 Joanne M. de Laat
Joanne M. de Laat Rachel S. van Leeuwaarde
Rachel S. van Leeuwaarde- Department of Endocrine Oncology, University Medical Center Utrecht, Utrecht, Netherlands
Multiple Endocrine Neoplasia type 1 (MEN1) is a rare autosomal dominant inherited condition, causing significant morbidity, and a reduction of life expectancy. A timely and accurate diagnosis of MEN1 is paramount to improve disease outcomes. This enables early identification of tumor manifestations allowing timely treatment for reducing morbidity and improving survival. Current management of MEN1 poses two challenges regarding the MEN1 diagnosis: diagnostic delay and the issue of phenocopies. A delay in diagnosis can be caused by a delay in identifying the index case, and by a delay in identifying affected family members of an index case. At present, lag time between diagnosis of MEN1 in index cases and genetic testing of family members was estimated to be 3.5 years. A subsequent delay in diagnosing affected family members was demonstrated to cause potential harm. Non-index cases have been found to develop clinically relevant tumor manifestations during the lag times. Centralized care, monitoring of patients outcomes on a national level and thereby improving awareness of physicians treating MEN1 patients, will contribute to improved care. The second challenge relates to “phenocopies.” Phenocopies refers to the 5–25% of clinically diagnosed patients with MEN1in whom no mutation can be found. Up to now, the clinical diagnosis of MEN1 is defined as the simultaneous presence of at least two of the three characteristic tumors (pituitary, parathyroids, or pancreatic islets). These clinically diagnosed patients undergo intensive follow up. Recent insights, however, challenge the validity of this clinical criterion. The most common mutation-negative MEN1 phenotype is the combination of primary hyperparathyroidism and a pituitary adenoma. This phenotype might also be caused by mutations in the CDKN1B gene, causing the recently described MEN4 syndrome. Moreover, primary hyperparathyroidism and pituitary adenoma are relatively common in the general population. Limiting follow-up in patients with a sporadic co-occurrence of pHPT and PIT could reduce exposure to radiation from imaging, healthcare costs and anxiety.
Introduction
Multiple Endocrine Neoplasia type 1 (MEN1) is a rare autosomal dominant inherited condition, with a prevalence estimated around 1–10/100 000 (1). MEN1 causes significant morbidity and reduction of life expectancy in those affected (2–4). Penetrance of MEN1 is high, over 90% of individuals carrying a MEN1 mutation will be affected (2, 3, 5). A timely and accurate diagnosis of index cases and their family members is key to management of MEN1. There is mounting evidence that intensive follow-up of MEN1 patients and screening for tumor manifestations reduces morbidity and improves survival (3, 6). Conversely, a delay in MEN1 diagnosis has been found to cause potential harm (7). In a recent study the lag time between diagnosis of MEN1 in index cases and genetic testing of family members was 3.5 years. Before or during this lag time several familial cases already had developed metastatic neuroendocrine tumor manifestations (7).
The diagnostic approach to patients in whom MEN1 is suspected has rapidly evolved since the discovery of the MEN1 gene in 1997 located at 11q13 (1, 8). Nowadays, genetic testing has a well-established role in confirming diagnosis of MEN1, identifying family members of index patients with MEN1 mutation who are at risk to develop tumor manifestation, and reassuring family members without a mutation.
Nevertheless, new diagnostic challenges have also emerged. In 5–25% of patients with a clinical diagnosis of MEN1 no mutation can be found, a phenomenon often referred to as “phenocopies” (9, 10). There is increasing debate if such patients are correctly diagnosed as having MEN1 since some of these patients might have a sporadic coincidence of two neuroendocrine tumors (3). However, there are also mutations in other genes that can cause a MEN-1 like phenotype. Mutations in the cyclin-dependent kinase inhibitor (CDNK1B), have been found to cause a syndrome of parathyroid and anterior pituitary tumors (11). Patients with a CDNK1B gene mutation have a clinical course different from patients with MEN1 mutations, and have a lower risk to develop the pancreatic neuro-endocrine tumors (pNET). For this reason the syndrome is now referred to as MEN4.
Another diagnostic challenge is to reduce a diagnostic delay of MEN1. A delay in diagnosis can be caused by a delay in identifying the index case, and by a delay in identifying affected family members after diagnosing an index case. Identifying index cases raises the question “who and when to screen for a MEN1 mutation?.” Incidence of some manifestations related to MEN1 is high in the general population. The incidence of primary hyperparathyroidism (pHPT) for example is estimated as high as 34 to 120 per 100,000 person-years among women, and from 13 to 36 among men (12). Current guidelines advice genetic screening for MEN1 in patients with pHPT if diagnosed at an age below the age of 30 years (or below the age of 40 years in multigland parathyroid disease). Concern has been raised that such policy might results in a significant delay in diagnosis of the index case (13–15). Reducing lag times in identification of MEN1 mutation carriers among family members of MEN1 patients requires awareness among treating physicians and organized structured program (7, 16, 17).
In this review we will discuss implications of recent epidemiological insights on clinical diagnostic criteria for MEN1 and the “phenocopies” phenomenon. Furthermore we will assess the impact of delay in diagnosis and discuss medical and organizational advances that can help reducing lag times in both index cases and their family members (Table 1).

Table 1. Review highlights.
Clinical Diagnosis of MEN1
In the early 20th century simultaneous occurrence of three tumors characteristic for MEN1 (pituitary, parathyroids, and pancreatic islets) had been reported as a pathological rarity (18, 19). In 1953, Underdahl et al. described a clinical diagnosis of MEN1 based on a case series of eight patients with a syndrome of multiple endocrine adenomas including a literature study comprising another fourteen cases (20). In addition to the tumors of the pituitary, parathyroids and pancreatic islets, three of his eight patients presented with peptic ulcers and one patient had an adrenal adenoma. Wermer et al. found that the syndrome was autosomal dominantly inherited, describing the cases of four sisters and their father with similar tumors (21).
Up to now, the simultaneous presence of at least two of the three characteristic tumors (pituitary, parathyroids or pancreatic islets) is still considered pathognomic for MEN1 (10). The current clinical practice guidelines describes three criteria for diagnosis of MEN1:(10)
- The genetic criterion: presence of a known MEN1 mutation, irrespective of clinical or biomechanical manifestation.
- The familial criterion: occurrence of at least one MEN1 associated tumor and a first-degree relative with MEN1.
- The clinical criterion: defined by the occurrence of at least two out of three characteristic tumors (pituitary, parathyroid, or pancreatic islets).
The clinical criterion, however, has now become subject to debate. Genetical testing in MEN1 patients meeting the clinical criterion is negative in up to 25% of cases (10). Negative mutation analysis is predominantly found in patients without a family history of MEN1 (14, 15, 22, 23). Till now, these patients were considered and treated as new index cases of MEN1. With the introduction of new highly sensitive genetic tests such as multiplex ligation-dependent probe amplification (MLPA), incidence of negative genetic testing in patients clinically diagnosed with MEN1 indeed declined, but still is around 10% (9, 24, 25). It is questionable whether all patients meeting the clinical criterion and a negative mutation analysis have MEN1 since clinical observations show that mutation negative patients often have a more favorable clinical course (3, 14, 22).
Negative mutation testing is most prevalent in the clinical MEN1 phenotype comprising a combination of tumors of the parathyroids and pituitary gland. In a study from Hai et al. reporting on 20 clinically diagnosed index cases of MEN1, frequency of MEN1 mutation was only 11% in nine patients presenting with a combination of tumors of parathyroids and pituitary glands vs. a 63% frequency in eleven patients with other tumor combinations (22). These results were confirmed in a study by Klein et al. (26). In twenty index cases undergoing mutational analysis in this study, none of the ten patients presenting with tumors of parathyroids and pituitary glands tested positive for MEN1 mutation, vs. 60% of patients in whom phenotype included a pancreatic tumor. Interestingly, frequency of MEN1 mutation was also only 25% in 32 pedigrees of familial occurrence of tumors of parathyroids and pituitary gland (26).
Alternative explanations for negative testing in patients with clinical diagnosed MEN1, or “phenocopies,” should be considered. There are either other syndromes that can cause a MEN1-like phenotype or sporadic co-incidence of two neuro-endocrine tumors.
Other Syndromes That Can Cause a Men1-Like Phenotype
Two syndromes that should be considered in mutation-negative MEN1 patients that are of particular interest are MEN4 and Familial Isolated Pituitary Adenomas (FIPA). MEN4 is a syndrome of parathyroid and anterior pituitary tumors that is caused by mutations in the CDKN1B gene, which encodes the cyclin-dependent kinase inhibitor p27kip1 (11). Mutations in this gene were first found to cause a syndrome of neuroendocrine tumors in rat studies, in patterns that could overlap both MEN1 and MEN2 (27). Pellegata et al. subsequently reported a CDKN1B gene mutation in a patient presenting with pituitary and parathyroid tumors, in whom analysis of his pedigree revealed MEN1-like phenotypes in multiple generations (28).
Several other reports of CDKN1B mutations have been made (3, 29–34). Most reported on cases of parathyroid and anterior pituitary tumors, besides one case of a pituitary tumor in combination with a well differentiated pancreatic neoplasm (32). Agarwal et al. screened 196 patients with MEN1 phenotype who scored negative for MEN1 mutation analysis for MEN4 and mutation in other cyclin-dependent kinase inhibitors (33). In their analyses, seven potentially pathological mutation in cyclin-dependent kinase inhibitors, 3 of which in p27 related to MEN4 are described. Compared to MEN1 patients, patients with MEN4 develop tumors at a relatively late age with a mean age for developing pHPT at 56 years (29–31, 33, 34). Little is known about long-term follow-up of MEN4 because of the small number of patients reported and limited follow-up.
FIPA is a syndrome of familial occurring isolated pituitary adenomas caused by a mutation in the AIP gene, most commonly resulting in prolactinomas and GH- secreting pituitary tumors (PIT) (35, 36). Patients with FIPA develop pituitary tumors at a relative younger age as compared to sporadic cases (36). FIPA syndrome in itself will not result in a complete MEN1- like phenotype. However patients with FIPA need only to develop one sporadic neuroendocrine manifestation (of which pHPT is not uncommon in the normal population) to meet clinical criteria for MEN1. In combination with the familial occurrence of PIT in patients FIPA, one can easily imagine that some patients with FIPA might mistakenly be diagnosed as having MEN1. Georgitsi et al. reported on screening for AIP mutation in 490 patients with PIT, 91 of whom were previously screened for MEN1 (37). Two of 91 patients previously suspected for MEN1 an AIP mutation was found. Both patients developed a GH-secreting PIT at young age (16 and 18 years respectively). The report did not mention if these patients had also developed other MEN1 related manifestations.
No specific guidelines for follow-up of patients with FIPA and MEN4 exist. Nonetheless, it seems safe to apply a much more limited screening program to patients with FIPA and MEN4 as compared to patients with true MEN1. For patients with FIPA, Korbonits et al. recommend yearly clinical assessment and pituitary function tests, accompanied by dynamic testing to evaluate for hormone excess or deficiency as needed, and follow-up pituitary MRI (38). In patients with MEN4 the need for thoracic imaging, as performed in patients MEN1 for early diagnosis of bronchopulmonary NET and thymic tumors, might be waived. Screening for pancreatic NET in MEN4 can be debated. Sporadic cases of pancreatic NET in patients with MEN4 have been reported, although the incidence seems much lower than in patients with MEN1 (32).
Syndromes such as MEN4 and FIPA predisposing for neuroendocrine tumors can cause a MEN1-like phenotype and need to be considered in mutation- negative patients. However, these syndromes might explain only a minority of the total of mutation- negative MEN1 phenotypes. Georgitsi et al. screened 106 patients to find one case of MEN4, as compared to Agarwal et al. who found 3 cases in 196 patients (29, 33). In a national cohort study from the Netherlands, de Laat et al. found only one CDKN1B mutations and no AIP mutations upon additional screening among 30 mutation-negative MEN1 patients (3). Igreja et al. found no CDKN1B or AIP mutation in 21 patients mutation-negative MEN1 patients (39). Several other papers testing mutation-negative patients for CDKN1B mutation, but not AIP, could not demonstrate a pathologic mutation (40, 41).
Sporadic Co-incidence of Two Neuroendocrine Tumors
Another explanation for mutation negative MEN1 phenotype is a sporadic co-incidence of two neuro-endocrine tumors. Such sporadic co-incidence might be much more common than generally perceived. The prevalence of pHPT is relatively high in the normal population, and has been rising over the past decades. In recent years, a prevalence of 233 per 100,000 women and 85 per 100,000 men has been reported (12). With improving imaging techniques, prevalence of incidental PIT is also increasing. The clinical significance of such incidentalomas is still uncertain. The current clinical practice guideline for pituitary incidentalomas advises lifelong radiological follow-up (42). In a healthy volunteer study including 100 subjects pituitary incidentalomas on MRI were prevalent in 10% (43). A systematic review even reported incidental pituitary adenomas in 22.5% of cerebral imaging studies (44). Reflecting these numbers, a co-incidence of pHPT and an incidental pituitary adenoma could occur without the necessity for a tumor predisposition that requires intensive follow-up.
The findings of a long-term follow-up study comparing between 293 mutation-positive and 30 mutation-negative MEN1 patients in the Netherlands indeed showed that the mutation-negative patients have a very mild natural course of disease with a life expectancy that is comparable to that of the general Dutch population (3). As in other studies, a combination of pHPT and PIT was the most prevalent phenotype (77%) among mutation-negative patients. At the time of this study all mutation-negative patients were index cases without a familial history of MEN1. Later a sibling of one of the mutation-negative patients also developed a clinically diagnosed MEN1 (7). None of the 30 mutation-negative patients developed a third MEN1 manifestation during a median of 8 years of follow-up. Also none of the mutation-negative patients died from a MEN1 related cause as opposed to the mutation positive group in whom 60% of mortality was related to MEN1. Finally, median survival in mutation-negative patients was 84.0 years (95% CI: NA) compared to 73.0 years (95% CI: 69.3-76.6) in mutation-positive patients (P = 0.013) (3). Both this Dutch cohort and a Japanese cohort showed that mutation-negative patients develop tumor manifestations at a significant later age, supporting the evidence for a milder natural course (3, 22).
It is impossible to exclude that clinically diagnosed mutation-negative MEN1 patients, might harbor a yet unknown mutation to either the MEN1 gene or any other gene that predisposes for neuroendocrine tumor development. Nevertheless, it seems reasonable that many such patients have a sporadic co-incidence of two tumors and that at least part of the mutation-negative MEN1 patients can be discharged from intensive follow-up. Hereby it is important to recognize that patients with genetically proven MEN1, who are systematically followed-up, have a high fear of disease occurrence which is associated with a lower quality of life (45). In addition, intensive follow-up in subjects without a high risk of tumor occurrence leads to overutilization of health care resources and costs.
A key issue remains how to select patients that can be discharged from follow-up. The first important modulating factor to consider is family history. A diligent pedigree analysis remains one the most important clinical tools to raise suspicion about a yet unknown inheritable disorder.
Because most mutation-negative patients will present with pHPT, it is necassary to consider modulating factors in the clinical presentation of pHPT, i.e., multiglandular vs. single gland disease. Hyperparathyroidism in true MEN1 patients typically present as “asymmetrical hyperplasia” affecting multiple glands, for this reason a subtotal hyperparathyroidectomy is considered the optimal treatment for pHPT in MEN1 (4, 46, 47). Although this has been criticized in a recent retrospective cohort 8/24 patient with pHPT was treated by unilateral clearance with a 87.5% success rate (48). The study unfortunately did not report on mutation status of these patients or further manifestations.
In an additional analysis of the Dutch cohort of mutation-negative patients the pathology and imaging results of 28/30 (93.3%) mutation-negative patients presenting with pHPT were reviewed (3). In 22 of the 28 mutation negative patients with pHPT an parathyreoidectomy was performed. The pathology showed a uniglandular adenoma in all cases. Six mutation negative patients with pHPT were not operated, however in 3 out of these 6 patients imaging results suggested uniglandular disease (ultrasound or Tc99m-sestamibi parathyroid scintigraphy). In conclusion, mutation-negative patients with pHPT not only present with a significantly better clinical course but also predominantly presented with a uniglandular pHPT. An uniglandular pHPT therefore seems to increase the likelihood of co-incidence of two sporadic neuroendocrine tumors instead of true MEN1.
Summarizing these epidemiological data, we propose an approach to clinically diagnosed MEN1 patients based upon co-occurrence of pHPT and PIT without a MEN1 mutation. A MEN4 or FIPA diagnosis should be considered in these patients. If such mutation is found, follow-up as appropriate for these conditions should be provided. If no mutation is found in these genes, the familial history and the presentation of pHPT should be carefully reviewed. Patients without family history of neuroendocrine tumors or other MEN1 manifestations and “only” uniglandular parathyroid disease might safely be discharged from further intensive follow-up. Limiting follow-up in patients with evidence for MEN4, FIPA, and sporadic co-occurrence of pHPT and PIT could reduce exposure to radiation from imaging, healthcare costs and anxiety.
Delay in Diagnosis of MEN1
There is broad consensus on the need for timely recognition of MEN1. First degree family members of MEN1 patients should be offered genetic testing at the most early account (10). Pediatric cases of MEN1 manifestations have been described as early as 5 years, and several cohort studies indicate that close follow-up and treatment improves survival in MEN1 gene mutation carriers (2–4, 10). For this reason, the clinical practice guidelines also recommend start of the follow-up program in early childhood. Pediatric manifestations of MEN1, however, are relatively rare, and physicians should weigh the potential benefits against potential harm from radiation by imaging studies, quality of life and costs in the very young patient. Because no apparent genotype/ phenotype relation have described for MEN1 it is difficult to recommend about individualization of MEN1 follow-up.
Delays in the diagnosis of MEN1 can occur by a delay in identifying an index case, or by lag time between diagnosing the index case and testing of family members. Because MEN1 is a rare disease, delays in identification can possibly occur by a lack of awareness about the disease and the indications for genetic screening by treating physicians. A recent review of the Italian MEN1 registry revealed that the average age of first MEN1 manifestation was 41.6 years, while the average age of MEN1 diagnosis was 55.1 years, suggesting a significant potential to improve the time between first manifestation and diagnosis of MEN1 (16).
Delay in Diagnosis of Index Case
According to the current practice guidelines genetic mutation analysis should be offered to all patients meeting the familial or clinical criteria for diagnosis of MEN1, and patients presenting with a MEN1 related tumor. Suspicion for MEN1 is described as: parathyroid adenoma below the age of 30 years (or multiglandular parathyroid disease at any age); gastrinoma or multiple pancreatic NET at any age or individuals who have two or more MEN1-associated tumors that are not part of the classical triad of parathyroid, pancreatic islet and anterior pituitary tumors (e.g., parathyroid tumor plus adrenal tumor) (10).
Indications for genetic testing in patients presenting with pHPT have been subject to debate and are slowly evolving. The incidence of pHPT in the general population is high, and genetic screening laborious and costly. Thus testing for MEN1 in all patients with pHPT is not considered a cost-effective approach. Previous guidelines recommended testing for MEN1 only in patients presenting with pHPT before the age of 30 (49). Lassen et al. reported on a case of a 31 year old woman presenting with multiglandular pHPT who was initially not tested under this previous guidelines. Diagnosis of MEN1 was only established 7 years later when she developed recurrent disease after subtotal parathyroidectomy (13). In the current clinical practice guidelines indication for MEN1 mutation analysis has therefore been expanded to forty years in case of multiglandular disease.
Concerns over delayed diagnosis in patients presenting with pHPT, however, are still not fully met. Analysis of referrals for genetic testing in both Sweden and the Netherlands revealed that physicians in both countries frequently referred patients that did not meet the criteria for genetic testing according to the clinical practice guidelines (64 and 81% of all patients referred for genetic counseling respectively) (15, 50). Moreover, in both groups MEN1 mutations were found among the patients who were tested but did not meet the clinical practice guideline criteria.
Concerns over delayed diagnosis have also been raised in index patients presenting with a pancreatic NET (51). The ENETS consensus guidelines for the management of pancreatic NET's recommend testing for MEN1 in patients presenting with insulinoma before the age of 20, in addition to testing of patients with multiple pancreatic NET's at any age (51). Two recent studies revealed that in 10–12% of MEN1 patients younger than 21 years insulinomas were the first presentation of MEN1(52, 53). This is in line with studies confirming that insulinomas in particular present at young age in patients with MEN1, as opposed to gastrinomas (54, 55). Type 2 well differentiated gastric neuoendocrine neoplasms have been associated with gastrinomas and MEN1 (56). Since nonfunctioning pancreatic NET's also occur from a young age, MEN1 testing should perhaps not only be limited to patients with insulinomas before the age of 20, but recommended for all patients with pancreatic NET's before this age.
Only a relative small percentage of bronchial NET are associated with MEN1 (< 5%). The ESMO and ENETS guideline statement on genetic testing for MEN1 in patients with bronchial NET are in agreement with the clinical practice guidelines for MEN1 (10, 57–59). Testing is adviced if the familial history is suggestive of MEN1 or a second MEN1 feature is present, e.g., hyperparathyroidism (57). Several cohorts report thymic NET in MEN1 patients to occur predominantly in men with a mean age around the fifth decade (60–62). Nevertheless, in a large Japanese cohort of MEN1 patients a relative high percentage (36%) of women was found among patients with thymic NET (63). Thus MEN1 should still be considered in female patients presenting with thymic NET.
Several authors have emphasized the role of family history as a prognostic factor (50, 64, 65). Not only a family history positive for MEN1 should be considered a risk factor for the disease, but also a family history positive for pHPT or any other neuro endocrine tumor up to third degree family members is significantly correlated with an increased risk of finding a MEN1 mutation.
Based on the Dutch cohort a prediction model was made which was validated in an independent Swedish cohort. This prediction model that allows clinicians in daily clinical practice to estimate the individual risk for a MEN1 mutation in their patients (50). Based on the risk factors for MEN1 in individual patients, the risk for having a positive MEN1 mutation risk can be calculated which can be used in counseling patients at higher risk for MEN1. Risk factors from this model include: recurrent or multiglandular pHPT; nonrecurrent pHPT; pancreatic and duodenal NET; pituitary tumor; NET of stomach, thymus or bronchus; and positive family history (up to third degree relatives) for any neuroendocrine tumor (Figure 1).
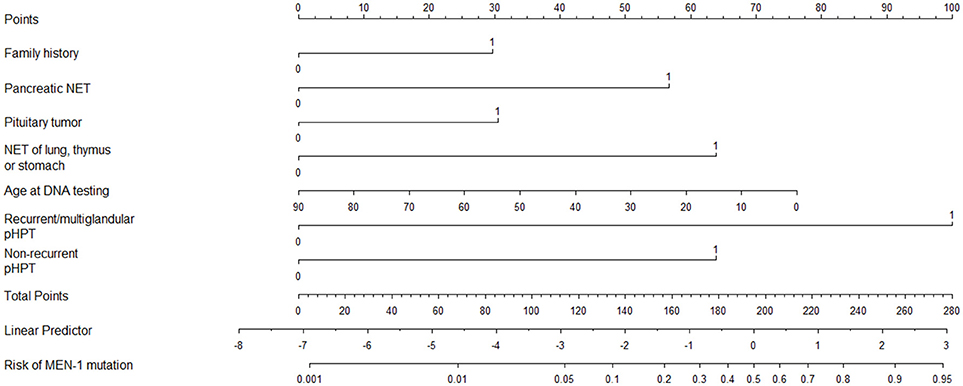
Figure 1. Nomogram for predicting the risk of a MEN1 mutation. NET, neuro- endocrine tumor; pHPT, primary hyperparathyroidism. How to use: Nomogram to calculate the risk of a MEN1 mutation. Draw a vertical line for each variable to the “points” axis at the top. Sum the points for the eight variables and locate this total score on the “total points” axis. Draw a vertical line from this through the bottom two scales to determine the linear predictor and the predicted risk of a MEN1 mutation. Example: a 54-year-old patient (score = 30 points) with the combination of a negative family history. (score = 0 points), a nonrecurrent and nonmultiglandular pHPT (score = 63 points), and a pNET (n = 57 points) has a sum score of 150 points, corresponding with a linear predictor of −0.50 and a risk of 38% of having a MEN1 mutation. Example: a 41-year-old patient (score = 42 points) with a positive family history (score = 29 points) and recurrent pHPT (score = 100 points) has a sum score of 171 points, corresponding with a linear predictor of 0.50 and a risk of 63% of having a MEN1 mutation. Originally appeared in: de Laat, J. M., et al. (50).
Delay in Diagnosis of Men1 in Family Members of Patients
In the nationwide Dutch MEN1 study, van Leeuwaarde et al. recently systematically described lag times between diagnosis of a MEN1 index case and testing of family members (7). Delayed genetic testing of family members appeared to be an important cause for avoidable morbidity and even mortality. MEN1 patients are prone for severe morbidity such as osteoporosis caused by Phpt (66, 67). Moreover, higher severity of bone involvement in comparison with sporadic pHPT has been reported (68). Complications due to bone mineral loss and urolithiasis caused by pHPT are early onset and thus have the potential to be progressive and severe (68, 69).
In the Dutch study, 304 MEN1 patients from 58 MEN1 families were included. The median lag time between diagnosis of the index cases and family members was 3.5 years. At the time of the subsequent MEN1 diagnosis in the family members of the index cases, 30 (12.1%) had a duodenopancreatic neuroendocrine tumor, of whom 20% already had metastatic disease. Mean lag time of patients with metastatic disease was 10.9 years, compared to 7.1 years in patients without metastases. Almost 40% of non-index cases had a pHPT at time of diagnosis. A total of five patients had a macroadenoma of whom two had compression of the optic chiasm. Ten non-index cases died because of a MEN1-related cause that might have been developed during the lag time.
This first report on lag time in diagnosis of non-index cases showed alarming outcomes. Reducing the delay in diagnosis of non-index cases requires national efforts to centralize care for MEN1 patients ensuring optimal quality of care. In this study a reduction in lag time from a median of 8 years before 1998 till 0.75 years after 2007 was found (7). During this period MEN1 care was centralized in the Netherlands. Centralized care and monitoring of outcomes in MEN1 patient care through national registries has been well established in a number of European countries as well, including the Group d'etude des Tumeurs Endocrine in France and the Italian network of MEN1 referral centers (16, 61). Centralization and collaboration on a national level in the Netherlands has improved awareness of MEN1 in physicians treating patients with MEN1, which probably has contributed to the reduction of lag times.
In conclusion, a timely MEN1 diagnosis in index cases and their affected family members should be pursued by physicians that treat patients with MEN1. Evidently, not all physicians are aware of the disease and the consequences arising from a delayed diagnosis. Ongoing (inter)national publications and scientific meetings in collaboration with patient advocacy groups will further increase this awareness. In spite of the rarity of the disease, the MEN1 landscape in respect of an accurate MEN1 diagnosis is evolving and still open for debate and improvement. Future cost effectiveness studies could be useful to enhance this discussion and a helpful tool for physicians and policy makers for both clinical and policy wise MEN1 decision-making.
Author Contributions
JdL conception of the work, analysis of available literature, drafting the article, final approval. RvL and GV conception of the work, analysis of available literature, critical revision of the article, final approval.
Funding
This research did not receive any specific grant from any funding agency in the 24 public, commercial or not-for-profit sector.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science (1997) 276:404–7. doi: 10.1126/science.276.5311.404
2. Lourenço DM Jr, Toledo RA, Coutinho FL, Margarido LC, Siqueira SA, dos Santos MA, et al. The impact of clinical and genetic screenings on the management of the multiple endocrine neoplasia type 1. Clinics (2007) 62:465–76. doi: 10.1590/S1807-59322007000400014
3. de Laat JM, van der Luijt RB, Pieterman CRC, Oostveen MP, Hermus AR, Dekkers OM, et al. MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med. (2016) 14:182. doi: 10.1186/s12916-016-0708-1
4. Pieterman CR, van Hulsteijn LT, den HM, van der Luijt RB, Bonenkamp JJ, Hermus AR, et al. Primary hyperparathyroidism in MEN1 patients: a cohort study with longterm follow-up on preferred surgical procedure and the relation with genotype. Ann Surg. (2012) 255:1171–8. doi: 10.1097/SLA.0b013e31824c5145
5. Pieterman CRC, Conemans EB, Dreijerink KMA, de Laat JM, Timmers HTM, Vriens MR, et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer (2014) 21:R121–42. doi: 10.1530/ERC-13-0482
6. Ramundo V, Milone F, Severino R, Savastano S, Di Somma C, Vuolo L, et al. Clinical and prognostic implications of the genetic diagnosis of hereditary net syndromes in asymptomatic patients. Horm Metab Res. (2011) 43:794–800. doi: 10.1055/s-0031-1286324
7. van Leeuwaarde RS, van Nesselrooij BPM, Hermus AR, Dekkers OM, de Herder WW, van der Horst-Schrivers AN, et al. Impact of delay in diagnosis in outcomes in MEN1: results from the Dutch MEN1 study group. J Clin Endocrinol Metab. (2016) 101:jc.2015-3766. doi: 10.1210/jc.2015-3766
8. Lemmens I, de Van V, Kas K, Zhang CX, Giraud S, Wautot V, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. (1997) 6:1177–83.
9. Lemos MC, Thakker R V. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. (2008) 29:22–32. doi: 10.1002/humu.20605
10. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. (2012) 97:2990–3011. doi: 10.1210/jc.2012-1230
11. Thakker R V. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol. (2014) 386:2–15. doi: 10.1016/j.mce.2013.08.002
12. Yeh MW, Ituarte PHG, Zhou HC, Nishimoto S, Liu I-LA, Harari A, et al. Incidence and prevalence of primary hyperparathyroidism in a racially mixed population. J Clin Endocrinol Metab. (2013) 98:1122–9. doi: 10.1210/jc.2012-4022
13. Lassen T, Friis-Hansen L, Rasmussen AK, Knigge U, Feldt-Rasmussen U. Primary hyperparathyroidism in young people. When should we perform genetic testing for multiple endocrine neoplasia 1 (MEN-1)? J Clin Endocrinol Metab. (2014) 99:3983–7. doi: 10.1210/jc.2013-4491
14. Ellard S, Hattersley AT, Brewer CM, Vaidya B. Detection of an MEN1 gene mutation depends on clinical features and supports current referral criteria for diagnostic molecular genetic testing. Clin Endocrinol. (2005) 62:169–75. doi: 10.1111/j.1365-2265.2005.02190.x
15. Tham E, Grandell U, Lindgren E, Toss G, Skogseid B, Nordenskjold M. Clinical testing for mutations in the MEN1 gene in Sweden: a report on 200 unrelated cases. J Clin Endocrinol Metab. (2007) 92:3389–95. doi: 10.1210/jc.2007-0476
16. Giusti F, Cianferotti L, Boaretto F, Cetani F, Cioppi F, Colao A, et al. Multiple endocrine neoplasia syndrome type 1: institution, management, and data analysis of a nationwide multicenter patient database. Endocrine (2017) 58:349–59. doi: 10.1007/s12020-017-1234-4
17. Marini F, Giusti F, Tonelli F, Brandi ML. Management impact: effects on quality of life and prognosis in MEN1. Endocr Relat Cancer (2017) 24:T227–42. doi: 10.1530/ERC-17-0203
18. Erdheim J. Zur normalen und pathologischen Histologie der Glandula thyreoidea, parathyreoidea und Hypophysis. Beitr Pathol Anat. (1903) 33:158–236.
19. Llyod PC. A case of hypophyseal tumor with associated tumorlike enlargement of the parathyroids and islands of Langerhans. Bull Johns Hopkins Hosp. (1929) 45:1–14.
20. Underdahl L, Woolner L, BMB. Multiple endocrine adenomas; report of 8 cases in which the parathyroids, pituitary and pancreatic islets were involved. J Clin Endocrinol Metab. (1953) 13:20–47. doi: 10.1210/jcem-13-1-20
21. Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med. (1954) 16:363–71. doi: 10.1016/0002-9343(54)90353-8
22. Hai N, Aokis N, Shlmatsu A, Mod T, Kosugi S. Clinical features of multiple endocrine neoplasia type 1 ( MENI ) phenocopy without germline MEN7 gene mutations : analysis of 20 Japanese sporadic cases with MEN1. Clin Endocrinol. (2000) 52:509–18. doi: 10.1046/j.1365-2265.2000.00966.x
23. Cebrian A, Ruiz-Llorente S, Cascon A, Pollan M, Diez JJ, Pico A, Telleria D, et al. Mutational and gross deletion study of the MEN1 gene and correlation with clinical features in Spanish patients. J Med Genet. (2003) 40:e72. doi: 10.1136/jmg.40.5.e72
24. Raef H, Zou M, Baitei EY, Al-Rijjal RA, Kaya N, Al-Hamed M, et al. A novel deletion of the MEN1 gene in a large family of multiple endocrine neoplasia type 1 (MEN1) with aggressive phenotype. Clin Endocrinol. (2011) 75:791–800. doi: 10.1111/j.1365-2265.2011.04134.x
25. Giacche M, Panarotto A, Mori L, Daffini L, Tacchetti MC, Pirola I, et al. A novel menin gene deletional mutation in a little series of Italian patients affected by apparently sporadic multiple endocrine neoplasia type 1 syndrome. J Endocrinol Invest. (2012) 35:124–8. doi: 10.1007/BF03345419
26. Klein RD, Salih S, Bessoni J, Bale AE. Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet Med. (2005) 7:131–8. doi: 10.1097/01.GIM.0000153663.62300.F8
27. Fritz A, Walch A, Piotrowska K, Rosemann M, Schäffer E, Weber K, et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. (2002) 62:3048–51. Available online at: http://cancerres.aacrjournals.org/content/62/11/3048.long
28. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, et al. Germ-line mutations in p27(Kip1) cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA. (2006) 103:15558–63. doi: 10.1073/pnas.0603877103
29. Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab. (2007) 92:3321–5. doi: 10.1210/jc.2006-2843
30. Molatore S, Marinoni I, Lee M, Pulz E, Ambrosio MR, degli Uberti EC, et al. A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Hum Mutat. (2010) 31:E1825–35. doi: 10.1002/humu.21354
31. Malanga D, De Gisi S, Riccardi M, Scrima M, De Marco C, Robledo M, et al. Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur J Endocrinol. (2012) 166:551–60. doi: 10.1530/EJE-11-0929
32. Occhi G, Regazzo D, Trivellin G, Boaretto F, Ciato D, Bobisse S, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet. (2013) 9:e1003350. doi: 10.1371/journal.pgen.1003350
33. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. (2009) 94:1826–34. doi: 10.1210/jc.2008-2083
34. Tonelli F, Giudici F, Giusti F, Marini F, Cianferotti L, Nesi G, et al. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. Eur J Endocrinol. (2014) 171:K7–K17. doi: 10.1530/EJE-14-0080
35. Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab. (2008) 93:2390–401. doi: 10.1210/jc.2007-2611
36. Daly AF, Jaffrain-Rea ML, Ciccarelli A, Valdes-Socin H, Rohmer V, Tamburrano G, et al. Clinical characterization of familial isolated pituitary adenomas. J Clin Endocrinol Metab. (2006) 91:3316–23. doi: 10.1210/jc.2005-2671
37. Georgitsi M, Raitila A, Karhu A, Tuppurainen K, Makinen MJ, Vierimaa O, et al. Molecular diagnosis of pituitary adenoma predisposition caused by aryl hydrocarbon receptor-interacting protein gene mutations. Proc Natl Acad Sci USA. (2007) 104:4101–5. doi: 10.1073/pnas.0700004104
38. Korbonits M, Kumar A. AIP -related familial isolated pituitary adenomas. Gene Rev. (2012) 2:1–21.
39. Igreja S, Chahal HS, Akker SA, Gueorguiev M, Popovic V, Damjanovic S, et al. Assessment of p27 (cyclin-dependent kinase inhibitor 1B) and aryl hydrocarbon receptor-interacting protein (AIP) genes in multiple endocrine neoplasia (MEN1) syndrome patients without any detectable MEN1 gene mutations. Clin Endocrinol. (2009) 70:259–64. doi: 10.1111/j.1365-2265.2008.03379.x
40. Ozawa A, Agarwal SK, Mateo CM, Burns AL, Rice TS, Kennedy PA, et al. The parathyroid/pituitary variant of multiple endocrine neoplasia type 1 usually has causes other than p27Kip1 mutations. J Clin Endocrinol Metab. (2007) 92:1948–51. doi: 10.1210/jc.2006-2563
41. Owens M, Stals K, Ellard S, Vaidya B. Germline mutations in the CDKN1B gene encoding p27 Kip1 are a rare cause of multiple endocrine neoplasia type 1. Clin Endocrinol. (2009) 70:499–500. doi: 10.1111/j.1365-2265.2008.03363.x
42. Freda PU, Beckers AM, Katznelson L, Molitch ME, Montori VM, Post KD, et al. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2011) 96:894–904. doi: 10.1210/jc.2010-1048
43. Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. (1994) 120:817–20.
44. Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, et al. The prevalence of pituitary adenomas: a systematic review. Cancer (2004) 101:613–9. doi: 10.1002/cncr.20412
45. van Leeuwaarde RS, Pieterman CRC, Bleiker EMA, Dekkers OM, van der Horst-Schrivers AN, Hermus AR, et al. High fear of disease occurrence is associated with low quality of life in patients with Multiple Endocrine Neoplasia type 1 (MEN1): results from the dutch MEN1 study group. J Clin Endocrinol Metab. (2018) 103:2354–61. doi: 10.1210/jc.2018-00259
46. Kebebew E, Hwang J, Reiff E, Duh Q-Y, Clark OH. Predictors of single-gland vs multigland parathyroid disease in primary hyperparathyroidism: a simple and accurate scoring model. Arch Surg. (2006) 141:777–82; discussion 782. doi: 10.1001/archsurg.141.8.777
47. Schreinemakers JMJ, Pieterman CRC, Scholten A, Vriens MR, Valk GD, Rinkes IHMB. The optimal surgical treatment for primary hyperparathyroidism in MEN1 patients: a systematic review. World J Surg. (2011) 35:1993–2005. doi: 10.1007/s00268-011-1068-9
48. Kluijfhout WP, Beninato T, Drake FT, Vriens MR, Gosnell J, Shen WT, et al. Unilateral clearance for primary hyperparathyroidism in selected patients with multiple endocrine neoplasia type 1. World J Surg. (2016) 40:2964–9. doi: 10.1007/s00268-016-3624-9
49. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. (2001) 86:5658–71. doi: 10.1210/jcem.86.12.8070
50. de Laat JM, Tham E, Pieterman CR, Vriens MR, Dorresteijn JA, Bots ML, et al. Predicting the risk of multiple endocrine neoplasia type 1 for patients with commonly occurring endocrine tumors. EurJ Endocrinol. (2012) 167:181–7. doi: 10.1530/EJE-12-0210
51. Falconi M, Eriksson B, Kaltsas G, Bartsch DK, Capdevila J, Caplin M, et al. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology (2016) 103:153–71. doi: 10.1159/000443171
52. Goudet P, Dalac A, Le Bras M, Cardot-Bauters C, Niccoli P, Lévy-Bohbot N, et al. MEN1 disease occurring before 21 years old: a 160-patient cohort study from the groupe d'étude des tumeurs endocrines. J Clin Endocrinol Metab. (2015) 100:1568–77. doi: 10.1210/jc.2014-3659
53. Goncalves TD, Toledo RA, Sekiya T, Matuguma SE, Maluf Filho F, Rocha MS, et al. Penetrance of functioning and nonfunctioning pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1 in the second decade of life. J Clin Endocrinol Metab. (2014) 99:89–96. doi: 10.1210/jc.2013-1768
54. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome - A prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (2004) 83:43–83. doi: 10.1097/01.md.0000112297.72510.32
55. Jensen RI, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer (2008) 113:1807–43. doi: 10.1002/cncr.23648
56. Fave GD, O'Toole D, Sundin A, Taal B, Ferolla P, Ramage JK, et al. ENETS consensus guidelines update for gastroduodenal neuroendocrine neoplasms. Neuroendocrinology (2016) 103:119–24. doi: 10.1159/000443168
57. Caplin ME, Baudin E, Ferolla P, Filosso P, Garcia-Yuste M, Lim E, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. (2015) 26:1604–20. doi: 10.1093/annonc/mdv041
58. Oberg K, Ferone D, Kaltsas G, Knigge UP, Taal B, Plockinger U, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: biotherapy. Neuroendocrinology (2009) 90:194–202. doi: 10.1159/000225948
59. Oberg K, Knigge U, Kwekkeboom D, Perren A. Neuroendocrine gastro-entero-pancreatic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2012) 23 (Suppl. 7):vii124–30. doi: 10.1093/annonc/mds295
60. Ferolla P, Falchetti A, Filosso P, Tomassetti P, Tamburrano G, Avenia N, et al. Thymic neuroendocrine carcinoma (carcinoid) in multiple endocrine neoplasia type 1 syndrome: the Italian series. J ClinEndocrinol Metab. (2005) 90:2603–9. doi: 10.1210/jc.2004-1155
61. Goudet P, Murat A, Cardot-Bauters C, Emy P, Baudin E, du Boullay CH, et al. Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1: a comparative study on 21 cases among a series of 761 MEN1 from the GTE (Groupe des Tumeurs Endocrines). World J Surg. (2009) 33:1197–1207. doi: 10.1007/s00268-009-9980-y
62. de Laat JM, Pieterman CR, van den Broek MF, Twisk JW, Hermus AR, Dekkers OM, et al. Natural course and survival of neuroendocrine tumors of thymus and lung in MEN1 patients. J Clin Endocrinol Metab. (2014) 99:3325–33. doi: 10.1210/jc.2014-1560
63. Sakurai A, Imai T, Kikumori T, Horiuchi K, Okamoto T, Uchino S, et al. Thymic neuroendocrine tumour in multiple endocrine neoplasia type 1: female patients are not rare exceptions. Clin Endocrinol. (2013) 78:248–54. doi: 10.1111/j.1365-2265.2012.04467.x
64. Karges W, Schaaf L, Dralle H, Boehm BO. Clinical and molecular diagnosis of multiple endocrine neoplasia type 1. Langenbeck's Arch Surg (2002) 386:547–52. doi: 10.1007/s00423-001-0268-4
65. Cardinal JW, Bergman L, Hayward N, Sweet A, Warner J, Marks L, et al. A report of a national mutation testing service for the MEN1 gene: clinical presentations and implications for mutation testing. J Med Genet. (2005) 42:69–74. doi: 10.1136/jmg.2003.017319
66. Burgess JR, Parameswaran V, Shepherd JJ. Osteoporosis in multiple endocrine neoplasia type 1. Arch Surg. (1999) 134:1119–23. doi: 10.1001/archsurg.134.10.1119
67. Lourenço DM, Toledo RA, Mackowiak II, Coutinho FL, Cavalcanti MG, Correia-Deur JEM, et al. Multiple endocrine neoplasia type 1 in Brazil: MEN1 founding mutation, clinical features, and bone mineral density profile. Eur J Endocrinol. (2008) 159:259–74. doi: 10.1530/EJE-08-0153
68. Eller-Vainicher C, Chiodini I, Battista C, Viti R, Mascia ML, Massironi S, et al. Sporadic and MEN1-related primary hyperparathyroidism: differences in clinical expression and severity. J Bone Miner Res. (2009) 24:1404–10. doi: 10.1359/jbmr.090304
69. Lourenço DM, Coutinho FL, Toledo RA, Montenegro FL, Correia-Deur JE, Toledo SP. Early-onset, progressive, frequent, extensive, and severe bone mineral and renal complications in multiple endocrine neoplasia type 1-associated primary hyperparathyroidism. J Bone Miner Res. (2010) 25:2382–91. doi: 10.1002/jbmr.125
Keywords: MEN1, diagnosis, genetic testing, epidemiology, delayed diagnosis
Citation: de Laat JM, van Leeuwaarde RS and Valk GD (2018) The Importance of an Early and Accurate MEN1 Diagnosis. Front. Endocrinol. 9:533. doi: 10.3389/fendo.2018.00533
Received: 18 April 2018; Accepted: 22 August 2018;
Published: 11 September 2018.
Edited by:
Delmar Muniz Lourenco Jr., Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, BrazilReviewed by:
Piero Ferolla, Azienda Ospedaliera Universitaria di Perugia Santa Maria della Misericordia, ItalyLuiz Eduardo Armondi Wildemberg, Instituto Estadual do Cérebro Paulo Niemeyer, Brazil
Copyright © 2018 de Laat, van Leeuwaarde and Valk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerlof D. Valk, Zy5kLnZhbGtAdW1jdXRyZWNodC5ubA==