 Konstantinos Lontos1
Konstantinos Lontos1 Juraj Adamik1
Juraj Adamik1 Anastasia Tsagianni2
Anastasia Tsagianni2 Deborah L. Galson1
Deborah L. Galson1 John M. Chirgwin3
John M. Chirgwin3 Attaya Suvannasankha3*
Attaya Suvannasankha3*
- 1Hematology-Oncology Division, Department of Medicine, UPMC Hillman Cancer Center, McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, PA, United States
- 2Department of Medicine, University of Pittsburgh Medical Center, Pittsburgh, PA, United States
- 3Hematology and Oncology Division, Department of Medicine, Indiana University School of Medicine, Richard L. Roudebush VA Medical Center, Indianapolis, IN, United States
Semaphorin 4D (Sema4D; CD100) is a transmembrane homodimer 150-kDa glycoprotein member of the Semaphorin family. Semaphorins were first identified as chemorepellants that guide neural axon growth. Sema4D also possesses immune regulatory activity. Recent data suggest other Sema4D functions: inactivation of platelets, stimulation of angiogenesis, and regulation of bone formation. Sema4D is a coupling factor expressed on osteoclasts that inhibits osteoblast differentiation. Blocking Sema4D may, therefore, be anabolic for bone. Sema4D and its receptor Plexin-B1 are commonly dysregulated in cancers, suggesting roles in cancer progression, invasion, tumor angiogenesis, and skeletal metastasis. This review focuses on Sema4D in bone and cancer biology and the molecular pathways involved, particularly Sema4D–Plexin-B1 signaling crosstalk between cancer cells and the bone marrow microenvironment—pertinent areas since a humanized Sema4D-neutralizing antibody is now in early phase clinical trials in cancers and neurological disorders.
Introduction
Semaphorin 4D (Sema4D; also known as CD100), is a member of class IV of the Semaphorin protein family, with established functions as an immune regulator. This review focuses on additional, emerging roles of Sema4D in bone biology and cancer bone metastases. Recent pivotal findings support the pertinence of Sema4D in bone and cancers: (1) Negishi-Koga et al. (1) identified Sema4D as a major coupling factor expressed on osteoclasts that inhibits osteoblast differentiation. They found that mice with a global knockout of Sema4d had increased bone volume. (2) Terpos et al. (2) reported increased soluble Sema4D in serum and bone marrow plasma of patients with myeloma, a bone marrow cancer with uncoupled osteoclast activation and osteoblast suppression, compared to controls. (3) Sema4D and its primary receptor Plexin-B1 are commonly overexpressed in cancers. (4) Yang et al. (3) found that shRNA knockdown of Sema4D in MDA-MB-231 breast cancer cells decreased bone metastases in a standard xenograft model. (5) A humanized antibody that neutralizes Sema4D has shown antitumor activity in animal models and is under clinical testing in early phase clinical trials (4).
SEMA4D Structure and Role in Human Physiology
Semaphorins form a highly conserved family of proteins that contain a signature amino-terminal sema domain. The semaphorin family contains more than 20 genes divided into seven classes, of which classes III–VII are expressed in vertebrates. They have diverse roles in human biology including regulation of tumor growth and metastasis, angiogenesis, axonal guidance, bone formation, tissue regeneration, and autoimmunity (5).
Sema4D belongs to class IV of the Semaphorin family. In addition to the signature sema domain, the C-terminal region of Sema4D includes an IgG-like domain, a transmembrane domain, and a short cytoplasmic tail that contains one tyrosine phosphorylation site and multiple sites for serine–threonine phosphorylation. Membrane-bound Sema4D forms a stable homodimer via a disulfide bond between cysteines 679 within the sema domain. Proteolytic shedding of Sema4D by membrane-type 1-matrix metalloproteinase (MT1-MMP/MMP14) gives rise to soluble, dimeric Sema4D (sSema4D) (6). Both membrane-bound and soluble Sema4D can activate Plexin-B1 signaling.
Sema4D is expressed by many tissues including brain, kidney, and heart. However, Sema4D knockout mice have immune defects without other obvious organ dysfunctions, suggesting that its major role is in immune regulation. Sema4D is expressed strongly by resting T cells and weakly on B and antigen-presenting cells. Expression is increased upon cellular activation (7). Engagement of Sema4D enhances its association with the membrane protein tyrosine phosphatase CD45, which is expressed broadly in hematopoietic cells (8). The complex becomes active and recruits further proteins to sustain B and T cell activation and aggregation.
Most work on Sema4D function has focused on its role as a ligand in soluble form after proteolytic shedding. Several receptors for Sema4D have been identified, including C-type lectin protein CD72, and three members of the plexin family; Plexin-B1, Plexin-B2, and Plexin-C1 (9–12). Target cells express different receptors, leading to a broad variety of potential responses to Sema4D in different tissues. CD72 is the main Sema4D receptor on immune cells, although monocytes and immature dendritic cells require Plexin-C1 and Plexin-B1, respectively. Plexins-B1 and -B2 mediate the Sema4D responses on non-immune cells. CD72 (also known as Lyb-2) is a 45-kDa type II transmembrane protein of the C-type lectin family (13) which is expressed throughout B-cell differentiation (14). The CD72 cytoplasmic domain contains two immune-receptor tyrosine-based inhibition motifs that recruit the tyrosine phosphatase SHP-1, resulting in inhibition of src family kinases and JNK and B cell inhibition (15). Sema4D engagement of CD72 triggers tyrosine dephosphorylation of CD72, leading to SHP-1 dissociation (10), thereby relieving CD72-mediated B cell inhibition. Since Sema4D and CD72 are expressed preferentially on T and B cell, respectively, their interaction couples T and B cells to dial the immune reaction up or down. Dendritic cells, macrophages, and some subpopulations of T cells express CD72 (16). Sema4D may, therefore, play an additional role in T cell communication via these other immune cells.
Plexins are transmembrane proteins with a sema ligand-binding domain in their extracellular domain. Upon ligand binding, Plexin-B1 and Plexin-B2 extracellular domains undergo proteolysis by subtilisin-like proprotein convertases to further increase their affinity for Sema4D (17). The highly conserved cytoplasmic region of plexins is devoid of enzymatic activity, but it can interact, directly or indirectly, with small G proteins for various functions (18). Transduction cascades downstream of the Sema4D/Plexin-B1 complex vary, dependent on the different membrane proteins and G proteins recruited to the complex.
Figure 1 details downstream signaling of Sema4D/Plexin-B1. Without engagement with Sema4D, the cytoplasmic tail of Plexin-B1 is in an inactive conformation. R-Ras is in a GTP-bound state and activates membrane integrin to control cellular adhesion to the extracellular matrix. Rac is not bound to Plexin-B1 and promotes activation of p21-activated kinase (PAK) to activate LIMK1 and cofilin to increase actin polymerization and microtubule assembly (19). Binding of Sema4D to Plexin-B1 alters Plexin-B1 conformation allowing recruitment of Rac1, which sequesters it from the PAK-LIMK1-cofilin signaling cascade, causing disassembly of actin fibers (20). Initiation of PlexinB1-GAP activity inhibits R-Ras-mediated integrin activation, which blocks cell adhesion and increase cell motility and invasion (21). Deactivation of R-Ras further decreases the PI3K–Akt–GSK3β pathway, which leads to deactivation of CRMP-2 and subsequent microtubule disassembly (22).
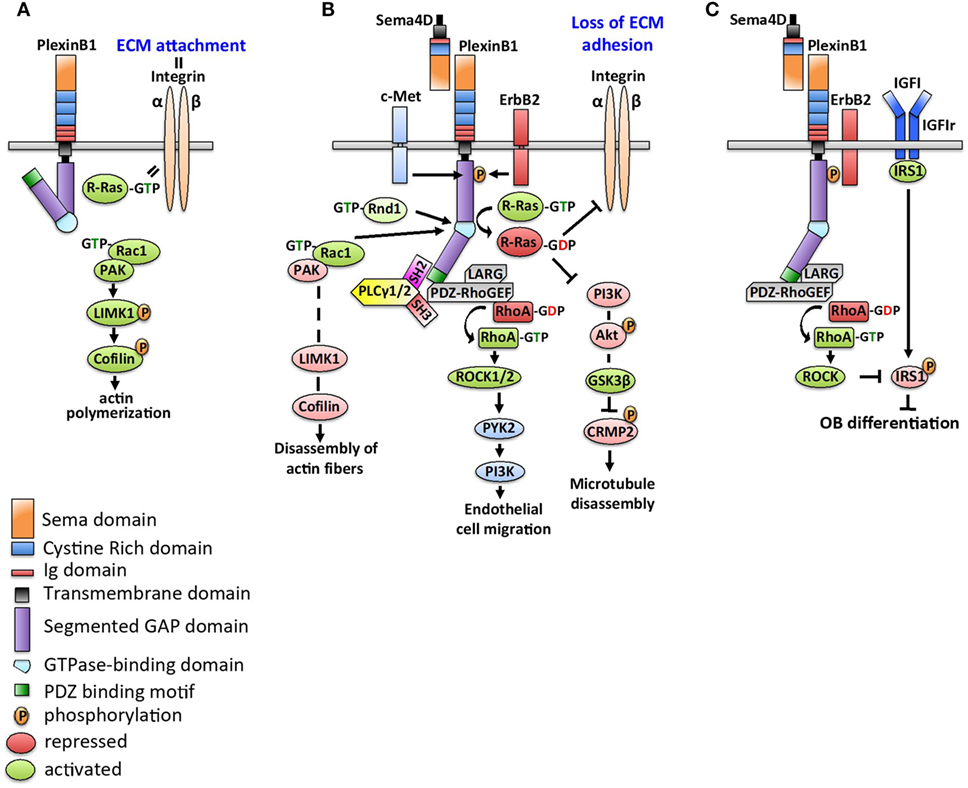
Figure 1. Schematics of Sema4D/Plexin-B1 signaling. (A) In the absence of Sema4D, the cytoplasmic tail of Plexin-B1 is in an inactive conformation. R-Ras is in a GTP-bound state to collaborate with integrin and control cellular adhesion to the extracellular matrix (ECM). With Plexin-B1 in an inactive state, Rac activates p21-activated kinase (PAK) to trigger LIMK1 signaling and downstream phosphorylation of cofilin, which results in increased actin polymerization and microtubule assembly. (B) Sema4D binding to Plexin-B1 alters the Plexin-B1 conformation, recruits Rac1 to the complex, and inhibits activation of the PAK–LIMK1–cofilin signaling cascade. In addition, R-Ras recruitment to the Sema4D/Plexin-B1 complex inhibits its ability to regulate integrin-mediated activation, resulting in increased cell motility. Deactivation of R-Ras further decreases PI3K-Akt GSK3β pathway, which leads to deactivation of CRMP-2 and subsequent microtubule disassembly. In an ErbB2-dependent signaling step, PLCγ1/2 is recruited to Plexin-B1 via its SH2 domain. Structural interaction of the PLCγ1/2 SH3 domain triggers PDZ-RhoGEF/LARG complex, resulting in RhoA activation. Active GTP-RhoA acts on Raf and Rho-associated kinases (ROCK1/2), which in turn stimulate PYK2 to induce cell invasiveness and migration. (C) Sema4D-Plexin-B1 signaling inhibits osteoblast differentiation. Plexin-B1/ErbB2-dependent RhoA activation stimulates activation of downstream kinase ROCK, which phosphorylates the insulin receptor substrate 1 (IRS1). This causes suppression of insulin like growth factor (IGF1)-dependent signaling and blocks osteoblast differentiation.
Whether Sema4D/Plexin-B1 complexes activate or inhibit downstream GTPases also depends on their interactions with specific receptor tyrosine kinases: ErbB-2 or c-Met (23). In ErbB2 expressing cells, binding of Sema4D to Plexin-B1 activates the intrinsic tyrosine kinase activity of ErbB-2, resulting in the phosphorylation of both Plexin-B1 and ErbB-2 (24), generating docking sites on Plexin-B1 for the SH2 domains of 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-1/2 (PLCγ1/2) (25). The SH3 domain of the recruited PLCγ1/2 activates the guanine nucleotide-exchange factors (GEFs) LARG (leukemia-associated RhoGEF) and PRG (PDZ-RhoGEF) that constitutively bind to the PDZ binding site of the carboxyl-terminal sequence of Plexin-B1 (26). Activated RhoGEFs LARG and PRG mediate activation of the small GTPase RhoA, which cooperates with Ras to activate the serine/threonine kinases Raf and Rho-associated kinases (e.g., ROCK1/2) to stimulate various pathways, including mitogen-activated phosphokinases (MAPKs) ERK1/2 and p38, protein kinases (PI3/Akt—phosphatidylinositol 3′-kinase) (27). Downstream activation of MAPKs and PI3K control dendritic and axonal morphogenesis by differentially regulating branching and extension (27). On the other hand, Sema4D/Plexin-B1 can interact with Rnd1 to downregulate the GTPase activity of R-Ras, inducing growth cone collapse in hippocampal neurons (28). Plexin-B1 also binds and competes for activated Rho GTPase Rac, thus preventing Rac from activating its downstream effector PAK to initiate actin polymerization. In endothelial cells, Sema4D/Plexin-B1 signals stimulate PI3K-Akt to activate PYK2 and Src to control endothelial cell migration (29). c-Met (HGF receptor tyrosine kinase) and macrophage stimulating protein (MSP, encoded by the Ron gene) are members of the Scatter Factor Receptors family. They share structural homology with Plexin-B1 (30, 31). In cells with c-Met expression but absent ErbB2, Sema4D/Plexin-B1 recruits and activates c-Met or Ron kinases to regulate cell motility and invasiveness. In normal fibroblasts, phosphorylation of c-Met creates a docking site for growth factor receptor bound-2 (Grb2), which then interacts with p190 RhoGAP and inactivates RhoA, causing inhibition of fibroblast motility (32).
SEMA4D in Bone Physiology
Successful crystallization of Sema4D and solution of its structure led to a recognition of the homology between the Sema4D homodimer and the αVβ3 integrin heterodimer, the first clue to a role for Sema4D in bone biology (33). αVβ3 integrin is an osteoclast regulator, and β3 integrin knockout mice become progressively osteosclerotic with age due to dysfunctional osteoclasts that fail to polarize correctly and display abnormal ruffled membranes (34).
Two groups reported an increased bone mass in Sema4D knockout animals and the expression of Sema4D in osteoclasts but not osteoblasts (1, 35). Negishi-Koga and coworkers comprehensively surveyed osteoclasts and osteoblasts for expression of candidate molecules to couple bone formation to resorption, including the members of the ephrin, netrin, semaphorin, and slit gene families. They found that only Sema4D was highly expressed in RANKL-activated osteoclasts. Utilizing a soluble Fc receptor-Sema4D fusion protein, they evaluated directly the effects of the Sema4D extracellular domain on osteoblasts. Fc-Sema4D inhibited the osteoblast differentiation markers alkaline phosphatase and osteocalcin, as well as the formation of mineralized nodules in culture, without a change in osteoblast proliferation. Co-immunoprecipitation confirmed a complex of Fc-Sema4D with Plexin-B1, which is highly expressed in osteoblasts. This complex leads to ErbB2 phosphorylation and downstream activation of RhoA. Plexin−/− animals, as well as mice expressing an osteoblast-targeted dominant negative RhoA, had a high bone mass due to enhanced osteoblastic bone formation, recapitulating the bone phenotype of the Sema4d−/− mice. A recent study used optoPlexin (optogenetic activation of Plexin-B1) in osteoblasts to show that Plexin-B1 activation results in retraction of the leading edge and induces distal membrane protrusions, causing the osteoblasts to migrate. Thus, osteoclast-produced Sema4D may cause repulsion of osteoblasts via activation of both the RhoA and rasGTP pathways (36). Overall, these findings support the function of the Sema4D/Plexin-B1/RhoA axis in osteoblast inhibition by osteoclasts (1). However, Sema4D’s regulation of bone mass may be more complicated. Dacquin et al (35) noted that the increased bone mass phenotype in Sema4d−/− mice primarily occurred in mature female mice. The increased bone mass phenotype in Sema4d−/− mice was not reversed after a transplantation of bone marrow cells from wild-type mice, as a source of new osteoclasts. Sema4d−/− mice have impaired ovarian function, small litter size, and decreased hypothalamic gonadotropin-hormone releasing hormone. The authors propose an indirect mechanism by which Sema4D regulates bone mass via the hypothalamic–pituitary–ovarian axis (35).
Serum Sema4D has been explored as a biomarker in osteoporosis, with conflicting results to date, likely reflecting different patient populations, the non-randomized nature of the trials and different time points for Sema4D analysis. In postmenopausal women, serum Sema4D was higher among those with osteoporosis (37). However, in an open label study of osteoporotic women receiving the osteoclast-targeting agents zoledronic acid or RANKL monoclonal antibody denosumab or the bone-anabolic agent teriparatide (PTH1-34), serum Sema4D decreased after 3 months of teriparatide treatment, while it increased with denosumab and zoledronic acid (38). There was no attempt to correlate the Sema4D levels with degree of osteoporosis, and serum Sema4D was assayed at only one time point, not allowing a determination of kinetics, which may differ among treatment groups.
Congenital defects due to mutation or loss of Sema4D have been little studied in humans. Segmental copy number loss in the region of the Sema4D gene was seen in one third of patients with acetabular dysplasia, which increases risk of osteoarthritis, but a causal relationship was not explored (39).
Sema4D has been implicated in bone and joint inflammation. In rheumatoid arthritis (RA), Sema4D was elevated in both serum and synovial fluid from RA patients, and disease activity markers were correlated with serum Sema4D levels. Sema4D-expressing cells also accumulated in RA synovium, and sSema4D-induced tumor necrosis factor α (TNFα) and interleukin-6 (IL-6) production from CD14+ monocytes (40). Movila and group explored treatment-related osteonecrosis of the jaw and showed increased Sema4D-expressing γδ-T cells in bone lesions, which were decreased by anti-Sema4D antibody (41). In osteoarthritis, bone tissue had low Plexin-B1 expression compared to age- and gender-matched cadaveric control bones, indicating that the loss of Sema4D/Plexin-B1 inhibition of osteoblast activity may lead to an increased bone volume fraction and decreased bone matrix mineralization, contributing to osteoarthritis (42).
Elevated Sema4D is observed in diseases unrelated to the musculoskeletal systems, including hemorrhagic fever with renal syndrome (43) and autoimmune diseases (44). Inflammation and increased shedding from peripheral blood mononuclear cells are possible sources. Serum Sema4D is also increased in non-inflammatory condition such as atrial fibrillation (45). The value of sSema4D as a biomarker in some of these diseases may warrant further investigation.
SEMA4D in Cancer
While other semaphorins such as Sema3B and 3F are tumor suppressors, Sema4D promotes tumor growth (46, 47). Sema4D and Plexin-B1 are abnormally expressed in various cancers and have been associated with invasive phenotypes and poor prognosis. The mechanisms by which Sema4D confers these features are complex and involve both tumor cells and their microenvironment. While not directly linked to malignant transformation, Sema4D/Plexin-B1 signaling contributes to many critical aspects of cancer progression, including proliferation, invasion, angiogenesis, immune escape, and metastasis. We summarize the role of Sema4D/Plexin-B1 in cancers based on overexpression of Sema4D or Plexin-B1 by the cancer cells or by other cells in the tumor microenvironment including immune cells, and the role of Sema4D/Plexin-B1 in distant metastasis.
Because of its known role in lymphoid cells, Sema4D expression in cancer was first evaluated in lymphoma and lymphoblastic leukemia. T cell lymphomas universally express Sema4D, while only a minority of B cell lymphomas are Sema4D positive (48), paralleling the expression of Sema4D by normal T and B cells. In chronic lymphocytic leukemia (CLL), CD38 is a known negative predictor of survival. CD38 binds to (49) stromal cells expressing CD31, leading to relocalization of Sema4D, facilitating its binding to Plexin-B1 on bone marrow cells. The increased expression of Plexin-B1 in stromal cells, follicular dendritic cells, and activated T-cells, enhances the complex interplay of CD38/CD31 and Sema4D/Plexin-B1 to sustain CLL growth (50, 51).
Sema4D is highly expressed by many solid tumors including prostate, glioma, lung, ovarian, sarcoma, and cutaneous squamous cell carcinoma. Expression is correlated with tumor aggressiveness and poor prognosis, but controversial data exist for some tumors. In an array of 888 genes, Sema4D mRNA was highest in early stage breast cancer compared to normal tissue and downregulated in advanced disease (52). Jiang and coworkers confirmed high Sema4D protein in breast cancer cell lines compared to normal breast epithelial cells. In addition, knockdown of Sema4D by shRNA inhibit breast cancer proliferation and tumor growth in xenografts (49). Malik et al., however, showed an opposite result of decreased Sema4D, Plexin-B1 and -B2 protein in primary breast tumors of patients who subsequently developed local recurrence, compared to the patients who remained disease-free (53). These studies used whole tumor tissues and may be confounded by different cell types within the tissue, since other cells in the microenvironment, including endothelial cells and macrophages, also express Sema4D. In whole-blood RNA, Sema4D was identified as one of six RNAs strongly predicting shorter survival among patients with castrate-resistant prostate cancer (54). Most data on Sema4D and clinical outcomes are based on small sample sizes with subjects receiving different treatments and not controlled for other prognostic markers. Prognostic value of Sema4D requires further validation in larger cohorts of patients, controlled for treatment types and other variables.
Overexpression of Plexin-B1 has been reported in skin, prostate and pancreatic cancers, and sarcoma (55–57). It correlates with lymph node metastasis, distant metastasis, and poor prognosis in patients with pancreas cancer (56). Plexin-B1 activation increases phosphorylation and translocation into the nucleus of the androgen receptor (AR), leading to activation of AR-regulated genes, which could play a role in castration resistance in prostate cancer (58). However, Rody et al. showed a rather opposite finding in breast cancer, in which loss of Plexin-B1 identifies a subgroup of estrogen receptor expressing breast cancer with high proliferative rate and hormone resistance (59). Cancer cell lines that express higher levels of Plexin-B1 exhibit increased perineural invasion. The mechanism is proposed to be the attraction of the cells due to Sema4D production from the nerves. Interestingly, higher nerve density in tumors expressing Sema4D (60) suggests that these tumors may use nerve-secreted factors for growth, pointing to a possible role of Sema4D in cancer pain.
While no Sema4D mutation has been reported in human cancers, Plexin-B1 mutations and copy number changes are noted commonly in various cancers, including melanomas, pancreas, breast, and prostate cancers (58, 61, 62). Thirteen somatic missense mutations in the cytoplasmic domain of Plexin-B1 were found in 46% of prostate cancers. Mutational hotspots mapped to the Rho GTPase binding domain in the cytoplasmic region of the receptor, causing loss of Rac and R-Ras binding and R-RasGAP activity. This resulted in an increase in cell motility, invasion, and adhesion, and could explain the metastatic phenotype (61). In some cancers, however, Plexin-B1 acts as a tumor suppressor. For example, Plexin-B1 is lost in deep and metastatic melanomas. Introducing Plexin-B1 into melanoma cells suppresses c-Met and hence proliferative responses to HGF. Increased Plexin-B1 confers resistance to cisplatin (63). A similar finding was noted in clear cell carcinoma (64). Sema4D/Plexin-B1 responses may vary among different cell lines of the same tumor type. Sema4D/Plexin-B1 increased the proliferative and invasive potential of LNCaP prostate cancer cells through the activation of ErbB2 and Akt, but decreased the motility and proliferation of PC3 prostate cells (65).
Different phenotypes of Sema4D/Plexin-B1-expressing tumors may depend on the expression of its partner proteins ErbB2 and c-Met. Since c-Met is one of the most commonly deregulated oncogene in cancers, its collaboration with Sema4D/Plexin-B1 is an alternative pathway to promote tumor invasion (66). Constitutive activation of Met in tumor cells with high Plexin-B1 can occur in the absence of Sema4D (66). T lymphoma invasion and metastasis 1 (Tiam1) is another Rac-specific guanine nucleotide-exchange factor that is activated by Sema4D/Plexin-B1 to stimulate Rac and promote proliferation, invasion, and metastasis in oral squamous cell carcinoma (67). In cancer cells that express both Sema4D and Plexin-B1, the pair could function in an autocrine/paracrine manner, although this requires future study.
In addition to direct proliferative actions in cancer cells, Sema4D/Plexin-B1 abnormalities within the tumor niche support cancer progression by promoting angiogenesis. Sema4D/Plexin-B1 function in angiogenesis was first described by Basile and coworkers (68), who showed that recombinant Sema4D-induced chemotaxis of endothelial cells, in vitro tubulogenesis and enhanced blood vessel formation in an in vivo mouse model. Sema4D/Plexin-B1 phosphorylation of c-Met promotes angiogenesis in a non-redundant manner from HGF (69). Sema4D can also stimulate angiogenesis via a c-Met-independent pathway through recruitment of PDZ-RhoGEF and LARG to the Sema4D/Plexin-B1 complex leading to Rho pathway activation, followed by downstream AKT and NF-κB activation and increased expression of proangiogenic IL-8 (70). Hypoxia induces Sema4D in a HIF-1-dependent manner. Sema4D then cooperates with VEGF to promote tumor growth and vascularity (71, 72). In addition, tumor-secreted Sema4D increases endothelial expression of platelet-derived growth factor-B and angiopoietin-like protein 4, which promote endothelial proliferation and vascular permeability (73). Lentiviral overexpression of Sema4D in colorectal cancer cell lines caused a proangiogenic response regardless of VEGF status (74). Sema4D may also be a biomarker for tumor angiogenesis, since its expression in ovarian cancer correlates with HIF-1, VEGF, and poor prognosis. Sema4D inhibition causes dissociation of endothelial cells from pericytes, a crucial step for successful antiangiogenic therapy (75). Both VEGF and Sema4D may cooperate in a multi-step process to reorganize the vasculature within the malignant niche. Tumor cells increase their Sema4D expression as an escape mechanism from anti-VEGF treatment (75). Concurrent targeting of VEGF and Sema4D may have additive or synergistic antiangiogenic effects. Sema4D may also support lymphangiogenesis, since Sema4D targeting with neutralizing antibody or shRNA suppressed VEGF-C and VEGF-D, key factors for lymphangiogenesis (76).
The role of Sema4D in immune regulation supports the importance of dysregulated Sema4D in immune escape of cancer cells. In a head and neck cancer model, tumor-secreted Sema4D promoted the expansion of myeloid-derived suppressor cells, which inhibit T-cell functions (77). Sema4D affects both the activities of immune cells and their recruitment to the tumor microenvironment. Delaire and coworkers noted that Sema4D inhibited both spontaneous and chemokine-induced migration of human monocytes (78). Strong expression of Sema4D at the invasive margins of actively growing tumors changed the infiltration and distribution of leukocytes within the tumor microenvironment. Neutralization of Sema4D by blocking antibodies disrupted this gradient of expression and enhanced recruitment of activated monocytes and lymphocytes into the tumor. This shifted the balance of cells and cytokines in a pro-inflammatory and antitumorigenic direction and was associated with durable tumor rejection in murine Colon26 and ERBB2+ mammary carcinoma models (4). These functions are at odds with the known roles of Sema4D in T and B cell activation. The dual nature of Sema4D between pro and antitumor action may depend on subsets of immune cells in the tumor niche, which in turn may depend on the plasticity of macrophages and T cells within the tumor. Classical activation of macrophages with interferon γ (M1) promotes the differentiation of cytotoxic T cells, which can improve antigen phagocytosis. However, alternative pathway of macrophage activation by IL-4, IL-14, or LPS gives rise to M2 macrophages. Tumor-associated monocytes are M2 and communicate effectively with regulatory T cells (Treg) to suppress antigen recognition and promote an inflammatory tumor microenvironment, angiogenesis, and tumor progression (79). In tumors with high M2 macrophages and Tregs, Sema4D may contribute to immune suppression, even though in normal physiology, Sema4D is required for T and B cell function.
Sema4D may promote metastasis at distant sites. Many cancer cells, including head and neck squamous cell carcinoma lines, express the membrane-tethered collagenase, MT1-MMP which cleaves Sema4D. sSema4D could thus promote angiogenesis and cell migration both locally and at distant sites, thereby promoting metastasis (80).
SEMA4D in the Bone Marrow Metastatic Niche
The involvement of Sema4D in bone biology and cancer progression suggests a role in bone metastasis. Bone is the most common site of distant metastases for prostate and breast cancers. Bone metastases are driven by complex interactions between cancer cells, bone marrow cells, and bone cells, often leading to increased osteoclast and suppressed osteoblast activities. Even in osteosclerotic metastasis, occurring in prostate and some breast cancers, the newly formed bone is poorly organized, and osteoclasts remain activated, leading to loss of bone quality. Osteolysis with osteoblast suppression is a hallmark of multiple myeloma, an incurable blood cancer that originates within bone (81, 82). Current treatments are limited to osteoclast-targeting agents, which provide palliative benefit with marginal effect on tumor control. Zoledronic acid modestly prevents bone metastases in breast and prostate cancer xenograft models, but its benefit as a bone metastasis prevention is limited to postmenopausal women (83). No survival advantage was seen in premenopausal women or men with prostate cancers. This evidence suggests that osteoclast targeting alone is inadequate for tumor control.
Sema4D is a coupling molecule of osteoclasts and osteoblasts (1). In the tumor niche, Sema4D from the tumor cells and activated osteoclasts inhibits osteoblast differentiation, while inducing IL-8 secretion to further promote osteoclast proliferation and activity (70). Because soluble Sema4D can reach distant bone sites, it could hypothetically prime the bone niche to support future metastasis. Since tumor cells with high Sema4D also have high motility and invasiveness and Sema4D promotes angiogenesis, Sema4D could contribute to metastasis at both at the primary tumor site and distant sites. This notion is supported by a decrease in skeletal metastasis when Sema4D was knocked down using shRNA in the MDA-MB-231 breast cancer model (3).
Terpos and coworkers (2) showed increased Sema4D in peripheral blood and bone marrow plasma from myeloma patients compared to controls. We have shown that myeloma cell lines and primary myeloma cells express Sema4D at higher levels than MDA-MB-131 cells. In addition, we found that coculture of myeloma cells with bone increased expression of Sema4D by both tumor cells and bone (84). Myeloma cells, as well as breast cancers, express MMP14 (MT1-MMP), which releases membrane-bound Sema4D by proteolytic cleavage (85). Sema4D can promote angiogenesis, which is required for myeloma progression. Sema4D is, therefore, a potential target in myeloma, as well as in cancers that metastasize to the skeleton.
Strategy for SEMA4D/Plexin-B1 Targeting
VX15/2503 is the first and only currently available humanized Sema4D blocking antibody in clinical testing. The antibody was generated in Sema4D−/− mice by using a panel of SEMA4D-specific hybridomas that react with murine, primate, and human SEMA4D (86). VX15/2503 bound with high affinity (1–5 nmol/L) to Sema4D and achieved complete Sema4D blockade in animal models, and its activity was subsequently confirmed in humans. No dose-limiting toxicity or maximal tolerated dose was observed at the dose required for complete Sema4D blockade in phase I clinical trials of multiple sclerosis and refractory solid malignancies (87, 88).
In the phase I study of 42 patients with refractory cancers receiving dose escalation of VX15/2503, one patient (2.4%) achieved partial response and 27 patients (64.3%) achieved long duration stable disease. Subjects with elevated baseline B and T lymphocytes exhibited longer progression-free survival, suggesting involvement of immune-mediated antitumor activity. Bone parameters and development of metastases were not endpoints. Tumor response was based on reduction in tumor size; therefore, patients with isolated bone metastases without other measurable tumor masses were not included.
The large, planar binding interface of the Sema4D/Plexin-B1 interaction makes it challenging to target with small molecules. Matsunaga and coworkers discovered a macrocyclic peptide, PB1m6, which binds Plexin-B1 with high affinity and specifically inhibits binding of the physiological ligand Sema4D and completely suppressed Sema4D-induced cell collapse in vitro (89). No data for this peptide in vivo or in cancer models are currently available.
Because of the central role of Sema4D in osteoblast inhibition, Zhang and group developed bone-specific drug delivery of Sema4D siRNA using N-(2-hydroxypropyl) methacrylamide copolymers with d-aspartic acid octapeptide (90). They showed that in an osteoporotic animal model induced by ovariectomy, weekly intravenous injections of this compound decreased osteoclast Sema4D expression and increased osteoblast differentiation. Treated animals showed higher femoral bone volume both in prevention and treatment models. Similar reduction of bone loss was seen in alveolar bone of the mandibles (91). Clinical testing of this molecule in patients has not been reported, but the study provides a proof of benefit for Sema4D targeting in bone.
While blocking the Sema4D/Plexin-B1 complex is of potential benefit in cancers and neuroinflammatory diseases, some cancers are growth inhibited by Sema4D signaling. In an acute myeloid leukemia cell line Kasumi-1, Sema4D binding of CD72, its preferred receptor in immune cells, leads to inhibition of growth and cell death, as a result of phosphorylation of CD72, the formation of the CD72–SHP-1 complex and dephosphorylation of src family kinases and JNK (15). Future development of Sema4D targeting should take into account the cellular context where Sema4D exerts its function.
Discussion
We highlight Sema4D as a novel regulator of bone homeostasis. Sema4D also supports crucial steps in tumor progression, ranging from invasion, migration, angiogenesis, and immune suppression, to the pathological alteration of the tumor niche to support metastasis. Targeting Sema4D in the clinic has become practical with the development of a specific neutralizing antibody, which has low toxicity and an impressive response rate in early clinical trials of patients with refractory cancers. Because of the potential role of Sema4D in bone metastasis, this molecule should be further explored in these specific disease groups, and bone metastases should be included in clinical endpoints. Our preliminary data also support the potential of Sema4D blockade in multiple myeloma. Baseline B and T cell profiles have been correlated to response in a preliminary cancer clinical trial and should be further developed as a screening tool to identify patients likely to respond, or as a biomarker of response. In addition, Sema4D targeting may have synergistic antitumor effects when combined with immunomodulatory agents, warranting further study.
A major open question is whether Sema4D from non-osteoclast sources plays a significant role to inhibit osteoblast activity in cancer/bone diseases. Sema4D is expressed on tumor cells that colonize bone, while cells of the microenvironment other than osteoclasts may also express Sema4D. Although the extracellular domain can be proteolytically shed, it seems unlikely that local sSema4D concentrations are sufficient to activate osteoblast Plexin-B1. Both Negishi-Koga et al. (1) and Yang et al. (3) showed osteoblast suppression with micrograms per milliliter amounts of sSema4D:Fc fusions, orders of magnitude higher than those found in bone marrow plasma from multiple myeloma by Terpos et al. (2). Non-osteoblast and tumor sources of Sema4D could still inhibit osteoblast activity by direct cell:cell contact. Direct evidence for such actions is lacking; they may occur only in the context of co-receptors specific to the osteoclast. Patient data from clinical trials with Sema4D-neutralizing antibody could provide answers to these questions. Patients receiving antibody treatment should have increased bone mineral density. If the osteoclast Sema4D axis is the only significant contributor to osteoblast suppression, then there may be little bone effect of treatment in patients also receiving anti-osteoclast drugs (bisphosphonates or denosumab), which are the standard of care for cancer bone diseases.
Author Contributions
All authors contributed to the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Work was supported by the VA merit award CX000977 and pilot research funding of the Indiana University Simon Cancer Center (AS and JC), NIH/NIAMS R01 AR059679 and Funding from the Hematology-Oncology Division, Department of Medicine, University of Pittsburgh (DG) and the American Society of Hematology HONORS award (KL).
References
1. Negishi-Koga T, Shinohara M, Komatsu N, Bito H, Kodama T, Friedel RH, et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med (2011) 17(11):1473–80. doi:10.1038/nm.2489
2. Terpos E, Kastritis E, Bagratuni T, Christoulas D, Papatheodorou A, Kanellias N, et al. Semaphorin-4D and plexin-B1 are elevated in multiple myeloma microenvironment and possibly contribute in the development of lytic bone disease. Blood (2012) 120:1819.
3. Yang YH, Buhamrah A, Schneider A, Lin YL, Zhou H, Bugshan A, et al. Semaphorin 4D promotes skeletal metastasis in breast cancer. PLoS One (2016) 11(2):e0150151. doi:10.1371/journal.pone.0150151
4. Evans EE, Jonason AS Jr, Bussler H, Torno S, Veeraraghavan J, Reilly C, et al. Antibody blockade of semaphorin 4D promotes immune infiltration into tumor and enhances response to other immunomodulatory therapies. Cancer Immunol Res (2015) 3(6):689–701. doi:10.1158/2326-6066.CIR-14-0171
5. Worzfeld T, Offermanns S. Semaphorins and plexins as therapeutic targets. Nat Rev Drug Discov (2014) 13(8):603–21. doi:10.1038/nrd4337
6. Elhabazi A, Delaire S, Bensussan A, Boumsell L, Bismuth G. Biological activity of soluble CD100. I. The extracellular region of CD100 is released from the surface of T lymphocytes by regulated proteolysis. J Immunol (2001) 166(7):4341–7. doi:10.4049/jimmunol.166.7.4341
7. Bougeret C, Mansur IG, Dastot H, Schmid M, Mahouy G, Bensussan A, et al. Increased surface expression of a newly identified 150-kDa dimer early after human T lymphocyte activation. J Immunol (1992) 148(2):318–23.
8. Delaire S, Elhabazi A, Bensussan A, Boumsell L. CD100 is a leukocyte semaphorin. Cell Mol Life Sci (1998) 54(11):1265–76. doi:10.1007/s000180050252
9. Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, et al. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell (1999) 99(1):71–80. doi:10.1016/S0092-8674(00)80063-X
10. Kumanogoh A, Watanabe C, Lee I, Wang X, Shi W, Araki H, et al. Identification of CD72 as a lymphocyte receptor for the class IV semaphorin CD100: a novel mechanism for regulating B cell signaling. Immunity (2000) 13(5):621–31. doi:10.1016/S1074-7613(00)00062-5
11. Chabbert-de Ponnat I, Marie-Cardine A, Pasterkamp RJ, Schiavon V, Tamagnone L, Thomasset N, et al. Soluble CD100 functions on human monocytes and immature dendritic cells require plexin C1 and plexin B1, respectively. Int Immunol (2005) 17(4):439–47. doi:10.1093/intimm/dxh224
12. Luque MC, Gutierrez PS, Debbas V, Kalil J, Stolf BS. CD100 and plexins B2 and B1 mediate monocyte-endothelial cell adhesion and might take part in atherogenesis. Mol Immunol (2015) 67(2 Pt B):559–67. doi:10.1016/j.molimm.2015.07.028
13. Biancone L, Bowen MA, Lim A, Aruffo A, Andres G, Stamenkovic I. Identification of a novel inducible cell-surface ligand of CD5 on activated lymphocytes. J Exp Med (1996) 184(3):811–9. doi:10.1084/jem.184.3.811
14. Gordon J. B-cell signalling via the C-type lectins CD23 and CD72. Immunol Today (1994) 15(9):411–7. doi:10.1016/0167-5699(94)90270-4
15. Kataoka TR, Kumanogoh A, Hirata M, Moriyoshi K, Ueshima C, Kawahara M, et al. CD72 regulates the growth of KIT-mutated leukemia cell line Kasumi-1. Sci Rep (2013) 3:2861. doi:10.1038/srep02861
16. Kumanogoh A, Kikutani H. Biological functions and signaling of a transmembrane semaphorin, CD100/Sema4D. Cell Mol Life Sci (2004) 61(3):292–300. doi:10.1007/s00018-003-3257-7
17. Artigiani S, Barberis D, Fazzari P, Longati P, Angelini P, van de Loo JW, et al. Functional regulation of semaphorin receptors by proprotein convertases. J Biol Chem (2003) 278(12):10094–101. doi:10.1074/jbc.M210156200
18. Gherardi E, Love CA, Esnouf RM, Jones EY. The sema domain. Curr Opin Struct Biol (2004) 14(6):669–78. doi:10.1016/j.sbi.2004.10.010
19. Wells CM, Jones GE. The emerging importance of group II PAKs. Biochem J (2010) 425(3):465–73. doi:10.1042/BJ20091173
20. Vikis HG, Li W, Guan KL. The plexin-B1/Rac interaction inhibits PAK activation and enhances Sema4D ligand binding. Genes Dev (2002) 16(7):836–45. doi:10.1101/gad.966402
21. Oinuma I, Katoh H, Negishi M. Semaphorin 4D/Plexin-B1-mediated R-Ras GAP activity inhibits cell migration by regulating beta(1) integrin activity. J Cell Biol (2006) 173(4):601–13. doi:10.1083/jcb.200508204
22. Ito Y, Oinuma I, Katoh H, Kaibuchi K, Negishi M. Sema4D/plexin-B1 activates GSK-3beta through R-Ras GAP activity, inducing growth cone collapse. EMBO Rep (2006) 7(7):704–9. doi:10.1038/sj.embor.7400737
23. Swiercz JM, Worzfeld T, Offermanns S. ErbB-2 and met reciprocally regulate cellular signaling via plexin-B1. J Biol Chem (2008) 283(4):1893–901. doi:10.1074/jbc.M706822200
24. Swiercz JM, Kuner R, Offermanns S. Plexin-B1/RhoGEF-mediated RhoA activation involves the receptor tyrosine kinase ErbB-2. J Cell Biol (2004) 165(6):869–80. doi:10.1083/jcb.200312094
25. Swiercz JM, Worzfeld T, Offermanns S. Semaphorin 4D signaling requires the recruitment of phospholipase C gamma into the plexin-B1 receptor complex. Mol Cell Biol (2009) 29(23):6321–34. doi:10.1128/MCB.00103-09
26. Aurandt J, Vikis HG, Gutkind JS, Ahn N, Guan KL. The semaphorin receptor plexin-B1 signals through a direct interaction with the Rho-specific nucleotide exchange factor, LARG. Proc Natl Acad Sci U S A (2002) 99(19):12085–90. doi:10.1073/pnas.142433199
27. Aurandt J, Li W, Guan KL. Semaphorin 4D activates the MAPK pathway downstream of plexin-B1. Biochem J (2006) 394(Pt 2):459–64. doi:10.1042/BJ20051123
28. Oinuma I, Ishikawa Y, Katoh H, Negishi M. The semaphorin 4D receptor plexin-B1 is a GTPase activating protein for R-Ras. Science (2004) 305(5685):862–5. doi:10.1126/science.1097545
29. Basile JR, Afkhami T, Gutkind JS. Semaphorin 4D/plexin-B1 induces endothelial cell migration through the activation of PYK2, Src, and the phosphatidylinositol 3-kinase-Akt pathway. Mol Cell Biol (2005) 25(16):6889–98. doi:10.1128/MCB.25.16.6889-6898.2005
30. Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science (1991) 251(4995):802–4. doi:10.1126/science.1846706
31. Gaudino G, Follenzi A, Naldini L, Collesi C, Santoro M, Gallo KA, et al. RON is a heterodimeric tyrosine kinase receptor activated by the HGF homologue MSP. EMBO J (1994) 13(15):3524–32.
32. Sun T, Krishnan R, Swiercz JM. Grb2 mediates semaphorin-4D-dependent RhoA inactivation. J Cell Sci (2012) 125(Pt 15):3557–67. doi:10.1242/jcs.101063
33. Love CA, Harlos K, Mavaddat N, Davis SJ, Stuart DI, Jones EY, et al. The ligand-binding face of the semaphorins revealed by the high-resolution crystal structure of SEMA4D. Nat Struct Biol (2003) 10(10):843–8. doi:10.1038/nsb977
34. McHugh KP, Hodivala-Dilke K, Zheng MH, Namba N, Lam J, Novack D, et al. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest (2000) 105(4):433–40. doi:10.1172/JCI8905
35. Dacquin R, Domenget C, Kumanogoh A, Kikutani H, Jurdic P, Machuca-Gayet I. Control of bone resorption by semaphorin 4D is dependent on ovarian function. PLoS One (2011) 6(10):e26627. doi:10.1371/journal.pone.0026627
36. Deb Roy A, Yin T, Choudhary S, Rodionov V, Pilbeam CC, Wu YI. Optogenetic activation of Plexin-B1 reveals contact repulsion between osteoclasts and osteoblasts. Nat Commun (2017) 8:15831. doi:10.1038/ncomms15831
37. Zhang Y, Feng E, Xu Y, Wang W, Zhang T, Xiao L, et al. Serum Sema4D levels are associated with lumbar spine bone mineral density and bone turnover markers in patients with postmenopausal osteoporosis. Int J Clin Exp Med (2015) 8(9):16352–7.
38. Anastasilakis AD, Polyzos SA, Makras P, Gkiomisi A, Sakellariou G, Savvidis M, et al. Circulating semaphorin-4D and plexin-B1 levels in postmenopausal women with low bone mass: the 3-month effect of zoledronic acid, denosumab or teriparatide treatment. Expert Opin Ther Targets (2015) 19(3):299–306. doi:10.1517/14728222.2014.983078
39. Sekimoto T, Ishii M, Emi M, Kurogi S, Funamoto T, Hamada H, et al. Segmental copy number loss in the region of semaphorin 4D gene in patients with acetabular dysplasia. J Orthop Res (2013) 31(6):957–61. doi:10.1002/jor.22310
40. Yoshida Y, Ogata A, Kang S, Ebina K, Shi K, Nojima S, et al. Semaphorin 4D contributes to rheumatoid arthritis by inducing inflammatory cytokine production: pathogenic and therapeutic implications. Arthritis Rheumatol (2015) 67(6):1481–90. doi:10.1002/art.39086
41. Movila A, Mawardi H, Nishimura K, Kiyama T, Egashira K, Kim JY, et al. Possible pathogenic engagement of soluble semaphorin 4D produced by gammadelta T cells in medication-related osteonecrosis of the jaw (MRONJ). Biochem Biophys Res Commun (2016) 480(1):42–7. doi:10.1016/j.bbrc.2016.10.012
42. Hopwood B, Gronthos S, Kuliwaba JS, Robey PG, Findlay DM, Fazzalari NL. Identification of differentially expressed genes between osteoarthritic and normal trabecular bone from the intertrochanteric region of the proximal femur using cDNA microarray analysis. Bone (2005) 36(4):635–44. doi:10.1016/j.bone.2005.02.003
43. Liu B, Ma Y, Yi J, Xu Z, Zhang YS, Zhang C, et al. Elevated plasma soluble Sema4D/CD100 levels are associated with disease severity in patients of hemorrhagic fever with renal syndrome. PLoS One (2013) 8(9):e73958. doi:10.1371/journal.pone.0073958
44. Wang X, Kumanogoh A, Watanabe C, Shi W, Yoshida K, Kikutani H. Functional soluble CD100/Sema4D released from activated lymphocytes: possible role in normal and pathologic immune responses. Blood (2001) 97(11):3498–504. doi:10.1182/blood.V97.11.3498
45. Xiang L, You T, Chen J, Xu W, Jiao Y. Serum soluble semaphorin 4D is associated with left atrial diameter in patients with atrial fibrillation. Med Sci Monit (2015) 21:2912–7. doi:10.12659/MSM.895441
46. Huang C, Wang Y, Huang JH, Liu W. Sema3A drastically suppresses tumor growth in oral cancer Xenograft model of mice. BMC Pharmacol Toxicol (2017) 18(1):55. doi:10.1186/s40360-017-0163-4
47. Liu MH, Fu WJ, Cui YH, Guo QN, Zhou Y. Downregulation of semaphorin-3F is associated with poor prognostic significance in osteosarcoma patients. Am J Cancer Res (2016) 6(10):2252–62.
48. Dorfman DM, Shahsafaei A, Nadler LM, Freeman GJ. The leukocyte semaphorin CD100 is expressed in most T-cell, but few B-cell, non-Hodgkin’s lymphomas. Am J Pathol (1998) 153(1):255–62. doi:10.1016/S0002-9440(10)65566-6
49. Jiang H, Chen C, Sun Q, Wu J, Qiu L, Gao C, et al. The role of semaphorin 4D in tumor development and angiogenesis in human breast cancer. Onco Targets Ther (2016) 9:5737–50. doi:10.2147/OTT.S114708
50. Deaglio S, Vaisitti T, Bergui L, Bonello L, Horenstein AL, Tamagnone L, et al. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood (2005) 105(8):3042–50. doi:10.1182/blood-2004-10-3873
51. Granziero L, Circosta P, Scielzo C, Frisaldi E, Stella S, Geuna M, et al. CD100/Plexin-B1 interactions sustain proliferation and survival of normal and leukemic CD5+ B lymphocytes. Blood (2003) 101(5):1962–9. doi:10.1182/blood-2002-05-1339
52. Gabrovska PN, Smith RA, Tiang T, Weinstein SR, Haupt LM, Griffiths LR. Semaphorin-plexin signalling genes associated with human breast tumourigenesis. Gene (2011) 489(2):63–9. doi:10.1016/j.gene.2011.08.024
53. Malik MF, Ye L, Jiang WG. Reduced expression of semaphorin 4D and plexin-B in breast cancer is associated with poorer prognosis and the potential linkage with oestrogen receptor. Oncol Rep (2015) 34(2):1049–57. doi:10.3892/or.2015.4015
54. Ross RW, Galsky MD, Scher HI, Magidson J, Wassmann K, Lee GS, et al. A whole-blood RNA transcript-based prognostic model in men with castration-resistant prostate cancer: a prospective study. Lancet Oncol (2012) 13(11):1105–13. doi:10.1016/S1470-2045(12)70263-2
55. Cao J, Zhang C, Chen T, Tian R, Sun S, Yu X, et al. Plexin-B1 and semaphorin 4D cooperate to promote cutaneous squamous cell carcinoma cell proliferation, migration and invasion. J Dermatol Sci (2015) 79(2):127–36. doi:10.1016/j.jdermsci.2015.05.002
56. Kato S, Kubota K, Shimamura T, Shinohara Y, Kobayashi N, Watanabe S, et al. Semaphorin 4D, a lymphocyte semaphorin, enhances tumor cell motility through binding its receptor, plexinB1, in pancreatic cancer. Cancer Sci (2011) 102(11):2029–37. doi:10.1111/j.1349-7006.2011.02053.x
57. Moriarity BS, Otto GM, Rahrmann EP, Rathe SK, Wolf NK, Weg MT, et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat Genet (2015) 47(6):615–24. doi:10.1038/ng.3293
58. Williamson M, de Winter P, Masters JR. Plexin-B1 signalling promotes androgen receptor translocation to the nucleus. Oncogene (2016) 35(8):1066–72. doi:10.1038/onc.2015.160
59. Rody A, Holtrich U, Gaetje R, Gehrmann M, Engels K, von Minckwitz G, et al. Poor outcome in estrogen receptor-positive breast cancers predicted by loss of plexin B1. Clin Cancer Res (2007) 13(4):1115–22. doi:10.1158/1078-0432.CCR-06-2433
60. Binmadi NO, Yang YH, Zhou H, Proia P, Lin YL, De Paula AM, et al. Plexin-B1 and semaphorin 4D cooperate to promote perineural invasion in a RhoA/ROK-dependent manner. Am J Pathol (2012) 180(3):1232–42. doi:10.1016/j.ajpath.2011.12.009
61. Wong OG, Nitkunan T, Oinuma I, Zhou C, Blanc V, Brown RS, et al. Plexin-B1 mutations in prostate cancer. Proc Natl Acad Sci U S A (2007) 104(48):19040–5. doi:10.1073/pnas.0702544104
62. Balakrishnan A, Penachioni JY, Lamba S, Bleeker FE, Zanon C, Rodolfo M, et al. Molecular profiling of the “plexinome” in melanoma and pancreatic cancer. Hum Mutat (2009) 30(8):1167–74. doi:10.1002/humu.21017
63. Stevens L, McClelland L, Fricke A, Williamson M, Kuo I, Scott G. Plexin B1 suppresses c-Met in melanoma: a role for plexin B1 as a tumor-suppressor protein through regulation of c-Met. J Invest Dermatol (2010) 130(6):1636–45. doi:10.1038/jid.2010.13
64. Gomez Roman JJ, Garay GO, Saenz P, Escuredo K, Sanz Ibayondo C, Gutkind S, et al. Plexin B1 is downregulated in renal cell carcinomas and modulates cell growth. Transl Res (2008) 151(3):134–40. doi:10.1016/j.trsl.2007.12.003
65. Damola A, Legendre A, Ball S, Masters JR, Williamson M. Function of mutant and wild-type plexinb1 in prostate cancer cells. Prostate (2013) 73(12):1326–35. doi:10.1002/pros.22678
66. Conrotto P, Corso S, Gamberini S, Comoglio PM, Giordano S. Interplay between scatter factor receptors and B plexins controls invasive growth. Oncogene (2004) 23(30):5131–7. doi:10.1038/sj.onc.1207650
67. Zhou H, Kann MG, Mallory EK, Yang YH, Bugshan A, Binmadi NO, et al. Recruitment of Tiam1 to semaphorin 4D activates Rac and enhances proliferation, invasion, and metastasis in oral squamous cell carcinoma. Neoplasia (2017) 19(2):65–74. doi:10.1016/j.neo.2016.12.004
68. Basile JR, Barac A, Zhu T, Guan KL, Gutkind JS. Class IV semaphorins promote angiogenesis by stimulating Rho-initiated pathways through plexin-B. Cancer Res (2004) 64(15):5212–24. doi:10.1158/0008-5472.CAN-04-0126
69. Conrotto P, Valdembri D, Corso S, Serini G, Tamagnone L, Comoglio PM, et al. Sema4D induces angiogenesis through Met recruitment by Plexin B1. Blood (2005) 105(11):4321–9. doi:10.1182/blood-2004-07-2885
70. Yang YH, Zhou H, Binmadi NO, Proia P, Basile JR. Plexin-B1 activates NF-kappaB and IL-8 to promote a pro-angiogenic response in endothelial cells. PLoS One (2011) 6(10):e25826. doi:10.1371/journal.pone.0025826
71. Zhou H, Yang YH, Binmadi NO, Proia P, Basile JR. The hypoxia-inducible factor-responsive proteins semaphorin 4D and vascular endothelial growth factor promote tumor growth and angiogenesis in oral squamous cell carcinoma. Exp Cell Res (2012) 318(14):1685–98. doi:10.1016/j.yexcr.2012.04.019
72. Sun Q, Zhou H, Binmadi NO, Basile JR. Hypoxia-inducible factor-1-mediated regulation of semaphorin 4D affects tumor growth and vascularity. J Biol Chem (2009) 284(46):32066–74. doi:10.1074/jbc.M109.057166
73. Zhou H, Yang YH, Basile JR. The semaphorin 4D-plexin-B1-RhoA signaling axis recruits pericytes and regulates vascular permeability through endothelial production of PDGF-B and ANGPTL4. Angiogenesis (2014) 17(1):261–74. doi:10.1007/s10456-013-9395-0
74. Ding X, Qiu L, Zhang L, Xi J, Li D, Huang X, et al. The role of semaphorin 4D as a potential biomarker for antiangiogenic therapy in colorectal cancer. Onco Targets Ther (2016) 9:1189–204. doi:10.2147/OTT.S98906
75. Zhou H, Binmadi NO, Yang YH, Proia P, Basile JR. Semaphorin 4D cooperates with VEGF to promote angiogenesis and tumor progression. Angiogenesis (2012) 15(3):391–407. doi:10.1007/s10456-012-9268-y
76. Liu H, Yang Y, Xiao J, Yang S, Liu Y, Kang W, et al. Semaphorin 4D expression is associated with a poor clinical outcome in cervical cancer patients. Microvasc Res (2014) 93:1–8. doi:10.1016/j.mvr.2014.02.007
77. Younis RH, Han KL, Webb TJ. Human head and neck squamous cell carcinoma-associated semaphorin 4D induces expansion of myeloid-derived suppressor cells. J Immunol (2016) 196(3):1419–29. doi:10.4049/jimmunol.1501293
78. Delaire S, Billard C, Tordjman R, Chedotal A, Elhabazi A, Bensussan A, et al. Biological activity of soluble CD100. II. Soluble CD100, similarly to H-SemaIII, inhibits immune cell migration. J Immunol (2001) 166(7):4348–54. doi:10.4049/jimmunol.166.7.4348
79. Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol (2012) 2012:948098. doi:10.1155/2012/948098
80. Basile JR, Holmbeck K, Bugge TH, Gutkind JS. MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J Biol Chem (2007) 282(9):6899–905. doi:10.1074/jbc.M609570200
81. Silbermann R, Roodman GD. Current controversies in the management of myeloma bone disease. J Cell Physiol (2016) 231(11):2374–9. doi:10.1002/jcp.25351
82. Galson DL, Silbermann R, Roodman GD. Mechanisms of multiple myeloma bone disease. Bonekey Rep (2012) 1:135. doi:10.1038/bonekey.2012.135
83. Beuzeboc P, Scholl S. Prevention of bone metastases in breast cancer patients. Therapeutic perspectives. J Clin Med (2014) 3(2):521–36. doi:10.3390/jcm3020521
84. Suvannasankha A, Crean CD, Tompkins DR, Delgado-Calle J, Bellido TM, David Roodman G, et al. Regulation of osteoblast function in myeloma bone disease by semaphorin 4D. Blood (2016) 128:4439.
85. Parmo-Cabanas M, Molina-Ortiz I, Matias-Roman S, Garcia-Bernal D, Carvajal-Vergara X, Valle I, et al. Role of metalloproteinases MMP-9 and MT1-MMP in CXCL12-promoted myeloma cell invasion across basement membranes. J Pathol (2006) 208(1):108–18. doi:10.1002/path.1876
86. Fisher TL, Reilly CA, Winter LA, Pandina T, Jonason A, Scrivens M, et al. Generation and preclinical characterization of an antibody specific for SEMA4D. MAbs (2016) 8(1):150–62. doi:10.1080/19420862.2015.1102813
87. LaGanke C, Samkoff L, Edwards K, Jung Henson L, Repovic P, Lynch S, et al. Safety/tolerability of the anti-semaphorin 4D Antibody VX15/2503 in a randomized phase 1 trial. Neurol Neuroimmunol Neuroinflamm (2017) 4(4):e367. doi:10.1212/NXI.0000000000000367
88. Patnaik A, Weiss GJ, Leonard JE, Rasco DW, Sachdev JC, Fisher TL, et al. Safety, pharmacokinetics, and pharmacodynamics of a humanized anti-semaphorin 4D antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res (2016) 22(4):827–36. doi:10.1158/1078-0432.CCR-15-0431
89. Matsunaga Y, Bashiruddin NK, Kitago Y, Takagi J, Suga H. Allosteric inhibition of a semaphorin 4D receptor plexin B1 by a high-affinity macrocyclic peptide. Cell Chem Biol (2016) 23(11):1341–50. doi:10.1016/j.chembiol.2016.09.015
90. Zhang Y, Wei L, Miron RJ, Shi B, Bian Z. Anabolic bone formation via a site-specific bone-targeting delivery system by interfering with semaphorin 4D expression. J Bone Miner Res (2015) 30(2):286–96. doi:10.1002/jbmr.2322
Keywords: semaphorin 4D, Sema4D, Plexin-B1, plexin, osteoclasts, osteoblasts, cancer
Citation: Lontos K, Adamik J, Tsagianni A, Galson DL, Chirgwin JM and Suvannasankha A (2018) The Role of Semaphorin 4D in Bone Remodeling and Cancer Metastasis. Front. Endocrinol. 9:322. doi: 10.3389/fendo.2018.00322
Received: 15 February 2018; Accepted: 28 May 2018;
Published: 19 June 2018
Edited by:
Julie A. Sterling, Vanderbilt University, United StatesReviewed by:
Jawed Akhtar Siddiqui, University of Nebraska Medical Center, United StatesChrista Maes, KU Leuven, Belgium
Copyright: © 2018 Lontos, Adamik, Tsagianni, Galson, Chirgwin and Suvannasankha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Attaya Suvannasankha, YXN1dmFubmFAaXUuZWR1