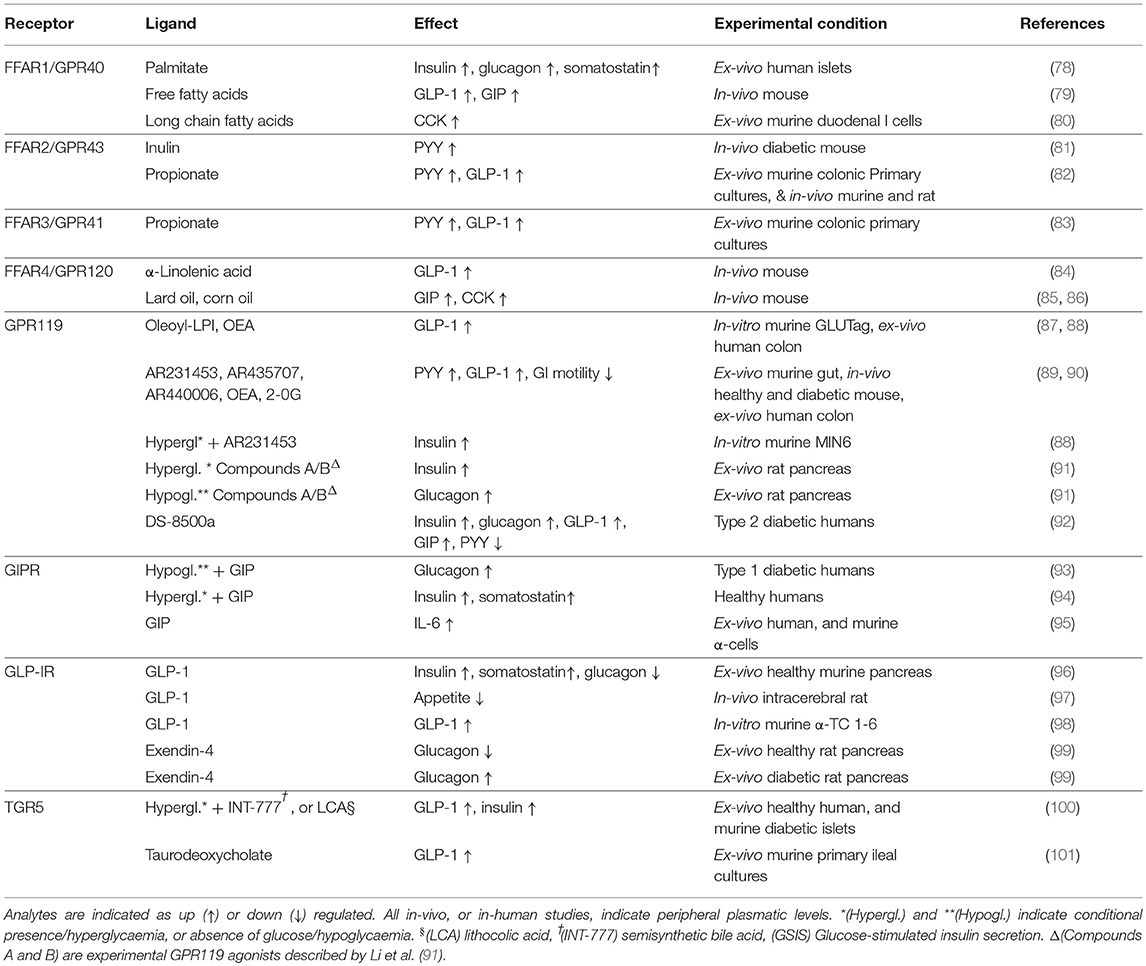
Table 1. Demonstrated primary effects of the major GLP-1-stimulating receptors.
A summary of the recognized main activities of all the major GLP-1-secreting receptors, including the GIPR (93, 94), is shown in Table 1.
Many of these chemosensors are also expressed by other enteroendocrine cells, so that the same dietary ligand traveling along the GI tract, leads to the release of multiple hormones.
There are some receptors, such as GPRC6A, with a pleiotropic distribution and still a limited understanding of its physiology. GPRC6A is highly expressed in GLUTag cells, and its activation by L-ornithine has shown to induce GLP-1 secretion (102). Nonetheless, mice deficient for the receptor, show no difference in responsiveness to both L-ornithine and L-arginine (103).
The Physiology of GLP-1
In the last three decades a major tenet seeing GLP1 (7-36)NH2, GLP1 (7-37) and the Gastric Insulinotropic Peptide (GIP) as the major contributors of the physiological incretin effect has reached widespread consensus (104). The remaining Glucose-stimulated insulin secretion (GSIS) appears to be enhanced by nutrients, hormones such as CCK, bile acids and endogenous ethanolamides. Animal models show compensatory mechanisms by which, in absence of a major incretin axis, other minor pathways are promoted in the β-cells to maintain their metabolic activity; namely proteins such as GPR119, or the CCK A receptor itself are upregulated, implying a highly plastic metabolic adaptation (105).
Multiple cell types found in the enteroendocrine cell system, the pancreatic islets or the brain have been shown to express the GCG product, a 180 aminoacids long peptide known as proglucagon (PG) (106, 107), which gets trimmed tissue-dependently into at least 6 different bio-active peptides, namely glicentin, oxyntomodulin, glucagon, miniglucagon, GLP-1 and GLP-2 (108, 109). The post-translational processing of the preproglucagon gene into the individual peptides is controlled by two distinct serine proteases, specifically prohormone convertases named Psck1/3 and Psck2, also known as PC1/3, or just PC1, and PC2 respectively (107, 108, 110). PC1/3 and PC2 are responsible for the metabolism of a plethora of peptide pro-hormones, including insulin and GCG among others (111). In particular PC1/3 expressing cells, such as intestinal L-cells and pancreatic β-cells, produce GLP-1, GLP-2 oxyntomodulin and glicentin (110, 112), while PC2 action on PG results in the production of glucagon and its active metabolite mini-glucagon (113, 114). Differential expression of PC genes regulates the hormonal output, and indeed it has been proven that both are expressed along the intestine, with PC1/3 positive cells found more distally than PC2 expressing cells (51), likely secreting glucagon (115). Indeed, the RYGB surgery removes the biggest pool of PC2/glucagon expressing cells from the exposure to nutrients, possibly contributing to the surgical success.
Active GLP-1(7-37), in human and mice is largely metabolized by the enzyme peptidyl-glycine α-amidating monooxygenase (PAM) into the equally active GLP-1(7-36)NH2 (49, 116). Both these peptide species are trimmed at their N-term, and inactivated by the ubiquitous protease dipeptidyl-peptidase-IV (DPP-IV), found in the intestinal capillaries, vena porta and liver. Indeed, it has been estimated that just 10-15% of the secreted GLP-1(7-36)NH2 reaches the systemic circulation (117), with some authors reporting meager peripheral meal-induced changes in both healthy and diabetic people (118). Furthermore, the DPP-IV product, GLP-1(9-36)NH2, is trimmed into GLP-1(28-36)NH2 and GLP-1(32-36)NH2 by another ubiquitous protease, known as NEP24.11, CD10 or also Neprilysin among other names (119, 120).
Indeed, these once thought inactive metabolites of the recognized GLP-1 receptor agonist GLP-1(7-36) NH2 have recently shown to possess multiple beneficial properties. The 9 aminoacids long GLP-1(28-36) protects β-cells from glucolipotoxicity (121), diet-induced steatosis of the liver (122), improves hepatic glucose tolerance in diabetic mice (122–124). Similarly, the 5 aminoacids long GLP-1(32-36)NH2 improves glucose disposal, increases energy expenditure and protects β-cells in a diabetic environment in vivo (125–127). Indeed GLP-1(9-36) pharmacodynamics studies in human might be partially explained by the activity of its metabolites (128).
These metabolites have possibly important implications for any future treatment of metabolic pathologies such as type 2 diabetes, where our understanding of the pharmacokinetic and pharmacodynamics in humans is virtually absent (128).
In healthy humans, intact GLP-1(7-36) NH2 is mainly released by intestinal EECs after the ingestion of food, especially meals rich in fat and proteins (14, 129). Other stimuli, such as physical activity, are also capable to raise its plasmatic levels for up to 90 min after exercise (130).
This hormone generates both short-term and long-term pleiotropic effects. GLP-1 stimulates the β-cells to produce Insulin, blocks pancreatic α-cells' glucagon release via somatostatin (96), slows down gastric emptying (131), improves peripheral glucose tolerance (132), suppresses appetite in the hypothalamus and amygdala (97), increases β-cell mass, GSIS, and elicits protection from glucolipotoxicity (133) and apoptosis (134). Curiously, it also regulates bone physiology (135), and shows anti-inflammatory properties (136).
On the other hand, the most abundant DPP-IV-processed metabolite GLP-1 (9-36)NH2, has also been reported to have biological activities, protecting human aortic endothelial cells and cardiomyocytes in vivo in dogs (137) and ex vivo in mice (138) and rats (139), even in the absence of a GLP-1 Receptor (139, 140). Some authors postulate the existance of an unknown GLP-1(9-36)NH2 receptor (141, 142), because indeed this cleaved peptide is found in peripheral blood at one order of magnitude higher concentrations than “active” GLP-1 (7-36)NH2 and shows cardioprotection, antioxidant properties (138) and appears capable to also inhibit hepatic neoglucogenesis (141).
GLP-1 (7-36)NH2 itself is known to have general protective and modulating cardiovascular effects (143), as shown by different commercial GLP-1 mimics with proven cardioprotection type 2 diabetes (144).
In healthy fasted individuals, it is recognized that peripheral plasmatic active GLP-1 (7-36)NH2 plasmatic levels hover around 5 pM, but within 5–10 min after an oral glucose load, they start to rise, up to a maximum of less than 10 pM after 40–90 min, and slowly descend back to baseline values in 150 min. On the other hand, the cleaved GLP-1 (9-36)NH2 summed to the GLP-1 (7-36)NH2 to give what is normally referred to as total GLP-1 levels, raise up to more than 40–60 pM (108). In perspective, GIP and Insulin show much broader dynamic ranges, with post meal levels reaching 300 and 400 pM respectively, from their baselines <20 pM within 30 min post glucose ingestion (108, 145). Curiously, some bariatric RYGB patients experience up to a 10-fold increase in post-meal active GLP-1 plasmatic levels (from fasting 5 pM to post-prandial 30–65 pM) (146), and have a 2- to 3-fold higher glucose-stimulated Insulin secretion (147), which in some diabetic patients results in GLP-1-mediated hyperinsulinemic hypoglycaemia that requires GLP-1 antagonism or surgical reversal of the intestinal anatomy (148).
Different authors consider the success of surgical intervention a consequence of a major change in gut hormonal profile, primarily a supra physiological post-prandial GLP-1 secretion (29, 30). This reasoning fits with the observation that type 2 diabetic patients display a shorter post-prandial peak of GLP-1, hence they are deficient for the longer response seen in healthy individuals. Multiple groups describe diabetic patients with lower plasmatic GLP-1 but heightened GIP levels and β-cell resistance to the stimulatory effect of both GLP-1 and GIP (18, 149–153).
Nonetheless, different animal models deficient for GLP-1 signaling, in addition to human studies, prove the dispensability of GLP-1 for surgical success (31–34), questioning the causative nature of GLP-1 for the reported metabolic benefits.
On the other hand, PYY has been proven to be upregulated, and necessary, for RYGB mediated restoration of the diabetic islets, and overall cure of diabetes in rats (35) and humans (154).
Another important source of endogenous GLP-1 is the brain, a tissue where it acts as a neurotransmitter. Indeed central GLP-1 production appears essential, since peripheral GLP-1 is assumed to not be able to cross the blood-brain barrier (BBB). In particular, neurons of the hindbrain found in the nucleus-tractus solitarius (NTS) secrete GLP-1 and activate hypothalamic neurons of the paraventricular nucleus (PVN), resulting in satiety (155, 156). Indeed it is clear that PC1/3 dominant neurons of the NTS express also other the PG peptides oxyntomodulin, glicentin, and GLP-2 together with GLP-1 (157). Although expressed at much lower levels, PC2 activity has also been recognized in these neurons, and traces amounts of glucagon might have important implications.
NTS neurons-derived GLP-1 appears to reach out to multiple locations within the central nervous system (CNS), which have been proven to express the receptor, and be activated after a central administration of GLP-1 receptor agonists. These areas include the NTS itself, the supraoptic nuclei, the arcuate nucleus (ARC) and the area postrema (AP) other than corticotropin-releasing hormone (CRH) PVN neurons (158, 159). Beyond satiety, this signaling appears to be a key factor for neuroprotection (160) insulin sensitivity and glucose metabolism (158).
Curiously, the feeling of satiety, is also achieved by another neurotransmitter, the Cocaine- and amphetamine-regulated transcript (CART) (161). This peptide, acts also as a hormone, and is expressed by both β-cells and intestinal GLP-1 and GIP producing cells causing GLP-1 secretion in vivo via a yet unknown GPCR (162).
It is not entirely clear to what extent endogenous GLP-1 activates all the reported GLP-1 receptor expressing neurons and to what extent it depends on the CART peptide especially in type 2 diabetes or obesity. Nonetheless, some commercial mimics of GLP-1, such as Liraglutide, even when administered peripherally, appear to cross the BBB and activate neurons within the ARC resulting in GABA dependent inhibition of neuropeptide Y (NPY) and agouti-related peptide (AgRP) secretion. This signaling has proven to be essential for the Liraglutide mediated weight loss in rats (163). GLP-1R expressing hypothalamic neurons have proven dispensable for the beneficial metabolic activity of both BBB permeable Liraglutide and Exending-4 (164).
Singularly, BBB impermeable mimics of GLP-1 have still shown to activate GLP-1 Receptor expressing neurons (165), but they require a functional gut-brain axis through the vagus nerve (166). In particular, vagal afferent neurons expressing the GLP-1R are necessary for GLP-1 mediated induction of satiety (167) but not glucose lowering effects (168).
The complex inter-organ pharmacokinetic of GLP-1, compounds into a convoluted pharmacodynamics encompassing multiple metabolic systems.
Indeed the GLP-1(7-36) NH2 receptor, a GPCR, is found to be expressed by a wide range of tissues and cells such as: α, β, and δ-cells (169), sinoatrial node myocytes, arterial smooth muscle cells of lungs and kidneys, megakaryocytes, macrophages, monocytes, lymphocytes, gastrointestinal tract mucosa [mainly Brunner's gland in the duodenum, but also in the parietal cells of the stomach, jejunum ileum and the nerve plexus around the small and large intestine (170, 171)], central nervous system [neocortex, cerebellum, thalamus, amygdala, area postrema, hypothalamus, hippocampus, nucleus tractus solitarius (158)], peripheral nervous system (myenteric plexus) and in the skin (14, 172–176).
Counterintuitively, mice completely defective for the GLP-1 receptor were reported to be protected from high-fat diet-induced peripheral Insulin resistance (177) and, consistently with this, central inhibition of GLP-1R signaling with the antagonist exendin 9-39 improves glucose tolerance and glycaemia (178). Conversely, mice defective for both the receptors for glucagon and GLP-1, or GLP-1 and GIP, show a highly plastic entero-pancreatic system that adapts and gives these animals no overt phenotype in terms of glucose homeostasis (105).
Nonetheless, the pharmacological activation of the GLP-1R is clinically beneficial (179), offering an improved glycaemic control with lower cardiovascular morbidity and without the risk of hypoglycaemia associated with some current anti diabetic drugs (173). Furthermore, being an appetite suppressant, GLP-1 signaling also helps to lose body weight, especially if in combination with metformin. Conversely, anti-diabetic drugs such as sulfonylureas, or Insulin, are known to induce not only weight gain (180, 181), but also an increased risk of hypoglycaemic events (182). Pharmacological activation of the GLP-1 Receptor has also shown to help exogenous insulin in the control of glycaemia in patients with type 1 diabetes, by slowing the gastric emptying and blocking glucagon secretion (183, 184).
Currently, six different peptide GLP1-Receptor agonists are on the market, with more in clinical trials. In particular, two short-acting formulations of Lixisenatide and Exenatide and four long acting preparations of Exenatide, Liraglutide, Dulaglutide and the most recent and successful Semaglutide, were approved in October, 2017 for the North American markets by FDA1 (25, 185). The first GLP-1 analog to be approved by FDA in 2005 for the management of Type 2 diabetes was the chemically synthesized Exenatide under the name of Byetta (186), a formulation of the DPP-IV resistant peptide discovered in the gila monster Heloderma suspectum saliva in 1992 (187). Despite the longer half-life in serum, Byetta needs to be injected twice a day. In the last decade, formulations with extended release entered the market with once-weekly self-administrations pens.
Pleiotropic beneficial effects have been reported for this class of drugs. Beyond the improved glycaemia control, essential for the short term treatment of diabetes (188), different GLP-1RAs are powerful clinical tools for the management of diabetic kidney disease (DKD) (28, 189) non-alcoholic steatohepatitis (NASH) (190), neuroinflammation (191), obesity and cardiovascular disease (192–195).
Although GLP-1RA are improving the lives of patients affected by type 2 diabetes or the metabolic syndrome (196), the physiology of GLP-1 is far from being clear.
More recent data suggest how the unimolecular co-activation of GLP-1 and GIP receptors, has powerful anti-diabetic effects superior to either agonism (197). Furthermore, oxyntomodulin is a natural dual-agonist of GLP-1 and glucagon receptors and displays anti-diabetic properties in humans (198, 199). Upon this finding, a tri-agonist peptide, targeting the receptors of GLP-1, GIP, and glucagon was created (200). The in vivo effects of this drug are unparalleled, even superior to what can be achieved with the dual agonists for either combination. The synergistic activation of these three important receptors is capable to revert diet-induced obesity, cognitive impairment and T2D in mice models, warranting future human studies (201, 202).
Expanding the Physiology of GLP-1
When examining the physiology of glucagon-like peptide-1, it is important to consider that there is an expanding body of evidence that questions its systemic endocrine physiology (203, 204). Pancreatic α-cells have been demonstrated to express and secrete not only GLP-1 (205, 206), but also PYY (35) GIP (207, 208) mini-glucagon (209) or even Xenin (210) together with glucagon (Figure 2). The key protease responsible for the processing of the proglucagon peptide into GLP-1 is Psck1/3, which has shown to be upregulated in α-cells during hyperglycaemic, hyperlipidemic, or inflammatory conditions to promote glucose-induced glucagon suppression, a compensatory response to a metabolic insult as in type 2 diabetes (205). Insulin itself has shown to modulate PC1/3 expression to possibly aid its own metabolic activity (211).
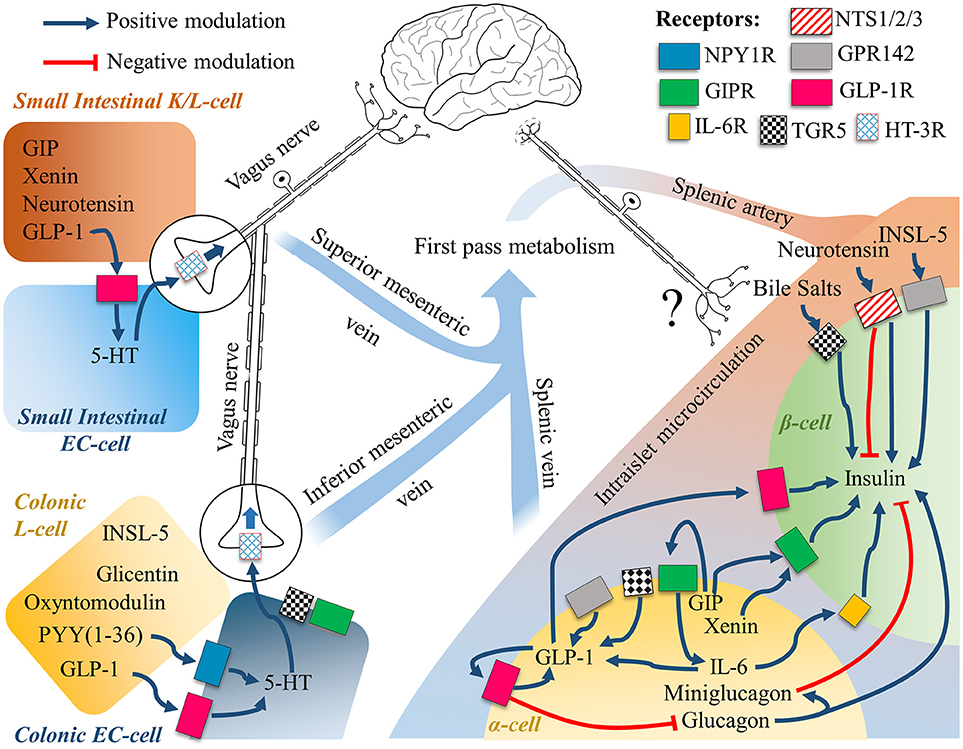
Figure 2. The gut-brain-islet axes of GLP-1. The intestinal EECs secretome is subject to first pass metabolism, while intraislet signaling relies on paracrine signaling. Intestinal cells are known to communicate with the Enteric Nervous System, and the Central Nervous System through the Vagus Nerve. Neuronal engagement between the gut lumen and the islets of Langerhans is a possible compounding explanation to the incretin effect, whereby the mechanistic of the single molecular players are still largely unknown. See text for further details.
Recently, the whole dogma of the role of intestinal GLP-1, envisioning the traveling from the gut to the liver and ultimately reaching the pancreatic β-cells to bind its GLP-1R has been questioned in transgenic mice (204). Indeed, since both DPP-IV degrades and NEP24.11 degrade GLP-1 within seconds, the possibilities of any intestinal GLP-1 to reach the system circulation and then the islet microcirculation are doubted. Besides, it is important to consider that intestinal GLP-1 has a local concentration in the nM range (10–100 pico moles per gram of tissue, see Figure 1), further advocating that the main action of this protein have evolved to be locally restricted.
Animals deficient for the GCG gene in the intestine, still experience a normal incretin effect disrupted with the GLP-1R antagonist Exendin (9-39) (204). This indicates that it is the intra islet, α-cell derived GLP-1 that shows the meal-induced insulinotropic properties. A critic to the use of a murine model deficient for intestinal GCG products, would be that other gut hormones might compensate for the lack of a functional GCG gene in that tissue, hence explaining the normalized incretin effect. Indeed other gut hormones such as GIP must be responsible for the incretin effect to a higher degree than once thought. Nonetheless, it is also clear that intra-islet GLP-1R signaling is essential for GSIS, with more evidence that an intra-islet paracrine GLP-1 signaling is physiologically present (212, 213) and necessary for β-cell health under metabolic (214).
In contrast, mice deficient for GLP-1R only in β-cells have a normal incretin response and oral glucose tolerance, indicating the dispensability of intra-islet signaling of GLP-1 for the incretin effect. Interestingly, these same animals have an improvement of their glucose tolerance in response to oral DPP-IV treatment, but not to subcutaneous GLP-1 mimics, indicating how the former relies completely on localized, non β-cell GLP-1R (215).
There are still multiple gaps into our understanding of how different GLP-1 producing tissues communicate, especially in the brain to islet axis. It is known that acute, but not chronic, central GLP-1 receptor activation directly modulates glucose-induced Insulin secretion implicating a direct brain to islet neuronal communication (61).
On the other hand, chronic GLP-1 activity in α-cells increases its own secretion, feeding an autocrine loop that gets overstimulated with the use of exogenous synthetic GLP-1R agonists [(98); Figure 2]. Curiously in diabetic rats, it has recently been shown that this loop might indeed induce the production of more glucagon than in healthy animals (99).
It has been known for more than two decades and has been confirmed more recently, that an infusion of GLP-1(7-36)NH2 has insulinotropic and glucagonostatic effects. This is seen when the plasmatic levels are above 50–60 pM, equivalent to more than five times the levels seen post-prandial in healthy individuals challenged with a bolus of glucose, or 10-fold their basal levels (153, 216), adding further doubt to the physiological hormonal dogma of intestinal GLP-1. Considering the mounting evidence, it is clear that we need to understand what hormonal and/or neuronal signals are bridging the gut luminal content to the insulin secretion explaining the incretin effect. Given that Intestinal oxyntomodulin, glicentin, glucagon and GLP-1 expression have proven to be dispensable in mice (204); other intestinal hormones such as GIP, PYY, Neurotensin, INSL-5 or the GIP co-secreted Xenin (217) might play an important role (Figure 2). Currently, not much is known about the physiology of Neurotensin, INSL-5 and Xenin. The first two have been reported to be co-expressed with GLP-1 in the small and large intestine respectively, with Neurotensin being reported also in pancreatic β-cells (210), while Xenin in a sub population of duodenal GIP positive cells and α-cells. Neurotensin levels are correlated with leptin (218), rise in response to fatty meals, signals through two different G-protein coupled receptors known as NTSR1 and 2, and a third single transmembrane receptor, NTSR3, also known as sortilin (219). All of these receptors are expressed by pancreatic β cells, where their activation appears to mediate insulin release at low glucose levels and blockage at high levels (219, 220), (see right side of Figure 2). On the other hand, INSL-5 targets a GPCR known as GPR142, also known as RXFP4, a receptor found to be expressed by the NCI-H716 cell line (54), and both α and β-cells in the pancreas, and its activation directly stimulates the expression of GLP-1 and insulin, representing a possible new pharmacological tool for the treatment of type 2 diabetes (77, 221), and supporting a possible role for INSL-5 in the incretin effect. Xenin is another gut-derived food-induced peptide known to potentiate GIP activity (222, 223). Considering that α and β-cells express GIPR (224) and that the GIP-potentiating activity of Xenin has been reported to be lost in human diabetics (223), it appears to be a critical player in this disease, likely involving the activity of GLP-1.
In addition, both in vitro and in vivo Interleukin-6 (IL-6) has shown to be a powerful GLP-1 secretagogue, capable to positively modulate both the proglucagon gene, and the expression of PC1/3 in α-cells and intestinal L-cells (225, 226). Indeed, GIP has shown to not only be co-expressed with GLP-1 and glucagon in α-cells (207); it also stimulates in an autocrine/paracrine fashion the expression of IL-6 in the same α-cells, thus indirectly acting as a GLP-1 secretagogue (95).
IL-6 has shown also to induce the secretion of intestinal GLP-1, indirectly via the release of adipocytes derived Leptin (227).
Curiously, it was recently reported that this pro-inflammatory cytokine, IL-6, similarly, but independently from GLP-1, slows gastric emptying (228). Furthermore an inflammatory status, as seen in pathologies such as type 2 diabetes, might compromise the gut mucosal permeability, leading to the exposure of intestinal EECs to luminal LPS, and a TLR4-mediated release of GLP-1 (229). This is consistent with the knowledge that GLP-1, as well as glucagon, has shown to possess powerful anti-inflammatory properties in vivo, an area that hold with vast therapeutical potential (136, 230).
Ghrelin is another possible player, since it has been proven to be expressed not only in the gut, but also in a distinct subpopulation of islet cells named ε-cells (231) and, being known to be a stress-induced (232) GLP-1 secretagogue (233, 234), it might play an important role in the intra-islet signaling.
Recently, it has been demonstrated that mice with a deletion of the GLP-1 receptor only in β-cells, are resistant to the beneficial anti-diabetic effect of a vertical sleeve-gastrectomy (36), suggesting how GLP-1 activity in β-cells is key to the bariatric surgery success. It is not known if intra-islet α-cells production of GLP-1 is affected by the surgical procedure or, more importantly, how this axis is impaired in the metabolic syndrome, type 2 diabetes and related pathologies.
It appears that only in RYGB and SG patients intestinal derived GLP-1 has a true endocrine role, while in healthy individuals, localized, paracrine and neuronal signals primarily define the GLP-1 physiology.
It is therefore clear that currently available GLP-1RAs, mimicking on the peripheral action of GLP-1 (7-36)NH2, not only ignore the yet unknown physiology of GLP-1 (9-36)NH2 or its metabolites, but they also fail to address the tissue specific physiology of GLP-1 (7-36)NH2, while pushing to supra-physiological limits the endocrine GLP-1 receptor axis, likely explaining the reported side-effects and only partial success in the treatment of T2D.
In addition, it is important to notice that the ubiquitous DPP-IV protease targets not only GLP-1 but also oxyntomodulin, GIP and PYY among other proteins (235). Specifically, the GLP-1 co-secreted cousin PYY(1-36), agonist of the vasoconstrictive Y(1) receptor, is physiologically trimmed by DPP-IV to give rise to the appetite-suppressant, anti-diabetic and blood-brain barrier permeable PYY(3-36) agonist of Y(2) receptor (220). It is therefore clear that pharmacological DPP-IV blockage disrupts this axis and induces hypertension (236).
Recent studies provide new evidence supporting the paracrine nature of intestinal GLP-1, whereby Serotonin-(5-HT)-secreting enterochromaffin (EC) cells are directly stimulated by locally produced GLP-1, which in turn stimulate afferent Vagal nerves (Figure 2) bridging the gut to brain axis. Accumulating evidence suggest that, especially in the colon, EC cells express multiple receptors for the microbiome metabolites, representing a new important link bridging the microbiome to the brain (237, 238).
A better way to amend the pathophysiology of GLP-1 reported in diabetes or other diseases, would be to induce tissue specific de novo GLP-1 production, leading to a more physiological and likely safer, short and medium distance signaling. Numerous attempts have been made with multiple GLP-1 secretagogues such as GPR119 agonists (239) but so far no compound has reached the market because of bioavailability issues and systemic off-target toxicity. One possible way to minimize the side-effects of the single drugs is to combine them to achieve synergistic effects, as reported recently with a combination of a DPP-IV inhibition, SSTR5 antagonism and GPR40 and TGR5 agonism, capable to raise circulatory active GLP-1(7-36)NH2 levels to more than 300-400 pM in mice (240).
Sweetness in the Gut
Studies in vitro and ex-vivo with isolated human primary cells suggest that there are two temporally distinct pathways that lead to the glucose-stimulated release of GLP-1, similarly to what happens in β-cells with the 1st or 2nd phase insulin release. A quick mechanism independent of the cell energetical state and a slower one, metabolism dependent, mediate the release of this incretin (53, 72).
The 1st phase in the pathway of glucose signaling, sees the electrogenic sodium-coupled glucose transporters 1 (SGLT1) mediated uptake of two Na+ ions for every internalized glucose molecule (53). This depolarization is propagated through voltage-dependent Calcium and Sodium channels, which currents lead to the discharge of the hormones containing vesicles (72).
The 2nd phase is exemplified by the absorption of simple sugars, such as Glucose or Fructose, via the facilitative transporters GLUT2 and GLUT5 respectively, which leads to an increased internal metabolism mirrored by intracellular ATP levels. This state leads to the blockage of ATP dependent potassium channels and the subsequent membrane depolarization, followed by the secretion of the hormonal cargo.
Mace et al. (241) demonstrated how diazoxide, a K+ATP channel opener, completely abolished the glucose-dependent incretin release while a channel blocker, tolbutamide, exacerbates it in terms of secreted GLP-1, GIP and PYY.
More recent data, question the first mechanism in enteroendocrine cells. Glucose mediated GLP-1 release happens in humans only in the proximal and distal small intestine and independently of ATP mediated potassium channels closure. Furthermore, concentrations of up to 300 mM glucose do not induce GLP-1 secretion from colonic human mucosa because GLP-1 producing L-cells barely express SGLT1 (43, 53, 58, 72).
Consistently, the use of α-methyl-D-glucopyranoside (MDG), an acaloric substrate of SGLT1, within 5 min triggers the release of GLP-1 as glucose does, demonstrating how it is the sodium current that triggers the release of the incretin, and not the metabolic ATP-driven arrest of potassium currents and following calcium spike (58).
The pharmacological blockage of SGLT-1 with phloridzin, in a rat small intestine perfused system, results in just a halved secretion of GIP, GLP-1, or PYY, and the addition of phloretin, a GLUT2 inhibitor, brings these values down to basal levels. In fact, this double blockage of SGLT1 and GLUT2, completely inhibits the responsiveness to other stimulants as well, such as sucralose, glycylsarcosine, OEA, propionate and taurocholate. The activity of the calcium channel CasR is also essential for the responsiveness to free aminoacids (241).
All these observations are challenged by longer term in vivo studies. Blockage of SGLT-1 markedly improves glucose-stimulated GLP-1 release if a 3-h long period is considered.
The rationale given by Oguma et al. (242) is that SGLT-1 is expressed mainly in the small intestine, hence its inactivation results in heightened luminal glucose that travels down to the colon where it someway stimulates GLP-1 release. Given the fact that SGLT-1 is barely detectable in colonic proglucagon positive cells and that potassium channels in this tissue are unresponsive to sulfonylureas, the molecular sensor(s) that causes the release of GLP-1 in vivo, remains elusive.
Another enigmatic G protein is α-gustducin, a key element in sweet-taste transduction pathways downstream of the heterodimer formed between the GPCRs Tas1R2 (T1R2) and Tas1R3 (T1R3).
Its expression has been reported in colonic L-cells and appears to be responsible for the glucose-stimulated release of incretins (243, 244). This is confirmed by the impaired glucose-stimulated release of GLP-1 in mice lacking either T1R3 or α-gustducin (244).
Interestingly, this axis is also activated by the disaccharide sucrose and by the non-metabolizable and therefore anergic sucralose (243). Of note also Aspartame, Acesulfame K, Glycyrrhizin and Saccharin bind the sweet receptor heterodimer Tas1R2/3 and they have shown to stimulate GLP-1 secretion in the human duodenal adenocarcinoma-derived HuTu-80 cell line (245, 246). Despite this report, other groups weren't able to replicate these results (53). Indeed, it was shown that proglucagon expressing cells, derived from the colon of Venus mice cultures, were not responding significantly to Sucralose (1 mM) in terms of both released GLP-1 and intracellular Calcium. Conversely, proglucagon negative cells responded to the sweetener. More doubts about the role of Tas1 receptors were raised after the demonstration that oral gavage with sucralose, saccharin, stevia, acesulfame potassium or tryptophan do not cause a gut incretin release in Zucker diabetic fatty rats (247).
Long and Middle Chain Fatty Acid Receptors
The study of the receptome of enteroendocrine cells, has provided invaluable pharmacological insight with the discovery of proteins capable to sense multiple compounds once thought to be only nutrients.
A prime example is given by two GPCRs, GPR40 and GPR120, also known as Free Fatty Acid Receptor 1 (FFAR1) and 4 (FFAR4) respectively. These chemosensors are two major molecular players in the detection of dietary, medium (C8-12) and long (C14-22) chain fatty acids (LCFA) (84, 248).
GPR40 is primarily expressed by the pancreatic β-cells, where it plays a pivotal role in FFA-mediated insulin secretion (249) but also in α-cells (78, 250), CCK (80), GIP (251), and GLP-1 (79) producing cells in the gut and in hypothalamic neurons (248, 252, 253). Animals deficient for this receptor are protected from obesity-induced hepatic steatosis, hyperinsulinemia, hypertriglyceridemia and hyperglycaemia. More than a decade ago a study showed that GPR40 mediates the long-term FFA-induced lipotoxicity seen in the diabetic islets (254); nonetheless, these findings are still under debate today. Recent data are still highly polarized, with some authors supporting (255), and others disproving this (256), or even indicating that GPR40 protects β-cells from lipotoxicity (257) rendering difficult to draw any conclusive mechanistic involvement in healthy and diabetic individuals. Nonetheless, the activation of this receptor with FFAs has demonstrated to induce the secretion of incretins (79, 258) glucagon (78, 250) and partially glucose-stimulated insulin (259, 260) reducing food intake, and lowering body weight in animals models (261). Mice without a functional GPR40 display an impaired CCK and GLP-1 secretion after an oil gavage, while surprisingly animals deficient for GPR120 display a normal corn oil-induced GLP-1 secretion (80, 262).
GPR40 is coupled to both Gq and Gs proteins and in vivo studies suggest how signaling through both these cascades elicits the most powerful GLP-1 secretion (258). Ligands that bind GPR40 and activate predominantly only the Gq pathway are not good GLP-1 secretagogues. Indeed recently it has been shown that dietary triglycerides appear to induce the secretion of GLP-1 via GPR40 in synergy with the Gs activating GPR119 (263). Nonetheless, chylomicrons have been reported to be powerful GPR40-Gq activators and GLP-1 secretagogues, acting from the basolateral side of the intestinal mucosa (264).
The two synthetic GPR40-specific compounds AM-1638 and AM-5262, have been found to act as double Gq and Gs agonists but also as positive allosteric modulators, capable to enhance the GLP-1-secreting capabilities of Gq-only agonists such as dietary docosahexaenoic (DHA) and α-linolenic acid (ALA), independently of the orthosteric site (265).
GPR120 shows very little sequence similarities to the other free fatty acid receptors but, likewise, is found to be expressed by the enteroendocrine cell system, especially in the colon (see Figure 3), but also in the lungs (267), white and brown adipose tissue (274, 275), hypothalamic microglia (253), macrophages and, contrarily to GPR40, not in β-cells but in somatostatin producing δ-cells (276). Both small intestinal GIP and colonic GLP-1 secreting cells express GPR120, and the molecular cascade triggered by this receptor has been shown to mediate dietary incretin release directly or indirectly through CCK (84–86). Interestingly, both of these two receptors are expressed only by a fraction of hormone positive EECs; in particular, it has been reported that only 3% of GLP-1 positive cells express GPR40, and 23% GPR120 (266).
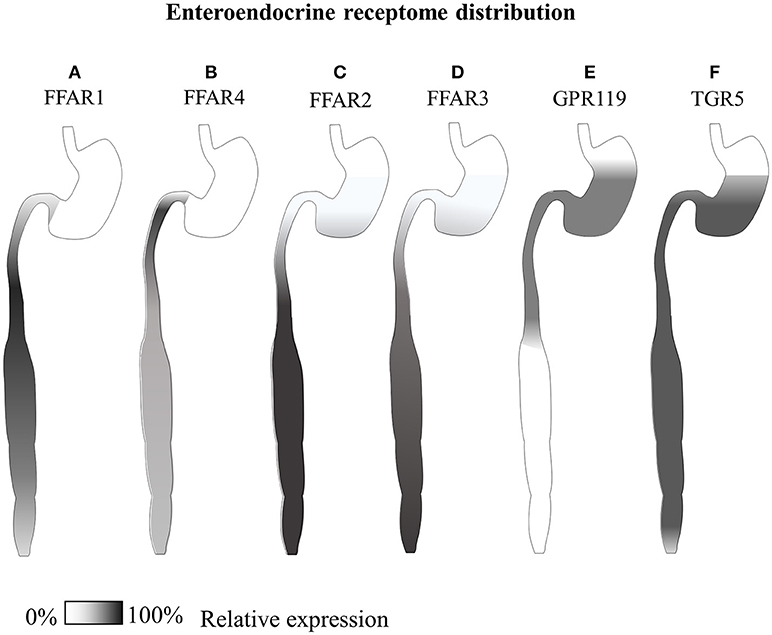
Figure 3. Gastrointestinal GLP-1-secreting receptome distribution. Summary of available expression studies of different GLP-1-secreting receptors along the gastrointestinal tract. (A) GPR40/FFAR1 has been reported to be expressed in the small intestine in different EECs, with overall higher transcript levels than GPR120, and superior co-localization with GIP In the distal small intestine (79, 80, 266). (B) GPR120/FFAR4 has shown co-expression with both proximal small intestinal GIP+ and large intestinal GLP-1+ cells (85, 266–268) (C) GPR43/FFAR2 and (D) GPR41/FFAR3 are co-expressed by all types of enteroendocrine cells, from the stomach to the rectum, especially in the colon (83, 269, 270). (E) Reports of comparative GPR119 transcript are contradictory, while immunohistochemical data indicate co-localization with a minor fraction of CCK and GLP-1 positive cells mainly in the stomach and small intestine (266, 271). (F) TGR5, has been reported equally distributed along the whole gastrointestinal tract of dogs (101, 272, 273).
GPR120 displays a ligand preference similar to GPR40; a broad range of long chain fatty acids signal through it, with some ligands eliciting more robust calcium responses than others (84). Multiple dietary compounds have shown to be powerful agonists of GPR120, such as pinolenic acid, a poly-unsaturated fatty acid (C18:3 trans, cis, cis Δ 5, 9, 12) found in pine nut oil (277), or the yeast derived phytosphingosine (278).
In macrophages and adipose tissue, GPR120 mediates ω-3-mediated anti-inflammatory and insulin sensitizing effects (279, 280). Contrarily to GPR40, the genetic deficiency of GPR120 is more dramatic. Knockout animals show hyperinsulinemia and insulin resistance, hyperglycaemia and osteoarthritis (281), hepatic steatosis and therefore obesity. Furthermore, an absence of GPR120, results in an overactive glucagon signaling, explaining the hyperglycaemia (282). Indeed, in humans, a single aminoacid mutation of the receptor that hampers its signaling is associated with obesity and insulin resistance (283). Expectedly, GPR120 agonism shows powerful anti-diabetic, anorexic, and hepatoprotective properties in multiple animal models (275, 284–287), at least partially mediated by GLP-1 (288).
Considering the overlap of natural ligands of GPR40 and GPR120, it has been difficult to study them individually and understand their individual physiology, while recent data indicate that indeed these two receptors work synergistically, to exert anti-diabetic activity in vivo from the gut (289), and the brain (253).
Despite these advancements, in clinics there are currently no available drugs targeting GPR40 and GPR120. TAK-875, the best candidate for GPR40 which showed promising GSIS capabilities up to Clinical Phase III for the treatment of T2D, had to be halted because of hepatotoxicity and alteration of bile salts composition (290).
Despite these setbacks, encouraging animal data warrant future efforts for the development of new drugs capable to activate synergistically both GPR40 and GPR120 and mediate, through GLP-1 and other intestinal, pancreatic and cerebral peptides, better treatments for multifactorial chronic metabolic diseases.
Short Chain Fatty Acid Receptors
In 1997, four 7 α-helixes transmembrane receptors, GPR 40, 41, 42, and 43 were mapped on the same locus found on the long arm of chromosome 19 (291). Soon after, different groups identified GPR 43 and 41 as the receptors for free fatty acids, which were then chronologically renamed FFAR2 and FFAR3 respectively (292–294).
Both these receptors are activated by similar types of short chain fatty acids (292), and both these signal through an inhibitory G type protein, but FFAR2 is also capable to signal through Gq/11 proteins (293) by which it has shown to mediate GLP-1 and PYY secretion in vitro and in vivo (82, 295).
Along the gastrointestinal tract, both GPR 41 and 43 have been reported to be co-expressed, with FFAR2/GPR43 at higher levels and overall number of cells, especially intraepithelial leukocytes, while FFAR3/GPR42 is found on submucosal neurons [see Figure 3, (83, 295–297)]. Indeed FFAR2 holds promise for the management of Inflammatory Bowel Disease (IBD) (298) a possible side-effect of anti-diabetic treatment with DPP-IV inhibitors (299).
Feeding rats with fructo-oligosaccharide as a source of SCFAs has also shown to upregulate FFAR2 (270). Recently, both the receptors have shown to heteromerize in vitro, eliciting synergistic signaling and β-arrestin-2 recruitment (300). Furthermore FFAR2 activation in vivo with an inulin-enriched diet in mice results in PYY release and proliferation of L cells in vitro (81). Nonetheless, there is still some controversy on the in vivo involvement of FFAR2 and FFAR3 in GLP-1 modulation (301, 302), with some reports indicating that blockade of GPR43 in vitro releases GLP-1 (303) and others indicating different mechanisms of action, with FFAR2 releasing PYY from intestinal L-cells (81), while FFAR3 restricted to submucosal neuronal activity (295) despite its apparent expression by the majority of enteroendocrine cells (83).
In pancreatic β-cells, both GPR43 and GPR41 are expressed, and the latter antagonizes GSIS (304).
Adding complexity to the study of these receptors, there is extensive species-specificity, so that animal findings result in poorly translatable data, requiring the generation of complex human-murine chimera currently under intense study (305, 306).
Nonetheless, considering that the half-maximal effective concentration (EC50) for Acetate, Propionate, and Butyrate is around 0.5 millimolar upon both GPR41 and GPR43 (292) and that the SCFA concentration in the human ileum and colon lumen is superior to 100 millimoles per kg (307–309), it is likely that both receptors are constitutively active. Obese patients, have been reported to produce more SCFAs in their intestines (310), but indeed meaningful diet-induced shifts in SCFA production fluxes have proven not sufficient to modulate peripheral levels of GLP-1 and PYY (311).
GPR42 is another G-Protein-Coupled-Receptor that was initially considered to be an inactive pseudogene derived from GPR41. In 2009, 29% of 202 human alleles of GPR42 were shown to have an inactivating single nucleotide polymorphism (SNP) at W174, and 61% with an arginine in like GPR41, resulting in a fully functional receptor, differing from it by only 5 aminoacids (312). A more recent study highlights how GPR42 is not only functional, but displays a pool of haplotypes in a great proportion of humans, with a distinct pharmacology (313).
GPR119
GPR119, also known among other names as glucose-dependent insulinotropic receptor (GDIR), was independently discovered less than two decades ago by several groups around the world and deorphanized soon after with the discovery of Oleoylethanolamide (OEA) as its first endogenous ligand (314–316).
Recently our group has demonstrated that indeed OEA is just a partial agonist of GPR119, and the biological ligand of this receptor is the lysophospholipid Oleoyl-Lysophosphatidylinositol (Oleoyl-LPI) (87). This bioactive lipid induces a powerful GPR119 mediated-GLP-1 secretion in vitro and ex-vivo from intestines of wild type, but not GPR119 deficient mice. This peculiarity is not shared by LPI species with different aliphatic chains, which have been described as the ligands of GPR55 (317).
This GPCR is primarily expressed in the pancreas by α-cells, β-cells and γ-cells (271, 318, 319), and is found at lower concentrations along the GI tract, especially in the stomach and duodenum, where counterintuitively only a minor fraction of CCK, and GLP-1 expressing duodenal enteroendocrine cells display GPR119 (266, 271). This receptor is also expressed, and hence can be studied, in vitro, by the human enteroendocrine cell model NCI-H716 or by the murine GLUTag cell line (320). Heterologous expression in vitro unveiled its constitutive activity capable to raise intracellular cAMP levels through Gαs (321) and lead to the secretion of GLP-1 and PYY (89). Rodents, contrarily to humans, express GPR119 also in some regions of the brain (316). The activation of this receptor is known to mediate glucose-stimulated insulin secretion and a glucose-independent release of incretin hormones by intestinal enteroendocrine cells (88).
Long-chain fatty acids and phospholipids like lysophosphatidylcholine (LPC), other compounds such as retinoic acid (RA) and multiple N-acylethanolamines (NAE) such as N-oleyldopamine (OLDA), palmitoylethanolamide (PEA), or oleylethanolamide (OEA), all act as endogenous ligands of GPR119. OEA is a more potent GPR119 agonist than its glycerol ester 2-Oleoyl Glycerol (2-OG) found in olive oil (322).
Indeed, oleic acid is internalized via CD36 and converted to OEA in the duodeno-jejunal enterocytes, which in turn causes satiety directly via PPAR-α (323) or indirectly through an incretin secretion mediated via GPR119 in the gut (324). Curiously, fat-induced OEA synthesis is a fairly conserved pathway in metazoan, being present in fish and extremely slow-metabolism reptiles such as pythons (325, 326).
Triglycerides, with medium length fatty acids such as 1,3 Dioctanoyl- 2 Oleoyl-glycerol, can also cause the release of GLP-1 in humans. However, this happens via the metabolized 2-OG component, since dietary medium chain fatty acid do not cause any appreciable release of incretins (322).
Counterintuitively, long term olive oil feeding does not improve glucose tolerance or insulin responses in diabetic rats (5). Indeed, more recently it has been reported that a high-fat diet enriched in oleic acid leads to an impaired endogenous OEA and other N-acylethanolamides intestinal production in mice (327), suggesting that a chronically resistance is taking place within the OEA synthesis pathway.
Surprisingly, a daily activation of GPR119 with OEA or other synthetic agonists, increases β-cell responsiveness in islets transplanted into STZ-induced diabetic mice (328).
The importance of GPR119 in the fat-induced incretin secretion is demonstrated by the impaired incretin signaling displayed by transgenic animals deficient for this protein only in PG expressing intestinal cells. Male and female mice, completely loose the GLP-1 response to an oral gavage of olive and corn oil (329).
More recently, it was reported that whole-body GPR119−knockout mice are protected from high-fat induced glucose intolerance and insulin insensitivity. Interestingly, the specific ablation of GPR119 only in β-cells does not affect glucose tolerance nor insulin secretion. In fact AR231453, a selective GPR119 agonist, improves glucose tolerance and insulin sensitivity in both WT and Gpr119βcell−/−, suggesting how insulin release is independent from pancreatic GPR119 but depends on gut incretin release (330).
Curiously, GPR119 activity appears to be directly dependent on the PYY receptor NPY1 (331). This phenomenon is independent of DPP-IV, the GLP-1 receptor, or the PYY related peptide NPY.
Furthermore, GPR40 also shows synergism with GPR119, mediating a more than additive GLP-1 response to triglycerides in the large intestine (263).
Agonism of GPR119 in both healthy or diabetic and obese mice, is known to improve glucose tolerance (90), or even prevent atherosclerosis in mice (332), while at the same time inducing the secretion of glucagon under low glucose levels avoiding hypoglycaemia (91); therefore since 2008, multiple agonists have been synthesized (239, 254, 333), as well as unimolecular dual DPP-4 inhibitors and GPR119 agonists (334). Despite the good results seen in rodents, species-specific pharmacology might be to blame (335).
Up to now all the prospective GPR119 agonists were plagued by low bioavailability, lack of efficacy and more importantly, cardiotoxicity which has stopped all human studies before any large scale Phase III clinical trials (239).
Despite the multiple failures, the compound DS-8500a is showing promising glucose lowering properties in Phase II clinical trials without any apparent toxicological issues in clinical trials (92).
TGR5
Bile acids (BAs) are cholesterol-derived molecules produced in the liver and temporarily stored in the gallbladder. When food is ingested, BAs are released into duodenum to solubilize dietary lipids under the form of micelles, a necessary step for the maximization of the surface-to-volume ratio of fat droplets, aiding interface-acting lipases.
Indeed the release of lipids from micelles has directly proven to release GLP-1 and GIP via the FFAR1 in the duodenum (264).
This release of bile acids, mainly cholic (CA) and chenodeoxycholic (CDCA) acid derivatives, happens through the relaxation of the smooth muscle sphincter upon CCK signaling (336) or indirectly through a similar VIP action on the sphincter of Oddi (337).
Historically described as mere fat-solubilizing agents, these amphipathic compounds were recently recognized as key signaling molecules capable to modulate the host metabolism directly acting as ligands of intestinal GPCRs (101, 338, 339), or after being metabolized by the colonic microbiota into secondary bile acids, mostly deoxycholic and lithocholic acid(340).
The chemosensor believed to be the main receptor of bile acids is TGR5, also known as GPR131 or GPBAR1 among other names. This receptor has been reported to be expressed by colonic GLP-1-secreting enteroendocrine cells and pancreatic α-and β-cells (100, 101), with some controversy regarding the presence in murine islets (339).
TGR5 activity appears to not have been lost in type 2 diabetic humans whereby the infusion of CCK, or rectal taurocholate, causes GLP-1 and insulin release via the TGR5 axis in colonic L-cells and pancreatic β-cells respectively (341, 342).
This notion is in stark contrast to the well-known anti-diabetic properties of BAs sequestrants, (343) and some, have proven to elicit GLP-1 secretion via TGR5 mediated PC1/3 upregulation (344). A likely explanation is that the BAs bound to a sequestrant into the intestinal lumen can't be absorbed and hence travel more distally in the GI tract where the complexes are still capable to activate the TGR5 expressing colonic L-cells. Furthermore, the lower systemic levels of bile salts prompt the liver to produce more bile, which in turn feeds more TGR5 agonism into the colon (343).
This chemosensor is expressed by the pancreatic α-cells where its signaling activates Gs proteins and induces the secretion of GLP-1 directly through Epac proteins and indirectly via CREB mediated expression of Psck1, while in β-cells mediates insulin release [(100); Figure 3].
TGR5 is the target of different BAs, but the most potent endogenous agonist has shown to be lithocholic acid (LCA) and its taurine conjugates with activity at nanomolar concentrations (273, 339). Secondary bile salts, metabolized by the microbiota, exhibit less potency toward this receptor.
Despite this promising anti-diabetic activity of TGR5 mediate by GLP-1 (345), its pharmacological activation in diabetic patients has shown side effects at the level of gallbladder and heart, hampering its clinical use (346).
Another bile salts chemosensor is the nuclear farnesoid X receptor (FXR) (347) which activation, contrarily to TGR5, blocks the release of GLP-1 in the colonic L-cells (348), while in the liver induces glycogenesis helping to improve glucose homeostasis. This counterintuitive pharmacology has been confirmed in vivo whereby the administration of the FXR agonist GW4064 by mouth drives hyperglycaemia and obesity (349) while intraperitoneal injection exerts protection from it (350). Consistently, an indirect inhibition of intestinal FXR through microbiota modulation, or genetic deletion of intestinal FXR, corroborate this phenome displaying protection from high-fat diets induced obesity and fatty liver disease (351).
This could explain why bile acid sequestrants support a positive glucometabolic homeostasis. Indeed, the insoluble complexes of bile salts can activate lumen-facing TGR5 receptors, while they cannot cross plasma membranes to activate intra-cellular GLP-1-suppressant FXR receptors.
FXR is a very important receptor, part of a negative feedback in the liver, whereby the binding of bile salts, especially chenodeoxycholic acid, represses the de-novo synthesis of bile salts (352, 353). Indeed, there are multiple primary or secondary bile acid chemosensors in the liver (348, 354) or scattered along the gastrointestinal tract (355), where they ensure a direct negative feedback aiding detoxification (356) and protecting from hepatotoxicity and carcinogenicity displayed by some secondary bile salt such as lithocholic acid.
Accumulated evidence, indicate how bile acids are important modulators of the whole body metabolism, bridging the microbiome to the brain, likely being key signaling molecules in the pathogenesis of obesity and type 2 diabetes. Indeed remittance from diabetes experienced by RYGB or SG patients, has been attributed to the elevation of circulating bile acids (37, 38, 357), warranting further investigation, especially the development of gut-restricted TGR5 agonists (358).
TRPV1 and the TRP Channel Family
The transient receptor potential vanilloid 1 (TRPV1) is a tetrameric non-specific cationic channel found in most of mammalian sensory neurons (359). Each of its constituting monomers crosses the plasma-membrane six times and both the N and C-term face the cytoplasmic side, where they make up 70% of the receptors' entire volume (360). This chemosensor, together with other 27 non-selective cationic channels, is part of a larger family named transient receptor potential (TRP) channel superfamily and is known to play an important role in the metabolic syndrome (361, 362).
TRPV1 is primarily activated by vanilloids and capsaicinoids including Capsaicin (360), eliciting the sensation of spiciness; multiple stress-related stimuli cause its activation and opening with subsequent membrane depolarization. For example cigarette smoke, excess of protons (pH < 5.9) (363), temperatures above 43° (360), certain animal toxins (364, 365), ATP (366) or even cannabinoids such as Anandamide (367) and cannabidiol (359, 368), are all stimuli known to activate this sensor. Indirect stimulation has also been demonstrated by bradykinin (366), NGF (366), PGE2 (369), PGI2 (369) and agonists of Protease-activated Receptors (PARs) (370).
TRPV1 has been shown to be expressed in the brain, β-cells (371), nociceptor C fibers, dorsal root ganglia, hepatocytes, spermatozoa (372), airway neurons (373), bladder and urothelium (374), blood vessels, and the whole gastrointestinal myenteric plexus (375), especially in colonic and rectal neurons (376). Consistently, TRPV1 is also found to be expressed by the murine enteroendocrine cell line model STC-1 and its agonism induces the release of GLP-1 in vivo (377).
This receptor has recently seen an increasing interest since its activation has been found to have pleiotropic beneficial metabolic effects (378).
Indeed, it has been known for more than a decade that capsaicin is capable to elicit a glucose-stimulated insulin release in vivo (379). A crossover study operated on 30 human healthy subjects (380), showed a slight increase in plasmatic GLP-1 and a slight decrease in ghrelin levels 30 min after a Capsaicin enriched meal (containing 1,030 mg of 80,000 Scoville heat units red pepper); Peptide YY changes were not statistically significant. Despite these promising results, TRPV1 knockout mice display contrasting phenotypes with the report of opposite phenotypes. One author describes an obese insulin and leptin resistant mouse (381), while another group report animal protected from diet-induced obesity (382).
Considering all the recent findings, drugs targeting TRPV1 would be beneficial for the management of obesity (383) metabolic syndrome (384) and type 2 diabetes (385). Nonetheless, considering the EECs receptome responsible for gut-peptide modulation, TRPV1 has received much less attention, with a yet largely unexplored physiology.
The Microbiota
Animals' GI tract is known to host a population of hundreds of different species of bacteria (386), viruses and fungi, estimated to equal in number the cells that constitute the human body (387). These microorganisms thrive in the colon's lumen, where they secrete small molecules ultimately affecting the host immunity (240) and metabolism (388).
The relative abundance of different microbial species is known to depend on the presence of specific nutrients (389); hence, considering that an imbalance in the microbiota correlates with chronic inflammation pathologies of the bowel, or even Type 2 diabetes, it is likely that dietary components indirectly influence the occurrence of these pathologies via the microbiota (390, 391).
The human colonic microflora is known to produce high concentrations of Short-Chain-Fatty acids (SCFAs), among other metabolites, from the anaerobic fermentation of dietary indigestible carbohydrates, or even derivatives of bile salts (389). In fact, the SCFAs Acetate, Propionate and Butyrate are the principal luminal anions in humans and other mammalian's colon (309, 392), with some inter-species variability. Rats show higher levels of fecal Acetate, 75 mM vs. human's 50 mM, Propionate, 27 vs. 11 mM and Butyrate, 16 vs. 5 mM respectively. On the other hand, surprisingly similarly to humans' colonic and fecal values, rumens of herbivores, such as sheep or cows, also contain high levels of acetate, propionate and butyrate, with reported concentrations of 65, 21, and 18 mM, respectively (308). These levels appear to be independent of dietary proteins or fibers; conversely, it is the caloric intake that affects the relative composition and concentrations of SCFAs (308). These metabolites have been found to target specific receptors among the repertoire expressed by the EECs, triggering a hormonal response. It is estimated that in humans almost all fermented SCFA are absorbed by the colonocytes and only 5% are excreted with stool, equivalent to 5–30 millimoles per day. Indeed, it is not practically feasible to measure intraluminal production fluxes of various metabolites in vivo in humans; therefore, most studies focus on the easiest but less informative quantification of fecal SCFA content (393).
Despite the most recent studies of transgenic and germ-free animals, it is still largely unknown by what degree hormones such as GLP-1, and all its related peptides, depend on the microflora, especially in pathologies such as type 2 diabetes.
Recent high-throughput pharmacogenomic studies have deepened our understanding of the molecular players in this human-microbiota relationship. Recently it was shown that a new class of N-acyl amides is produced by the microbiota, and target GPCRs expressed by the enteroendocrine cells, modulating GLP-1 expression and overall glucose metabolism. In particular, N-oleoyl serinol (N-OS) is described as a potent GPR119 agonist, acting in the lower micromolar range with twice the efficacy of the endogenous ligand OEA (394).
From the evolutionary perspective, dietary components, together with the microbiota-fermented products, have activated the enteroendocrine system for billions of years, since the dawn of metazoan. Considering the vast and continuous pool of metabolites produced and modulated by the microbiota, the distinction between orthosteric and allosteric ligand becomes blurred; different molecules are likely working in synergy to elicit a specific hormonal response.
Modulation of the microbiome has shown promising results in the treatment of type 2 diabetes. For example, recently it was reported that a rhubarb extract, Rhein, increasing the intestinal population of Bacteroidetes, mediates an increase in ileal GLP-1 producing cells, peripheral GLP-1(7-36)NH2 levels and improved glucose tolerance in diabetic db/db mice (395). Consistently, STZ-treated rats, are protected from oxidative and inflammatory stress when treated with Liraglutide, and Bacteroides, as well as Lactobacilli strain populations appear to be restored (396).
In the last decade, the scientific community has just started to unveil the molecular pathways produced by this long-lasting symbiosis. It appears that SCFAs not only induce the release of GLP-1, they also represent a mitogenic signal. Rats fed oligofructose, a substrate for the colonic microbiota which leads to higher SCFAs levels, possess an increased number of colonic L-cells (397). This has been confirmed ex-vivo in human and mouse small intestinal crypts organoids (398).
Other compounds such as bile salts and xenobiotics (399), are known to be metabolized and excreted by the microbiota, affecting the host physiology. Indeed, the pharmacokinetic and pharmacodynamics of any drug taken by mouth should be appraised considering the role of the microbiota, as the varied efficacy of some chemotherapeutics such as 5-FU has been proven to directly depend on this host-microbiota metabolism (400). Even though the anatomical intestinal rearrangement of RYGB and SG patients is known to affect the microbiota, this doesn't appear to result in a different bile acid metabolism in a rat model (401).
We are at the beginning of a new branch of medical practice, tailored not only to the single person genome, but also to the microbiome.
Future human studies will help us to better understand the big picture of this relationship, to hopefully provide mechanistic knowledge upon which new treatments could be created, such as microbiome-directed gene-therapies for the management of metabolic diseases.
Conclusion and Perspective
GLP-1R-independent signaling of GLP-1, its intra-islet axis, and its once-thought inactive metabolites, all represent new important additions to our understanding of this peptide in health and disease.
Omnivores' gastrointestinal tract has co-evolved in strict relationship with a dynamic microbiota and a complex seasonal and regional diet, resulting into a robust and flexible system tightly interconnected via multiple neuroendocrine axes with different organs.
In nature, dietary fats are scarce energy-dense nutrients primarily found in fish and meat. This evolutionary pressure over millions of years has shaped a system for the attentive sensation, assimilation and storage of precious bioactive molecules in all superior animals.
Sensation happens at multiple levels with a plethora of somewhat redundant intestinal receptors (402), specifically in the enteroendocrine cell system. This redundancy can be seen in transgenic animals, whereby the genetic absence of a single chemosensor doesn't always result in a phenotype, probably due to metabolic compensation from similar and overlapping pathways.
Virtually all macronutrients are absorbed in the small intestine, where maximal activity of the EECs is ensured, while the colonic and rectal GLP-1 secretion is enforced in response to secondary metabolites even hours after the meal ingestion. This pattern is disrupted in bariatric patients undergoing RYGB surgery, where a remodeled GI tract delivers more nutrients to the large intestine, and changes gut-secretome, including its microflora.
Attempts to mimic this altered meal processing, such as proximal blockage of nutrient absorption resulting in increased delivery of nutrients in the distal intestine, have shown some promising results in healthy and diabetic humans (403). Although this is more challenging with fats because dietary lipids require partial digestion by lipases to become efficient secretagogues (404, 405). However, distant delivery of free fatty acids, or even Oleoyl-Glycerol and sodium taurocholate have shown negligible effects on peripheral levels of GLP-1 or PYY, satiety and glucose tolerance (311, 406, 407). Similarly, distal delivery of the best known aminoacidic GLP-1-secretagogue, glutamine, has proven ineffective at ameliorating glucose tolerance in both healthy and diabetic subjects (407–409).
Furthermore, a recent report (410) examined the effect of RYGB on lean pigs, and indicates how it is the post-operative GLP-1 (9-36)NH2 levels that raise, while surprisingly the “active” (7-36)NH2 peripheral levels were reduced.
Indeed, most authors focus only on the peripheral levels of only one of these two peptide species, vastly excluding GLP-1(28-36) NH2 and (32-36)NH2 activity, rendering the overall understanding of each individual GLP-1 species, in both health and disease, difficult to discern.
Technical advances ELISA, capable to specifically dissect these peptide species locally and peripherally, will help us to shed new light into this complex physiology (411).
Conclusively, bearing in mind that insulinotropic or incretinotropic effects are not secondary to any single receptor modulation, whereby pools of different luminal stimuli act synergistically on tens of different chemosensors during their intestinal transit and absorption, while interacting with the microflora metabolism, rendering the restoration of a healthy physiology in diabetic patients with the pharmacological correction of a single axis, highly improbable.
The final dissection of the molecular axis causative of either metabolic syndrome will need more evidence regarding the localized and inter-neuronal physiology of GLP-1 in physiological and pathological statuses. To ultimately tease apart any possible cause from secondary events, species-specific biology will also need to be carefully dissected and interpreted.
Author Contributions
SP researched and interpreted all the data from available scientific literature on the PUBMED database, organized, wrote and revised the whole manuscript. SP also conceptualized and drew all the figures assembling the final formatted review. MF conceived, organized, wrote and revised the whole manuscript.
Funding
Work in the Falasca lab is supported by Diabetes Australia. SP is supported by the Curtin University Health Sciences Faculty International Research Scholarships.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the infrastructure and staff support provided by the School of Biomedical Sciences and CHRI, Faculty of Health Sciences Curtin University.
Footnotes
1. ^http://press.novonordisk-us.com/2017-12-5-Novo-Nordisk-Receives-FDA-Approval-of-OZEMPIC-R-semaglutide-Injection-For-the-Treatment-of-Adults-with-Type-2-Diabetes
References
1. Roth K, Kim S, Gordon J. Immunocytochemical studies suggest two pathways for enteroendocrine cell differentiation in the colon. Am J Physiol. (1992) 263(2 Pt 1):G174–G180
2. Gribble FM, Reimann F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol. (2016) 78:277–99. doi: 10.1146/annurev-physiol-021115-105439
3. Hansen CF, Vrang N, Sangild PT, Jelsing J. Novel insight into the distribution of L-cells in the rat intestinal tract. Am J Transl Res. (2013) 5:347–358.
4. Habib A, Richards P, Rogers G, Reimann F, Gribble F. Co-localisation and secretion of glucagon-like peptide 1 and peptide YY from primary cultured human L cells. Clin Exp Diabetes Metabol. (2013) 56:1413–6. doi: 10.1007/s00125-013-2887-z
5. Cancelas J, Prieto PG, Villanueva-Peñacarrillo ML, Valverde I, Malaisse W. Effects of an olive oil-enriched diet on glucagon-like peptide 1 release and intestinal content, plasma insulin concentration, glucose tolerance and pancreatic insulin content in an animal model of type 2 diabetes. Horm Metabol Res. (2006) 38:98–105. doi: 10.1055/s-2006-925126
6. Mace OJ, Tehan B, Marshall F. Pharmacology and physiology of gastrointestinal enteroendocrine cells. Pharmacol Res Perspect. (2015) 3:e00155. doi: 10.1002/prp2.155
7. Piomelli D. A fatty gut feeling. Trends Endocrinol Metabol. (2013) 24:332–41. doi: 10.1016/j.tem.2013.03.001
8. Drucker D. Glucagon-like peptides: regulators of cell proliferation, differentiation, and apoptosis. Mol Endocrinol. (2003) 17: 161–71. doi: 10.1210/me.2002-0306
9. Moran GW, Leslie FC, Levison SE, Worthington J, McLaughlin JT. Enteroendocrine cells: neglected players in gastrointestinal disorders? Therap Adv Gastroenterol. (2008) 1:51–60. doi: 10.1177/1756283x08093943
10. Bayliss WM, Starling EH. O n the causation of the so-called'peripheral reflex secretion'of the pancreas.(Preliminary Communication.). Proc R Soc Lond I (1901) 69:352–353.
11. Moore B. On the treatment of Diabetus mellitus by acid extract of Duodenal Mucous Membrane. Biochem J. (1906) 1:28.
12. La Barre J. Sur les possibilite's d'un traitement du diabète par l'incrétine. Bull Acad R Med Belg. (1932) 12:620–34.
13. Perley MJ, Kipnis DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic sujbjects. J Clin Invest. (1967) 46:1954–1962.
14. Drucker DJ. The biology of incretin hormones. Cell Metabol. (2006) 3:153–65. doi: 10.1016/j.cmet.2006.01.004
15. Vilsbøll T, Krarup T, Deacon CF, Madsbad S, Holst JJ. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes (2001) 50:609–13. doi: 10.2337/diabetes.50.3.609
16. Manell H, Staaf J, Manukyan L, Kristinsson H, Cen J, Stenlid R, et al. Altered plasma levels of glucagon, GLP-1 and glicentin during OGTT in adolescents with obesity and Type 2 diabetes. J Clin Endocrinol Metabol. (2016) 101:1181–9. doi: 10.1210/jc.2015-3885
17. Wewer Albrechtsen NJ, Hornburg D, Albrechtsen R, Svendsen B, Torang S, Jepsen SL, et al. Oxyntomodulin identified as a marker of type 2 diabetes and gastric bypass surgery by mass-spectrometry based profiling of human plasma. EBioMed. (2016) 7:112–20. doi: 10.1016/j.ebiom.2016.03.034
18. Meier J, Hucking K, Holst J, Deacon C. Reduced insulinotropic effect of gastric inhibitory polypeptide in first-degree relatives of patients with type 2 diabetes. Diabetes (2001) 50:2497–2504. doi: 10.2337/diabetes.50.11.2497
19. Nauck MA, Meier JJ. Incretin hormones: Their role in health and disease. Diabetes Obesity Metabol. (2018) 20(Suppl 1):5–21. doi: 10.1111/dom.13129
20. Holst JJ, Pedersen J, Wewer Albrechtsen NJ, Knop FK. The Gut: a key to the pathogenesis of type 2 diabetes? Metabol Syndr Relat Disord. (2017) 15:259–62. doi: 10.1089/met.2017.0015
21. Gil-Lozano M, Mingomataj EL, Wu WK, Ridout SA, Brubaker PL. Circadian secretion of the intestinal hormone GLP-1 by the rodent L cell. Diabetes (2014) 63:3674–85. doi: 10.2337/db13-1501
22. Martchenko A, Oh RH, Wheeler SE, Gurges P, Chalmers JA, Brubaker PL. Suppression of circadian secretion of glucagon-like peptide-1 by the saturated fatty acid, palmitate. Acta Physiol (Oxford, England) (2018) 222:e13007. doi: 10.1111/apha.13007
23. Nannipieri M, Baldi S, Mari A, Colligiani D, Guarino D, Camastra S, et al. Roux-en-Y gastric bypass and sleeve gastrectomy: mechanisms of diabetes remission and role of gut hormones. J Clin Endocrinol Metabol. (2013) 98:4391–9. doi: 10.1210/jc.2013-2538
24. Choi YY, Noh SH, An JY. A randomized controlled trial of Roux-en-Y gastrojejunostomy vs. gastroduodenostomy with respect to the improvement of type 2 diabetes mellitus after distal gastrectomy in gastric cancer patients. PLoS ONE (2017) 12:e0188904. doi: 10.1371/journal.pone.0188904
25. Dalsgaard N, Brønden A, Vilsbøll T, Knop F. Cardiovascular safety and benefits of GLP-1 receptor agonists. Expert Opin Drug Saf . (2017) 16:351–63. doi: 10.1080/14740338.2017.1281246
26. Bajaj HS, Al-Jabri B, Verma S. Glucagon-like peptide-1 receptor agonists and cardiovascular protection in type 2 diabetes: a pathophysiology-based review of clinical implications. Curr Opin Cardiol. (2018). doi: 10.1097/hco.0000000000000562. [Epub ahead of print].
27. Billeter A, Kopf S, Zeier M, Scheurlen KFL, Schulte TM, Kenngott HG, et al. Renal function in type 2 diabetes following gastric bypass. Dtsch Arztebl Int. (2016) 113:827–833. doi: 10.3238/arztebl.2016.0827
28. Dieter BP, Alicic RZ, Tuttle KR. GLP-1 Receptor agonists in diabetic kidney disease: from the Patient-Side to the Bench-Side. Am J Physiol Renal Physiol. (2018). doi: 10.1152/ajprenal.00211.2018. [Epub ahead of print].
29. Ten Kulve JS, Veltman DJ, Gerdes VEA, van Bloemendaal L, Barkhof F, Deacon CF, et al. RG IJ elevated postoperative endogenous GLP-1 levels mediate effects of roux-en-Y Gastric bypass on neural responsivity to food cues. Diabetes care (2018) 40:1522–1529. doi: 10.2337/dc16-2113
30. Jirapinyo P, Jin DX, Qazi T, Mishra N, Thompson CC. A meta-analysis of GLP-1 after roux-en-y gastric bypass: impact of surgical technique and measurement strategy. Obesity Surg. (2018) 28:615–26. doi: 10.1007/s11695-017-2913-1
31. Mokadem M, Zechner JF, Margolskee RF, Drucker DJ, Aguirre V. Effects of Roux-en-Y gastric bypass on energy and glucose homeostasis are preserved in two mouse models of functional glucagon-like peptide-1 deficiency. Mol Metabol. (2014) 3:191–201. doi: 10.1016/j.molmet.2013.11.010
32. Vetter ML, Wadden TA, Teff KL, Khan ZF, Carvajal R, Ritter S, et al. GLP-1 plays a limited role in improved glycemia shortly after Roux-en-Y gastric bypass: a comparison with intensive lifestyle modification. Diabetes (2015) 64:434–46. doi: 10.2337/db14-0558
33. Wilson-Perez HE, Chambers AP, Ryan KK, Li B, Sandoval DA, Stoffers D, et al. Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like Peptide 1 receptor deficiency. Diabetes (2013) 62:2380–5. doi: 10.2337/db12-1498
34. Ye J, Hao Z, Mumphrey MB, Townsend RL, Patterson LM, Stylopoulos N, et al. GLP-1 receptor signaling is not required for reduced body weight after RYGB in rodents. Am J Physiol Regulat Integr Compar Physiol. (2014) 306:R352–362. doi: 10.1152/ajpregu.00491.2013
35. Guida C, Stephen S, Guitton R, Ramracheya RD. The Role of PYY in pancreatic islet physiology and surgical control of diabetes. Trends Endocrinol Metabol. (2017) 28:626–36. doi: 10.1016/j.tem.2017.04.005
36. Garibay D, McGavigan AK, Lee SA, Ficorilli JV, Cox AL, Michael MD, et al. beta-cell glucagon-like peptide-1 receptor contributes to improved glucose tolerance after vertical sleeve gastrectomy. Endocrinology (2016) 157:3405–9. doi: 10.1210/en.2016-1302
37. Albaugh VL, Banan B, Ajouz H, Abumrad NN, Flynn CR. Bile acids and bariatric surgery. Mol Aspects Med. (2017) 56:75–89. doi: 10.1016/j.mam.2017.04.001
38. Patti ME, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, et al. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity (Silver Spring, Md) (2009) 17:1671–7. doi: 10.1038/oby.2009.102
39. Moreno-Arciniegas A, Falckenheiner-Soria J, Bancalero-de Los Reyes J, Camacho-Ramirez A, de Los Angeles Mayo-Ossorio M, Pacheco-Garcia JM, et al. The main participation of the enterohormone GLP-1 after bariatric surgery. Minerva Chir. (2018). doi: 10.23736/s0026-4733.18.07681-2. [Epub ahead of print].
40. Meier JJ, Nauck MA. Incretin-based therapies: where will we be 50 years from now? Diabetologia (2015) 58:1745–50. doi: 10.1007/s00125-015-3608-6
41. Creutzfeldt W. Origin, chemistry, physiology, and pathophysiology of the gastrointestinal hormones. In: International Symposium. Wiesbaden: Schattauer (1970).
42. Eissele R, Göke R, Willemer S, Harthus HP, Vermeer H, Arnold R, et al. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur J Clin Invest. (1992) 22:283.
43. Spreckley E, Murphy KG. The L-cell in nutritional sensing and the regulation of appetite. Front Nutr. (2015) 2:23. doi: 10.3389/fnut.2015.00023
44. Thanasupawat T, Hammje K, Adham I, Ghia JE, Del Bigio MR, Krcek J, et al. INSL5 is a novel marker for human enteroendocrine cells of the large intestine and neuroendocrine tumours. Oncol Rep. (2013) 29:149–54. doi: 10.3892/or.2012.2119
45. Billing LJ, Smith CA, Larraufie P, Goldspink DA, Galvin S, Kay RG, et al. Co-storage and release of insulin-like peptide-5, glucagon-like peptide-1 and peptideYY from murine and human colonic enteroendocrine cells. Mol Metabol. (2018) 16:65–75. doi: 10.1016/j.molmet.2018.07.011
46. Larraufie P, Martin-Gallausiaux C, Lapaque N, Dore J, Gribble FM, Reimann F, et al. SCFAs strongly stimulate PYY production in human enteroendocrine cells. Sci Rep. (2018) 8:74. doi: 10.1038/s41598-017-18259-0
47. Arora T, Akrami R, Pais R, Bergqvist L, Johansson BR, Schwartz TW, et al. Microbial regulation of the L cell transcriptome. Sci Rep. (2018) 8:1207. doi: 10.1038/s41598-017-18079-2
48. Cho HJ, Robinson ES, Rivera LR, McMillan PJ, Testro A, Nikfarjam M, et al. Glucagon-like peptide 1 and peptide YY are in separate storage organelles in enteroendocrine cells. Cell Tissue Res. (2014) 357:63–9. doi: 10.1007/s00441-014-1886-9
49. Kuhre RE, Albrechtsen NW, Windelov JA, Svendsen B, Hartmann B, Holst JJ. GLP-1 amidation efficiency along the length of the intestine in mice, rats and pigs and in GLP-1 secreting cell lines. Peptides (2014) 55:52–7. doi: 10.1016/j.peptides.2014.01.020
50. Buffa R, Capella C, Fontana P, Usellini L, Solcia E. Types of endocrine cells in the human colon and rectum. Cell Tissue Res. (1978) 192:227–40.
51. Jorsal T, Rhee NA, Pedersen J, Wahlgren CD, Mortensen B, Jepsen SL, et al. Enteroendocrine K and L cells in healthy and type 2 diabetic individuals. Diabetologia (2018) 61:284–94. doi: 10.1007/s00125-017-4450-9
52. Wynne K, Bloom SR. The role of oxyntomodulin and peptide tyrosine-tyrosine (PYY) in appetite control. Nat Clin Pract Endocrinol Metab. (2006) 2:612–20. doi: 10.1038/ncpendmet0318
53. Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM. Glucose sensing in l cells: a primary cell study. Cell Metabol. (2008) 8:532–9. doi: 10.1016/j.cmet.2008.11.002
54. Ang SY, Evans BA, Poole DP, Bron R, DiCello JJ, Bathgate RAD, et al. INSL5 activates multiple signalling pathways and regulates GLP-1 secretion in NCI-H716 cells. J Mol Endocrinol. (2018) 60:213–24. doi: 10.1530/jme-17-0152
55. Verhoeckx K, Cotter P, López-Expósito I, Kleiveland C, Lea T, Mackie A, et al. The Impact of Food Bioactives on Health: In vitro and Ex Vivo Models. Springer International Publishing (2016). doi: 10.1007/978-3-319-16104-4
56. Kuhre RE, Wewer Albrechtsen NJ, Deacon CF, Balk-Moller E, Rehfeld JF, Reimann F, et al. Peptide production and secretion in GLUTag, NCI-H716, and STC-1 cells: a comparison to native L-cells. J Mol Endocrinol. (2016) 56:201–11. doi: 10.1530/jme-15-0293
57. Grosse J, Heffron H, Burling K, Hossain MA, Habib AM, Rogers GJ, et al. Insulin-like peptide 5 is an orexigenic gastrointestinal hormone. Proc Natl Acad Sci. USA. (2014) 111:11133–8. doi: 10.1073/pnas.1411413111
58. Moriya R, Shirakura T, Ito J, Mashiko S, Seo T. Activation of sodium-glucose cotransporter 1 ameliorates hyperglycemia by mediating incretin secretion in mice. Am J Physiol Endocrinol Metab. (2009) 297:E1358–65. doi: 10.1152/ajpendo.00412.2009
59. Bohórquez DV, Shahid RA, Erdmann A, Kreger AM, Wang Y, Calakos N, et al. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. J Clin Invest. (2015) 125:782. doi: 10.1172/JCI78361
60. Anini Y, Fu-Cheng X, Cuber JC, Kervran A, Chariot J, Roz C. Comparison of the postprandial release of peptide YY and proglucagon-derived peptides in the rat. Pflugers Arch. (1999) 438:299–306.
61. Tuduri E, Beiroa D, Porteiro B, Lopez M, Dieguez C, Nogueiras R. Acute but not chronic activation of brain glucagon-like peptide-1 receptors enhances glucose-stimulated insulin secretion in mice. Diabetes Obesity Metabol. (2015) 17:789–99. doi: 10.1111/dom.12488
62. Attwood MM, Krishnan A, Almen MS, Schioth HB. Highly diversified expansions shaped the evolution of membrane bound proteins in metazoans. Sci Rep. (2017) 7:12387. doi: 10.1038/s41598-017-11543-z
63. Kenakin T. New lives for seven transmembrane receptors as drug targets. Trends Pharmacol Sci. (2015) 36:705–6. doi: 10.1016/j.tips.2015.09.004
64. Milligan G, Shimpukade B, Ulven T, Hudson BD. Complex pharmacology of free fatty acid receptors. Chem Rev. (2017) 117:67–110. doi: 10.1021/acs.chemrev.6b00056
65. Kessenbrock M, Groth G. Circular dichroism and fluorescence spectroscopy to study protein structure and protein-protein interactions in ethylene signaling. Methods Mol Biol. (2017) 1573:141–59. doi: 10.1007/978-1-4939-6854-1_12
66. Safdari HA, Pandey S, Shukla AK, Dutta S. Illuminating GPCR Signaling by Cryo-EM. Trends Cell Biol. (2018) 28:591–4. doi: 10.1016/j.tcb.2018.06.002
67. Stauch B, Cherezov V. Serial femtosecond crystallography of G protein-coupled receptors. Annu Rev Biophys. (2018) 47:377–97. doi: 10.1146/annurev-biophys-070317-033239
68. Kahsai AW, Pani B, Lefkowitz RJ. GPCR signaling: conformational activation of arrestins. Cell Res. (2018) 28:783–4. doi: 10.1038/s41422-018-0067-x
69. de Graaf C, Donnelly D, Wootten D, Lau J, Sexton P, Miller L, et al. Glucagon-like peptide-1 and its class B G protein-coupled receptors: a long march to therapeutic successes. Pharmacol Rev. (2016) 68:954–1013. doi: 10.1124/pr.115.011395
70. Zhang X, Cai C, Winters M, Wells M, Wall M, Lanter J, et al. Design, synthesis and SAR of a novel series of heterocyclic phenylpropanoic acids as GPR120 agonists. Bioorganic Medic. Chem. Lett. (2017) 27:3272–8. doi: 10.1016/j.bmcl.2017.06.028
71. Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-mediated signaling of metabolites. Cell Metab. (2017) 25:777–96. doi: 10.1016/j.cmet.2017.03.008
72. Sun EW, de Fontgalland D, Rabbitt P, Hollington P, Sposato L, Due SL, et al. Mechanisms controlling glucose-induced GLP-1 secretion in human small intestine. Diabetes (2017) 66:2144–9. doi: 10.2337/db17-0058
73. Ding X, Hu CA, Huang P, Li Y, He S, Yang H, et al. Intestinal enteroendocrine L cells in amino acid sensing and diseases. Front Biosci. (2018) 23:1740–53. doi: 10.2741/4670
74. Lindqvist A, Shcherbina L, Fischer AT, Wierup N. Ghrelin is a regulator of glucagon-like peptide 1 secretion and transcription in mice. Front Endocrinol. (2017) 8:135. doi: 10.3389/fendo.2017.00135
75. Feng R, Qian C, Liu Q, Jin Y, Liu L, Li S, et al. Expression of sweet taste receptor and gut hormone secretion in modelled type 2 diabetes. General Compar Endocrinol. (2017) 252:142–9. doi: 10.1016/j.ygcen.2017.08.008
76. Reimann F, Gribble F. G protein-coupled receptors as new therapeutic targets for type 2 diabetes. Clin Exp Diabetes Metabol. (2016) 59:229–33. doi: 10.1007/s00125-015-3825-z
77. Lin HV, Wang J, Wang J, Li W, Wang X, Alston JT, et al. GPR142 prompts glucagon-like Peptide-1 release from islets to improve beta cell function. Mol Metabol. (2018) 11:205–11. doi: 10.1016/j.molmet.2018.02.008
78. Kristinsson H, Sargsyan E, Manell H, Smith DM, Gopel SO, Bergsten P. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci Rep. (2017) 7:4657. doi: 10.1038/s41598-017-04730-5
79. Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes (2008) 57:2280–87. doi: 10.2337/db08-0307
80. Liou AP, Lu X, Sei Y, Zhao X, Pechhold S, Carrero RJ, et al. The G-Protein– Coupled Receptor GPR40 Directly Mediates Long-Chain Fatty Acid– Induced Secretion of Cholecystokinin. Gastroenterology (2011) 140:903–12. e904. doi: 10.1053/j.gastro.2010.10.012
81. Brooks L, Viardot A, Tsakmaki A, Stolarczyk E, Howard JK, Cani PD, et al. Fermentable carbohydrate stimulates FFAR2-dependent colonic PYY cell expansion to increase satiety. Mol Metab. (2017) 6:48–60. doi: 10.1016/j.molmet.2016.10.011
82. Psichas A, Sleeth ML, Murphy KG, Brooks L, Bewick GA, Hanyaloglu AC, et al. The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int. J. Obes. (Lond). (2015) 39:424–9. doi: 10.1038/ijo.2014.153
83. Nohr MK, Pedersen MH, Gille A, Egerod KL, Engelstoft MS, Husted AS, et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology (2013) 154:3552–64. doi: 10.1210/en.2013-1142
84. Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. (2005) 11:90–4. doi: 10.1038/nm1168
85. Iwasaki K, Harada N, Sasaki K, Yamane S, Iida K, Suzuki K, et al. Free fatty acid receptor GPR120 is highly expressed in enteroendocrine K cells of the upper small intestine and has a critical role in GIP secretion after fat ingestion. Endocrinology (2015) 156:837–46. doi: 10.1210/en.2014-1653
86. Sankoda A, Harada N, Iwasaki K, Yamane S, Murata Y, Shibue K, et al. Long-Chain Free Fatty Acid Receptor GPR120 Mediates Oil-Induced GIP Secretion Through CCK in Male Mice. Endocrinology (2017) 158:1172–80. doi: 10.1210/en.2017-00090
87. Arifin SA, Paternoster S, Carlessi R, Casari I, Ekberg JH, Maffucci T, et al. Oleoyl-lysophosphatidylinositol enhances glucagon-like peptide-1 secretion from enteroendocrine L-cells through GPR119. Biochim Biophys. Acta (2018) 1863:1132–41. doi: 10.1016/j.bbalip.2018.06.007
88. Lan H, Lin HV, Wang CF, Wright MJ, Xu S, Kang L, et al. Agonists at GPR119 mediate secretion of GLP-1 from mouse enteroendocrine cells through glucose-independent pathways. Br J Pharmacol. (2012) 165:2799–807. doi: 10.1111/j.1476-5381.2011.01754.x
89. Tough IR, Forbes S, Herzog H, Jones RM, Schwartz TW, Cox HM. Bidirectional GPR119 agonism requires peptide YY and glucose for activity in mouse and human colon mucosa. Endocrinology (2018) 159:1704–17. doi: 10.1210/en.2017-03172
90. Patel S, Mace OJ, Tough IR, White J, Cock TA, Warpman Berglund U, et al. Gastrointestinal hormonal responses on GPR119 activation in lean and diseased rodent models of type 2 diabetes. Int J Obes. (2014) 38:1365. doi: 10.1038/ijo.2014.10
91. Li NX, Brown S, Kowalski T, Wu M, Yang L, Dai G, et al. GPR119 Agonism increases glucagon secretion during insulin-induced hypoglycemia. Diabetes (2018) 67:1401–13. doi: 10.2337/db18-0031
92. Yamada Y, Terauchi Y, Watada H, Nakatsuka Y, Shiosakai K, Washio T, et al. Efficacy and Safety of GPR119 Agonist DS-8500a in Japanese Patients with Type 2 Diabetes: a Randomized, Double-Blind, Placebo-Controlled, 12-Week Study. Adv Ther. (2018) 35:367–81. doi: 10.1007/s12325-018-0668-2
93. Christensen MB. Glucose-dependent insulinotropic polypeptide: effects on insulin and glucagon secretion in humans. Danish Med J. (2016) 63:B5230. Available online at: https://pdfs.semanticscholar.org/b19d/0b5fbd402a2929e2cfc6f01d6cccdd7ea676.pdf
94. Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metabol. (1973) 37:826–8. doi: 10.1210/jcem-37-5-826
95. Timper K, Dalmas E, Dror E, Rutti S, Thienel C, Sauter NS, et al. Glucose-dependent insulinotropic peptide stimulates glucagon-like Peptide 1 production by pancreatic islets via interleukin 6, Produced by alpha Cells. Gastroenterology (2016) 151:165–79. doi: 10.1053/j.gastro.2016.03.003
96. Orgaard A, Holst JJ. The role of somatostatin in GLP-1-induced inhibition of glucagon secretion in mice. Diabetologia (2017) 60:1731–9. doi: 10.1007/s00125-017-4315-2
97. Turton M, Shea D, Gunn I, Beak S. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature (1996) 379:69.
98. Piro S, Mascali LG, Urbano F, Filippello A, Malaguarnera R, Calanna S, et al. Chronic exposure to GLP-1 increases GLP-1 synthesis and release in a pancreatic alpha cell line (alpha-TC1): evidence of a direct effect of GLP-1 on pancreatic alpha cells. PLoS ONE (2014) 9:e90093. doi: 10.1371/journal.pone.0090093
99. Nakashima K, Kaneto H, Shimoda M, Kimura T, Kaku K. Pancreatic alpha cells in diabetic rats express active GLP-1 receptor: Endosomal co-localization of GLP-1/GLP-1R complex functioning through intra-islet paracrine mechanism. Sci Rep. (2018) 8:3725. doi: 10.1038/s41598-018-21751-w
100. Kumar DP, Asgharpour A, Mirshahi F, Park SH, Liu S, Imai Y, et al. Activation of transmembrane bile acid receptor TGR5 modulates pancreatic islet alpha cells to promote glucose homeostasis. J Biol Chem. (2016) 291:6626–40. doi: 10.1074/jbc.M115.699504
101. Brighton CA, Rievaj J, Kuhre RE, Glass LL, Schoonjans K, Holst JJ, et al. Bile Acids Trigger GLP-1 release predominantly by accessing basolaterally located G protein-coupled bile acid receptors. Endocrinology (2015) 156:3961–70. doi: 10.1210/en.2015-1321
102. Oya M, Kitaguchi T, Pais R, Reimann F, Gribble F, Tsuboi T. The G protein-coupled receptor family C group 6 subtype A (GPRC6A) receptor is involved in amino acid-induced glucagon-like peptide-1 secretion from GLUTag cells. J Biol Chem. (2013) 288:4513–21. doi: 10.1074/jbc.M112.402677
103. Clemmensen C, Jorgensen CV, Smajilovic S, Brauner-Osborne H. Robust GLP-1 secretion by basic L-amino acids does not require the GPRC6A receptor. Diabetes Obesity Metabol. (2017) 19:599–603. doi: 10.1111/dom.12845
104. Gupta V. (2012). Pleiotropic effects of incretins. Ind J Endocrinol Metabol. 16 (Suppl. 1):S47–56. doi: 10.4103/2230-8210.94259
105. Ali S, Lamont BJ, Charron MJ, Drucker DJ. Dual elimination of the glucagon and GLP-1 receptors in mice reveals plasticity in the incretin axis. J Clin Invest. (2011) 121:1917–1929. doi: 10.1172/JCI43615
106. White JW, Saunders GF. Structure of the human glucagon gene. Nucleic Acids Res. (1986) 14:4719–4730.
107. Holst JJ, Bersani M, Johnsen AH, Kofod H, Hartmann B, Orskov C. Proglucagon processing in porcine and human pancreas. J Biol Chem. (1994) 269:18827–18833.
108. Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. (2007) 87:1409–39. doi: 10.1152/physrev.00034.2006
109. Bataille D, Dalle S. The forgotten members of the glucagon family. Diabetes Res Clin Pract. (2014) 106:1–10. doi: 10.1016/j.diabres.2014.06.010
110. Sandoval DA, D'Alessio DA. Physiology of proglucagon peptides: role of glucagon and GLP-1 in health and disease. Physiol Rev. (2015) 95:513–48. doi: 10.1152/physrev.00013.2014
111. Creemers JW, Jackson RS, Hutton JC. Molecular and cellular regulation of prohormone processing. Seminars Cell Dev Biol. (1998) 9:3–10. doi: 10.1006/scdb.1997.0195
112. Holst JJ, Albrechtsen NJW, Gabe MBN, Rosenkilde MM. Oxyntomodulin: Actions and role in diabetes. Peptides (2018) 100:48–53. doi: 10.1016/j.peptides.2017.09.018
113. Bataille D, Fontes G, Costes S, Longuet C, Dalle S. The glucagon-miniglucagon interplay: a new level in the metabolic regulation. Ann N Y Acad Sci. (2006) 1070:161–6. doi: 10.1196/annals.1317.005
114. Bataille D, Blache P, Bergeron F. Endoprotease regulation of miniglucagon production. Ann N Y Acad Sci. (1996) 805:1–8; discussion 8–9.
115. Lund A, Bagger JI, Wewer Albrechtsen NJ, Christensen M, Grondahl M, Hartmann B, et al. Evidence of extrapancreatic glucagon secretion in man. Diabetes (2016) 65:585–97. doi: 10.2337/db15-1541
116. Orskov C, Rabenhøj L, Wettergren A, Kofod H, Holst J. Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes (1994) 43:535–539.
117. Deacon CF, Pridal L, Klarskov L, Olesen M, Holst JJ. Glucagon-like peptide 1 undergoes differential tissue-specific metabolism in the anesthetized pig. Am J Physiol. (1996) 271:E458–464. doi: 10.1152/ajpendo.1996.271.3.E458
118. Yabe D, Kuroe A, Lee S, Watanabe K, Hyo T, Hishizawa M, et al. Little enhancement of meal-induced glucagon-like peptide 1 secretion in Japanese: Comparison of type 2 diabetes patients and healthy controls. J Diabetes Invest. (2010) 1:56–9. doi: 10.1111/j.2040-1124.2010.00010.x
119. Hupe-Sodmann K, McGregor GP, Bridenbaugh R, Goke R, Goke B, Thole H, et al. Characterisation of the processing by human neutral endopeptidase 24.11 of GLP-1(7-36) amide and comparison of the substrate specificity of the enzyme for other glucagon-like peptides. Regul Peptides (1995) 58:149–156.
120. Plamboeck A, Holst JJ, Carr RD, Deacon CF. Neutral endopeptidase 24.11 and dipeptidyl peptidase IV are both involved in regulating the metabolic stability of glucagon-like peptide-1 in vivo. Adv Exp Med Biol. (2003) 524:303–312. doi: 10.1007/0-306-47920-6_36
121. Liu Z, Stanojevic V, Brindamour LJ, Habener JF. GLP1-derived nonapeptide GLP1(28-36) amide protects pancreatic beta-cells from glucolipotoxicity. J Endocrinol. (2012) 213:143–54. doi: 10.1530/joe-11-0328
122. Tomas E, Wood JA, Stanojevic V, Habener JF. GLP-1-derived nonapeptide GLP-1(28-36) amide inhibits weight gain and attenuates diabetes and hepatic steatosis in diet-induced obese mice. Regulat Peptides (2011) 169:43–8. doi: 10.1016/j.regpep.2011.04.006
123. Shao W, Wang Z, Ip W, Chiang YT, Xiong X, Chai T, et al. GLP-1(28-36) improves beta-cell mass and glucose disposal in streptozotocin-induced diabetic mice and activates cAMP/PKA/beta-catenin signaling in beta-cells in vitro. Am J Physiol Endocrinol Metab. (2013) 304:E1263–72. doi: 10.1152/ajpendo.00600.2012
124. Ip W, Shao W, Chiang YT, Jin T. GLP-1-derived nonapeptide GLP-1(28-36)amide represses hepatic gluconeogenic gene expression and improves pyruvate tolerance in high-fat diet-fed mice. Am J Physiol Endocrinol Metab. (2013) 305:E1348–58. doi: 10.1152/ajpendo.00376.2013
125. Sun L, Dai Y, Wang C, Chu Y, Su X, Yang J, et al. Novel Pentapeptide GLP-1 (32-36) amide inhibits beta-cell apoptosis in vitro and improves glucose disposal in streptozotocin-induced diabetic mice. Chem Biol Drug Design (2015) 86:1482–90. doi: 10.1111/cbdd.12615
126. Elahi D, Angeli FS, Vakilipour A, Carlson OD, Tomas E, Egan JM, et al. GLP-1(32-36)amide, a novel pentapeptide cleavage product of GLP-1, modulates whole body glucose metabolism in dogs. Peptides (2014) 59:20–4. doi: 10.1016/j.peptides.2014.06.004
127. Tomas E, Stanojevic V, McManus K, Khatri A, Everill P, Bachovchin WW, et al. GLP-1(32-36)amide pentapeptide increases basal energy expenditure and inhibits weight gain in obese mice. Diabetes (2015) 64:2409–19. doi: 10.2337/db14-1708
128. Guglielmi V, Sbraccia P. GLP-1 receptor independent pathways: emerging beneficial effects of GLP-1 breakdown products. Eating Weight Disorders (2017) 22:231–40. doi: 10.1007/s40519-016-0352-y
129. Parvaresh Rizi E, Loh TP, Baig S, Chhay V, Huang S, Caleb Quek J, et al. A high carbohydrate, but not fat or protein meal attenuates postprandial ghrelin, PYY and GLP-1 responses in Chinese men. PLoS ONE (2018) 13:e0191609. doi: 10.1371/journal.pone.0191609
130. Hallworth JR, Copeland JL, Doan J, Hazell TJ. The effect of exercise intensity on total PYY and GLP-1 in healthy females: a pilot study. J Nutr Metabol. (2017) 2017:4823102. doi: 10.1155/2017/4823102
131. Schirra J, Nicolaus M, Roggel R, Katschinski M, Storr M, Woerle HJ, et al. Endogenous glucagon-like peptide 1 controls endocrine pancreatic secretion and antro-pyloro-duodenal motility in humans. Gut (2006) 55:243–51. doi: 10.1136/gut.2004.059741
132. Vella A, Shah P, Basu R, Basu A, Camilleri M, Schwenk FW, et al. Effect of glucagon-like peptide-1 (7-36)-amide on initial splanchnic glucose uptake and insulin action in humans with type 1 diabetes. Diabetes (2001) 50:565–72. doi: 10.2337/diabetes.50.3.565
133. Zummo FP, Cullen KS, Honkanen-Scott M, Shaw JA, Lovat PE, Arden C. Glucagon-like peptide 1 protects pancreatic β-cells from death by increasing autophagic flux and restoring lysosomal function. Diabetes (2017) 66:1272–85. doi: 10.2337/db16-1009
134. Cornu M, Yang J-Y, Jaccard E, Poussin C, Widmann C, Thorens B. Glucagon-like peptide-1 protects β-cells against apoptosis by increasing the activity of an IGF-2/IGF-1 receptor autocrine loop. Diabetes (2009) 58:1816–25. doi: 10.2337/db09-0063
135. Ramsey W, Isales CM. Intestinal incretins and the regulation of bone physiology. Adv Exp Med Biol. (2017) 1033:13–33. doi: 10.1007/978-3-319-66653-2_2
136. Insuela DBR, Carvalho VF. Glucagon and glucagon-like peptide-1 as novel anti-inflammatory and immunomodulatory compounds. Eur J Pharmacol. (2017) 812:64–72. doi: 10.1016/j.ejphar.2017.07.015
137. Nikolaidis LA, Elahi D, Shen YT, Shannon RP. Active metabolite of GLP-1 mediates myocardial glucose uptake and improves left ventricular performance in conscious dogs with dilated cardiomyopathy. Am J Physiol Heart Circul Physiol. (2005) 289:H2401–8. doi: 10.1152/ajpheart.00347.2005
138. Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation (2008) 117:2340–50. doi: 10.1161/circulationaha.107.739938
139. Ossum A, van Deurs U, Engstrom T, Jensen JS, Treiman M. The cardioprotective and inotropic components of the postconditioning effects of GLP-1 and GLP-1(9-36)a in an isolated rat heart. Pharmacol Res. (2009) 60:411–7. doi: 10.1016/j.phrs.2009.06.004
140. Ban K, Kim K-H, Cho C-K., Sauve M, Diamandis EP, Backx PH, et al. Glucagon-like peptide (GLP)-1 (9-36) amide-mediated cytoprotection is blocked by exendin (9-39) yet does not require the known GLP-1 receptor. Endocrinology (2010) 151:1520–1531. doi: 10.1210/en.2009-1197
141. Tomas E, Habener JF. Insulin-like actions of glucagon-like peptide-1: a dual receptor hypothesis. Trends Endocrinol Metabol. (2010) 21:59–67. doi: 10.1016/j.tem.2009.11.007
142. Sonne DP, Engstrom T, Treiman M. Protective effects of GLP-1 analogues exendin-4 and GLP-1(9-36) amide against ischemia-reperfusion injury in rat heart. Regulat Peptides (2008) 146:243–9. doi: 10.1016/j.regpep.2007.10.001
143. Gardiner S, March J, Kemp P, Bennett T, Baker D. Possible involvement of GLP-1 (9–36) in the regional haemodynamic effects of GLP-1 (7–36) in conscious rats. Br J Pharmacol. (2010) 161:92–102. doi: 10.1111/j.1476-5381.2010.00867.x
144. Bailey CJ, Marx N. Cardiovascular protection in type 2 diabetes: insights from recent outcome trials. Diabetes Obesity Metabol. (2018). doi: 10.1111/dom.13492
145. Meier JJ, Nauck MA. Glucagon-like peptide 1 (GLP-1) in biology and pathology. Diabetes Metabol Res Rev. (2005) 21:91–117. doi: 10.1002/dmrr.538
146. Yousseif A, Emmanuel J, Karra E, Millet Q, Elkalaawy M, Jenkinson AD, et al. Differential effects of laparoscopic sleeve gastrectomy and laparoscopic gastric bypass on appetite, circulating acyl-ghrelin, peptide YY3-36 and active GLP-1 levels in non-diabetic humans. Obesity Surg. (2014) 24:241–52. doi: 10.1007/s11695-013-1066-0
147. Salehi M, Prigeon RL, D'Alessio DA. Gastric bypass surgery enhances glucagon-like peptide 1-stimulated postprandial insulin secretion in humans. Diabetes (2011) 60:2308–14. doi: 10.2337/db11-0203
148. Davis DB, Khoraki J, Ziemelis M, Sirinvaravong S, Han JY, Campos GM. Roux en Y gastric bypass hypoglycemia resolves with gastric feeding or reversal: Confirming a non-pancreatic etiology. Mol Metabol. (2018) 9:15–27. doi: 10.1016/j.molmet.2017.12.011
149. Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet (2006) 368:1696–705. doi: 10.1016/S0140-6736(06)69705-5
150. Meier J, Nauck M. Incretins and the development of type 2 diabetes. Curr Diab Rep. (2006) 6:194–201.
151. Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in Type 2 (non-insulin-dependent) diabetes. Clin Exp Diabet Metabol. (1986) 29:46–52. doi: 10.1007/BF02427280
152. Holst J. (2006). Glucagon-like peptide-1: from extract to agent. The Claude Bernard Lecture, 2005. Clin Exp Diab Metabol. 49:253–260. doi: 10.1007/s00125-005-0107-1
153. Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 7-36 amide but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. (1993) 91:301. doi: 10.1172/JCI116186
154. Guida C, McCulloch LJ, Godazgar M, Stephen SD, Baker C, Basco D, et al. Sitagliptin and Roux-en-Y gastric bypass modulate insulin secretion via regulation of intra-islet PYY. Diabetes Obesity Metabol. (2018) 20:571–81. doi: 10.1111/dom.13113
155. Lefort S, Tschop MH, Garcia-Caceres C. A synaptic basis for GLP-1 action in the brain. Neuron (2017) 96:713–5. doi: 10.1016/j.neuron.2017.10.034
156. Liu J, Conde K, Zhang P, Lilascharoen V, Xu Z, Lim BK, et al. Enhanced AMPA Receptor trafficking mediates the anorexigenic effect of endogenous glucagon-like peptide-1 in the paraventricular hypothalamus. Neuron (2017) 96:897–909.e895. doi: 10.1016/j.neuron.2017.09.042
157. Vrang N, Larsen PJ. Preproglucagon derived peptides GLP-1, GLP-2 and oxyntomodulin in the CNS: role of peripherally secreted and centrally produced peptides. Progr Neurobiol. (2010) 92:442–62. doi: 10.1016/j.pneurobio.2010.07.003
158. Sandoval D, Sisley SR. Brain GLP-1 and insulin sensitivity. Mol Cell Endocrinol. (2015) 418(Pt 1):27–32. doi: 10.1016/j.mce.2015.02.017
159. Larsen PJ, Tang-Christensen M, Jessop DS. Central administration of glucagon-like peptide-1 activates hypothalamic neuroendocrine neurons in the rat. Endocrinology (1997) 138:4445–55. doi: 10.1210/endo.138.10.5270
160. Holscher C. Central effects of GLP-1: new opportunities for treatments of neurodegenerative diseases. J Endocrinol. (2014) 221:T31–41. doi: 10.1530/joe-13-0221
161. Rogge G, Jones D, Hubert GW, Lin Y, Kuhar MJ. CART peptides: regulators of body weight, reward and other functions. Nat Rev Neurosci. (2008) 9:747–58. doi: 10.1038/nrn2493
162. Shcherbina L, Lindqvist A, Thoren Fischer AH, Ahlqvist E, Zhang E, Falkmer SE, et al. Intestinal CART is a regulator of GIP and GLP-1 secretion and expression. Mol Cell Endocrinol. (2018) 476:8–16. doi: 10.1016/j.mce.2018.04.002
163. Secher A, Jelsing J, Baquero AF, Hecksher-Sorensen J, Cowley MA, Dalboge LS, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest. (2014) 124:4473–88. doi: 10.1172/jci75276
164. Burmeister MA, Ayala JE, Smouse H, Landivar-Rocha A, Brown JD, Drucker DJ, et al. The hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes (2017) 66:372–84. doi: 10.2337/db16-1102
165. Baggio LL, Huang Q, Brown TJ, Drucker DJ. A recombinant human glucagon-like peptide (GLP)-1-albumin protein (albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis. Diabetes (2004) 53:2492–500. doi: 10.2337/diabetes.53.9.2492
166. Charpentier J, Waget A, Klopp P, Magnan C, Cruciani-Guglielmacci C, Lee SJ, et al. Lixisenatide requires a functional gut-vagus nerve-brain axis to trigger insulin secretion in controls and type 2 diabetic mice. Am J Physiol Gastrointest Liver Physiol. (2018). doi: 10.1152/ajpgi.00348.2017. [Epub ahead of print].
167. Kanoski SE, Fortin SM, Arnold M, Grill HJ, Hayes MR. Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology (2011) 152:3103–12. doi: 10.1210/en.2011-0174
168. Sisley S, Gutierrez-Aguilar R, Scott M, D'Alessio DA, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide's anorectic but not glucose-lowering effect. J Clin Invest. (2014) 124:2456–63. doi: 10.1172/jci72434
169. Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S, et al. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes (2014) 63:1224–33. doi: 10.2337/db13-1440
170. Broide E, Bloch O, Ben-Yehudah G, Cantrell D, Shirin H, Rapoport MJ. GLP-1 receptor is expressed in human stomach mucosa: analysis of its cellular association and distribution within gastric glands. J Histochem Cytochem. (2013) 61:649–58. doi: 10.1369/0022155413497586
171. Wismann P, Barkholt P, Secher T, Vrang N, Hansen HB, Jeppesen PB, et al. The endogenous preproglucagon system is not essential for gut growth homeostasis in mice. Mol Metabol. (2017) 6:681–92. doi: 10.1016/j.molmet.2017.04.007
172. List JF, He H, Habener JF. Glucagon-like peptide-1 receptor and proglucagon expression in mouse skin. Regulat Peptides (2006) 134:149–57. doi: 10.1016/j.regpep.2006.02.007
173. Cameron-Vendrig A, Reheman A, Siraj MA, Xu XR, Wang Y, Lei X, et al. Glucagon-like peptide 1 receptor activation attenuates platelet aggregation and thrombosis. Diabetes (2016) 65:1714–23. doi: 10.2337/db15-1141
174. Pyke C, Heller RS, Kirk RK, Orskov C, Reedtz-Runge S, Kaastrup P, et al. GLP-1 receptor localization in monkey and human tissue: novel distribution revealed with extensively validated monoclonal antibody. Endocrinology (2014) 155:1280–90. doi: 10.1210/en.2013-1934
175. Yusta B, Baggio LL, Koehler J, Holland D, Cao X, Pinnell LJ, et al. GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R. Diabetes (2015) 64:2537–49. doi: 10.2337/db14-1577
176. Baggio LL, Yusta B, Mulvihill EE, Cao X, Streutker CJ, Butany J, et al. GLP-1 Receptor expression within the human heart. Endocrinology (2018) 159:1570–84. doi: 10.1210/en.2018-00004
177. Ayala JE, Bracy DP, James FD, Burmeister MA, Wasserman DH, Drucker DJ. Glucagon-like peptide-1 receptor knockout mice are protected from high-fat diet-induced insulin resistance. Endocrinology (2010) 151:4678–87. doi: 10.1210/en.2010-0289
178. Knauf C, Cani PD, Ait-Belgnaoui A, Benani A, Dray C, Cabou C, et al. Brain glucagon-like peptide 1 signaling controls the onset of high-fat diet-induced insulin resistance and reduces energy expenditure. Endocrinology (2008) 149:4768–77. doi: 10.1210/en.2008-0180
179. Cheang JY, Moyle PM. Glucagon-like peptide-1 (GLP-1)-based therapeutics: current status and future opportunities beyond type 2 diabetes. ChemMed Chem. (2018) 13:662–71. doi: 10.1002/cmdc.201700781
180. Defronzo R. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes (2009) 58:773–95. doi: 10.2337/db09-9028
181. Demir S, Temizkan S, Sargin M. C-peptide levels predict the effectiveness of dipeptidyl peptidase-4 inhibitor therapy. J Diabetes Res. (2016) 2016:4509603. doi: 10.1155/2016/4509603
182. Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, Lafont S, Bergeonneau C, Kassai B, et al. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. BMJ (2011) 343:d4169. doi: 10.1136/bmj.d4169
183. Behme MT, Dupré J, McDonald TJ. Glucagon-like peptide I improved glycemic control in type I diabetes. BMC Endocrine Disorders (2003) 3:3. doi: 10.1186/1472-6823-3-3
184. Frandsen CS, Dejgaard TF, Madsbad S, Holst JJ. Non-insulin pharmacological therapies for treating type 1 diabetes. Expert Opin Pharmacother. (2018) 19:947–60. doi: 10.1080/14656566.2018.1483339
186. Bond A. Exenatide (Byetta) as a novel treatment option for type 2 diabetes mellitus. Proceedings (Baylor University Medical Center) (2006) 19:281–4. doi: 10.1080/08998280.2006.11928181
188. Tahrani AA, Bellary S, Barnett AH. Once-weekly GLP-1R agonists: moving the goal posts. Lancet Diabetes Endocrinol. (2018) 6:260–1. doi: 10.1016/s2213-8587(18)30049-4
189. Julia M, Mara KB, Julia O, Robert S, Vera J, Joachim J, et al. Glucagon-Like Peptide-1 and its cleavage products are renoprotective in murine diabetic nephropathy. Diabetes (2018) 67:db171212. doi: 10.2337/db17-1212
190. Ipsen DH, Rolin B, Rakipovski G, Skovsted GF, Madsen A, Kolstrup S, et al. Liraglutide decreases hepatic inflammation and injury in advanced lean non-alcoholic steatohepatitis. Basic Clin Pharmacol Toxicol. (2018). doi: 10.1111/bcpt.13082. [Epub ahead of print].
191. Bae CS, Song J. The role of glucagon-like peptide 1 (GLP1) in type 3 diabetes: GLP-1 controls insulin resistance, neuroinflammation and neurogenesis in the brain. Int J Mol Sci. (2017) 18:11. doi: 10.3390/ijms18112493
192. Rask Larsen J, Dima L, Correll CU, Manu P. The pharmacological management of metabolic syndrome. Expert Rev Clin Pharmacol. (2018) 11:397–410. doi: 10.1080/17512433.2018.1429910
193. Dhir G, Cusi K. Glucagon like peptide-1 receptor agonists for the management of obesity and non-alcoholic fatty liver disease: a novel therapeutic option. J Invest Med. (2018) 66:7–10. doi: 10.1136/jim-2017-000554
194. Khat DZ, Husain M. Molecular mechanisms underlying the cardiovascular benefits of SGLT2i and GLP-1RA. Curr Diabetes Rep. (2018) 18:45. doi: 10.1007/s11892-018-1011-7
195. Drucker DJ. The ascending GLP-1 road from clinical safety to reduction of cardiovascular complications. Diabetes (2018) 67:1710–9. doi: 10.2337/dbi18-0008
196. Mensberg P, Nyby S, Jorgensen PG, Storgaard H, Jensen MT, Sivertsen J, et al. Near-normalization of glycaemic control with glucagon-like peptide-1 receptor agonist treatment combined with exercise in patients with type 2 diabetes. Diabetes Obesity Metabol. (2017) 19:172–80. doi: 10.1111/dom.12797
197. Finan B, Ma T, Ottaway N, Muller TD, Habegger KM, Heppner KM, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. (2013) 5:209ra151. doi: 10.1126/scitranslmed.3007218
198. Pocai A. Unraveling oxyntomodulin, GLP1's enigmatic brother. J Endocrinol. (2012) 215:335–46. doi: 10.1530/joe-12-0368
199. Wynne K, Park AJ, Small CJ, Patterson M, Ellis SM, Murphy KG, et al. Subcutaneous oxyntomodulin reduces body weight in overweight and obese subjects: a double-blind, randomized, controlled trial. Diabetes (2005) 54:2390–5. doi: 10.2337/diabetes.54.8.2390
200. Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. (2015) 21:27–36. doi: 10.1038/nm.3761
201. Jall S, Sachs S, Clemmensen C, Finan B, Neff F, DiMarchi RD, et al. Monomeric GLP-1/GIP/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol Metabol. (2017) 6:440–6. doi: 10.1016/j.molmet.2017.02.002
202. Li T, Jiao JJ, Holscher C, Wu MN, Zhang J, Tong JQ, et al. A novel GLP-1/GIP/Gcg triagonist reduces cognitive deficits and pathology in the 3xTg mouse model of Alzheimer's disease. Hippocampus (2018) 28:358–72. doi: 10.1002/hipo.22837
203. D'Alessio D. Is GLP-1 a hormone: Whether and When? J Diabetes Invest. (2016) 7 (Suppl. 1):50–5. doi: 10.1111/jdi.12466
204. Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim KS, et al. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab. (2017) 25:927–934.e923. doi: 10.1016/j.cmet.2017.02.008
205. Sancho V, Daniele G, Lucchesi D, Lupi R, Ciccarone A, Penno G, et al. Metabolic regulation of GLP-1 and PC1/3 in pancreatic alpha-cell line. PLoS ONE (2017) 12:e0187836. doi: 10.1371/journal.pone.0187836
206. Kilimnik G, Kim A, Steiner DF, Friedman TC, Hara M. Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic α-cells in mouse models of β-cell regeneration. Islets (2010) 2:149–55. doi: 10.4161/isl.2.3.11396
207. Vasu S, Moffett RC, Thorens B, Flatt PR. Role of endogenous GLP-1 and GIP in beta cell compensatory responses to insulin resistance and cellular stress. PLoS ONE (2014) 9:e101005. doi: 10.1371/journal.pone.0101005
208. Fujita Y, Wideman RD, Asadi A, Yang GK, Baker R, Webber T, et al. Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet alpha-cells and promotes insulin secretion. Gastroenterology (2010) 138:1966–75. doi: 10.1053/j.gastro.2010.01.049
209. Dalle S, Fontés G, Lajoix AD, LeBrigand L, Gross R, Ribes G, et al. Miniglucagon (glucagon 19-29): a novel regulator of the pancreatic islet physiology. Diabetes (2002) 51:406–12. doi: 10.2337/diabetes.51.2.406
210. Khan D, Vasu S, Moffett RC, Gault VA, Flatt PR, Irwin N. Locally produced xenin and the neurotensinergic system in pancreatic islet function and beta-cell survival. Biol Chem. (2017) 399:79–92. doi: 10.1515/hsz-2017-0136
211. Liu P, Song J, Liu H, Yan F, He T, Wang L, et al. Insulin regulates glucagon-like peptide-1 secretion by pancreatic alpha cells. Endocrine (2018). doi: 10.1007/s12020-018-1684-3. [Epub ahead of print].
212. Fava GE, Dong EW, Wu H. Intra-islet glucagon-like peptide 1. J Diab Complic. (2016) 30:1651–8. doi: 10.1016/j.jdiacomp.2016.05.016
213. Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA, et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia (2012) 55:3262–72. doi: 10.1007/s00125-012-2716-9
214. Traub S, Meier DT, Schulze F, Dror E, Nordmann TM, Goetz N, et al. Pancreatic alpha cell-derived glucagon-related peptides are required for beta cell adaptation and glucose homeostasis. Cell Rep. (2017) 18:3192–203. doi: 10.1016/j.celrep.2017.03.005
215. Smith EP, An Z, Wagner C, Lewis AG, Cohen EB, Li B, et al. The role of beta cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell Metab. (2014) 19:1050–7. doi: 10.1016/j.cmet.2014.04.005
216. Ritzel R, Orskov C, Holst JJ, Nauck MA. Pharmacokinetic, insulinotropic, and glucagonostatic properties of GLP-1 [7-36 amide] after subcutaneous injection in healthy volunteers. Dose-response-relationships. Diabetologia (1995) 38:720–5
217. Anlauf M, Weihe E, Hartschuh W, Hamscher G, Feurle GE. Localization of xenin-immunoreactive cells in the duodenal mucosa of humans and various mammals. J Histochem Cytochem. (2000) 48:1617–26. doi: 10.1177/002215540004801205
218. Barchetta I, Ciccarelli G, Cimini FA, Ceccarelli V, Orho-Melander M, Melander O, et al. Association between systemic leptin and neurotensin concentration in adult individuals with and without type 2 diabetes mellitus. J Endocrinol Invest. (2018) 41:1159–63. doi: 10.1007/s40618-018-0845-9
219. Mazella J, Beraud-Dufour S, Devader C, Massa F, Coppola T. Neurotensin and its receptors in the control of glucose homeostasis. Front Endocrinol. (2012) 3:143. doi: 10.3389/fendo.2012.00143
220. Price SL, Bloom SR. Protein PYY and its role in metabolism. Front Horm Res. (2014) 42:147–54. doi: 10.1159/000358343
221. Luo X, Li T, Zhu Y, Dai Y, Zhao J, Guo ZY, et al. The insulinotrophic effect of insulin-like peptide 5 in vitro and in vivo. Biochem J. (2015) 466:467–73. doi: 10.1042/bj20141113
222. Hasib A, Ng MT, Khan D, Gault VA, Flatt PR, Irwin N. A novel GLP-1/xenin hybrid peptide improves glucose homeostasis, circulating lipids and restores GIP sensitivity in high fat fed mice. Peptides (2018) 100:202–11. doi: 10.1016/j.peptides.2017.10.015
223. Wang S, Oestricker LZ, Wallendorf MJ, Sterl K, Dunai J, Kilpatrick CR, et al. Cholinergic signaling mediates the effects of xenin-25 on secretion of pancreatic polypeptide but not insulin or glucagon in humans with impaired glucose tolerance. PLoS ONE (2018) 13:e0192441. doi: 10.1371/journal.pone.0192441
224. Lyssenko V, Eliasson L, Kotova O, Pilgaard K, Wierup N, Salehi A, et al. Pleiotropic effects of GIP on islet function involve osteopontin. Diabetes (2011) 60:2424–33. doi: 10.2337/db10-1532
225. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. (2011) 17:1481–9. doi: 10.1038/nm.2513
226. Donath MY, Burcelin R. GLP-1 effects on islets: hormonal, neuronal, or paracrine? Diabetes care (2013) 36 (Suppl 2):S145–148. doi: 10.2337/dcS13-2015
227. Wueest S, Laesser CI, Boni-Schnetzler M, Item F, Lucchini FC, Borsigova M, et al. IL-6-Type cytokine signaling in adipocytes induces intestinal GLP-1 secretion. Diabetes (2018) 67:36–45. doi: 10.2337/db17-0637
228. Lang Lehrskov L, Lyngbaek MP, Soederlund L, Legaard GE, Ehses JA, Heywood SE, et al. Interleukin-6 delays gastric emptying in humans with direct effects on glycemic control. Cell Metab. (2018) 27:1201–11.e1203. doi: 10.1016/j.cmet.2018.04.008
229. Lebrun LJ, Lenaerts K, Kiers D, Pais de Barros JP, Le Guern N, Plesnik J, et al. Enteroendocrine L cells sense LPS after gut barrier injury to enhance GLP-1 secretion. Cell Rep. (2017) 21:1160–8. doi: 10.1016/j.celrep.2017.10.008
230. Chen T, Tian P, Huang Z, Zhao X, Wang H, Xia C, et al. Engineered commensal bacteria prevent systemic inflammation-induced memory impairment and amyloidogenesis via producing GLP-1. Appl Microbiol Biotechnol. (2018) 102:7565–75. doi: 10.1007/s00253-018-9155-6
231. Wierup N, Sundler F, Heller RS. The islet ghrelin cell. J. Mol. Endocrinol. (2014) 52:R35–49. doi: 10.1530/jme-13-0122
232. Rostamkhani F, Zardooz H, Goshadrou F, Baveisi M, Hedayati M. Stress increased ghrelin secretion from pancreatic isolated islets in male rats. Gen Physiol Biophys. (2016) 35:109–17. doi: 10.4149/gpb_2015037
233. Xu G, Hong X, Tang H, Jiang S, Liu F, Shen Z, et al. Ghrelin regulates GLP-1 production through mTOR signaling in L cells. Mol Cell Endocrinol. (2015) 416:9–18. doi: 10.1016/j.mce.2015.08.016
234. Gagnon J, Baggio LL, Drucker DJ, Brubaker PL. Ghrelin is a novel regulator of GLP-1 secretion. Diabetes (2015) 64:1513–21. doi: 10.2337/db14-1176
235. Chen X. Biochemical properties of recombinant prolyl dipeptidases DPP-IV and DPP8. Adv Exp Med Biol. (2006) 575:27–32. doi: 10.1007/0-387-32824-6_3
236. Jackson EK, Dubinion JH, Mi Z. Effects of dipeptidyl peptidase iv inhibition on arterial blood pressure. Clin Exp Pharmacol Physiol. (2008) 35:29–34. doi: 10.1111/j.1440-1681.2007.04737.x
237. Lund ML, Egerod KL, Engelstoft MS, Dmytriyeva O, Theodorsson E, Patel BA, et al. Enterochromaffin 5-HT cells - A major target for GLP-1 and gut microbial metabolites. Mol Metab. (2018) 11:70–83. doi: 10.1016/j.molmet.2018.03.004
238. Martin CR, Osadchiy V, Kalani A, Mayer EA. The brain-gut-microbiome axis. Cell Mol Gastroenterol Hepatol. (2018) 6:133–48. doi: 10.1016/j.jcmgh.2018.04.003
239. Ritter K, Buning C, Halland N, Poverlein C, Schwink L. G protein-coupled receptor 119 (GPR119) agonists for the treatment of diabetes: recent progress and prevailing challenges. J Med Chem. (2016) 59:3579–92. doi: 10.1021/acs.jmedchem.5b01198
240. Guo CJ, Chang FY, Wyche TP, Backus KM, Acker TM, Funabashi M, et al. Discovery of reactive microbiota-derived metabolites that inhibit host proteases. Cell (2017) 168:517–26.e518. doi: 10.1016/j.cell.2016.12.021
241. Mace OJ, Schindler M, Patel S. The regulation of K- and L-cell activity by GLUT2 and the calcium-sensing receptor CasR in rat small intestine. J Physiol. (2012) 590:2917–36. doi: 10.1113/jphysiol.2011.223800
242. Oguma T, Nakayama K, Kuriyama C, Matsushita Y, Yoshida K, Hikida K, et al. Intestinal sodium glucose cotransporter 1 inhibition enhances glucagon-like peptide-1 secretion in normal and diabetic rodents. J Pharmacol Exper Therapeut. (2015) 354:279–89. doi: 10.1124/jpet.115.225508
243. Jang HJ, Kokrashvili Z, Theodorakis MJ, Carlson OD, Kim BJ, Zhou J, et al. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc Natl Acad Sci USA. (2007) 104:15069–74. doi: 10.1073/pnas.0706890104
244. Kokrashvili Z, Mosinger B, Margolskee RF. T1r3 and alpha-gustducin in gut regulate secretion of glucagon-like peptide-1. Ann NY Acad Sci. (2009) 1170:91–4. doi: 10.1111/j.1749-6632.2009.04485.x
245. Li X, Staszewski L, Xu H, Durick K, Zoller M, Adler E. Human receptors for sweet and umami taste. Proc Natl Acad Sci USA. (2002) 99:4692–6. doi: 10.1073/pnas.072090199
246. Ohtsu Y, Nakagawa Y, Nagasawa M, Takeda S, Arakawa H, Kojima I. Diverse signaling systems activated by the sweet taste receptor in human GLP-1-secreting cells. Mol Cell Endocrinol. (2014) 394:70–9. doi: 10.1016/j.mce.2014.07.004
247. Fujita Y, Wideman RD, Speck M, Asadi A, King DS, Webber TD, et al. Incretin release from gut is acutely enhanced by sugar but not by sweeteners in vivo. Am J Physiol Endocrinol Metab. (2009) 296:E473–9. doi: 10.1152/ajpendo.90636.2008
248. Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. (2003) 278:11303–11. doi: 10.1074/jbc.M211495200
249. Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature (2003) 422:173–176. doi: 10.1038/nature01478
250. Flodgren E, Olde B, Meidute-Abaraviciene S, Winzell MS, Ahren B, Salehi A. GPR40 is expressed in glucagon producing cells and affects glucagon secretion. Biochem Biophys Res Commun. (2007) 354:240–5. doi: 10.1016/j.bbrc.2006.12.193
251. Parker HE, Habib AM, Rogers GJ, Gribble FM, Reimann F. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia (2009) 52:289–98. doi: 10.1007/s00125-008-1202-x
252. Nakamoto K. A new pain regulatory system via the brain long chain fatty acid receptor GPR40/FFA1 signal. Yakugaku Zasshi (2017) 137:199–204. doi: 10.1248/yakushi.16-00208
253. Dragano NRV, Solon C, Ramalho AF, de Moura RF, Razolli DS, Christiansen E, et al. Polyunsaturated fatty acid receptors, GPR40 and GPR120, are expressed in the hypothalamus and control energy homeostasis and inflammation. J Neuroinflammation (2017) 14:91. doi: 10.1186/s12974-017-0869-7
254. Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. (2005) 1:245–58. doi: 10.1016/j.cmet.2005.03.007
255. Kristinsson H, Smith DM, Bergsten P, Sargsyan E. FFAR1 is involved in both the acute and chronic effects of palmitate on insulin secretion. Endocrinology (2013) 154:4078–88. doi: 10.1210/en.2013-1352
256. Lan H, Hoos LM, Liu L, Tetzloff G, Hu W, Abbondanzo SJ, et al. Lack of FFAR1/GPR40 does not protect mice from high-fat diet-induced metabolic disease. Diabetes (2008) 57:2999–3006. doi: 10.2337/db08-0596
257. Panse M, Gerst F, Kaiser G, Teutsch CA, Dolker R, Wagner R, et al. Activation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) by free fatty acid receptor 1 (FFAR1/GPR40) protects from palmitate-induced beta cell death, but plays no role in insulin secretion. Cell Physiol Biochemi. (2015) 35:1537–45. doi: 10.1159/000373969
258. Ho JD, Chau B, Rodgers L, Lu F, Wilbur KL, Otto KA, et al. Structural basis for GPR40 allosteric agonism and incretin stimulation. Nat Commun. (2018) 9:1645. doi: 10.1038/s41467-017-01240-w
259. Latour MG, Alquier T, Oseid E, Tremblay C, Jetton TL, Luo J, et al. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes (2007) 56:1087–94. doi: 10.2337/db06-1532
260. Pachanski MJ, Kirkland ME, Kosinski DT, Mane J, Cheewatrakoolpong B, Xue J, et al. GPR40 partial agonists and AgoPAMs: Differentiating effects on glucose and hormonal secretions in the rodent. PLoS ONE (2017) 12:e0186033. doi: 10.1371/journal.pone.0186033
261. Gorski JN, Pachanski MJ, Mane J, Plummer CW, Souza S, Thomas-Fowlkes BS, et al. GPR40 reduces food intake and body weight through GLP-1. Am J Physiol Endocrinol Metab. (2017) 313:E37–e47. doi: 10.1152/ajpendo.00435.2016
262. Xiong Y, Swaminath G, Cao Q, Yang L, Guo Q, Salomonis H, et al. Activation of FFA1 mediates GLP-1 secretion in mice. Evidence for allosterism at FFA1. Mol Cell Endocrinol. (2013) 369:119–129.
263. Ekberg JH, Hauge M, Kristensen LV, Madsen AN, Engelstoft MS, Husted AS, et al. GPR119, a major enteroendocrine sensor of dietary triglyceride metabolites coacting in synergy with FFA1 (GPR40). Endocrinology (2016) 157:4561–9. doi: 10.1210/en.2016-1334
264. Psichas A, Larraufie PF, Goldspink DA, Gribble FM, Reimann F. Chylomicrons stimulate incretin secretion in mouse and human cells. Diabetologia (2017) 60:2475–85. doi: 10.1007/s00125-017-4420-2
265. Lin DC-H, Guo Q, Luo J, Zhang J, Nguyen K, Chen M, et al. Identification and pharmacological characterization of multiple allosteric binding sites on the free fatty acid 1 receptor. Mol Pharmacol. (2012) 82:843–59. doi: 10.1124/mol.112.079640
266. Little TJ, Isaacs NJ, Young RL, Ott R, Nguyen NQ, Rayner CK, et al. Characterization of duodenal expression and localization of fatty acid-sensing receptors in humans: relationships with body mass index. Am J Physiol Gastroint Liver Physiol. (2014) 307:G958–67. doi: 10.1152/ajpgi.00134.2014
267. Miyauchi S, Hirasawa A, Iga T, Liu N, Itsubo C, Sadakane K, et al. Distribution and regulation of protein expression of the free fatty acid receptor GPR120. Naunyn Schmiedeberg Arch Pharmacol. (2009) 379:427–34. doi: 10.1007/s00210-008-0390-8
268. van der Wielen N, van Avesaat M, de Wit NJ, Vogels JT, Troost F, Masclee A, et al. Cross-species comparison of genes related to nutrient sensing mechanisms expressed along the intestine. PLoS ONE (2014) 9:e107531. doi: 10.1371/journal.pone.0107531
269. Tazoe H, Otomo Y, Kaji I, Tanaka R, Karaki SI, Kuwahara A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J Physiol Pharmacol. (2008) 59 (Suppl. 2):251–62.
270. Kaji I, Karaki S, Tanaka R, Kuwahara A. Density distribution of free fatty acid receptor 2 (FFA2)-expressing and GLP-1-producing enteroendocrine L cells in human and rat lower intestine, and increased cell numbers after ingestion of fructo-oligosaccharide. J Mol Histol. (2011) 42:27–38. doi: 10.1007/s10735-010-9304-4
271. Odori S, Hosoda K, Tomita T, Fujikura J, Kusakabe T, Kawaguchi Y, et al. GPR119 expression in normal human tissues and islet cell tumors: evidence for its islet-gastrointestinal distribution, expression in pancreatic beta and alpha cells, and involvement in islet function. Metab Clin Exp. (2013) 62:70–8. doi: 10.1016/j.metabol.2012.06.010
272. Giaretta PR, Suchodolski JS, Blick AK, Steiner JM, Lidbury JA, Rech RR. Distribution of bile acid receptor TGR5 in the gastrointestinal tract of dogs. Histol Histopathol. (2018). doi: 10.14670/HH-18-025. [Epub ahead of print].
273. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. (2003) 278:9435–40. doi: 10.1074/jbc.M209706200
274. Hasan AU, Ohmori K, Hashimoto T, Kamitori K, Yamaguchi F, Noma T, et al. GPR120 in adipocytes has differential roles in the production of pro-inflammatory adipocytokines. Biochem Biophys Res Commun. (2017) 486:76–82. doi: 10.1016/j.bbrc.2017.03.001
275. Schilperoort M, van Dam AD, Hoeke G, Shabalina IG, Okolo A, Hanyaloglu AC, et al. The GPR120 agonist TUG-891 promotes metabolic health by stimulating mitochondrial respiration in brown fat. EMBO Mol Med. (2018) 10:e8047. doi: 10.15252/emmm.201708047
276. Stone VM, Dhayal S, Brocklehurst KJ, Lenaghan C, Sorhede Winzell M, Hammar M, et al. GPR120 (FFAR4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia (2014) 57:1182–91. doi: 10.1007/s00125-014-3213-0
277. Christiansen E, Watterson KR, Stocker CJ, Sokol E, Jenkins L, Simon K, et al. Activity of dietary fatty acids on FFA1 and FFA4 and characterisation of pinolenic acid as a dual FFA1/FFA4 agonist with potential effect against metabolic diseases. Br. J. Nutr. (2015) 113:1677–88. doi: 10.1017/s000711451500118x
278. Nagasawa T, Nakamichi H, Hama Y, Higashiyama S, Igarashi Y, Mitsutake S. Phytosphingosine is a novel activator of GPR120. J. Biochem. (2018) 164:27–32. doi: 10.1093/jb/mvy017
279. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell (2010) 142:687–98. doi: 10.1016/j.cell.2010.07.041
280. Anbazhagan AN, Priyamvada S, Gujral T, Bhattacharyya S, Alrefai WA, Dudeja PK, et al. A novel anti-inflammatory role of GPR120 in intestinal epithelial cells. Am J Physiol Cell Physiol. (2016) 310:C612–21. doi: 10.1152/ajpcell.00123.2015
281. Chen Y, Zhang D, Ho KW, Lin S, Suen WC, Zhang H, et al. GPR120 is an important inflammatory regulator in the development of osteoarthritis. Arthr Res. Ther. (2018) 20:163. doi: 10.1186/s13075-018-1660-6
282. Suckow AT, Polidori D, Yan W, Chon S, Ma JY, Leonard J, et al. Alteration of the glucagon axis in GPR120 (FFAR4) knockout mice: a role for GPR120 in glucagon secretion. J Biol Chem. (2014) 289:15751–63. doi: 10.1074/jbc.M114.568683
283. Ichimura A, Hirasawa A, Poulain-Godefroy O, Bonnefond A, Hara T, Yengo L, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature (2012) 483:350–4. doi: 10.1038/nature10798
284. Sheng R, Yang L, Zhang Y, Xing E, Shi R, Wen X, et al. Discovery of novel selective GPR120 agonists with potent anti-diabetic activity by hybrid design. Bioorg Med Chem Lett. (2018) 28:2599–604. doi: 10.1016/j.bmcl.2018.06.047
285. Winters MP, Sui Z, Wall M, Wang Y, Gunnet J, Leonard J, et al. Discovery of N-arylpyrroles as agonists of GPR120 for the treatment of type II diabetes. Bioorg Med Chem Lett. (2018) 28:841–6. doi: 10.1016/j.bmcl.2018.02.013
286. Nakamoto K, Shimada K, Harada S, Morimoto Y, Hirasawa A, Tokuyama S. DHA supplementation prevent the progression of NASH via GPR120 signaling. Eur J Pharmacol. (2018) 820:31–8. doi: 10.1016/j.ejphar.2017.11.046
287. Kang S, Huang J, Lee BK, Jung YS, Im E, Koh JM, et al. Omega-3 polyunsaturated fatty acids protect human hepatoma cells from developing steatosis through FFA4 (GPR120). Biochim Biophys Acta (2018) 1863:105–16. doi: 10.1016/j.bbalip.2017.11.002
288. Sundstrom L, Myhre S, Sundqvist M, Ahnmark A, McCoull W, Raubo P, et al. The acute glucose lowering effect of specific GPR120 activation in mice is mainly driven by glucagon-like peptide 1. PLoS ONE (2017) 12:e0189060. doi: 10.1371/journal.pone.0189060
289. Satapati S, Qian Y, Wu MS, Petrov A, Dai G, Wang SP, et al. GPR120 suppresses adipose tissue lipolysis and synergizes with GPR40 in antidiabetic efficacy. J Lipid Res. (2017) 58:1561–78. doi: 10.1194/jlr.M075044
290. Wolenski FS, Zhu AZX, Johnson M, Yu S, Moriya Y, Ebihara T, et al. Fasiglifam (TAK-875) Alters Bile Acid Homeostasis in Rats and Dogs: a Potential Cause of Drug Induced Liver Injury. Toxicol Sci. (2017) 157:50–61. doi: 10.1093/toxsci/kfx018
291. Sawzdargo M, George SR, Nguyen T, Xu S, Kolakowski LF, O'Dowd BF. A cluster of four novel human G protein-coupled receptor genes occurring in close proximity to CD22 gene on chromosome 19q13.1. Biochem Biophys Res Commun. (1997) 239:543–547. doi: 10.1006/bbrc.1997.7513
292. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. (2003) 278:11312–9. doi: 10.1074/jbc.M211609200
293. Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. (2003) 278:25481–9. doi: 10.1074/jbc.M301403200
294. Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem Biophys Res Commun. (2003) 303:1047–52. doi: 10.1016/s0006-291x(03)00488-1
295. Tough IR, Forbes S, Cox HM. Signaling of free fatty acid receptors 2 and 3 differs in colonic mucosa following selective agonism or coagonism by luminal propionate. Neurogastroenterol Motil. (2018). doi: 10.1111/nmo.13454. [Epub ahead of print].
296. Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes (2012) 61:364–71. doi: 10.2337/db11-1019
297. Karaki S, Tazoe H, Hayashi H, Kashiwabara H, Tooyama K, Suzuki Y, et al. Expression of the short-chain fatty acid receptor, GPR43, in the human colon. J Mol Histol. (2008) 39:135–42. doi: 10.1007/s10735-007-9145-y
298. Yang G, Chen S, Deng B, Tan C, Deng J, Zhu G, et al. Implication of G protein-coupled receptor 43 in intestinal inflammation: a mini-review. Front Immunol. (2018) 9:1434. doi: 10.3389/fimmu.2018.01434
299. Abrahami D, Douros A, Yin H, Yu OHY, Renoux C, Bitton A, et al. Dipeptidyl peptidase-4 inhibitors and incidence of inflammatory bowel disease among patients with type 2 diabetes: population based cohort study. BMJ (2018) 360:k872. doi: 10.1136/bmj.k872
300. Ang Z, Xiong D, Wu M, Ding JL. FFAR2-FFAR3 receptor heteromerization modulates short-chain fatty acid sensing. FASEB J. (2018) 32:289–303. doi: 10.1096/fj.201700252RR
301. Forbes S, Stafford S, Coope G, Heffron H, Real K, Newman R, et al. Selective FFA2 agonism appears to act via intestinal PYY to reduce transit and food intake but does not improve glucose tolerance in mouse models. Diabetes (2015) 64:3763–71. doi: 10.2337/db15-0481
302. Christiansen CB, Gabe MBN, Svendsen B, Dragsted LO, Rosenkilde MM, Holst JJ. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am J Physiol Gastrointest Liver Physiol. (2018) 315:G53–65. doi: 10.1152/ajpgi.00346.2017
303. Park BO, Kim SH, Kong GY, Kim DH, Kwon MS, Lee SU, et al. Selective novel inverse agonists for human GPR43 augment GLP-1 secretion. Eur J Pharmacol. (2016) 771:1–9. doi: 10.1016/j.ejphar.2015.12.010
304. Tang C, Ahmed K, Gille A, Lu S, Grone HJ, Tunaru S, et al. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat Med. (2015) 21:173–7. doi: 10.1038/nm.3779
305. Milligan G, Alvarez-Curto E, Hudson BD, Prihandoko R, Tobin AB. FFA4/GPR120: pharmacology and therapeutic opportunities. Trends Pharmacol Sci. (2017) 38:809–21. doi: 10.1016/j.tips.2017.06.006
306. Hudson BD, Tikhonova IG, Pandey SK, Ulven T, Milligan G. Extracellular ionic locks determine variation in constitutive activity and ligand potency between species orthologs of the free fatty acid receptors FFA2 and FFA3. J Biol Chem. (2012) 287:41195–209. doi: 10.1074/jbc.M112.396259
307. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut (1987) 28:1221–7.
309. Topping DL, Clifton P. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev. (2001) 81:1031–64. doi: 10.1152/physrev.2001.81.3.1031
310. Rahat-Rozenbloom S, Fernandes J, Gloor GB, Wolever TMS. Evidence for greater production of colonic short-chain fatty acids in overweight than lean humans. Int J Obes. (2014) 38:1525. doi: 10.1038/ijo.2014.46
311. Rahat-Rozenbloom S, Fernandes J, Cheng J, Wolever TMS. Acute increases in serum colonic short-chain fatty acids elicited by inulin do not increase GLP-1 or PYY responses but may reduce ghrelin in lean and overweight humans. Eur J Clin Nutr. (2017) 71:953–8. doi: 10.1038/ejcn.2016.249
312. Liaw CW, Connolly DT. Sequence polymorphisms provide a common consensus sequence for GPR41 and GPR42. DNA Cell Biol. (2009) 28:555–60. doi: 10.1089/dna.2009.0916
313. Puhl HL III, Won YJ, Lu VB, Ikeda SR. Human GPR42 is a transcribed multisite variant that exhibits copy number polymorphism and is functional when heterologously expressed. Sci Rep. (2015) 5:12880. doi: 10.1038/srep12880
314. Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett. (2002) 520:97–101. doi: 10.1016/s0014-5793(02)02775-8
315. Fredriksson R, Höglund PJ, Gloriam DE, Lagerström MC, Schiöth HB. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. (2003) 554:381–8. doi: 10.1016/s0014-5793(03)01196-7
316. Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. (2006) 3:167–75. doi: 10.1016/j.cmet.2006.02.004
317. Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun. (2007) 362:928–34. doi: 10.1016/j.bbrc.2007.08.078
318. Sakamoto Y, Inoue H, Kawakami S, Miyawaki K, Miyamoto T, Mizuta K, et al. Expression and distribution of Gpr119 in the pancreatic islets of mice and rats: predominant localization in pancreatic polypeptide-secreting PP-cells. Biochem Biophys Res Commun. (2006) 351:474–80. doi: 10.1016/j.bbrc.2006.10.076
319. Chu ZL, Jones RM, He H, Carroll C, Gutierrez V, Lucman A, et al. A role for beta-cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucose-dependent insulin release. Endocrinology (2007) 148:2601–9. doi: 10.1210/en.2006-1608
320. Lauffer LM, Iakoubov R, Brubaker PL. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes (2009) 58:1058–66. doi: 10.2337/db08-1237
321. Engelstoft MS, Norn C, Hauge M, Holliday ND, Elster L, Lehmann J, et al. Structural basis for constitutive activity and agonist-induced activation of the enteroendocrine fat sensor GPR119. Br J Pharmacol. (2014) 171:5774–89. doi: 10.1111/bph.12877
322. Mandoe MJ, Hansen KB, Hartmann B, Rehfeld JF, Holst JJ, Hansen HS. The 2-monoacylglycerol moiety of dietary fat appears to be responsible for the fat-induced release of GLP-1 in humans. Am J Clin Nutr. (2015) 102:548–55. doi: 10.3945/ajcn.115.106799
323. Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De Fonseca F, et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature (2003) 425:90–3. doi: 10.1038/nature01921
324. Schwartz GJ, Fu J, Astarita G, Li X, Gaetani S, Campolongo P, et al. The lipid messenger OEA links dietary fat intake to satiety. Cell Metab. (2008) 8:281–8. doi: 10.1016/j.cmet.2008.08.005
325. Astarita G, Rourke BC, Andersen JB, Fu J, Kim JH, Bennett AF, et al. Postprandial increase of oleoylethanolamide mobilization in small intestine of the Burmese python (Python molurus). Am J Physiol Regul Integr Comparat Physiol. (2006) 290:R1407–12. doi: 10.1152/ajpregu.00664.2005
326. Tinoco AB, Armirotti A, Isorna E, Delgado MJ, Piomelli D, de Pedro N. Role of oleoylethanolamide as a feeding regulator in goldfish. J Exper Biol (2014) 217 (Pt 15):2761–9. doi: 10.1242/jeb.106161
327. Diep TA, Madsen AN, Krogh-Hansen S, Al-Shahwani M, Al-Sabagh L, Holst B, et al. Dietary non-esterified oleic Acid decreases the jejunal levels of anorectic N-acylethanolamines. PLoS ONE (2014) 9:e100365. doi: 10.1371/journal.pone.0100365
328. Gao J, Tian L, Weng G, O'Brien TD, Luo J, Guo Z. Stimulating beta-cell replication and improving islet graft function by AR231453, A GPR119 agonist. Transplant Proc. (2011) 43:3217–20. doi: 10.1016/j.transproceed.2011.10.021
329. Moss CE, Glass LL, Diakogiannaki E, Pais R, Lenaghan C, Smith DM, et al. Lipid derivatives activate GPR119 and trigger GLP-1 secretion in primary murine L-cells. Peptides (2016) 77:16–20. doi: 10.1016/j.peptides.2015.06.012
330. Panaro BL, Flock GB, Campbell JE, Beaudry JL, Cao X, Drucker DJ. beta-Cell Inactivation of Gpr119 Unmasks Incretin Dependence of GPR119-Mediated Glucoregulation. Diabetes (2017) 66:1626–35. doi: 10.2337/db17-0017
331. Cox HM, Tough IR, Woolston AM, Zhang L, Nguyen AD, Sainsbury A, et al. Peptide YY is critical for acylethanolamine receptor Gpr119-induced activation of gastrointestinal mucosal responses. Cell Metab. (2010) 11:532–42. doi: 10.1016/j.cmet.2010.04.014
332. Hu YW, Yang JY, Ma X, Chen ZP, Hu YR, Zhao JY, et al. A lincRNA-DYNLRB2-2/GPR119/GLP-1R/ABCA1-dependent signal transduction pathway is essential for the regulation of cholesterol homeostasis. J Lipid Res. (2014) 55:681–97. doi: 10.1194/jlr.M044669
333. Koshizawa T, Morimoto T, Watanabe G, Watanabe T, Yamasaki N, Sawada Y, et al. Optimization of a novel series of potent and orally bioavailable GPR119 agonists. Bioorg Med Chem Lett. (2017) 27:3249–53. doi: 10.1016/j.bmcl.2017.06.034
334. Huan Y, Jiang Q, Li G, Bai G, Zhou T, Liu S, et al. The dual DPP4 inhibitor and GPR119 agonist HBK001 regulates glycemic control and beta cell function ex and in vivo. Sci. Rep. (2017) 7:4351. doi: 10.1038/s41598-017-04633-5
335. Scott JS, Brocklehurst KJ, Brown HS, Clarke DS, Coe H, Groombridge SD, et al. Conformational restriction in a series of GPR119 agonists: differences in pharmacology between mouse and human. Bioorg Med Chem Lett. (2013) 23:3175–9. doi: 10.1016/j.bmcl.2013.04.006
336. Schjoldager B, Shaw MJ, Powers SP, Schmalz PF, Szurszewski J, Miller LJ. Bovine gallbladder muscularis: source of a myogenic receptor for cholecystokinin. Am J Physiol. (1988) 254:G294–9. doi: 10.1152/ajpgi.1988.254.3.G294
337. Wiley JW, O'Dorisio TM, Owyang C. Vasoactive intestinal polypeptide mediates cholecystokinin-induced relaxation of the sphincter of Oddi. J Clin Invest. (1988) 81:1920–4. doi: 10.1172/jci113539
338. Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. (2005) 329:386–90. doi: 10.1016/j.bbrc.2005.01.139
339. Kuhre RE, Wewer Albrechtsen NJ, Larsen O, Jepsen SL, Balk-Moller E, Andersen DB, et al. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol Metab (2018) 11:84–95. doi: 10.1016/j.molmet.2018.03.007
340. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. (2009) 89:147–91. doi: 10.1152/physrev.00010.2008
341. Bronden A, Alber A, Rohde U, Gasbjerg LS, Rehfeld JF, Holst JJ, et al. The bile acid-sequestering resin sevelamer eliminates the acute GLP-1 stimulatory effect of endogenously released bile acids in patients with type 2 diabetes. Diabetes Obes Metab. (2017) 20:362–9. doi: 10.1111/dom.13080
342. Adrian TE, Gariballa S, Parekh KA, Thomas SA, Saadi H, Al Kaabi J, et al. Rectal taurocholate increases L cell and insulin secretion, and decreases blood glucose and food intake in obese type 2 diabetic volunteers. Diabetologia (2012) 55:2343–7. doi: 10.1007/s00125-012-2593-2
343. Sonne DP, Hansen M, Knop FK. Bile acid sequestrants in type 2 diabetes: potential effects on GLP1 secretion. Eur J Endocrinol. (2014) 171:R47–65. doi: 10.1530/eje-14-0154
344. Morimoto K, Watanabe M, Sugizaki T, Irie J, Itoh H. Intestinal bile acid composition modulates prohormone convertase 1/3 (PC1/3) expression and consequent GLP-1 production in male mice. Endocrinology (2016) 157:1071–81. doi: 10.1210/en.2015-1551
345. Lasalle M, Hoguet V, Hennuyer N, Leroux F, Piveteau C, Belloy L, et al. Topical Intestinal Aminoimidazole Agonists of G-Protein-Coupled Bile Acid Receptor 1 Promote Glucagon Like Peptide-1 Secretion and Improve Glucose Tolerance. J Med Chem. (2017) 60:4185–211. doi: 10.1021/acs.jmedchem.6b01873
346. Duan H, Ning M, Zou Q, Ye Y, Feng Y, Zhang L, et al. Discovery of Intestinal Targeted TGR5 Agonists for the Treatment of Type 2 Diabetes. J Med Chem. (2015) 58:3315–28. doi: 10.1021/jm500829b
347. Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell (1995) 81:687–93.
348. Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J Biol Chem. (2004) 279:49517–22. doi: 10.1074/jbc.M409041200
349. Watanabe M, Horai Y, Houten SM, Morimoto K, Sugizaki T, Arita E, et al. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J Biol Chem. (2011) 286:26913–20. doi: 10.1074/jbc.M111.248203
350. Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA. (2006) 103:1006–11. doi: 10.1073/pnas.0506982103
351. Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun. (2013) 4:2384. doi: 10.1038/ncomms3384
352. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science (1999) 284:1362–5.
353. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science (1999) 284:1365–8.
354. Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA. (2001) 98:3369–74. doi: 10.1073/pnas.051551698
355. Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science (2002) 296:1313–6. doi: 10.1126/science.1070477
356. Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab. Rev. (2004) 36:703–22. doi: 10.1081/dmr-200033475
357. Tian J, Huang S, Sun S, Ding L, Zhang E, Huang W. Bile acid signaling and bariatric surgery. Liver Res. (2017) 1:208–13. doi: 10.1016/j.livres.2017.12.007
358. Chen T, Reich NW, Bell N, Finn PD, Rodriguez D, Kohler J, et al. Design of gut-restricted thiazolidine agonists of g protein-coupled bile acid receptor 1 (GPBAR1, TGR5). J Med Chem. (2018) 61:7589–613. doi: 10.1021/acs.jmedchem.8b00308
359. De Petrocellis L, Vellani V, Schiano-Moriello A, Marini P, Magherini PC, Orlando P, et al. Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J Pharmacol Exp Ther. (2008) 325:1007–15. doi: 10.1124/jpet.107.134809
360. Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature (1997) 389:816–24. doi: 10.1038/39807
361. Zhu Z, Luo Z, Ma S, Liu D. TRP channels and their implications in metabolic diseases. Pflug Arch. (2011) 461:211–23. doi: 10.1007/s00424-010-0902-5
362. Liu D, Zhu Z, Tepel M. The role of transient receptor potential channels in metabolic syndrome. Hyperten Res. (2008) 31:1989–95. doi: 10.1291/hypres.31.1989
363. Jordt SE, Tominaga M, Julius D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc Natl Acad Sci USA. (2000) 97:8134–9. doi: 10.1073/pnas.100129497
364. Bohlen CJ, Priel A, Zhou S, King D, Siemens J, Julius D. A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell (2010) 141:834–45. doi: 10.1016/j.cell.2010.03.052
365. Min JW, Liu WH, He XH, Peng BW. Different types of toxins targeting TRPV1 in pain. Toxicon (2013) 71:66–75. doi: 10.1016/j.toxicon.2013.05.016
366. Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature (2001) 411:957–62. doi: 10.1038/35082088
367. Jia Y, McLeod RL, Wang X, Parra LE, Egan RW, Hey JA. Anandamide induces cough in conscious guinea-pigs through VR1 receptors. Br J Pharmacol. (2002) 137:831–6. doi: 10.1038/sj.bjp.0704950
368. Bisogno T, Hanus L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. (2001) 134:845–52. doi: 10.1038/sj.bjp.0704327
369. Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto Y, et al. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain (2005) 1:3. doi: 10.1186/1744-8069-1-3
370. Amadesi S, Nie J, Vergnolle N, Cottrell GS, Grady EF, Trevisani M, et al. Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J Neurosci. (2004) 24:4300–12. doi: 10.1523/jneurosci.5679-03.2004
371. Akiba Y, Kato S, Katsube K, Nakamura M, Takeuchi K, Ishii H, et al. Transient receptor potential vanilloid subfamily 1 expressed in pancreatic islet beta cells modulates insulin secretion in rats. Biochem Biophys Res Commun. (2004) 321:219–25. doi: 10.1016/j.bbrc.2004.06.149
372. De Toni L, Garolla A, Menegazzo M, Magagna S, Di Nisio A, Sabovic I, et al. Heat Sensing Receptor TRPV1 is a mediator of thermotaxis in human spermatozoa. PLoS ONE (2016) 11:e0167622. doi: 10.1371/journal.pone.0167622
373. Lieu TM, Myers AC, Meeker S, Undem BJ. TRPV1 induction in airway vagal low-threshold mechanosensory neurons by allergen challenge and neurotrophic factors. Am J Physiol Lung Cell Mol Physiol. (2012) 302:L941–8. doi: 10.1152/ajplung.00366.2011
374. Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, Kanai AJ, et al. Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat Neurosci. (2002) 5:856–60. doi: 10.1038/nn902
375. Ward SM, Bayguinov J, Won KJ, Grundy D, Berthoud HR. Distribution of the vanilloid receptor (VR1) in the gastrointestinal tract. J Comp Neurol. (2003) 465:121–35. doi: 10.1002/cne.10801
376. Matsumoto K, Kurosawa E, Terui H, Hosoya T, Tashima K, Murayama T, et al. Localization of TRPV1 and contractile effect of capsaicin in mouse large intestine: high abundance and sensitivity in rectum and distal colon. Am J Physiol Gastrointest Liver Physiol. (2009) 297:G348–60. doi: 10.1152/ajpgi.90578.2008
377. Wang P, Yan Z, Zhong J, Chen J, Ni Y, Li L, et al. Transient receptor potential vanilloid 1 activation enhances gut glucagon-like peptide-1 secretion and improves glucose homeostasis. Diabetes (2012) 61:2155–65. doi: 10.2337/db11-1503
378. Kang JH, Goto T, Han IS, Kawada T, Kim YM, Yu R. Dietary capsaicin reduces obesity-induced insulin resistance and hepatic steatosis in obese mice fed a high-fat diet. Obesity (Silver Spring) (2010) 18:780–7. doi: 10.1038/oby.2009.301
379. Tolan I, Ragoobirsingh D, Morrison EY. The effect of capsaicin on blood glucose, plasma insulin levels and insulin binding in dog models. Phytother Res. PTR (2001) 15:391–4. doi: 10.1002/ptr.750
380. Smeets AJ, Westerterp-Plantenga MS. The acute effects of a lunch containing capsaicin on energy and substrate utilisation, hormones, and satiety. Eur J Nutr. (2009) 48:229–34. doi: 10.1007/s00394-009-0006-1
381. Lee E, Jung DY, Kim JH, Patel PR, Hu X, Lee Y, et al. Transient receptor potential vanilloid type-1 channel regulates diet-induced obesity, insulin resistance, and leptin resistance. FASEB J. (2015) 29:3182–92. doi: 10.1096/fj.14-268300
382. Motter AL, Ahern GP. TRPV1-null mice are protected from diet-induced obesity. FEBS Lett. (2008) 582:2257–62. doi: 10.1016/j.febslet.2008.05.021
383. Zhang LL, Yan Liu D, Ma LQ, Luo ZD, Cao TB, Zhong J, et al. Activation of transient receptor potential vanilloid type-1 channel prevents adipogenesis and obesity. Circ Res. (2007) 100:1063–70. doi: 10.1161/01.RES.0000262653.84850.8b
384. Panchal SK, Bliss E, Brown L. Capsaicin in metabolic syndrome. Nutrients (2018) 10:630. doi: 10.3390/nu10050630
385. Derbenev AV, Zsombok A. Potential therapeutic value of TRPV1 and TRPA1 in diabetes mellitus and obesity. Semin Immunopathol. (2016) 38:397–406. doi: 10.1007/s00281-015-0529-x
386. Greiner TU, Backhed F. Microbial regulation of GLP-1 and L-cell biology. Mol Metab. (2016) 5:753–8. doi: 10.1016/j.molmet.2016.05.012
387. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. (2016) 14:1002533. doi: 10.1371/journal.pbio.1002533
388. Lukovac S, Belzer C, Pellis L, Keijser BJ, de Vos WM, Montijn RC, et al. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. MBio (2014) 5:e01438-14. doi: 10.1128/mBio.01438-14
389. Nicholson J, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science (2012) 336:1262–7. doi: 10.1126/science.1223813
390. Frank DN St. Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. (2007) 104:13780–5. doi: 10.1073/pnas.0706625104
391. Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature (2012) 489:242–9. doi: 10.1038/nature11552
392. Cook S. Review article: short chain fatty acids in health and disease. Aliment Pharmacol Ther. (1998) 12:499–507. doi: 10.1046/j.1365-2036.1998.00337.x
393. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. (2013) 54:2325–40. doi: 10.1194/jlr.R036012
394. Cohen LJ, Esterhazy D, Kim SH, Lemetre C, Aguilar RR, Gordon EA, et al. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature (2017) 549:48–53. doi: 10.1038/nature23874
395. Wang R, Zang P, Chen J, Wu F, Zheng Z, Ma J, et al. Gut Microbiota Play an Essential Role in the Antidiabetic Effects of Rhein. Evid Based Complemen Alter Med. (2018) 2018:6093282. doi: 10.1155/2018/6093282
396. Yuan X, Ni H, Chen X, Feng X, Wu Q, Chen J. Identification of therapeutic effect of glucagon-like peptide 1 in the treatment of STZ-induced diabetes mellitus in rats by restoring the balance of intestinal flora. J Cell Biochem. (2018). doi: 10.1002/jcb.27343. [Epub ahead of print].
397. Cani PD, Hoste S, Guiot Y, Delzenne NM. Dietary non-digestible carbohydrates promote L-cell differentiation in the proximal colon of rats. Br J Nutr. (2007) 98:32–7. doi: 10.1017/s0007114507691648
398. Petersen N, Reimann F, Bartfeld S, Farin HF, Ringnalda FC, Vries RG, et al. Generation of L cells in mouse and human small intestine organoids. Diabetes (2014) 63:410–20. doi: 10.2337/db13-0991
399. Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol. (2016) 14:273–87. doi: 10.1038/nrmicro.2016.17
400. Scott TA, Quintaneiro LM, Norvaisas P, Lui PP, Wilson MP, Leung KY, et al. Host-microbe co-metabolism dictates cancer drug efficacy in C. elegans. Cell (2017) 169:442–56.e418. doi: 10.1016/j.cell.2017.03.040
401. Duboc H, Nguyen CC, Cavin JB, Ribeiro-Parenti L, Jarry AC, Rainteau D, et al. Roux-en-Y Gastric-Bypass and sleeve gastrectomy induces specific shifts of the gut microbiota without altering the metabolism of bile acids in the intestinal lumen. Int J Obes. (2018). doi: 10.1038/s41366-018-0015-3. [Epub ahead of print].
402. Thompson GL, Canals M, Poole DP. Biological redundancy of endogenous GPCR ligands in the gut and the potential for endogenous functional selectivity. Front Pharmacol. (2014) 5:262. doi: 10.3389/fphar.2014.00262
403. Narita T, Yokoyama H, Yamashita R, Sato T, Hosoba M, Morii T, et al. Comparisons of the effects of 12-week administration of miglitol and voglibose on the responses of plasma incretins after a mixed meal in Japanese type 2 diabetic patients. Diabetes Obes Metab. (2012) 14:283–7. doi: 10.1111/j.1463-1326.2011.01526.x
404. Ellrichmann M, Kapelle M, Ritter PR, Holst JJ, Herzig K-H, Schmidt WE, et al. Orlistat inhibition of intestinal lipase acutely increases appetite and attenuates postprandial glucagon-like peptide-1-(7–36)-Amide-1, Cholecystokinin, and Peptide YY Concentrations. J Clin Endocrinol Metab. (2008) 93:3995–8. doi: 10.1210/jc.2008-0924
405. Knop FK, Vilsboll T, Larsen S, Hojberg PV, Volund A, Madsbad S, et al. Increased postprandial responses of GLP-1 and GIP in patients with chronic pancreatitis and steatorrhea following pancreatic enzyme substitution. Am J Physiol Endocrinol Metab. (2007) 292:E324–30. doi: 10.1152/ajpendo.00059.2006
406. Rahat-Rozenbloom S, Fernandes J, Cheng J, Gloor GB, Wolever TM. The acute effects of inulin and resistant starch on postprandial serum short-chain fatty acids and second-meal glycemic response in lean and overweight humans. Eur J Clin Nutr. (2017) 71:227–33. doi: 10.1038/ejcn.2016.248
407. Gribble FM, Meek CL, Reimann F. Targeted intestinal delivery of incretin secretagogues—towards new diabetes and obesity therapies. Peptides (2018) 100:68–74. doi: 10.1016/j.peptides.2017.11.008
408. Chang J, Wu T, Greenfield JR, Samocha-Bonet D, Horowitz M, Rayner CK. Effects of intraduodenal glutamine on incretin hormone and insulin release, the glycemic response to an intraduodenal glucose infusion, and antropyloroduodenal motility in health and type 2 diabetes. Diabetes Care (2013) 36:2262–5. doi: 10.2337/dc12-1663
409. Meek CL, Lewis HB, Vergese B, Park A, Reimann F, Gribble F. The effect of encapsulated glutamine on gut peptide secretion in human volunteers. Peptides (2016) 77:38–46. doi: 10.1016/j.peptides.2015.10.008
410. Lindqvist A, Ekelund M, Pierzynowski S, Groop L, Hedenbro J, Wierup N. Gastric bypass in the pig increases GIP levels and decreases active GLP-1 levels. Peptides (2017) 90:78–82. doi: 10.1016/j.peptides.2017.02.009
Keywords: glucagon-like peptide-1, metabolic disease, type 2 diabetes, enteroendocrine cell system, GPCR, L-cells, microbiome, α-cells
Citation: Paternoster S and Falasca M (2018) Dissecting the Physiology and Pathophysiology of Glucagon-Like Peptide-1. Front. Endocrinol. 9:584. doi: 10.3389/fendo.2018.00584
Received: 04 June 2018; Accepted: 14 September 2018;
Published: 11 October 2018.
Edited by:
Gary Sweeney, York University, CanadaReviewed by:
Kyoung-Han Kim, University of Ottawa, CanadaGiulia Cantini, Università degli Studi di Firenze, Italy
Copyright © 2018 Paternoster and Falasca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Falasca, bWFyY28uZmFsYXNjYUBjdXJ0aW4uZWR1LmF1