 Chen Xu1*
Chen Xu1* Peng Lin1
Peng Lin1 Luni Sun1
Luni Sun1 Hongmei Chen2
Hongmei Chen2 Wei Xing1
Wei Xing1 Manoj Kamalanathan3
Manoj Kamalanathan3 Patrick G. Hatcher2
Patrick G. Hatcher2 Maureen H. Conte4
Maureen H. Conte4 Antonietta Quigg3
Antonietta Quigg3 Peter H. Santschi1
Peter H. Santschi1- 1Department of Marine Science, Texas A&M University at Galveston, Galveston, TX, United States
- 2Department of Chemistry and Biochemistry, Old Dominion University, Norfolk, VA, United States
- 3Department of Marine Biology, Texas A&M University at Galveston, Galveston, TX, United States
- 4Bermuda Institute of Ocean Sciences, St. George’s, Bermuda
Marine sinking particulate organic matter (POM), acting as a link between surface primary production and burial of organic matter in marine sediments, undergoes a variety of physical and biochemical alterations on its way to the deep ocean, resulting in an increase in its un-characterizable proportion with diagenesis. Further, the binding ligands in POM for iron, an essential nutrient to marine life and tightly coupled with organic matter, has rarely been studied. In the current study, we employed an approach combining sequential extraction with ultrahigh resolution mass spectrometry (ESI-FTICRMS), in order to explore and unravel the chemical characteristics of organic matter compounds relevant to marine particle flux within the mesopelagic and deep ocean, with a focus on the potential iron-carrying molecules. With increasing depth, POM increases in aliphaticity, and decreases in intensity-normalized O/C ratios, aromatics, and carboxylic-rich alicyclic molecules (CRAM)-like compounds. The potential iron-carrying molecules account for ∼14% of total identified molecules, and appear to have been incorporated into the marine particles via ion complexation, hydrophobic interaction, and/or interlayered “occlusion.” The relative abundance of iron-binding organic molecules in these three operationally-defined categories changes with depth: “surficially-complexed” fraction decreases with depth, the “interlayered-occluded” fraction increases to a comparable extent and “hydrophobic interaction” fraction occurs at all depths. Collectively, the potential iron-carrying organic molecules exhibit a set of unique molecular characteristics: a relatively lower average H/C ratio and a higher O/C ratio compared to bulk POM, a dominance of aromatics, black carbon-like compounds and CRAM-like compounds, and minor amounts of aliphatics. These molecules exhibit partial similar molecular features as precursors formed from photochemical reactions in the surface ocean, but they have been greatly modified by flux processes. Noticeably, a minor fraction of these iron-carrying molecules (<1%) was identified to contain hydroxamate-like moieties [N(OH)-COOH], the key functionality of one of the strongest iron-binding ligands in the dissolved phase. This agrees with improved spectrophotometric results and corroborates their presence in the POM. These hydroxamate-like moieties play an important role in controlling the distributions and fluxes of Fe and particle-reactive radionuclides with similar chemical complexing properties as Fe (e.g., thorium) in the ocean.
Introduction
Sinking particles, composed of organic (sinking particulate organic matter, POM) and inorganic material, are the critical conduit for delivering photosynthetically-fixed organic carbon, food and energy from the surface ocean to the deep sea (Verdugo et al., 2004; Boeuf et al., 2019). They vary in size and shape, consisting of a suit of dead phytoplankton, zooplankton carcasses, larvacean houses, fecal pellet, etc. (Omand et al., 2020). Typically accounting for <20% of the surface primary production, sinking POM forms through a variety of biological, physical and chemical processes that alter its composition and attenuate the flux on its way to the sea floor (Volkman and Tanoue, 2002). Despite its paramount role in global carbon cycling, sinking POM is still a poorly understood organic matter pool in terms of the physical and chemical processes it undergoes while sinking (Lee, 2002).
Iron, as a trace element in the dissolved phase (dFe: 0.02–1.0 nmol L–1; Bruland et al., 1991) at the surface ocean, is tightly coupled with organic carbon in both of their biogeochemical cycles and also regulates the export of POM flux from the euphotic zone to the deep ocean. For example, in high-nutrient, low-chlorophyll (HNLC) regions, the low surface ocean dFe concentration can limit primary productivity by as much as 30–40% (Marchetti and Maldonado, 2016). In response to the scarcity of Fe, microorganisms produce a variety of low-molecular-weight (LMW, <1 kDa) organic compounds, i.e., siderophores, with strong binding affinity to metals, especially Fe(III) (Reid et al., 1993; Neilands, 1995; Butler, 1998, 2005; Winkelmann, 2002; Vraspir and Butler, 2009; Butler and Theisen, 2010). Siderophores are typically multidentate, oxygen-donor ligands that include hydroxamate, catecholate and α-hydroxy-carboxylate types. Out of these three types of siderophores, hydroxamate has been the focus of many studies on marine dFe binding ligands (Macrellis et al., 2001; Mawji et al., 2008; Velasquez et al., 2011; Boiteau et al., 2016). First, dissolved hydroxamate siderophores (300–1000 Da) are ubiquitous in oceanic systems. For example, in surface waters and water below the photic zone of the Central California upwelling, more than half of the Fe-binding compounds have been characterized as either hydroxamate type or catecholate type siderophores (Macrellis et al., 2001). In contrast, in the non-Fe-limiting regions (e.g., the North Atlantic Ocean), the characterizable siderophores exclusively consist of hydroxamate siderophores, which are produced by heterotrophic bacteria and fungi (Raines et al., 2015) in the concentration range of 3–20 pM (Mawji et al., 2008). In the eastern Pacific Ocean, two types of hydroxamate siderophores (hydrophilic siderophore Ferrioxamine B and amphiphilic amphibactins) dominate the siderophore pool and alternate in their abundance across a transect from coastal waters to the HNLC region to the oligotrophic ocean (Boiteau et al., 2016). Second, dissolve hydroxamate siderophores have the highest stability constants (as logK) that range from 16 to 62 at 25°C for complexation of Fe(H2O)63+ with the fully deprotonated ligand of hydroxamate functionality (N(OH)-COOH) (Boukhalfa and Crumbliss, 2002). The tri-hydroxamate siderophores have an average conditional stability constant for Fe of 30.6 (Kraemer et al., 2005; Ahmed and Holmstrom, 2014). Third, hydroxamate siderophores are hydrophilic or amphiphilic, and they have relatively higher resistance towards photolysis compared to other types of Fe-binding ligands (Barbeau et al., 2001, 2003). These properties likely enhance their transport and stability in the upper ocean.
Most current studies have focused on the iron-carrying ligands in the dissolved phase (Macrellis et al., 2001; Hunter and Boyd, 2007; Vraspir and Butler, 2009; Bundy et al., 2016), and only recently it was suggested that hydroxamate-like moieties also existed in the particles/colloids (Chuang et al., 2013, 2015a). Yet the mechanisms by which these hydroxamate-like moieties are formed/incorporated in the particulate or colloidal phases, their molecular characteristics, their micro-molecular environment, and whether these potential Fe-binding sites play a significant role in Fe biogeochemical cycling remain largely unknown. The reason lies in the analytical difficulty of qualitatively and quantitatively assessing these iron-carrying ligands in the particulate phase, especially those hydroxamate-like moieties, the latter of which are a minor component of the particulate organic carbon (POC) pool [determined as <1.5% of POC in Chuang et al. (2013, 2015a)], and thus “masked” when applying techniques like NMR, FTIR or C K-edge X-ray absorption spectroscopy, and/or gas and liquid chromatography.
In the present study, electrospray ionization (ESI) Fourier-transform ion cyclotron resonance mass spectrometry (FTICRMS) was applied to elucidate the composition of organic matter from marine settling particles, using a sequential extraction method. Our objectives were to: (1) examine the overall molecular information of the settling particulate organic matter (POM) with depth, i.e., what the micro-molecular environment is like for the iron-complexing moieties; (2) characterize the molecular features of iron-complexing sites in fractions of the marine particles that were probed by different extraction reagents, thus representing possible mechanisms for their incorporation into the settling particles; and (3) investigate their distributions at three depths (500, 1500, and 3200 m) in the northwestern Sargasso Sea, where there have been several studies in the particle and elemental fluxes with the Bermuda Time-Series Site (Conte et al., 2019).
Materials and Methods
Here we apply an optimized sequential extraction method (Chuang et al., 2015b) followed by ESI-FTICRMS, to characterize the molecular features of the Fe-complexing sites in sinking particles (500, 1500, and 3200 m) in the northwestern Sargasso Sea near Bermuda. Using Fe(III) as the tracer, we identified potentially iron-complexing ligands in different operationally-defined fractions: an EDTA (Ethylenediaminetetraacetic acid disodium salt dihydrate) fraction of “surficial metal-complexed” POM (EDTA is used to chelate the surficial cations, dissociate the POM-metal complex, and thus the OM will be released into the solution); a SDS (sodium dodecyl sulfate) fraction of molecules that are bound to particles via hydrophobic interactions, i.e., “hydrophobically-sorbed POM;” and a HF (hydrofluoric acid) fraction of residual “interlayered and interstitial” mineral complexed/occluded POM.
Sample Collection
Particle flux material was collected by the Bermuda Oceanic Flux Program (OFP) time-series, located at 31°50′N, 64°10′W, 3000 m) brine (40 ppt) and amended with ultra–high purity HgCl2 (200 mg/L) to minimize microbial degradation of the POM during sample collection. Details of sample collection and analytical methods are described elsewhere (Conte et al., 2019). Samples analyzed herein were collected in 2005 and 2006, and stored refrigerated. Additional sample information is provided in Supplementary Table S1.
Sequential Extraction of Marine Particles
The bulk hydroxamate concentration was determined using samples collected at different time points (n = 3 for each depth, see below section “Hydroxamate–Like Functionalities Determination in Marine Particles” and Supplementary Table S1). Due to the limited sample material, the sequential extraction required pooling of three discrete samples collected at different time points of the same depth (Supplementary Table S1). The sequential extraction procedure is shown in Figure 1. Briefly, ∼10 mg of particles from each depth were first incubated with 1 mL 100 mM EDTA solution (pH 8.0) at 4°C overnight on an end–to–end orbital shaker. The extractant soluble phase was separated from the particles via centrifugation and filtration with a syringe filter (PTFE membrane, diameter 25 mm, pore size 0.45 μm). The filtrate was extensively diafiltered (10–12 times) against ultrapure water with an Amicon Ultra–4 centrifugal filter unit with a membrane cutoff of 3 kDa. This retentate (>3 kDa and <0.45 μm) is defined as “EDTA extract.” The residual particles were then re–suspended with 1 mL 1% SDS in 0.01 M Tris buffer (pH 6.8) at 95°C for 1 h. This extractant was processed the same way as described for “EDTA extract” and defined as the “SDS extract.” The residual pellet from SDS extraction was then washed three times with ultrapure water and subsequently treated with 1 mL 52% HF on ice for 1 h. The slurry was evaporated to dryness under a N2 stream. After this, the residual particle material was re–suspended in 1 mL Tris–HCl (250 mM, pH 8.0) and centrifuged at 5000 × g for 15 min to separate the particles from the solution, the latter of which was processed as described previously by ultrafiltration to isolate the “HF extract” (Chuang et al., 2015b).
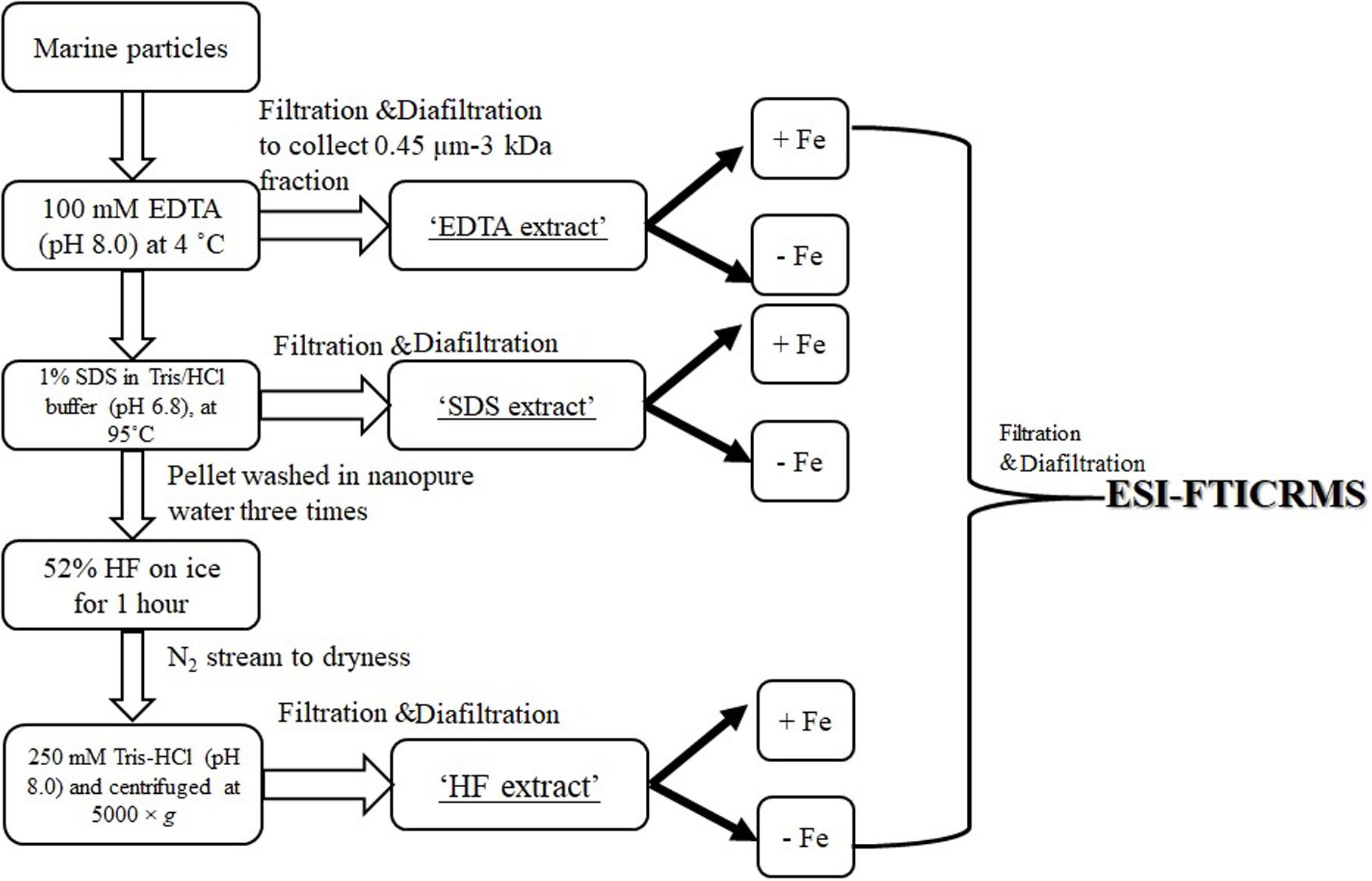
Figure 1. Flow chart of sequential extraction and electrospray ionization (ESI) Fourier–transform ion cyclotron resonance mass spectrometry (FTICRMS) analysis.
In order to better understand the stepwise extraction efficiency of POM from the marine particles, particles collected at different time points from the three depths (Supplementary Table S1) were well mixed (∼5 mg each) with a spatula and four replicate particles were extracted in parallel to obtain the particles from each sequential step for elemental analysis, using a Perkin–Elmer CHNS/O 2400 analyzer. The mass loss and organic carbon contents of the residual particles after each extraction step were determined and the analytical error of these four replicates were reported. For elemental analysis, the pre–weighed sample was directly acidified in a silver capsule three times and was heated at 60°C overnight to remove any residual acid, prior to the analysis. Acetanilide (71.09% C, 6.71% H, 10.36% N) was used as the calibration standard.
Incubation of Fe (III) and Sequentially-Extracted POM Fractions
Sequential extractants, in the matrix of ultrapure water, from marine particles (section “Sequential Extraction of Marine Particles”) were split into two aliquots, one amended with FeCl3 (10 μM) and one without (control). The pH difference between these two treatments [i.e., Fe (+) and Fe (-)] were within 0.5 and both were around neutral. These were incubated at 4°C on an end-to-end orbital shaker in the dark for 3 days. After that, excess FeCl3 was removed by extensively diafiltering the sample against ultrapure water with an Amicon Ultra-4 centrifugal filter unit with a membrane cutoff of 3 kDa (Figure 1), and the retentate (>3 kDa) was kept for further ESI-FTICRMS analysis. The sample of the Fe (-) treatment was processed the same manner (with diafiltration) for comparison purpose.
ESI-FTICRMS Analysis
Marine particle extracts were diluted with methanol (1:1, v/v) immediately before injection into a Bruker Daltonics 12 Tesla Apex Qe ESI-FTICRMS (College of Sciences Major Instrumentation Cluster, COSMIC, Old Dominion University, Virginia). Positive ion mode was used, as the molecules of interest are iron-carrying molecules, which have better ionization efficiency in this mode (Waska et al., 2016). Samples were continuously injected by a syringe pump (infusion rate of 120 μL/h), and electrospray voltages were optimized for each sample. Ions were accumulated in a hexapole for 2.0 s before being transferred to the ICR cell and 300 transients were co-added. Fourier transformation and magnitude calculation of the free induction decay signal (FID) was accomplished using the Bruker Daltonics Data Analysis software (Version 4.0 SP 2). A total of 18 samples (three depths × three extractions × two treatments), as well as six procedure blanks (three extractions × two treatments) following the same steps as the samples (one for Fe addition and the other for no Fe addition), were included for FTICRMS analysis. Selected samples were analyzed multiple times to ensure the analytical reproducibility and all samples were analyzed within the same week to ensure stable instrumentation parameters and conditions for all samples (Sleighter et al., 2012).
Mass spectra were externally calibrated with a polyethylene glycol standard and then internally calibrated using a set of m/z values within the samples. Several criteria were met for picking out the calibration peak list: (1) peaks have a signal-to-noise (S/N) ratio ≥5, (2) peaks that have intermediate intensities to minimize instrumental artifacts that yield peaks with too high or too low intensities, (3) peaks have an error (i.e., the difference between the exact mass of calculated formula and the measured m/z) of <0.5 ppm, and (4) peaks can generate only one possible formula containing only C, H, O and/or Na elements, and all formulas have their corresponding structures found in Pubchem database. There was at least one calibration point every 15–20 m/z units (i.e., every other CH2), across the entire m/z region (200–1400 m/z). Linear mode calibration was performed on each dataset.
All m/z lists, with an S/N ≥ 4, were first searched for 13C peaks and confirmed they were mostly singly-charged. Those 13C peaks were not included in the formula calculations. Moreover, the m/z values present in the procedure blank spectra were excluded from the sample mass list. A molecular formula calculator (Molecular Formula Calc Version 1.2.3 NHMFL, 2016) generated empirical formula matches within 1.0 ppm using elemental ranges of C(≥ 5)H(≥ 5)N(0–10)O(≥ 0)S(0–2)Na(0–1)Fe(0–1). All formulas were screened to eliminate those that are unlikely to occur in OM, according to a list of selection criteria (Neubert et al., 2002; Kind and Fiehn, 2007; Stubbins et al., 2010; Walker et al., 2016). Kendrick mass defect (KMD) analyses were then performed to look for homologous series of CH2, CH2O, COO, O, H2O, H2, etc., respectively, formed by at least two unique formulas (i.e., only one possible formula corresponds to a single m/z value after the formula rule’s screening). If there are multiple possible formulas for a single m/z value (usually those with m/z values >400 Da), the one that fell within a homologous series was kept as the more likely formula (Sleighter and Hatcher, 2007). Subsequently, multiple formulas of one m/z value were screened by searching chemical structures in the Pubchem database. Formulas with no chemical structures identified were excluded from the final formula assignment. Fe-carrying molecules were also examined if their 54Fe isotopic peak could be found (i.e., the singly-charged 56Fe and 54Fe-carrying peak m/z difference is 1.99533 ± 0.00070 and the isotopic ratio of 54Fe/56Fe as 0.063 ± 0.030) (Boiteau and Repeta, 2015; Walker et al., 2016). Adduct of iron hydroxide with methanol is likely irrelevant, since the mass list of the procedure blank (with the addition of FeCl3 yet without the addition of particle extracts) had been subtracted from the sample mass lists. Besides, this issue should occur much less likely at μM FeCl3 concentrations (Waska et al., 2016).
Molecules in each sample were categorized by both elemental classes and compound classification. The double bond equivalent (DBE) was calculated as DBE = 1+ 0.5 (2C-H+N). Aliphatic compounds were assigned to formulas with DBE: C < 0.3 and H:C = 1.0–3.0. Aromatic compounds and condensed aromatic compounds were assigned to formulas with aromaticity indices [AI, calculated by (1+C-O-S-0.5(N+H))/(C-O-S-N)] >0.5 and >0.67, respectively. A modified AI, which was suggested to be more inclusive by dividing the possible number of carbonyl unsaturation by two: AImod = (1+C-0.5O-S-0.5(H+N))/(C-0.5O-S-N) (Stubbins et al., 2010; Koch and Dittmar, 2016), was also calculated. Black carbon was defined as molecules with O:C 0.3–0.6, H:C 0.5–0.8 (Kim et al., 2004). Carboxylic-rich alicyclic molecules (CRAM) were defined as molecules having DBE: C 0.30–0.68, H 0.20–0.95, and O 0.77–1.75 (Hertkorn et al., 2006).
To unambiguously identify a specific functional group, a tandem mass spectrometry analysis (MS/MS) would be required (Sleighter and Hatcher, 2007, 2008). However, MS/MS analysis would have great difficulty to identify a trace component, i.e., a hydroxamate functional group (<1% of POC, see section “Bulk Hydroxamate Concentrations in Marine Particles”) using the bulk OM without any extensive pre-separation and concentration. Instead, a KMD (Kendrick Mass Defect) analysis was conducted to search the whole mass list for molecules that fall in some homologous series of the key functionality of hydroxamate, N(OH)-COOH (with the formula total H number being corrected for Na adducts or Fe complexation). A homologous series of N(OH)-COOH was formed, if at least two formulas differ only by [N(OH)-COOH]n.
Hydroxamate-Like Functionalities Determination in Marine Particles
Bulk hydroxamate concentration of the particles was quantified using the classical “Csaky” spectrophotometric method (Gillam et al., 1981), which required a hydrolysis step to first release the bound hydroxamic acid. The un-hydrolyzed particle yielded no response to the reactions, suggesting it is hydroxamic acid rather than hydroxylamine that is being detected. We noticed a rapid “browning” effect of this method when analyzing particle samples, likely caused by sulfuric acid. This effect was corrected by subtracting the absorbance of a control sample without the addition of the final coloring reagent from that of a sample aliquot with the addition of the final coloring reagent. The net absorbance was used to calculate hydroxamate concentration from the calibration curve using acetohydroxamic acid (AHA) as the standard. This modified method was validated with siderophore standards added to marine particles or soil particles (Xu et al., 2015). In addition, we also critically scrutinized if there could be any other interferences by the following biomolecule classes: (1) neutral polysaccharides (e.g., dextran); (2) acidic polysaccharides and monosaccharides (e.g., alginic acid and glucuronic acid); (3) proteins/amino acid (e.g., cytochrome c and phenylalanine); (4) fatty acids (e.g., palmitic acid); (5) humic acid (e.g., 1S104H from IHSS); (6) fulvic acid (2S101F from IHSS); (7) deoxyribonuclease. We verified that none of these compounds caused any significant interferences to this method (i.e., their absorbance is the same as in ultrapure water) (Xu et al., data not shown).
The presence of N(OH)-COOH in the marine particles, either detected as “positive” with the “Csaky” method, or calculated by KMD analysis to fall in the homologous N(OH)-COOH series, are defined as “hydroxamate-like” molecules below.
Results
Bulk Hydroxamate Concentrations in Marine Particles
The average hydroxamate concentration of the settling particles at 500, 1500, and 3200 m depth was 19.4 (± 12.4), 8.7 (± 1.4), and 7.3 (± 0.1) μM-AHA eq./g-particle (n = 3 for each depth, Supplementary Table S1 and Figure 2A). While particulate hydroxamate decreased with water depth, the POC-normalized hydroxamate concentrations decreased only slightly or remained steady (Figure 2B, ∼0.2–0.6% POC).
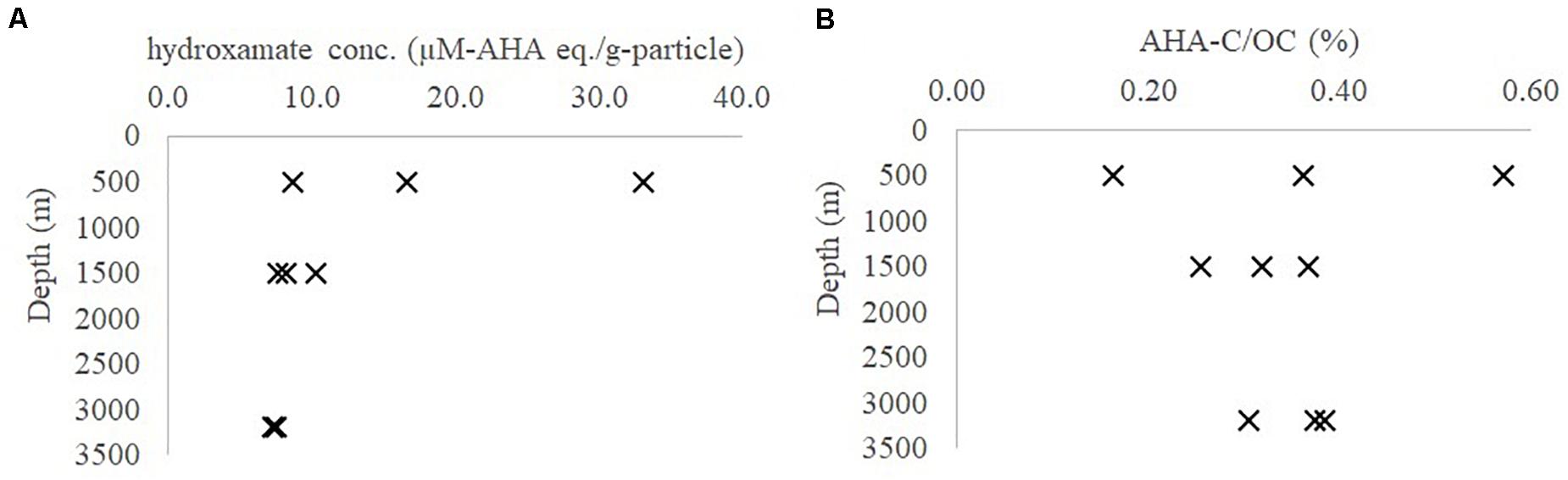
Figure 2. Hydroxamate concentration in (A) μM-AHA eq./g-particle; (B) AHA-C/OC (%) in marine particles from the northern Atlantic Ocean at different seasons (AHA, acetohydroxamic acid; OC, organic carbon).
Relative Distribution of POM in Sequential Extracts With Depth
The mass and organic carbon changes after each sequential extraction step are shown in Supplementary Table S2. The organic carbon (%, residual mass) generally increased with each extraction step, from the original value of (5.55% ± 0.05) to (45.48% ± 1.90) after HF extraction. EDTA extraction released (67% ± 2) of the particle mass and (39.74% ± 7.52) of the total original organic carbon into the solution. SDS and HF extraction released similar amounts of the original mass (13% ± 2) and (17% ± 3), yet quite different amounts of original organic carbon: (30.17% ± 2.35) and (7.46% ± 0.95), respectively. Their respective extraction efficiencies, which were calculated as the loss of particulate mass or organic carbon (the part being extracted into the solution) to those in the previous particles/residues being extracted, change in the following order: HF (86% ± 3) > EDTA (67% ± 2) > SDS (41% ± 7) by mass, and SDS: (49.92% ± 2.34) > EDTA (39.74% ± 7.52) > HF (24.68% ± 2.74) based on organic carbon. These rankings are likely to be different if one changes the sequential extraction order, which is not the focus of the current study. These three consecutive steps removed substantial amounts of original mass (98%) and of original organic carbon (77%) from the marine sinking particles, supporting the idea that ESI-FTICRMS analysis of these fractions represents the overall composition of OM in the marine particles.
The ESI-FTICRMS intensity percentages of the three extracts with depth is given in Figure 3. For each depth, the mass lists of the three extract (EDTA, SDS and HF) of the non-FeCl3-amended samples were combined. If there are common m/z values shared by different samples within the same depth, the average intensity was adopted. At 500 m, the POM consists mostly of SDS-extractable material (87%), followed by EDTA-extractable compounds (12%), and the HF-extractable material is only a very minor fraction (1%). At 1500 m depth, the SDS fraction decreases to 51% of the total extractable POM, while the EDTA and HF fractions increase (22 and 27%, respectively). At 3200 m, POM compounds are more evenly distributed and comprise 34, 46, and 20% for the EDTA, SDS and HF fractions, respectively.
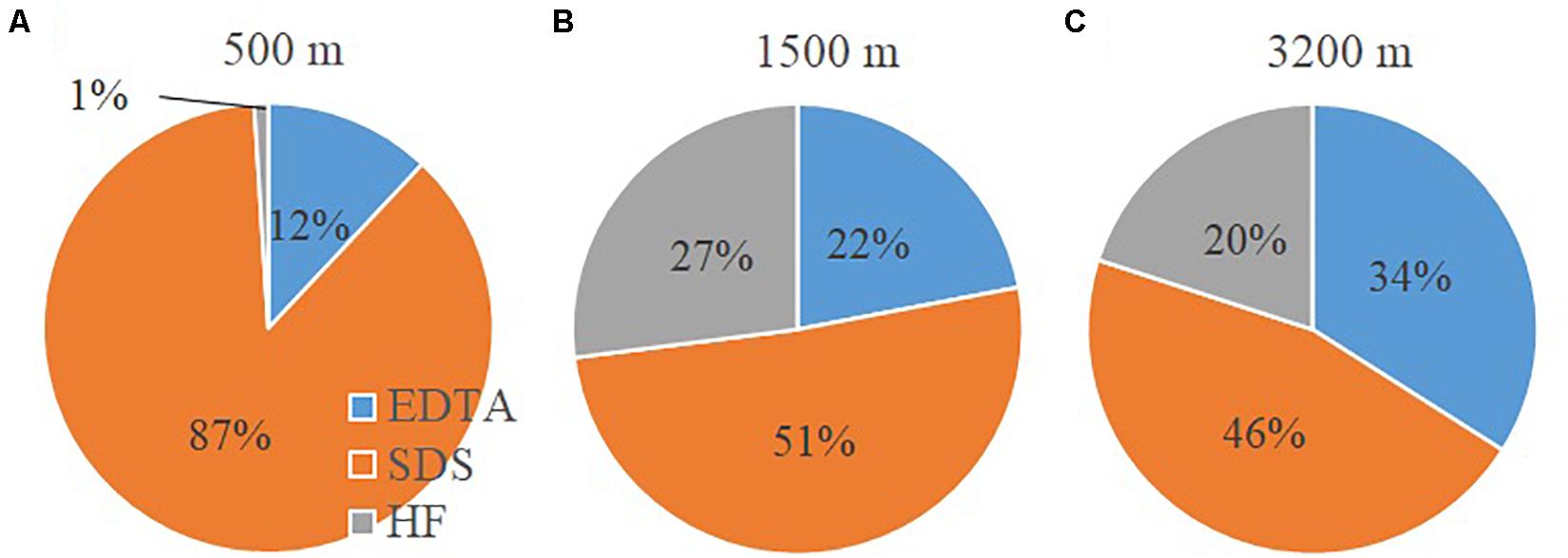
Figure 3. Intensity percentages of the three sequential fractions determined by electrospray ionization (ESI) Fourier-transform ion cyclotron resonance mass spectrometry (FTICRMS) in the settling particles: (A) 500 m; (B) 1500 m; (C) 3200 m.
Molecular Characteristics of Sequential Extracts of Marine Particles
Each sequential extractant (the non-FeCl3-amended) recovered a distinct set of compounds (in terms of elemental groupings and compound classifications) from the particles. The formula elemental groupings are listed in Table 1. CHO, CHON, and CHONS formulas are three major groups, all together accounting for 96, 91, and 85% of the total formulas for 500, 1500, and 3200 m, respectively. The number of peaks shared among POM at the different depths is shown in Supplementary Table S3 (773 formulas between 500 and 1500 m; 790 formulas between 1500 and 3200 m; 877 formulas between 500 and 3200 m). CHO formulas increased with depth, from 27% at 500 m to 35% at 1500 m, and to 41% at 3200 m. In contrast, the CHON formulas slightly decreased with depth, from 38% at 500 m to 39% at 1500 m, and to 34% at 3200 m. CHONS formulas decreased more rapidly with depth, from 31% at 500 m to 17% at 1500 m, and to 9% at 3200 m. CHOS, a minor fraction of the total identified formulas (within 5%) increased by twofold from 1.5% at 500 m to 3.7% at 1500 m, and stayed steady as 3.4% at 3200 m. Notably, deoxy-compounds increased from 2% at 500 m to 5% at 1500 m, and to 12% at 3200 m.
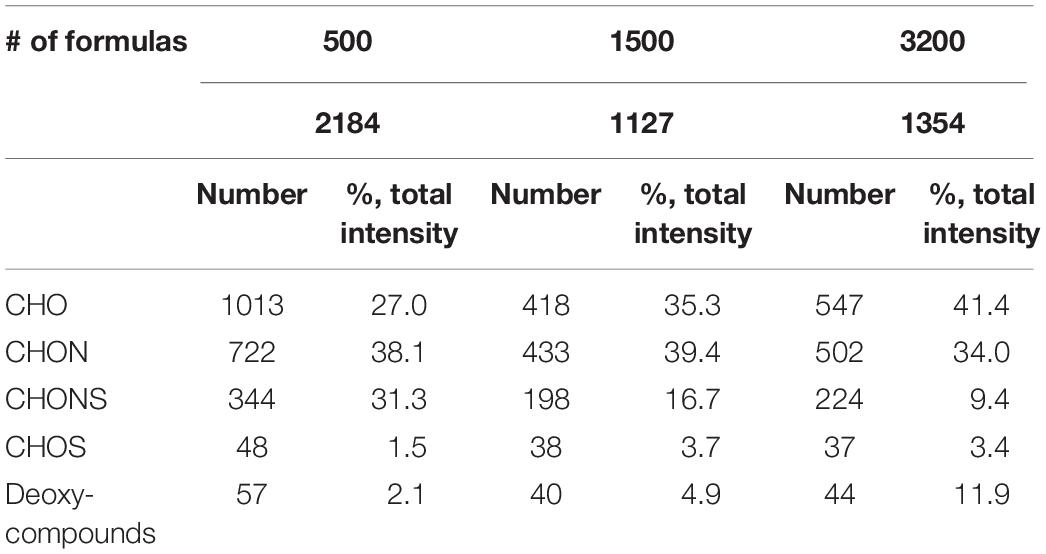
Table 1. Elemental grouping of assignable peaks at different depths.
The compound classifications are displayed in van Krevelen diagrams (Figure 4) and also listed in Table 2. From 500 to 3200 m, aliphatics steadily increased from 41.8 to 67.9%, while the N-containing aliphatics, which mostly consist of protein, peptides, and amino sugars, first increased from 15.4% at 500 m to 33.1% at 1500 m, and then decreased to 17.6% at 3200 m. Aromatics, condensed aromatics and black-carbon-like compounds decreased from 27.2 to 8.6%, 7.5 to 3.7%, and 3.1 to 0.7%, respectively. Similarly, with increasing depth the intensity-normalized H/C values (Sleighter and Hatcher, 2008) (calculated by summing up the product of individual intensity and the corresponding H/C ratio of each formula, and then being divided by the total intensity) increased from 1.31 to 1.62 whereas O/C values decreased from 0.20 to 0.16. CRAM-like compounds were only a minor fraction (5.0% at 500 m to 4.4% at 3200 m).
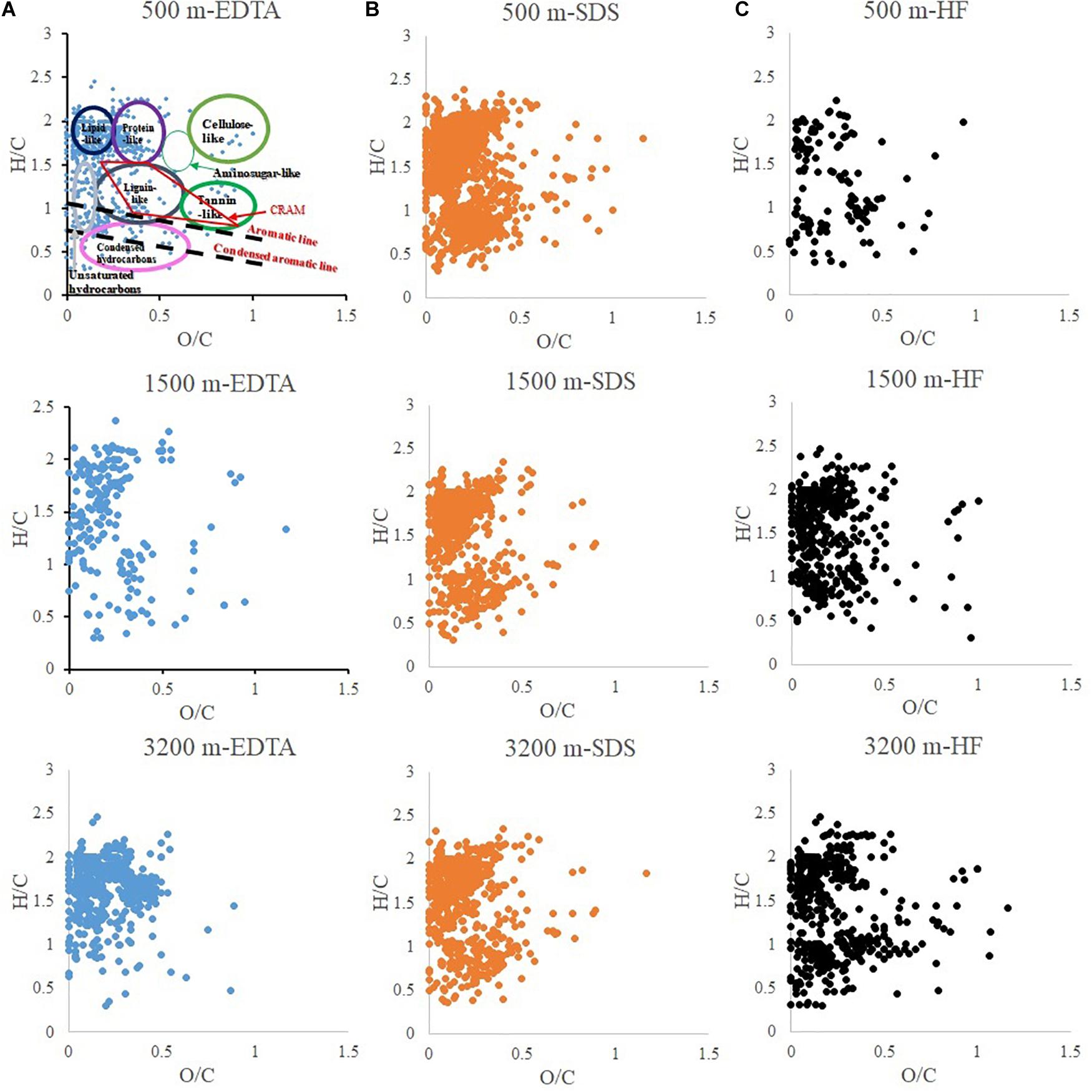
Figure 4. van-Krevelen diagram of particulate organic matter extracted by different reagents: (A) EDTA (ethylenediaminetetraacetic acid disodium salt dihydrate); (B) SDS (sodium dodecyl sulfate); (C) HF (hydrofluoric acid).
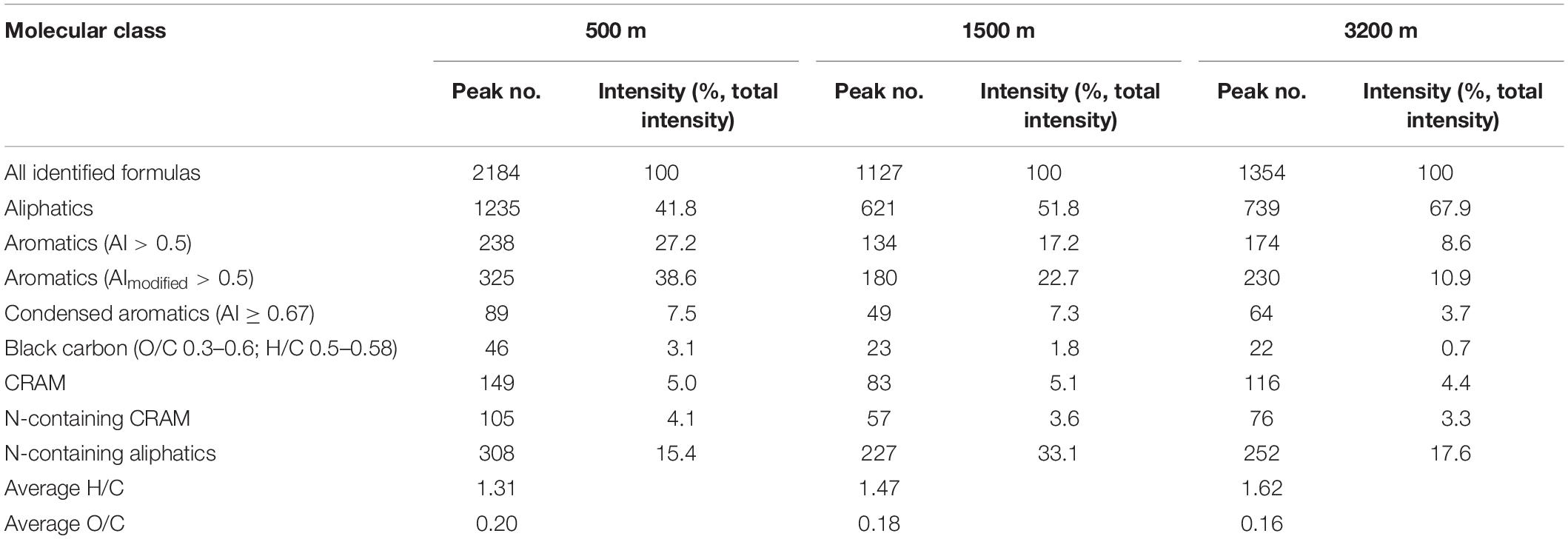
Table 2. Compound classification of assignable peaks at different depths (CRAMs were defined as molecules having DBE: C 0.30–0.68, DBE: H 0.20–0.95, and DBE: O 0.77–1.75; average H/C and O/C are calculated as intensity-normalized average values).
Iron-Carrying Molecules
In total, 745 iron-carrying molecules were identifiable, accounting for ∼14% of total assignable molecules including all depths, FeCl3-amended and non-FeCl3-amended treatments (Figure 5A). The majority of the Fe-carrying molecules were found in the FeCl3-amended samples, and only 33 formulas were found in the non-FeCl3-amended samples. These iron-carrying molecules could have been corroborated by adding another trivalent metal (e.g., Ga) at the same concentration (10 μM) and identifying if the same apo molecule forms the similar complex with the other metal, which can be applied in future study.
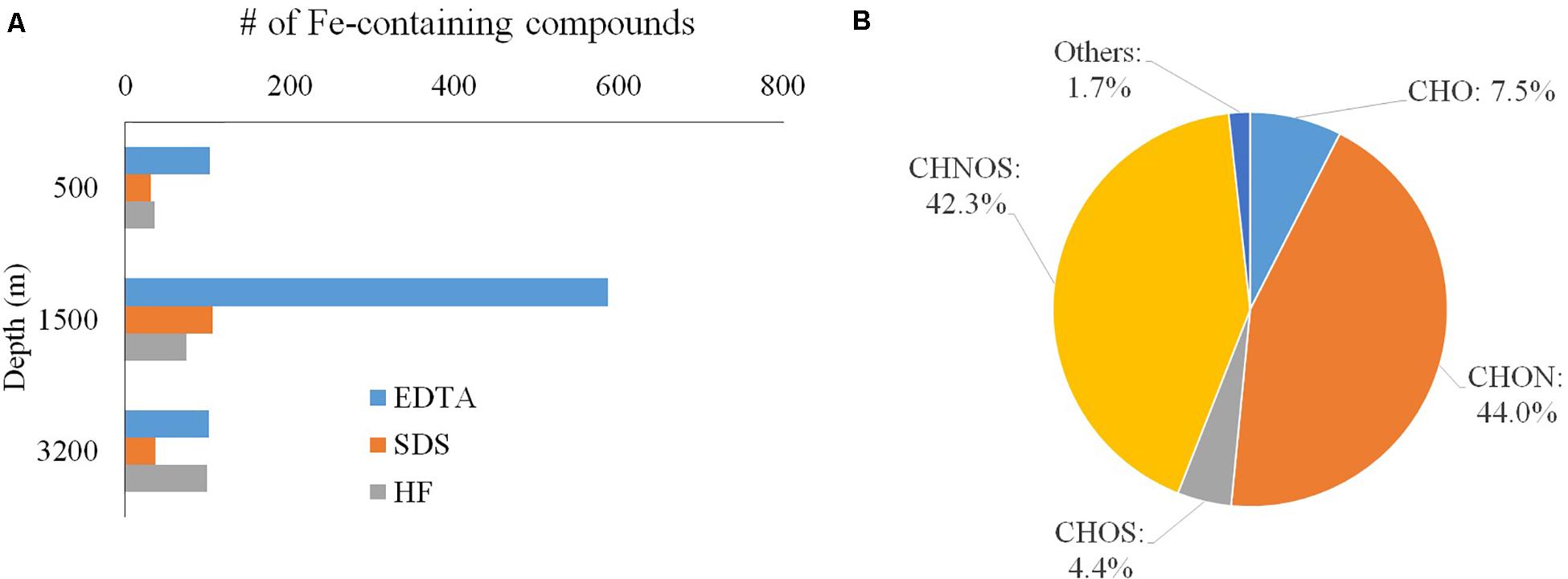
Figure 5. (A) Depth distribution; (B) elemental grouping of Fe-carrying compounds.
The absolute number of iron-carrying molecules in the “EDTA extract” increased from 103 at 500 m to 587 at 1500 m and then dropped to 101 at 3200 m; those in the “SDS extract” increased from 31 at 500 m to 106 at 1500 m and then dropped to 37 at 3200 m; those in the “HF extract” increased from 35 at 500 m to 74 at 1500 m and to 99 at 3200 m (Figure 5A). The EDTA fraction ranked highest in the number of iron-carrying molecules at both the 500 m and 1500 m depths, while at 3200 m depth the EDTA and HF fractions have a similar number of Fe-carrying molecules.
CHON and CHONS were the dominant elemental groups and accounted for 86.3% of total assignable Fe-carrying formulas (Figure 5B). CHO and CHOS were both minor elemental groups, and accounted for 7.5 and 4.4% of total assignable Fe-carrying formulas, respectively. The compound classification of the Fe-carrying formulas is distinct from that of the bulk POM in that these Fe-carrying molecules consist mainly of aromatics (57.6% if using AI, and 61.7% if using modified AI), black carbon (14.8%), CRAM-like compounds (11.5%), and very minor amount of aliphatics (1.7%) (Table 3 and Figure 6). The intensity-normalized H/C and O/C ratios are 0.75 and 0.44 (Figure 6), respectively, indicating an unsaturated/aromatic and highly-oxygenated composition.
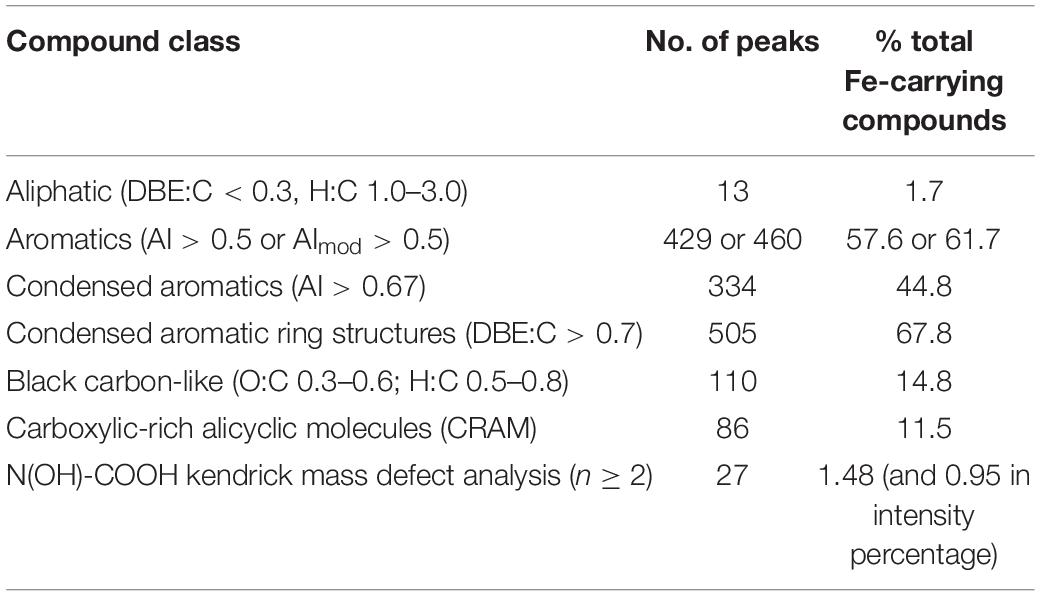
Table 3. Compound classification of assignable Fe-containing peaks at all depths (CRAMs were defined as molecules having DBE: C 0.30–0.68, DBE: H 0.20–0.95, and DBE: O 0.77–1.75, percentage is calculated by number of each class to the total number of formulas, i.e., 745).
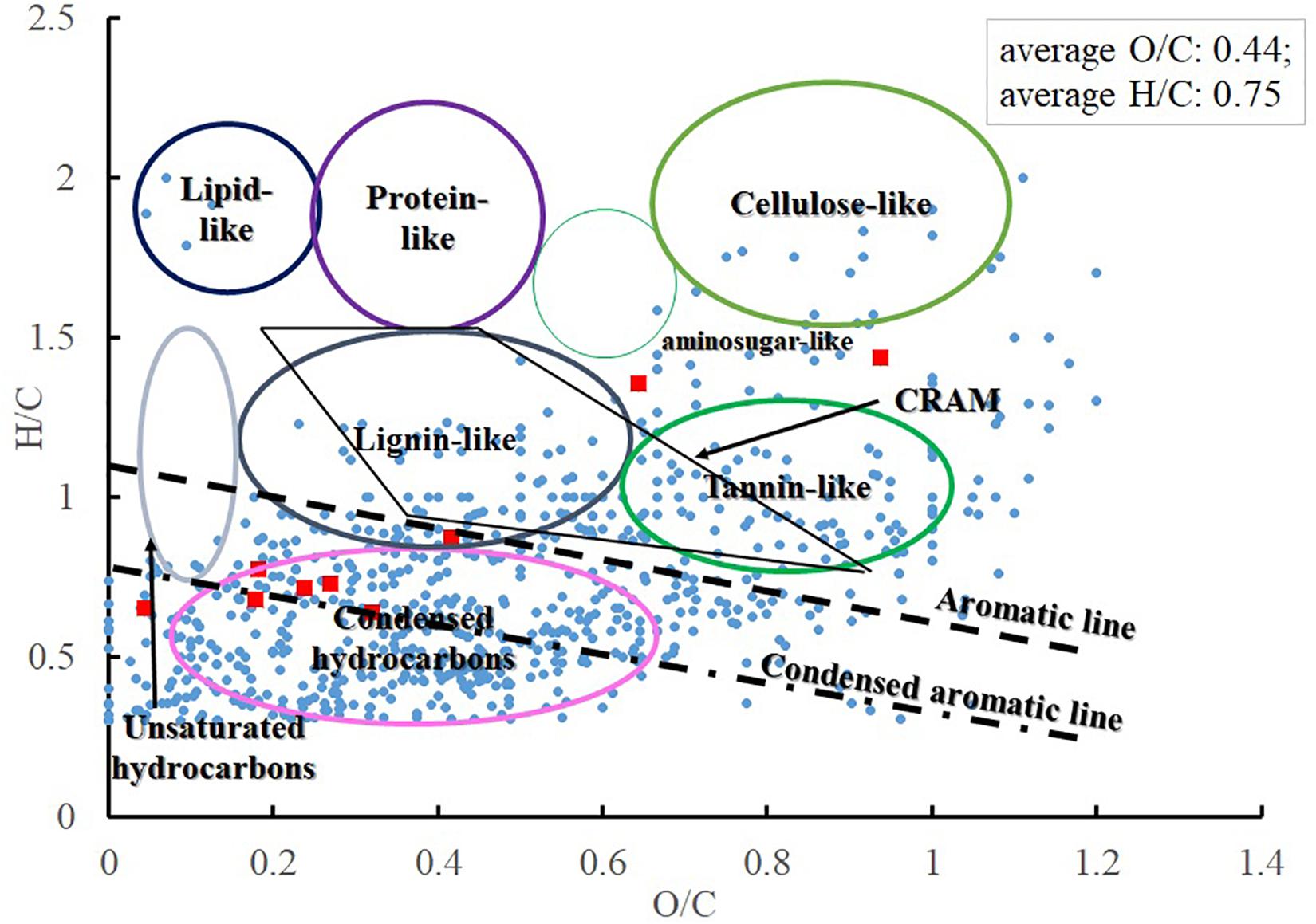
Figure 6. Compound classification of Fe-carrying compounds from three extractants (blue circle, Fe-containing formulas; red square, Fe, hydroxamate-like containing formulas).
Direct Evidence for the Presence of Hydroxamate-Like Molecules
Eleven formulas were found that belong to some N(OH)-COOH homologous series (Supplementary Table S4). Their m/z ranged from 431.98069 to 684.99753, mostly falling into the regions of aromatics and condensed aromatics (Figure 6). All these hydroxamate-like molecules accounted for 0.20 and 0.41% of all the assignable formulas in number and intensity, respectively, and 1.48 and 0.95% of the total Fe-carrying formulas in number and intensity, respectively (Table 3). These values provide a lower limit of hydroxamate-like moieties because not all these molecules may necessarily fall into any N(OH)-COOH homologous series. In the CH2-Kendrick-Mass-Defect (KMD)-Kendrick-Mass (KM) plot, which categorizes the molecules according to their mass defect (or the elemental composition), Kendrick mass defect of the Fe-containing molecules shifted below that of their corresponding apo molecules, the latter of which were found in the Fe (-) treatment (Supplementary Figure S1).
Discussion
Characteristics of the Molecular Micro-Environment of the Iron-Carrying Molecules in the Settling Particles
The changing molecular characteristics of the sinking particles with depth can be contrasted with trends observed for dissolved organic matter (DOM) (Chen et al., 2014b; Timko et al., 2015). First, higher average H/C values of DOM have been reported for the surface water of both North Pacific and Atlantic oceans, compared to those values of their respective deep water (Chen et al., 2014b; Timko et al., 2015), whereas the sinking particles exhibit an opposite trend with depth in our study (i.e., increasing H/C values with depth, Table 2). Second, the contents of aliphatic and carbohydrate-like compounds in DOM decrease with depth, likely due to the selective biodegradation of these molecules with increasing depth (Chen et al., 2014b). In samples collected at the same Sargasso Sea location, Timko et al. (2015) found higher concentrations of aliphatic compounds in the surface DOM pool than deep waters, while conversely those of aromatic/unsaturated and polyphenolic-like compounds were higher in deep waters. In contrast, we observe that particulate aliphatics (Figure 4 and Table 2) contents increased with depth. In addition, though carbohydrates are not well ionized in ESI (Shen and Perreault, 1998; Chen et al., 2011), spectrophotometric measurements by Chuang et al. (2013) provide evidence for the carbohydrate preservation in the sinking particles: 30% of OC at 500 m, 33% at 1500 m, and 26% at 3200 m, compared to the percentage of POC in particle mass as 11% at 500 m, 7% at 1500 m, and 5% at 3200 m. Third, higher percentages of CRAM-like formulas were observed in the intermediate and deep waters compared to surface water DOM in both North Pacific and Atlantic oceans, generally accounting for a substantial portion of the total mass spectrum intensity (40–65%) (Chen et al., 2014b). However, the particulate CRAM-like compounds are not a significant component of the POM (<6%) (Figure 4 and Table 2).
The results show that aliphatic compounds, and carbohydrate-like molecules become more predominant in sinking particles with increasing depth while aromatics, condensed aromatics, black carbon-like molecules, and CRAM-like compounds are preferentially “removed.” One hypothetical explanation for this pattern is the microbial community attached on particles actively modifies POM and thus produces labile material (Boeuf et al., 2019; Omand et al., 2020). The other alternative explanation is these compounds might be condensation products resulting from a Maillard-type reaction (Brandes et al., 2004) and thus become less biodegradable (Hedges et al., 2001). The differences in the molecular data for the three sequential extraction fractions provide some hints for understanding the molecular interactions within the organic phase of the sinking particles. Hydrophobic interactions (as probed by SDS extraction) appear to be the dominant mechanism for surface-active organic materials to associate with particles at mesopelagic depths (500 m) (Figure 3). With increasing depth there is, however, a greater importance of metal complexation and cation bridging. It could be due to the fact that more intact (dead or senescent) cells are present in the sinking particles at the 500 m than the other two deeper depths (1500 and 3200 m), and thus more cellular and membrane material became available for SDS extraction. However, at the other two depths, the cells could have mostly broken down, and the resulting OM either has been degraded or resulted in the metal complexed fraction. Lastly, more organic materials appear to partition into particle interstices (as probed by HF extraction) with depth (1500 and 3200 m vs. 500 m, Figure 3), which could provide a physical protective mechanism for organic molecules to become less accessible to microbial activity.
Refractory Nature of Particulate Hydroxamate-Like Moieties Revealed by Their Depth Distribution
In general, the absolute hydroxamate concentration in the settling particles decreased with depth in the Sargasso Sea while the POC-normalized concentration did not. This could indicate that hydroxamate-like moieties in the settling particles might be relatively less labile, compared to the bulk POC (Figures 2A,B). These hydroxamate concentrations are 10 to 20-fold lower than what was reported by Chuang et al. (2013). The source of this discrepancy is likely because Chuang et al. (2013) did not correct for the “browning effect” caused by the sulfuric acid step (see above).
The particulate hydroxamate concentrations determined at different seasons in the upper mesopelagic depth (500 m) has a larger variation compared to those in the intermediate (1500 m) and bathypelagic (3200 m) depths (Figure 2 and Supplementary Table S1). This suggests that abundances of iron and microbially-derived iron-binding ligands are more seasonally-driven in upper mesopelagic (e.g., seasonality of atmospheric dust input to surface waters) than at the intermediate and deep depths (Sedwick et al., 2005; Bundy et al., 2016). Once incorporated into the settling particles and transported to depth, hydroxamate-like molecules could have been stabilized via metal complexation (as probed by EDTA) (Iskrenova-Tchoukova et al., 2010), or hydrophobic sorption (as probed by SDS) or mineral interlayered occlusion (as probed by HF), and thus they may have been protected against enzymatic reaction (see below discussion).
While one needs to be cautious about applying the ESI-FTICRMS data for any quantitative purpose given the ionization efficiencies of ESI are variable for different types of compound groups, our results are in good agreement with the spectrophotometric results (a range of 0.2–0.6% of POC contributed by the carbon of N(OH)-COOH, in a comparison to 0.41% of total peak intensity contributed by hydroxamate-like molecules, Table 3 and Supplementary Table S1). Both results show that the N(OH)-COOH functionality is only a trace component compared to the bulk POM. These two independent lines of evidence collectively support the presence of N(OH)-COOH functional group in the sinking particles of the open ocean, at a level < 1% of the POC.
Using the lower limit of particle mass concentration and flux [10 μg/L and 10 mg/m2/d reported in Chuang et al. (2013)] and a median value of 10 μM-AHA eq./g-particle for particulate hydroxamate concentration, the particulate hydroxamate concentration (in overall volume) and flux at the studied site can be estimated to be 100 pM and 0.1 μmol-AHA eq./m2/d, respectively. This is on the same order of magnitude as the dissolved equivalent hydroxamate flux in the same area which is estimated to be 0.2 μmol-AHA eq. /m2/d, assuming a dissolved organic carbon flux of 15 mg/m2/d (Giering et al., 2014) and a hydroxamate-C to dissolved organic carbon ratio of 10–6. Meanwhile, Mawji et al. (2008) reported the dissolved hydroxamate siderophore in the surface water of the Atlantic Ocean was 3–20 pM. Thus, our results indicate that while hydroxamate-like moieties appear to be a trace component of POM, they could be comparable to their dissolved counterparts in the concentrations and fluxes. However, one has to be cautious that a direct comparison between the hydroxamate-like moieties in the particulate phase and the dissolved hydroxamate siderophores deserves more scrutiny. The reason is that there are still quantitative uncertainties based on the assumptions of relative concentrations of hydroxamate moieties and organic carbon in particles and water, and lack of evidence to connect dissolved hydroxamate siderophore as the source of the hydroxamate-like moieties in the particulate phase.
Possible Mechanisms for Incorporation and Preservation of Iron-Carrying Molecules and the Subset Hydroxamate-Like Molecules
We observed distinct molecular differences between Fe-carrying molecules with likely Fe-chelating properties and the bulk POM. This could reflect differences in biogeochemical sources and respective biosynthetic pathways. For example, Chen et al. (2014a) showed that POM produced from photo-flocculated DOM was enriched with both aliphatic and aromatic components, yet no direct evidence supported whether the co-precipitated Fe was complexed with both types of compounds or preferentially complexed with one or the other. This Fe-rich photo-produced black carbon pool (∼10% of the POC) was estimated to be nearly equivalent to the estimated global flux of dissolved black carbon from land to the ocean (Chen et al., 2014a). In a study that was not conducted in the oceanic context, plutonium (an A-type or hard metal (i.e., ion with low polarizability) that was often referred to having similar chemical complexing properties as Fe due to their similar ionic potential Z/r) (Chuang et al., 2013) was found to be predominantly associated with nitrogen-enriched, low average H/C (∼0.64), high DBE (∼16), aromatic and condensed aromatic compounds (DiDonato et al., 2017). In the present study, we did observe the Fe-carrying molecules are enriched in aromatic and condensed aromatics, and also contain black carbon with a content similar to what was reported in Chen et al. (2014a), however, these particulate Fe-carrying molecules were deplete in aliphatic (only 1.7%, Table 3), and also distinctly different from the bulk POM (41.8–67.9%, Table 2). A hypothetical explanation is as follows: Below 500 m where photochemical reactions are minor, the aliphatic moieties of these molecules are preferentially removed via microbial activity. Meanwhile, both the black carbon-like and CRAM-like compounds are preserved, compared to the bulk of the POM, and the latter exhibit a totally opposite pattern (i.e., aliphatic-enriched, aromatic and CRAM-depleted and deoxygenation) with increasing depth (Table 2 vs. Table 3).
While the hydroxamate-like moieties are only a minor fraction of the potential Fe-binding ligands of the POM, they may play an important role in both carbon and Fe cycling. As shown here, a substantial portion of the POM (∼77% of POC) can be removed by our sequential extraction method and saturated with dissolved Fe3+, which would otherwise not likely be exposed and accessible to Fe3+ under natural conditions. Furthermore, the flux of the hydroxamate-like moieties in the sinking particles is the same order of magnitude as their dissolved counterparts. We adopt here the hydroxamate flux of 0.1 μmol-AHA eq./m2/d as we calculated in section “Refractory Nature of Particulate Hydroxamate-Like Moieties Revealed by Their Depth Distribution.” Considering hydroxamate and Fe usually forms hexadentate or octahedral complexes via three bidentate ligands, this particulate hydroxamate flux has a theoretical maximum capacity of carrying an Fe flux of 0.03 μmol-Fe/m2/d. This is to be compared to an average particulate Fe flux of 4 μmol-Fe/m2/d (Chuang et al., 2013). Uncertainties in this assessment are possibly large, yet these rough calculations suggest that particulate hydroxamate could be one of the strongest organic Fe-binding ligands in natural sinking particles, considering their high binding affinity to Fe(III) over other known ligands (Reid et al., 1993; Neilands, 1995; Winkelmann, 2002; Butler, 2005; Butler and Theisen, 2010).
In addition to the above mentioned general molecular features of the iron-carrying molecules (Figure 6 and Table 3), the prevalence of sulfur with these hydroxamate-like functionalities (Supplementary Table S4) is noticeable, as dissolved S-containing hydroxamates were previously reported yet in a low proportion to the total dissolved hydroxamate siderophores in natural environments (Gledhill et al., 2019). How sulfur was incorporated into these hydroxamate-like moieties in the marine particles is equivocal, with one option being via abiotic pathways (Sleighter and Hatcher, 2008; Raven et al., 2016; Pohlabeln et al., 2017), or biological processes (Boeuf et al., 2019). Another molecular feature is that these hydroxamate-like molecules have relatively low H/C ratio (average H/C as 0.83, Supplementary Table S4) compared to those of the dissolved hydroxamate siderophores which usually exceed 1.0 (e.g., average H/C in the dissolved hydroxamate siderophore is 1.49; Gledhill et al., 2019). It is possible that dissolved hydroxamate siderophores produced by particle-attached bacteria or fungi are a direct source to be incorporated into the particles. However, these molecular characteristics suggest that these hydroxamate-like moieties are by no means similar in chemical composition or structure to their dissolved counterparts, thus they likely have experienced complex biogeochemical processes yet to be defined (Figure 7).
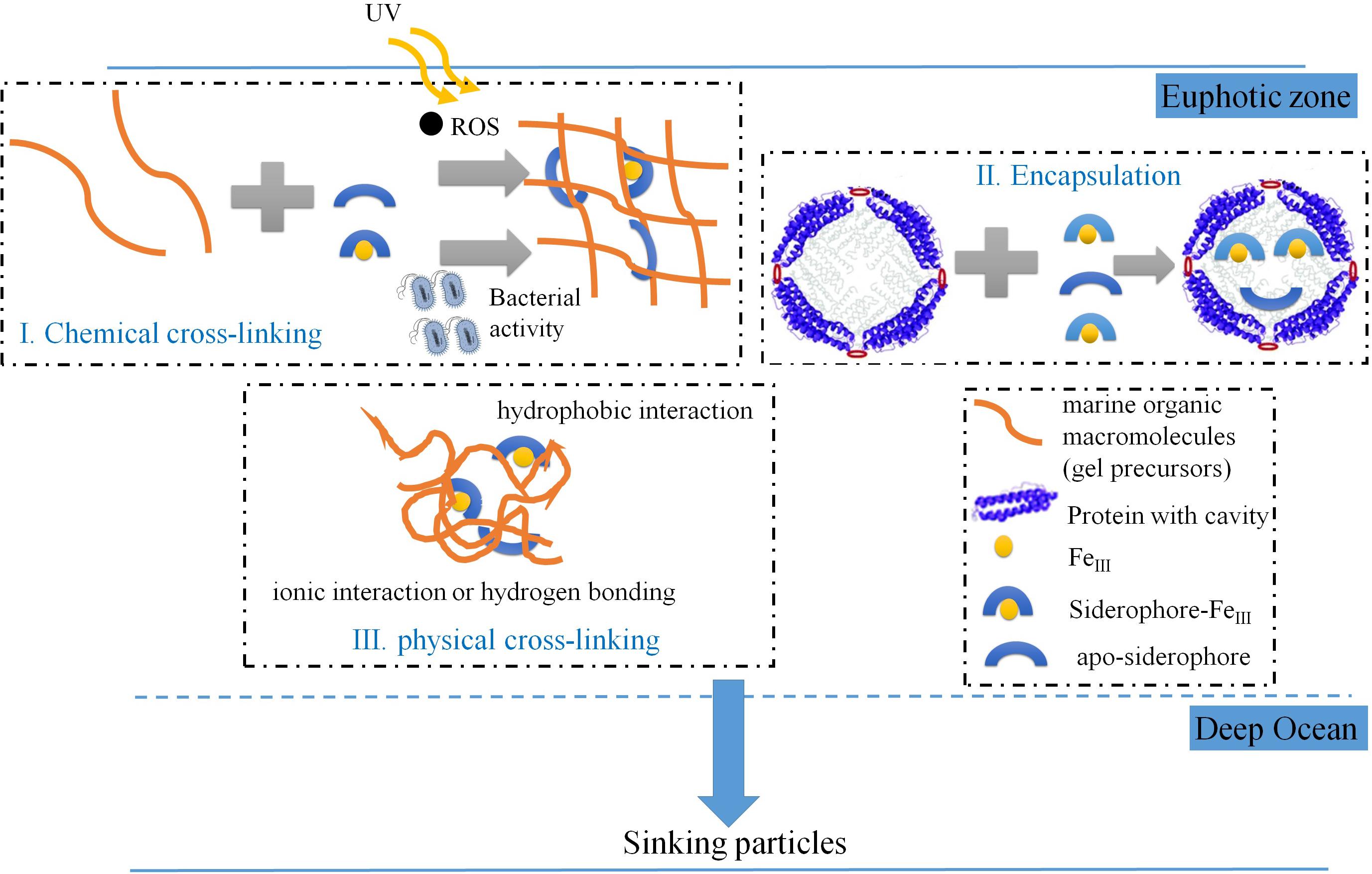
Figure 7. Conceptual scheme showing the possible mechanisms for hydroxamate-like moieties incorporation into marine settling particle.
First, hydroxamate-like moieties might be incorporated into the particles via sunlight-induced reactive oxygen species (ROS) mediated protein aggregation (Sun et al., 2017, 2018, 2019) in surface waters. In support of this mechanism, many previous studies have shown, although indirectly, that more nitrogen than carbon could accumulate into particles during irradiation (Helms et al., 2013; Chen et al., 2014a; Sun et al., 2019). We have observed N-containing aliphatics, consisting mostly of proteins, peptides, and amino sugars (Chen et al., 2018), accounting for 15.4–33.1% of the POM (in intensity %, Table 2). In comparison Chuang et al. (2013) found that about 45% of POC in particles consisted of protein (Chuang et al., 2013). Thus, it is compelling to believe that proteinaceous components are a significant component of the settling particles. The photo-reaction-induced and ROS-mediated crosslinking process in surface waters would generate precursors including aliphatic and condensed aromatic-like molecules (Chen et al., 2014a) that would be further modified as particles are processed in the water column (see above in this section).
A protein encapsulating mechanism could be an alternative pathway for hydroxamate-like moieties incorporation into marine particles. For example, ferritin, a large protein primarily storing iron mineral internally (Frankel, 1991) by marine microbes (Marchetti et al., 2009; Shire and Kustka, 2015), has been demonstrated to also encapsulate siderophores into cavity-like structures within the apoferritin (Dominguez-Vera, 2004). This ferritin-encapsulating mechanism has been exploited in health sciences to design slow-release drugs (Dominguez-Vera, 2004; Zhang et al., 2006; He and Marles-Wright, 2015).
Finally, some labile organic molecules, e.g., amino acids or simple sugars, have been shown to be preserved in marine sediments via sorption mechanisms (Keil et al., 1994; Barber et al., 2017). This likely involves relatively “weak” forces such as hydrophobic, ionic interactions or hydrogen bonding that are associated with mineral phases. Some bacteria (e.g., Pseudomonas aeruginosa) have been shown to utilize pyoverdine (a siderophore that contains both catecholate- and hydroxamate-like ligands) to complex the metal oxide surface for attachment (Upritchard et al., 2007). Thus, it is possible that some hydroxamate-like moieties in particles might be located with bacteria. In addition, the apparent LMW hydroxamate-like moieties observed in ESI-FTICRMS (Supplementary Table S4) could also be ionically bound as they were reversibly desorbed by the EDTA reagent. LMW molecules that are held together via hydrophobic interactions, hydrogen bonding and ionic interactions (complexation) can form supramolecular (“physical cross-linking”) associations (Sutton and Sposito, 2005) with an apparent molecular weight over 3 kDa (either present in the marine particles captured by sediment trap or a 0.45 μm membrane, or an ultrafiltration membrane). These supramolecular associations can dissociate within the electrospray interface, releasing LMW molecules.
Regardless of the exact mechanism of incorporation, it is clearly evident from our work that hydroxamate-like moieties exist in the particulate phase, potentially equivalent in concentrations and fluxes to their counterparts in the dissolved phase. The Fe-carrying compounds which represent all the potential organic Fe-binding ligands of the settling particles, have molecular features in a contrast to the bulk POM, the latter of which is then different from the vast DOM pool. The next step would be to identify the exact mechanism of hydroxamate-like molecule formation/incorporation, as this would be of great value for better understanding of the cycling of Fe, organic matter and organic nitrogen in the ocean. While our study only analyzed particulate Fe-carrying molecules in the oligotrophic North Atlantic gyre, our results strongly suggest the relative importance of hydroxamate-like molecules, which possibly have been incorporated via various physical and chemical mechanisms. These, along with the major fractions of Fe-binding compounds identified here, likely play an important role in controlling Fe and other A-type particle-reactive radionuclides (e.g., thorium) distributions and flux in the ocean.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
CX and PS conceived and designed the experiment. CX and WX performed the particle extraction experiment. CX performed the iron amendment incubation experiment. PL, CX, and HC performed the ESI-FTICRMS experiment. CX, PL, LS, HC, WX, and MK performed the ESI-FTICRMS data analysis. CX, PL, HC, WX, MK, PH, MC, AQ, and PS wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The early part of our work was funded by the NSF-OCE Grant No. 1356453. The OFP time-series was funded by the NSF-OCE Grant No. 0623505 to MC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the crew of the R/V Weatherbird II and R/V Atlantic Explorer for assistance with OFP ship operations, and JC Weber, S. Huang, and J. Barkman for assistance with sample processing. We greatly appreciate two reviewers for their very constructive comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2020.00266/full#supplementary-material
References
Ahmed, E., and Holmstrom, S. J. M. (2014). The effect of soil horizon and mineral type on the distribution of siderophores in soil. Geochim. Cosmochim. Acta 131, 184–195. doi: 10.1016/j.gca.2014.01.031
Barbeau, K., Rue, E. L., Bruland, K. W., and Butler, A. (2001). Photochemical cycling of iron in the surface ocean mediated by microbial iron(III)-binding ligands. Nature 413, 409–413. doi: 10.1038/35096545
Barbeau, K., Rue, E. L., Trick, C. G., Bruland, K. T., and Butler, A. (2003). Photochemical reactivity of siderophores produced by marine heterotrophic bacteria and cyanobacteria based on characteristic Fe(III) binding groups. Limnol. Oceanogr. 48, 1069–1078. doi: 10.4319/lo.2003.48.3.1069
Barber, A., Brandes, J., Leri, A., Lalonde, K., Balind, K., Wirick, S., et al. (2017). Preservation of organic matter in marine sediments by inner-sphere interactions with reactive iron. Scie. Rep. 7:366.
Boeuf, D., Edwards, B. R., Eppley, J. M., Hu, S. K., Poff, K. E., Romano, A. E., et al. (2019). Biological composition and microbial dynamics of sinking particulate organic matter at abyssal depths in the oligotrophic open ocean. Proc. Natl. Acad. Sci. U.S.A. 116, 11824–11832. doi: 10.1073/pnas.1903080116
Boiteau, R. M., Mende, D. R., Hawco, N. J., McIlvin, M. R., Fitzsimmons, J. N., Saito, M. A., et al. (2016). Siderophore-based microbial adaptations to iron scarcity across the eastern Pacific Ocean. Proc. Natl. Acad. Scie. U.S.A. 113, 14237–14242. doi: 10.1073/pnas.1608594113
Boiteau, R. M., and Repeta, D. J. (2015). An extended siderophore suite from Synechococcus sp PCC 7002 revealed by LC-ICPMS-ESIMS. Metallomics 7, 877–884. doi: 10.1039/c5mt00005j
Boukhalfa, H., and Crumbliss, A. L. (2002). Chemical aspects of siderophore mediated iron transport. Biometals 15, 325–339. doi: 10.1023/A:1020218608266
Brandes, J. A., Lee, C., Wakeham, S., Peterson, M., Jacobsen, C., Wirick, S., et al. (2004). Examining marine particulate organic matter at sub-micron scales using scanning transmission X-ray microscopy and carbon X-ray absorption near edge structure spectroscopy. Mar. Chem. 92, 107–121. doi: 10.1016/j.marchem.2004.06.020
Bruland, K. W., Donat, J. R., and Hutchins, D. A. (1991). Interactive influences of bioactive trace-metals on biological production in oceanic waters. Limnol. Oceanogr. 36, 1555–1577. doi: 10.4319/lo.1991.36.8.1555
Bundy, R. M., Jiang, M., Carter, M., and Barbeau, K. A. (2016). Iron-binding ligands in the Southern California current system: mechanistic studies. Front. Mar. Sci. 3:27. doi: 10.3389/fmars.2016.00027
Butler, A. (1998). Acquisition and utilization of transition metal ions by marine organisms. Science 281, 207–210. doi: 10.1126/science.281.5374.207
Butler, A. (2005). Marine Siderophores and microbial iron mobilization. Biometals 18, 369–374. doi: 10.1007/s10534-005-3711-0
Butler, A., and Theisen, R. M. (2010). Iron(III)–siderophore coordination chemistry: reactivity of marine siderophores. Coord. Chem. Rev. 254, 288–296. doi: 10.1016/j.ccr.2009.09.010
Chen, H., Stubbins, A., and Hatcher, P. G. (2011). A mini-electrodialysis system for desalting small volume saline samples for Fourier transform ion cyclotron resonance mass spectrometry. Limnol. Oceanogr. Methods 9, 582–592. doi: 10.4319/lom.2011.9.582
Chen, H. M., Abdulla, H. A. N., Sanders, R. L., Myneni, S. C. B., Mopper, K., and Hatcher, P. G. (2014a). Production of black carbon-like and aliphatic molecules from terrestrial dissolved organic matter in the presence of sunlight and Iron. Environ. Sci. Technol. Lett. 1, 399–404. doi: 10.1021/ez5002598
Chen, H. M., Stubbins, A., Perdue, E. M., Green, N. W., Helms, J. R., Mopper, K., et al. (2014b). Ultrahigh resolution mass spectrometric differentiation of dissolved organic matter isolated by coupled reverse osmosis-electrodialysis from various major oceanic water masses. Mar. Chem. 164, 48–59. doi: 10.1016/j.marchem.2014.06.002
Chen, H. M., Yang, Z. M., Chu, R. K., Tolic, N., Liang, L. Y., Graham, D. E., et al. (2018). Molecular Insights into arctic soil organic matter degradation under warming. Environ. Sci. Technol. 52, 4555–4564. doi: 10.1021/acs.est.7b05469
Chuang, C. Y., Ho, Y. F., Conte, M. H., Guo, L. D., Schumann, D., Ayranov, M., et al. (2013). Role of biopolymers as major carrier phases of Th, Pa, Po, Pb and Be radionuclides in settling particles from the Atlantic Ocean. Mar. Chem. 157, 131–143. doi: 10.1016/j.marchem.2013.10.002
Chuang, C. Y., Santschi, P. H., Wen, L. S., Guo, L., Xu, C., Zhang, S., et al. (2015a). Binding of Th, Pa, Pb, Po and Be radionuclides to marine colloidal macromolecular organic matter. Mar. Chem. 173, 320–329. doi: 10.1016/j.marchem.2014.10.014
Chuang, C. Y., Santschi, P. H., Xu, C., Jiang, Y. L., Ho, Y. F., Quigg, A., et al. (2015b). Molecular level characterization of diatom-associated biopolymers that bind Th-234, Pa-233, Pb-210, and Be-7 in seawater: a case study with Phaeodactylum tricornutum. J. Geophys. Res. Biogeosci. 120, 1858–1869. doi: 10.1002/2015jg002970
Conte, M. H., Carter, A. M., Koweek, D. A., Huang, S., and Weber, J. C. (2019). The elemental composition of the deep particle flux in the Sargasso Sea. Chem. Geol. 511, 279–313. doi: 10.1016/j.chemgeo.2018.11.001
DiDonato, N., Xu, C., Santschi, P. H., and Hatcher, P. G. (2017). Substructural components of organic colloids from a pu-polluted soil with implications for pu mobilization. Environ. Sci. Technol. 51, 4803–4811. doi: 10.1021/acs.est.6b04955
Dominguez-Vera, J. M. (2004). Iron(III) complexation of Desferrioxamine B encapsulated in apoferritin. J. Inorganic Biochem. 98, 469–472. doi: 10.1016/j.jinorgbio.2003.12.015
Frankel, R. B. (1991). “Iron biominerals: an overview,” in Iron Biominerals, eds R. B. Frankel and R. P. Blakemore (Boston, MA: Springer), 1–6. doi: 10.1007/978-1-4615-3810-3_1
Giering, S. L. C., Sanders, R., Lampitt, R. S., Anderson, T. R., Tamburini, C., Boutrif, M., et al. (2014). Reconciliation of the carbon budget in the ocean’s twilight zone. Nature 507, 480–483. doi: 10.1038/nature13123
Gillam, A. H., Lewis, A. G., and Andersen, R. J. (1981). Quantitative determination of hydroxamic acids. Anal. Chem. 53, 841–844. doi: 10.1021/ac00229a023
Gledhill, M., Basu, S., and Shaked, Y. (2019). Metallophores associated with Trichodesmium erythraeum colonies from the Gulf of Aqaba. Metallomics 11, 1547–1557. doi: 10.1039/c9mt00121b
He, D. D., and Marles-Wright, J. (2015). Ferritin family proteins and their use in bionanotechnology. New Biotechnol. 32, 651–657. doi: 10.1016/j.nbt.2014.12.006
Hedges, J. I., Baldock, J. A., Gelinas, Y., Lee, C., Peterson, M., and Wakeham, S. G. (2001). Evidence for non-selective preservation of organic matter in sinking marine particles. Nature 409, 801–804. doi: 10.1038/35057247
Helms, J. R., Mao, J., Schmidt-Rohr, K., Abdulla, H., and Mopper, K. (2013). Photochemical flocculation of terrestrial dissolved organic matter and iron. Geochim. Cosmochim. Acta 121, 398–413. doi: 10.1016/j.gca.2013.07.025
Hertkorn, N., Benner, R., Frommberger, M., Schmitt-Kopplin, P., Witt, M., Kaiser, K., et al. (2006). Characterization of a major refractory component of marine dissolved organic matter. Geochim. Cosmochim. Acta 70, 2990–3010. doi: 10.1016/j.gca.2006.03.021
Hunter, K. A., and Boyd, P. W. (2007). Iron-binding ligands and their role in the ocean biogeochemistry of iron. Environ. Chem. 4, 221–232.
Iskrenova-Tchoukova, E., Kalinichev, A. G., and Kirkpatrick, R. J. (2010). Metal cation complexation with natural organic matter in aqueous solutions: molecular dynamics simulations and potentials of mean force. Langmuir 26, 15909–15919. doi: 10.1021/la102535n
Keil, R. G., Montluçon, D. B., Prahl, F. G., and Hedges, J. I. (1994). Sorptive preservation of labile organic matter in marine sediments. Nature 370, 549–552. doi: 10.1038/370549a0
Kim, S. W., Kaplan, L. A., Benner, R., and Hatcher, P. G. (2004). Hydrogen-deficient molecules in natural riverine water samples - evidence for the existence of black carbon in DOM. Mar. Chem. 92, 225–234. doi: 10.1016/j.marchem.2004.06.042
Kind, T., and Fiehn, O. (2007). Seven golden rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics 8:105. doi: 10.1186/1471-2105-8-105
Koch, B. P., and Dittmar, T. (2016). From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter (vol 20, pg 926, 2006). Rapid Commun. Mass Spectrom. 30, 250–250. doi: 10.1002/rcm.7433
Kraemer, S. M., Butler, A., Borer, P., and Cervini-Silva, J. (2005). Siderophores and the dissolution of iron-bearing minerals in marine systems. Rev. Mineral. Geochem. 59, 53–84. doi: 10.1515/9781501509551-008
Lee, C. (2002). “Particulate organic matter composition and fluxes in the Sea,” in Chemistry of Marine Water and Sediments, eds A. Gianguzza, E. Pelizzetti, and S. Sammartano (Berlin: Springer), 125–146. doi: 10.1007/978-3-662-04935-8_5
Macrellis, H. M., Trick, C. G., Rue, E. L., Smith, G., and Bruland, K. W. (2001). Collection and detection of natural iron-binding ligands from seawater. Mar. Chem. 76, 175–187. doi: 10.1016/s0304-4203(01)00061-5
Marchetti, A., and Maldonado, M. T. (2016). Iron. Physiol. Microal. 6, 233–279. doi: 10.1007/978-3-319-24945-2_11
Marchetti, A., Parker, M. S., Moccia, L. P., Lin, E. O., Arrieta, A. L., Ribalet, F., et al. (2009). Ferritin is used for iron storage in bloom-forming marine pennate diatoms. Nature 457, 467–470. doi: 10.1038/nature07539
Mawji, E., Gledhill, M., Milton, J. A., Tarran, G. A., Ussher, S., Thompson, A., et al. (2008). hydroxamate siderophores: occurrence and importance in the Atlantic Ocean. Environ. Sci. Technol. 42, 8675–8680. doi: 10.1021/es801884r
Neilands, J. B. (1995). Siderophores - structure and function of microbial iron transport compounds. J. Biol. Chem. 270, 26723–26726. doi: 10.1074/jbc.270.45.26723
Neubert, H., Hider, R. C., and Cowan, D. A. (2002). Speciation of Fe(III)-chelate complexes by electrospray ionization ion trap and laser desorption/ionization Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 16, 1556–1561. doi: 10.1002/rcm.756
Omand, M. M., Govindarajan, R., He, J., and Mahadevan, A. (2020). Sinking flux of particulate organic matter in the oceans: sensitivity to particle characteristics. Sci. Rep. 10:5582. doi: 10.1038/s41598-020-60424-5
Pohlabeln, A. M., Gomez-Saez, G. V., Noriega-Ortega, B. E., and Dittmar, T. (2017). Experimental evidence for abiotic sulfurization of marine dissolved organic matter. Front. Mar. Sci. 4:364. doi: 10.3389/fmars.2017.00364
Raines, D. J., Sanderson, T. J., Wilde, E. J., and Duhme-Klair, A. K. (2015). Siderophores, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering. Amsterdam: Elsevier.
Raven, M. R., Sessions, A. L., Adkins, J. F., and Thunell, R. C. (2016). Rapid organic matter sulfurization in sinking particles from the Cariaco Basin water column. Geochim. Cosmochim. Acta 190, 175–190. doi: 10.1016/j.gca.2016.06.030
Reid, R. T., Live, D. H., Faulkner, D. J., and Butler, A. (1993). A siderophore from a marine bacterium with an exceptional ferric Ion Affinity Constant. Nature 366, 455–458. doi: 10.1038/366455a0
Sedwick, P. N., Church, T. M., Bowie, A. R., Marsay, C. M., Ussher, S. J., Achilles, K. M., et al. (2005). Iron in the Sargasso Sea (Bermuda Atlantic Time-series Study region) during summer: eolian imprint, spatiotemporal variability, and ecological implications. Global Biogeochem. Cycles 19:GB4006. doi: 10.1029/2004gb002445
Shen, X., and Perreault, H. (1998). Characterization of carbohydrates using a combination of derivatization, high-performance liquid chromatography and mass spectrometry. J. Chromatogr. A 811, 47–59. doi: 10.1016/S0021-9673(98)00238-6
Shire, D. M., and Kustka, A. B. (2015). Luxury uptake, iron storage and ferritin abundance in Prochlorococcus marinus (Synechococcales) strain MED4. Phycologia 54, 398–406. doi: 10.2216/14-109.1
Sleighter, R. L., Chen, H. M., Wozniak, A. S., Willoughby, A. S., Caricasole, P., and Hatcher, P. G. (2012). Establishing a measure of reproducibility of ultrahigh-resolution mass spectra for complex mixtures of natural organic matter. Anal. Chem. 84, 9184–9191. doi: 10.1021/ac3018026
Sleighter, R. L., and Hatcher, P. G. (2007). The application of electrospray ionization coupled to ultrahigh resolution mass spectrometry for the molecular characterization of natural organic matter. J. Mass Spectrom. 42, 559–574. doi: 10.1002/jms.1221
Sleighter, R. L., and Hatcher, P. G. (2008). Molecular characterization of dissolved organic matter (DOM) along a river to ocean transect of the lower chesapeake bay by ultrahigh resolution electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Mar. Chem. 110, 140–152. doi: 10.1016/j.marchem.2008.04.008
Stubbins, A., Spencer, R. G. M., Chen, H., Hatcher, P. G., Mopper, K., Hernes, P. J., et al. (2010). Illuminated darkness: molecular signatures of Congo River dissolved organic matter and its photochemical alteration as revealed by ultrahigh precision mass spectrometry. Limnol. Oceanogr. 55, 1467–1477. doi: 10.4319/lo.2010.55.3.1467
Sun, L., Chin, W.-C., Chiu, M.-H., Xu, C., Lin, P., Schwehr, K. A., et al. (2019). Sunlight induced aggregation of dissolved organic matter: role of proteins in linking organic carbon and nitrogen cycling in seawater. Sci. Total Environ. 654, 872–877. doi: 10.1016/j.scitotenv.2018.11.140
Sun, L., Chiu, M.-H., Xu, C., Lin, P., Schwehr, K. A., Bacosa, H., et al. (2018). The effects of sunlight on the composition of exopolymeric substances and subsequent aggregate formation during oil spills. Mar. Chem. 203, 49–54. doi: 10.1016/j.marchem.2018.04.006
Sun, L. N., Xu, C., Zhang, S. J., Lin, P., Schwehr, K. A., Quigg, A., et al. (2017). Light-induced aggregation of microbial exopolymeric substances. Chemosphere 181, 675–681. doi: 10.1016/j.chemosphere.2017.04.099
Sutton, R., and Sposito, G. (2005). Molecular structure in soil humic substances: the new view. Environ. Sci. Technol. 39, 9009–9015. doi: 10.1021/es050778q
Timko, S., Maydanov, A., Pittelli, S., Conte, M., Cooper, W., Koch, B., et al. (2015). Depth-dependent photodegradation of marine dissolved organic matter. Front. Mar. Sci. 2:66. doi: 10.3389/fmars.2015.00066
Upritchard, H. G., Yang, J., Bremer, P. J., Lamont, I. L., and McQuillan, A. J. (2007). Adsorption to metal oxides of the Pseudomonas aeruginosa siderophore pyoverdine and implications for bacterial biofilm formation on metals. Langmuir 23, 7189–7195. doi: 10.1021/la7004024
Velasquez, I., Nunn, B. L., Ibisanmi, E., Goodlett, D. R., Hunter, K. A., and Sander, S. G. (2011). Detection of hydroxamate siderophores in coastal and Sub-Antarctic waters off the South Eastern Coast of New Zealand. Mar. Chem. 126, 97–107. doi: 10.1016/j.marchem.2011.04.003
Verdugo, P., Alldredge, A. L., Azam, F., Kirchman, D. L., Passow, U., and Santschi, P. H. (2004). The oceanic gel phase: a bridge in the DOM–POM continuum. Mar. Chem. 92, 67–85. doi: 10.1016/j.marchem.2004.06.017
Volkman, J. K., and Tanoue, E. (2002). Chemical and biological studies of particulate organic matter in the ocean. J. Oceanogr. 58, 265–279.
Vraspir, J. M., and Butler, A. (2009). Chemistry of marine ligands and siderophores. Annu. Rev. Mar. Sci. 1, 43–63. doi: 10.1146/annurev.marine.010908.163712
Walker, L. R., Hoyt, D. W., Walker, S. M., Ward, J. K., Nicora, C. D., and Bingol, K. (2016). Unambiguous metabolite identification in high-throughput metabolomics by hybrid 1D H-1 NMR/ESI MS1 approach. Magn. Reson. Chem. 54, 998–1003. doi: 10.1002/mrc.4503
Waska, H., Koschinsky, A., and Dittmar, T. (2016). Fe- and cu-complex formation with Artificial ligands investigated by ultra-high resolution fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR-MS): implications for natural metal-organic complex studies. Front. Mar. Sci. 3:119. doi: 10.3389/fmars.2016.00119
Winkelmann, G. (2002). Microbial siderophore-mediated transport. Biochem. Soc. Trans. 30, 691–696. doi: 10.1042/bst0300691
Xu, C., Zhang, S. J., Kaplan, D. I., Ho, Y. F., Schwehr, K. A., Roberts, K. A., et al. (2015). Evidence for hydroxamate siderophores and other n-containing organic compounds controlling (PU)-P-239,240 immobilization and remobilization in a wetland sediment. Environ. Sci. Technol. 49, 11458–11467. doi: 10.1021/acs.est.5b02310
Zhang, B., Watt, R. K., Galvez, N., Dominguez-Vera, J. M., and Watt, G. D. (2006). Rate of iron transfer through the horse spleen ferritin shell determined by the rate of formation of Prussian Blue and Fe-desferrioxamine within the ferritin cavity. Biophys. Chem. 120, 96–105. doi: 10.1016/j.bpc.2005.10.005
Keywords: particulate organic matter, ESI-FTICR mass spectrometry, iron-carrying molecules, marine particles, hydroxamate-like molecules
Citation: Xu C, Lin P, Sun L, Chen H, Xing W, Kamalanathan M, Hatcher PG, Conte MH, Quigg A and Santschi PH (2020) Molecular Nature of Marine Particulate Organic Iron-Carrying Moieties Revealed by Electrospray Ionization Fourier-Transform Ion Cyclotron Resonance Mass Spectrometry (ESI-FTICRMS). Front. Earth Sci. 8:266. doi: 10.3389/feart.2020.00266
Received: 22 February 2020; Accepted: 15 June 2020;
Published: 08 July 2020.
Edited by:
Gilad Antler, Ben-Gurion University of the Negev, IsraelReviewed by:
Martha Gledhill, GEOMAR Helmholtz Center for Ocean Research Kiel, GermanyMina Bižic, Leibniz-Institute of Freshwater Ecology and Inland Fisheries (IGB), Germany
Copyright © 2020 Xu, Lin, Sun, Chen, Xing, Kamalanathan, Hatcher, Conte, Quigg and Santschi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chen Xu, eHVjaGVuNjZAdGFtdS5lZHU=