 Annunziata G. Cicatiello
Annunziata G. Cicatiello Daniela Di Girolamo
Daniela Di Girolamo Monica Dentice
Monica Dentice- Department of Clinical Medicine and Surgery, University of Naples Federico II, Naples, Italy
Thyroid hormones (THs) are key determinants of cellular metabolism and regulate a variety of pathways that are involved in the metabolism of carbohydrates, lipids and proteins in several target tissues. Notably, hyperthyroidism induces a hyper-metabolic state characterized by increased resting energy expenditure, reduced cholesterol levels, increased lipolysis and gluconeogenesis followed by weight loss, whereas hypothyroidism induces a hypo-metabolic state characterized by reduced energy expenditure, increased cholesterol levels, reduced lipolysis and gluconeogenesis followed by weight gain. Thyroid hormone is also a key regulator of mitochondria respiration and biogenesis. Besides mirroring systemic TH concentrations, the intracellular availability of TH is potently regulated in target cells by a mechanism of activation/inactivation catalyzed by three seleno-proteins: type 1 and type 2 iodothyronine deiodinase (D1 and D2) that convert the biologically inactive precursor thyroxine T4 into T3, and type 3 iodothyronine deiodinase (D3) that inactivates TH action. Thus, the pleiotropic effects of TH can fluctuate among tissues and strictly depend on the cell-autonomous action of the deiodinases. Here we review the mechanisms of TH action that mediate metabolic regulation. This review traces the critical impact of peripheral regulation of TH by the deiodinases on the pathways that regulate energy metabolism and the balance among energy intake, expenditure and storage in specific target tissues.
Introduction
Thyroid hormones (TH) have long been known to regulate energy metabolism (1). Patients with TH dysfunction often have symptoms of metabolic dysregulation, including fatigue and weight changes (2). Indeed, pathological excess of THs in humans raises the basal metabolic rate (BMR) while TH deficiency is accompanied by a decreased BMR (2). TSH and TRH levels are also critical determinants of whole body energy metabolism. In fact, they exert thyroidal and non-thyroidal effects and thus integrate signals from nutritional status and the adrenergic nervous system with a fine regulation of THs production (3). The wide spectrum of THs effects on body metabolism is exerted mainly by stimulating catabolic and anabolic reactions and by regulating turnover of fats, carbohydrates and proteins (1). A peculiar feature of TH-dependent metabolic regulation is the acceleration of the rates of anabolic and catabolic reactions (4). For instance, TH increases fat mobilization thereby leading to increased concentrations of fatty acids in plasma as well as to enhanced oxidation of fatty acids. THs stimulate insulin-dependent glucose uptake, and both gluconeogenesis and glycogenolysis. Therefore, the action of THs culminates in promoting futile cycles that contribute significantly to the increase oxygen consumption seen in thyrotoxicosis (“hyperthyroidism”). Thyroid hormones also stimulate ion cycling by altering membrane permeability, the expression of ion pumps and the characteristics of these pumps (5–8).
The classic endocrine view of TH biology is that THs are produced and secreted by the thyroid gland for transport to target tissues. Accordingly, TH concentrations determine the extent of hormonal regulation and generate downstream effects in peripheral cells. Classical regulation of the thyroid gland involves the hypothalamic–pituitary–thyroid axis, whereas low TH concentrations trigger a negative feedback that results in the release of both thyroid releasing hormone (TRH) from the hypothalamus and thyroid stimulating hormone (TSH) from the pituitary gland (9, 10). However, besides the capacity of the thyroid gland to produce the correct amount of THs, the periphery can modify the TH signal in time and space. Indeed, while plasma concentrations of TH are relatively stable, tissues can coordinate TH levels through the cell-autonomous regulation of TH transporters, deiodinases and TH receptors (11). The iodothyronine deiodinase family of selenoproteins is constituted by three enzymes, D1, D2, and D3. These enzymes are present in specific tissues, and regulate TH activation and inactivation (12). The differential expression of deiodinases enables close control of T3 and its prohormone, T4, by removing iodine moieties (“deiodination”) at different sites of the phenolic or tyrosylic ring of the TH hormones (13). T4 has a long half-life and is converted to the active form, T3, within cells by the activating deiodinases (D1 and D2) that catalyze outer ring deiodination. The third deiodinase, D3, terminates TH action by inactivating T3 and T4 by removing the iodine at the inner ring (13). The local regulation of TH at intracellular level enables wide fluctuations of TH in local tissues and is a powerful tool with which to modulate TH action without perturbing systemic TH levels.
The correlation of a Thr92Ala polymorphism in the DIO2 gene, encoding protein D2, with altered glycemic control, obesity and type 2 diabetes mellitus (T2DM) (14–16), as well as the association of genetic variants of the DIO1 gene, encoding protein D1, with insulin resistance (17), reinforces the clinical relevance of the peripheral T4-to-T3 conversion in metabolic control.
In this review we summarize the role of the local control of TH by the deiodinases in the metabolic program of cells in the context of the tissue-specific impact of deiodination on energy metabolism, and discuss the effect of local alteration of TH on the metabolic functions of the body.
Metabolic Role of TH and the Deiodinases
Although each cell of the body is virtually a TH target, the TH signal is differentially integrated in each tissue depending on the cell-autonomous machinery. Therefore, the action of TH on whole body metabolism is best evaluated by examining the specific contribution of TH and its modulating enzymes to energy metabolism in the context of each target tissue. The relative roles of most components of the TH signaling pathways have been assessed in mouse models of inducible, tissue-specific activation or inactivation of deiodinases, receptors and transporters (1). These studies revealed how different TH-induced processes contribute to regulating metabolic homeostasis in humans (Figure 1).
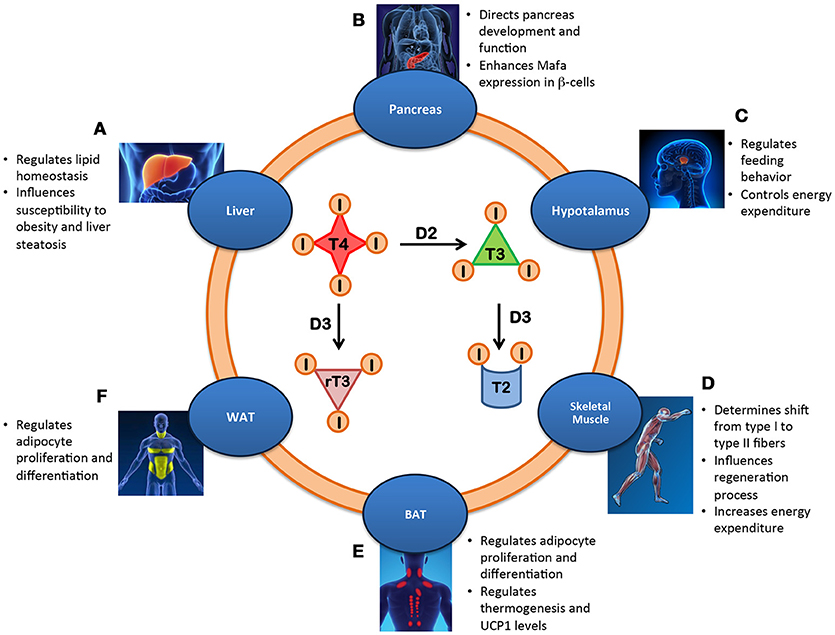
Figure 1. Metabolic effects of the intracellular regulation of thyroid hormone in different tissues. (A) Liver: Lipid homeostasis is regulated by local T3 level thereby influencing susceptibility to obesity and liver steatosis. (B) Pancreas: The balance between the deiodinases controls the development and function of β-cells by enhancing Mafa transcription factor and inducing insulin secretion. (C) Hypothalamus: Local availability of TH regulates feeding behavior and controls energy expenditure. (D) Skeletal Muscle: Increased T3 levels in skeletal muscle promote the shift from type I to type II fibers, influence regeneration and increase energy expenditure. (E) BAT: D2-mediated TH activation regulates UCP1 expression and thermogenesis, adipocyte proliferation and differentiation and body weight. (F) WAT: Local T3 metabolism regulates adipocyte proliferation/differentiation.
Liver
There is an intricate relationship between TH metabolism and liver (1, 18, 19). Thyroid hormones regulate hepatic function by modulating the basal metabolic rate of hepatocytes; the liver in turn metabolizes the THs and regulates their systemic endocrine effects (20). In the liver, TH regulates lipid metabolism mainly through the T3-TRβ (TH receptor beta) (1), and the downstream regulation of cholesterol homeostasis (synthesis and efflux), bile acid synthesis and fatty acid metabolism (1). The local control of TH metabolism in liver is mediated by the expression of all three deiodinases. D1 is highly expressed in liver, where it contributes to plasmatic T3 homeostasis and mediates the clearance of rT3 from the circulation (21). D1 expression is highly sensitive to T3 levels to such an extent that it is an indicator of the thyroid state of the liver (22). Despite the high D1 levels in the liver, the intracellular T3 level in hepatocytes is not mediated by D1, but by the other TH-activating deiodinase, D2 (23). The liver is the paradigm of spatio-temporal regulated expression of D2 that is transiently turned on in the neonatal mouse liver between the first and the fifth post-natal day. During this short time, a peak of hepatic D2 expression occurs and rapidly declines to background levels (24). This brief peak of D2 produces an excess of T3 that modifies the methylation and the expression pattern of thousands of hepatic genes thereby increasing future susceptibility to diet-induced obesity and liver steatosis (24). Mouse models of hepatocyte-specific D2 inactivation (Alb-D2KO) do not undergo this physiological increase of liver T3 at birth, with a consequent delay in neonatal expression of lipid-related genes and a phenotype of resistance to obesity and liver steatosis (24). These fundamental changes during perinatal life indicate that the thyroid state of specific tissue impacts on whole body metabolism thereby affecting the phenotype in adult life (25). Finally, D3 is almost undetectable in the liver of healthy individuals, but robust re-activation of D3 has been found in regenerating liver tissue, in certain hepatic tumors and in sera and liver samples from critically ill humans, thus influencing the systemic thyroid status (26). These results suggest that D3 plays a role in the tissue response to injury and in the imbalance of TH homeostasis commonly observed during critical illness.
Pancreas
Thyroid hormone plays a critical role in the development, maturation, and function of pancreatic cells, where T3 is required for the physiological maturation of pancreatic β-cells to glucose-stimulated insulin-secreting cells (27). Pancreatic cells express both TRα and TRβ isoforms and the activated T3-TR complex directly bind to the promoter of islet transcription factor Mafa thereby resulting in its activation (27, 28). However, the exact physiological role of TH in glucose homeostasis remains controversial (29, 30). Although numerous in vitro and ex vivo studies have demonstrated that T3 mediates positive effects on β-cell function, exposure to high doses of TH results in a phenotype of glucose intolerance. Indeed, hyperthyroidism is associated with glucose intolerance consequent to decreased insulin secretion (31, 32) and to stimulation of hepatic gluconeogenesis (33). Probably, in hyperthyroid conditions, impaired insulin secretion is not sufficient to suppress high hepatic glucose production. Accordingly, the prevalence of diabetes mellitus in hyperthyroid patients is approximately double that of non-affected subjects (34). In contrast, systemic hypothyroidism is associated with reduced hepatic gluconeogenesis and enhanced insulin sensitivity, as demonstrated by the onset of a hypoglycemic state after an insulin injection (35). While during vertebrate development, reduced TH levels are important for normal function and for glucose homeostasis of pancreatic β-cells, exposure to high TH doses induces apoptosis of pancreatic β-cells (36). In this context, the TH hormone-inactivating deiodinase D3 plays a fundamental role in lineage fate decisions and endocrine cell specification (34). Indeed, studies in D3KO mice demonstrated that the reduction D3-mediated of TH action is critical for normal maturation and function of pancreatic β-cells (34). D3KO mice exhibited a glucose intolerant phenotype due to impaired glucose-stimulated insulin secretion, reduced size, and absolute mass of pancreatic islet and β-cells, decreased insulin content, and reduced expression of key genes involved in glucose sensing, insulin synthesis, and exocytosis (34). The pancreatic phenotype of the D3KO mice is proof that attenuation of TH-signaling via D3 activation is essential for normal development.
Hypothalamus
Peripheral TH signals are integrated within the hypothalamus and processed into coordinated responses to regulate energy balance. The center for regulation of food intake and of body weight is the melanocortin system, constituted by three neuronal populations: the pro-opiomelanocortin (POMC)-expressing neurons, the neuropeptide Y (NPY) and agouti-related peptide (AgRP)-co-expressing neurons and the melanocortin 4 receptor (MC4R)-expressing neurons (37, 38). The POMC neurons exert an anorexigenic function by activating MC4R neurons, which induce a reduction of food intake and increased energy expenditure. On contrary, NPY/AgRP neurons are the orexigenic neurons: by antagonizing the action exerted by POMC on MC4R, they increase food intake and decrease energy expenditure. All these neurons are sensitive to the TH signal that can either activate or inhibit melanocortin neurons, and thus, it is not surprising that local TH metabolism plays a critical role in appetite and feeding regulation. Changes in central T3 levels occur in various metabolic conditions (39), for example elevated T3 levels have been found in the hypothalamus during fasting (40). Fasting induces alterations in the thyroid state, namely, a reduction in pituitary D2 levels and liver D1 levels correlated with low peripheral T3 levels in the presence of increased hypothalamic D2 activity. The high D2 activity in the hypothalamus causes an increase of local T3 concentrations, which in turn activate orexigenic NPY/AgRP neurons and inhibit anorexigenic POMC neurons, thereby inducing hyperfagia (1). The molecular mechanism underlying TH-mediated NPY/AgRP activation resembles that in brown adipose tissue (BAT) in which T3 increases uncoupling protein 1 (UCP1) activity. In fact, high T3 levels in the hypothalamus during fasting, consequent to D2 activation, promote UCP2 expression and stimulate mitochondrial proliferation in orexigenic NPY/AgRP neurons, so promoting their activity and stimulating rebound feeding upon food deprivation. The increase of T3 in the hypothalamus also causes TRH mRNA suppression (40, 41). Therefore, under food deprivation, despite a reduction in peripheral TH levels, there is a localized increase in T3 within the hypothalamus, which in turn increases orexigenic signals and decreases TRH production. The hypothalamus probably maintains low TH levels to preserve energy stores, which would be dissipated in hyperthyroid condition.
The fundamental role of deiodinases in the regulation of energy balance in brain has been demonstrated in mouse models of deiodinases depletion (42). Despite the low TH circulating levels in adult Dio3−/− mice, their central nervous system is in a hyperthyroid state (42). The enhanced TH levels alter the functioning of the hypothalamic circuitries, including the leptin-melanocortin system, thereby regulating energy balance and adiposity. In detail, Dio3−/− mice have decreased adiposity, but an abnormally functioning leptin-melanocortin system associated with leptin resistance (43). The hypothalamic D2-mediated T4 to T3 conversion is important for the photoperiodic response of the gonads (44) in which fine-tuned D2 and D3 expression tightly regulates LH stimulation (45).
Skeletal Muscle
Skeletal muscle represents 40–50% of the total body mass in humans and is crucial for metabolism, heat generation and maintenance of posture. TH influences skeletal muscle contraction, regeneration and metabolism (46). All components of the TH signaling process, from TR to TH transporters (MCT8 and MCT10), and D2 and D3, are expressed in the skeletal muscle of rodents and humans (47). During skeletal muscle development, D2 is up-regulated, particularly during the first postnatal days, and decreases at day 30, although its activity returns to high levels during differentiation of muscle stem cells (12, 48, 49). In particular, during post-injury regeneration processes, D2 mRNA is up-regulated to enable correct myoblast differentiation (50). D2 is a target of FOXO3, which is a protein involved in myocyte fusion and metabolism as well as in atrophy and autophagy (12). Loss of D2 impairs stem cell differentiation and prevents up-regulation of myogenic transcription factor MyoD thereby increasing the proliferative potential of muscle stem cells. D2-mediated TH in skeletal muscle influences also muscle fibers. High TH levels induce a shift from type I fibers (slow) to type II fibers (fast), which results in up-regulation of sarcoendoplasmic reticulum Ca2+ATPase, of glucose transporter 4 (GLUT4) and of uncoupling protein 3 (UCP3) thereby producing heat and increasing energy expenditure (51). D2-dependent T3 activation influences insulin response in skeletal muscle (52). Indeed, D2KO mice are insulin-resistant, which demonstrates the relevance of D2 in glucose homeostasis. In humans, a common polymorphism of the Dio2 gene, the Thr92Ala substitution in protein D2, which partially impairs enzymatic activity, has been correlated with insulin resistance and diabetes (53, 54). Furthermore, muscle fibers respond to cold through TH-related mechanisms, namely increased glucose uptake, activation of oxidative pathways and increased mitochondria biogenesis (55, 56). Interestingly, D2 is up-regulated in muscle after 4 h of cold exposure (57). Moreover, D2 is up-regulated in response to such metabolic signals as bile acids and insulin (1, 58) and during exercise under β-adrenergic stimulus in order to amplify TH signaling and regulate PGC-1α expression (59, 60). Coordinated D2-D3 expression is required to fine-tune intracellular TH availability during muscle stem cell differentiation, and in vivo, during muscle regeneration (47). While D2 is essential for a correct T3 surge and the subsequent differentiation of muscle stem cells, D3 fosters muscle stem cell proliferation by lowering TH availability during the early phases of the myogenic program (47). This dual regulation is so critical that D3-depletion in vivo causes massive apoptosis of proliferating satellite cells and drastically impairs a full regeneration process. These studies highlight the pivotal role of the intracellular TH coordination by the deiodinases in muscle physiology.
Brown Adipose Tissue
Brown adipose tissue is characterized by multilocular lipid droplets and numerous mitochondria, and governs heat production (61). In fact, BAT is activated in response to a high fat diet or cold exposure in order to protect the organism from weight gain and hypothermia. Thyroid hormone critically influences BAT activity (62). The most obvious metabolic role of D2 is the regulation of energy expenditure in the BAT of small mammals, including human newborns. During cold exposure, the sympathetic nervous system induces D2 expression in brown adipocytes, thereby promoting local T4-to-T3 conversion, and activation of the transcription of target genes involved in the thermogenic program (63). Loss of function of D2 reduces the level of UCP-1, which is normally up-regulated at RNA level by TH. D2 is thus considered a marker of BAT activity (1, 57). Interestingly, global D2KO mice are resistant to diet-induced obesity, highly tolerant to glucose, and have a deficit in respiratory quotient at 22°C, while at 30°C they become more susceptible to obesity and develop intolerance to glucose (64, 65). T3 regulates the expression of several genes during adipogenic differentiation, among which GPD, ME, PEPCK, S14, FAS, and GLUT4 (66, 67). While D2 activity is important during differentiation, D3 is considered a mitogenic marker in brown pre-adipocytes. In fact, D3 mRNA and activity are induced by bFGF and aFGF in proliferating brown pre-adipocytes (68). In BAT, T3 also accelerates fatty acid oxidation and lipogenesis through the action of the ACC and ME lipogenic enzymes. Consequently, D2KO mice have reduced fatty acid oxidation and lipogenesis (4).
White Adipose Tissue
The primary function of white adipose tissue (WAT) is to store energy in the form of single large lipid droplets, although it also secretes the leptin and adiponectin adipokines. White adipocytes differ anatomically and physiologically from brown adipocytes. However, the latter may appear at sites corresponding to WAT, in a so-called process of WAT “browning” caused by a thermogenic stimulus, such as prolonged cold exposure (69), or treatment with β3-adrenergic receptor activators (70). The brown adipocytes in WAT are often called “inducible,” “beige,” or “brite.” D1 and D2 are barely expressed in epidermal WAT, the adipose tissue that, contrary to inguinal WAT, is never converted in BAT. All TR isoforms and the TH transporter MCT8 are expressed in human subcutaneous adipose tissue (71). D1 expression in epidermal WAT is only 1% of the D1 found in the liver. Similarly, the D2 mRNA level is 7% of the D2 in BAT (72). Interestingly, D1 expression and activity are increased in the subcutaneous and visceral WAT of obese subjects (71). On the other hand, D2 is up-regulated in beige/brite adipocytes and its expression is correlated to increased energy expenditure (73). A high-fat diet stimulates D1 and leptin expression, while caloric restriction decreases D1 activity as well as leptin levels, and increases levels of the leptin mediator SCD-1. Leptin overexpression increases D1 activity and down-regulates SCD-1 expression (74). Similar to brown adipocytes, in white adipocytes, D2 plays an important role in lipogenesis and in the regulation of the expression of genes related to adipocyte differentiation, while D3 sustains the proliferation of white adipocytes (61). Interestingly, thyroidectomized mice have an increased level of both D1 and D2 (72). Moreover, D2 is expressed also in human pre-adipocytes although its role is unclear (75).
Future Directions and Conclusions
Monodeiodination is quantitatively the most important pathway of TH activation. Within peripheral tissue, multiple pathways modulate TH availability. These pathways govern the action and regulation of deiodinase expression, the action of TH transporters, and the expression and crosstalk of TH receptors with multiple partners. This intricate network of TH modifiers increases the sensitivity and the speed of responses to changes induced in the internal and external environment by the thyroid signal. The price to be paid for this is an intricate regulation of each component in time and space. Given the vast spectrum of metabolic body functions regulated by the TH signal, the deiodinases represent a powerful tool with which to modulate cellular metabolism in specific tissues without perturbing systemic levels of THs. Consequently, the development of drugs that target deiodinase action is the next challenge in this field. Extensive work is still required to delineate the kinetics and regulation of the deiodinase enzymes in specific tissues to understand the full spectrum of their biological roles. Thus, pharmacological research is poised to develop deiodinase modulators aimed at driving specific metabolic outcomes. Targeting tissue-specific TH actions may result in novel and safe therapeutic options for metabolic dysfunctions.
Author Contributions
AC and DD wrote the manuscript. MD wrote and supervised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the European Research Council under the European Union's Horizon 2020 Programme–ERCStG2014 (STARS−639548) to MD. and from AIRC to MD. (IG 20766). We thank Jean Ann Gilder (Scientific Communication srl., Naples, Italy) for writing assistance.
References
1. Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. (2014) 94:355–82. doi: 10.1152/physrev.00030.2013
2. Iwen KA, Schroder E, Brabant G. Thyroid hormones and the metabolic syndrome. Eur Thyroid J. (2013) 2:83–92. doi: 10.1159/000351249
3. Lopez M, Alvarez CV, Nogueiras R, Dieguez C. Energy balance regulation by thyroid hormones at central level. Trends Mol Med. (2013) 19:418–27. doi: 10.1016/j.molmed.2013.04.004
4. Oppenheimer JH, Schwartz HL, Lane JT, Thompson MP. Functional relationship of thyroid hormone-induced lipogenesis, lipolysis, and thermogenesis in the rat. J Clin Invest. (1991) 87:125–32. doi: 10.1172/JCI114961
5. Freake HC, Schwartz HL, Oppenheimer JH. The regulation of lipogenesis by thyroid hormone and its contribution to thermogenesis. Endocrinology (1989) 125:2868–74. doi: 10.1210/endo-125-6-2868
6. Haber RS, Loeb JN. Stimulation of potassium efflux in rat liver by a low dose of thyroid hormone: evidence for enhanced cation permeability in the absence of Na,K-ATPase induction. Endocrinology (1986) 118:207–11. doi: 10.1210/endo-118-1-207
7. Silva JE. Thermogenic mechanisms and their hormonal regulation. Physiol Rev. (2006) 86:435–64. doi: 10.1152/physrev.00009.2005
8. Clausen T, Van Hardeveld C, Everts ME. Significance of cation transport in control of energy metabolism and thermogenesis. Physiol Rev. (1991) 71:733–74. doi: 10.1152/physrev.1991.71.3.733
9. Fekete C, Lechan RM. Central regulation of hypothalamic-pituitary-thyroid axis under physiological and pathophysiological conditions. Endocr Rev. (2014) 35:159–94. doi: 10.1210/er.2013-1087
10. Ortiga-Carvalho TM, Chiamolera MI, Pazos-Moura CC, Wondisford FE. Hypothalamus-pituitary-thyroid axis. Compr Physiol. (2016) 6:1387–428. doi: 10.1002/cphy.c150027
11. Gereben B, Zeold A, Dentice M, Salvatore D, Bianco AC. Activation and inactivation of thyroid hormone by deiodinases: local action with general consequences. Cell Mol Life Sci. (2008) 65:570–90. doi: 10.1007/s00018-007-7396-0
12. Dentice M, Salvatore D. Deiodinases: the balance of thyroid hormone: local impact of thyroid hormone inactivation. J Endocrinol. (2011) 209:273–82. doi: 10.1530/JOE-11-0002
13. Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. (2008) 29:898–938. doi: 10.1210/er.2008-0019
14. Zhang X, Sun J, Han W, Jiang Y, Peng S, Shan Z, et al. The Type 2 Deiodinase Thr92Ala polymorphism is associated with worse glycemic control in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. J Diabetes Res. (2016) 2016:5928726. doi: 10.1155/2016/5928726
15. Canani LH, Capp C, Dora JM, Meyer EL, Wagner MS, Harney JW, et al. The type 2 deiodinase A/G (Thr92Ala) polymorphism is associated with decreased enzyme velocity and increased insulin resistance in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. (2005) 90:3472–8. doi: 10.1210/jc.2004-1977
16. Nair S, Muller YL, Ortega E, Kobes S, Bogardus C, Baier LJ. Association analyses of variants in the DIO2 gene with early-onset type 2 diabetes mellitus in Pima Indians. Thyroid (2012) 22:80–7. doi: 10.1089/thy.2010.0455
17. Bos MM, Smit RAJ, Trompet S, van Heemst D, Noordam R. Thyroid signaling, insulin resistance, and 2 diabetes mellitus: a mendelian randomization study. J Clin Endocrinol Metab. (2017) 102:1960–70. doi: 10.1210/jc.2016-2816
18. Singh BK, Sinha RA, Zhou J, Tripathi M, Ohba K, Wang ME, et al. Hepatic FOXO1 target genes are co-regulated by thyroid hormone via RICTOR protein deacetylation and MTORC2-AKT protein inhibition. J Biol Chem. (2016) 291:198–214. doi: 10.1074/jbc.M115.668673
19. Singh BK, Sinha RA, Zhou J, Xie SY, You SH, Gauthier K, et al. FoxO1 deacetylation regulates thyroid hormone-induced transcription of key hepatic gluconeogenic genes. J Biol Chem. (2013) 288:30365–72. doi: 10.1074/jbc.M113.504845
20. Malik R, Hodgson H. The relationship between the thyroid gland and the liver. QJM (2002) 95:559–69. doi: 10.1093/qjmed/95.9.559
21. Schneider MJ, Fiering SN, Thai B, Wu SY, St Germain E, Parlow AF, et al. Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology (2006) 147:580–9. doi: 10.1210/en.2005-0739
22. Jakobs TC, Schmutzler C, Meissner J, Kohrle J. The promoter of the human type I 5′-deiodinase gene–mapping of the transcription start site and identification of a DR+4 thyroid-hormone-responsive element. Eur J Biochem. (1997) 247:288–97.
23. Christoffolete MA, Doleschall M, Egri P, Liposits Z, Zavacki AM, Bianco AC, et al. Regulation of thyroid hormone activation via the liver X-receptor/retinoid X-receptor pathway. J Endocrinol. (2010) 205:179–86. doi: 10.1677/JOE-09-0448
24. Fonseca TL, Fernandes GW, McAninch EA, Bocco BM, Abdalla SM, Ribeiro MO, et al. Perinatal deiodinase 2 expression in hepatocytes defines epigenetic susceptibility to liver steatosis and obesity. Proc Natl Acad Sci USA. (2015) 112:14018–23. doi: 10.1073/pnas.1508943112
25. Fernandes GW, Bocco B, Fonseca TL, McAninch EA, Jo S, Lartey LJ, et al. The foxo1-inducible transcriptional repressor Zfp125 causes hepatic steatosis and hypercholesterolemia. Cell Rep. (2018) 22:523–34. doi: 10.1016/j.celrep.2017.12.053
26. Rodriguez-Perez A, Palos-Paz F, Kaptein E, Visser TJ, Dominguez-Gerpe L, Alvarez-Escudero J, et al. Identification of molecular mechanisms related to nonthyroidal illness syndrome in skeletal muscle and adipose tissue from patients with septic shock. Clin Endocrinol. (Oxf) (2008) 68:821–7. doi: 10.1111/j.1365-2265.2007.03102.x
27. Aguayo-Mazzucato C, Zavacki AM, Marinelarena A, Hollister-Lock J, El Khattabi I, Marsili A, et al. Thyroid hormone promotes postnatal rat pancreatic beta-cell development and glucose-responsive insulin secretion through MAFA. Diabetes (2013) 62:1569–80. doi: 10.2337/db12-0849
28. Furuya F, Shimura H, Yamashita S, Endo T, Kobayashi T. Liganded thyroid hormone receptor-alpha enhances proliferation of pancreatic beta-cells. J Biol Chem. (2010) 285:24477–86. doi: 10.1074/jbc.M109.100222
29. Lenzen S, Bailey CJ. Thyroid hormones, gonadal and adrenocortical steroids and the function of the islets of Langerhans. Endocr Rev. (1984) 5:411–34. doi: 10.1210/edrv-5-3-411
30. Chidakel A, Mentuccia D, Celi FS. Peripheral metabolism of thyroid hormone and glucose homeostasis. Thyroid (2005) 15:899–903. doi: 10.1089/thy.2005.15.899
31. Lenzen S, Panten U, Hasselblatt A. Thyroxine treatment and insulin secretion in the rat. Diabetologia (1975) 11:49–55.
32. Ximenes HM, Lortz S, Jorns A, Lenzen S. Triiodothyronine (T3)-mediated toxicity and induction of apoptosis in insulin-producing INS-1 cells. Life Sci. (2007) 80:2045–50. doi: 10.1016/j.lfs.2007.03.001
33. Klieverik LP, Janssen SF, van Riel A, Foppen E, Bisschop PH, Serlie MJ, et al. Thyroid hormone modulates glucose production via a sympathetic pathway from the hypothalamic paraventricular nucleus to the liver. Proc Natl Acad Sci USA. (2009) 106:5966–71. doi: 10.1073/pnas.0805355106
34. Medina MC, Molina J, Gadea Y, Fachado A, Murillo M, Simovic G, et al. The thyroid hormone-inactivating type III deiodinase is expressed in mouse and human beta-cells and its targeted inactivation impairs insulin secretion. Endocrinology (2011) 152:3717–27. doi: 10.1210/en.2011-1210
35. Taguchi Y, Tasaki Y, Terakado K, Kobayashi K, Machida T, Kobayashi T. Impaired insulin secretion from the pancreatic islets of hypothyroidal growth-retarded mice. J Endocrinol. (2010) 206:195–204. doi: 10.1677/JOE-09-0465
36. Jorns A, Tiedge M, Lenzen S. Thyroxine induces pancreatic beta cell apoptosis in rats. Diabetologia (2002) 45:851–5. doi: 10.1007/s00125-002-0842-5
37. Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron (1999) 24:155–63.
38. Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron (1999) 22:221–32.
39. van Haasteren GA, Linkels E, Klootwijk W, van Toor H, Rondeel JM, Themmen AP, et al. Starvation-induced changes in the hypothalamic content of prothyrotrophin-releasing hormone (proTRH) mRNA and the hypothalamic release of proTRH-derived peptides: role of the adrenal gland. J Endocrinol. (1995) 145:143–53.
40. Coppola A, Hughes J, Esposito E, Schiavo L, Meli R, Diano S. Suppression of hypothalamic deiodinase type II activity blunts TRH mRNA decline during fasting. FEBS Lett. (2005) 579:4654–8. doi: 10.1016/j.febslet.2005.07.035
41. Vella KR, Ramadoss P, Lam FS, Harris JC, Ye FD, Same PD, et al. NPY and MC4R signaling regulate thyroid hormone levels during fasting through both central and peripheral pathways. Cell Metab. (2011) 14:780–90. doi: 10.1016/j.cmet.2011.10.009
42. Hernandez A, Quignodon L, Martinez ME, Flamant F, St Germain DL. Type 3 deiodinase deficiency causes spatial and temporal alterations in brain T3 signaling that are dissociated from serum thyroid hormone levels. Endocrinology (2010) 151:5550–8. doi: 10.1210/en.2010-0450
43. Wu Z, Martinez ME, St Germain DL, Hernandez A. Type 3 deiodinase role on central thyroid hormone action affects the leptin-melanocortin system and circadian activity. Endocrinology (2017) 158:419–30. doi: 10.1210/en.2016-1680
44. Dunn IC, Wilson PW, Shi Y, Burt DW, Loudon ASI, Sharp PJ. Diurnal and photoperiodic changes in thyrotrophin-stimulating hormone beta expression and associated regulation of deiodinase enzymes (DIO2, DIO3) in the female juvenile chicken hypothalamus. J Neuroendocrinol. (2017) 29. doi: 10.1111/jne.12554
45. Nakao N, Ono H, Yamamura T, Anraku T, Takagi T, Higashi K, et al. Thyrotrophin in the pars tuberalis triggers photoperiodic response. Nature (2008) 452:317–22. doi: 10.1038/nature06738
46. Salvatore D, Simonides WS, Dentice M, Zavacki AM, Larsen PR. Thyroid hormones and skeletal muscle–new insights and potential implications. Nat Rev Endocrinol. (2014) 10:206–14. doi: 10.1038/nrendo.2013.238
47. Dentice M, Ambrosio R, Damiano V, Sibilio A, Luongo C, Guardiola O, et al. Intracellular inactivation of thyroid hormone is a survival mechanism for muscle stem cell proliferation and lineage progression. Cell Metab. (2014) 20:1038–48. doi: 10.1016/j.cmet.2014.10.009
48. Dentice M, Marsili A, Zavacki A, Larsen PR, Salvatore D. The deiodinases and the control of intracellular thyroid hormone signaling during cellular differentiation. Biochim Biophys Acta (2013) 1830:3937–45. doi: 10.1016/j.bbagen.2012.05.007
49. Ambrosio R, Damiano V, Sibilio A, De Stefano MA, Avvedimento VE, Salvatore D, et al. Epigenetic control of type 2 and 3 deiodinases in myogenesis: role of Lysine-specific Demethylase enzyme and FoxO3. Nucleic Acids Res. (2013) 41:3551–62. doi: 10.1093/nar/gkt065
50. Dentice M, Marsili A, Ambrosio R, Guardiola O, Sibilio A, Paik JH, et al. The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J Clin Invest. (2010) 120:4021–30. doi: 10.1172/JCI43670
51. Simonides WS, van Hardeveld C. Thyroid hormone as a determinant of metabolic and contractile phenotype of skeletal muscle. Thyroid (2008) 18:205–16. doi: 10.1089/thy.2007.0256
52. Bloise FF, Cordeiro A, Ortiga-Carvalho TM. Role of thyroid hormone in skeletal muscle physiology. J Endocrinol. (2018) 236:R57–68. doi: 10.1530/JOE-16-0611
53. Mentuccia D, Proietti-Pannunzi L, Tanner K, Bacci V, Pollin TI, Poehlman ET, et al. Association between a novel variant of the human type 2 deiodinase gene Thr92Ala and insulin resistance: evidence of interaction with the Trp64Arg variant of the beta-3-adrenergic receptor. Diabetes (2002) 51:880–3. doi: 10.2337/diabetes.51.3.880
54. Castagna MG, Dentice M, Cantara S, Ambrosio R, Maino F, Porcelli T, et al. DIO2 Thr92Ala reduces deiodinase-2 activity and serum-T3 levels in thyroid-deficient patients. J Clin Endocrinol Metab. (2017) 102:1623–30. doi: 10.1210/jc.2016-2587
55. Louzada RA, Santos MC, Cavalcanti-de-Albuquerque JP, Rangel IF, Ferreira AC, Galina A, et al. Type 2 iodothyronine deiodinase is upregulated in rat slow- and fast-twitch skeletal muscle during cold exposure. Am J Physiol Endocrinol Metab. (2014) 307:E1020–9. doi: 10.1152/ajpendo.00637.2013
56. McAninch EA, Bianco AC. Thyroid hormone signaling in energy homeostasis and energy metabolism. Ann N Y Acad Sci. (2014) 1311:77–87. doi: 10.1111/nyas.12374
57. Ramadan W, Marsili A, Larsen PR, Zavacki AM, Silva JE. Type-2 iodothyronine 5'deiodinase (D2) in skeletal muscle of C57Bl/6 mice. II. Evidence for a role of D2 in the hypermetabolism of thyroid hormone receptor alpha-deficient mice. Endocrinology (2011) 152:3093–102. doi: 10.1210/en.2011-0139
58. Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature (2006) 439:484–9. doi: 10.1038/nature04330
59. Silva JE, Bianco SD. Thyroid-adrenergic interactions: physiological and clinical implications. Thyroid (2008) 18:157–65. doi: 10.1089/thy.2007.0252
60. Bocco BM, Louzada RA, Silvestre DH, Santos MC, Anne-Palmer E, Rangel IF, et al. Thyroid hormone activation by type 2 deiodinase mediates exercise-induced peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression in skeletal muscle. J Physiol. (2016) 594:5255–69. doi: 10.1113/JP272440
61. Obregon MJ. Adipose tissues and thyroid hormones. Front Physiol. (2014) 5:479. doi: 10.3389/fphys.2014.00479
62. Shu L, Hoo RL, Wu X, Pan Y, Lee IP, Cheong LY, et al. A-FABP mediates adaptive thermogenesis by promoting intracellular activation of thyroid hormones in brown adipocytes. Nat Commun. (2017) 8:14147. doi: 10.1038/ncomms14147
63. de Jesus LA, Carvalho SD, Ribeiro MO, Schneider M, Kim SW, Harney JW, et al. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J Clin Invest. (2001) 108:1379–85. doi: 10.1172/JCI13803
64. Castillo M, Hall JA, Correa-Medina M, Ueta C, Kang HW, Cohen DE, et al. Disruption of thyroid hormone activation in type 2 deiodinase knockout mice causes obesity with glucose intolerance and liver steatosis only at thermoneutrality. Diabetes (2011) 60:1082–9. doi: 10.2337/db10-0758
65. Fonseca TL, Werneck-De-Castro JP, Castillo M, Bocco BM, Fernandes GW, McAninch EA, et al. Tissue-specific inactivation of type 2 deiodinase reveals multilevel control of fatty acid oxidation by thyroid hormone in the mouse. Diabetes (2014) 63:1594–604. doi: 10.2337/db13-1768
66. Kinlaw WB, Church JL, Harmon J, Mariash CN. Direct evidence for a role of the “spot 14” protein in the regulation of lipid synthesis. J Biol Chem. (1995) 270:16615–8.
67. Bianco AC, Carvalho SD, Carvalho CR, Rabelo R, Moriscot AS. Thyroxine 5′-deiodination mediates norepinephrine-induced lipogenesis in dispersed brown adipocytes. Endocrinology (1998) 139:571–8. doi: 10.1210/endo.139.2.5737
68. Garcia B, Obregon MJ. Growth factor regulation of uncoupling protein-1 mRNA expression in brown adipocytes. Am J Physiol Cell Physiol. (2002) 282:C105–112. doi: 10.1152/ajpcell.01396.2000
69. Young P, Arch JR, Ashwell M. Brown adipose tissue in the parametrial fat pad of the mouse. FEBS Lett. (1984) 167:10–14
70. Cousin B, Cinti S, Morroni M, Raimbault S, Ricquier D, Penicaud L, et al. Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. J Cell Sci. (1992) 103:931–42.
71. Ortega FJ, Jilkova ZM, Moreno-Navarrete JM, Pavelka S, Rodriguez-Hermosa JI, Kopeck Ygrave J, et al. Type I iodothyronine 5'-deiodinase mRNA and activity is increased in adipose tissue of obese subjects. Int J Obes (Lond) (2012) 36:320–4. doi: 10.1038/ijo.2011.101
72. Calvo RM, Obregon MJ. Presence and regulation of D1 and D2 deiodinases in rat white adipose tissue. Metabolism (2011) 60:1207–10. doi: 10.1016/j.metabol.2011.01.014
73. Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. (2013) 19:1252–63. doi: 10.1038/nm.3361
74. Macek Jilkova Z, Pavelka S, Flachs P, Hensler M, Kus V, Kopecky J. Modulation of type I iodothyronine 5′-deiodinase activity in white adipose tissue by nutrition: possible involvement of leptin. Physiol Res. (2010) 59:561–9.
Keywords: Thyroid hormone, Deiodinases, energy metabolism, Thyroid hormone receptors, local regulation of thyroid function
Citation: Cicatiello AG, Di Girolamo D and Dentice M (2018) Metabolic Effects of the Intracellular Regulation of Thyroid Hormone: Old Players, New Concepts. Front. Endocrinol. 9:474. doi: 10.3389/fendo.2018.00474
Received: 04 June 2018; Accepted: 01 August 2018;
Published: 11 September 2018.
Edited by:
Pieter de Lange, Università degli Studi della Campania “Luigi Vanvitelli” Caserta, ItalyReviewed by:
Cheng Han, Albert Einstein College of Medicine, United StatesWarner Simonides, VU University Medical Center, Netherlands
Copyright © 2018 Cicatiello, Di Girolamo and Dentice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monica Dentice, bW9uaWNhLmRlbnRpY2VAdW5pbmEuaXQ=