 Jing Ma1,2
Jing Ma1,2 Adrian Vella2*
Adrian Vella2*
- 1Division of Endocrinology and Metabolism, Shanghai Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China
- 2Division of Endocrinology, Diabetes and Metabolism, Mayo Clinic College of Medicine, Rochester, NY, United States
The interaction between the upper gastrointestinal tract and the endocrine system is important in the regulation of metabolism and of weight. The gastrointestinal tract has a heterogeneous cellular content and comprises a variety of cells that elaborate paracrine and endocrine mediators that collectively form the entero-endocrine system. The advent of therapy that utilizes these pathways as well as the association of bariatric surgery with diabetes remission has (re-)kindled interest in the role of the gastrointestinal tract in glucose homeostasis. In this review, we will use the changes wrought by bariatric surgery to provide insights into the various gut–pancreas interactions that maintain weight, regulate satiety, and limit glucose excursions after meal ingestion.
Background
In the United States, the prevalence of obesity is rapidly increasing with 65% of adults and 17% of adolescents and children classified as being overweight or obese (1). Obesity is associated with multiple diseases, such as type 2 diabetes, non-alcoholic steatohepatitis, and osteoarthritis, as well as being associated with an increased frequency of the risk factors for cardiovascular disease (2). Approximately 9% of national health-care costs have been attributed to excess weight (3). Because of the evidence that weight reduction ameliorates or corrects the comorbidities of obesity, the US Preventive Services Task Force has recommended that body mass index (BMI) is routinely assessed and weight management recommended for obese patients (4).
Behavioral intervention with lifestyle and dietary modification usually achieves modest weight loss (4). While generally safe, most regain the weight lost within 5 years. Pharmacotherapy for obesity is considered for patients who have failed efforts at lifestyle modification and who have a BMI ≥ 30 kg/m2 or a BMI ≥ 27kg/m2 in the presence of comorbidities such as diabetes (5). However, there have been significant concerns about the long-term safety of such medications and many of the currently available medications have limited efficacy (6).
Bariatric surgery, sometimes referred to as metabolic surgery, is usually considered for patients who have a BMI ≥ 40 kg/m2 or a BMI ≥ 35 kg/m2 associated with comorbidities such as type 2 diabetes (5). Restrictive surgeries such as adjustable gastric banding (AGB) and sleeve gastrectomy (SG) limit the capacitance of the stomach. Roux-en-Y gastric bypass (RYGB) is the most commonly performed bypass procedure and produces gastric restriction together with selective malabsorption. RYGB involves creation of a gastric pouch by separating the stomach across the fundus. Drainage of this 10–30 ml pouch is achieved by a gastrojejunostomy. The distal end of the jejunum is anastomosed ~150 cm below the gastrojejunostomy effectively bypassing the distal stomach, duodenum, and proximal jejunum. Duodenal switch (DS) is a variation of biliopancreatic diversion and involves a SG with division of the duodenum below the pylorus. The distal ileum is anastomosed to the short stump of the duodenum producing a ~100 cm channel for nutrient absorption. The other end of the duodenum is closed and the remaining small bowel connected onto the enteral limb 75–100 cm from the ileocecal valve (2).
Observational and prospective studies have suggested that bariatric surgery is the most effective intervention for weight loss producing an average weight loss of 30–35% that is maintained in ~60% of patients at 5 years (7). This has led to a dramatic increase in the number of procedures performed annually from 13,365 in 1998 (8) to 216,000 in 2016 according to the data released by American Society for Metabolic and Bariatric Surgery (9). In a meta-analysis of 136 studies of bariatric surgery, which included a total of 22,094 patients, Buchwald et al. reported that within studies examining type 2 diabetes after bariatric surgery, 1,417 of 1,846 (76%) patients experienced complete resolution. When categorized by operative procedure, there were clear differences in efficacy. Diabetes resolved in 98.9% of patients undergoing biliopancreatic diversion or DS. In contrast, the rate was 83.7% for RYGB and 47.9% for AGB (10). A retrospective review of 257 patients who underwent the long-limb modification of RYGB (400–500 cm Roux limb length) at our institution reported resolution of type 2 diabetes in 94% of patients (11). Recent prospective, randomized controlled trials have, however, reported lower remission rates for diabetes with RYGB, although it remains superior to medical therapy (12–14). Setting aside the superiority of one procedure over the other in terms of inducing diabetes remission [which is likely related to residual β-cell function at the time of the procedure (15, 16) as well as the magnitude of weight loss (17)], obvious differences between procedures can be used to explore the role of the gastrointestinal tract in metabolism. RYGB is sometimes complicated by the occurrence of hyperinsulinemic hypoglycemia (18). Its incidence is uncertain although it has been suggested that excessive glucagon-like peptide-1 (GLP-1) secretion after RYGB (19) may be the cause of this phenomenon, but this is unlikely (20). The condition has been the subject of an extensive review recently (21) (Figure 1).
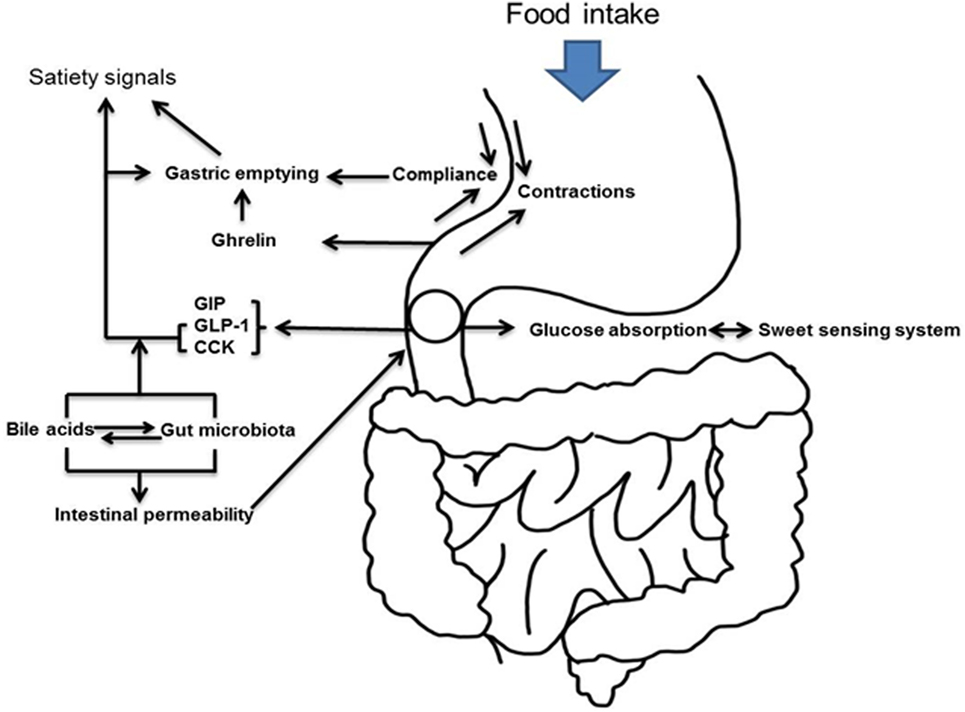
Figure 1. Schematic representation of upper gastrointestinal tract and hyperglycemia.
The anatomic differences among bariatric procedures result in differences in enteroendocrine secretion (Table 1): postprandial GLP-1 concentrations are lower after SG compared to RYGB in the comparative studies undertaken in humans (22–26). On the other hand, a liquid meal, especially after gastric restriction, may not recreate conditions present after a solid meal (27). Indeed, liquid emptying especially after restrictive gastric surgery is dependent on fasting gastric volume (28, 29). SG removes the capacitance function of the stomach and decreases ghrelin concentrations to a greater extent than does RYGB (24, 25, 30). This is more apparent when acyl-ghrelin is measured (22, 31, 32). Whether these differences can explain metabolic outcomes will be explored in detail below (Figure 2).
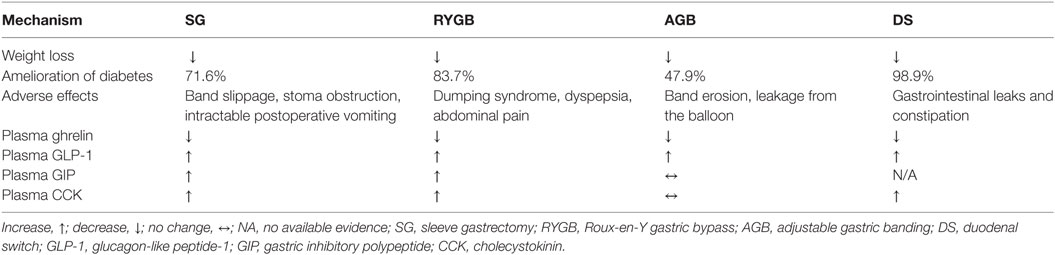
Table 1. Efficacy of different bariatric surgeries.
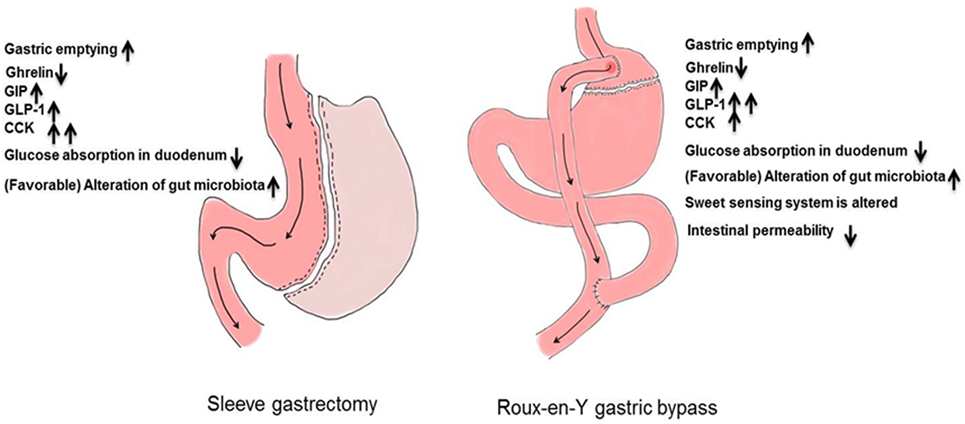
Figure 2. Upper intestinal adaptions after bariatric surgery.
Caloric Restriction—Changes in Gastric Volume, Compliance, and Accommodation
Restrictive procedures reduce gastric volume—indeed, the postoperative period of any form of bariatric surgery is characterized by a significant degree of caloric restriction (33). Fasting blood glucose and insulin resistance improves within 6 days of gastric bypass and occurs before any weight loss (34). This had been observed previously with very low-calorie diets outside of bariatric surgery (35), suggesting that caloric restriction at least partially, explains the acute improvement in glucose control after bariatric surgery (14, 36, 37). Six weeks of caloric restriction (700–900 kcal/day) decreases fasting and postprandial glycemia by lowering fasting endogenous glucose production and improving β-cell function (38). Jackness et al. showed that very low caloric restriction (500 kcal/day) produced a similar improvement in β-cell function as those who underwent post-RYGB (39). Indeed, caloric restriction after RYGB outweighs the effect of GLP-1 on glucose metabolism [as studied by the use of a competitive antagonist of GLP-1 at its receptor (40)]. Of course outside of the mechanical restrictions induced by bariatric surgery, there are difficulties with long-term compliance with regimens of caloric restriction. However, in an open-label, cluster-randomized trial 24% patients achieved weight losses of 15 kg or more in 12 months, and 86% experienced diabetes remission (41).
Multiple factors influence satiation including stomach capacitance and emptying. The stomach increases in volume in anticipation of food ingestion (42). In the normal stomach, this volume expansion is not associated with an increase in gastric wall stiffness (or increased intraluminal pressure) suggesting a change in the mechanical properties of the stomach wall (increased compliance) to accommodate the capacitive function of the stomach (43). This function is primarily fulfilled by the proximal stomach, which serves as a food reservoir, while the distal stomach triturates food to a size that can pass the pylorus. The physical nature, particle size, fat, and caloric content of food alter emptying rate (27). Although nutrient and non-caloric liquids empty rapidly, solids are initially retained in the stomach while antral contractions propel particles toward the closed pylorus. Food particles are emptied once they are ~2 mm in diameter (27). Restrictive procedures eliminate the function of the proximal stomach displacing food to the distal stomach and accelerating emptying. It is uncertain if restriction of accommodation alone alters gastric emptying (44). In SG, a functioning pylorus is retained and gastric volume is usually larger than the pouch created after RYGB. Post-RYGB it has been assumed that there is little neuromuscular control on gastric emptying since the pylorus is bypassed [although this may not be correct (45)]. Surgical vagotomy (which occurs in RYGB when the gastric pouch is created) alters gastric accommodation but may not change emptying (46) and typically does not have durable effects on weight (47).
Gastric emptying plays an important role in determining the magnitude of change in glucose concentrations after nutrient ingestion (48). Indeed, variation in the rate of gastric emptying alters peak insulin response after 75 g oral glucose, in both healthy subjects and patients with type 2 diabetes (49, 50). Fasting gastric volume affects the rate of emptying of a liquid challenge (51). Interventions that delay gastric emptying have the potential to regulate glycemia in patients with diabetes. Accelerating nutrient flow to the small intestine with erythromycin increases the postprandial glycemic response (52), whereas slowing gastric emptying with Xenin-25, a 25-amino acid neurotensin-related peptide, reduces postprandial blood glucose (53). Although it is important to match the rate of gastric emptying and the onset and offset of insulin action, significantly lower insulin requirements are observed in patients with type 1 diabetes with gastroparesis than those without, during the first hour of the postprandial period (54). Delaying gastric emptying is also a mechanism of action of some antidiabetic medications, such as GLP-1 analogs and pramlintide (55, 56).
Gut hormones can modulate food intake over and above that caused by mechanical restriction after bariatric surgery (22). Ghrelin increases food intake after esophagectomy or gastrectomy (57, 58). Neuronal GLP-1R mediates the anorectic effects of GLP-1 (59). Inhibition of GLP-1 action with Exendin-9,39 after RYGB accelerates gastric emptying (45). Taken together, these observations suggest that factors other than anatomy contribute to the upper gastrointestinal response to food ingestion. The attraction of certain foods decreases after RYGB (60) and appetite may be altered by enteroendocrine secretion (61, 62). A potential mechanism is via GLP-1, which alters gastrointestinal transit, gastric accommodation (45, 46, 63), and has direct effects on hypothalamic nuclei outside of the blood–brain barrier (64). GLP-1 and GLP-1 receptor agonists decrease food intake and cause weight loss (65, 66). GLP-1 also modulates taste sensitivity in rodents (67–70). The peripheral concentrations of GLP-1 observed in the early postprandial period in subjects post-RYGB, exceed concentrations observed after infusion at 0.75 pmol/kg/min, and are similar to those observed after infusion at 1.5 pmol/kg/min—both infusion rates that alter gastrointestinal function (71). It is, therefore, reasonable to consider that the postprandial rise in GLP-1 might affect feeding behavior after RYGB, and to a lesser extent SG, where the increase in GLP-1 is less marked (22–26). The elevated postprandial concentrations of GLP-1 observed after RYGB are unlikely to be the cause of diabetes remission after bariatric surgery. We (45) and others (72) have shown that inhibition of GLP-1 actions in the postprandial period has limited effects on glucose concentrations in people after RYGB. This is in agreement with data from mice deficient in the GLP-1 receptor that lost the same amount of weight as wild-type mice (73). This is also the case after SG in humans (74) and in mice deficient in the GLP-1 receptor (75). On the other hand, SG decreases acyl-ghrelin concentrations, presumably due to excision of a large part of the ghrelin-secreting stomach, which should decrease appetite (22, 31, 32). Fasting after SG is not associated with a rise in (low) ghrelin concentrations, in contrast to RYGB (22, 24, 25, 30–32).
In an effort to circumvent the costs and complications associated with bariatric surgery, various attempts have been made to develop endoscopically placed devices that might cause weight loss. One such device is a synthetic sleeve placed post-pylorus under endoscopic control. The rationale underlying such a device is to ensure that nutrients are prevented from coming in contact with the absorptive surfaces of the proximal small bowel (76). Unfortunately, such a device is prone to migration, bleeding, and bolus obstruction. A placebo-controlled study utilizing the device as treatment for type 2 diabetes was terminated prematurely because of a ~3% incidence of hepatic abscess in subjects using the device (77). Other devices such as intra-gastric balloons to induce early satiety are under study.
Ghrelin
Ghrelin is a 28-amino acid peptide and is the only orexigenic hormone recognized in humans. It is secreted from the gastric mucosa and hypothalamus in both rodents and humans. There are two forms of circulating ghrelin, unacylated and acylated ghrelin (AG) (78). In the fasting state, AG is elevated (~110 pM) and decreases (~70 pM) in response to food ingestion. Patients with Prader–Willi syndrome—a syndrome characterized by excessive feeding behavior—have high concentrations of circulating ghrelin (79). Fasting and postprandial acyl-ghrelin levels are decreased following SG, compared to Roux-en-Y gastric bypass (RYGB), which may play a role in weight loss (26). SG involves removal of the gastric fundus—the primary source of ghrelin synthesis and secretion. Exogenous ghrelin administration increases energy intake in both rodents (80) and humans (81). Infusion of ghrelin in patients after esophagectomy (58) or gastrectomy (57) increases caloric intake and appetite. Although the contribution of ghrelin to normal physiology is unclear, it has been demonstrated that ghrelin can directly inhibit insulin secretion (82). Pharmacologic concentrations of ghrelin or ghrelin receptor agonists accelerate gastric emptying, suppress insulin secretion, and increase glucagon secretion (83). In a randomized controlled phase Ib clinical trial, ghrelin accelerated gastric emptying and improved gastrointestinal symptoms in patients with type 2 diabetes (84). Ghrelin receptor agonists are being developed as potential therapies for gastroparesis (85).
Incretin Hormones
The incretin effect is a phenomenon first observed several decades ago when intravenous glucose produced lower insulin concentrations, despite higher glucose concentrations than observed after ingestion of an equivalent amount of glucose (86). This observation has subsequently been confirmed with isoglycemic infusion studies (87). The subsequent discovery of glucagon-like immunoreactivity in the gut led to the realization that proglucagon is synthesized in enteroendocrine cells intercalated between enterocytes and distributed throughout the intestine. GLP-1 and gastric inhibitory polypeptide (GIP) are two incretin hormones, which stimulate postprandial insulin secretion (88). GLP-1 is released from L-cells, most densely located in the distal small intestine and colon, although they are also located more proximally in the duodenum and jejunum (89). There is some evidence in rodent models of paracrine GLP-1 secretion within pancreatic islets (90). GIP is secreted from K cells (which reside mainly in the duodenum and upper jejunum) in response to nutrient ingestion. The early secretion of GLP-1 might involve an indirect neural or hormonal mechanism (91). The later secretion of GLP-1 is dependent on direct contact of nutrients in the small intestine with L-cells (92). Targeted delivery of lauric acid in enteric-coated pellets to the ileum and colon can stimulate substantial endogenous GLP-1 release and attenuate postprandial glycemia (93). To stimulate its receptor, GLP-1 requires the presence of 2 N-terminal amino acids, which are cleaved by the enzyme, dipeptidyl peptidase-4 (DPP-4), rendering the truncated form (GLP-1-9,36) inactive. Because of the widespread distribution of DPP-4, the active form of GLP-1 has a short half-life in the circulation (94). GLP-1 receptor agonists that are not substrates of DPP-4 and DPP-4 inhibitors are approved for the treatment of type 2 diabetes. They lower fasting and postprandial glucose concentrations (66). In addition to stimulating insulin secretion, pharmacologic concentrations of GLP-1 (and GLP-1 receptor agonists) inhibit gastric emptying, and suppress glucagon secretion. Moreover, GLP-1 and GLP-1 receptor agonists increase satiety, leading to a reduction in weight (95).
Although GIP secretion is preserved, the insulinotropic effect of GIP is diminished in type 2 diabetes. Unlike GLP-1, GIP stimulates glucagon secretion during hypoglycemia (96, 97) and has no effect on gastric emptying. Circulating concentrations of GIP are related to BMI (98), which suggests a role of GIP in energy metabolism. In mice, high GIP concentrations promote obesity and insulin resistance (99). However, recent study shows that there is a synergistic effect of GIP and GLP-1 co-agonists in weight lowering (100) and glycemic improvement in patients with type 2 diabetes than mono-agonist (101). Addition of a dual GIP/GLP-1 receptor agonist (NNC0090-2746) to metformin improved glycemic control with accompanying reductions in body weight and circulating cholesterol (102). The molecular mechanism underneath the metabolic improvements is not known. The effects of GIP on glucose metabolism are an area of ongoing investigation, which will hopefully be accelerated by the development of a specific GIP receptor antagonist (103).
Cholecystokinin (CCK)
Cholecystokinin is secreted from the I-cells by exposure to nutrients in the duodenum and upper jejunum. Fat is a strong stimulus for CCK secretion, followed by protein, whereas carbohydrate is a weaker stimulus of CCK secretion. CCK concentrations increase from fivefold to tenfold after ingestion of a mixed meal and inhibit gastric emptying through activation of CCK-1 receptors (104). Physiological concentrations of CCK delay entry of glucose into the duodenum, reducing postprandial glucose excursions (105). In rats, CCK decreased hepatic glucose production to maintain glucose homeostasis by inhibiting CCK-A receptors and triggering a gut–brain–liver neuronal axis (106). In humans, CCK dose-dependently presents early satiety and reduces the energy intake at a buffet style meal, which was attenuated by the CCK-1 antagonist, loxiglumide (107). However, the long-term effects of CCK administration in humans and its role in obesity therapy are not clear.
Role of the Vagus—Vagal Blockade/Vagotomy
The gastrointestinal tract is innervated by the parasympathetic and sympathetic divisions of the autonomic nervous system. The parasympathetic innervation originates from the dorsal motor nucleus of the vagus (DMV) in the medulla (108), while the sympathetic supply derives from the prevertebral ganglia (109). Gastric motility is partially controlled by the vagus nerve, a mixed motor, and sensory nerve. The sensory axons of the vagus receive afferent inputs from gastrointestinal receptors and then project to the nucleus of the solitary tract (110). Nucleus of the solitary tract (NTS) neurons activate vagal motor neurons in the nucleus ambiguus and the dorsomedial nucleus to regulate the smooth muscle contractions in the stomach and duodenum, with these neural loops being known as vagovagal reflexes (111). Bilateral truncal vagotomy (112), aiming for treating of peptic ulcer surgery, and electrical vagal blockade (113) results in delayed gastric emptying, and weight loss—at least in the short term. The gastric vagal branches are often damaged during bariatric surgery (114). It remains controversial whether vagal innervation of the portal hepatis contributes to the beneficial effects of RYGB on food intake, energy expenditure, and body weight (115). Electrical vagal blockade does not seem to have significant effects on glucose metabolism (116).
Obese subjects exhibit decreased heart rate variability likely due to an imbalance of sympathetic and parasympathetic activity (117). Overactivity of the sympathetic nervous system is more significant in obese subjects with type 2 diabetes than in those subjects without diabetes (118). Weight reduction following RYGB and AGB in severely obese patients is associated with an increase in heart rate variability (119). The underlying mechanism(s) remain unknown but the improvement in autonomic function does not appear to be related to improved insulin action (120). It has been posited that these changes in autonomic function could arise from crosstalk between the gastrointestinal tract and the central nervous system (121) generated by a neuro-inflammatory reflex (122) arising from the gut microflora.
The Taste Signaling System
It is increasingly recognized that bariatric surgery may alter food preference and taste, in particular, the perception of sweet taste. This likely contributes to the reduction in energy intake after surgery (123). Both SG and RYGB result in a reduction of the frequency of food craving and the hedonic component of taste perception (124). Subjects experience a decreased desire to consume sweet and fatty flavors after RYGB (60) and SG (125).
The sweet taste signaling system includes heterodimeric G protein-coupled receptors, composed of the taste receptors (TRs), T1R2 + T1R3 heterodimers, which are activated by the binding of sweet compounds such as monosaccharides and disaccharides (126). These receptors are G-protein coupled (gustducin), and activation increases phospholipase C-β2 activity, which ultimately results in the release of Ca2+ from intracellular stores and the opening of a transient receptor potential ion channel TRPM5. The resulting membrane depolarization activates gustatory afferents (127). Sweet TRs are found in the tongue, gastrointestinal tract, pancreas, adipose tissue, brain, and bone (128). Expression of T1R2 + T1R3 also occurs in the entero-endocrine L cells (129), suggesting that the sweet sensing system in the gut is involved in incretin secretion. T1R3 knockout mice exhibit impaired GLP-1 secretion and glucose intolerance (130). Intragastric infusion of nutrients with lactisole, a T1R2/T1R3 blocker, attenuates GLP-1 and peptide YY secretion in humans (131, 132). The expression of sweet taste receptors and downstream molecule transcripts are disordered in models of type 2 diabetes (133). T1R2 expression is reciprocally regulated by luminal glucose in health, but not in patients with type 2 diabetes; during acute hyperglycemia, T1R2 transcript levels decrease in response to duodenal glucose infusion in healthy subjects, but increase in subjects with type 2 diabetes (134).
In addition to changes in oral taste sensitivity, the expression of T1R2 and T1R3 is decreased in the small intestine of rats after bariatric surgery; this occurs in parallel with elevation of GLP-1 (135). Functional magnetic resonance imaging or positron emission tomography demonstrates a decrease in neural activity in the brain reward areas in response to high-calorie foods (136).
Permeability and Glucose Transport
The proximal small intestine initiates carbohydrate absorption after digestion. Glucose absorption is mediated by the sodium glucose co-transporter-1 (SGLT1) across the apical cell membrane and partially by the glucose transporter 2 (GLUT-2) at high glucose concentrations (137). The small intestine has a maximal capacity of glucose absorption of about 0.5 g/min (or 2 kcal/min) per 30 cm (138). The absorptive rate depends on the exposure rate of glucose, region, and length of the small intestine, and the expression of glucose transporters (139). The inhibition of motility and blood flow in the small intestine also attenuates glucose absorption (140). Plasma concentration of 3-O-methyl-glucose, a glucose analog that is not metabolized, is normally used to measure the absorption rate of glucose. Physiologically, enhanced glucose absorption in the proximal gut would increase blood glucose concentrations; acute hyperglycemia itself appears to enhance glucose absorption (141). Rodent models of diabetes exhibit small intestinal hyperplasia and increased absorption of glucose from intestinal mucosa (142). It is unclear to what extent inhibition of SGLT-1 can alter glucose absorption in a way that is relevant to postprandial glycemic control in diabetes.
Active glucose transport and intestinal permeability are increased in obesity and diabetes. For a given caloric intake, this could alter the nutrient load entering the portal circulation (143–146). Changes in intestinal thickness and transcription of SGLT-1 and GLUT-2 occur after RYGB (147, 148). Foregut exclusion decreases glucose absorption in rodents (149). However, it is currently not known, and if so, the extent to which RYGB and SG alter the rate of active intestinal glucose absorption or the rate of passive intestinal permeability.
Intestinal integrity provides a physical barrier to luminal bacteria, toxins, and antigens from the external environment. In health, it allows the passage of water and nutrients. Increased paracellular permeability, following disruption of the intestinal tight junctions enables bacteria to leak out of the intestinal lumen into the blood stream (150). Factors that influence permeability include the gut microbiome and fatty acids (whether ingested directly or as products of bacterial fermentation) (151). Bile acids could alter gut permeability through the G-protein-coupled bile acid receptor (TGR5), a cell surface receptor, which occurs at a high level expression in the human placenta and spleen and is also found in multiple tissues such as the lung, liver, adipocytes, and the gastrointestinal tract (152). A systematic review of 14 studies suggests that that fasting and postprandial lipopolysaccharide (LPS) are increased in patients with diabetes (153). LPS is the core component of the outer membrane of Gram-negative bacteria. Metabolic endotoxemia is defined by a twofold to threefold increase in plasma LPS concentration (154). Rosiglitazone is the most effective in the lowering the LPS in patients with type 2 diabetes, but the extent to which this contributes to the glucose-lowering effects of this compound are unknown (155).
Bile Acid Metabolism
Bile acids are synthesized in hepatocytes via cytochrome P450-mediated oxidation of cholesterol and then secreted into the intestinal lumen through the biliary system. 95% of intestinal bile acids are reabsorbed in the distal gut and transported back to the liver by the enterohepatic circulation (156, 157). CCK induces production of bile, contraction of the gall bladder, and relaxation of the sphincter of Oddi, to deliver bile into the duodenum (158). Bile acids promote digestion and absorption of lipids in the gastrointestinal tract as well as participate in the regulation of glucose and energy homeostasis (159), acting through two specific receptors, the farnesoid X receptor (FXR) and TGR5.
FXR is expressed in the liver and the intestine in humans and is a member of the nuclear receptor super-family. It can be activated by both primary and secondary conjugated bile acids (160, 161). Similar to other nuclear receptors, FXR translocates to the cell nucleus and subsequently induces expression of the small heterodimer partner (SHP). SHP is involved in bile acids synthesis by downregulating the gene transcription of cholesterol 7 alpha-hydroxylase (CYP7A1), a rate-limiting enzyme in bile acid synthesis. The activation of TGR5 triggers the production of intracellular cAMP and secondary active the mitogen activated protein kinase signaling pathway to perform different functions in various organs. For instance, TGR5 is expressed in rodent and human pancreatic islets and regulates insulin secretion (162). TGR5 in enteroendocrine L cells stimulates secretion of GLP-1 (163). In addition, TGR5 may regulate energy homeostasis through activating deiodinases to convert the prohormone thyroxine (T4) into the active hormone triiodothyronine (T3) (164, 165).
Circulating bile acids’ concentrations after meal ingestion are decreased in obese subjects compared to lean controls (166). This difference is no longer significant after bariatric surgery (167, 168). The effects of SG on body weight and glucose tolerance are attenuated in the absence of FXR (169) and in TGR5 knock-out mice (170). In a diet-induced obesity mouse model, diversion of bile flow to the ileum produces similar metabolic benefits to RYGB (171), while the ability of RYGB to decrease body weight and improve glucose tolerance is substantially reduced in the absence of FXR. Bile acids may stimulate insulin secretion via activation of FXR and inhibition of ATP-dependent K + channels (172). It has been suggested that the changes in bile acid composition and concentrations induced by bariatric surgery can contribute to metabolic changes via FXR and TGR5-signaling pathways.
However, in humans, the contribution of bile acid changes to metabolic improvements after bariatric procedures is less clear. One study reported that total plasma bile acid concentrations increased twofold after RYGB but decreased after AGB, despite similar weight loss (173). Longitudinal study suggests that there are two phasic increases in plasma bile acid concentrations in a cohort of RYGB patients at 1 month and up to 24 months after surgery (168). This time course differs from the time course of metabolic resolution suggesting that they are unrelated phenomena.
The Gut Microbiome
The human gut microbiome consists of 10–100 trillion of microorganisms, primarily bacteria, in the digestive tract (174). The composition of the gut microbiome influences digestion, absorption, inflammation, and intestinal motility. Over the past decade, several studies have demonstrated that gut microbial populations are closely associated with metabolic disorders such as dyslipidemia, obesity, and diabetes (175). The gut microbiome is established early in life (176). Exposure to antibiotics alters the normal distribution of intestinal flora and is associated with changes in metabolism in some (177) but not all studies (178). Diet and lifestyle and geography are the primary influencers of the distribution of intestinal flora (179).
In humans, gut microbiota produce glycoside hydrolases and polysaccharide lyases, which facilitate digestion of sucrose, lactose, and starch (180). Undigested polysaccharides are subject to fermentation by intestinal bacterial leading to the production of short-chain fatty acids, which can provide 5–10% energy consumption (181). Gut microbiota is also involved in signaling of FXR and TGR5 by modifying the bile acid pool (182). In fact, bile acids interact with gut microbiota by direct effects on the mucosal defense, membrane integrity, oxidative and pH stress to increase the growth of bile acid-metabolizing bacteria (183).
d-lactate acidosis is a rare complication of jejuno-ileal bypass surgery or patients with short bowel syndrome (SBS) (184). d-lactate production is mainly dependent on the colonic microbiome (184). Notably, in patients with SBS or after jejuno-ileal bypass surgery, delivery of an increased amount of undigested carbohydrates to the colon can result in excess d-lactate accumulation (185, 186). Bacteroides thetaiotaomicron abundance is decreased in obese subjects compared to lean individuals (187). Patients with type 2 diabetes may exhibit decreased abundance of butyrate-producing bacteria and an increase in various opportunistic pathogens (188). Dietary fiber intake in patients with type 2 diabetes increases acetate and butyrate-producing bacteria improves glycemic control (189). Use of metformin is accompanied by increased abundance of Escherichia and a decrease of Intestinibacter (190). Impaired glucose tolerance is reversed after the transfer of metformin-altered microbiota to germ-free mice (191). Acarbose alters bile acid metabolism through changes in gut microbial populations (192). This results in great interest in microbiota alteration on improvement of metabolic parameters. Recently, transplantation of fecal microbiota or “bacteriotherapy” seems a promising therapeutic method for metabolic syndrome (193).
Individuals with obesity exhibit markedly decreased abundance of B. thetaiotaomicron. However, the abundance of this microbe increased after SG despite similar metabolic outcomes suggesting that this is incidental to the improvements in glucose metabolism after bariatric surgery (187). Randomized trials are warranted in the future to further assess the gut mechanism after bariatric surgeries in humans.
Conclusion
The upper gastrointestinal tract plays a primary role in the regulation of glucose excursions in response to meal ingestion by determining the rate of gastric emptying and indirectly by regulating appetite and satiation, barrier integrity, and nutrient absorption. Bariatric surgery has helped improve our knowledge of the mechanisms underlying gut–pancreas interactions and may enable development of effective dietary or pharmacological strategies in the management of diabetes.
Author Contributions
JM wrote the paper with input from AV regarding content and layout. AV edited the draft for clarity and content.
Conflict of Interest Statement
AV is an investigator in an investigator-initiated study sponsored by Novo Nordisk. He has consulted for vTv Therapeutics, XOMA, Sanofi-Aventis, Novartis, and Bayer in the past 5 years. JM has no relevant disclosures.
Acknowledgments
AV’s research is supported by the Mayo Clinic General Clinical Research Center (UL1 TR000135) and by the National Institutes of Health (DK78646, DK116231).
References
1. Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA (2006) 295(13):1549–55. doi:10.1001/jama.295.13.1549
2. Kushner RF. Obesity management. Gastroenterol Clin North Am (2007) 36(1):191–210, viii. doi:10.1016/j.gtc.2007.01.004
3. Elmer PJ, Brown JB, Nichols GA, Oster G. Effects of weight gain on medical care costs. Int J Obes Relat Metab Disord (2004) 28(11):1365–73. doi:10.1038/sj.ijo.0802774
4. U.S. Preventive Services Task Force. Screening for obesity in adults: recommendations and rationale. Ann Intern Med (2003) 139(11):930–2. doi:10.7326/0003-4819-139-11-200312020-00012
5. National Institutes of Health. Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults – the evidence report. Obes Res (1998) 6(Suppl 2):51S–209S.
6. Bachman KH. Obesity, weight management, and health care costs: a primer. Dis Manag (2007) 10(3):129–37. doi:10.1089/dis.2007.103643
7. Maggard MA, Shugarman LR, Suttorp M, Maglione M, Sugerman HJ, Livingston EH, et al. Meta-analysis: surgical treatment of obesity. Ann Intern Med (2005) 142(7):547–59. doi:10.7326/0003-4819-142-7-200504050-00013
8. Santry HP, Gillen DL, Lauderdale DS. Trends in bariatric surgical procedures. JAMA (2005) 294(15):1909–17. doi:10.1001/jama.294.15.1909
9. English WJ, DeMaria EJ, Brethauer SA, Mattar SG, Rosenthal RJ, Morton JM. American Society for Metabolic and Bariatric Surgery estimation of metabolic and bariatric procedures performed in the United States in 2016. Surg Obes Relat Dis (2018) 14(3):259–63. doi:10.1016/j.soard.2017.12.013
10. Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA (2004) 292(14):1724–37. doi:10.1001/jama.292.14.1724
11. Nelson WK, Fatima J, Houghton SG, Thompson GB, Kendrick ML, Mai JL, et al. The malabsorptive very, very long limb Roux-en-Y gastric bypass for super obesity: results in 257 patients. Surgery (2006) 140(4):517–22, discussion 22–3. doi:10.1016/j.surg.2006.06.020
12. Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Leccesi L, et al. Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med (2012) 366(17):1577–85. doi:10.1056/NEJMoa1200111
13. Schauer PR, Kashyap SR, Wolski K, Brethauer SA, Kirwan JP, Pothier CE, et al. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med (2012) 366(17):1567–76. doi:10.1056/NEJMoa1200225
14. Ikramuddin S, Korner J, Lee WJ, Connett JE, Inabnet WB, Billington CJ, et al. Roux-en-Y gastric bypass vs intensive medical management for the control of type 2 diabetes, hypertension, and hyperlipidemia: the Diabetes Surgery Study randomized clinical trial. JAMA (2013) 309(21):2240–9. doi:10.1001/jama.2013.5835
15. Still CD, Wood GC, Benotti P, Petrick AT, Gabrielsen J, Strodel WE, et al. Preoperative prediction of type 2 diabetes remission after Roux-en-Y gastric bypass surgery: a retrospective cohort study. Lancet Diabetes Endocrinol (2014) 2(1):38–45. doi:10.1016/S2213-8587(13)70070-6
16. Nguyen KT, Billington CJ, Vella A, Wang Q, Ahmed L, Bantle JP, et al. Preserved insulin secretory capacity and weight loss are the predominant predictors of glycemic control in patients with type 2 diabetes randomized to Roux-en-Y gastric bypass. Diabetes (2015) 64(9):3104–10. doi:10.2337/db14-1870
17. Schauer PR, Bhatt DL, Kirwan JP, Wolski K, Brethauer SA, Navaneethan SD, et al. Bariatric surgery versus intensive medical therapy for diabetes – 3-year outcomes. N Engl J Med (2014) 370(21):2002–13. doi:10.1056/NEJMoa1401329
18. Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med (2005) 353(3):249–54. doi:10.1056/NEJMoa043690
19. Goldfine AB, Mun EC, Devine E, Bernier R, Baz-Hecht M, Jones DB, et al. Patients with neuroglycopenia after gastric bypass surgery have exaggerated incretin and insulin secretory responses to a mixed meal. J Clin Endocrinol Metab (2007) 92(12):4678–85. doi:10.1210/jc.2007-0918
20. Vella A, Service FJ. Incretin hypersecretion in post-gastric bypass hypoglycemia – primary problem or red herring? J Clin Endocrinol Metab (2007) 92(12):4563–5. doi:10.1210/jc.2007-2260
21. Salehi M, Vella A, McLaughlin T, Patti ME. Hypoglycemia after gastric bypass surgery: current concepts and controversies. J Clin Endocrinol Metab (2018). doi:10.1210/jc.2018-00528
22. Yousseif A, Emmanuel J, Karra E, Millet Q, Elkalaawy M, Jenkinson AD, et al. Differential effects of laparoscopic sleeve gastrectomy and laparoscopic gastric bypass on appetite, circulating acyl-ghrelin, peptide YY3-36 and active GLP-1 levels in non-diabetic humans. Obes Surg (2014) 24(2):241–52. doi:10.1007/s11695-013-1066-0
23. Kashyap SR, Bhatt DL, Wolski K, Watanabe RM, Abdul-Ghani M, Abood B, et al. Metabolic effects of bariatric surgery in patients with moderate obesity and type 2 diabetes: analysis of a randomized control trial comparing surgery with intensive medical treatment. Diabetes Care (2013) 36(8):2175–82. doi:10.2337/dc12-1596
24. Peterli R, Steinert RE, Woelnerhanssen B, Peters T, Christoffel-Courtin C, Gass M, et al. Metabolic and hormonal changes after laparoscopic Roux-en-Y gastric bypass and sleeve gastrectomy: a randomized, prospective trial. Obes Surg (2012) 22(5):740–8. doi:10.1007/s11695-012-0622-3
25. Peterli R, Wolnerhanssen B, Peters T, Devaux N, Kern B, Christoffel-Courtin C, et al. Improvement in glucose metabolism after bariatric surgery: comparison of laparoscopic Roux-en-Y gastric bypass and laparoscopic sleeve gastrectomy: a prospective randomized trial. Ann Surg (2009) 250(2):234–41. doi:10.1097/SLA.0b013e3181ae32e3
26. Lee WJ, Chong K, Ser KH, Lee YC, Chen SC, Chen JC, et al. Gastric bypass vs sleeve gastrectomy for type 2 diabetes mellitus: a randomized controlled trial. Arch Surg (2011) 146(2):143–8. doi:10.1001/archsurg.2010.326
27. Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med (2007) 356(8):820–9. doi:10.1056/NEJMcp062614
28. Maddern GJ, Jamieson GG. Fundoplication enhances gastric emptying. Ann Surg (1985) 201(3):296–9. doi:10.1097/00000658-198503000-00006
29. Jamieson GG, Maddern GJ, Myers JC. Gastric emptying after fundoplication with and without proximal gastric vagotomy. Arch Surg (1991) 126(11):1414–7. doi:10.1001/archsurg.1991.01410350108017
30. Cummings DE, Overduin J. Gastrointestinal regulation of food intake. J Clin Invest (2007) 117(1):13–23. doi:10.1172/JCI30227
31. Lee WJ, Chen CY, Chong K, Lee YC, Chen SC, Lee SD. Changes in postprandial gut hormones after metabolic surgery: a comparison of gastric bypass and sleeve gastrectomy. Surg Obes Relat Dis (2011) 7(6):683–90. doi:10.1016/j.soard.2011.07.009
32. Malin SK, Samat A, Wolski K, Abood B, Pothier CE, Bhatt DL, et al. Improved acylated ghrelin suppression at 2 years in obese patients with type 2 diabetes: effects of bariatric surgery vs standard medical therapy. Int J Obes (Lond) (2014) 38(3):364–70. doi:10.1038/ijo.2013.196
33. Riccioppo D, Santo MA, Rocha M, Buchpiguel CA, Diniz MA, Pajecki D, et al. Small-volume, fast-emptying gastric pouch leads to better long-term weight loss and food tolerance after Roux-en-Y gastric bypass. Obes Surg (2018) 28(3):693–701. doi:10.1007/s11695-017-2922-0
34. Wickremesekera K, Miller G, Naotunne TD, Knowles G, Stubbs RS. Loss of insulin resistance after Roux-en-Y gastric bypass surgery: a time course study. Obes Surg (2005) 15(4):474–81. doi:10.1381/0960892053723402
35. Kelley DE, Wing R, Buonocore C, Sturis J, Polonsky K, Fitzsimmons M. Relative effects of calorie restriction and weight loss in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab (1993) 77(5):1287–93. doi:10.1210/jc.77.5.1287
36. Steven S, Hollingsworth KG, Small PK, Woodcock SA, Pucci A, Aribasala B, et al. Calorie restriction and not glucagon-like peptide-1 explains the acute improvement in glucose control after gastric bypass in type 2 diabetes. Diabet Med (2016) 33(12):1723–31. doi:10.1111/dme.13257
37. Perry RJ, Peng L, Cline GW, Wang Y, Rabin-Court A, Song JD, et al. Mechanisms by which a very-low-calorie diet reverses hyperglycemia in a rat model of type 2 diabetes. Cell Metab (2017) 27(1):210–7.e3. doi:10.1016/j.cmet.2017.10.004
38. Sathananthan M, Shah M, Edens KL, Grothe KB, Piccinini F, Farrugia LP, et al. Six and 12 weeks of caloric restriction increases beta cell function and lowers fasting and postprandial glucose concentrations in people with type 2 diabetes. J Nutr (2015) 145(9):2046–51. doi:10.3945/jn.115.210617
39. Jackness C, Karmally W, Febres G, Conwell IM, Ahmed L, Bessler M, et al. Very low-calorie diet mimics the early beneficial effect of Roux-en-Y gastric bypass on insulin sensitivity and beta-cell function in type 2 diabetic patients. Diabetes (2013) 62(9):3027–32. doi:10.2337/db12-1762
40. Lips MA, de Groot GH, van Klinken JB, Aarts E, Berends FJ, Janssen IM, et al. Calorie restriction is a major determinant of the short-term metabolic effects of gastric bypass surgery in obese type 2 diabetic patients. Clin Endocrinol (Oxf) (2014) 80(6):834–42. doi:10.1111/cen.12254
41. Lean ME, Leslie WS, Barnes AC, Brosnahan N, Thom G, McCombie L, et al. Primary care-led weight management for remission of type 2 diabetes (DiRECT): an open-label, cluster-randomised trial. Lancet (2017) 391:541–51. doi:10.1016/S0140-6736(17)33102-1
42. Bredenoord AJ, Chial HJ, Camilleri M, Mullan BP, Murray JA. Gastric accommodation and emptying in evaluation of patients with upper gastrointestinal symptoms. Clin Gastroenterol Hepatol (2003) 1(4):264–72. doi:10.1053/S1542-3565(03)00130-7
43. Delgado-Aros S, Kim DY, Burton DD, Thomforde GM, Stephens D, Brinkmann BH, et al. Effect of GLP-1 on gastric volume, emptying, maximum volume ingested, and postprandial symptoms in humans. Am J Physiol Gastrointest Liver Physiol (2002) 282(3):G424–31. doi:10.1152/ajpgi.2002.282.3.G424
44. Camilleri M, Breen M, Ryks M, Burton D. Proximal and overall gastric emptying of solids in patients with reduced gastric volume accommodation compared to matched controls. Dig Dis Sci (2011) 56(6):1729–34. doi:10.1007/s10620-011-1615-0
45. Shah M, Law JH, Micheletto F, Sathananthan M, Dalla Man C, Cobelli C, et al. Contribution of endogenous glucagon-like peptide 1 to glucose metabolism after Roux-en-Y gastric bypass. Diabetes (2014) 63(2):483–93. doi:10.2337/db13-0954
46. Delgado-Aros S, Vella A, Camilleri M, Low PA, Burton DD, Thomforde GM, et al. Effects of glucagon-like peptide-1 and feeding on gastric volumes in diabetes mellitus with cardio-vagal dysfunction. Neurogastroenterol Motil (2003) 15(4):435–43. doi:10.1046/j.1365-2982.2003.00422.x
47. Kral JG. Effects of truncal vagotomy on body weight and hyperinsulinemia in morbid obesity. Am J Clin Nutr (1980) 33(2 Suppl):416–9. doi:10.1093/ajcn/33.2.416
48. Ma J, Pilichiewicz AN, Feinle-Bisset C, Wishart JM, Jones KL, Horowitz M, et al. Effects of variations in duodenal glucose load on glycaemic, insulin, and incretin responses in type 2 diabetes. Diabet Med (2012) 29(5):604–8. doi:10.1111/j.1464-5491.2011.03496.x
49. Marathe CS, Rayner CK, Lange K, Bound M, Wishart J, Jones KL, et al. Relationships of the early insulin secretory response and oral disposition index with gastric emptying in subjects with normal glucose tolerance. Physiol Rep (2017) 5(4):e13122. doi:10.14814/phy2.13122
50. Marathe CS, Horowitz M, Trahair LG, Wishart JM, Bound M, Lange K, et al. Relationships of early and late glycemic responses with gastric emptying during an oral glucose tolerance test. J Clin Endocrinol Metab (2015) 100(9):3565–71. doi:10.1210/JC.2015-2482
51. Vazquez-Roque MI, Camilleri M, Vella A, Carlson P, Laugen J, Zinsmeister AR. Association of TCF7L2 allelic variations with gastric function, satiation, and GLP-1 levels. Clin Transl Sci (2011) 4(3):183–7. doi:10.1111/j.1752-8062.2011.00284.x
52. Parthasarathy G, Kudva YC, Low PA, Camilleri M, Basu A, Bharucha AE. Relationship between gastric emptying and diurnal glycemic control in type 1 diabetes mellitus: a randomized trial. J Clin Endocrinol Metab (2017) 102(2):398–406. doi:10.1210/jc.2016-2809
53. Chowdhury S, Reeds DN, Crimmins DL, Patterson BW, Laciny E, Wang S, et al. Xenin-25 delays gastric emptying and reduces postprandial glucose levels in humans with and without type 2 diabetes. Am J Physiol Gastrointest Liver Physiol (2014) 306(4):G301–9. doi:10.1152/ajpgi.00383.2013
54. Ishii M, Nakamura T, Kasai F, Onuma T, Baba T, Takebe K. Altered postprandial insulin requirement in IDDM patients with gastroparesis. Diabetes Care (1994) 17(8):901–3. doi:10.2337/diacare.17.8.901
55. Meier JJ, Rosenstock J, Hincelin-Mery A, Roy-Duval C, Delfolie A, Coester HV, et al. Contrasting effects of lixisenatide and liraglutide on postprandial glycemic control, gastric emptying, and safety parameters in patients with type 2 diabetes on optimized insulin glargine with or without metformin: a randomized, open-label trial. Diabetes Care (2015) 38(7):1263–73. doi:10.2337/dc14-1984
56. Hinshaw L, Schiavon M, Dadlani V, Mallad A, Dalla Man C, Bharucha A, et al. Effect of pramlintide on postprandial glucose fluxes in type 1 diabetes. J Clin Endocrinol Metab (2016) 101(5):1954–62. doi:10.1210/jc.2015-3952
57. Adachi S, Takiguchi S, Okada K, Yamamoto K, Yamasaki M, Miyata H, et al. Effects of ghrelin administration after total gastrectomy: a prospective, randomized, placebo-controlled phase II study. Gastroenterology (2010) 138(4):1312–20. doi:10.1053/j.gastro.2009.12.058
58. Yamamoto K, Takiguchi S, Miyata H, Adachi S, Hiura Y, Yamasaki M, et al. Randomized phase II study of clinical effects of ghrelin after esophagectomy with gastric tube reconstruction. Surgery (2010) 148(1):31–8. doi:10.1016/j.surg.2009.11.026
59. Sisley S, Gutierrez-Aguilar R, Scott M, D’Alessio DA, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J Clin Invest (2014) 124(6):2456–63. doi:10.1172/JCI72434
60. Miras AD, Jackson RN, Jackson SN, Goldstone AP, Olbers T, Hackenberg T, et al. Gastric bypass surgery for obesity decreases the reward value of a sweet-fat stimulus as assessed in a progressive ratio task. Am J Clin Nutr (2012) 96(3):467–73. doi:10.3945/ajcn.112.036921
61. Stefater MA, Wilson-Perez HE, Chambers AP, Sandoval DA, Seeley RJ. All bariatric surgeries are not created equal: insights from mechanistic comparisons. Endocr Rev (2012) 33(4):595–622. doi:10.1210/er.2011-1044
62. Shah M, Vella A. Effects of GLP-1 on appetite and weight. Rev Endocr Metab Disord (2014) 15(3):181–7. doi:10.1007/s11154-014-9289-5
63. Vella A, Camilleri M, Rizza RA. The gastrointestinal tract and glucose tolerance. Curr Opin Clin Nutr Metab Care (2004) 7(4):479–84. doi:10.1097/01.mco.0000134375.01310.97
64. Williams DL, Grill HJ, Cummings DE, Kaplan JM. Vagotomy dissociates short- and long-term controls of circulating ghrelin. Endocrinology (2003) 144(12):5184–7. doi:10.1210/en.2003-1059
65. Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet (2002) 359(9309):824–30. doi:10.1016/S0140-6736(02)07952-7
66. Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet (2006) 368(9548):1696–705. doi:10.1016/S0140-6736(06)69705-5
67. Dotson CD, Geraedts MC, Munger SD. Peptide regulators of peripheral taste function. Semin Cell Dev Biol (2013) 24(3):232–9. doi:10.1016/j.semcdb.2013.01.004
68. Elson AE, Dotson CD, Egan JM, Munger SD. Glucagon signaling modulates sweet taste responsiveness. FASEB J (2010) 24(10):3960–9. doi:10.1096/fj.10-158105
69. Martin B, Dotson CD, Shin YK, Ji S, Drucker DJ, Maudsley S, et al. Modulation of taste sensitivity by GLP-1 signaling in taste buds. Ann N Y Acad Sci (2009) 1170:98–101. doi:10.1111/j.1749-6632.2009.03920.x
70. Shin YK, Martin B, Golden E, Dotson CD, Maudsley S, Kim W, et al. Modulation of taste sensitivity by GLP-1 signaling. J Neurochem (2008) 106(1):455–63. doi:10.1111/j.1471-4159.2008.05397.x
71. Sathananthan A, Man CD, Micheletto F, Zinsmeister AR, Camilleri M, Giesler PD, et al. Common genetic variation in GLP1R and insulin secretion in response to exogenous GLP-1 in nondiabetic subjects: a pilot study. Diabetes Care (2010) 33(9):2074–6. doi:10.2337/dc10-0200
72. Vetter ML, Wadden TA, Teff KL, Khan ZF, Carvajal R, Ritter S, et al. GLP-1 plays a limited role in improved glycemia shortly after Roux-en-Y gastric bypass: a comparison with intensive lifestyle modification. Diabetes (2015) 64(2):434–46. doi:10.2337/db14-0558
73. Ye J, Hao Z, Mumphrey MB, Townsend RL, Patterson LM, Stylopoulos N, et al. GLP-1 receptor signaling is not required for reduced body weight after RYGB in rodents. Am J Physiol Regul Integr Comp Physiol (2014) 306(5):R352–62. doi:10.1152/ajpregu.00491.2013
74. Jimenez A, Mari A, Casamitjana R, Lacy A, Ferrannini E, Vidal J. GLP-1 and glucose tolerance after sleeve gastrectomy in morbidly obese subjects with type 2 diabetes. Diabetes (2014) 63(10):3372–7. doi:10.2337/db14-0357
75. Douros JD, Lewis AG, Smith EP, Niu J, Capozzi M, Wittmann A, et al. Enhanced glucose control following vertical sleeve gastrectomy does not require a beta-cell glucagon-like peptide 1 receptor. Diabetes (2018). doi:10.2337/db18-0081
76. de Jonge C, Rensen SS, Verdam FJ, Vincent RP, Bloom SR, Buurman WA, et al. Endoscopic duodenal-jejunal bypass liner rapidly improves type 2 diabetes. Obes Surg (2013) 23(9):1354–60. doi:10.1007/s11695-013-0921-3
77. Cohen R, Galvao Neto MP, Roux CL. Another look at the (endoscopic duodenal liner) ENDO trial, or how to avoid the burial of a valuable antidiabetic tool. Surg Obes Relat Dis (2016) 12(3):702–4. doi:10.1016/j.soard.2015.08.503
78. Spiegel K, Tasali E, Leproult R, Scherberg N, Van Cauter E. Twenty-four-hour profiles of acylated and total ghrelin: relationship with glucose levels and impact of time of day and sleep. J Clin Endocrinol Metab (2011) 96(2):486–93. doi:10.1210/jc.2010-1978
79. Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE, Frayo RS, et al. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med (2002) 8(7):643–4. doi:10.1038/nm0702-643
80. Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature (2000) 407(6806):908–13. doi:10.1038/35038090
81. Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab (2001) 86(12):5992. doi:10.1210/jcem.86.12.8111
82. Dezaki K, Damdindorj B, Sone H, Dyachok O, Tengholm A, Gylfe E, et al. Ghrelin attenuates cAMP-PKA signaling to evoke insulinostatic cascade in islet beta-cells. Diabetes (2011) 60(9):2315–24. doi:10.2337/db11-0368
83. Yanagi S, Sato T, Kangawa K, Nakazato M. The homeostatic force of ghrelin. Cell Metab (2018) 27(4):786–804. doi:10.1016/j.cmet.2018.02.008
84. Shin A, Camilleri M, Busciglio I, Burton D, Stoner E, Noonan P, et al. Randomized controlled phase Ib study of ghrelin agonist, RM-131, in type 2 diabetic women with delayed gastric emptying: pharmacokinetics and pharmacodynamics. Diabetes Care (2013) 36(1):41–8. doi:10.2337/dc12-1128
85. Shin A, Camilleri M, Busciglio I, Burton D, Smith SA, Vella A, et al. The ghrelin agonist RM-131 accelerates gastric emptying of solids and reduces symptoms in patients with type 1 diabetes mellitus. Clin Gastroenterol Hepatol (2013) 11(11):1453–9.e4. doi:10.1016/j.cgh.2013.04.019
86. Elrick H, Stimmler L, Hlad CJ Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab (1964) 24:1076–82. doi:10.1210/jcem-24-10-1076
87. Perley MJ, Kipnis DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic subjects. J Clin Invest (1967) 46(12):1954–62. doi:10.1172/JCI105685
88. Vilsboll T, Krarup T, Madsbad S, Holst JJ. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept (2003) 114(2–3):115–21. doi:10.1016/S0167-0115(03)00111-3
89. Eissele R, Goke R, Willemer S, Harthus HP, Vermeer H, Arnold R, et al. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur J Clin Invest (1992) 22(4):283–91. doi:10.1111/j.1365-2362.1992.tb01464.x
90. Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim KS, et al. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab (2017) 25(4):927–34.e3. doi:10.1016/j.cmet.2017.02.008
91. Brubaker PL, Anini Y. Direct and indirect mechanisms regulating secretion of glucagon-like peptide-1 and glucagon-like peptide-2. Can J Physiol Pharmacol (2003) 81(11):1005–12. doi:10.1139/y03-107
92. Little TJ, Doran S, Meyer JH, Smout AJ, O’Donovan DG, Wu KL, et al. The release of GLP-1 and ghrelin, but not GIP and CCK, by glucose is dependent upon the length of small intestine exposed. Am J Physiol Endocrinol Metab (2006) 291(3):E647–55. doi:10.1152/ajpendo.00099.2006
93. Ma J, Checklin HL, Wishart JM, Stevens JE, Jones KL, Horowitz M, et al. A randomised trial of enteric-coated nutrient pellets to stimulate gastrointestinal peptide release and lower glycaemia in type 2 diabetes. Diabetologia (2013) 56(6):1236–42. doi:10.1007/s00125-013-2876-2
94. Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology (1995) 136(8):3585–96. doi:10.1210/endo.136.8.7628397
95. Burcelin R, Gourdy P. Harnessing glucagon-like peptide-1 receptor agonists for the pharmacological treatment of overweight and obesity. Obes Rev (2017) 18(1):86–98. doi:10.1111/obr.12465
96. Christensen M, Vedtofte L, Holst JJ, Vilsboll T, Knop FK. Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes (2011) 60(12):3103–9. doi:10.2337/db11-0979
97. Christensen M, Calanna S, Sparre-Ulrich AH, Kristensen PL, Rosenkilde MM, Faber J, et al. Glucose-dependent insulinotropic polypeptide augments glucagon responses to hypoglycemia in type 1 diabetes. Diabetes (2015) 64(1):72–8. doi:10.2337/db14-0440
98. Calanna S, Christensen M, Holst JJ, Laferrere B, Gluud LL, Vilsboll T, et al. Secretion of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes: systematic review and meta-analysis of clinical studies. Diabetes Care (2013) 36(10):3346–52. doi:10.2337/dc13-0465
99. Nasteska D, Harada N, Suzuki K, Yamane S, Hamasaki A, Joo E, et al. Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high-fat diet conditions. Diabetes (2014) 63(7):2332–43. doi:10.2337/db13-1563
100. Finan B, Ma T, Ottaway N, Muller TD, Habegger KM, Heppner KM, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med (2013) 5(209):209ra151. doi:10.1126/scitranslmed.3007218
101. Schmitt C, Portron A, Jadidi S, Sarkar N, DiMarchi R. Pharmacodynamics, pharmacokinetics and safety of multiple ascending doses of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 in people with type 2 diabetes mellitus. Diabetes Obes Metab (2017) 19(10):1436–45. doi:10.1111/dom.13024
102. Frias JP, Bastyr EJ III, Vignati L, Tschop MH, Schmitt C, Owen K, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab (2017) 26(2):343–52.e2. doi:10.1016/j.cmet.2017.07.011
103. Gasbjerg LS, Gabe MBN, Hartmann B, Christensen MB, Knop FK, Holst JJ, et al. Glucose-dependent insulinotropic polypeptide (GIP) receptor antagonists as anti-diabetic agents. Peptides (2018) 100:173–81. doi:10.1016/j.peptides.2017.11.021
104. Chandra R, Liddle RA. Cholecystokinin. Curr Opin Endocrinol Diabetes Obes (2007) 14(1):63–7. doi:10.1097/MED.0b013e3280122850
105. Liddle RA, Rushakoff RJ, Morita ET, Beccaria L, Carter JD, Goldfine ID. Physiological role for cholecystokinin in reducing postprandial hyperglycemia in humans. J Clin Invest (1988) 81(6):1675–81. doi:10.1172/JCI113505
106. Cheung GW, Kokorovic A, Lam CK, Chari M, Lam TK. Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metab (2009) 10(2):99–109. doi:10.1016/j.cmet.2009.07.005
107. Beglinger C, Degen L, Matzinger D, D’Amato M, Drewe J. Loxiglumide, a CCK-A receptor antagonist, stimulates calorie intake and hunger feelings in humans. Am J Physiol Regul Integr Comp Physiol (2001) 280(4):R1149–54. doi:10.1152/ajpregu.2001.280.4.R1149
108. Travagli RA, Hermann GE, Browning KN, Rogers RC. Brainstem circuits regulating gastric function. Annu Rev Physiol (2006) 68:279–305. doi:10.1146/annurev.physiol.68.040504.094635
109. Furness JB. Novel gut afferents: intrinsic afferent neurons and intestinofugal neurons. Auton Neurosci (2006) 125(1–2):81–5. doi:10.1016/j.autneu.2006.01.007
110. Chang HY, Mashimo H, Goyal RK. Musings on the wanderer: what’s new in our understanding of vago-vagal reflex? IV. Current concepts of vagal efferent projections to the gut. Am J Physiol Gastrointest Liver Physiol (2003) 284(3):G357–66. doi:10.1152/ajpgi.00478.2002
111. Broussard DL, Altschuler SM. Brainstem viscerotopic organization of afferents and efferents involved in the control of swallowing. Am J Med (2000) 108(Suppl 4a):79S–86S. doi:10.1016/S0002-9343(99)00343-5
112. Smith DK, Sarfeh J, Howard L. Truncal vagotomy in hypothalamic obesity. Lancet (1983) 1(8337):1330–1. doi:10.1016/S0140-6736(83)92437-6
113. Sarr MG, Billington CJ, Brancatisano R, Brancatisano A, Toouli J, Kow L, et al. The EMPOWER study: randomized, prospective, double-blind, multicenter trial of vagal blockade to induce weight loss in morbid obesity. Obes Surg (2012) 22(11):1771–82. doi:10.1007/s11695-012-0751-8
114. Browning KN, Fortna SR, Hajnal A. Roux-en-Y gastric bypass reverses the effects of diet-induced obesity to inhibit the responsiveness of central vagal motoneurones. J Physiol (2013) 591(9):2357–72. doi:10.1113/jphysiol.2012.249268
115. Shin AC, Zheng H, Berthoud HR. Vagal innervation of the hepatic portal vein and liver is not necessary for Roux-en-Y gastric bypass surgery-induced hypophagia, weight loss, and hypermetabolism. Ann Surg (2012) 255(2):294–301. doi:10.1097/SLA.0b013e31823e71b7
116. Sathananthan M, Ikramuddin S, Swain JM, Shah M, Piccinini F, Dalla Man C, et al. The effect of vagal nerve blockade using electrical impulses on glucose metabolism in nondiabetic subjects. Diabetes Metab Syndr Obes (2014) 7:305–12. doi:10.2147/DMSO.S65733
117. Karason K, Molgaard H, Wikstrand J, Sjostrom L. Heart rate variability in obesity and the effect of weight loss. Am J Cardiol (1999) 83(8):1242–7. doi:10.1016/S0002-9149(99)00066-1
118. Lips MA, de Groot GH, De Kam M, Berends FJ, Wiezer R, Van Wagensveld BA, et al. Autonomic nervous system activity in diabetic and healthy obese female subjects and the effect of distinct weight loss strategies. Eur J Endocrinol (2013) 169(4):383–90. doi:10.1530/EJE-13-0506
119. Maser RE, Lenhard MJ, Irgau I, Wynn GM. Impact of surgically induced weight loss on cardiovascular autonomic function: one-year follow-up. Obesity (Silver Spring) (2007) 15(2):364–9. doi:10.1038/oby.2007.554
120. Maser RE, Lenhard MJ, Peters MB, Irgau I, Wynn GM. Effects of surgically induced weight loss by Roux-en-Y gastric bypass on cardiovascular autonomic nerve function. Surg Obes Relat Dis (2013) 9(2):221–6. doi:10.1016/j.soard.2011.11.014
121. Casellini CM, Parson HK, Hodges K, Edwards JF, Lieb DC, Wohlgemuth SD, et al. Bariatric surgery restores cardiac and sudomotor autonomic C-fiber dysfunction towards normal in obese subjects with type 2 diabetes. PLoS One (2016) 11(5):e0154211. doi:10.1371/journal.pone.0154211
122. Parekh PJ, Nayi VR, Johnson DA, Vinik AI. The role of gut microflora and the cholinergic anti-inflammatory neuroendocrine system in diabetes mellitus. Front Endocrinol (2016) 7:55. doi:10.3389/fendo.2016.00055
123. Kenler HA, Brolin RE, Cody RP. Changes in eating behavior after horizontal gastroplasty and Roux-en-Y gastric bypass. Am J Clin Nutr (1990) 52(1):87–92. doi:10.1093/ajcn/52.1.87
124. Nance K, Eagon JC, Klein S, Pepino MY. Effects of sleeve gastrectomy vs. Roux-en-Y gastric bypass on eating behavior and sweet taste perception in subjects with obesity. Nutrients (2017) 10(1):E18. doi:10.3390/nu10010018
125. Van Vuuren MAJ, Strodl E, White KM, Lockie PD. Taste, enjoyment, and desire of flavors change after sleeve gastrectomy-short term results. Obes Surg (2017) 27(6):1466–73. doi:10.1007/s11695-016-2497-1
126. Nelson G, Hoon MA, Chandrashekar J, Zhang Y, Ryba NJ, Zuker CS. Mammalian sweet taste receptors. Cell (2001) 106(3):381–90. doi:10.1016/S0092-8674(01)00451-2
127. Liu D, Liman ER. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc Natl Acad Sci U S A (2003) 100(25):15160–5. doi:10.1073/pnas.2334159100
128. Laffitte A, Neiers F, Briand L. Functional roles of the sweet taste receptor in oral and extraoral tissues. Curr Opin Clin Nutr Metab Care (2014) 17(4):379–85. doi:10.1097/MCO.0000000000000058
129. Young RL, Sutherland K, Pezos N, Brierley SM, Horowitz M, Rayner CK, et al. Expression of taste molecules in the upper gastrointestinal tract in humans with and without type 2 diabetes. Gut (2009) 58(3):337–46. doi:10.1136/gut.2008.148932
130. Kokrashvili Z, Mosinger B, Margolskee RF. T1r3 and alpha-gustducin in gut regulate secretion of glucagon-like peptide-1. Ann N Y Acad Sci (2009) 1170:91–4. doi:10.1111/j.1749-6632.2009.04485.x
131. Steinert RE, Gerspach AC, Gutmann H, Asarian L, Drewe J, Beglinger C. The functional involvement of gut-expressed sweet taste receptors in glucose-stimulated secretion of glucagon-like peptide-1 (GLP-1) and peptide YY (PYY). Clin Nutr (2011) 30(4):524–32. doi:10.1016/j.clnu.2011.01.007
132. Gerspach AC, Steinert RE, Schonenberger L, Graber-Maier A, Beglinger C. The role of the gut sweet taste receptor in regulating GLP-1, PYY, and CCK release in humans. Am J Physiol Endocrinol Metab (2011) 301(2):E317–25. doi:10.1152/ajpendo.00077.2011
133. Feng R, Qian C, Liu Q, Jin Y, Liu L, Li S, et al. Expression of sweet taste receptor and gut hormone secretion in modelled type 2 diabetes. Gen Comp Endocrinol (2017) 252:142–9. doi:10.1016/j.ygcen.2017.08.008
134. Young RL, Chia B, Isaacs NJ, Ma J, Khoo J, Wu T, et al. Disordered control of intestinal sweet taste receptor expression and glucose absorption in type 2 diabetes. Diabetes (2013) 62(10):3532–41. doi:10.2337/db13-0581
135. Bueter M, Miras AD, Chichger H, Fenske W, Ghatei MA, Bloom SR, et al. Alterations of sucrose preference after Roux-en-Y gastric bypass. Physiol Behav (2011) 104(5):709–21. doi:10.1016/j.physbeh.2011.07.025
136. Ochner CN, Laferrere B, Afifi L, Atalayer D, Geliebter A, Teixeira J. Neural responsivity to food cues in fasted and fed states pre and post gastric bypass surgery. Neurosci Res (2012) 74(2):138–43. doi:10.1016/j.neures.2012.08.002
137. Kellett GL. The facilitated component of intestinal glucose absorption. J Physiol (2001) 531(Pt 3):585–95. doi:10.1111/j.1469-7793.2001.0585h.x
138. Duchman SM, Ryan AJ, Schedl HP, Summers RW, Bleiler TL, Gisolfi CV. Upper limit for intestinal absorption of a dilute glucose solution in men at rest. Med Sci Sports Exerc (1997) 29(4):482–8. doi:10.1097/00005768-199704000-00009
139. Holst JJ, Gribble F, Horowitz M, Rayner CK. Roles of the gut in glucose homeostasis. Diabetes Care (2016) 39(6):884–92. doi:10.2337/dc16-0351
140. Thazhath SS, Marathe CS, Wu T, Chang J, Khoo J, Kuo P, et al. The glucagon-like peptide 1 receptor agonist exenatide inhibits small intestinal motility, flow, transit, and absorption of glucose in healthy subjects and patients with type 2 diabetes: a randomized controlled trial. Diabetes (2016) 65(1):269–75. doi:10.2337/db15-0893
141. Fischer E, Lauterbach F. Effect of hyperglycaemia on sugar transport in the isolated mucosa of guinea-pig small intestine. J Physiol (1984) 355:567–86. doi:10.1113/jphysiol.1984.sp015439
142. Adachi T, Mori C, Sakurai K, Shihara N, Tsuda K, Yasuda K. Morphological changes and increased sucrase and isomaltase activity in small intestines of insulin-deficient and type 2 diabetic rats. Endocr J (2003) 50(3):271–9. doi:10.1507/endocrj.50.271
143. Nguyen NQ, Debreceni TL, Bambrick JE, Chia B, Wishart J, Deane AM, et al. Accelerated intestinal glucose absorption in morbidly obese humans: relationship to glucose transporters, incretin hormones, and glycemia. J Clin Endocrinol Metab (2015) 100(3):968–76. doi:10.1210/jc.2014-3144
144. Seimon RV, Brennan IM, Russo A, Little TJ, Jones KL, Standfield S, et al. Gastric emptying, mouth-to-cecum transit, and glycemic, insulin, incretin, and energy intake responses to a mixed-nutrient liquid in lean, overweight, and obese males. Am J Physiol Endocrinol Metab (2013) 304(3):E294–300. doi:10.1152/ajpendo.00533.2012
145. Damci T, Nuhoglu I, Devranoglu G, Osar Z, Demir M, Ilkova H. Increased intestinal permeability as a cause of fluctuating postprandial blood glucose levels in type 1 diabetic patients. Eur J Clin Invest (2003) 33(5):397–401. doi:10.1046/j.1365-2362.2003.01161.x
146. Horton F, Wright J, Smith L, Hinton PJ, Robertson MD. Increased intestinal permeability to oral chromium (51 Cr)-EDTA in human type 2 diabetes. Diabet Med (2014) 31(5):559–63. doi:10.1111/dme.12360
147. Nguyen NQ, Debreceni TL, Bambrick JE, Chia B, Deane AM, Wittert G, et al. Upregulation of intestinal glucose transporters after Roux-en-Y gastric bypass to prevent carbohydrate malabsorption. Obesity (Silver Spring) (2014) 22(10):2164–71. doi:10.1002/oby.20829
148. Camilleri M, Madsen K, Spiller R, Greenwood-Van Meerveld B, Verne GN. Intestinal barrier function in health and gastrointestinal disease. Neurogastroenterol Motil (2012) 24(6):503–12. doi:10.1111/j.1365-2982.2012.01921.x
149. Pal A, Rhoads DB, Tavakkoli A. Foregut exclusion disrupts intestinal glucose sensing and alters portal nutrient and hormonal milieu. Diabetes (2015) 64(6):1941–50. doi:10.2337/db14-1578
150. Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci (2013) 70(4):631–59. doi:10.1007/s00018-012-1070-x
151. Camilleri M, Lasch K, Zhou W. Irritable bowel syndrome: methods, mechanisms, and pathophysiology. The confluence of increased permeability, inflammation, and pain in irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol (2012) 303(7):G775–85. doi:10.1152/ajpgi.00155.2012
152. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem (2003) 278(11):9435–40. doi:10.1074/jbc.M209706200
153. Gomes JMG, Costa JA, Alfenas RCG. Metabolic endotoxemia and diabetes mellitus: a systematic review. Metabolism (2017) 68:133–44. doi:10.1016/j.metabol.2016.12.009
154. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56(7):1761–72. doi:10.2337/db06-1491
155. Al-Attas OS, Al-Daghri NM, Al-Rubeaan K, da Silva NF, Sabico SL, Kumar S, et al. Changes in endotoxin levels in T2DM subjects on anti-diabetic therapies. Cardiovasc Diabetol (2009) 8:20. doi:10.1186/1475-2840-8-20
156. Kuipers F, Bloks VW, Groen AK. Beyond intestinal soap – bile acids in metabolic control. Nat Rev (2014) 10(8):488–98. doi:10.1038/nrendo.2014.60
157. Staels B, Fonseca VA. Bile acids and metabolic regulation: mechanisms and clinical responses to bile acid sequestration. Diabetes Care (2009) 32(Suppl 2):S237–45. doi:10.2337/dc09-S355
158. Reeve JR Jr, Eysselein VE, Rosenquist G, Zeeh J, Regner U, Ho FJ, et al. Evidence that CCK-58 has structure that influences its biological activity. Am J Physiol (1996) 270(5 Pt 1):G860–8.
159. Vella A, Camilleri M. The gastrointestinal tract as an integrator of mechanical and hormonal response to nutrient ingestion. Diabetes (2017) 66(11):2729–37. doi:10.2337/dbi17-0021
160. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science (1999) 284(5418):1362–5. doi:10.1126/science.284.5418.1362
161. Teodoro JS, Rolo AP, Palmeira CM. Hepatic FXR: key regulator of whole-body energy metabolism. Trends Endocrinol Metab (2011) 22(11):458–66. doi:10.1016/j.tem.2011.07.002
162. Kumar DP, Rajagopal S, Mahavadi S, Mirshahi F, Grider JR, Murthy KS, et al. Activation of transmembrane bile acid receptor TGR5 stimulates insulin secretion in pancreatic beta cells. Biochem Biophys Res Commun (2012) 427(3):600–5. doi:10.1016/j.bbrc.2012.09.104
163. Camilleri M, Vazquez-Roque MI, Carlson P, Burton D, Wong BS, Zinsmeister AR. Association of bile acid receptor TGR5 variation and transit in health and lower functional gastrointestinal disorders. Neurogastroenterol Motil (2011) 23(11):995–9, e458. doi:10.1111/j.1365-2982.2011.01772.x
164. Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature (2006) 439(7075):484–9. doi:10.1038/nature04330
165. Baxter JD, Webb P. Metabolism: bile acids heat things up. Nature (2006) 439(7075):402–3. doi:10.1038/439402a
166. Haeusler RA, Camastra S, Nannipieri M, Astiarraga B, Castro-Perez J, Xie D, et al. Increased bile acid synthesis and impaired bile acid transport in human obesity. J Clin Endocrinol Metab (2016) 101(5):1935–44. doi:10.1210/jc.2015-2583
167. Jansen PL, van Werven J, Aarts E, Berends F, Janssen I, Stoker J, et al. Alterations of hormonally active fibroblast growth factors after Roux-en-Y gastric bypass surgery. Dig Dis (2011) 29(1):48–51. doi:10.1159/000324128
168. Albaugh VL, Flynn CR, Cai S, Xiao Y, Tamboli RA, Abumrad NN. Early increases in bile acids post Roux-en-Y gastric bypass are driven by insulin-sensitizing, secondary bile acids. J Clin Endocrinol Metab (2015) 100(9):E1225–33. doi:10.1210/jc.2015-2467
169. Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature (2014) 509(7499):183–8. doi:10.1038/nature13135
170. McGavigan AK, Garibay D, Henseler ZM, Chen J, Bettaieb A, Haj FG, et al. TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut (2017) 66(2):226–34. doi:10.1136/gutjnl-2015-309871
171. Flynn CR, Albaugh VL, Cai S, Cheung-Flynn J, Williams PE, Brucker RM, et al. Bile diversion to the distal small intestine has comparable metabolic benefits to bariatric surgery. Nat Commun (2015) 6:7715. doi:10.1038/ncomms8715
172. Dufer M, Horth K, Wagner R, Schittenhelm B, Prowald S, Wagner TF, et al. Bile acids acutely stimulate insulin secretion of mouse beta-cells via farnesoid X receptor activation and K(ATP) channel inhibition. Diabetes (2012) 61(6):1479–89. doi:10.2337/db11-0815
173. Kohli R, Bradley D, Setchell KD, Eagon JC, Abumrad N, Klein S. Weight loss induced by Roux-en-Y gastric bypass but not laparoscopic adjustable gastric banding increases circulating bile acids. J Clin Endocrinol Metab (2013) 98(4):E708–12. doi:10.1210/jc.2012-3736
174. Ursell LK, Metcalf JL, Parfrey LW, Knight R. Defining the human microbiome. Nutr Rev (2012) 70(Suppl 1):S38–44. doi:10.1111/j.1753-4887.2012.00493.x
175. Hartstra AV, Bouter KE, Backhed F, Nieuwdorp M. Insights into the role of the microbiome in obesity and type 2 diabetes. Diabetes Care (2015) 38(1):159–65. doi:10.2337/dc14-0769
176. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature (2012) 486(7402):222–7. doi:10.1038/nature11053
177. Schulfer A, Blaser MJ. Risks of antibiotic exposures early in life on the developing microbiome. PLoS Pathog (2015) 11(7):e1004903. doi:10.1371/journal.ppat.1004903
178. Reijnders D, Goossens GH, Hermes GD, Neis EP, van der Beek CM, Most J, et al. Effects of gut microbiota manipulation by antibiotics on host metabolism in obese humans: a randomized double-blind placebo-controlled trial. Cell Metab (2016) 24(1):63–74. doi:10.1016/j.cmet.2016.06.016
179. Gomez A, Petrzelkova KJ, Burns MB, Yeoman CJ, Amato KR, Vlckova K, et al. Gut microbiome of coexisting BaAka pygmies and Bantu reflects gradients of traditional subsistence patterns. Cell Rep (2016) 14(9):2142–53. doi:10.1016/j.celrep.2016.02.013
180. El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol (2013) 11(7):497–504. doi:10.1038/nrmicro3050
181. McNeil NI. The contribution of the large intestine to energy supplies in man. Am J Clin Nutr (1984) 39(2):338–42. doi:10.1093/ajcn/39.2.338
182. Fiorucci S, Distrutti E. Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol Med (2015) 21(11):702–14. doi:10.1016/j.molmed.2015.09.001
183. Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab (2016) 24(1):41–50. doi:10.1016/j.cmet.2016.05.005
184. Kowlgi NG, Chhabra L. D-lactic acidosis: an underrecognized complication of short bowel syndrome. Gastroenterol Res Pract (2015) 2015:476215. doi:10.1155/2015/476215
185. Vella A, Farrugia G. D-lactic acidosis: pathologic consequence of saprophytism. Mayo Clin Proc (1998) 73(5):451–6. doi:10.4065/73.5.451
186. Boccia S, Torre I, Santarpia L, Iervolino C, Del Piano C, Puggina A, et al. Intestinal microbiota in adult patients with short bowel syndrome: preliminary results from a pilot study. Clin Nutr (2017) 36(6):1707–9. doi:10.1016/j.clnu.2016.09.028
187. Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu Y, et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat Med (2017) 23(7):859–68. doi:10.1038/nm.4358
188. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature (2012) 490(7418):55–60. doi:10.1038/nature11450
189. Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science (2018) 359(6380):1151–6. doi:10.1126/science.aao5774
190. Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature (2015) 528(7581):262–6. doi:10.1038/nature15766
191. Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Manneras-Holm L, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med (2017) 23(7):850–8. doi:10.1038/nm.4345
192. Gu Y, Wang X, Li J, Zhang Y, Zhong H, Liu R, et al. Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment. Nat Commun (2017) 8(1):1785. doi:10.1038/s41467-017-01682-2
Keywords: incretin hormones, bariatric surgery, gastric emptying, gastric accommodation, insulin secretion, insulin action, vagus nerve
Citation: Ma J and Vella A (2018) What Has Bariatric Surgery Taught Us About the Role of the Upper Gastrointestinal Tract in the Regulation of Postprandial Glucose Metabolism? Front. Endocrinol. 9:324. doi: 10.3389/fendo.2018.00324
Received: 19 April 2018; Accepted: 31 May 2018;
Published: 26 June 2018
Edited by:
Derek LeRoith, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Yukihiro Fujita, Asahikawa Medical University, JapanBurkhard Göke, Medical School Hamburg, Germany
Copyright: © 2018 Ma and Vella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrian Vella, dmVsbGEuYWRyaWFuQG1heW8uZWR1