 Nicholas J. Bouskill1*
Nicholas J. Bouskill1* Mark E. Conrad1
Mark E. Conrad1 Markus Bill1
Markus Bill1 Eoin L. Brodie1
Eoin L. Brodie1 Yiwei Cheng1
Yiwei Cheng1 Chad Hobson1
Chad Hobson1 Matthew Forbes2Karen L. Casciotti2
Matthew Forbes2Karen L. Casciotti2 Kenneth H. Williams1
Kenneth H. Williams1- 1Earth and Environmental Sciences Area, Lawrence Berkeley National Laboratory, Berkeley, CA, United States
- 2Department of Environmental Earth System Science, Stanford University, Stanford, CA, United States
The capillary fringe is a subsurface terrestrial-aquatic interface that can be a significant hotspot for biogeochemical cycling of terrestrially derived organic matter and nutrients. However, pathways of nitrogen (N) cycling within this environment are poorly understood, and observations of temporal fluctuations in nitrate (NO3−) concentrations lack the necessary resolution to partition between biotic or abiotic mechanisms. At discrete sampling points we measured NO3−, nitrite (NO2−), ammonium (NH4+), gaseous nitrous oxide (N2O), and nitrogen (N2), and the corresponding isotopic composition of NO3− within floodplain sediments at Rifle, Colorado. Coincident with an annually reoccurring spring/summer excursion in groundwater elevation driven by snowmelt, we observed a rapid decline in NO3− followed by transient peaks in NO2−, at three depths (2, 2.5, and 3 m) below the ground surface. Isotopic measurements (δ15N and δ18O of NO3−) suggest an immediate onset of biological N loss at 2 m. At 2.5 and 3 m, NO3− concentrations declined initially with no observable isotopic response, indicating dilution of NO3− as the NO3−-deficient groundwater rose, followed by denitrification after prolonged saturation. A simple Rayleigh model further supports this depth-dependent variability in the significance of actively fractionating mechanisms (i.e., NO3− reduction) relative to non-fractionating mechanisms (mixing and dilution). NO3− reduction was calculated to be responsible for 64% of the NO3− decline at 2 m, 28% at 2.5 and 47% at 3 m, respectively. Finally, by accounting for previous molecular and geochemical analysis at this site, and comparing the trajectories between Δδ18O: Δδ15N, we conclude that biological NO3− consumption at the two deeper and frequently saturated depths (2.5 and 3 m) can be attributed to heterotrophic denitrification. However, the Δδ18O: Δδ15N trajectory at the shallower, irregularly saturated site at 2 m shows a more complicated relationship best explained by the cyclic production of NO3− via aerobic oxidation, and consumption via NO3− reduction.
1. Introduction
Subsurface terrestrial-aquatic interfaces are hotspots for biogeochemical cycling of organic matter and nutrients (McClain et al., 2003; Lohse et al., 2009), and particularly nitrogen (Hefting et al., 2004; Heffernan et al., 2012; Zhu et al., 2013; Gonneea and Charette, 2014; Smith et al., 2015). Seasonal event-driven fluctuations in water table height can alter trace gas dynamics (Haberer et al., 2012), substrate availability (Persson et al., 2015), and the distribution of microbial metabolisms (Berkowitz et al., 2004). These activities can promote the formation of sharp oxic/anoxic gradients, which facilitate the spatial and temporal coupling of aerobic and anaerobic metabolisms. Previous work characterizing the subsurface nitrogen cycle has observed the accumulation and dissipation of NO3− at the capillary fringe concomitant with the rise and fall of the water table (Hefting et al., 2004; Abit et al., 2008; Sorensen et al., 2015). However, the mechanistic basis for subsurface NO3− dynamics are still largely unknown beyond the inference of several interacting and interdependent abiotic and biotic processes.
Herein, we examine seasonal NO3− dynamics around the capillary fringe in an alluvial floodplain at a field site in Rifle, Colorado, USA. Above the water table, near-atmospheric concentrations of oxygen provide a niche for diverse groups of aerobic and facultative metabolisms responsible for the turnover of specific carbon pools (Stegen et al., 2016) and the release of NH4+ that can be nitrified to NO3− (Smith et al., 2006). Nitrification is traditionally recognized to be a two-step process (although complete nitrification was recently shown to occur in Nitrospira bacteria (Daims et al., 2015, 2016). The rate-limiting first step, the oxidation of NH4+ to NO2− via a hydroxylamine (NH2OH) intermediate, is carried out by obligately aerobic chemolithoautotrophic bacteria and archaea (Ward, 2011), that have previously observed to be present in high abundance within the vadose zone (Hug et al., 2015b; Anantharaman et al., 2016). NO2− can be subsequently oxidized to NO3− via a diverse group of bacteria, some of which are mixotrophic (Daims et al., 2016; Le Roux et al., 2016). Nitrification, driven by NH4+ released by organic matter mineralization, under oxic conditions can lead to the accumulation of high concentrations of NO3−, which can be augmented by atmospheric deposition and infiltration of NO3− into the vadose zone (Einsiedl and Mayer, 2006).
At the Rifle site spring snowmelt drives the incursion of the water table into the vadose zone, altering redox through reduced gaseous exchange (Haberer et al., 2012; Jost et al., 2015). Such event driven changes in geochemical conditions have previously been observed to accompany a decline in NO3− concentrations (Abit et al., 2008), including at Rifle (Williams et al., unpublished), however, the mechanisms that lead to the observed decline in NO3− concentrations are unclear. The anoxic groundwater at Rifle contains very little NO3− (ranging from undetectable to 80 μM, Zachara et al., 2013; Yabusaki et al., 2017), and dilution during groundwater rise is a plausible explanation for an observed drop in measurable NO3− concentrations. However, rapid fluctuations in redox can also select for different suites of microbial traits (Hug et al., 2015a; Anantharaman et al., 2016) and the expression of anaerobic metabolisms (Heffernan et al., 2012; Zhu et al., 2013). Therefore, measured declines in NO3− concentrations could also reflect biological NO3− reduction preceding gaseous nitrogen loss (as either N2 or N2O). Diverse nitrogen cycling metabolisms, including denitrification (NO3− → NO2− → NO → N2O → N2), by facultative aerobic heterotrophic bacteria, dissimilatory nitrate reduction to ammonium, NO3−-dependent sulfide and iron oxidation and anammox, the anaerobic oxidation of NH4+ to N2 using NO2− as an electron acceptor, have all been shown to occur within the Rifle floodplain (Hug et al., 2015a; Anantharaman et al., 2016; Jewell et al., 2016).
In the current study we seek to characterize the nitrogen biogeochemistry of the Rifle subsurface as snowmelt driven fluctuations in water table depth change the saturation profile of the vadose zone soils, and to determine the role abiotic and biological mechanisms play in the fate of NO3−. The accumulation and dissipation of NO3− occurs annually at this site (Williams et al., unpublished data), however, we focus our efforts on 1 year, 2014, and measured different inorganic nitrogen species from pore water samples (NO3−, NO2−, NH4+), and gaseous measurements of nitrous oxide concentrations. Furthermore, we measured the corresponding isotopic composition, δ15N and δ18O, of NO3−. The stable isotopes of NO3− are an ideal tool for characterizing the contribution of different pathways to the formation and loss of NO3− as the water table rises and falls. The fractionation of δ15N and δ18O, of NO3− associated with bacterial NO3− reduction (Granger et al., 2008) has previously been used to identify biological activity in subsurface environments (Böhlke et al., 2006; Kendall et al., 2007; Frey et al., 2014; Clague et al., 2015). During NO3− reduction via denitrification, the active fractionation of δ15N and δ18O of NO3− has been shown to enrich the residual NO3− pool between + 5 and +25‰ (Granger and Wankel, 2016). Conversely, as the groundwater at Rifle tends to be NO3−-deficient (Zachara et al., 2013), rising water-table height could dilute capillary fringe NO3− concentrations. Because dilution imparts no isotopic fractionation on the NO3− pool, it is possible to broadly separate abiotic and biotic mechanisms by measuring shifts in δ15N and δ18O of NO3−.
2. Materials and Methods
2.1. Field Site Description
The Rifle field site is a small (~9 ha) floodplain lying adjacent to the Colorado River (Figure S1). The site hosted a former uranium mill processing facility, and has been intensely studied in the decades since closure (Yabusaki et al., 2007; Williams et al., 2011; Hug et al., 2015a). The unconfined aquifer under the floodplain is composed of unconsolidated sands, silts, clays and gravel deposited by the river, sitting atop a relatively impermeable layer of the Miocene Wasatch formation (Williams et al., 2011). The groundwater typically lies 3.5 m below the ground surface, but fluctuates annually. During snowmelt (generally beginning in April), the groundwater table can rise by 1–1.5 m into the unsaturated zone. This groundwater rise can persist for a period of weeks, with apparent interannual variability related to the magnitude of discharge in the Colorado River adjoining the site on its southern boundary. The subsurface hydrology of the Rifle floodplain maintains a strong south-southwest gradient (i.e., toward the Colorado River) and gradient reversals are very rare (Zachara et al., 2013; Yabusaki et al., 2017). There is no evidence to suggest lateral flow of river water reaches the experimental plots or has any influence on groundwater chemistry (Zachara et al., 2013). This semiarid site receives 292 mm of precipitation annually (Tokunaga et al., 2016), however, precipitation was above average (~331 mm) during 2014, and the water table rose higher than normal and persisted for longer.
The TT03 monitoring location at the Rifle field site consists of a series of vertically resolved suction lysimeters and gas sampler ports installed at the following depths: 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, and 3.14 meters below surface depth (bsd). Installation and well-sampling have recently been described in full (Tokunaga et al., 2016). Briefly, lysimeters and gas sampler ports were installed as part of a drilling operation from the ground surface to the water table (~3.25 m at the time of drilling). Porous ceramic cup lysimeters (Soilmoisture Equipment, Corp.; Tucson, AZ) were placed at the aforementioned depths. A groundwater monitoring well-adjacent to the vertically resolved lysimeters was used to track variations in groundwater quality parameters (including temperature, pH, specific conductivity, oxidation reduction potential, and dissolved oxygen). Samples for the present study were taken on a nearly weekly basis between March and September of 2014, a time period during which measurements from previous years have shown a decline in NO3− during groundwater incursion into the unsaturated zone. Fluids recovered from each lysimeter were filtered (0.45 μM) and immediately analyzed for NO3− and NO2− concentrations via anion chromatography (Dionex, Corp. ICS-2100, Sunnyvale, CA) using an AS-18 anion exclusion column. Field measurements of NO3− were further corroborated in the laboratory by colorimetric reduction of NO3− to NO2− via vanadium(III) chloride through a previously described protocol (Bouskill et al., 2013). Porewater NH4+ concentrations were also measured colorimetrically via reduction by sodium salicylate (Allison et al., 2008). Samples for isotope analysis were frozen in the field and shipped to Stanford University, where they were stored at −80°C prior to analysis.
2.2. Dual-Isotope Measurements
The isotope ratios of NO3− (δ15NNO3 and δ18ONO3), where δ(‰) = (RNO3/Rstd − 1)*1000, R indicates either 15N/14N or 18O/16O and “std” refers to a standard reference material, either N2 in air for δ15N or Vienna standard mean ocean water (VSMOW) for δ18O, were measured by the denitrifier method (Sigman et al., 2001; Casciotti et al., 2002). NO2−, which interferes with the analysis, was initially removed from the porewater samples prior to analysis using sulfamic acid, according to a previously published method (Granger and Sigman, 2009). Water samples were injected into a suspension of Pseudomonas aureofaciens, which lacks the N2O reductase, and quantitatively converts the NO3− to N2O. The N2O was analyzed on Finnigan DeltaPLUS XP isotope ratio mass spectrometer connected to a Finnigan GasBench. Individual N2O injection samples were standardized by comparison to NO3− isotope standards USGS32, USGS34, and USGS35 (Böhlke et al., 2003). Samples were measured in triplicate and the δ15N and δ18O of NO3− reported using the notation ‰ relative to atmospheric N2 and VSMOW, respectively. Typical reproducibility was ±0.2 ‰ for both δ15N and δ18O.
2.2.1. NO3− Sources
A simple isotope mixing model (Wexler et al., 2014) was used to estimate the contribution of different sources (i.e., atmospheric deposition and infiltration of NO3− or nitrification) to the NO3− accumulating in the unsaturated zone prior to groundwater rise. Using literature values for two different end members (nitrification and snowmelt) we estimated the source of well NO3− prior to the onset of denitrification (toward the beginning of May) as follows,
which can be rearranged to give f,
Values for δ18ONO3 from snowmelt were taken from previously published values (Kendall et al., 2007), estimated to be ~+67 ‰ (with a range of +40 to +70 ‰). We used two approaches in order to estimate the δ18ONO3 value likely imparted by nitrification (Fang et al., 2012). These two approaches are used because they differ in their consideration of O-exchange between NO2− and δ18OH2O during the first step of nitrification, while still accounting for kinetic isotopic effects. Both approaches are used in the above calculation (Equation 2), to calculate an upper and lower bound for the contribution of nitrification to the accumulation of NO3−. The first approach follows the assumption that nitrification occurs with no exchange between the nitrification intermediates and water, though isotopic fractionation during oxygen atom incorporation is accounted for (Buchwald et al., 2012):
Here we used a fixed value of 23.5 ‰ for the δ18OO2, and measurements of δ18OH2O from the Rifle groundwater, which spans a range of −13.3 to −14.7‰ between the 2 and 3 m depths considered in this study (Williams, personal communication). 18εK,O2 and 18εK,H2O, 1 represents the isotopic fractionation associated with 18O incorporation from O2, and H2O during the first step of nitrification, ammonia oxidation. Similarly, 18εK,H2O, 2 represent the isotopic fractionation associated with 18O incorporation into NO3 from H2O during NO2− oxidation. Values for 18εK,O2, 18εK,H2O, 1, and 18εK,H2O, 2 were derived from a previously published range of values (Buchwald and Casciotti, 2010; Casciotti et al., 2010), where 18εK,O2 + 18εK,H2O, 1 was estimated as 17.9–37.6 to ‰ (Casciotti et al., 2010), while 18εK,H2O, 2 has been estimated to be 12.8–18.2 to ‰ (Buchwald and Casciotti, 2010). The range of nitrification values obtained through this first approach is -20.3 to -11.2‰.
The second approach allows full exchange of oxygen atoms between NO2− and H2O during nitrification (Buchwald and Casciotti, 2010; Casciotti et al., 2010):
where 18εeq is the equilibrium isotope effect between NO2− and H2O, which is ~14–15‰ at room temperature (Casciotti et al., 2007). The range of nitrification values obtained through this second approach is -11.5 to -7.7‰. From these two approaches, we used the upper and lower range in δ18ONO3 values to parameterize the simple mixing model.
2.2.2. NO3− Sinks
We used a simple Rayleigh fractionation equation (δ15N = εlnf + δ15Ninitial) to calculate the fraction of NO3− loss attributable to NO3−-fractionating mechanisms, such as NO3− reduction. We took the δ15N measured for the highest NO3− concentration at each depth, subtracted it from the maximum δ15N and divided that by an ϵ value of +15‰ that represents the average value for denitrification (Granger and Wankel, 2016). This would give the fraction of NO3− remaining if NO3− reduction was the only process occurring. Knowing the initial concentration of NO3− prior to loss, and the fraction purportedly lost due to NO3− reduction, we calculated the NO3− concentration utilized by this fractionating process. We compared this value to the total change in NO3− in order to derive a value of NO3− lost due to reduction. Finally, we examined how the ϵ value affected our conclusion of NO3− lost to NO3− reduction by varying this value between 10 and 20. This range is also reported below.
2.3. N2O and N2 Concentrations
Gas samples from 2 m bsd were collected every 2 weeks or every 2 months from April to November, 2014 (except when groundwater rise saturated the lower depth intervals). Samples were drawn from the subsurface using a peristaltic pump (flow rate 2 cm3 s−1). Following purging of at least three volumes of the sampling apparatus, the effluent end of the tubing from the peristaltic pump was attached to a 60 ml syringe and allowed to fill. The resulting gas sample was then injected into a pre-evacuated serum vials sealed with 14 mm-thick chlorobutyl septa (Bellco Glass, Inc.) that were then shipped to Lawrence Berkeley National Laboratory for analyses. Concentrations of N2O and N2 were analyzed using a Shimadzu Gas Chromatograph (GC-2014). 4.5 ml of gas from the sample bottles was flushed through a 1 ml stainless steel loop and injected into the GC where the gases were separated on a HayeSep-D packed column (4 m × 1/8, 25 mL/min, and 75 °C) and analyzed using an electron capture detector. The detection limit is 0.2 ppmv, with calibration reference material values of 1.02 and 10.1 ppmv and the precision of the measurements ~±10% of the measured value.
3. Results and Discussion
Sharp redoxclines created at terrestrial-aquatic interfaces are potential hotspots of nitrogen loss (Lohse et al., 2009). Previous observations have recorded the annual accumulation and dissipation of NO3− at the capillary fringe of the Rifle site (Williams et al., unpublished data). However, the unusually high water table incursion in 2014 facilitated an excellent opportunity to examine the mechanistic basis of nitrogen loss over the year, and our efforts here focused solely on this event. Measurements presented here imply a combination of both abiotic (i.e., NO3− dilution by rising groundwater) and biotic processes (autotrophic nitrification and heterotrophic denitrification) are responsible for the nitrogen dynamics observed at the capillary fringe and are driven by the changing water table levels.
3.1. Pore Water Chemistry
Over the course of the water table excursion from March to October 2014, mean values of groundwater temperature, pH, specific conductivity, oxidation reduction potential, and dissolved oxygen were 13.5 °C, 7.1, 2.3 mS cm−1, -23.1 mV, and 16 μM, respectively. Sampling from an adjacent well at 3 m bsd showed that over the course of the year, and rise in the water table, pH and specific conductivity showed little variability (between 6.9 and 7.1, and 2,200–2,400 μS s−1, respectively). Dissolved oxygen declined from atmospheric concentrations under unsaturated conditions to <0.1 μM as the water table rose, before increasing monotonically as the water table fell to background levels again. High resolution dissolved oxygen data from this well has also been published recently (Yabusaki et al., 2017).
3.2. Sequential Nitrification and Denitrification Drive Subsurface Nitrogen Cycling
3.2.1. NO3− Accumulation in the Vadose Zone
At the outset of the study, just prior to snowmelt, NO3− concentrations ranged with depth between 2 μM and 1.8 mM in the first 1.5 m. At 2, 2.5, and 3 m bsd NO3− accumulated to 5.6, 6.1, and 4.5 mM, respectively, but showed clear seasonal trends concomitant with changes in water table height (Figure 1). At this point δ15NNO3 and δ18ONO3 averaged -1.8 ± 0.1 and −8.1 ± 0.3‰ at 2 m bsd, 3.5 ± 0.3 and −6.3 ± 1.2‰ at 2.5 m, and 3.8 ± 0.01 and −7.3 ± 0.1‰ at 3 m, respectively (Figure 2A). We used a simple a two end-member mixing model (Wexler et al., 2014) to determine the contribution of different sources to NO3− accumulation. This model estimates between 82.6 and 99% of the NO3− accumulating under oxygenated conditions in the vadose zone was attributable to coupled ammonia and NO2− oxidation (nitrification). The source of the variance in this estimate stems from the range of potential δ18ONO3. The lower range of this estimate arises when not accounting for the full exchange of oxygen atoms between NO2− and H2O. This high attribution of NO3− accumulation due to nitrification is plausible given the low rates of net recharge from surface precipitation (~3 cm yr−1, Yabusaki et al., 2017).
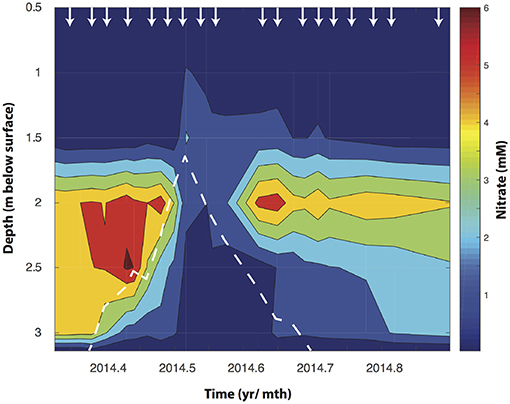
Figure 1. Depth resolved NO3− concentrations changes in the Rifle floodplain well (TT-03) during an annual rise watertable depth (shown by the dotted white line). The arrows indicate the time points at which water samples were collected from each depth.
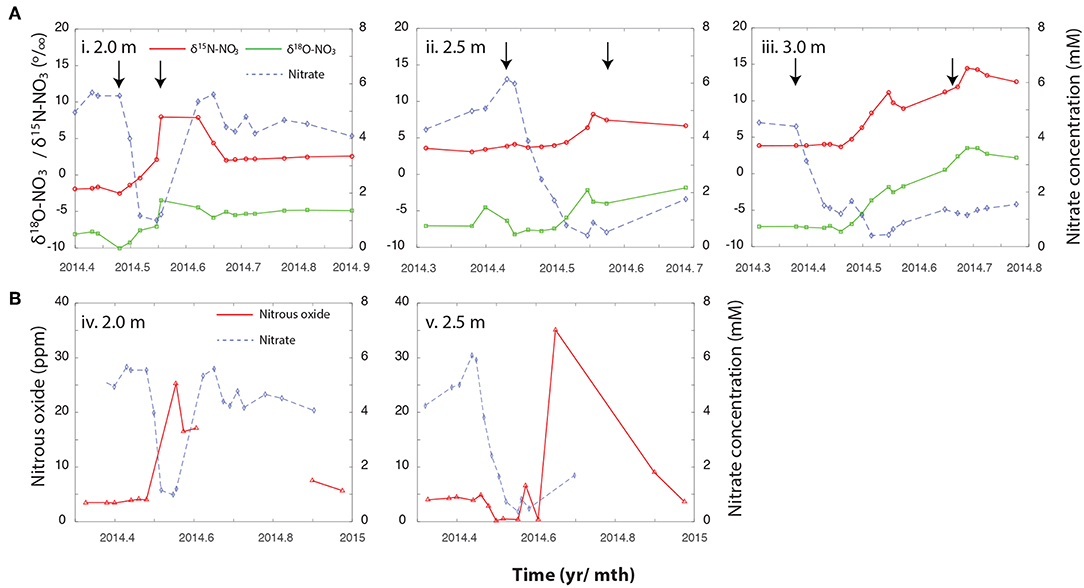
Figure 2. (A) Changes in NO3− concentrations, and the associated δ15NNO3 and δ18ONO3 over the Spring and Summer of 2014 at (i) 2 m, (ii) 2.5 m, and (iii) 3 m bsd. (B) Trajectory of N2O trace gas measurements against NO3− concentrations across the same depth profile, (iv) 2 m, and (v) 2.5 m. These measurements were not taken as frequently than the NO3− isotopic measurements but encompass the period depicted in the corresponding panels above. Arrows indicate the period during which the water table saturated the relevant depth. Please note that the x-axis is different between the upper panel (A) and lower panel (B).
Metagenomic surveys of the Rifle floodplain indicate that ammonia-oxidation is likely carried out by the Thaumarchaeota (AOA) (Castelle et al., 2013; Hug et al., 2015a). This is perhaps unsurprising given the low NH4+ concentrations in the Rifle subsurface (which were typically below method detection limits, but when measured ranged from 5 to 85 μM), as the AOA have previously been characterized as having half-saturation constants in the nM range (Martens-Habbena et al., 2009) and dominate nitrification in ecosystems where NH4+ concentrations are similarly low (Beman et al., 2012). The source of NH4+ supporting nitrification at the capillary fringe has yet to be identified, and while sediment adsorbed NH4+ (Böhlke et al., 2006) and biological nitrogen fixation (Swanner and Templeton, 2011; Lau et al., 2014) might be a source of subsurface nitrogen it is likely that OM mineralization contributes the bulk of NH4+. Rifle sediments can be carbon rich due to the advective downward transport of DOM (Tokunaga et al., 2016), and heterogeneously distributed fine-grained sediment lenses (Janot et al., 2016). These lenses are enriched in organic carbon that likely represent the deep burial of soil horizons (Janot et al., 2016), common within floodplain sediments (Blazejewski et al., 2009; Hill, 2010).
3.2.2. NO3− Loss During Groundwater Rise
During the spring snowmelt the Colorado River rises increasing the height of the groundwater table in the adjacent Rifle floodplain. As the water table rose (during May at 2 and 2.5 m and April for 3 m), NO3− concentrations declined. The onset of NO3− loss was temporally offset between the different depths, occurring first at 3 m bsd (around mid to late April), then at 2.5 m (early May), and finally at 2 m by mid May. This is consistent with the timing of groundwater incursion into the unsaturated zone at these depths (Figure 1). The temporal offset in NO3− decline between 3, 2.5, and 2 m bsd (Figure 1) indicates that the groundwater rise and resultant change in O2 availability are primarily responsible for hot moments of biogeochemical activity.
To further characterize how biotic and abiotic pathways contribute to the observed NO3− dynamics we measured the and δ15NNO3 of NO3−. The enrichment of the and δ15NNO3 alongside changes in NO3− concentration is indicative of enzymatic fractionating mechanisms, including metabolisms producing and consuming NO3−. Conversely, dilution imparts no isotopic effect on NO3− despite a decline in NO3− concentrations. At the 2 m depth, a steady increase in the δ15NNO3 from initial values (−1.8 ± 0.1) to 7.9 ± 0.3‰ and from −8.1 ± 0.3 to −3.5‰, accompany the rapid fall in NO3− concentrations, and is indicative of NO3− cycling by an actively fractionating process (e.g., enzymatic NO3− reduction, further discussed below). As NO3− begins to accumulate again, δ15NNO3 and δ18ONO3 dropped to ~ 2.3 ± 0.21 and −5.1 ± 0.27 ‰, respectively (Figure 2A). At both 2.5 and 3 m, the onset of NO3− loss precedes the enrichment of δ15NNO3 and , indicative of dilution of NO3− as the NO3−-depleted groundwater mixes with NO3− enriched porewater (Figure 2A). While the groundwater at Rifle is anoxic, as it rises into the unsaturated zone it sequentially entrains O2 at the interface of the groundwater and the unsaturated zone (Yabusaki et al., 2017). This subsequent oxygenation can inhibit denitrification, and delay its onset at the deeper depths. The 2 m depth does not follow this trajectory despite the likelihood of O2 entrainment at this depth. It is plausible that a higher rate of activity at this depth is responsible for the rapid consumption of O2 upon saturation stimulating the onset of NO3− reduction, however, further work is required to verify this.
Further evidence for biological NO3− reduction comes from the timing of NO2− production at the different depths. While variable throughout the year, NO2− peaked during mid-May at both 2.5 and 3 m, at 0.23 and 0.22 mM, respectively, These peaks are temporally lagged relative to the observed decline in NO3− (Figure 3), and likely represents the product of NO3− reduction at these depths.
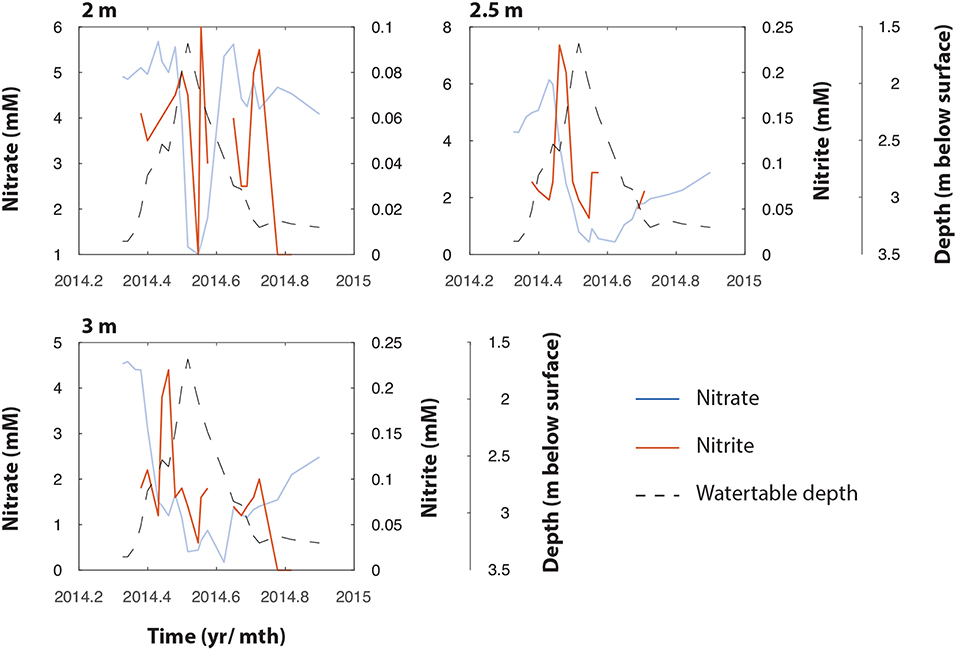
Figure 3. NO3− (right axis) and NO2− (left axis) measurements at 2.0, 2.5, and 3.0 m bgs in TT-03 during 2014. The plots show coincidental and temporally lagged production of NO2− as NO3− declines, followed by apparent loss of NO2−. The water table depth is represented by the black dotted line.
A drop in NO3− at 3 m bsd after May 23rd was accompanied by an increase in δ15NNO3 from 3.5 ± 0.3 to 11.1 ± 0.3‰ and δ18ONO3 from −6.3 ± 1.2 to −1.8 ± 0.4‰, which could be the result of NO3− reduction as this depth becomes anoxic. The 2.5 m bsd shows further evidence of dilution/NO3− reduction being responsible for the decline in NO3−. At this depth NO3− concentrations declined with no initial impact on δ15NNO3 and . However, after June 6th isotope enrichment was observed in the residual NO3− increasing from ~ 4.0 ± 0.1 to 8.4 ± 0.2‰ and, from −7.2 ± 0.1 to −2.8 ± 0.1‰ for δ15NNO3 and , respectively. A simple Rayleigh model was used to estimate the contribution of actively fractionating processes to the observed drop in NO3− concentrations at each depth. At 2 m bsd, a shift in δ15NNO3 of +10‰ suggests NO3− reduction is responsible for ~64% (with a range of 52–83% when varying the ϵ value between 10 and 20‰) of the drop in NO3− concentrations. By contrast, dilution appeared to dominate at 2.5 bsd and NO3− reduction was calculated to be responsible for only ~ 28% (22–39%) of the observed decline in NO3− concentrations. Finally, at 3 m bsd the initial decline in NO3− was followed by a second period of NO3− decrease. Approximately 91% (86–93%) of the NO3− loss during this first event was estimated to be attributable to dilution. Conversely, the contribution of dilution declines to 53% (46–63%) during the second event with the rest attributed to NO3− reduction.
Plotting the Δδ18O against Δδ15N values from all three depths revealed distinct dual isotope dynamics at the different depths. At 2 m, the slope of the dual isotope regression was 0.58 (Figure 4), while both 2.5 and 3 m had slopes of ~1 (1.08 and 1.05 at 2.5 and 3 m, respectively). Evaluating the change in fractionation of δ18ONO3 relative to δ15NNO3 provides information on the sources and transformations of NO3− within this aquifer (Granger and Wankel, 2016). The relative isotopic enrichment for denitrification has previously been shown to range from a ratio of 0.6 in freshwater aquifers, representative of sequential nitrite oxidation/ denitrification or anammox (Lehmann et al., 2003; Granger and Wankel, 2016), to a ratio of 1 within the marine environment (Sigman et al., 2005) and bacterial cultures (Granger et al., 2008), characteristic of heterotrophic denitrification. These relationships provide further support for heterotrophic NO3− reduction as the dominant biological NO3− loss mechanism at 2.5 m and 3 m depth. However, the lower ratio (0.6) at 2 m suggests additional metabolisms contributing to the isotopic signal at this depth, such as NO3− production via anammox or aerobic NO2− oxidation (Granger and Wankel, 2016). Both processes produce NO3− from NO2− with an inverse N isotope effect, yielding NO3− with a relatively high δ15N value (Casciotti, 2009; Brunner et al., 2013; Kobayashi et al., 2019). Both processes also incorporate an O atom from water with isotopic fractionation (Buchwald and Casciotti, 2010; Kobayashi et al., 2019). A Δδ18O: Δδ15N ratio of 0.6 within freshwater sediments has previously been attributed to the in situ activity of anammox bacteria (Smith et al., 2015). The relatively high δ15NNO3 and low δ18ONO3 produced during anammox (Brunner et al., 2013; Kobayashi et al., 2019), means that anammox bacteria only have to produce a small amount of NO3− to force a deviation the Δδ18O: Δδ15N trajectory. Indeed, anammox has been identified to occur deeper within the Rifle sediment (Jewell et al., 2016), and could, therefore, be contributing to the deviation from 1 observed at the 2 m depth. However, we believe this is unlikely. The 2 m depth at this site is saturated only during years of high snowpack that lead to groundwater depths shallower than 2.5 m. Therefore, the highly reducing conditions that form the niche of anammox bacteria at the Rifle site are not replicated at the predominantly aerobic 2 m depth.
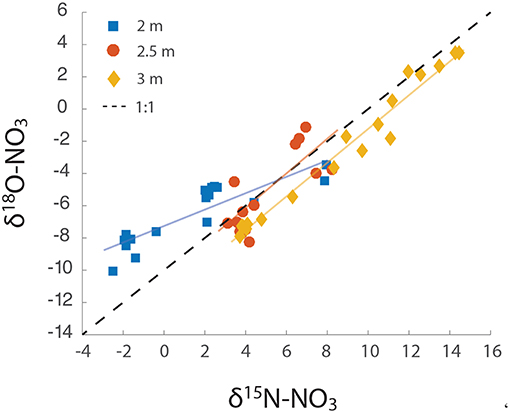
Figure 4. Δδ18O: Δδ15N over the course of the groundwater rise. The slope of each line was calculated as 2 m = 0.58, 2.5 m = 1.08, and 3 m = 1.05.
We propose that the observed deviation in Δδ18O: Δδ15N at the 2 m depth is attributable to two main factors: (1) the cyclic production of NO3−, via aerobic oxidation and consumption via NO3− reduction, and (2) a relatively low value for δ18OH2O, of ~ −14‰, incorporated during NO2− oxidation. Granger and Wankel (2016), using a model of NO3− isotope dynamics, demonstrated that the ratio of NO2− oxidation to dissimilatory NO3− reduction to NO2− is a critical determinant of the δ15NNO3 and δ18ONO3 trajectory. Increasing ratios of NO2− oxidation to NO3− consumption (>0.5) shift this relationship below a slope of 1 (Granger and Wankel, 2016). A further increase in NO3− production relative to its consumption (>0.8) can shift the Δδ18O: Δδ15N relationship above 1. However, high NO2− oxidation with a low δ18OH2O (> − 10‰) results in a slope significantly below 1, and similar to that found at the 2 m depth in this study. In summary, rapid redox dynamics at the shallowest and irregularly saturated depth provide an ideal niche for NO2− oxidizers and facultative heterotrophic denitrifiers that likely act sequentially to produce and remove NO3−.
The metabolisms responsible for the turnover of NO3− are difficult to distinguish through conventional molecular methods. Several recent studies have highlighted the predominance of chemolithoautotrophic metabolisms within the groundwater, including NO3−-dependent iron oxidation, via the Gallionellaceae, and anammox (Hug et al., 2015a; Jewell et al., 2016). A question therefore remains as to whether these metabolisms move as the water table rises, and contribute to NO3− and NO2− turnover, or whether NO3− is consumed at the capillary fringe by heterotrophic denitrifiers. As the water table rises and oxygen availability declines, we propose that heterotrophic denitrifiers catalyze the bulk of biological NO3− turnover, with an uncertain, but likely minor, role for chemolithoautotrophic metabolisms (i.e., NO3− reduction coupled to anammox). There are several reasons to hypothesize in this way. Recent studies of the molecular microbial diversity of the Rifle subsurface demonstrate a broad distribution of heterotrophs capable of carrying out NO3− reduction and additional denitrification pathways from the vadose zone into the groundwater (Hug et al., 2015b; Anantharaman et al., 2016), but a general restriction of anaerobic chemolithoautotrophs to the groundwater and naturally reduced zones (NRZs) within the floodplain (Jewell et al., 2016).
The likely distribution of N-cycling organisms can be explained by the interaction between the traits of different functional guilds and their environment. The fluctuating aerobic conditions at the capillary fringe likely favors facultative denitrifying aerobes with the metabolic flexibility to switch from respiration via oxygen (O2) as an electron acceptor, to NO3− (NO3−). On other hand, the anammox bacteria are obligate anaerobes, with a low tolerance of O2 (Oshiki et al., 2016), and characterized by a slow growth rate (~0.0026–0.0041 h−1) and a thermodynamically limiting metabolism (Kartal et al., 2007, 2011), impinging on the rate at which these organisms respond to changing environmental conditions. It is, therefore, unlikely that the N-cycling metabolisms characterized within the NRZs are responsible for the observed rapid biological loss of NO3− and NO2−.
Furthermore, the peak in N2O at 2 m bsd coincides with the most intense period of NO3− and NO2− removal (Figures 2B, 3), and an increasing trend in N2 gas (Figure S2). Concentrations of N2O measured in the unsaturated zone above the water table were elevated relative to atmospheric concentrations throughout the study period. At 2 m bsd, N2O peaked at 25 ppm between late May and early June, concomitant with the most intense period of NO3− decline (Figure 2B). The N2O concentration at the 2.5 m depth peaked at 35 ppm in late July shortly after the water table dropped below this depth and during maximal enrichment of the δ15N of the NO3−. The 3 m interval was saturated during the period from late-April through late-October and no N2O samples were taken during this period of intense denitrification at this depth. N2O is produced as an intermediate during denitrification or nitrifier denitrification. To our knowledge, N2O is not an intermediate produced during anammox. The conditions that partition N2O flux between heterotrophic denitrification and autotrophic denitrification are not well-defined, however, concurrent measurements of the δ15NN2O and δ18ON2O point to heterotrophic denitrification as the principal N2O source in the unsaturated zone (Bill et al., in prep.).
4. Conclusion
We show here the annually observed build up and dissipation of NO3− at the capillary fringe of the Rifle site is attributable to abiotic and biotic mechanisms. Furthermore, we conclude here that biological nitrogen cycling around the capillary fringe within the Rifle floodplain was predominantly attributable to sequential nitrification-denitrification. We offer the following conceptual model (Figure 5) for the distribution of N-cycling organisms. Around the capillary fringe high organic matter concentrations (either within or proximate to the NRZs, Janot et al., 2016) support sequential nitrification and denitrification, oxidizing reduced nitrogen, released during organic matter mineralization, to NO3−, which can be reduced to N2O or N2. Within the NRZs, high iron and sulfide concentrations support chemolithoautotrophic NO3−-reduction to NO2− that can be coupled to anammox activity (Jewell et al., 2016). It is unlikely these metabolisms move with the rising water table, and we propose that the bulk of nitrogen loss can be attributable to heterotrophic denitrification. It is likely that such a cycle is characteristic of mountainous floodplains in the Rockies, however, further work is required to verify this. Finally, we highlight during the most intense periods of heterotrophic denitrification, N2O concentrations emitted are significantly larger than have been previously associated with subsurface aquifers (McMahon et al., 2000; Hiscock et al., 2003; Weymann et al., 2008), thus arguing for a greater consideration of such regions within global N2O budgets (Davidson and Kanter, 2014).
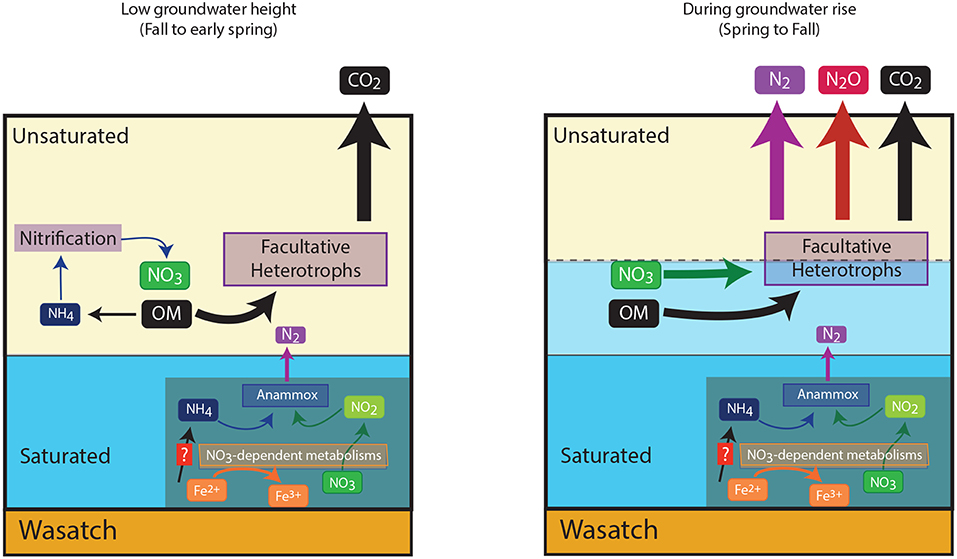
Figure 5. Conceptual nitrogen cycle within the Rifle aquifer based on measurements made in the present study, and recent molecular based studies. The figure shows the switch in metabolic processes at the capillary fringe, and also the likely positioning of anaerobic chemolithoautotrophs in the naturally reduced zones of the aquifer. This aquifer is underlain by an impermeable Wasatch layer indicated in the figure.
Author Contributions
NB, MC, and KW designed and carried out the research. KW collected samples and provided ancillary data. MF and KC performed isotopic analysis. NB, MC, and KC analyzed the data. MB measured N2O and N2 in gas samples. NB, MC, and KC wrote the manuscript with contribution from all co-authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was performed as part of the Lawrence Berkeley National Laboratory's Sustainable Systems Scientific Focus Area funded by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under contract DE-AC02-05CH11231, and in part from a Laboratory Directed Research and Development Grant from the Office of the Director at Lawrence Berkeley National Laboratory. KC acknowledges support from the Stanford University Terman Fellowship. Thismanuscript was previously released as a preprint (doi: 10.5194/bg-2017-212), prior to being withdrawn. We are grateful to Boris Faybishenko, Tetsu Tokunaga, and Peter Nico for ancillary data and discussion during the preparation of this manuscript. We also acknowledge the comments of two reviewers which improved this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2019.00189/full#supplementary-material
References
Abit, S. M., Amoozegar, A., Vepraskas, M. J., and Niewoehner, C. P. (2008). Fate of NO3− in the capillary fringe and shallow groundwater in a drained sandy soil. Geoderma 146, 209–215. doi: 10.1016/j.geoderma.2008.05.015
Allison, S. D., Czimczik, C. I., and Treseder, K. K. (2008). Microbial activity and soil respiration under nitrogen addition in Alaskan boreal forest. Glob. Change Biol. 14, 1156–1168. doi: 10.1111/j.1365-2486.2008.01549.xs
Anantharaman, K., Brown, C. T., Hug, L. A., Sharon, I., Castelle, C. J., Probst, A. J., et al. (2016). Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 7:13219. doi: 10.1038/ncomms13219
Beman, J. M., Popp, B. N., and Alford, S. E. (2012). Quantification of ammonia oxidation rates and ammonia-oxidizing archaea and bacteria at high resolution in the Gulf of California and eastern tropical North Pacific Ocean. Limnol. Oceangr. 57, 711–726. doi: 10.4319/lo.2012.57.3.0711
Berkowitz, B., Silliman, S. E., and Dunn, A. M. (2004). Impact of the capillary fringe on local flow, chemical migration, and microbiology. Vadose Zone J. 3, 534–548. doi: 10.2136/vzj2004.0534
Blazejewski, G. A., Stolt, M. H., and Gold, A. J. (2009). Spatial distribution of carbon in the subsurface of riparian zones. Soil Sci. Soc. Am. J. 73:1733. doi: 10.2136/sssaj2007.0386
Böhlke, J. K., Mroczkowski, S. J., and Coplen, T. B. (2003). Oxygen isotopes in NO3−: new reference materials for 18O:17O:16O measurements and observations on NO3−-water equilibration. Rapid Commun. Mass Spectrom. 17, 1835–1846. doi: 10.1002/rcm.1123
Böhlke, J. K., Smith, R. L., and Miller, D. N. (2006). NH4+ transport and reaction in contaminated groundwater: application of isotope tracers and isotope fractionation studies. Water Resourc. Res. 42, 1–19. doi: 10.1029/2005WR004349
Bouskill, N. J., Lim, H. C., Borglin, S., Salve, R., Wood, T. E., Silver, W. L., et al. (2013). Pre-exposure to drought increases the resistance of tropical forest soil bacterial communities to extended drought. ISME J. 7, 384–394. doi: 10.1038/ismej.2012.113
Brunner, B., Contreras, S., Lehmann, M. F., Matantseva, O., Rollog, M., Kalvelage, T., et al. (2013). Nitrogen isotope effects induced by anammox bacteria. Proc. Natl. Acad. Sci. U.S.A. 110, 18994–18999. doi: 10.1073/pnas.1310488110
Buchwald, C., and Casciotti, K. L. (2010). Oxygen isotopic fractionation and exchange during bacterial NO2− oxidation. Limnol. Oceangr. 55, 1064–1074. doi: 10.4319/lo.2010.55.3.1064
Buchwald, C., Santoro, A. E., McIlvin, M. R., and Casciotti, K. L. (2012). Oxygen isotopic composition of NO3− and NO2− produced by nitrifying co-cultures and natural marine assemblages. Limnol. Oceanogr. 47, 1361–1375. doi: 10.4319/lo.2012.57.5.1361
Casciotti, K. L. (2009). Inverse kinetic isotope fractionation during bacterial nitrite oxidation. Geochim. Cosmochim. Acta 73, 2061–2076. doi: 10.1016/j.gca.2008.12.022
Casciotti, K. L., Böhlke, J. K., McIlvin, M. R., Mroczkowski, S. J., and Hannon, J. E. (2007). Oxygen isotopes in NO3−: analysis, calibration and equilibration. Anal. Chem. 79, 2427–2436. doi: 10.1021/ac061598h
Casciotti, K. L., McIlvin, M. R., and Buchwald, C. (2010). Oxygen isotopic exchange and fractionation during bacterial ammonia oxidation. Limnol. Oceanogr. 55, 753–762. doi: 10.4319/lo.2010.55.2.0753
Casciotti, K. L., Sigman, D. M., Hastings, M. G., Böhlke, J. K., and Hilkert, A. (2002). Measurement of the oxygen isotopic composition of NO3− in seawater and freshwater using the denitrifier method. Anal. Chem. 74, 4905–4912. doi: 10.1021/ac020113w
Castelle, C. J., Hug, L. A., Wrighton, K. C., Thomas, B. C., Williams, K. H., Wu, D., et al. (2013). Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat. Commun. 4:2120. doi: 10.1038/ncomms3120
Clague, J. C., Stenger, R., and Clough, T. J. (2015). Evaluation of the stable isotope signatures of NO3− to detect denitrification in a shallow groundwater system in New Zealand. Agric. Ecosyst. Environ. 202, 188–197. doi: 10.1016/j.agee.2015.01.011
Daims, H., Lebedeva, E. V., Pjevac, P., Han, P., Herbold, C., Albertsen, M., et al. (2015). Complete nitrification by nitrospira bacteria. Nature 528, 504–509. doi: 10.1038/nature16461
Daims, H., Lücker, S., and Wagner, M. (2016). A new perspective on microbes formerly Known as NO2−-oxidizing bacteria. Trends Microbiol. 24, 699–712. doi: 10.1016/j.tim.2016.05.004
Davidson, E. A., and Kanter, D. (2014). Inventories and scenarios of nitrous oxide emissions. Environ. Res. Lett. 9:105012. doi: 10.1088/1748-9326/9/10/105012
Einsiedl, F., and Mayer, B. (2006). Hydrodynamic and microbial processes controlling NO3− in a fissured-porous Karst aquifer of the Franconian Alb, Southern Germany. Environ. Sci. Technol. 40, 6697–6702. doi: 10.1021/es061129x
Fang, Y., Koba, K., Makabe, A., Zhu, F., Fan, S., Liu, X., et al. (2012). Low δ18O Values of NO3− produced from nitrification in temperate forest soils. Environ. Sci. Technol. 46, 8723–8730. doi: 10.1021/es300510r
Frey, C., Hietanen, S., Jürgens, K., Labrenz, M., and Voss, M. (2014). N and O isotope fractionation in NO3− during chemolithoautotrophic denitrification by sulfurimonas gotlandica. Environ. Sci. Technol. 48:22. doi: 10.1021/es503456g
Gonneea, M. E., and Charette, M. A. (2014). Hydrologic controls on nutrient cycling in an unconfined coastal aquifer. Environ. Sci. Technol. 48, 14178–14185. doi: 10.1021/es503313t
Granger, J., and Sigman, D. M. (2009). Removal of NO2− with sulfamic acid for NO3− N and O isotope analysis with the denitrifier method. Rapid Commun. Mass Spectrom. 23, 3753–3762. doi: 10.1002/rcm.4307
Granger, J., Sigman, D. M., Lehmann, M. F., and Tortell, P. D. (2008). Nitrogen and oxygen isotope fractionation during dissimilatory NO3− reduction by denitrifying bacteria. Limnol. Oceangr. 53, 2533–2545. doi: 10.4319/lo.2008.53.6.2533
Granger, J., and Wankel, S. D. (2016). Isotopic overprinting of nitrification on denitrification as a ubiquitous and unifying feature of environmental nitrogen cycling. Proc. Natl. Acad. Sci. U.S.A. 113, E6391–E6400. doi: 10.1073/pnas.1601383113
Haberer, C. M., Rolle, M., Cirpka, O. A., and Grathwohl, P. (2012). Oxygen transfer in a fluctuating capillary fringe. Vadose Zone J. 11:vzj2014.04.0039. doi: 10.2136/vzj2011.0056
Heffernan, J. B., Albertin, A. R., Fork, M. L., Katz, B. G., and Cohen, M. J. (2012). Denitrification and inference of nitrogen sources in the karstic Floridan aquifer. Biogeosciences 9, 1671–1690. doi: 10.5194/bg-9-1671-2012
Hefting, M., Clement, J. C., Dowrick, D., and Cosandey, A. C. (2004). Water table elevation controls on soil nitrogen cycling in riparian wetlands along a European climatic gradient. Biogeochemistry 67, 113–134. doi: 10.1023/b:biog.0000015320.69868.33
Hill, A. R. (2010). Buried organic-rich horizons: their role as nitrogen sources in stream riparian zones. Biogeochemistry 104, 347–363. doi: 10.1007/s10533-010-9507-5
Hiscock, K. M., Bateman, A. S., Mühlherr, I. H., Fukada, T., and Dennis, P. F. (2003). Indirect emissions of nitrous oxide from regional aquifers in the United Kingdom. Environ. Sci. Technol. 37, 3507–3512. doi: 10.1021/es020216w
Hug, L. A., Thomas, B. C., Brown, C. T., Frischkorn, K. R., Williams, K. H., Tringe, S. G., et al. (2015a). Aquifer environment selects for microbial species cohorts in sediment and groundwater. ISME J. 9, 1846–1856. doi: 10.1038/ismej.2015.2
Hug, L. A., Thomas, B. C., Sharon, I., Brown, C. T., Sharma, R., Hettich, R. L., et al. (2015b). Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ. Microbiol. 18, 159–173. doi: 10.1111/1462-2920.12930
Janot, N., Lezama Pacheco, J. S., Pham, D. Q., O'Brien, T. M., Hausladen, D., Noël, V., et al. (2016). Physico-chemical heterogeneity of organic-rich sediments in the Rifle aquifer, CO: impact on uranium biogeochemistry. Environ. Sci. Technol. 50, 46–53. doi: 10.1021/acs.est.5b03208
Jewell, T. N., Karaoz, U., Brodie, E. L., Williams, K. H., and Beller, H. R. (2016). Metatranscriptomic evidence of pervasive and diverse chemolithoautotrophy relevant to C, S, N and Fe cycling in a shallow alluvial aquifer. ISME J. 10, 2106–2117. doi: 10.1038/ismej.2016.25
Jost, D., Haberer, C. M., and Grathwohl, P. (2015). Oxygen transfer in a fluctuating capillary fringe: impact of microbial respiratory activity. Vadose Zone J. 14:vzj2014.04.0039. doi: 10.2136/vzj2014.04.0039
Kartal, B., Geerts, W., and Jetten, M. S. (2011). Cultivation, detection, and ecophysiology of anaerobic NH4+-oxidizing bacteria. Meth. Enzymol. 486, 89–108. doi: 10.1016/S0076-6879(11)86004-0
Kartal, B., Kuypers, M. M. M., Lavik, G., Schalk, J., Op den Camp, H. J. M., Jetten, M. S. M., et al. (2007). Anammox bacteria disguised as denitrifiers: nitrate reduction to dinitrogen gas via nitrite and ammonium. Environ. Microbiol. 9, 635–642. doi: 10.1111/j.1462-2920.2006.01183.x
Kendall, C., Elliott, E. M., and Wankel, S. D. (2007). “Chapter 12: Tracing anthropogenic inputs of nitrogen to ecosystems,” in Stable Isotopes in Ecology and Environmental Science, 2nd Edn, eds R. H. Michener and K. Lajtha (Malden, MA: Blackwell Publishing), 375–449. doi: 10.1002/9780470691854.ch12
Kobayashi, K., Makabe, A., Yano, M., Oshiki, M., Kindaichi, T., Casciotti, K. L., et al. (2019). Dual nitrogen and oxygen isotope fractionation during anaerobic ammonium oxidation by anammox bacteria. ISME J. doi: 10.1038/s41396-019-0440-x. [Epub ahead of print].
Lau, M. C., Cameron, C., Magnabosco, C., Brown, C. T., Schilkey, F., Grim, S., et al. (2014). Phylogeny and phylogeography of functional genes shared among seven terrestrial subsurface metagenomes reveal N-cycling and microbial evolutionary relationships. Front. Microbiol. 5:531. doi: 10.3389/fmicb.2014.00531
Le Roux, X., Bouskill, N. J., Niboyet, A., Barthes, L., Dijkstra, P., Field, C. B., et al. (2016). Predicting the responses of soil NO2−-oxidizers to multi-factorial global change: a trait-based approach. Front. Microbiol. 7:628. doi: 10.3389/fmicb.2016.00628
Lehmann, M. F., Reichert, P., Bernasconi, S. M., Barbieri, A., and McKenzie, J. A. (2003) Modeling nitrogen oxygen isotope fractionation during denitrification in a lacustrine redox-transition zone. Geochim. Cosmochim. Acta 67, 2529–2542. doi: 10.1016/S0016-7037(03)00085-1
Lohse, K. A., Brooks, P. D., McIntosh, J. C., Meixner, T., and Huxman, T. E. (2009). Interactions between biogeochemistry and hydrologic systems. Annu. Rev. Environ. Resourc. 34, 65–96. doi: 10.1146/annurev.environ.33.031207.111141
Martens-Habbena, W., Berube, P. M., Urakawa, H., de la Torre, J. R, and Stahl, D. A. (2009). Ammonia oxidation kinetics determine niche separation of nitrifying archaea and bacteria. Nature 461, 976–979. doi: 10.1038/nature08465
McClain, M. E., Boyer, E. W., Dent, C. L., Gergel, S. E., Grimm, N. B., Groffman, P. M., et al. (2003). Biogeochemical hot spots and hot moments at the interface of terrestrial and aquatic ecosystems. Ecosystems 6, 301–312. doi: 10.1007/s10021-003-0161-9
McMahon, P. B., Bruce, B. W., Becker, M. F., Pope, L. M., and Dennehy, K. F. (2000). Occurrence of nitrous oxide in the central high plains aquifer, 1999. Environ. Sci. Technol. 34, 4873–4877. doi: 10.1021/es001233t
Oshiki, M., Satoh, H., and Okabe, S. (2016). Ecology and physiology of anaerobic NH4+ oxidizing bacteria. Environ. Microbiol. 18, 2784–2796. doi: 10.1111//1462-2920.13134
Persson, M., Dahlin, T., and Günther, T. (2015). Observing solute transport in the capillary fringe using image analysis and electrical resistivity tomography in laboratory experiments. Vadose Zone J. 14, 1–12. doi: 10.2136/vzj2014.07.0085
Sigman, D. M., Casciotti, K. L., Andreani, M., Barford, C., Galanter, M., and Böhlke, J. K. (2001). A bacterial method for the nitrogen isotopic analysis of NO3− in seawater and freshwater. Anal. Chem. 73, 4145–4153. doi: 10.1021/ac010088e
Sigman, D. M., Granger, J., DiFiore, P. J., Lehmann, M. M., Ho, R., Cane, G., et al. (2005). Coupled nitrogen and oxygen isotope measurements of NO3− along the eastern North Pacific margin. Glob. Biogeochem. Cycles 19:GB4022. doi: 10.1029/2005GB002458
Smith, R. L., Baumgartner, L. K., Miller, D. N., Repert, D. A., and Böhlke, J. K. (2006). Assessment of nitrification potential in ground water using short term, single-well injection experiments. Microb. Ecol. 51, 22–35. doi: 10.1007/s00248-004-0159-7
Smith, R. L., Böhlke, J. K., Song, B., and Tobias, C. R. (2015). Role of anaerobic NH4+ oxidation (anammox) in nitrogen removal from a freshwater aquifer. Environ. Sci. Technol. 49, 12169–12177. doi: 10.1021/acs.est.5b02488
Sorensen, J. P. R., Butcher, A. S., Stuart, M. E., and Townsend, B. R. (2015). NO3− fluctuations at the water table: implications for recharge processes and solute transport in the Chalk aquifer. Hydrol. Process. 29, 3355–3367. doi: 10.1002/hyp.10447
Stegen, J. C., Fredrickson, J. K., Wilkins, M. J., Konopka, A. E., Nelson, W. C., Arntzen, E. V., et al. (2016). Groundwater-surface water mixing shifts ecological assembly processes and stimulates organic carbon turnover. Nat. Commun. 7:11237. doi: 10.1038/ncomms11237
Swanner, E. D., and Templeton, A. S. (2011). Potential for nitrogen fixation and nitrification in the granite-hosted subsurface at Henderson Mine, CO. Front. Microbiol. 2:254. doi: 10.3389/fmicb.2011.00254
Tokunaga, T. K., Kim, Y., Conrad, M. E., Bill, M., Hobsona, C., Williams, K. H., et al. (2016). Deep vadose zone respiration contributions to carbon dioxide fluxes from a semiarid floodplain. Vadose Zone J. 15, 1–14. doi: 10.2136/vzj2016.02.0014
Ward, B. B. (2011). “Nitrification in the ocean,” in Nitrification, eds B. B. Ward, M. G. Klotz, and D. A. Arp (Washington, DC: ASM Press), 325–345.
Wexler, S. K., Goodale, C. L., McGuire, K. J., Bailey, S. W., and Groffman, P. M. (2014). Isotopic signals of summer denitrification in a northern hardwood forested catchment. Proc. Natl. Acad. Sci. U.S.A. 111, 16413–16418. doi: 10.1073/pnas.1404321111
Weymann, D., Well, R., Flessa, H., von der Heide, C., Deurer, M., Meyer, K., et al. (2008). Assessment of excess N2 and groundwater N2O emission factors of NO3−-contaminated aquifers in northern Germany. Biogeosciences 5, 1215–1226. doi: 10.5194/bg-5-1215-2008
Williams, K. H., Long, P. E., Davis, J. A., Wilkins, M. J., N'Guessan, A. L., Steefel, C. I., et al. (2011). Acetate availability and its influence on sustainable bioremediation of uranium-contaminated groundwater. Geomicrobiol. J. 28, 519–539. doi: 10.1080/01490451.2010.520074
Yabusaki, S. B., Fang, Y., Long, P. E., Resch, C. T., Peacock, A. D., Komlos, J., et al. (2007). Uranium removal from groundwater via in situ biostimulation: field-scale modeling of transport and biological processes. J. Contam. Hydrol. 93, 216–235. doi: 10.1016/j.jconhyd.2007.02.005
Yabusaki, S. B., Wilkin, M. J., Fang, Y., Williams, K. H., Arora, B, Bargar, J., et al. (2017). Water table dyanmics and biogeochemical cycling in a shallow, variably-saturated floodplain. Environ. Sci. Technol. 51:3307. doi: 10.1021/acs.est.6b04873
Zachara, J. M., Long, P. E., Bargar, J., Davis, J. A., Fox, P., Fredrickson, J. K., et al. (2013). Persistence of uranium groundwater plumes: contrasting mechanisms at two DOE sites in the groundwater-river interaction zone. J. Contam. Hydrol. 147, 45–72. doi: 10.1016/j.jconhyd.2013.02.001
Keywords: nitrate cycling, nitrogen isotopes, subsurface aquifer, terrestrial aquatic interface, microbial modeling
Citation: Bouskill NJ, Conrad ME, Bill M, Brodie EL, Cheng Y, Hobson C, Forbes M, Casciotti KL and Williams KH (2019) Evidence for Microbial Mediated NO3− Cycling Within Floodplain Sediments During Groundwater Fluctuations. Front. Earth Sci. 7:189. doi: 10.3389/feart.2019.00189
Received: 13 August 2018; Accepted: 04 July 2019;
Published: 31 July 2019.
Edited by:
Timothy Ferdelman, Max Planck Institute for Marine Microbiology (MPG), GermanyReviewed by:
Hannah Karen Marchant, Max Planck Institute for Marine Microbiology (MPG), GermanyLaura Anne Bristow, University of Southern Denmark, Denmark
Copyright © 2019 Bouskill, Conrad, Bill, Brodie, Cheng, Hobson, Forbes, Casciotti and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas J. Bouskill, bmpib3Vza2lsbEBsYmwuZ292