 Viviana L. Vedder
Viviana L. Vedder
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 30 June 2020
Sec. Atherosclerosis and Vascular Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.00109
Cardiovascular diseases, such as atherosclerosis, are the leading cause of death worldwide. Although mice are currently the most commonly used model for atherosclerosis, zebrafish are emerging as an alternative, especially for inflammatory and lipid metabolism studies. Here, we review the history of in vivo atherosclerosis models and highlight the potential for future studies on inflammatory responses in lipid deposits in zebrafish, based on known immune reactions in humans and mice, in anticipation of new zebrafish models with more advanced atherosclerotic plaques.
Cardiovascular diseases (CVDs) remain the leading cause of death worldwide (1). Research on atherosclerosis is of great clinical importance, because it increases the risk of CVDs.
Atherosclerosis is a chronic inflammatory disease that leads to myocardial infarction and cerebrovascular events. In addition to independent risk factors, such as blood pressure, hypertriglyceridemia, diet, and smoking, genetic disorders can have major pro-atherosclerotic effects. Atherosclerotic plaques develop when low density lipoproteins (LDL) accumulate in the sub-endothelial space of the arterial wall, where they are oxidized, leading to inflammatory responses (Figure 1). Then, dendritic cells, T-cells, and monocyte-derived macrophages are recruited to the affected area. Macrophages absorb oxidized LDL (oxLDL), which can accumulate and convert macrophages into foam cells, which in turn stimulate the proliferation and migration of vascular smooth muscle cells (VSMCs) into the developing plaque. As the plaque grows, its stability may change. Decreasing stability potentially leads to plaque rupture, which triggers a thrombosis cascade. The consequences of rupture include occluded arteries, ischemic events, myocardial infarction, stroke, and sudden death. Several animal models have been developed to further understanding of the pathomechanisms underlying CVD, especially atherosclerosis. The ideal disease model should develop various stages of atherosclerotic plaques and have human-like lipid metabolism and immune responses. In this review, we discuss rabbit, rat, mouse, and zebrafish, four of the most commonly used animal models for atherosclerosis. Primarily, we compared mice and zebrafish as models for human patients.
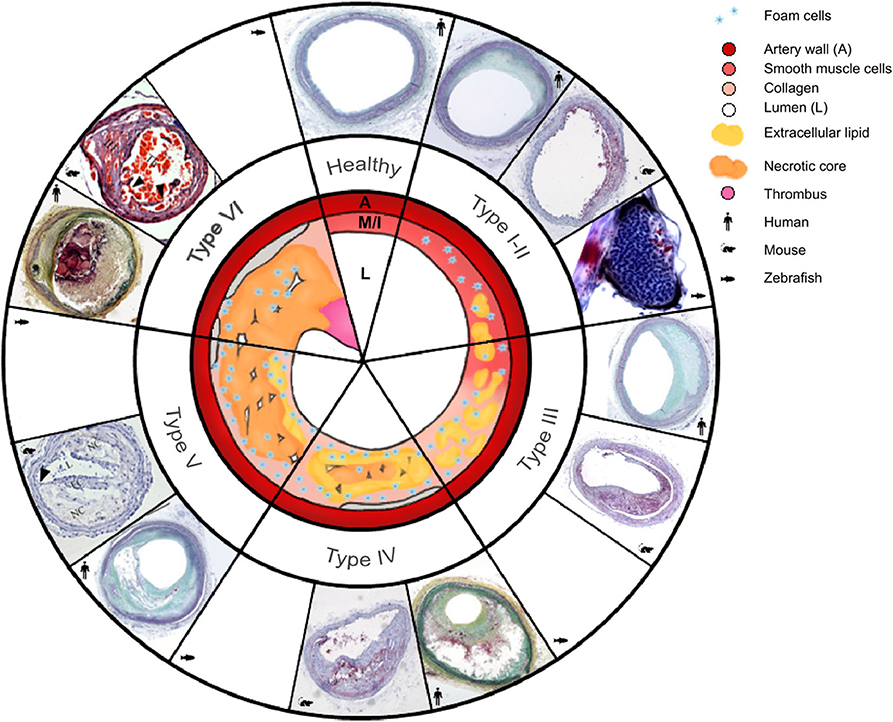
Figure 1. Morphology of the six types of atherosclerosis in human, mouse, and zebrafish: a schematic overview with a comparison of micrographs. Healthy arteries have an adventitia surrounding the media (M), a very thin intima (I) layer, and a large lumen (L). As atherosclerosis initiates and progresses, different types are classified as follows: Type I, intima thickening; type II, fatty streaks, with or without macrophages; type III, intermediate lesion; type IV, advanced atheroma; type V, fibroatheroma; type VI, complicated plaques with surface defects, leading to plaque rupture. The type VI mouse lesion image shows an unstable plaque in the right common carotid artery in the tandem stenosis mouse model, and it is used to represent future mouse plaque rupture models. Micrographs representing human atherosclerosis, are from Yahagi et al. (2); micrographs of ApoE−/− mouse type I–IV atherosclerosis in the brachiocephalic artery and zebrafish belong to the Institute for Cardiogenetics. A set of micrographs of mouse types V and VI were taken from Chen et al. (3) Permission was granted for the use of all micropgraphs.
Atherosclerosis pathogenesis comprises a complex interaction of hemodynamics, lipid metabolism, immune response and vascular injury response. Human atherosclerosis was classified into six fluent developmental stages, Type I-VI [Figure 1; (4)]. Lesions usually develop in regions of the arterial tree with local flow field disturbances leading to low shear stress, that induces endothelial cell (EC) activation and thereby the development of type I atherosclerosis (intimal thickening) (5, 6). Additionally, flow field changes contribute to accumulation of excess LDL in the sub-endothelial space of the arterial wall (LDL concentration polarization), where LDL is oxidized to oxLDL (5, 7). This reaction induces a T-helper type 1 (TH1)-driven inflammatory response, which stimulates expression of adhesion proteins, such as vascular cell adhesion molecule (VCAM)-1, E-selectin, P-selectin, and chemokines, including CCL2, CCL5, and CX3CL1 (8). One of the key players in plaque development is interferon-γ of TH1, which elevates chemokine secretion and upregulates adhesion molecule levels. Subsequently, dendritic cells, T-cells, and monocytes are recruited to the affected area in the arterial wall. Endothelial cells of normal and atherosclerotic arteries, as well as monocyte-derived macrophages, express various pattern recognition receptors (PRRs), including toll-like receptor (TLR) 1, TLR2, TLR3, TLR4, TLR5, TLR7, and TLR9 and inflammasomes [e.g., nucleotide oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing protein-3 (NLRP3)] (8, 9). The latter are damage-associated molecular pattern (DAMP)-activated intracellular innate immune signaling complexes, that activate pro-inflammatory transcription factors such as NF-κB and p53 (6, 10). Dendritic cells in healthy arterial walls silence T-cells, whereas in plaques, activation of danger signals, such as DAMPs [e.g., oxLDL, necrotic cell debris, cholesterol crystals; (11, 12)], promotes dendritic cell tolerance to T-cell antigens. Concurrently, monocytes differentiate into macrophages that absorb oxLDL through their scavenger receptors (SRs), such as CD36, MARCO, SRA-1 and -2, and SR-B1. SRs mediate the import of oxLDL via fluid phase uptake, CD36-dependent endocytosis, or micropinocytosis (13). Atherogenesis can be mediated by cholesterol crystals that directly and indirectly prime and activate macrophages via neutrophil extracellular traps (NETs) and NLRP3s (6, 14). When oxLDL accumulates as cytosolic droplets, it converts macrophages into foam cells (15). Newest in vitro findings, that are still under discussion, indicate that oxLDL activates NLRP3 inflammasomes and restricts autophagy, thereby reducing the inflammatory response. One explanation is, that low level oxLDL stimulation dampens the inflammatory response, whereas hyperlipidemia ultimately leads to chronic inflammation (12, 16). The transition from type I to type II atherosclerosis, the fatty streak, is very fluid; accordingly, we chose to merge these types together in Figure 1.
In type III atherosclerosis (intermediate lesions), extracellular lipid droplets are scattered throughout the intima. Foam cells present antigens [e.g., heat-shock protein 60 (Hsp60), interleukin-6 (IL-6) and IL-1ß] to immune cells, such as monocytes and T-cells, thereby stimulating the proliferation of VSMCs in the developing plaque (17). Eventually, extracellular lipids concentrate into a growing lipid core (type IV). Concurrently, apoptotic foam cell membranes stimulate endothelial cells to recruit additional monocytes, creating an inflammatory positive-feedback loop that leads to the formation of a necrotic core (type V) (6, 14). Additionally, migrating VSMCs contribute to the development of a fibrous cap. Lesion growth eventually restricts blood circulation and thereby increases blood pressure, which in turn can lead to hypertension and thrombus formation.
In type VI atherosclerosis, the complicated plaque, lesions grow further until the artery is sealed and blood flow is prevented, resulting in myocardial infarction. Low shear stress is not only an induction, but also a progression factor of atherogenesis, that reduces collagen fibers, increases the necrotic core and causes thinning of the fibrous cap. Taken together, this makes the fibrous cap more susceptible to tensile stress and can lead to rupture, which triggers a thrombosis cascade that occludes the artery and causes ischemic events, myocardial infarction, unstable angina, stroke, acute coronary syndrome, and sudden death (18).
Over the last 100 years, many processes involved in the pathogenesis of atherosclerosis have been revealed; however, many aspects of this disease still require clarification. In 1908, Ignatowski discovered the potential of rabbits as an atherosclerosis model by describing the thickening of the intima accompanied by the formation of large cells in the aorta of rabbits fed an animal protein-enriched diet (19–21). In 1926, Clarkson and Newburgh were the first to publish on atherosclerosis using rabbits. They evaluated the effect of different diets varying in cholesterol and protein concentration and discovered that high-cholesterol diet (HCD) as well as high protein diet led to atherosclerosis and hypercholesterolemia (22). Further research on diet-induced modifications of arteries was performed from 1926 to 1935. After World War II ended in 1945, new animal models for CVD emerged; first the rat, later the mouse, and in the last 20 years, the zebrafish (Figure 2).
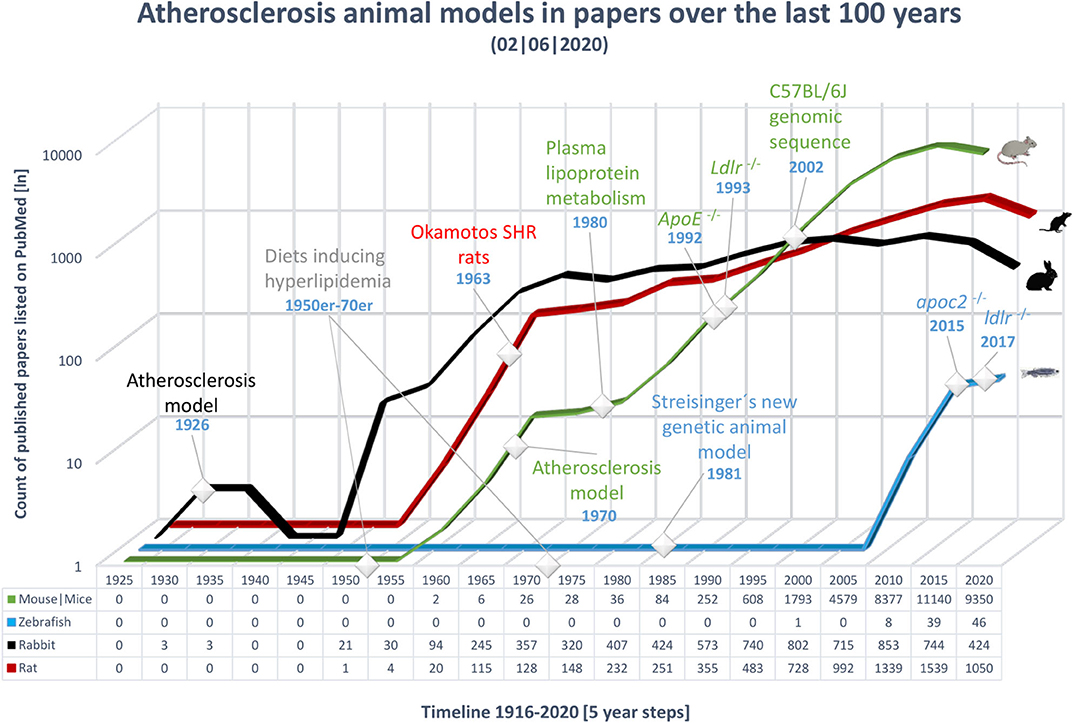
Figure 2. Overview of publications over the last 100 years on the topic of atherosclerosis in various animal models. The x-axis shows time, from 1921 to 2018, in 5 year bins; the last time point includes only 3 years. The most important events in the history of atherosclerosis research have been marked. The y-axis shows the number of publications in PubMed, on a logarithmic scale; an exact count is shown below the timeline for each animal model. Results were gathered using the MeSH term atherosclerosis in combination with the model to include a wide range of publications. Black, rabbit; red, rat; green, mouse; blue, zebrafish.
From the 1950s to 1970s, various diets capable of inducing hyperlipidemia were developed and tested in rats and rabbits (21). Two examples of prominent diets used to experimentally induce atherosclerosis include the Paigen diet (PD) (15% fat, 1.25% cholesterol, and 0.5% cholic acid) and the Western-type diet (WTD) (21% fat by weight, 0.15% cholesterol, and no cholic acid) (21). Hence, investigations of diet-inducible atherosclerosis have made critical contributions to the understanding of the pathogenesis of this condition.
In the 1970s and 1980s, intensive investigations of atherosclerosis began in mice. The characterization of plasma lipoprotein metabolism in the 1980s, in combination with the emergence of transgenic technologies in the 1990s, led to the development of the transgenic knockout mouse lines: ApoE−/− in 1992 (23) and Ldlr−/− in 1993 (24). Further, the complete DNA sequence of the diet-sensitive mouse strain, C57BL/6, was published in 2002 (25).
In 1981, Streisinger et al. were the first to show that forward genetics can be performed in zebrafish (26). The first zebrafish study to mention atherosclerosis, in the context of loss of the C-terminal end of the mammalian lipoprotein lipase protein, was published in 1996 (27). Starting in 2007, the number of studies published has increased, including research that led to the first genetic model of hyperlipidemia, the apoc2−/− zebrafish mutant (28). Recently, Liu et al. reported the first Ldlr−/− zebrafish mutant, a genetic model of hypercholesterolemia (29). Lipid metabolism and lipid trafficking genes are conserved in zebrafish, making this mutant an important tool for atherosclerosis studies (30).
We observed that the focus on animal models for CVDs has shifted over the last 100 years: from rabbit and rat, to mouse, and, possibly soon, to zebrafish. As mice and zebrafish need less space than rabbits and rats, their husbandry is cheaper. Furthermore, they are easier to genetically manipulate. Therefore, in the following sections we critically evaluate the mouse and zebrafish as the most prominent animal models for atherosclerosis.
Due to its low associated costs, easy maintenance, and breeding, extensive tools for genetic manipulation, and the ability to control its diet to rapidly induce atherosclerosis in various mutants, the mouse (Mus musculus) is a very useful model. Physiological similarities based on its phylogenetic relationship to humans, in addition to other factors mentioned below, make the mouse an important model for scientific experiments and the development of therapies (31). Therefore, we described the advantage and disadvantage of using genetically modified models in research and drug development.
Wild-type (WT) rodents are mostly resistant to atherosclerosis when fed a HCD (15); however, the development of inbred strains, such as C57BL/6J, C57L/J, and DBA/2J (32), has enabled atherosclerosis research in mice. Forward and reverse genetics play crucial roles in the development of atherosclerosis models. Knockouts, knockins, knockdowns, and transgenic modifications can be generated by various methods, including the CRISPR/Cas9 system, the Cre-loxP-system, Vivo-Morpholinos (MOs), lentiviral infection, and ethylnitrosourea (ENU) mutagenesis (33, 34). Genetic manipulation yielded the two most commonly used mouse models of atherosclerosis, namely Ldlr−/− and ApoE−/− (Table 1).
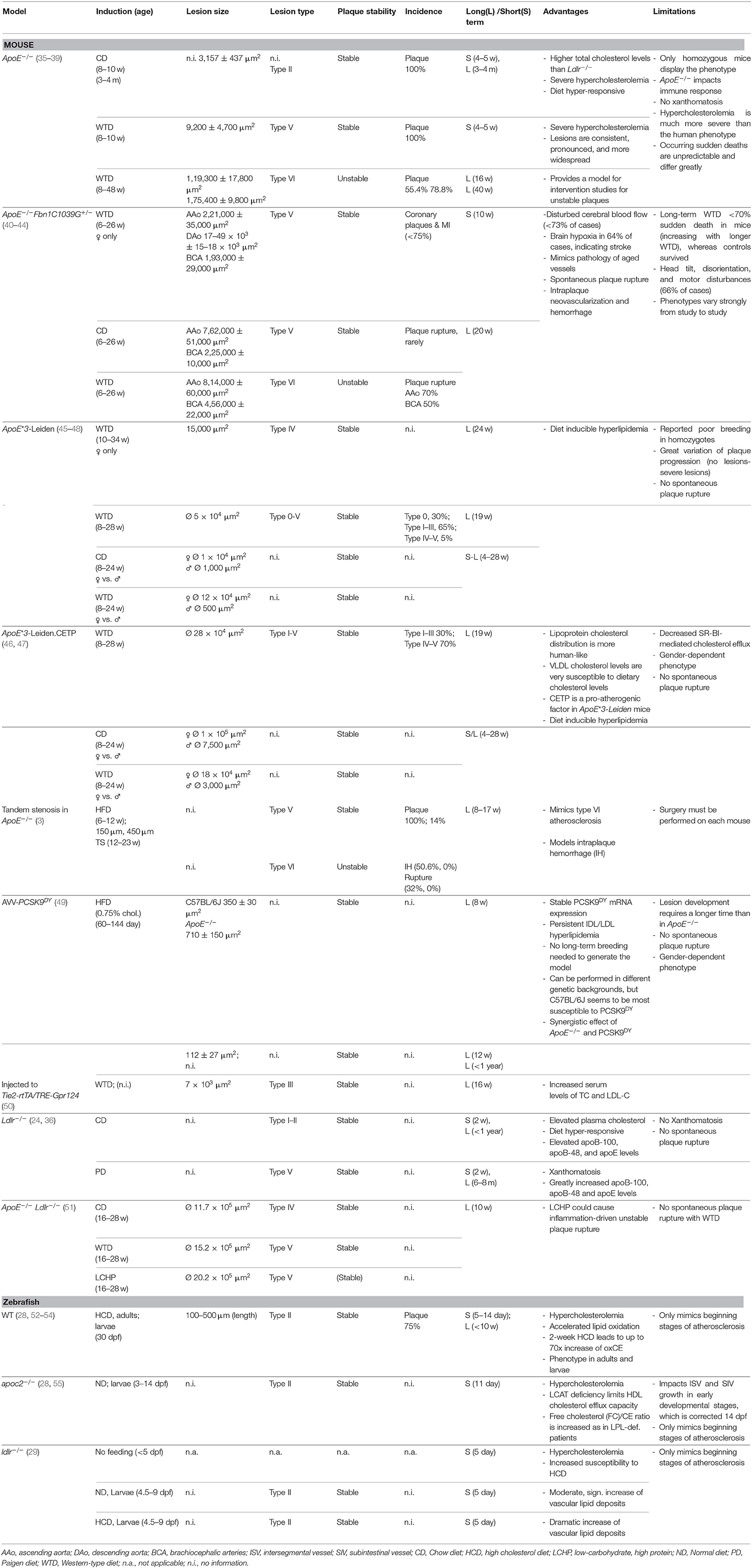
Table 1. Phenotypes and traits of mouse and zebrafish atherosclerosis models.
The major apolipoprotein in chylomicrons, Apolipoprotein E (ApoE), can serve as a ligand for the LDL receptor (LDLR). Synthesis of ApoE occurs in the brain, liver, and other tissues in both humans and mice, although humans have high-density lipoprotein (HDL) subsets and ApoE isoforms that mice lack (56, 57). In addition to its effects on lipoprotein metabolism, ApoE has roles in macrophage biology, immune function, and adipose tissue biology (56, 58). Mutations in mouse ApoE result in human-like phenotypes, such as hyperlipoproteinemia type III, xanthomatosis, or dysbetalipoproteinemia (35). At the age of 3 months on a normal diet, ApoE-deficient mice show signs of type II fatty streaks (Figure 1). Type III diffusely intimal foam cell deposits in the aortic sinus can be observed at 5 months. Finally, at 8 months, the atherosclerotic plaques in ApoE−/− mice seal the artery almost completely. The relatively early development of lesions and the anthropomorphous lipid metabolism are major advantages of the mouse as an animal model (35). Yet, lesion rupture is usually only observed under severe stress (30).
Along with LDL, ApoE is a structural component of all lipoproteins and a critical ligand for hepatic clearance of plasma lipoproteins, and it is mediated by LDLR (59). Hepatic clearance may explain why mice are naturally resistant to atherosclerosis (36). Deficiency of LDLR, more common in humans than non-functional ApoE, leads to reduced uptake and clearance of lipoproteins, resulting in the predominance of LDL as a cholesterol carrier and to familial hypercholesterolemia, which in turn increases the risk of CVDs (59). The human-like lipid metabolism of Ldlr−/− mice enables the study of macrophages loaded with cholesterol creating xanthomas in the skin and subcutaneous tissue, as well as atherosclerotic lesions in arteries (36). In 2017 Emini Veseli et al. confirmed the plaque rupture in 20-week-old ApoE−/−Fbn1C1039G+/− and Il1r1−/−ApoE−/− mice, that mimicked human type VI atherosclerosis (17). Fragmentation of elastic fibers in the vessel wall, increases arterial stiffness and thereby leads to plaque rupture in 50% of the brachiocephalic arteries (40, 41). Inactivation of IL-1 signaling in atherosclerotic plaques decreased plaque collagen levels in Il1r1−/−ApoE−/− mice, subsequently destabilizing the lesion (60); however, 50% of the mice die suddenly after receiving a HCD for 16–23 weeks (40). Lack of natural type VI atherosclerosis (i.e., lesion rupture; Figure 1), remained a detractor of the mouse model for many years. This changed not only with development of the ApoE−/−Fbn1C1039G+/− and Il1r1−/−ApoE−/− but also the tandem stenosis (TS) mouse model (3), which can mimic various stages of human atherosclerosis, including type VI lesion rupture (Figure 1). Chen et al. demonstrated that treatment with atorvastatin increased collagen content, and they concluded that plaque stability increased, although they observed no difference in total plaque area. Earlier studies showed that statins, combined with ezetimibe, could reduce lesion size by 17%, and had anti-inflammatory effects in mice, although there was no effect on serum oxLDL (61).
As an alternative approach Roche-Molina et al. (49) and Bjørklund et al. (62), established the AAV-PCSK9DY model with adeno-associated virus (AAV) vectors serotype 8 resp. 9 carrying the human gain-of-function mutation Asp374Tyr in protein convertase subtilisin/kexin type 9 (PCSK9DY), that developed atherosclerosis and hyperlipidemia in the timespan of 84 day [Table 1; (49)]. Injection of PCSK9DY into 30 day-old mice resulted in stable PCSK9DY mRNA expression and persistent intermediate density lipoprotein (IDL)/LDL hyperlipidemia up to 1 year (49). Bjørklund et al. reported a lesion progression up to type V atherosclerosis (62). Since development of this approach it has been widely used, e.g., for highly efficient PCSK9-targeted genome editing via zinc-finger nuclease (ZFN) mRNAs, that were delivered to the liver by lipid nanoparticles (63); and to demonstrate the gender-dependent role of the EC-mineralocorticoid receptor (MR) for atherosclerosis and vascular inflammation, revealing the potential of EC-MR inhibitor therapy in male patients (64). Transexpression of human disease-causing genes in mice is a refined concept, that reduces time and number of animals required for the generation of mutants (65) and could provide a new effective and sustainable approach to study long-term atherosclerosis and hyperlipidemia (49) as well as to perform high-content experiments, in which it would reduce time and costs (62).
A particular disadvantage of genetically modified mouse models is that transport of cholesterol is mediated by HDL, rather than LDL (56), as lipid metabolism plays a crucial role in the development of lesions in the vascular wall. Recent publications report the use of very low density lipoprotein (VLDL) as the major plasma cholesterol carrier in ApoE−/− mutants, as well as having atheroprotective properties (59). Another point to consider is passenger gene effects, which can arise due to various types of incestuous pairings (66). Passenger gene effects do not influence the fitness of an individual but can lead to subtle changes in the genetic background. This situation arises when a donor strain is bred to a recipient strain that carries the mutation of interest. The F1 generation is then bred again with the recipient strain, and repetition of this process for ten generations leads to a statistically 99.8% pure strain; however, 0.2% contamination from the donor strain remains. Considering crossover during meiosis, a small contamination can lead to additional variation in features such as cellular composition, calcification, and lesion size in the newly-developed strain (67, 68).
Very few animal model studies lead to new therapies and drugs, as mice and humans differ not only in size, maturation, and metabolism per gram of tissue (and in nutrient requirements), but also in telomere length and microbiome (31). For example, mice with human inflammation-associated diseases, such as lupus, psoriasis, and rheumatoid arthritis, develop more atherosclerotic lesions than patients with these conditions. Thus, promotion of vascular inflammation by atherosclerosis is more likely to result from general inflammation than from shared risk genes (69, 70). Among 25,000 compounds investigated in labs, only 25 will be tested in humans, of which five will come to market (71), corresponding to a success rate of 0.02% for individual compounds. One rare example of success is the development of PCSK9 monoclonal antibodies (mAbs) (alirocumab and evolocumab) in mice, which contributes to proven therapy for high LDL cholesterol (LDLc) levels and atherosclerosis in humans (56). While these mAbs are approved vaccines, their short in vivo half-time makes them expensive and thereby frequent administration is required (72). In recent years, two further PCSK9-based drugs were investigated in a clinical trial (ClinicalTrials.gov NCT02508896), the phase I of which was already completed in August 2017, although no results have yet been published. In ApoE*3-Leiden.CETP mice immunization using this peptide-based vaccine elicits antibodies against PCSK9, that reduced lesion size, especially decreased necrotic core-formation and macrophage inflammation and lowered plasma lipid levels through humoral response (72, 73). This approach promises to be more cost effective and might offer a long-term LDLc management (72).
The advantages of mice, such as easy care and breeding, and the ability to control the diet to induce atherosclerotic lesions, are shared by zebrafish (Danio rerio), making them another promising candidate animal model to study atherosclerosis.
The translucency of zebrafish larvae until about 30 days post-fertilization (dpf) permits observation of vascular development in vivo in real time (74, 75), which is augmented by the existence of transgenic lines such as fli1:eGFP and lyz:DsRed2 (52, 76). The zebrafish is especially fecund, with females laying 200–300 eggs/day (77); however, excessive inbreeding leads to infertility (78), impeding the development of purer inbred lines. Although the vascular system of the zebrafish is different from that of mammals, its development and anatomy are similar (79).
During embryonic and first larval stages, zebrafish must rely on their innate immune system, since the adaptive immune system is not yet mature. Both adaptive and innate immune systems are highly conserved between zebrafish and mammals. The dominant leucocyte in zebrafish is the neutrophil, which responds to H2O2 gradients in injured tissues, while eosinophils provide an important host defense against parasites. Furthermore, macrophages are the key phagocytic cells that regulate cytokine-mediated immunity (80). In addition, zebrafish are likely to have dendritic and other antigen-presenting cells (81).
Zebrafish are ideal models for large-scale screens; the early larval stages are only 2–3 mm long, they develop rapidly, and their results are representative for mammals (82). Various genetic tools, such as MOs, targeting-induced local lesions in genes (TILLING), RNA caging, transgenic reporter lines expressing fluorescent marker proteins (i.e., the UAS/GAL4-system), ENU mutagenesis, the CRISPR/Cas9 system, and transcription activator-like effector nuclease (TALEN), facilitate evaluation of gene functions in zebrafish (83, 84). Like mammals, zebrafish process lipid throughout the intestine and hepatobiliary system (79, 83). Thus, they are often used to study vascular lipid accumulation and lipoprotein oxidation. In contrast to mammals, zebrafish cannot synthesize their own vitamin C and must obtain it from their diet; consequently, vitamin C levels can be manipulated to trigger oxidative stress [Table 2; (79)].
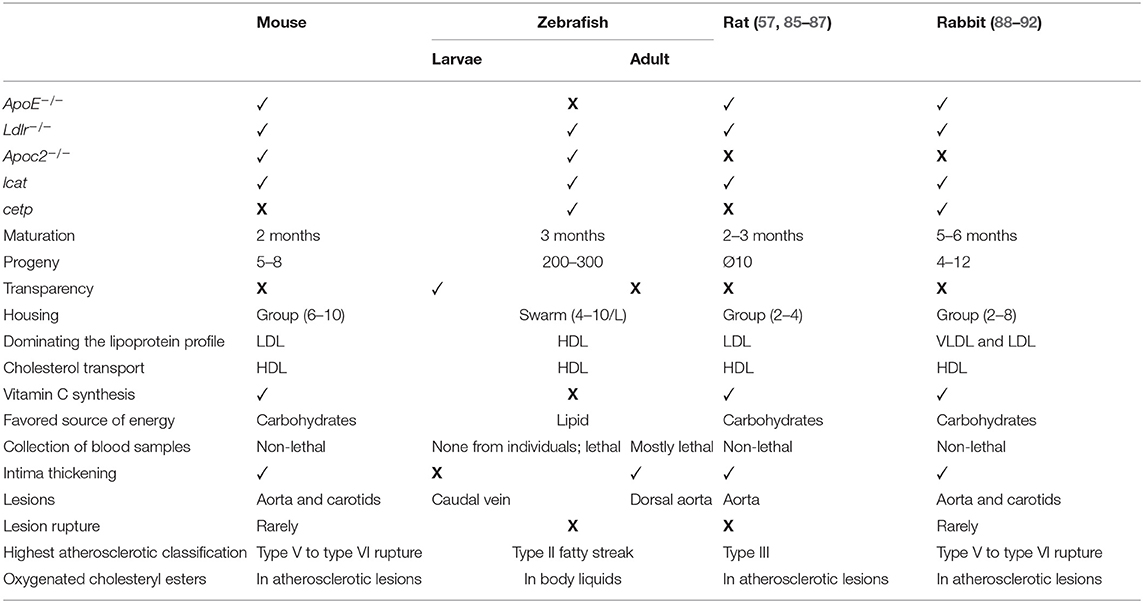
Table 2. Comparison of animal models for atherosclerosis.
In recent years zebrafish larvae, younger than 5 dpf, which are not considered animals until they start independent feeding according to the Directive 2010/63/EU of the European Parliament, have become a valuable animal model of drug discovery (93). Effects of administered compounds on vital developmental processes, such as vasculogenesis and organogenesis can be assessed in the whole organism during embryogenesis in a cost-effective and time-saving manner (93, 94). Today two approaches in terms of drug discovery are being pursued: Molecular target-based screens and high-throughput phenotypical screens. While performing target-based screens is fast and efficient, the risk that the compounds are eliminated downstream in the pipeline of preclinical and clinical trials is significantly higher than with phenotypic screens, as efficacy is lacking (95–97). Automated imaging systems and script-based evaluation tools improve the efficiency of phenotypic screens and thereby create a platform with enhanced data quality output, disease relevance, and reduced risk of elimination (95, 98).
Zebrafish animal models also have limitations. Collecting blood samples from juveniles from 40 to 45 dpf is possible, but lethal (52), and the larvae are so small that homogenates of several individuals are required for analysis. Another disadvantage of zebrafish is that poikilothermic vertebrates favor lipids as a source of energy, whereas carbohydrates are favored by homoeothermic mammals (52). Furthermore, mammals and fish differ not only in terms of favored energy source but also in their lipoprotein profile and LDL makeup. In zebrafish, their lipoprotein profile is dominated by HDL and their LDL contains more triglycerides and fewer cholesteryl esters (CEs) relative to human LDL (52). This is because zebrafish HDL functions as the cholesterol transporter, while in mammals it transports excess cholesterol from peripheral cells back to the liver, where it is recycled and excreted (99). Feeding a HCD to WT zebrafish causes their lipoprotein profiles to more closely resemble those of humans (74) and results in hypercholesterolemia, vascular lipid accumulation, and myeloid cell recruitment, among other symptoms (100). In both zebrafish and human, cholesteryl ester transfer proteins (CETPs) are involved in the transfer of CE from HDL to other lipoproteins (30, 101), which may explain why a HCD results in hypercholesterolemia in WT zebrafish.
All major classes of apolipoproteins (ApoA, ApoB, ApoC, and ApoE) are expressed in zebrafish and are highly similar to human apolipoproteins (79). As mentioned above, hypertriglyceridemia is an independent CVD risk factor caused by single mutations in the genes encoding lipoprotein lipase (LPL) or in those encoding LPL cofactors, such as APOC2 (28, 55). Liu et al. demonstrated that apoc2−/− zebrafish exhibit properties of human patients, including dyslipidemia, specifically hypertriglyceridemia as early as 14 dpf, which resulted in type II atherosclerosis. To examine whether vascular lipid deposits accumulated in macrophages, they crossed apoc2−/− with Tg (mpeg1-eGFP), that expresses eGFP in macrophages and emerged them in BODIPY to fluorescently stain neutral circulating lipoproteins and intracellular lipid droplets in vivo. They were able to show a co-localization of vascular lipid deposits with macrophages in apoc2−/− mpeg1-eGFP 14 dpf as well as IK17 co-localization in selected areas of the larval fatty streaks, which is observed in early atherosclerotic lesions in human and mice (28, 100). In concordance other studies demonstrated a significant increase of LDL concentrations at vascular bifurcations (LDL concentration polarization) in vivo in larvae 52 hpf after fluorescent DiI-LDL-injection 48 hpf (5) and after feeding larvae fluorescence-labeled HCD short term (10 day) and long term (<10 weeks) (53).
Recently, Liu et al. reported the first ldlr−/− zebrafish mutant, created using the CRISPR/Cas9 system, thus introducing a new model for atherosclerosis and hypercholesterolemia, that resembles the corresponding model in mice (24, 36). This mutant exhibits elevated susceptibility to HCDs and vascular lipid accumulation at 9 dpf, after 5 days of feeding a HCD (29).
Overall, studies conducted on zebrafish larvae may only be able to complement mammalian studies (102). Nevertheless, eight compounds discovered in zebrafish drug screens are being tested in clinical trials (93) and a promising approach showed that persistent expression of the human monoclonal antibody IK17 prevents vascular lipid accumulation and reduces existing lipid deposits in zebrafish (100). It is suspected that IK17 accumulates in vascular lesions and neutralizes oxidation-specific epitopes (74). Further, reduction in oxLDL, mediated by IK17, is predicted to decrease foam cell formation.
Very few studies have characterized the whole immune response involved in atherosclerosis in zebrafish, hence our comparisons of them with mice and humans are also based on non-CVD focused studies (Table 3).
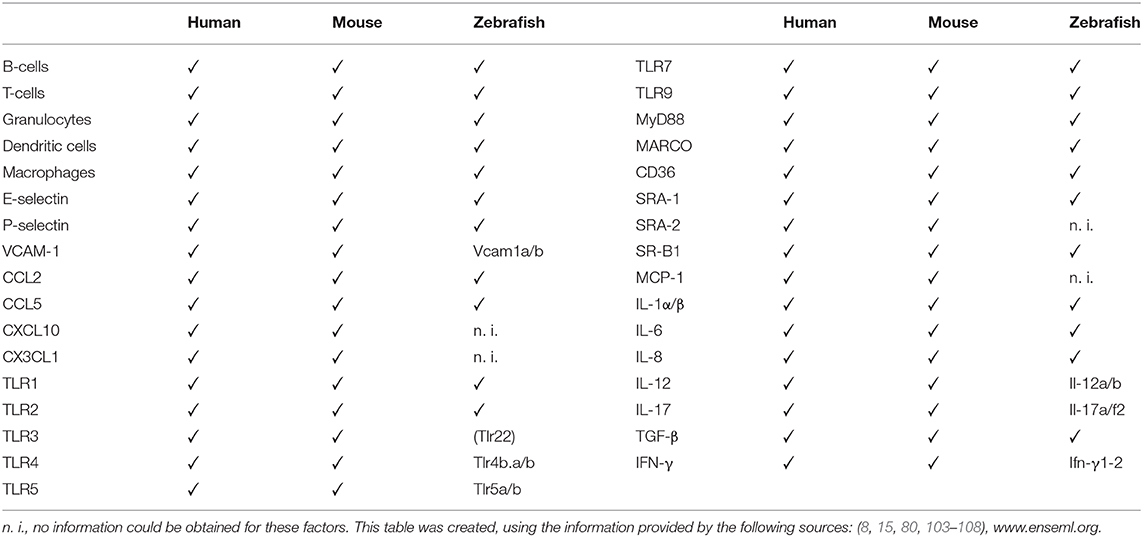
Table 3. Immune response factors in human atherosclerotic lesions.
In the early stages of atherosclerosis, TH1-induced adhesion molecules, such as P-selectin and E-selectin, are highly conserved from zebrafish to mammals and have important roles in the inflammatory response (109). Mammalian chemokines, such as CXCL11, have homologs in zebrafish (CXC-64), although initial functional analyses revealed differences in their release from monocytes following stimulation by bacteria (103). The TLR family is conserved from insects to mammals; however, their signaling pathways can differ. Li et al. analyzed the PRRs and their corresponding homologs in mammals and zebrafish, and found that some TLRs have two counterparts in zebrafish, due to gene duplication events during evolution (104). They identified the TLR22 of zebrafish as a homolog of mammalian TLR3. Although mice lack TLR10, they have three additional TLRs: TLR11, TLR12, and TLR13 (110). TLR4 is required for efficient lipid uptake by macrophages in humans, mice, and zebrafish (52). Myeloid differentiation primary response 88 (MyD88) is a key pro-atherosclerotic adaptor protein in signaling cascades mediated by IL-1R and various TLRs (105).
The phylogenetically-conserved autoantigens, Hsp60 and LDL, stand out as important promoters of atherosclerosis. Hsp60 functions as a chaperone at the cell surface, folding freshly-synthesized proteins, and protecting them under stress conditions such as heat shock. In some cases, Hsp60 can trigger adaptive and innate immune responses in vivo (111). Studies of Hsps in mice lead to preclinical trials of DiaPep277, a vaccine, that had the potential to change Hsp-mediated immune regulation and was intended to treat type-I diabetes (112), but was terminated due to scientific misconduct (113).
In mice, immunization against oxLDL attenuates atherosclerosis. Phospholipids are ubiquitous molecules that form integral parts of cell membranes and lipoproteins such as LDL. SRs of macrophages, such as CD36, TLR2, and TLR4, take up oxidized phospholipids (oxPLs) and oxLDL, thereby promoting inflammation because they are recognized by the innate immune system. Knockout of SRs that recognize oxPLs leads to abatement of atherosclerosis in both models. oxPLs are found in all types of atherosclerotic lesions, indicating their contribution to atherogenesis (114). Accessory proteins found in humans are also present in zebrafish, indicating similarities in the activation and folding of zebrafish TLRs. The SR accessory protein, CD36, fine-tunes TLR assembly, especially in response to TLR2/6 ligands (106), and drives atherosclerosis through the aforementioned interaction with oxLDL (107, 115). It was shown, that while highly conserved, alternative splicing may not occur and differences in C-terminal amino acids of Cd36 may alter the function of the protein to being a co-receptor in contrast to mammalian CD36 (107).
In this review, we compared the most important aspects of mouse and zebrafish models for human atherosclerosis. In the last decade, models mimicking human atherogenesis, different diets, and tools for forward and reverse genetics were also developed, accelerating, and increasing the efficiency of research progress.
Currently, mice are the most commonly used atherosclerosis model because mutant strains are susceptible to dietary interventions, and plaques progress to type V lesions in the aorta and carotids (35). Nevertheless, the development of late-stage lesions takes months, even when mice are fed a HCD and rupture is usually only observed under severe stress (30). Since mouse immune responses and pathways are similar to those of humans, they are a good model for studying atherosclerosis-related inflammatory responses, whereas the HDL-based lipid metabolism and the lack of CETP in mice are clear disadvantages, that are only abolished in ApoE*3-Leiden.CETP (Table 1). Increased use of the relatively new PCSK9DY mouse model negates the need to generate inbred strains. One recent example is the use of PCSK9DY-injections into mice expressing GPR124 under control of the Tie-2 promoter. Gong et al. found that GPR124, a receptor known to increase angiogenesis in the brain, influences the pathogenesis of atherosclerosis by activating nitrosative stress and NLRP3 inflammasome signaling (50). Nevertheless, while PCSK9DY itself is suitable to create atherosclerosis models user errors and gender-dependent susceptibility to PCSK9DY could lead to great variations of study outcomes between laboratories (64, 65).
Zebrafish have recently emerged as models for atherosclerosis and are also susceptible to diet, and they have the added advantage of their amenability to high-throughput screenings. The adaptive and innate immune systems are highly conserved between zebrafish and mammals. Use of transgenic zebrafish lines that can track macrophages and neutrophils in vivo has recently provided new insights into the early development of atherosclerosis. Chronological in vivo imaging over a 10-day period revealed, that neutrophils rather than macrophages may accumulate in the sub-endothelial space before LDL concentration polarization (53). These results illustrate the importance of appropriate in vivo models for studying the pathophysiology of complex diseases such as atherosclerosis.
Similarities with humans in lipid metabolism and expression of CETP, along with the development of the ldlr−/− and apoc2−/− mutant, demonstrate other advantages of this new model. Furthermore, the data from the apoc2−/− mutant reported by Liu et al. suggests, that zebrafish suitably and even more favorably than mice, model human dyslipidemia, a major risk factor for atherosclerosis (28, 116, 117). While mutant mice can exhibit atheroprotective properties, which can bias experimental results, passenger gene effects may not affect atherogenic zebrafish models because WT animals are already affected by HCD, negating the for inbred strains.
With advancing technology, zebrafish could also be treated with a TS approach to test whether the phenotype of atherosclerotic plaques will also be enhanced in this model. Haemodynamic forces in the arterial wall of zebrafish can be measured using transgenic lines, BODIPY, imaging systems and analysis tools such as ZebraLab (5, 28). The recent creation of the ldlr−/− zebrafish mutant will enhance the importance of zebrafish as an animal model for atherosclerosis. ldlr−/− mutants on a regular diet develop moderate hypercholesterolemia, whereas even short-term consumption of a HCD results in hypercholesterolemia and aggravated accumulation of lipids in the vascular wall. Therefore, ldlr−/− as well as apoc2−/− zebrafish could enable high-content screens for novel therapeutics and drug repositioning for atherosclerosis. Research on zebrafish atherosclerosis is heavily leaning on the lipid metabolism as the lesions are very small, due to the models size and disease progression limitations. On one hand, this could be a drawback for the model depending on the hypothesis, but on the other hand its easy genetic manipulation and opacity offers an opportunity to study early atherosclerosis pathomechanisms using e.g., transgenic lines, BODIPY, or injections with fluorescent dyes, into the bloodstream of larvae 48 dpf.
Considering the very low costs associated with a model that can be used at just 9 dpf, even laboratories with low budgets could drive research on atherosclerosis forward using zebrafish. The development of ApoE−/− zebrafish would create a new tool for atherosclerosis research that could be compared with mouse models. To date, there is no mouse model with consistent plaque instability, while the ApoE−/− mouse mutant appears to be key to the development of a transgenic model that completely mirrors human atherosclerosis. Feeding ApoE−/− mice a HCD is sufficient for them to exhibit complex and multi-layered lesions, but loss of ApoE makes this model much more complex, as it's an integral part of the immune response and lipid metabolism (118). Nevertheless, the mortality rate of this mouse line varies greatly for the same diet and length of the experiment (Table 1). While Van der Donckt et al. reported no dead animals, which is consistent with our own experience, Stöhr et al. published a 20% mortality and Johnson et al. a mortality rate of 70% after 10 months on WTD (37, 38, 40). Other reports of WTD for shorter periods show the same variability in mortality rates (42, 43). It is unclear whether this variation is a consequence of the ApoE KO or husbandry differences.
Studies of the atheroprotective characteristics of oxLDL antibodies, which were demonstrated in mice and rabbits, could also be performed in zebrafish, followed by immunization studies (53, 119). Furthermore, functional analysis of chemokines in growing plaques and regulation of their promoters in zebrafish could further drive atherosclerosis research.
The use of new imaging systems and tools, such as magnetic resonance imaging (MRI), Ultrasound biomicroscopy (UBM) and improved contrast enhancers for computerized tomography (CT) will provide more reliable information about plaque size and position in vivo. Currently gold nanoparticles (AuNP) are most commonly used as contrast agent for CTs, as they can attenuate X-rays, are rarely toxic for humans and can target specific tissues when combined with fluorescent dyes and surface molecules (120). Several toxicity studies in zebrafish embryos revealed size- and chemical-dependent biocompatibility and toxicity (121), resulting in small, under-pigmented eyes, neurological defects, and abnormal behavior activity (122–124). Studies in mice showed, that the use of AuNP, such as AuroVist™ can visualize macrophage infiltration in vascular bifurcations in mice in vivo. However, scans have to be performed pre- and post-contrast injection, which increases radiation time for each animal and thus may increase the mortality rate if an animal has to be examined several times within a study. Additionally animals must be fixed in place to reduce repositioning effects, that impact readout of the scans (125, 126). Taken together, a contrast agent for CT is needed that reduces the burden on animals to enable a time-lapse recording of plaque progression in vivo with reduced influence on the survival rate and pathogenesis for a reasonable price. Another imaging technique that has been used for decades in humans and mice is MRI. It enables differentiation of plaque components based on their biophysical and biochemical composition without radiation in vivo (127). In recent years new techniques have been developed, that not only increased the resolution, but enable MRI of adult zebrafish as well (128). Repeated MRIs with and without use of non-toxic, gadolinium-based contrast agents, were performed without side-effects in mice and zebrafish (128–131). Altogether, MRI is a well-established imaging technique, that provides in depth information for several animal models as well as for humans, but interpretation of the data still needs a lot of experience (127) and the images are static and cannot display hemodynamics. Another imaging technique, that allows to calculate blood flow based on Doppler blood velocity and vessel diameter as well as detection of atherosclerotic plaques is the multifrequency UBM combined with Doppler ultrasound (132). Readout quality from UBMs is frequently increasing, enabling reliable diagnosis of small animals with higher heart rates and smaller vessel sizes (133–136). Nevertheless, interpretation of the acquired data requires even more experience. To date, the best imaging techniques with the highest resolution, such as microCT and light-sheet microscopy, are still lethal or only applicable to one model (137–139). While in vivo imaging of mice can be achieved by adapting human imaging technologies and vice versa, imaging of zebrafish larvae can be achieved with simple microscopes. Automated microscopes enable high-throughput imaging of the small fish with high quality data output. Therefore, zebrafish are more commonly used for drug discovery. However, they also have limitations, that do not apply to murine models. Drug screening in zebrafish only succeeds if the compounds to be tested are predominantly water-soluble or are injected into the fish. Additionally, in-water dosing could yield unpredictable exposures (93). Furthermore, protocols to conduct high-content screens in zebrafish are very diverse, therefore results in different laboratories may vary. Taken together, zebrafish are a good model for primary drug screens, provided that all limitations have been considered. Moreover, primary screens in zebrafish reduce the number of mice needed for secondary drug screening.
Overall, mice will continue to be pioneers to study atherosclerosis pathogenesis until the majority of the mechanisms underlying atherosclerosis in zebrafish have been elucidated and mutant animals have been established. Zebrafish have clear advantages in studying the influence of lipid metabolism on atherosclerosis. Therefore, the model should be chosen according to the hypothesis and a combination of mouse and zebrafish experiments should be considered for in depth studies. Additionally, it is important to find an in vivo imaging method that meets the challenges of different models and at the same time creates a platform that delivers consistent, reliable, and above all comparable results, regardless of the model chosen.
ZA and VV contributed conception and design of the review. VV collected data, organized the database, and wrote the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This review was supported by the DZHK (German Centre for Cardiovascular Research). This work was supported by the Institute for Cardiogenetics and the University of Osnabrück.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Anna Schmidt and Karim Tarhbalouti for providing their micrographs and Loreto Muñoz Venegaz (https://orcid.org/0000-0003-2308-4158) for proofreading.
1. World Health Organization. Cardiovascular Diseases (CVDs). World Health Organization (2017). Available online at: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed November 27, 2017).
2. Yahagi K, Kolodgie FD, Otsuka F, Finn AV, Davis HR, Joner M, et al. Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat Rev Cardiol. (2016) 13:79–98. doi: 10.1038/nrcardio.2015.164
3. Chen Y-C, Bui AV, Diesch J, Manasseh R, Hausding C, Rivera J, et al. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ Res. (2013) 113:252–65. doi: 10.1161/CIRCRESAHA.113.301562
4. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol. (1995) 15:1512–31. doi: 10.1161/01.ATV.15.9.1512
5. Xie X, Tan J, Wei D, Lei D, Yin T, Huang J, et al. In vitro and in vivo investigations on the effects of low-density lipoprotein concentration polarization and haemodynamics on atherosclerotic localization in rabbit and zebrafish. J R Soc Interface. (2013) 10:20121053. doi: 10.1098/rsif.2012.1053
6. Wolf MP, Hunziker P. Atherosclerosis: insights into vascular pathobiology and outlook to novel treatments. J Cardiovasc Transl Res. (2020). doi: 10.1007/s12265-020-09961-y. [Epub ahead of print].
7. Marchio P, Guerra-Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. Targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxid Med Cell Longev. (2019) 2019:1–32. doi: 10.1155/2019/8563845
8. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. (2011) 12:204–12. doi: 10.1038/ni.2001
9. O. Krogmann A, Lüsebrink E, Lahrmann C, Flender A, Nickenig G, Zimmer S. Toll-like receptor 7 stimulation promotes the development of atherosclerosis in apolipoprotein E-deficient mice. Int Heart J. (2020) 61:364–72. doi: 10.1536/ihj.19-365
10. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
11. Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-α. Circulation. (2006) 114:2482–9. doi: 10.1161/CIRCULATIONAHA.106.642801
12. Zhou Z, Zhu X, Yin R, Liu T, Yang S, Zhou L, et al. K63 ubiquitin chains target NLRP3 inflammasome for autophagic degradation in ox-LDL-stimulated THP-1 macrophages. Aging. (2020) 12:1747–59. doi: 10.18632/aging.102710
13. Miller YI, Choi S-H, Fang L, Harkewicz R. Toll-like receptor-4 and lipoprotein accumulation in macrophages. Trends Cardiovasc Med. (2009) 19:227–32. doi: 10.1016/j.tcm.2010.02.001
14. Thålin C, Hisada Y, Lundström S, Mackman N, Wallén H. Neutrophil extracellular traps: villains and targets in arterial, venous, and cancer-associated thrombosis. Arterioscler Thromb Vasc Biol. (2019) 39:1724–38. doi: 10.1161/ATVBAHA.119.312463
15. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. (2005) 352:1685–95. doi: 10.1056/NEJMra043430
16. Miller YI, Shyy JYJ. Context-dependent role of oxidized lipids and lipoproteins in inflammation. Trends Endocrinol Metab. (2017) 28:143–52. doi: 10.1016/j.tem.2016.11.002
17. Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, Van der Donckt C, Martinet W, et al. Animal models of atherosclerosis. Eur J Pharmacol. (2017) 816:3–13. doi: 10.1016/j.ejphar.2017.05.010
18. Moore KJ, Tabas I. The cellular biology of macrophages in atherosclerosis. Cell. (2011) 145:341–55. doi: 10.1016/j.cell.2011.04.005
19. Ignatowski AC. Influence of animal food on the organism of rabbits. Izv Imp Voyenno-Med Akad Peter. (1908) 16:154–73.
20. Leong X-F, Ng C-Y, Jaarin K. Animal models in cardiovascular research: hypertension and atherosclerosis. BioMed Res Int. (2015) 2015:528757. doi: 10.1155/2015/528757
21. Jawien J, Nastałek P, Korbut R. Mouse models of experimental atherosclerosis. J Physiol Pharmacol. (2004) 55:503–17.
22. Clarkson S, Newburgh LH. The relation between atherosclerosis and ingested cholesterol in the rabbit. J Exp Med. (1926) 43:595–612. doi: 10.1084/jem.43.5.595
23. Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. (1992) 89:4471–5. doi: 10.1073/pnas.89.10.4471
24. Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. (1993) 92:883–93. doi: 10.1172/JCI116663
25. Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E. Initial sequencing and comparative analysis of the mouse genome. Nature. (2002) 420:520–62. doi: 10.1038/nature01262
26. Streisinger G, Walker C, Dower N, Knauber D, Singer F. Production of clones of homozygous diploid zebrafish (Brachydanio rerio). Nature. (1981) 291:293–6. doi: 10.1038/291293a0
27. Arnault F, Etienne J, Noé L, Raisonnier A, Brault D, Harney JW, et al. Human lipoprotein lipase last exon is not translated, in contrast to lower vertebrates. J Mol Evol. (1996) 43:109–15. doi: 10.1007/BF02337355
28. Liu C, Gates KP, Fang L, Amar MJ, Schneider DA, Geng H, et al. Apoc2 loss-of-function zebrafish mutant as a genetic model of hyperlipidemia. Dis Model Mech. (2015) 8:989–98. doi: 10.1242/dmm.019836
29. Liu C, Kim YS, Kim J, Pattison J, Kamaid A, Miller YI. Modeling hypercholesterolemia and vascular lipid accumulation in LDL receptor mutant zebrafish. J Lipid Res. (2017) 59:391–9. doi: 10.1194/jlr.D081521
30. Schlegel A. Zebrafish models for dyslipidemia and atherosclerosis research. Front Endocrinol. (2016) 7:159. doi: 10.3389/fendo.2016.00159
31. Perlman RL. Mouse models of human disease: an evolutionary perspective. Evol Med Public Health. (2016) 2016:170–6. doi: 10.1093/emph/eow014
32. Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. (1985) 57:65–73. doi: 10.1016/0021-9150(85)90138-8
33. Svenson KL, Ahituv N, Durgin RS, Savage H, Magnani PA, Foreman O, et al. A new mouse mutant for the LDL receptor identified using ENU mutagenesis. J Lipid Res. (2008) 49:2452–62. doi: 10.1194/jlr.M800303-JLR200
34. Miano JM, Zhu QM, Lowenstein CJ. A crispr path to engineering new genetic mouse models for cardiovascular research. Arterioscler Thromb Vasc Biol. (2016) 36:1058–75. doi: 10.1161/ATVBAHA.116.304790
35. Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. (1992) 258:468–71. doi: 10.1126/science.1411543
36. Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest. (1994) 93:1885–93. doi: 10.1172/JCI117179
37. Stöhr R, Kappel BA, Carnevale D, Cavalera M, Mavilio M, Arisi I, et al. TIMP3 interplays with apelin to regulate cardiovascular metabolism in hypercholesterolemic mice. Mol Metab. (2015) 4:741–52. doi: 10.1016/j.molmet.2015.07.007
38. Johnson J, Carson K, Williams H, Karanam S, Newby A, Angelini G, et al. Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: model characterization and effects of pravastatin treatment. Circulation. (2005) 111:1422–30. doi: 10.1161/01.CIR.0000158435.98035.8D
39. Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. (1992) 71:343–53. doi: 10.1016/0092-8674(92)90362-G
40. Van der Donckt C, Van Herck JL, Schrijvers DM, Vanhoutte G, Verhoye M, Blockx I, et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur Heart J. (2015) 36:1049–58. doi: 10.1093/eurheartj/ehu041
41. Van Herck JL, De Meyer GRY, Martinet W, Van Hove CE, Foubert K, Theunis MH, et al. Impaired fibrillin-1 function promotes features of plaque instability in apolipoprotein E–deficient mice. Circulation. (2009) 120:2478–87. doi: 10.1161/CIRCULATIONAHA.109.872663
42. Roth L, Van Dam D, Van der Donckt C, Schrijvers DM, Lemmens K, Van Brussel I, et al. Impaired gait pattern as a sensitive tool to assess hypoxic brain damage in a novel mouse model of atherosclerotic plaque rupture. Physiol Behav. (2015) 139:397–402. doi: 10.1016/j.physbeh.2014.11.047
43. Roth L, Van der Donckt C, Emini Veseli B, Van Dam D, De Deyn PP, Martinet W, et al. Nitric oxide donor molsidomine favors features of atherosclerotic plaque stability and reduces myocardial infarction in mice. Vascul Pharmacol. (2019) 118–119:106561. doi: 10.1016/j.vph.2019.05.001
44. Van der Veken B, De Meyer GRY, Martinet W. Axitinib attenuates intraplaque angiogenesis, haemorrhages and plaque destabilization in mice. Vascul Pharmacol. (2018) 100:34–40. doi: 10.1016/j.vph.2017.10.004
45. Wong MC, van Diepen JA, Hu L, Guigas B, de Boer HC, van Puijvelde GH, et al. Hepatocyte-specific IKKβ expression aggravates atherosclerosis development in APOE*3-Leiden mice. Atherosclerosis. (2012) 220:362–8. doi: 10.1016/j.atherosclerosis.2011.06.055
46. Westerterp M, van der Hoogt CC, de Haan W, Offerman EH, Dallinga-Thie GM, Jukema JW, et al. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden Mice. Arterioscler Thromb Vasc Biol. (2006) 26:2552–9. doi: 10.1161/01.ATV.0000243925.65265.3c
47. Bennett BJ, Davis RC, Civelek M, Orozco L, Wu J, Qi H, et al. Genetic architecture of atherosclerosis in mice: a systems genetics analysis of common inbred strains. PLOS Genet. (2015) 11:e1005711. doi: 10.1371/journal.pgen.1005711
48. van den Maagdenberg AM, Hofker MH, Krimpenfort PJ, de Bruijn I, van Vlijmen B, van der Boom H, et al. Transgenic mice carrying the apolipoprotein E3-Leiden gene exhibit hyperlipoproteinemia. J Biol Chem. (1993) 268:10540–5.
49. Roche-Molina M, Sanz-Rosa D, Cruz FM, García-Prieto J, López S, Abia R, et al. Induction of sustained hypercholesterolemia by single adeno-associated virus–mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. (2015) 35:50–9. doi: 10.1161/ATVBAHA.114.303617
50. Gong D-M, Zhang Y-L, Chen D-Y, Hong L-J, Han F, Liu Q-B, et al. Endothelial GPR124 exaggerates the pathogenesis of atherosclerosis by activating inflammation. Cell Physiol Biochem. (2018) 45:547–57. doi: 10.1159/000487032
51. Kostogrys RB, Franczyk-Zarów M, Maślak E, Gajda M, Mateuszuk Ł, Jackson CL, et al. Low carbohydrate, high protein diet promotes atherosclerosis in apolipoprotein E/low-density lipoprotein receptor double knockout mice (apoE/LDLR–/–). Atherosclerosis. (2012) 223:327–31. doi: 10.1016/j.atherosclerosis.2012.05.024
52. Stoletov K, Fang L, Choi S-H, Hartvigsen K, Hansen LF, Hall C, et al. Vascular lipid accumulation, lipoprotein oxidation, and macrophage lipid uptake in hypercholesterolemic zebrafish. Circ Res. (2009) 104:952–60. doi: 10.1161/CIRCRESAHA.108.189803
53. Luo H, Li Q-Q, Wu N, Shen Y-G, Liao W-T, Yang Y, et al. Chronological in vivo imaging reveals endothelial inflammation prior to neutrophils accumulation and lipid deposition in HCD-fed zebrafish. Atherosclerosis. (2019) 290:125–35. doi: 10.1016/j.atherosclerosis.2019.09.017
54. Fang L, Harkewicz R, Hartvigsen K, Wiesner P, Choi S-H, Almazan F, et al. Oxidized cholesteryl esters and phospholipids in zebrafish larvae fed a high cholesterol diet: macrophage binding and activation. J Biol Chem. (2010) 285:32343–51. doi: 10.1074/jbc.M110.137257
55. Liu C, Gaudet D, Miller YI. Deficient cholesterol esterification in plasma of apoc2 knockout zebrafish and familial chylomicronemia patients. PLoS ONE. (2017) 12:e0169939. doi: 10.1371/journal.pone.0169939
56. Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, García-Cardeña G, et al. Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association. Circ Res. (2017) 121:e53–79. doi: 10.1161/RES.0000000000000169
57. Gao M, Xin G, Qiu X, Wang Y, Liu G. Establishment of a rat model with diet-induced coronary atherosclerosis. J Biomed Res. (2017) 31:47–55. doi: 10.7555/JBR.31.20160020
58. Martínez-Martínez AB, Torres-Perez E, Devanney N, Del Moral R, Johnson LA, Arbones-Mainar JM. Beyond the CNS: the many peripheral roles of APOE. Neurobiol Dis. (2020) 138:104809. doi: 10.1016/j.nbd.2020.104809
59. Lee YT, Lin HY, Chan YWF, Li KHC, To OTL, Yan BP, et al. Mouse models of atherosclerosis: a historical perspective and recent advances. Lipids Health Dis. (2017) 16:12. doi: 10.1186/s12944-016-0402-5
60. Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL, et al. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest. (2012) 122:70–9. doi: 10.1172/JCI43713
61. Tie C, Gao K, Zhang N, Zhang S, Shen J, Xie X, et al. Ezetimibe attenuates atherosclerosis associated with lipid reduction and inflammation inhibition. PLoS ONE. (2015) 10:e0142430. doi: 10.1371/journal.pone.0142430
62. Bjørklund MM, Hollensen AK, Hagensen MK, Dagnæs-Hansen F, Christoffersen C, Mikkelsen JG, et al. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. (2014) 114:1684–9. doi: 10.1161/CIRCRESAHA.114.302937
63. Conway A, Mendel M, Kim K, McGovern K, Boyko A, Zhang L, et al. Non-viral delivery of zinc finger nuclease mRNA enables highly efficient in vivo genome editing of multiple therapeutic gene targets. Mol Ther J Am Soc Gene Ther. (2019) 27:866–77. doi: 10.1016/j.ymthe.2019.03.003
64. Moss ME, Lu Q, Iyer SL, Engelbertsen D, Marzolla V, Caprio M, et al. Endothelial mineralocorticoid receptors contribute to vascular inflammation in atherosclerosis in a sex-specific manner. Arterioscler Thromb Vasc Biol. (2019) 39:1588–601. doi: 10.1161/ATVBAHA.119.312954
65. Jarrett KE, Lee C, De Giorgi M, Hurley A, Gillard BK, Doerfler AM, et al. Somatic editing of Ldlr with adeno-associated viral-CRISPR is an efficient tool for atherosclerosis research. Arterioscler Thromb Vasc Biol. (2018) 38:1997–2006. doi: 10.1161/ATVBAHA.118.311221
66. von Scheidt M, Zhao Y, Kurt Z, Pan C, Zeng L, Yang X, et al. Applications and limitations of mouse models for understanding human atherosclerosis. Cell Metab. (2017) 25:248–61. doi: 10.1016/j.cmet.2016.11.001
67. Ackert-Bicknell CL, Rosen CJ. Passenger gene mutations: unwanted guests in genetically modified mice. J Bone Miner Res. (2016) 31:270–3. doi: 10.1002/jbmr.2772
68. Allayee H. Using mice to dissect genetic factors in atherosclerosis. Arterioscler Thromb Vasc Biol. (2003) 23:1501–9. doi: 10.1161/01.ATV.0000090886.40027.DC
69. Gabriel SE. Cardiovascular morbidity and mortality in rheumatoid arthritis. Am J Med. (2008) 121(10 Suppl. 1):S9–14. doi: 10.1016/j.amjmed.2008.06.011
70. Karbach S, Croxford AL, Oelze M, Schüler R, Minwegen D, Wegner J, et al. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler Thromb Vasc Biol. (2014) 34:2658–68. doi: 10.1161/ATVBAHA.114.304108
71. Torjesen I. Drug development: the journey of a medicine from lab to shelf. The Pharmaceutical Journal. (2015). Available online at: http://www.pharmaceutical-journal.com/publications/tomorrows-pharmacist/drug-development-the-journey-of-a-medicine-from-lab-to-shelf/20068196.article (accessed November 13, 2017).
72. Landlinger C, Pouwer MG, Juno C, van der Hoorn JWA, Pieterman EJ, Jukema JW, et al. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. Eur Heart J. (2017) 38:2499–507. doi: 10.1093/eurheartj/ehx260
73. Roy P, Ali AJ, Kobiyama K, Ghosheh Y, Ley K. Opportunities for an atherosclerosis vaccine: from mice to humans. Vaccine. (2020) 38:4495–506. doi: 10.1016/j.vaccine.2019.12.039
74. Fang L, Miller YI. Emerging applications for zebrafish as a model organism to study oxidative mechanisms and their roles in inflammation and vascular accumulation of oxidized lipids. Free Radic Biol Med. (2012) 53:1411–20. doi: 10.1016/j.freeradbiomed.2012.08.004
75. Wilkinson RN, van Eeden FJM. The zebrafish as a model of vascular development and disease. Prog Mol Biol Transl Sci. (2014) 124:93–122. doi: 10.1016/B978-0-12-386930-2.00005-7
76. Asnani A, Peterson RT. The zebrafish as a tool to identify novel therapies for human cardiovascular disease. Dis Model Mech. (2014) 7:763–7. doi: 10.1242/dmm.016170
77. Santoriello C, Zon LI. Hooked! Modeling human disease in zebrafish. J Clin Invest. (2012) 122:2337–43. doi: 10.1172/JCI60434
78. Trede NS, Langenau DM, Traver D, Look AT, Zon LI. The use of zebrafish to understand immunity. Immunity. (2004) 20:367–79. doi: 10.1016/S1074-7613(04)00084-6
79. Fang L, Liu C, Miller YI. Zebrafish models of dyslipidemia: relevance to atherosclerosis and angiogenesis. Transl Res J Lab Clin Med. (2014) 163:99–108. doi: 10.1016/j.trsl.2013.09.004
80. Renshaw SA, Trede NS. A model 450 million years in the making: zebrafish and vertebrate immunity. Dis Model Mech. (2012) 5:38–47. doi: 10.1242/dmm.007138
81. Lugo-Villarino G, Balla KM, Stachura DL, Bañuelos K, Werneck MBF, Traver D. Identification of dendritic antigen-presenting cells in the zebrafish. Proc Natl Acad Sci USA. (2010) 107:15850–5. doi: 10.1073/pnas.1000494107
82. Peterson RT, Macrae CA. Systematic approaches to toxicology in the zebrafish. Annu Rev Pharmacol Toxicol. (2012) 52:433–53. doi: 10.1146/annurev-pharmtox-010611-134751
83. Patton EE, Zon LI. The art and design of genetic screens: zebrafish. Nat Rev Genet. (2001) 2:956–66. doi: 10.1038/35103567
84. Hwang WY, Peterson RT, Yeh JRJ. Methods for targeted mutagenesis in zebrafish using TALENs. Methods San Diego Calif. (2014) 69:76–84. doi: 10.1016/j.ymeth.2014.04.009
85. Wang HY, Quan C, Hu C, Xie B, Du Y, Chen L, et al. A lipidomics study reveals hepatic lipid signatures associating with deficiency of the LDL receptor in a rat model. Biol Open. (2016) 5:979–86. doi: 10.1242/bio.019802
86. Ou L, Li X, Chen B, Ge Z, Zhang J, Zhang Y, et al. Recombinant human cytoglobin prevents atherosclerosis by regulating lipid metabolism and oxidative stress. J Cardiovasc Pharmacol Ther. (2018) 23:162–73. doi: 10.1177/1074248417724870
87. Kaliste E, Mering S. The welfare of laboratory rats. In: Kaliste E, editor. The Welfare of Laboratory Animals. Dordrecht: Springer (2007) p. 153–80. doi: 10.1007/978-1-4020-2271-5_8
88. Lu R, Yuan T, Wang Y, Zhang T, Yuan Y, Wu D, et al. Spontaneous severe hypercholesterolemia and atherosclerosis lesions in rabbits with deficiency of low-density lipoprotein receptor (LDLR) on exon 7. EBioMedicine. (2018) 36:29–38. doi: 10.1016/j.ebiom.2018.09.020
89. Lidfors L, Edström T, Lindberg L. The welfare of laboratory rabbits. In: Kaliste E, editor. The Welfare of Laboratory Animals. Dordrecht: Springer (2007) p. 211–43. doi: 10.1007/978-1-4020-2271-5_10
90. Shiomi M, Ishida T, Kobayashi T, Nitta N, Sonoda A, Yamada S, et al. Vasospasm of atherosclerotic coronary arteries precipitates acute ischemic myocardial damage in myocardial infarction–prone strain of the Watanabe Heritable Hyperlipidemic rabbits. Arterioscler Thromb Vasc Biol. (2013) 33:2518–23. doi: 10.1161/ATVBAHA.113.301303
91. Chalmers AD, Bursill CA, Myerscough MR. Nonlinear dynamics of early atherosclerotic plaque formation may determine the efficacy of high density lipoproteins (HDL) in plaque regression. PLoS ONE. (2017) 12:e0187674. doi: 10.1371/journal.pone.0187674
92. Niimi M, Yang D, Kitajima S, Ning B, Wang C, Li S, et al. ApoE knockout rabbits: a novel model for the study of human hyperlipidemia. Atherosclerosis. (2016) 245:187–93. doi: 10.1016/j.atherosclerosis.2015.12.002
93. Cassar S, Adatto I, Freeman JL, Gamse JT, Iturria I, Lawrence C, et al. Use of zebrafish in drug discovery toxicology. Chem Res Toxicol. (2020) 33:95–118. doi: 10.1021/acs.chemrestox.9b00335
94. Williams CH, Hong CC. Zebrafish small molecule screens: taking the phenotypic plunge. Comput Struct Biotechnol J. (2016) 14:350–6. doi: 10.1016/j.csbj.2016.09.001
95. Aulner N, Danckaert A, Ihm J, Shum D, Shorte SL. Next-generation phenotypic screening in early drug discovery for infectious diseases. Trends Parasitol. (2019) 35:559–70. doi: 10.1016/j.pt.2019.05.004
96. Vincent F, Loria P, Pregel M, Stanton R, Kitching L, Nocka K, et al. Developing predictive assays: the phenotypic screening “rule of 3.” Sci Transl Med. (2015) 7:293ps15. doi: 10.1126/scitranslmed.aab1201
97. van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O'Collins V, et al. Can animal models of disease reliably inform human studies? PLoS Med. (2010) 7:e1000245. doi: 10.1371/journal.pmed.1000245
98. Moffat JG. Turning the light on in the phenotypic drug discovery black box. Cell Chem Biol. (2017) 24:545–7. doi: 10.1016/j.chembiol.2017.05.005
99. Fang L, Miller YI. Targeted cholesterol efflux. Cell Cycle Georget Tex. (2013) 12:3345–6. doi: 10.4161/cc.26401
100. Fang L, Green SR, Baek JS, Lee S-H, Ellett F, Deer E, et al. In vivo visualization and attenuation of oxidized lipid accumulation in hypercholesterolemic zebrafish. J Clin Invest. (2011) 121:4861–9. doi: 10.1172/JCI57755
101. CETP Gene - GeneCards. (2017). Available online at: http://www.genecards.org/cgi-bin/carddisp.pl?gene=CETP (accessed December 16, 2017).
102. Brown DR, Samsa LA, Qian L, Liu J. Advances in the study of heart development and disease using zebrafish. J Cardiovasc Dev Dis. (2016) 3:13. doi: 10.3390/jcdd3020013
103. Chen L-C, Wu J-L, Shiau C-Y, Chen J-Y. Organization and promoter analysis of the zebrafish (Danio rerio) chemokine gene (CXC-64) promoter. Fish Physiol Biochem. (2010) 36:511–21. doi: 10.1007/s10695-009-9321-y
104. Li Y, Li Y, Cao X, Jin X, Jin T. Pattern recognition receptors in zebrafish provide functional and evolutionary insight into innate immune signaling pathways. Cell Mol Immunol. (2017) 14:80–9. doi: 10.1038/cmi.2016.50
105. van der Vaart M, van Soest JJ, Spaink HP, Meijer AH. Functional analysis of a zebrafish myd88 mutant identifies key transcriptional components of the innate immune system. Dis Model Mech. (2013) 6:841–54. doi: 10.1242/dmm.010843
106. Kanwal Z, Wiegertjes GF, Veneman WJ, Meijer AH, Spaink HP. Comparative studies of Toll-like receptor signalling using zebrafish. Dev Comp Immunol. (2014) 46:35–52. doi: 10.1016/j.dci.2014.02.003
107. Fink IR, Benard EL, Hermsen T, Meijer AH, Forlenza M, Wiegertjes GF. Molecular and functional characterization of the scavenger receptor CD36 in zebrafish and common carp. Mol Immunol. (2015) 63:381–93. doi: 10.1016/j.molimm.2014.09.010
108. Svensson S, Abrahamsson A, Rodriguez GV, Olsson A-K, Jensen L, Cao Y, et al. CCL2 and CCL5 are novel therapeutic targets for estrogen-dependent breast cancer. Clin Cancer Res. (2015) 21:3794–805. doi: 10.1158/1078-0432.CCR-15-0204
109. Sun G, Pan J, Liu K, Wang X, Wang S. Molecular cloning and expression analysis of P-selectin from zebrafish (Danio rerio). Int J Mol Sci. (2010) 11:4618–30. doi: 10.3390/ijms11114618
110. Pietretti D, Spaink HP, Falco A, Forlenza M, Wiegertjes GF. Accessory molecules for Toll-like receptors in teleost fish. Identification of TLR4 interactor with leucine-rich repeats (TRIL). Mol Immunol. (2013) 56:745–56. doi: 10.1016/j.molimm.2013.07.012
111. Tsan M-F, Gao B. Heat shock proteins and immune system. J Leukoc Biol. (2009) 85:905–10. doi: 10.1189/jlb.0109005
112. Wick C. Tolerization against atherosclerosis using heat shock protein 60. Cell Stress Chaperones. (2016) 21:201–11. doi: 10.1007/s12192-015-0659-z
113. Hyperion Therapeutics Terminates DiaPep277(R) Program | FierceBiotech. Available online at: https://www.fiercebiotech.com/biotech/hyperion-therapeutics-terminates-diapep277-r-program (accessed April 6, 2020).
114. Berliner JA, Watson AD. A role for oxidized phospholipids in atherosclerosis. N Engl J Med. (2005) 353:9–11. doi: 10.1056/NEJMp058118
115. Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. (2014) 46:e99. doi: 10.1038/emm.2014.38
116. Yin W, Carballo-Jane E, McLaren DG, Mendoza VH, Gagen K, Geoghagen NS, et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J Lipid Res. (2012) 53:51–65. doi: 10.1194/jlr.M019927
117. Andreadou I, Schulz R, Badimon L, Adameová A, Kleinbongard P, Lecour S, et al. Hyperlipidaemia and cardioprotection: animal models for translational studies. Br J Pharmacol. (2020). doi: 10.1111/bph.14931. [Epub ahead of print].
118. Chen Y, Wen S, Jiang M, Zhu Y, Ding L, Shi H, et al. Atherosclerotic dyslipidemia revealed by plasma lipidomics on ApoE –/– mice fed a high-fat diet. Atherosclerosis. (2017) 262:78–86. doi: 10.1016/j.atherosclerosis.2017.05.010
119. Nilsson J, Hansson GK, Shah PK. Immunomodulation of atherosclerosis: implications for vaccine development. Arterioscler Thromb Vasc Biol. (2005) 25:18–28. doi: 10.1161/01.ATV.0000149142.42590.a2
120. Mahan MM, Doiron AL. Gold nanoparticles as X-ray, CT, and multimodal imaging contrast agents: formulation, targeting, and methodology. J Nanomater. (2018) 2018:1–15. doi: 10.1155/2018/5837276
121. Browning LM, Huang T, Xu X-HN. Real-time in vivo imaging of size-dependent transport and toxicity of gold nanoparticles in zebrafish embryos using single nanoparticle plasmonic spectroscopy. Interface Focus. (2013) 3:20120098. doi: 10.1098/rsfs.2012.0098
122. Kim K-T, Zaikova T, Hutchison JE, Tanguay RL. Gold nanoparticles disrupt zebrafish eye development and pigmentation. Toxicol Sci. (2013) 133:275–88. doi: 10.1093/toxsci/kft081
123. Chakraborty C, Sharma AR, Sharma G, Lee S-S. Zebrafish: A complete animal model to enumerate the nanoparticle toxicity. J Nanobiotechnol. (2016) 14:65. doi: 10.1186/s12951-016-0217-6
124. Cordeiro M, Carvalho L, Silva J, Saúde L, Fernandes A, Baptista P. Gold nanobeacons for tracking gene silencing in zebrafish. Nanomaterials. (2017) 7:10. doi: 10.3390/nano7010010
125. De Wilde D, Trachet B, Van der Donckt C, Vandeghinste B, Descamps B, Vanhove C, et al. Vulnerable plaque detection and quantification with gold particle–enhanced computed tomography in atherosclerotic mouse models. Mol Imaging. (2015) 14:9–19. doi: 10.2310/7290.2015.00009
126. Schürmann C, Gremse F, Jo H, Kiessling F, Brandes RP. Micro-CT technique is well suited for documentation of remodeling processes in murine carotid arteries. PLoS ONE. (2015) 10:e0130374. doi: 10.1371/journal.pone.0130374
127. Corti R, Fuster V. Imaging of atherosclerosis: magnetic resonance imaging. Eur Heart J. (2011) 32:1709–19. doi: 10.1093/eurheartj/ehr068
128. Koth J, Maguire ML, McClymont D, Diffley L, Thornton VL, Beech J, et al. High-resolution magnetic resonance imaging of the regenerating adult zebrafish heart. Sci Rep. (2017) 7:2917. doi: 10.1038/s41598-017-03050-y
129. Parant M, Sohm B, Flayac J, Perrat E, Chuburu F, Cadiou C, et al. Impact of gadolinium-based contrast agents on the growth of fish cells lines. Ecotoxicol Environ Saf. (2019) 182:109385. doi: 10.1016/j.ecoenv.2019.109385
130. Hockings PD, Roberts T, Galloway GJ, Reid DG, Harris DA, Vidgeon-Hart M, et al. Repeated three-dimensional magnetic resonance imaging of atherosclerosis development in innominate arteries of low-density lipoprotein receptor-knockout mice. Circulation. (2002) 106:1716–21. doi: 10.1161/01.CIR.0000030188.50326.8D
131. Jung C, Christiansen S, Kaul MG, Koziolek E, Reimer R, Heeren J, et al. Quantitative and qualitative estimation of atherosclerotic plaque burden in vivo at 7T MRI using Gadospin F in comparison to en face preparation evaluated in ApoE KO mice. PLoS ONE. (2017) 12:e0180407. doi: 10.1371/journal.pone.0180407
132. Zhou YQ, Foster FS, Qu DW, Zhang M, Harasiewicz KA, Adamson SL. Applications for multifrequency ultrasound biomicroscopy in mice from implantation to adulthood. Physiol Genomics. (2002) 10:113–26. doi: 10.1152/physiolgenomics.00119.2001
133. Li R-J, Yang Y, Wang Y-H, Xie J-J, Song L, Wang Z, et al. Micro-ultrasonographic imaging of atherosclerotic progression and correlation with inflammatory markers in apolipoprotein-E knockout mice. Tex Heart Inst J. (2011) 38:364–70. doi: 10.7555/JBR.31.20160020
134. Zhou X, Sun L, Yu Y, Qiu W, Lien C-L, Shung KK, et al. Ultrasound bio-microscopic image segmentation for evaluation of zebrafish cardiac function. IEEE Trans Ultrason Ferroelectr Freq Control. (2013) 60:718–26. doi: 10.1109/TUFFC.2013.2620
135. Benslimane FM, Alser M, Zakaria ZZ, Sharma A, Abdelrahman HA, Yalcin HC. Adaptation of a mice Doppler echocardiography platform to measure cardiac flow velocities for embryonic chicken and adult zebrafish. Front Bioeng Biotechnol. (2019) 7:96. doi: 10.3389/fbioe.2019.00096
136. Guo Y, Liu X-C, Wang Y-J, Li Q, Yang Q, Weng X-G, et al. Effects of Shenlian extract on experimental atherosclerosis in ApoE-deficient mice based on ultrasound biomicroscopy. BMC Complement Altern Med. (2016) 16:469. doi: 10.1186/s12906-016-1449-6
137. Becher T, Riascos-Bernal DF, Kramer DJ, Almonte VM, Chi J, Tong T, et al. Three-dimensional imaging provides detailed atherosclerotic plaque morphology and reveals angiogenesis after carotid artery ligation. Circ Res. (2020) 126:619–32. doi: 10.1161/CIRCRESAHA.119.315804
138. Salman HE, Yalcin HC. Advanced blood flow assessment in zebrafish via experimental digital particle image velocimetry and computational fluid dynamics modeling. Micron. (2020) 130:102801. doi: 10.1016/j.micron.2019.102801
Keywords: atherosclerosis, animal models, mouse, zebrafish, immune response, human disease, APOE, LDLR
Citation: Vedder VL, Aherrahrou Z and Erdmann J (2020) Dare to Compare. Development of Atherosclerotic Lesions in Human, Mouse, and Zebrafish. Front. Cardiovasc. Med. 7:109. doi: 10.3389/fcvm.2020.00109
Received: 06 February 2020; Accepted: 26 May 2020;
Published: 30 June 2020.
Edited by:
Ming-Hui Zou, Georgia State University, United StatesReviewed by:
Ying Hu Shen, Baylor College of Medicine, United StatesCopyright © 2020 Vedder, Aherrahrou and Erdmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viviana L. Vedder, viviana.vedder@uni-luebeck.de
†ORCID: Jeanette Erdmann orcid.org/0000-0002-4486-6231
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.