 Alexandre Rebelo-Marques1,2,3*
Alexandre Rebelo-Marques1,2,3* Adriana De Sousa Lages1,4
Adriana De Sousa Lages1,4 Renato Andrade2,3,5Carlos Fontes Ribeiro1
Renato Andrade2,3,5Carlos Fontes Ribeiro1 Anabela Mota-Pinto1Francisco Carrilho4João Espregueira-Mendes2,3,6,7,8
Anabela Mota-Pinto1Francisco Carrilho4João Espregueira-Mendes2,3,6,7,8
- 1Faculty of Medicine, University of Coimbra, Coimbra, Portugal
- 2Clínica do Dragão, Espregueira-Mendes Sports Centre – FIFA Medical Centre of Excellence, Porto, Portugal
- 3Dom Henrique Research Centre, Porto, Portugal
- 4Endocrinology, Diabetes and Metabolism Department, Coimbra Hospital and University Center, Coimbra, Portugal
- 5Faculty of Sports, University of Porto, Porto, Portugal
- 63B’s Research Group—Biomaterials, Biodegradables and Biomimetics, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, Guimarães, Portugal
- 7ICVS/3B’s–PT Government Associate Laboratory, Guimarães, Braga, Portugal
- 8Orthopaedics Department of Minho University, Minho, Portugal
World population has been continuously increasing and progressively aging. Aging is characterized by a complex and intraindividual process associated with nine major cellular and molecular hallmarks, namely, genomic instability, telomere attrition, epigenetic alterations, a loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication. This review exposes the positive antiaging impact of physical exercise at the cellular level, highlighting its specific role in attenuating the aging effects of each hallmark. Exercise should be seen as a polypill, which improves the health-related quality of life and functional capabilities while mitigating physiological changes and comorbidities associated with aging. To achieve a framework of effective physical exercise interventions on aging, further research on its benefits and the most effective strategies is encouraged.
Introduction
Aging is a complex and intraindividual process, usually defined as a time-dependent progressive loss of the individual’s physiological integrity, which eventually leads to deteriorated physical function (1). Within this line, the accumulated molecular and cellular damage across the individual’s life span often leads to age-associated pathological conditions and thus makes them more prone to death (2–5). Understanding the specific cellular and molecular mechanisms implicit in aging still represents one of the most complex and integral issues that biological research has yet to overcome.
Despite hundreds of explored and developed theories, not a single one fully and comprehensively explains the process of aging (6). Traditionally, aging was not seen as an adaptation or genetically programmed phenomenon. More recently, biologic currents point to two main theories: the programmed aging and the damage or error-based theories. The first suggests an intrinsic biologic programmed deterioration of the structural and functional capacity of the human cells (7). The latter highlights the cumulative damage to living organisms leading to intrinsic aging (8). Nonetheless, a combination of these theories is usually preferred. In this sense, López-Otín et al. (1), in a state-of-the-art review, proposed nine cellular and molecular hallmarks that contribute to the process of aging, including (1) genomic instability, (2) telomere attrition, (3) epigenetic alterations, (4) loss of proteostasis, (5) deregulated nutrient sensing, (6) mitochondrial dysfunction, (7) cellular senescence, (8) stem cell exhaustion, and (9) altered intercellular communication (Figure 1). These hallmarks should be expressed during normal aging, with their experimental aggravation speeding up the aging process, and in contrast, their experimental amelioration retards the normal aging process, thus increasing a healthy life span.
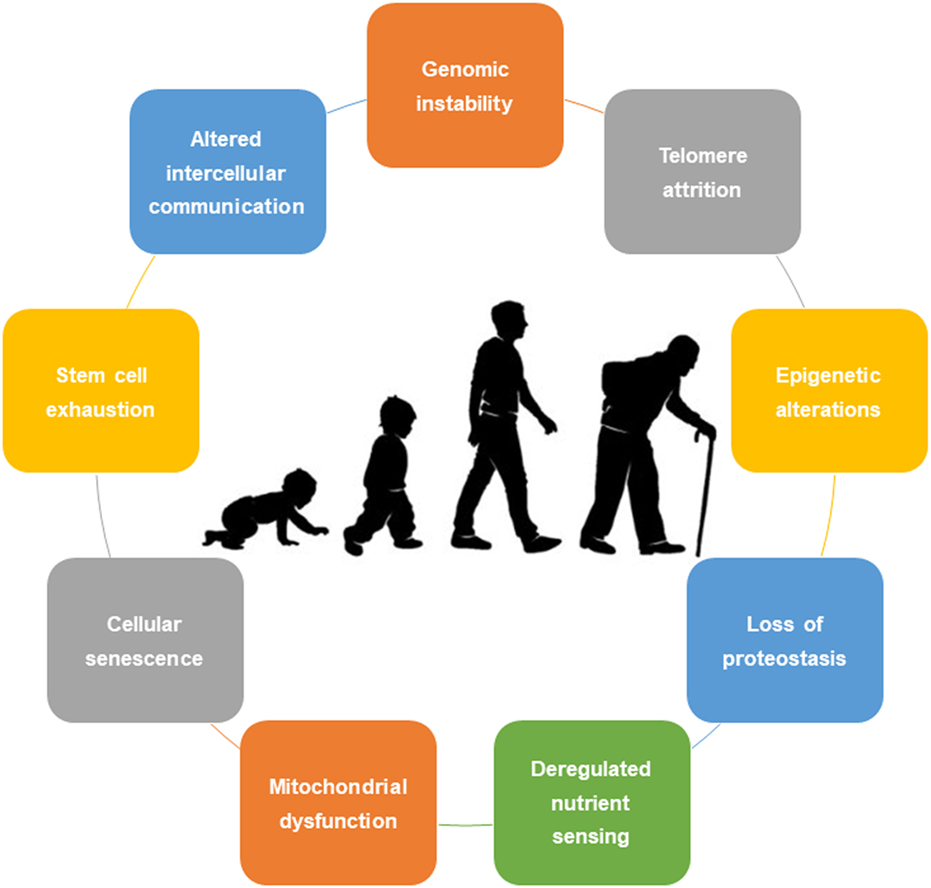
Figure 1. Nine cellular hallmarks contributing to aging.
The world population has been progressively aging and thus raising the average life expectancy. In 2015, an estimated 8.5% (617.1 million) of the world population were aged 65 and older. Within this line, the older population percentage is expected to keep increasing, with an average annual increase of 27.1 million, representing 12 and 16.7% of world population by the years 2030 and 2050, respectively (9). With this life span increase and its associated aging comorbidities, a growing challenge has arisen to make older people physically active and functionally independent until the rest of their lives. Along with the nine cellular and molecular hallmarks stated above, aging is known to be correlated with several cardiovascular, cardiorespiratory, musculoskeletal, metabolic, and cognitive impairments (10). In this sense, regular physical activity in the older population—especially aerobic and resistance training—plays an important role at a multisystem level, preventing severe muscle atrophy, maintaining cardiorespiratory fitness and cognitive function, boosting metabolic activity, and improving or maintaining functional independence (10–13). Within this line, Garatachea et al. (10) and Nelson et al. (13) made an outstanding summary of the specific antiaging effects of physical exercise at the multilevel system, including (1) increasing neurogenesis and attenuating neurodegeneration and cognitive alterations; (2) decreasing blood pressure levels and increasing numerous cardiovascular functions, such as maximal cardiac output, regional blood flow and blood volume, body fluid regulation, endothelial and autonomic function, vagal tone and heart rate variability, and cardiac preconditioning; (3) improving respiratory function by increasing ventilation and gas exchange; (4) enhancing metabolic function in raising the resting metabolic rate, muscle protein synthesis, and fat oxidation; and (5) augmenting muscle function and, subsequently, body composition by improving muscle strength and endurance, maintaining or regaining balance, motor control, and joint mobility, as well as reducing weight and regional adiposity and increasing muscle mass and bone density. In addition, physical exercise has a positive antiaging impact at the cellular level, and its specific role in each aging hallmark is described below.
Effect of Physical Exercise on Each Cellular Hallmark
Genomic Instability
The accumulation of genetic damage throughout one’s life span takes a major role in the aging process (14). Genomic instability, caused by exogenous (physical, chemical, and biological) and endogenous factors [deoxyribonucleic acid (DNA) replication errors, spontaneous hydrolytic reactions, and reactive oxygen species (ROS)], often results in mutations, translocations, chromosomal gains and losses, telomere shortening, and gene disruption (15). In this sense, several cellular events contribute to genomic instability and subsequently to aging, including somatic mutations of nuclear DNA, mutations and deletions in aged mitochondrial DNA (mtDNA), and defects in the nuclear lamina (14, 16, 17). Increased genomic damage has been linked to aging, highlighted by the DNA repair deficits found in accelerated mice models, translated into many human progeroid syndromes (15, 18, 19). Likewise, mtDNA mutations in aged subjects appear to be induced by early replication errors rather than later cumulative oxidative damage (20). In addition, the mutation of the nuclear lamina protein gene encoding and disturbances on its maturation or dynamics leads to a few progeria syndromes (21–23). This premature aging is supported by the delayed onset of progeroid features and extended life span after decreasing prelamin A or progerin levels (24–26).
In the face of genomic instability, the organism has developed a panoply of DNA repair mechanisms that skirmish altogether to overcome DNA nuclear damage (27). Within the same line, genomic stability systems hold specific mechanisms to maintain proper telomere length and function and mtDNA integrity (28, 29). Pharmacological and biologic strategies (mainly hormonal or genetic therapies) have been developed (30–32); howbeit, further research is required to validate nuclear architecture reinforcement for delaying normal aging (1).
Exercise plays a role in maintaining genomic stability. In rodent models, aerobic exercise improves DNA repair mechanisms and nuclear factor kappa B (NF-kB) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) signaling (33–35). Moreover, it augments DNA repair (36) and decreases the number of DNA adducts (up to 77%), related to aging and several risk factors for cardiovascular diseases (37). In addition, a six-month resistance training program in an institutionalized elderly population showed a tendency to reduce cell frequency with micronuclei (~15%) and the total number of micronuclei (~20%), leading to a higher resistance against genomic instability (38). In a meta-analysis comprising data of 478 genetic elements (387 unique genes) associated with exercise from 1,580 individuals, 238 out of 387 genes decreased in DNA methylation percentage after physical exercise among older people. More specifically, the genes that presented DNA methylation decreases were associated with a cancer-suppressing micro-ribonucleic acid (miRNA) gene network (39).
Telomere Attrition
Telomeres are ribonucleoprotein complex structures that protect the integrity of information-carrying DNA throughout cell cycle, preventing the base pair loss of chromosomal DNA during cellular division. Over consecutive cellular divisions, telomere length naturally decreases till a minimum critical size, which precludes further cell division, causing cellular senescence or apoptosis, also known as the end replication problem (40). Telomerase, as an enzyme with a catalytic unit, answers to the end replication problem, promoting telomere lengthening (41). Nevertheless, the fact that most mammalian somatic cells did not express this enzyme explains the progressive loss of the terminal chromosomes’ ends as well as the limited proliferative capacity observed in some in vitro-cultured cells (42, 43). This enzyme deficiency in humans has been linked to premature manifestations of chronic diseases specially related to scarce tissue regenerative ability (such as pulmonary fibrosis, congenital dyskeratosis, or aplastic anemia) (44). Telomere shortening is described during normal aging in human and mice cells (45–50). The fact that telomere length decreases with aging, contributing to the normal cell senescence process, suggested that this could be a potential marker for biological aging (51). Moreover, the leukocyte telomere length is also positively associated with a number of healthy living years, associated with numerous chronic conditions and their complications and with the mortality risk mainly at younger ages (52, 53). The association of chronic inflammation and elevation of pro-inflammatory cytokines [cytokine tumor necrosis factor (TNF-α) and interleukin (IL)-6] and shortened leukocyte telomeres has been proposed by several authors (54–57). Regarding TNF-α, this cytokine seems to have a specific role in downregulating telomerase activity, causing telomere shortening (55).
Interestingly, recent evidence supports that telomerase activation can revert aging, namely, in the premature aging of telomerase-deficient mice when the enzyme is genetically reactivated (58). The relationship between physical activity and healthy aging is well recognized, but the association between physical activity and telomere length remains unclear. Aging induces DNA damage accumulation, especially in some particularly sensitive chromosomal regions such as telomeres, and recent data suggest that physical activity may play a protective role against stress-related telomere attrition (28, 59). Although the potential mechanism is unclear, exercise exhibits a favorable impact on telomere length, especially on a chronic pattern and particularly in older individuals antagonizing the typical age-induced decrements in telomere attrition. Several potential mechanisms have been discussed linking exercise and telomere length decrements to changes in telomerase activity, inflammation, oxidative stress, and decreased skeletal muscle satellite cell content (60).
Exercise has been associated with an upregulation of protective proteins (such as telomeric repeat-binding factor 2) and DNA repair pathway proteins (such as Ku proteins) as well as a downregulation of negative regulator proteins of cell cycle progression (such as p16) in middle-aged athletes supporting this relationship (61). Additionally, although it increases oxidative stress, continuous physical exercise is associated with antioxidant activity and inferior levels of ROS, favoring the REDOX balance, protecting from DNA damage and subsequently shorter telomere attrition (62–64).
Satellite cells are specific skeletal muscle cell precursors activated during muscle regeneration processes or in response to muscle injuries. A positive correlation exists between the number of satellite cells and skeletal muscle telomere length in older adults (61). This pool of cells decreases normally during aging and appears to connect physical activity and skeletal muscle preservation since both resistance and aerobic exercise act as satellite cell pool stimulators, equalizing the decline related to aging (65).
Epigenetic Alterations
The relationship between epigenetic regulation and aging is controversial and complex (1). Epigenetics studies the mitotically and/or meiotically heritable changes within genetic function that cannot be explained by DNA sequence changes (66). Although epigenome refers to the combination of chemical changes to DNA and histone proteins in a cell, epigenetic changes include alterations in DNA methylation patterns, the posttranslational modification of histones, and chromatin remodeling (such as miRNA expression changes) (67).
A multiplicity of epigenetic modifications affects all tissues and cells throughout life (68). Increased histone H4K16 acetylation, H3K4 trimethylation, or H4K20 trimethylation, as well as decreased H3K27 trimethylation or H3K9 methylation, make up age-associated epigenetic marks (69, 70). In mammals, at least three members of the sirtuin (SIRT) family—SIRT1, SIRT3, and SIRT6—contribute to healthy aging (71–73). For example, the transgenic overexpression of mammalian SIRT1 (closest homolog to invertebrate Sir2) improves health aspects during aging despite not increasing longevity (73). Losing the function of SIRT6 reduces longevity, and the gain extends longevity in mice (71, 74).
Actually, the literature clearly reveals that the epigenetic response is highly dynamic and influenced by different environmental and biological factors, such as aging, nutrient availability, and physical exercise. Regular aerobic exercise can change the human genome through DNA methylation (75). Transient hypoxia conditions are a good example (76). Thus, by using epigenetic mechanisms, aerobic exercise can induce the transcription of genes encoding telomere-stabilizing proteins and telomerase activity not only in animals (61, 77, 78) but also in humans (61). Losing promoter methylation and histone H4 deacetylation is associated with the changed gene expression profile in adaptation to aerobic exercise (63–67, 75–79). Also, the class II HDACs 4 and 5 (transcriptional repressors) can translocate from the nucleus to the sarcoplasm of muscle fibers in response to aerobic exercise (79). In human and mouse muscles, the mitochondrial transcription factor (TFAM), PGC-1a methylation, citrate synthase, MEF2A, gene promoters, and pyruvate dehydrogenase kinase isozyme (PDK4) decrease after acute aerobic exercise (80). In addition, aerobic exercise-induced SIRT-1 regulates the tumor suppressor PGC-1a, p53, NF-jB, and other transcription factors via its deacetylase activity (3, 81). Exercise effects are blocked by the overexpression of HDAC5 in transgenic mice, suggesting that histones are important in the transcriptomic response to muscle contraction (82).
Acute exercise is also associated with increases in messenger RNA (mRNA) expression by transient DNA hypomethylation of gene-specific promoter regions (76, 81). In turn, during the recovery period, this mRNA elevation enables protein synthesis and induces gradual structural remodeling and long-term functional modifications (83). Also, calcium and insulin signaling was recently found to be differentially methylated in skeletal muscle after aerobic exercise (83). As far as we know, physical exercise, either aerobic or resistance, can influence miRNAs’ histone modifications or DNA methylation in various tissues. These adaptations can occur in at least the brain, muscle, or cardiovascular system and are intrinsic to the skeletal muscle response during exercise (e.g., mitochondrial respiratory capacity, substrate delivery, and contractile function) (84, 85).
Chronic moderate aerobic exercise increases the methylation levels of the pro-inflammatory apoptosis-associated speck-like protein caspase gene, which modulates IL-18 and IL-1b in the aged leukocytes, thereby contributing to reduced age-related pro-inflammatory cytokines (86). In addition, several myogenic regulatory factors—e.g., myoblast determination protein 1, myogenin, or myogenic factors 5 (Myf5) and 6 (Myf6, also known as myogenic regulatory factor 4, Mrf4, or herculin)—can help fight age-related sarcopenia and frailty, all of them modulated by aerobic or resistance exercise (87–89). Exercise can also upregulate brain-derived neurotrophic factor (BDNF) induction and promote the remodeling of the chromatin containing the BDNF gene (90).
The effect of physical exercise on epigenetic changes is just the beginning. However, studies so far show that an important modulation of exercise exists on the epigenetics mechanisms, particularly in DNA methylation, specially of regular physical exercise.
Loss of Proteostasis
Aging and some aging-related diseases are linked to impaired protein homeostasis, also known as proteostasis (91). The array of quality control is guaranteed through distinct mechanisms that involve location, concentration, conformation, and the turnover of individual proteins, such as autophagy, proteasomal degradation, or chaperone-mediated folding (92). These functions prevent the aggregation of damage components and ensure the continuous renewal of intracellular proteins, degrading altered proteins.
A loss of function or incoordination of these processes leads to accumulated damaged proteins and thus aging-associated deleterious effects (93) and neurological age-related conditions such as Alzheimer’s or Parkinson’s disease (91).
Aging impairs the autophagy–lysosomal and ubiquitin–proteasome systems, which play a central role in cellular proteostatic mechanisms (94, 95). Conversely, physical activity induces brain, muscle, and cardiac autophagy (96). A joint program of moderate-intensity leg-resistance exercises and walking demonstrated to upregulate autophagy muscle markers in old women (97), despite that these data are still restricted to aged subjects, and scarce evidence is still available in humans.
In mouse models, this effect seems to be mediated by the activation of BCL-2–beclin 1 complex (98). Acute resistance exercise programs induced muscle protein synthesis and decreased protein degradation through the activation of class 3 phosphatidylinositol 3OH kinase Vps34 mVps34, which forms an autophagy regulator complex with beclin-1 (99, 100). Moreover, in transgenic mice, the beclin-1 disruption reduces autophagy, leading to neurodegeneration (101).
Aerobic exercise induces autophagy, thus preventing the loss of strength and muscle mass through the modulation of IGF-1, protein kinase B (Akt)/mammalian target of rapamycin (mTOR), and Akt/Forkhead box O3A signaling pathways (102, 103), decreasing cardiovascular disease risk (97) and eliminating damaging proteins triggering neurodegeneration (98).
Similarly, the aging human brain exhibits a downregulation of beclin-1 (104). Higher basal levels of autophagy were related to healthy human exceptional longevity, and healthy centenarians have higher serum levels of beclin-1 compared with young controls (105).
Atrogin-1 (MAFbx) is a muscle-specific ubiquitin ligase involved in muscle atrophy through FoxO signaling (106). The atrogin-1 upregulation is associated with cardiac and skeletal muscle atrophy, and atrogin-1 knockout mouse models corroborate its association with autophagy dysfunction, cardiomyopathy, and premature death (107, 108). Within the same line, comparing aged atrogin-1 knockout mice with age-matched controls, the former shows a reduced tolerance to treadmill exercise and shortened life span (106).
The synthesis of cytosolic and organelle-specific chaperones is impaired in aging (109). Thus, chaperone associated functions, such as folding and protein stability, are conditioned throughout one’s life span (110, 111). In animal models, the upregulation of cochaperone of the heat-shock proteins (HSPs) was associated with prolonged life-span phenotypes (112), and HSF-1 activation, the heat-shock response regulator, was linked to longevity and thermotolerance (113, 114). Despite limited comparison studies, evidence supports that acute endurance- and resistance-type exercise protocols are associated with increased HSPs transcription not only during activity but also immediately postexercise or several hours following exercise, which points out the possible favorable impact of physical activity on proteostasis (115).
Deregulated Nutrient Sensing
The growth hormone (GH) is produced by the anterior pituitary gland and is regulated by the growth hormone-releasing hormone, acting mainly in the hepatocytes to induce insulin-like growth factor 1 (IGF-1) secretion. IGF-1 is also produced in distinct tissues, such as osteocytes, chondrocytes, and muscle, to act in an autocrine or paracrine pattern (116).
Insulin and IGF-1 share the same intracellular signaling pathway, an important aging-controlling route highly conserved during evolution. In this sense, enhanced longevity has been associated with the reduced functions of GH, IGF-1, and insulin receptors and their intracellular effectors (such as Akt and mTOR complexes) (117–119). Within this scope, several authors associated dietary restriction with an increased life or health span probably mediated by an attenuation of insulin and IGF-1 signaling pathway (118, 120–132).
Regarding the intracellular effectors downstream, in animal models, the transcription factor FOXO represents the most relevant alteration linked to longevity (123, 124). The tumor-suppressor gene PTEN has also been associated with an antiaging impact on this signaling pathway, promoting energy expenditure and improving mitochondrial oxidative metabolism (125, 126). This balance between molecules with antiaging properties, emphasized by nutrient scarcity [FOXO, 5’-adenosine monophosphate-activated protein kinase (AMPK), and PTEN] against those that favor the aging process (GH, IGF-1, Akt, and mTOR), shows the relevance of deregulated nutrient sensing as a hallmark of aging (123, 124, 127).
Aging is also physiologically associated with somatopause, which represents a progressive decline in the GH secretory rate starting in the third decade of life, as reflected in decreasing IGF-1 levels (117, 128). In mouse models of premature aging, this GH-IGF-1 axis decline is also noted, highlighting this common denominator in normal and sped-up aging processes (129). This paradoxical observation can be integrated as a defensive response that downmodulates the GH-IGF-1 axis, promoting lower cell growth and metabolism, reducing cellular damage and aiming to extend life span (130).
Besides the glucose sensing related to GH-IGF-1 axis activity, the interest on other nutrient-sensing systems is increasing—mTOR, amino acid concentrations and anabolic metabolism; AMPK and adenosine monophosphate levels; and SIRTs and NAD+ levels. These last two nutrient sensors, AMPK and SIRTs (SIRT 1–7), arise as alternative markers to low-energy states opposite mTOR (117, 131, 132).
Exercise plays an important role in not only the glucose-sensing somatotrophic axis but also the three nutrient-sensing systems referred above, promoting a beneficial anabolic cellular state (133–136). The effect of exercise on glucose metabolism through increased glucose transporter type 4 production is another well-known mechanism of improved insulin sensibility associated with physical activity (137). Additionally, exercise-induced GH and IGF-1 levels are influenced by exercise intensity, duration, and type (higher in intense interval protocols and resistance exercise) (138–140). Thus, the increased muscle protein synthesis associated with resistance exercise is pointed out as a successful strategy to prevent age-related sarcopenia (141–143).
Mitochondrial Dysfunction
The clear causal relationship between mitochondrial dysfunction and aging has long been a target of great discussion; however, the specific mechanisms involved remain unrevealed. Initially, the mitochondrial free radical aging theory proposed that with increasing age came a progressive mitochondrial dysfunction, increasing ROS levels and subsequently further mitochondrial deterioration and generalized cellular damage (144). Nevertheless, dysfunctional mitochondria may contribute to the increase in aging process, independent of ROS levels (145, 146). Interestingly, increased ROS levels may extend the life span of yeast and Caenorhabditis elegans (147–149). In addition, a genetically manipulated impaired mitochondrial function that does not increase ROS levels does not seem to accelerate the aging process (145, 146, 150–152). Within the same lines, mice with genetically manipulated increased ROS levels, greater oxidative damage, or higher antioxidant defense levels do not seem to age quickly or possess extended life spans (145, 146, 150–154). In fact, the main ROS effect is to activate compensatory homeostatic responses. However, with the increased cellular stress and damage present in the aging process, the ROS levels, when exceeding a determined threshold, may even deepen age-related damage (155). Altogether, the findings led to the reconsideration of the ROS role in aging (156).
With increasing age comes a decline in mitochondrial integrity and biogenesis because of alterations in mitochondrial dynamics and mitophagy inhibition, impairing dysfunctional mitochondria removal (156). Several mechanisms seem to be related to mitochondrial integrity and biogenesis, including mitochondrial deficiencies that increase their predisposition to permeabilize in the presence of stress, resulting in activated ROS-mediated and permeabilization-facilitated inflammatory reactions (157, 158). Within this line, lower biogenesis and reduced clearance often lead to a combination of increased damage and reduce mitochondrial turnover, which also accelerate the aging process (1).
With the aging process comes an accumulation of many mtDNA mutations, mostly deletions, which affect many tissues, including nervous and skeletal muscle tissues (159–162). Within this line, the respiratory system efficacy declines with increasing age, which leads to increased electron leakage and lower adenosine triphosphate generation levels (163). The accumulation of oxidative stress-induced mtDNA mutations leads to the progressive decay of the mitochondrial function (164, 165). Moreover, mtDNA is more vulnerable to oxidative damage than nuclear DNA as it lacks histones protection, DNA repair capacity, and non-coding introns (166). Additionally, mtDNA damage may proliferate as cells multiply, resulting in expanded physiologic damage (167).
The regular practice of physical exercise has a positive impact in the mitochondrial function. In this sense, endurance-trained humans presented higher levels of mitochondrial proteins expression, mtDNA, and TFAMs (168). When considering sedentary mtDNA mutator mice, which displayed symptoms of accelerated aging, a 5-month aerobic exercise program induced systemic mitochondrial biogenesis in the mtDNA and increased multiorgan oxidative capacity, thus providing phenotypic protection and reducing multisystem pathology and the risk for premature mortality (169). Hence, regular physical exercise may maintain a pool of bioenergetically functional mitochondria that, by improving the systemic mitochondrial function, contribute to morbidity and mortality risk reduction throughout one’s life span (169–172). Similarly, in the elderly population, the resistance exercise through the PGC-1 and SIRT regulators (173) has decreased DNA oxidative damage (by stimulating their endogenous antioxidant defenses) (174), mitochondrial alterations induced by aging (37), and the improved oxidative capacity of muscle fibers (175).
Aged individuals often show a deficiency in cytochrome c oxidase within the sarcopenic skeletal muscles’ fibers with higher levels of mtDNA mutations (176–180). Moreover, the decreased mitochondrial enzyme activity frequently seen in aged individuals (181, 182) is accompanied with a downregulation of mRNAs encoding mitochondrial proteins (183–185). In this sense, resistance training has the potential to shift the mtDNA of skeletal muscle from healthy aged individuals, augmenting the mitochondrial function (180). Within the same line, a 6-month resistance exercise-training program reversed aging transcriptional signature levels approaching the ones from younger adults, thus enhancing the mitochondrial function (185).
Cellular Senescence
Cellular senescence is the stable arrest of the cell cycle combined with stereotyped phenotypic modifications (186–188). Initially, cellular senescence was associated with telomere attrition, but other age-related triggers were posteriorly identified, namely, non-telomeric DNA damage, and derepression of the INK4/ARF locus (42, 187, 189). Some authors have directly used quantification of senescence-associated β-galactosidase (SABG) to identify senescent cells in different-aged tissues (190, 191). Surprisingly, they identified higher SABG values in hepatic, skin, lung, and spleen tissues in older mice compared to younger mice. However, this differential was not observed in heart, skeletal muscle, and kidney tissues. Based on these facts, the concept that cellular senescence is not a generalized property of all tissues in aged organisms was raised (191). Beyond that, senescent cell accumulation in different tissues seems to be dependent, in one hand, on an increased rate of senescent cell generation and, in other hand, on a decreased rate of clearance (192–194).
The cell’s senescence process is usually associated with a deleterious purpose from senescent cell proliferation with aging. Nevertheless, its primary purpose is to prevent damaged cell proliferation and trigger their demise by the immune system, resulting in a beneficial cell compensatory response, contributing to tissue homeostasis. When tissues exhaust their regenerative capacity, the compensatory response to damage becomes harmful and accelerates aging (195).
Exercise, specifically aerobic, induces the secretion of antitumorigenic myokines and greater natural killer cell activity, contributing to a decreased incidence of oncologic disease and improved cancer prognosis (196). Moreover, the increased expression of telomere repeat-binding factor 2 and Ku70 and the reduced expression of apoptosis regulators (such as cell cycle-checkpoint kinase 2, p16INK4a, and p53 or survival regulators) are associated with the beneficial impact of exercise on cellular senescence (61).
A part of DNA damage, excessive mitogenic signaling, is strongly associated with the senescence (197). The main mechanisms reported that implement senescence in response to this variety of oncogenic insults were p16INK4a/Rb and p19ARF/p53 pathways (1, 198). Senescent cells present upregulated p16INK4a and p21 cell cycle inhibitors. p21 is a downstream target of p53 and telomere dysfunction (199, 200). Aerobic exercise has been inversely correlated with p16INK4a mRNA levels in peripheral blood T lymphocytes, which might promote protective outcomes against age-dependent alterations (201).
The secretome of a senescent cell presents dramatic alterations becoming particularly enriched in pro-inflammatory cytokines and matrix metalloproteinases that may contribute to aging (10, 202, 203). This senescence-associated secretory phenotype (SASP) as a DNA damage response explains how senescent cells alter tissue microenvironments (204). Once again, aerobic exercise suppresses liver senescence markers and downregulates inflammatory mediators (reducing gamma glutamyltranspeptidase activity and levels of p21, p53, and IL-6) (205).
Exercise is capable of upregulating cardiac telomere-stabilizing proteins, providing protection against doxorubicin-induced cardiomyopathy and promoting antisenescent effects (77). Within the same line, the same research group (61) showed that besides improving telomere biology in the thoracic aorta and in mononuclear cells, exercise could also reduce the vascular expression of apoptosis regulators. Moreover, it allows endurance athletes to increase telomerase activity and downregulate cell-cycle inhibitors compared with sedentary individuals. In addition, Song et al. (206) found that in humans, the practice of aerobic exercise reduced the expression of DNA damage biomarkers and correlated negatively with telomere length in peripheral blood T lymphocytes and positively with p16INK4a expression, supporting previous findings.
Stem Cell Exhaustion
In aging, the declining regenerative potential of tissues is obvious (1). A good example is the age-related decline in hematopoiesis, causing a diminished production of adaptive immune cell, a process designated as immunosenescence (207). Similar processes were found in adult stem cell compartments, including the mouse forebrain (208), muscle fibers (209), or bone (210). An overall decrease in cell-cycle activity of hematopoietic stem cells has been revealed (211), connected with the overexpression of cell cycle-inhibitory proteins such as p16INK4a (212) and DNA damage accumulation (211).
For the long-term maintenance of the organism, the deficient proliferation of stem and progenitor cells is harmful, but an excessive proliferation can also be deleterious by speeding up the exhaustion of stem cell niches (1). Within this line, physical exercise is one of the most potent stimuli for the migration/proliferation of the stem cell subsets from their home tissue to impaired tissues for later engraftment and regeneration (95). In this sense, regular physical exercise attenuates age-associated reduction in the endothelium reparative capacity of endothelial progenitor cells (213). In addition, exercise activates pluripotent cells’ progenitors, including mesenchymal and neural stem cells, which improve brain regenerative capacity and cognitive ability (95).
The stem cells more affected by aging are myogenic, known as satellite cells (214). Satellite cell alterations manage the reduced replacement and repair efficiency potential in human skeletal muscle tissue myofibers. Age-reduced functionality or the number of these cells inhibits proper muscle-mass maintenance (214–217). Age-related atrophy by type II muscle fibers is influenced by the decline in the content of type II muscle fiber satellite cells (214). Sarcopenia is related to these cells’ atrophy and so are its pathophysiological mechanisms (218). Besides that, aging reductions in strength and muscle mass are directly connected with myonuclear content, the muscle fiber type-specific cross-sectional area, and satellite cell content (219). Animal studies showed that aerobic exercise not only promotes satellite cell pool expansion in young and old mice (220) but also potentiates myofibers with greater numbers of satellite cells in young and old rats (221). Thus, skeletal muscle regulation depends on the satellite cells (222). This contribution to skeletal muscle regeneration is well documented (223, 224) and involves several mechanisms, including neurotrophic and vascular factors (IGF-1 and other growth factors), immune response, neurotransmitters, and cytokines (such as IL-6, testosterone, or nitric oxide), most of which are modulated by exercise (167). Resistance training can induce, in young adults and during aging, the hypertrophy of type II fibers (225, 226) by skeletal muscle satellite cell proliferation and differentiation, which attenuates prosarcopenic physiological and age-related events (214, 218, 219). Moreover, muscular hypertrophy generated by resistance exercise training is related to satellite cells (227). Myostatin, a protein that inhibits muscle differentiation and growth in the myogenesis process, is also involved in the process (228). Probably, the same localization of myostatin and satellite cells explains the worsened myogenic capacity of the aged skeletal muscle (229). In fact, Snijders et al. (228) revealed an aging-blunted activation of type II muscle fiber satellite cells in response to acute stimuli of resistance exercise.
Interestingly, pharmacological interventions are also being explored to increase stem cell function. Using rapamycin for the inhibition of mTORC1 can delay aging by improving proteostasis and affecting energy sensing, which may improve stem cell function, in the hematopoietic system and intestine (230–232). The pharmacological inhibition of the GTPase CDC42, whose activity is increased in aged hematopoietic stem cells, may also rejuvenate human senescent cells (233).
Altered Intercellular Communication
The physiological aging process implicates several alterations on intracellular communication mechanisms, namely, in neuroendocrine, endocrine, and neuronal levels (234–237). Inflammation plays a central role in this age-related alteration, contributing to a predominant pro-inflammatory phenotype associated with progressive aging, also known as “inflammaging” (238). This inflammaging is caused by distinct mechanisms: pro-inflammatory tissue damage accumulation, additional cumulative dysfunction of the immune system, elevated levels of pro-inflammatory cytokines’ secretion by senescent cells, altered autophagy response, and the increased activation of the NF-kB transcription factor (163, 238–240). All these mechanisms will promote the activation of different pro-inflammatory pathways leading to increased levels of IL-1b, TNF, and interferons (163, 238). In this sense, different endocrine axes (renin–angiotensin, adrenergic, and insulin-GH), as part of neurohormonal signaling, are altered with aging because of increased inflammatory reaction levels, the decline of immunosurveillance against pathogenic agents and premalignant cells, and composition changes in the peri- and extracellular environment (238). Additionally, the decay factor AU-binding factor 1 (AUF1 or heterogeneous nuclear ribonucleoprotein D) has also been linked to inflammaging as an important factor in inflammatory response cessation, conditioning cytokine mRNA degradation, and contributes also to maintaining telomere length by activating the expression of the telomerase catalytic subunit telomerase reverse transcriptase, linking this same factor to different hallmarks discussed above (241). Chronic muscle contractile activity upgraded different AUF1 isoforms secretion (p37, p40, and p45) in the muscle of healthy rats, resulting in improved muscle plasticity (242).
Pro-inflammatory state and stress activate hypothalamic NF-kB expression, which downregulates gonadotropin-releasing hormone neurons and subsequently gonadotropins by the anterior pituitary, explaining some age-related comorbidities such as bone fragility, muscle weakness, and reduced neurogenesis (237). The enhanced activation of the NF-kB transcription factor is referred to as one of the transcriptional signatures of aging, and its expression restriction has been associated with skin rejuvenation in animal models (243). Besides, the use of genetic or pharmacological inhibitors of the NF-kB signaling was associated with the prevention of age-associated features in distinct sped-up aging mouse models (32, 244). The SIRT family (SIRT 1–7), in the same point of view, seems to downregulate inflammation-related genes acting as a protective factor to aging and many other inflammatory pathological conditions (245–248).
The cross-talk interorgan may explain the correlation between aging-related changes in different tissues. Cellular senescence influences neighbor cells during aging via gap–junction contacts, growth factors, interleukins, and ROS, highlighting the importance of microenvironment contribution during the process and offering the possibility to modulate aging in different levels (249). Chronic exercise, especially aerobic type, may restore defective intercellular communication, decreasing mitochondrial ROS production and upregulating the endogenous antioxidant profile (33).
Muscle contraction is traditionally associated with myokine secretion (proteins, growth factors, cytokines, or metallopeptidases) elevated during and after exercise (97). Interestingly, the muscle-released IL-6 creates a healthy influence, inducing the production of anti-inflammatory cytokines, IL-1 receptor antagonist, IL-10, or TNF soluble receptors, while restraining pro-inflammatory cytokine TNF-α production (250, 251). Within these lines, several authors associated lifelong aerobic exercise training with lower inflammatory levels, especially with lower levels of C-reactive protein, IL-6, and TNF-α, particularly in advanced decades of life (252–254).
Physical Activity Recommendations
Older individuals must practice physical exercise to maintain the health-related quality of life and functional capabilities that mitigate physiological changes and comorbidities associated with aging. Recommendations made herein are based on the most recent American College of Sports Medicine Guidelines (255) (Table 1). Physical exercise should include aerobic exercise, muscle strengthening and endurance training, and flexibility and neuromotor exercises. Physical exercise difficulty and intensity progression should be tailored to the individual’s tolerance, preference, and specific needs. Thus, lighter intensity and duration are recommended at earlier stages, especially for those who are deconditioned or functionally impaired or present chronic conditions that preclude the performance of more demanding physical tasks. More debilitated and frail individuals may initially require aerobic training activities to improve their physical fitness before proceeding to more demanding tasks. In opposition, individuals presenting sarcopenic muscles may need to improve their muscular strength and endurance before engaging in aerobic training. When the chronic conditions or comorbidities preclude accomplishing the recommended minimum of physical exercise, older individuals should not remain sedentary, and physical training should be performed as tolerated to provide a therapeutic benefit. Training sessions should be supervised by a qualified health professional and finish with an adequate cooldown (gradual reduction of physical intensity complemented with flexibility exercises), especially among individuals with cardiovascular disease.
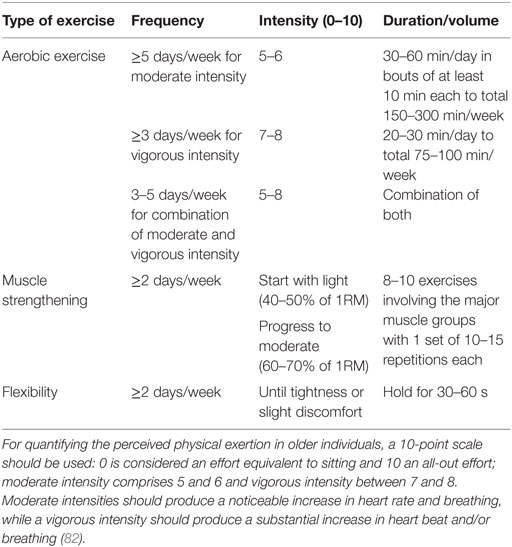
Table 1. General guidelines for exercise prescription in older individuals.
Individuals with increased fall risk or present functional and mobility limitations benefit from the addition of neuromotor exercise training (2–3 days/week), comprising balance, agility, and proprioceptive training (255–257). General recommendations to progressively increase the exercises’ difficulty include (i) more challenging postures by gradually shortening the support base, (ii) more exercises comprising more dynamic movement that perturb the center of gravity, (iii) higher focus on the postural muscle groups exercises, and (iv) progressive reduction of the sensory input.
Overview and Take-Home Message
Cellular aging hallmarks are codependent and co-occur with the aging process. Understanding their causal network enables the conception of a framework to develop novel interventions to attenuate the aging process. As López-Otín et al. (1) referred in their review, cellular hallmarks may have beneficial or deleterious effects and may be subclassified into three main categories: primary (genomic instability, telomere attrition, epigenetic alterations, and proteostasis loss), antagonistic (deregulated nutrient sensing, mitochondrial dysfunction, and cellular senescence), and integrative (stem cell exhaustion and altered intercellular communication) hallmarks. The primary hallmarks are the initiating triggers and are always associated with deleterious effects, such as DNA damage from chromosomal aneuploidies, mtDNA mutations, telomere loss, epigenetic drift, and defective proteostasis. On the opposite, the antagonistic hallmarks have a beneficial responsive effect to attenuate damage when present in lower levels; however, when these are exacerbated or present at chronic levels, especially when promoted by the primary hallmarks, they have a progressive harmful effect, inducing cellular damage and promoting the aging process. The integrative hallmarks result from the accumulated damage from the primary and antagonistic hallmarks and directly interfere with tissue homeostasis and account for age-associated functional decline.
To face the increase in average life expectancy, many therapeutic interventions aiming at the life-span expansion have emerged (1). Nevertheless, many of these therapeutic interventions comprise expensive pharmacologic agents associated with an increased complication risk because of adverse events and polymedication. On the other hand, physical exercise is free, reduces the risk of many potentially lethal diseases, and helps strike the increasing sedentary behavior and physical-inactivity pandemic. Within this line, although exercise does not mitigate the aging process, it attenuates many of the deleterious systemic and cellular effects and improves the function of most of the mechanisms involved in aging. In this sense, further research on its most effective benefits in elderly people is warranted.
Looking at the big picture, although many paths lead to Rome, the safest and most triumphant route should extensively rely on physical exercise. This should be seen as a polypill, and the elderly community should be encouraged to engage in the continuous and regular practice of healthy physical activities. The motto is “Move for your life,” and remember, exercise is medicine.
Author Contributions
AR-M, AL, and RA conceived the presented idea, performed the literature review and drafted the manuscript. CR, AM-P, FC, and JE-M provided advice on key aspects of the manuscript and critically revised the manuscript for important intellectual content. All the authors discussed the final manuscript and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with one of the authors RA.
Abbreviations
ACSM, American College of Sports Medicine; Akt, protein kinase B; AMP, adenosine monophosphate; AMPK, 5’-adenosine monophosphate-activated protein kinase; ASC, speck-like protein caspase; ATP, adenosine triphosphate; AUF1, AU-binding factor 1; BDNF, brain-derived neurotrophic factor; CRP, C-reactive protein; DNA, deoxyribonucleic acid; FoxO3, Forkhead box O3; GH, growth hormone; GHRH, growth hormone-releasing hormone; Glut 4, glucose transporter type 4; GnRH, gonadotropin-releasing hormone; GTP, guanosine triphosphate; hnRNP D, heterogeneous nuclear ribonucleoprotein D; HSPs, heat-shock proteins; IGF-1, insulin-like growth factor 1; IL, interleukin; MEF2, myocyte enhancer factor-2; miRNA, microRNA; mRNA, messenger RNA; mtDNA, mitochondrial DNA; mTOR, mammalian target of rapamycin; Myf, myogenic factors; MyoD, myoblast determination protein; NAD, nicotinamide adenine dinucleotide; NF-kB, nuclear factor kappa B; NK, natural killer; NO, nitric oxide; PDK4, pyruvate dehydrogenase kinase isozyme; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PI3K, class 3 phosphatidylinositol 3OH kinase; RNA, ribonucleic acid; ROS, reactive oxygen species; SABG, senescence-associated β-galactosidase; SASP, senescence-associated secretory phenotype; SIRT, sirtuin; sTNF-R, TNF soluble receptors; TERT, telomerase reverse transcriptase; TFAM, mitochondrial transcription factor; TNF, tumor necrosis factor; TRF-2, telomeric repeat-binding factor 2.
References
1. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi:10.1016/j.cell.2013.05.039
2. Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol (2013) 75:621–44. doi:10.1146/annurev-physiol-030212-183712
3. Kaliman P, Párrizas M, Lalanza JF, Camins A, Escorihuela RM, Pallàs M. Neurophysiological and epigenetic effects of physical exercise on the aging process. Ageing Res Rev (2011) 10(4):475–86. doi:10.1016/j.arr.2011.05.002
4. Kirkwood TB. Understanding the odd science of aging. Cell (2005) 120(4):437–47. doi:10.1016/j.cell.2005.01.027
5. Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature (2008) 454(7208):1065. doi:10.1038/nature07216
7. Fragala MS. The physiology of aging and exercise. In: Sullivan G, Pomidor A, editors. Exercise for Aging Adults. Springer (2015). p. 1–11.
8. Piedrafita G, Keller MA, Ralser M. The impact of non-enzymatic reactions and enzyme promiscuity on cellular metabolism during (oxidative) stress conditions. Biomolecules (2015) 5(3):2101–22. doi:10.3390/biom5032101
9. He W, Goodkind D, Kowal P. An Aging World: 2015 International Population Reports. United States Census Bureau (2016). p. 9–1.
10. Garatachea N, Pareja-Galeano H, Sanchis-Gomar F, Santos-Lozano A, Fiuza-Luces C, Morán M, et al. Exercise attenuates the major hallmarks of aging. Rejuvenation Res (2015) 18(1):57–89. doi:10.1089/rej.2014.1623
11. Arnold P, Bautmans I. The influence of strength training on muscle activation in elderly persons: a systematic review and meta-analysis. Exp Gerontol (2014) 58:58–68. doi:10.1016/j.exger.2014.07.012
12. Demontis F, Piccirillo R, Goldberg AL, Perrimon N. The influence of skeletal muscle on systemic aging and lifespan. Aging Cell (2013) 12(6):943–9. doi:10.1111/acel.12126
13. Nelson ME, Rejeski WJ, Blair SN, Duncan PW, Judge JO, King AC, et al. Physical activity and public health in older adults: recommendation from the American College of Sports Medicine and the American Heart Association. Circulation (2007) 116(9):1094. doi:10.1161/CIRCULATIONAHA.107.185650
14. Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Yanai H, et al. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev (2013) 12(2):661–84. doi:10.1016/j.arr.2012.02.001
15. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med (2009) 361(15):1475–85. doi:10.1056/NEJMra0804615
16. Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev (2008) 22(7):832–53. doi:10.1101/gad.1652708
17. Park CB, Larsson N-G. Mitochondrial DNA mutations in disease and aging. J Cell Biol (2011) 193(5):809–18. doi:10.1083/jcb.201010024
18. Gregg SQ, Gutiérrez V, Rasile Robinson A, Woodell T, Nakao A, Ross MA, et al. A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology (2012) 55(2):609–21. doi:10.1002/hep.24713
19. Murga M, Bunting S, Montaña MF, Soria R, Mulero F, Cañamero M, et al. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet (2009) 41(8):891–8. doi:10.1038/ng.420
20. Ameur A, Stewart JB, Freyer C, Hagström E, Ingman M, Larsson N-G, et al. Ultra-deep sequencing of mouse mitochondrial DNA: mutational patterns and their origins. PLoS Genet (2011) 7(3):e1002028. doi:10.1371/journal.pgen.1002028
21. Cabanillas R, Cadiñanos J, Villameytide JA, Pérez M, Longo J, Richard JM, et al. Néstor–Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A (2011) 155(11):2617–25. doi:10.1002/ajmg.a.34249
22. De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. Lamin a truncation in Hutchinson–Gilford progeria. Science (2003) 300(5628):2055. doi:10.1126/science.1084125
23. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature (2003) 423(6937):293–8. doi:10.1038/nature01629
24. Osorio FG, Varela I, Lara E, Puente XS, Espada J, Santoro R, et al. Nuclear envelope alterations generate an aging-like epigenetic pattern in mice deficient in Zmpste24 metalloprotease. Aging Cell (2010) 9(6):947–57. doi:10.1111/j.1474-9726.2010.00621.x
25. Varela I, Pereira S, Ugalde AP, Navarro CL, Suárez MF, Cau P, et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med (2008) 14(7):767–72. doi:10.1038/nm1786
26. Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, et al. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson–Gilford progeria syndrome mutation. J Clin Invest (2006) 116(8):2115. doi:10.1172/JCI28968
27. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature (2012) 481(7381):287–94. doi:10.1038/nature10760
28. Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med (2006) 12(10):1133–8. doi:10.1038/nm1006-1133
29. Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol (2012) 13(10):659–71. doi:10.1038/nrm3439
30. Liu G-H, Suzuki K, Qu J, Sancho-Martinez I, Yi F, Li M, et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell (2011) 8(6):688–94. doi:10.1016/j.stem.2011.04.019
31. Mariño G, Ugalde AP, Fernández ÁF, Osorio FG, Fueyo A, Freije JM, et al. Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proc Natl Acad Sci U S A (2010) 107(37):16268–73. doi:10.1073/pnas.1002696107
32. Osorio FG, Bárcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, et al. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev (2012) 26(20):2311–24. doi:10.1101/gad.197954.112
33. Gomez-Cabrera M-C, Domenech E, Viña J. Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic Biol Med (2008) 44(2):126–31. doi:10.1016/j.freeradbiomed.2007.02.001
34. Leick L, Lyngby SS, Wojtasewski JF, Pilegaard H. PGC-1α is required for training-induced prevention of age-associated decline in mitochondrial enzymes in mouse skeletal muscle. Exp Gerontol (2010) 45(5):336–42. doi:10.1016/j.exger.2010.01.011
35. Radák Z, Naito H, Kaneko T, Tahara S, Nakamoto H, Takahashi R, et al. Exercise training decreases DNA damage and increases DNA repair and resistance against oxidative stress of proteins in aged rat skeletal muscle. Pflügers Arch (2002) 445(2):273–8. doi:10.1007/s00424-002-0918-6
36. Cash SW, Beresford SA, Vaughan TL, Heagerty PJ, Bernstein L, White E, et al. Recent physical activity in relation to DNA damage and repair using the comet assay. J Phys Act Health (2014) 11(4):770–6. doi:10.1123/jpah.2012-0278
37. Izzotti A. Genomic biomarkers and clinical outcomes of physical activity. Ann N Y Acad Sci (2011) 1229(1):103–14. doi:10.1111/j.1749-6632.2011.06091.x
38. Franzke B, Halper B, Hofmann M, Oesen S, Pierson B, Cremer A, et al. The effect of six months of elastic band resistance training, nutritional supplementation or cognitive training on chromosomal damage in institutionalized elderly. Exp Gerontol (2015) 65:16–22. doi:10.1016/j.exger.2015.03.001
39. Brown WM. Exercise-associated DNA methylation change in skeletal muscle and the importance of imprinted genes: a bioinformatics meta-analysis. Br J Sports Med (2015) 49(24):1567–78. doi:10.1136/bjsports-2014-094073
40. Oeseburg H, de Boer RA, van Gilst WH, van der Harst P. Telomere biology in healthy aging and disease. Pflügers Arch (2010) 459(2):259–68. doi:10.1007/s00424-009-0728-1
41. Hohensinner PJ, Goronzy JJ, Weyand CM. Telomere dysfunction, autoimmunity and aging. Aging Dis (2011) 2(6):524.
42. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res (1961) 25(3):585–621. doi:10.1016/0014-4827(61)90192-6
43. Olovnikov AM. Telomeres, telomerase, and aging: origin of the theory. Exp Gerontol (1996) 31(4):443–8. doi:10.1016/0531-5565(96)00005-8
44. Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet (2012) 13(10):693–704. doi:10.1038/nrg3246
45. Armanios M, Alder JK, Parry EM, Karim B, Strong MA, Greider CW. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet (2009) 85(6):823–32. doi:10.1016/j.ajhg.2009.10.028
46. Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol (2007) 3(10):640–9. doi:10.1038/nchembio.2007.38
47. Blasco MA, Lee H-W, Hande MP, Samper E, Lansdorp PM, DePinho RA, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell (1997) 91(1):25–34. doi:10.1016/S0092-8674(01)80006-4
48. Herrera E, Samper E, Martín-Caballero J, Flores JM, Lee HW, Blasco MA. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J (1999) 18(11):2950–60. doi:10.1093/emboj/18.11.2950
49. Rudolph KL, Chang S, Lee H-W, Blasco M, Gottlieb GJ, Greider C, et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell (1999) 96(5):701–12. doi:10.1016/S0092-8674(00)80580-2
50. Tomás-Loba A, Flores I, Fernández-Marcos PJ, Cayuela ML, Maraver A, Tejera A, et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell (2008) 135(4):609–22. doi:10.1016/j.cell.2008.09.034
51. Müezzinler A, Zaineddin AK, Brenner H. A systematic review of leukocyte telomere length and age in adults. Ageing Res Rev (2013) 12(2):509–19. doi:10.1016/j.arr.2013.01.003
52. Boonekamp JJ, Simons MJ, Hemerik L, Verhulst S. Telomere length behaves as biomarker of somatic redundancy rather than biological age. Aging Cell (2013) 12(2):330–2. doi:10.1111/acel.12050
53. Njajou OT, Hsueh W-C, Blackburn EH, Newman AB, Wu S-H, Li R, et al. Association between telomere length, specific causes of death, and years of healthy life in health, aging, and body composition, a population-based cohort study. J Gerontol A Biol Sci Med Sci (2009) 64(8):860–4. doi:10.1093/gerona/glp061
54. Khan S, Chuturgoon AA, Naidoo DP. Telomeres and atherosclerosis. Cardiovasc J Afr (2012) 23(10):563–71. doi:10.5830/CVJA-2012-056
55. Nimmo M, Leggate M, Viana J, King J. The effect of physical activity on mediators of inflammation. Diabetes Obes Metab (2013) 15(s3):51–60. doi:10.1111/dom.12156
56. O’Donovan A, Pantell MS, Puterman E, Dhabhar FS, Blackburn EH, Yaffe K, et al. Cumulative inflammatory load is associated with short leukocyte telomere length in the Health, Aging And Body Composition Study. PLoS One (2011) 6(5):e19687. doi:10.1371/journal.pone.0019687
57. You T, Arsenis NC, Disanzo BL, LaMonte MJ. Effects of exercise training on chronic inflammation in obesity. Sports Med (2013) 43(4):243–56. doi:10.1007/s40279-013-0023-3
58. Jaskelioff M, Muller FL, Paik J-H, Thomas E, Jiang S, Adams AC, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature (2011) 469(7328):102–6. doi:10.1038/nature09603
59. Puterman E, Lin J, Blackburn E, O’Donovan A, Adler N, Epel E. The power of exercise: buffering the effect of chronic stress on telomere length. PLoS One (2010) 5(5):e10837. doi:10.1371/journal.pone.0010837
60. Arsenis NC, You T, Ogawa EF, Tinsley GM, Zuo L. Physical activity and telomere length: impact of aging and potential mechanisms of action. Oncotarget (2017) 8(27):45008–19. doi:10.18632/oncotarget.16726
61. Werner C, Fürster T, Widmann T, Pöss J, Roggia C, Hanhoun M, et al. Physical exercise prevents cellular senescence in circulating leukocytes and in the vessel wall. Circulation (2009) 120(24):2438–47. doi:10.1161/CIRCULATIONAHA.109.861005
62. Radak Z, Chung HY, Koltai E, Taylor AW, Goto S. Exercise, oxidative stress and hormesis. Ageing Res Rev (2008) 7(1):34–42. doi:10.1016/j.arr.2007.04.004
63. Gomes EC, Silva AN. Oliveira MRd. Oxidants, antioxidants, and the beneficial roles of exercise-induced production of reactive species. Oxid Med Cell Longev (2012) 2012:756132. doi:10.1155/2012/756132
64. Samjoo I, Safdar A, Hamadeh M, Raha S, Tarnopolsky M. The effect of endurance exercise on both skeletal muscle and systemic oxidative stress in previously sedentary obese men. Nutr Diabetes (2013) 3(9):e88. doi:10.1038/nutd.2013.30
65. Kadi F, Ponsot E. The biology of satellite cells and telomeres in human skeletal muscle: effects of aging and physical activity. Scand J Med Sci Sports (2010) 20(1):39–48. doi:10.1111/j.1600-0838.2009.00966.x
66. Russo VE, Martienssen RA, Riggs AD. Epigenetic Mechanisms of Gene Regulation. Cold Spring: Harbor Laboratory Press (1996).
67. Grazioli E, Dimauro I, Mercatelli N, Wang G, Pitsiladis Y, Di Luigi L, et al. Physical activity in the prevention of human diseases: role of epigenetic modifications. BMC Genomics (2017) 18(8):802. doi:10.1186/s12864-017-4193-5
68. Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell (2012) 11(4):694–703. doi:10.1111/j.1474-9726.2012.00835.x
69. Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet (2007) 23(8):413–8. doi:10.1016/j.tig.2007.05.008
70. Han S, Brunet A. Histone methylation makes its mark on longevity. Trends Cell Biol (2012) 22(1):42–9. doi:10.1016/j.tcb.2011.11.001
71. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell (2006) 124(2):315–29. doi:10.1016/j.cell.2005.11.044
72. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell (2010) 143(5):802–12. doi:10.1016/j.cell.2010.10.002
73. Herranz D, Muñoz-Martin M, Cañamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, et al. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun (2010) 1:3. doi:10.1038/ncomms1001
74. Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature (2012) 483(7388):218–21. doi:10.1038/nature10815
75. Ling C, Rönn T. Epigenetic adaptation to regular exercise in humans. Drug Discov Today (2014) 19(7):1015–8. doi:10.1016/j.drudis.2014.03.006
76. Pareja-Galeano H, Sanchis-Gomar F, García-Giménez JL. Physical exercise and epigenetic modulation: elucidating intricate mechanisms. Sports Med (2014) 44(4):429–36. doi:10.1007/s40279-013-0138-6
77. Werner C, Hanhoun M, Widmann T, Kazakov A, Semenov A, Pöss J, et al. Effects of physical exercise on myocardial telomere-regulating proteins, survival pathways, and apoptosis. J Am Coll Cardiol (2008) 52(6):470–82. doi:10.1016/j.jacc.2008.04.034
78. Wolf SA, Melnik A, Kempermann G. Physical exercise increases adult neurogenesis and telomerase activity, and improves behavioral deficits in a mouse model of schizophrenia. Brain Behav Immun (2011) 25(5):971–80. doi:10.1016/j.bbi.2010.10.014
79. McGee SL, Fairlie E, Garnham AP, Hargreaves M. Exercise-induced histone modifications in human skeletal muscle. J Physiol (2009) 587(24):5951–8. doi:10.1113/jphysiol.2009.181065
80. Barres R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab (2012) 15(3):405–11. doi:10.1016/j.cmet.2012.01.001
81. Ntanasis-Stathopoulos J, Tzanninis J, Philippou A, Koutsilieris M. Epigenetic regulation on gene expression induced by physical exercise. J Musculoskelet Neuronal Interact (2013) 13(2):133–46.
82. Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, et al. Histone deacetylase degradation andMEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest (2007) 117(9):2459. doi:10.1172/JCI31960
83. Perry CG, Lally J, Holloway GP, Heigenhauser GJ, Bonen A, Spriet LL. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J Physiol (2010) 588(23):4795–810. doi:10.1113/jphysiol.2010.199448
84. Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol (1984) 56(4):831–8. doi:10.1152/jappl.1984.56.4.831
85. Booth FW, Thomason DB. Molecular and cellular adaptation of muscle in response to exercise: perspectives of various models. Physiol Rev (1991) 71(2):541–85. doi:10.1152/physrev.1991.71.2.541
86. Nakajima K, Takeoka M, Mori M, Hashimoto S, Sakurai A, Nose H, et al. Exercise effects on methylation of ASC gene. Int J Sports Med (2010) 31(09):671–5. doi:10.1055/s-0029-1246140
87. Sanchis-Gomar F, Garcia-Gimenez JL, Perez-Quilis C, Gomez-Cabrera MC, Pallardo FV, Lippi G. Physical exercise as an epigenetic modulator: eustress, the “positive stress” as an effector of gene expression. J Strength Cond Res (2012) 26(12):3469–72. doi:10.1519/JSC.0b013e31825bb594
88. Perdiguero E, Sousa-Victor P, Ballestar E, Muñoz-Cánoves P. Epigenetic regulation of myogenesis. Epigenetics (2009) 4(8):541–50. doi:10.4161/epi.4.8.10258
89. Raue U, Slivka D, Jemiolo B, Hollon C, Trappe S. Myogenic gene expression at rest and after a bout of resistance exercise in young (18–30 yr) and old (80–89 yr) women. J Appl Physiol (2006) 101(1):53–9. doi:10.1152/japplphysiol.01616.2005
90. Gomez-Pinilla F, Zhuang Y, Feng J, Ying Z, Fan G. Exercise impacts brain-derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation. Eur J Neurosci (2011) 33(3):383–90. doi:10.1111/j.1460-9568.2010.07508.x
91. Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem (2009) 78:959–91. doi:10.1146/annurev.biochem.052308.114844
92. Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science (2008) 319(5865):916–9. doi:10.1126/science.1141448
93. Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res Rev (2011) 10(2):205–15. doi:10.1016/j.arr.2010.02.001
94. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell (2011) 146(5):682–95. doi:10.1016/j.cell.2011.07.030
95. Tomaru U, Takahashi S, Ishizu A, Miyatake Y, Gohda A, Suzuki S, et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol (2012) 180(3):963–72. doi:10.1016/j.ajpath.2011.11.012
96. Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal (2011) 14(11):2201–14. doi:10.1089/ars.2010.3482
97. Fiuza-Luces C, Delmiro A, Soares-Miranda L, González-Murillo Á, Martínez-Palacios J, Ramírez M, et al. Exercise training can induce cardiac autophagy at end-stage chronic conditions: insights from a graft-versus-host-disease mouse model. Brain Behav Immun (2014) 39:56–60. doi:10.1016/j.bbi.2013.11.007
98. He C, Sumpter R Jr, Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy (2012) 8(10):1548–51. doi:10.4161/auto.21327
99. MacKenzie MG, Hamilton DL, Murray JT, Taylor PM, Baar K. mVps34 is activated following high-resistance contractions. J Physiol (2009) 587(1):253–60. doi:10.1113/jphysiol.2008.159830
100. Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol (2010) 45(2):138–48. doi:10.1016/j.exger.2009.11.002
101. Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest (2008) 118(6):2190. doi:10.1172/JCI33585
102. Kim YA, Kim YS, Oh SL, Kim H-J, Song W. Autophagic response to exercise training in skeletal muscle with age. J Physiol Biochem (2013) 69(4):697–705. doi:10.1007/s13105-013-0246-7
103. Luo L, Lu A-M, Wang Y, Hong A, Chen Y, Hu J, et al. Chronic resistance training activates autophagy and reduces apoptosis of muscle cells by modulating IGF-1 and its receptors, Akt/mTOR and Akt/FOXO3a signaling in aged rats. Exp Gerontol (2013) 48(4):427–36. doi:10.1016/j.exger.2013.02.009
104. Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A (2010) 107(32):14164–9. doi:10.1073/pnas.1009485107
105. Emanuele E, Minoretti P, Sanchis-Gomar F, Pareja-Galeano H, Yilmaz Y, Garatachea N, et al. Can enhanced autophagy be associated with human longevity? Serum levels of the autophagy biomarker beclin-1 are increased in healthy centenarians. Rejuvenation Res (2014) 17(6):518–24. doi:10.1089/rej.2014.1607
106. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell (2004) 117(3):399–412. doi:10.1016/S0092-8674(04)00400-3
107. Zaglia T, Milan G, Franzoso M, Bertaggia E, Pianca N, Piasentini E, et al. Cardiac sympathetic neurons provide trophic signal to the heart via β2-adrenoceptor-dependent regulation of proteolysis. Cardiovasc Res (2012) 97(2):240–50. doi:10.1093/cvr/cvs320
108. Zaglia T, Milan G, Ruhs A, Franzoso M, Bertaggia E, Pianca N, et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J Clin Invest (2014) 124(6):2410. doi:10.1172/JCI66339
109. Calderwood SK, Murshid A, Prince T. The shock of aging: molecular chaperones and the heat shock response in longevity and aging—a mini-review. Gerontology (2009) 55(5):550–8. doi:10.1159/000225957
110. Morrow G, Samson M, Michaud S, Tanguay RM. Overexpression of the small mitochondrial Hsp22 extends Drosophila life span and increases resistance to oxidative stress. FASEB J (2004) 18:598–9. doi:10.1096/fj.03-0860fje
111. Walker GA, Lithgow GJ. Lifespan extension in C. elegans by a molecular chaperone dependent upon insulin-like signals. Aging Cell (2003) 2:131–9. doi:10.1046/j.1474-9728.2003.00045.x
112. Swindell WR, Masternak MM, Kopchick JJ, Conover CA, Bartke A, Miller RA. Endocrine regulation of heat shock protein mRNA levels in long-lived dwarf mice. Mech Ageing Dev (2009) 130:393–400. doi:10.1016/j.mad.2009.03.004
113. Chiang WC, Ching TT, Lee HC, Mousigian C, Hsu AL. HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell (2012) 148:322–34. doi:10.1016/j.cell.2011.12.019
114. Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science (2003) 300:1142–5. doi:10.1126/science.1083701
115. Morton JP, Kayani AC, McArdle A, Drust B. The exercise-induced stress response of skeletal muscle, with specific emphasis on humans. Sports Med (2009) 39:643. doi:10.2165/00007256-200939080-00003
116. Lazar MA, Birnbaum MJ. Principles of hormone action. 13th ed. In: Melmed S, editor. Williams Textbook of Endocrinology. Philadelphia: Elsevier Health Sciences (2016). p. 18–48.
117. Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes (2012) 61(6):1315–22. doi:10.2337/db11-1300
118. Fontana L, Partridge L, Longo VD. Extending healthy life span—from yeast to humans. Science (2010) 328(5976):321–6. doi:10.1126/science.1172539
120. Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science (2009) 325(5937):201–4. doi:10.1126/science.1173635
121. Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature (2012) 489(7415):318–21. doi:10.1038/nature11432
122. Masoro EJ. Overview of caloric restriction and ageing. Mech Ageing Dev (2005) 126(9):913–22. doi:10.1016/j.mad.2005.03.012
123. Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature (1993) 366(6454):461–4. doi:10.1038/366461a0
124. Slack C, Giannakou ME, Foley A, Goss M, Partridge L. dFOXO-independent effects of reduced insulin-like signaling in Drosophila. Aging Cell (2011) 10(5):735–48. doi:10.1111/j.1474-9726.2011.00707.x
125. Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, De Boer VC, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell (2012) 149(1):49–62. doi:10.1016/j.cell.2012.02.030
126. Ortega-Molina A, Efeyan A, Lopez-Guadamillas E, Muñoz-Martin M, Gómez-López G, Cañamero M, et al. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab (2012) 15(3):382–94. doi:10.1016/j.cmet.2012.02.001
127. Yamaza H, Komatsu T, Wakita S, Kijogi C, Park S, Hayashi H, et al. FoxO1 is involved in the antineoplastic effect of calorie restriction. Aging Cell (2010) 9(3):372–82. doi:10.1111/j.1474-9726.2010.00563.x
128. Lamberts SWJ, Beld AWVD. Endocrinology and aging. 13th ed. In: Melmed S, editor. Williams Textbook of Endocrinology. Philadelphia: Elsevier Health Sciences (2016). p. 1234–51.
129. Schumacher B, Van Der Pluijm I, Moorhouse MJ, Kosteas T, Robinson AR, Suh Y, et al. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet (2008) 4(8):e1000161. doi:10.1371/journal.pgen.1000161
130. Garinis GA, Van der Horst GT, Vijg J, Hoeijmakers JH. DNA damage and ageing: new-age ideas for an age-old problem. Nat Cell Biol (2008) 10(11):1241–7. doi:10.1038/ncb1108-1241
131. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol (2012) 13(4):225–38. doi:10.1038/nrm3293
132. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol (2012) 32(1):2–11. doi:10.1128/MCB.06159-11
133. Coiro V, Volpi R, Gramellini D, Maffei ML, Volta E, Melani A, et al. Effect of physical training on age-related reduction of GH secretion during exercise in normally cycling women. Maturitas (2010) 65(4):392–5. doi:10.1016/j.maturitas.2009.12.020
134. Fujita S, Rasmussen BB, Cadenas JG, Drummond MJ, Glynn EL, Sattler FR, et al. Aerobic exercise overcomes the age-related insulin resistance of muscle protein metabolism by improving endothelial function and Akt/mammalian target of rapamycin signaling. Diabetes (2007) 56(6):1615–22. doi:10.2337/db06-1566
135. Mayhew DL, Kim J-S, Cross JM, Ferrando AA, Bamman MM. Translational signaling responses preceding resistance training-mediated myofiber hypertrophy in young and old humans. J Appl Physiol (2009) 107(5):1655–62. doi:10.1152/japplphysiol.91234.2008
136. Pasiakos SM. Exercise and amino acid anabolic cell signaling and the regulation of skeletal muscle mass. Nutrients (2012) 4(7):740–58. doi:10.3390/nu4070740
137. Mann S, Beedie C, Balducci S, Zanuso S, Allgrove J, Bertiato F, et al. Changes in insulin sensitivity in response to different modalities of exercise: a review of the evidence. Diabetes Metab Res Rev (2014) 30(4):257–68. doi:10.1002/dmrr.2488
138. Karagiorgos A, Garcia JF, Brooks GA. Growth hormone response to continuous and intermittent exercise. Med Sci Sports (1979) 11(3):302–7.
139. Häkkinen K, Pakarinen A, Alen M, Kauhanen H, Komi P. Neuromuscular and hormonal responses in elite athletes to two successive strength training sessions in one day. Eur J Appl Physiol Occup Physiol (1988) 57(2):133–9. doi:10.1007/BF00640652
140. Schwarz AJ, Brasel J, Hintz RL, Mohan S, Cooper D. Acute effect of brief low-and high-intensity exercise on circulating insulin-like growth factor (IGF) I, II, and IGF-binding protein-3 and its proteolysis in young healthy men. J Clin Endocrinol Metab (1996) 81(10):3492–7. doi:10.1210/jcem.81.10.8855791
141. Hartman JW, Moore DR, Phillips SM. Resistance training reduces whole-body protein turnover and improves net protein retention in untrained young males. Appl Physiol Nutr Metab (2006) 31(5):557–64. doi:10.1139/h06-031
142. Yarasheski KE, Zachwieja JJ, Bier DM. Acute effects of resistance exercise on muscle protein synthesis rate in young and elderly men and women. Am J Physiol Endocrinol Metab (1993) 265(2):E210–4. doi:10.1152/ajpendo.1993.265.2.E210
143. Yarasheski KE, Pak-Loduca J, Hasten DL, Obert KA, Brown MB, Sinacore DR. Resistance exercise training increases mixed muscle protein synthesis rate in frail women and men ≥ 76 yr old. Am J Physiol Endocrinol Metab (1999) 277(1):E118–25. doi:10.1152/ajpendo.1999.277.1.E118
144. Harman D. The free radical theory of aging: effect of age on serum copper levels. J Gerontol (1965) 20(2):151–3. doi:10.1093/geronj/20.2.151
145. Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA, Nijtmans L, et al. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab (2009) 10(2):131–8. doi:10.1016/j.cmet.2009.06.010
146. Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One (2010) 5(7):e11468. doi:10.1371/journal.pone.0011468
147. Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, et al. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev (2008) 22(23):3236–41. doi:10.1101/gad.504808
148. Mesquita A, Weinberger M, Silva A, Sampaio-Marques B, Almeida B, Leão C, et al. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc Natl Acad Sci U S A (2010) 107(34):15123–8. doi:10.1073/pnas.1004432107
149. Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet (2009) 5(2):e1000361. doi:10.1371/journal.pgen.1000361
150. Kujoth G, Hiona A, Pugh T, Someya S, Panzer K, Wohlgemuth S, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science (2005) 309(5733):481–4. doi:10.1126/science.1112125
151. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature (2004) 429(6990):417–23. doi:10.1038/nature02517
152. Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet (2008) 40(4):392–4. doi:10.1038/ng.95
153. Zhang Y, Ikeno Y, Qi W, Chaudhuri A, Li Y, Bokov A, et al. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J Gerontol A Biol Sci Med Sci (2009) 64(12):1212–20. doi:10.1093/gerona/glp132
154. Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics (2003) 16(1):29–37. doi:10.1152/physiolgenomics.00122.2003
155. Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol (2011) 21(10):569–76. doi:10.1016/j.tcb.2011.06.008
156. Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med (2011) 51(2):327–36. doi:10.1016/j.freeradbiomed.2011.05.010
157. Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int (2014) 2014:238463. doi:10.1155/2014/238463
158. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev (2007) 87(1):99–163. doi:10.1152/physrev.00013.2006
159. Linnane A, Baumer A, Maxwell R, Preston H, Zhang C, Marzuki S. Mitochondrial gene mutation: the ageing process and degenerative diseases. Biochem Int (1990) 22(6):1067–76.
160. Cortopassi GA, Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res (1990) 18(23):6927–33. doi:10.1093/nar/18.23.6927
161. Cortopassi G, Shibata D, Soong N, Arnheim N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Natl Acad Sci U S A (1992) 89(16):7370–4. doi:10.1073/pnas.89.16.7370
162. McKenzie D, Bua E, McKiernan S, Cao Z, Aiken JM, Jonathan Wanagat. Mitochondrial DNA deletion mutations: a causal role in sarcopenia. Eur J Biochem (2002) 269(8):2010–5. doi:10.1046/j.1432-1033.2002.02867.x
163. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science (2011) 333(6046):1109–12. doi:10.1126/science.1201940
164. Linnane A, Ozawa T, Marzuki S, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet (1989) 333(8639):642–5. doi:10.1016/S0140-6736(89)92145-4
165. Cottrell D, Blakely E, Johnson M, Borthwick G, Ince P, Turnbull D. Mitochondrial DNA mutations in disease and ageing. In: Bock G, Goode JA, editors. Ageing Vulnerability: Causes and Interventions: Novartis Foundation Symposium. Wiley Online Library (2001). 235 p.
166. Wei Y-H, Lee H-C. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging 1. Exp Biol Med (2002) 227(9):671–82. doi:10.1177/153537020222700901
167. Kong Y, Trabucco SE, Zhang H. Oxidative stress, mitochondrial dysfunction and the mitochondria theory of aging. Aging (2014) 39:86–107.
168. Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, et al. Endurance exercise as a countermeasure for aging. Diabetes (2008) 57(11):2933–42. doi:10.2337/db08-0349
169. Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A (2011) 108(10):4135–40. doi:10.1073/pnas.1019581108
170. Johannsen DL, Ravussin E. The role of mitochondria in health and disease. Curr Opin Pharmacol (2009) 9(6):780–6. doi:10.1016/j.coph.2009.09.002
171. Stessman J, Hammerman-Rozenberg R, Cohen A, Ein-Mor E, Jacobs JM. Physical activity, function, and longevity among the very old. Arch Intern Med (2009) 169(16):1476–83. doi:10.1001/archinternmed.2009.248
172. Chakravarty EF, Hubert HB, Lingala VB, Fries JF. Reduced disability and mortality among aging runners: a 21-year longitudinal study. Arch Intern Med (2008) 168(15):1638–46. doi:10.1001/archinte.168.15.1638
173. Fiuza-Luces C, Garatachea N, Berger NA, Lucia A. Exercise is the real polypill. Physiology (Bethesda) (2013) 28(5):330–58. doi:10.1152/physiol.00019.2013
174. Parise G, Brose AN, Tarnopolsky MA. Resistance exercise training decreases oxidative damage to DNA and increases cytochrome oxidase activity in older adults. Exp Gerontol (2005) 40(3):173–80. doi:10.1016/j.exger.2004.09.002
175. Howald H, Hoppeler H, Claassen H, Mathieu O, Straub R. Influences of endurance training on the ultrastructural composition of the different muscle fiber types in humans. Pflügers Arch (1985) 403(4):369–76. doi:10.1007/BF00589248
176. Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol (1998) 43(2):217–23. doi:10.1002/ana.410430212
177. Wanagat J, Cao Z, Pathare P, Aiken JM. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J (2001) 15(2):322–32. doi:10.1096/fj.00-0320com
178. Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet (2006) 79(3):469–80. doi:10.1086/507132
179. Bua EA, McKiernan SH, Wanagat J, McKenzie D, Aiken JM. Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J Appl Physiol (2002) 92(6):2617–24. doi:10.1152/japplphysiol.01102.2001
180. Tarnopolsky M. Mitochondrial DNA shifting in older adults following resistance exercise training this paper article is one of a selection of papers published in this special issue, entitled 14th international biochemistry of exercise conference–muscles as molecular and metabolic machines, and has undergone the journal’s usual peer review process. Appl Physiol Nutr Metab (2009) 34(3):348–54. doi:10.1139/H09-022
181. Coggan AR, Spina RJ, King DS, Rogers MA, Rogers MA, Brown M, et al. Histochemical and enzymatic comparison of the gastrocnemius muscle of young and elderly men and women. J Gerontol (1992) 47(3):B71–6. doi:10.1093/geronj/47.3.B71
182. Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci U S A (1996) 93(26):15364–9. doi:10.1073/pnas.93.26.15364
183. Welle S, Bhatt K, Thornton CA. High-abundance mRNAs in human muscle: comparison between young and old. J Appl Physiol (2000) 89(1):297–304. doi:10.1152/jappl.2000.89.1.297
184. Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R, et al. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet (2006) 2(7):e115. doi:10.1371/journal.pgen.0020115
185. Melov S, Tarnopolsky MA, Beckman K, Felkey K, Hubbard A. Resistance exercise reverses aging in human skeletal muscle. PLoS One (2007) 2(5):e465. doi:10.1371/journal.pone.0000465
186. Campisi J, di Fagagna FDA. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi:10.1038/nrm2233
187. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell (2007) 130(2):223–33. doi:10.1016/j.cell.2007.07.003
188. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev (2010) 24(22):2463–79. doi:10.1101/gad.1971610
189. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu C-P, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science (1998) 279(5349):349–52. doi:10.1126/science.279.5349.349
190. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A (1995) 92(20):9363–7. doi:10.1073/pnas.92.20.9363
191. Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, Von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell (2009) 8(3):311–23. doi:10.1111/j.1474-9726.2009.00481.x
192. Hoenicke L, Zender L. Immune surveillance of senescent cells—biological significance in cancer-and non-cancer pathologies. Carcinogenesis (2012) 33(6):1123–6. doi:10.1093/carcin/bgs124
193. Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature (2011) 479(7374):547–51. doi:10.1038/nature10599
194. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature (2007) 445(7128):656–60. doi:10.1038/nature05529
195. Van Deursen JM. The role of senescent cells in ageing. Nature (2014) 509(7501):439–46. doi:10.1038/nature13193
196. Bigley AB, Spielmann G, LaVoy EC, Simpson RJ. Can exercise-related improvements in immunity influence cancer prevention and prognosis in the elderly? Maturitas (2013) 76(1):51–6. doi:10.1016/j.maturitas.2013.06.010
197. Gorgoulis VG, Halazonetis TD. Oncogene-induced senescence: the bright and dark side of the response. Curr Opin Cell Biol (2010) 22(6):816–27. doi:10.1016/j.ceb.2010.07.013
198. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16 INK4a. Cell (1997) 88(5):593–602. doi:10.1016/S0092-8674(00)81902-9
199. Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21 CIP1, but not p16 INK4a. Mol Cell (2004) 14(4):501–13. doi:10.1016/S1097-2765(04)00256-4
200. Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature (2014) 506(7488):316–21. doi:10.1038/nature13013
201. Pareja-Galeano H, Sanchis-Gomar F, Lucia A. p16INK4a, NAD+, and sestrins: new targets for combating aging-related chronic illness? J Cell Physiol (2014) 229(11):1575–6. doi:10.1002/jcp.24627
202. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol (2011) 192(4):547–56. doi:10.1083/jcb.201009094
203. Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer (2009) 9(2):81–94. doi:10.1038/nrc2560
204. Tchkonia T, Zhu Y, Van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest (2013) 123(3):966. doi:10.1172/JCI64098
205. Huang C-C, Chiang W-D, Huang W-C, Huang C-Y, Hsu M-C, Lin W-T. Hepatoprotective effects of swimming exercise against d-galactose-induced senescence rat model. Evid Based Complement Alternat Med (2013) 2013:275431. doi:10.1155/2013/275431
206. Song Z, Von Figura G, Liu Y, Kraus JM, Torrice C, Dillon P, et al. Lifestyle impacts on the aging-associated expression of biomarkers of DNA damage and telomere dysfunction in human blood. Aging Cell (2010) 9(4):607–15. doi:10.1111/j.1474-9726.2010.00583.x
207. Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Curr Opin Immunol (2010) 22(4):507–13. doi:10.1016/j.coi.2010.05.003
208. Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature (2006) 443(7110):448–52. doi:10.1038/nature05091
209. Conboy IM, Rando TA. Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches. Cell Cycle (2012) 11(12):2260–7. doi:10.4161/cc.20437
210. Gruber R, Koch H, Doll BA, Tegtmeier F, Einhorn TA, Hollinger JO. Fracture healing in the elderly patient. Exp Gerontol (2006) 41(11):1080–93. doi:10.1016/j.exger.2006.09.008
211. Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature (2007) 447(7145):725–9. doi:10.1038/nature05862
212. Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature (2006) 443(7110):421–6. doi:10.1038/nature05159
213. Xia WH, Li J, Su C, Yang Z, Chen L, Wu F, et al. Physical exercise attenuates age-associated reduction in endothelium-reparative capacity of endothelial progenitor cells by increasing CXCR4/JAK-2 signaling in healthy men. Aging Cell (2012) 11(1):111–9. doi:10.1111/j.1474-9726.2011.00758.x
214. Verdijk LB, Snijders T, Drost M, Delhaas T, Kadi F, van Loon LJ. Satellite cells in human skeletal muscle; from birth to old age. Age (Dordr) (2014) 36(2):545–57. doi:10.1007/s11357-013-9583-2
215. Jang Y, Sinha M, Cerletti M, Dall’Osso C, Wagers AJ. Skeletal muscle stem cells: effects of aging and metabolism on muscle regenerative function. Cold Spring Harb Symp Quant Biol (2011) 76:101–11. doi:10.1101/sqb.2011.76.010652
216. Renault V, Thorne LE, Eriksson PO, Butler-Browne G, Mouly V. Regenerative potential of human skeletal muscle during aging. Aging Cell (2002) 1(2):132–9. doi:10.1046/j.1474-9728.2002.00017.x
217. Gopinath SD, Rando TA. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell (2008) 7(4):590–8. doi:10.1111/j.1474-9726.2008.00399.x
218. Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab (2007) 292(1):E151–7. doi:10.1152/ajpendo.00278.2006
219. Verdijk LB, Snijders T, Beelen M, Savelberg HH, Meijer K, Kuipers H, et al. Characteristics of muscle fiber type are predictive of skeletal muscle mass and strength in elderly men. J Am Geriatr Soc (2010) 58(11):2069–75. doi:10.1111/j.1532-5415.2010.03150.x
220. Shefer G, Rauner G, Stuelsatz P, Benayahu D, Yablonka-Reuveni Z. Moderate-intensity treadmill running promotes expansion of the satellite cell pool in young and old mice. FEBS J (2013) 280(17):4063–73. doi:10.1111/febs.12228
221. Shefer G, Rauner G, Yablonka-Reuveni Z, Benayahu D. Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS One (2010) 5(10):e13307. doi:10.1371/journal.pone.0013307
222. Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab (2010) 11(6):554–65. doi:10.1016/j.cmet.2010.04.001
223. Kawiak J, Brzóska E, Grabowska I, Hoser G, Stremińska W, Wasilewska D, et al. Contribution of stem cells to skeletal muscle regeneration. Folia Histochem Cytobiol (2006) 44(2):75–9.
224. Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol (2001) 91(2):534–51. doi:10.1152/jappl.2001.91.2.534
225. Cermak NM, Snijders T, McKay BR, Parise G, Verdijk LB, Tarnopolsky MA, et al. Eccentric exercise increases satellite cell content in type II muscle fibers. Med Sci Sports Exerc (2013) 45(2):230–7. doi:10.1249/MSS.0b013e318272cf47
226. Verdijk LB, Gleeson BG, Jonkers RA, Meijer K, Savelberg HH, Dendale P, et al. Skeletal muscle hypertrophy following resistance training is accompanied by a fiber type-specific increase in satellite cell content in elderly men. J Gerontol A Biol Sci Med Sci (2009) 64(3):332–9. doi:10.1093/gerona/gln050
227. Bellamy LM, Joanisse S, Grubb A, Mitchell CJ, McKay BR, Phillips SM, et al. The acute satellite cell response and skeletal muscle hypertrophy following resistance training. PLoS One (2014) 9(10):e109739. doi:10.1371/journal.pone.0109739
228. Snijders T, Verdijk LB, Smeets JS, McKay BR, Senden JM, Hartgens F, et al. The skeletal muscle satellite cell response to a single bout of resistance-type exercise is delayed with aging in men. Age (Dordr) (2014) 36(4):9699. doi:10.1007/s11357-014-9699-z
229. McKay BR, Ogborn DI, Bellamy LM, Tarnopolsky MA, Parise G. Myostatin is associated with age-related human muscle stem cell dysfunction. FASEB J (2012) 26(6):2509–21. doi:10.1096/fj.11-198663
230. Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell (2009) 5(3):279–89. doi:10.1016/j.stem.2009.06.017
231. Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal (2009) 2(98):ra75. doi:10.1126/scisignal.2000559
232. Yilmaz ÖH, Katajisto P, Lamming DW, Gültekin Y, Bauer-Rowe KE, Sengupta S, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature (2012) 486(7404):490–5. doi:10.1038/nature11163
233. Florian MC, Dörr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell (2012) 10(5):520–30. doi:10.1016/j.stem.2012.04.007
234. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi:10.1016/j.cell.2012.03.017
235. Rando TA, Chang HY. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell (2012) 148(1):46–57. doi:10.1016/j.cell.2012.01.003
236. Russell SJ, Kahn CR. Endocrine regulation of ageing. Nat Rev Mol Cell Biol (2007) 8(9):681–91. doi:10.1038/nrm2234
237. Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, et al. Hypothalamic programming of systemic ageing involving IKK-[bgr], NF-[kgr] B and GnRH. Nature (2013) 497(7448):211–6. doi:10.1038/nature12143
238. Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) (2012) 4(3):166. doi:10.18632/aging.100444
239. Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol (2011) 27:585–610. doi:10.1146/annurev-cellbio-092910-154234
240. Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science (2012) 337(6102):1678–84. doi:10.1126/science.1224922
241. Pont AR, Sadri N, Hsiao SJ, Smith S, Schneider RJ. mRNA decay factor AUF1 maintains normal aging, telomere maintenance, and suppression of senescence by activation of telomerase transcription. Mol Cell (2012) 47(1):5–15. doi:10.1016/j.molcel.2012.04.019
242. Lai RY, Ljubicic V, D’souza D, Hood DA. Effect of chronic contractile activity on mRNA stability in skeletal muscle. Am J Physiol Cell Physiol (2010) 299(1):C155–63. doi:10.1152/ajpcell.00523.2009
243. Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev (2007) 21(24):3244–57. doi:10.1101/gad.1588507
244. Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, et al. NF-κB inhibition delays DNA damage–induced senescence and aging in mice. J Clin Invest (2012) 122(7):2601. doi:10.1172/JCI45785
245. Xie J, Zhang X, Zhang L. Negative regulation of inflammation by SIRT1. Pharmacol Res (2013) 67(1):60–7. doi:10.1016/j.phrs.2012.10.010
246. Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, et al. SirT1 regulates adipose tissue inflammation. Diabetes (2011) 60(12):3235–45. doi:10.2337/db11-0616
247. Yao H, Chung S, Hwang J-W, Rajendrasozhan S, Sundar IK, Dean DA, et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest (2012) 122(6):2032. doi:10.1172/JCI60132
248. Zhang Z, Lowry SF, Guarente L, Haimovich B. Roles of SIRT1 in the acute and restorative phases following induction of inflammation. J Biol Chem (2010) 285(53):41391–401. doi:10.1074/jbc.M110.174482
249. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell (2012) 11(2):345–9. doi:10.1111/j.1474-9726.2012.00795.x
250. Pedersen BK. Muscular interleukin-6 and its role as an energy sensor. Med Sci Sports Exerc (2012) 44(3):392–6. doi:10.1249/MSS.0b013e31822f94ac
251. Ball D. Metabolic and endocrine response to exercise: sympathoadrenal integration with skeletal muscle. J Endocrinol (2015) 224(2):R79–95. doi:10.1530/JOE-14-0408
252. Mikkelsen U, Couppe C, Karlsen A, Grosset J, Schjerling P, Mackey A, et al. Life-long endurance exercise in humans: circulating levels of inflammatory markers and leg muscle size. Mech Ageing Dev (2013) 134(11):531–40. doi:10.1016/j.mad.2013.11.004
253. Nicklas BJ, Brinkley TE. Exercise training as a treatment for chronic inflammation in the elderly. Exerc Sport Sci Rev (2009) 37(4):165. doi:10.1097/JES.0b013e3181b7b3d9
254. Beavers KM, Brinkley TE, Nicklas BJ. Effect of exercise training on chronic inflammation. Clin Chim Acta (2010) 411(11):785–93. doi:10.1016/j.cca.2010.02.069
255. American College of Sports Medicine (ACSM). ACSM’s Guidelines for Exercise Testing and Prescription. Philadelphia: Lippincott Williams & Wilkins (2018).
256. Colbert LH, Visser M, Simonsick EM, Tracy RP, Newman AB, Kritchevsky SB, et al. Physical activity, exercise, and inflammatory markers in older adults: findings from the Health, Aging and Body Composition Study. J Am Geriatr Soc (2004) 52(7):1098–104. doi:10.1111/j.1532-5415.2004.52307.x
257. Garber CE, Blissmer B, Deschenes MR, Franklin BA, Lamonte MJ, Lee I-M, et al. American College of Sports medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc (2011) 43(7):1334–59. doi:10.1249/MSS.0b013e318213fefb
Keywords: aging, physical exercise, hallmarks, cellular, molecular, life span
Citation: Rebelo-Marques A, De Sousa Lages A, Andrade R, Ribeiro CF, Mota-Pinto A, Carrilho F and Espregueira-Mendes J (2018) Aging Hallmarks: The Benefits of Physical Exercise. Front. Endocrinol. 9:258. doi: 10.3389/fendo.2018.00258
Received: 30 December 2017; Accepted: 03 May 2018;
Published: 25 May 2018
Edited by:
Romeu Mendes, University Porto, PortugalReviewed by:
Vladimir I. Titorenko, Concordia University, CanadaJ. A. Taylor, Harvard Medical School, United States
Copyright: © 2018 Rebelo-Marques, De Sousa Lages, Andrade, Ribeiro, Mota-Pinto, Carrilho and Espregueira-Mendes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre Rebelo-Marques, alexRmarques@gmail.com