 Shafeeq A. Mohammed
Shafeeq A. Mohammed Samuele Ambrosini
Samuele Ambrosini Thomas Lüscher1,2
Thomas Lüscher1,2 Francesco Paneni
Francesco Paneni Sarah Costantino
Sarah Costantino
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 04 March 2020
Sec. Cardiovascular Metabolism
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.00028
This article is part of the Research Topic Mitochondrial Dysfunction and Cardiovascular Diseases View all 10 articles
The molecular signatures of epigenetic regulation and chromatin architecture are emerging as pivotal regulators of mitochondrial function. Recent studies unveiled a complex intersection among environmental factors, epigenetic signals, and mitochondrial metabolism, ultimately leading to alterations of vascular phenotype and increased cardiovascular risk. Changing environmental conditions over the lifetime induce covalent and post-translational chemical modification of the chromatin template which sensitize the genome to establish new transcriptional programs and, hence, diverse functional states. On the other hand, metabolic alterations occurring in mitochondria affect the availability of substrates for chromatin-modifying enzymes, thus leading to maladaptive epigenetic signatures altering chromatin accessibility and gene transcription. Indeed, several components of the epigenetic machinery require intermediates of cellular metabolism (ATP, AcCoA, NADH, α-ketoglutarate) for enzymatic function. In the present review, we describe the emerging role of epigenetic modifications as fine tuners of gene transcription in mitochondrial dysfunction and vascular disease. Specifically, the following aspects are described in detail: (i) mitochondria and vascular function, (ii) mitochondrial ROS, (iii) epigenetic regulation of mitochondrial function; (iv) the role of mitochondrial metabolites as key effectors for chromatin-modifying enzymes; (v) epigenetic therapies. Understanding epigenetic routes may pave the way for new approaches to develop personalized therapies to prevent mitochondrial insufficiency and its complications.
Mitochondria, defined as semi-autonomous, membrane-bound organelle localized in the cytoplasm of eukaryotic cells, are emerging as a pivotal player in health, disease, and aging by regulating reactive oxygen species (ROS) production and contributing to retrograde redox signalling from the organelle to the cytosol and nucleus (Figure 1) (1, 2). Mitochondria play an important role in the overall cellular network formed by metabolic signalling and epigenetic pathways. Indeed, mitochondria drive catabolic and anabolic reactions supplying energy and metabolites with biosynthetic and signalling roles (3). They also maintain a bidirectional signalling crosstalk with the nucleus that generates reciprocal activation-repression patterns of gene expression (3–5). Finally, mitochondria can determine apoptotic and necrotic cell death mediated by Ca2+ overload and opening of the permeability transition pore (PTP) (6, 7).
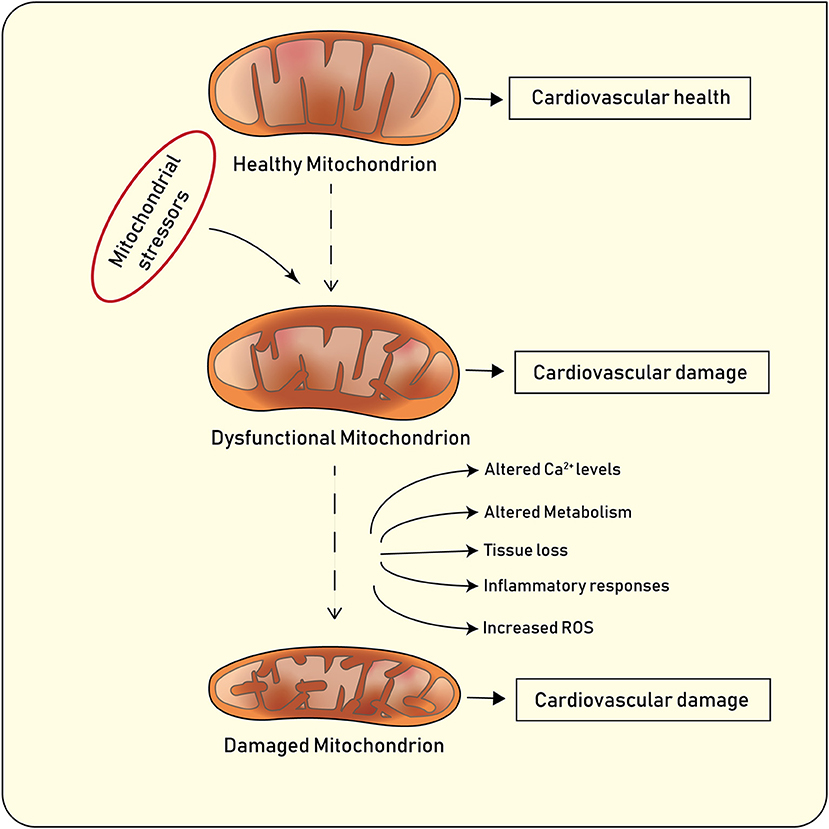
Figure 1. Main features of healthy and diseased mitochondria, and implications for cardiovascular disease.
Under physiological conditions, mitochondria undergo highly coordinated cycles of fission (division of a single organelle into two or more independent structures) or fusion (the opposing reaction) (8). Fission and fusion are active processes which require many specialized proteins, including mechanical enzymes that physically alter mitochondrial membranes, and adaptor proteins that regulate the interaction of these mechanical proteins with organelles. The balance between these two processes regulates the overall morphology of mitochondria within any given cell (8–10). The content of mitochondria in the cytoplasm of eukaryotic cells depend on two major processes known as mitochondrial biogenesis and mitophagy (11). Mitochondrial biogenesis is an intricate and not fully understood process which leads to an increased mitochondrial mass mainly via replication of mitochondrial DNA (mtDNA) and expression of nuclear and mitochondrial genes (12). PGC-1α (Peroxisome proliferator-activated receptor gamma coactivator−1α) plays a prominent role in mitochondrial biogenesis by activating the nuclear respiratory factor (Nrf)-1 and −2 to promote the expression nuclear genes. PGC-1α also activates transcription factors A and B which regulate the expression of mitochondrial genes (13, 14). Following mitochondrial damage, the organelles are being selectively degraded according to a well-known biological process called mitophagy, which promotes organelle turnover while preventing accumulation of dysfunctional mitochondria (Figure 1) (11). In addition to the selective removal of damaged mitochondria, mitophagy is also required to adjust mitochondrial numbers to changing cellular metabolic needs, for steady-state mitochondrial turnover, and during certain cellular developmental stages, such as during cellular differentiation of red blood cells (10). Mitochondrial content may vary based on the cell type and its function. For example, in endothelial cells mitochondria occupy around 6% of cytoplasm whereas in cardiomyocytes this reaches 32% (15). Notably, the blood brain barrier which consist of highly active endothelial cells has higher mitochondrial content as compared with endothelial cells present in capillary beds (15). Mitochondria play a pivotal role in endothelial cells. Several biological processes including mitochondrial biogenesis, fission and fusion as well as mitophagy, have shown to clearly affect endothelial cell function and metabolism. Several stimuli including hypoxia, calorie restriction or exercise induce mitochondrial biogenesis in endothelial cells by increasing the expression of the peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α). Induction of PGC-1α is associated with a favorable transcriptional profile which protects endothelial cells from oxidative damage and apoptosis (16). In line with this notion, endothelial-specific overexpression of PGC-1α protects against angiotensin II–induced hypertension (17). By contrast, loss of endothelial PGC-1α impairs endothelial NO bioactivity eventually leading to endothelial dysfunction (18). Alterations of mitochondrial dynamics also contribute to endothelial cell phenotype. Endothelial cells from patients with diabetes display mitochondrial fragmentation and increased expression of fission-1 protein (Fis1) and dynamin-related protein-1 (Drp1). Of note, in vitro experiments showed that gene silencing Fis1 or Drp1 expression blunted hyperglycemia-induced alterations in mitochondrial networks, ROS production, endothelial nitric oxide synthase activation, and cGMP production (19). Alterations of mitophagy as the result of disturbed Ucp2/PTEN signaling were also associated with inadequate mitochondrial biosynthesis and increased apoptosis in endothelium (20). Altered mitochondrial clearance may also contribute to age-dependent endothelial dysfunction. Indeed, senescent cells display altered mitochondrial dynamics and loss of membrane potential (21). Interestingly enough, overexpression of proteins involved in the autophagosome formation (ATG5 and ATG12) was associated with improved mitochondrial performance, as evidenced by higher membrane potential, increased ATP production, and decreased damage to mtDNA (22, 23).
Although several cytosolic enzymes (i.e., NADPH, cyclooxygenases, and xanthine oxidase) are implicated in redox balance, ROS generated from mitochondrial oxidative phosphorylation represent the most important source of oxidative stress in vascular cells (i.e., endothelial cells) (24, 25).
Mitochondrial ROS are responsible for peroxidation of polyunsaturated fatty acids (PUFAs) present in the cellular membrane as well as DNA (causing single and double strand breaks) and protein damage via oxidation of sulfhydryl and aldehyde groups, protein-protein interactions and fragmentation (26). In addition, damage of mtDNA may lead to decreased expression of electron transport chain components or expression of defective components that produce more ROS, thus creating a detrimental vicious cycle. mtDNA disruption also correlates with the extent of atherosclerosis in mouse models and human tissues. Despite the highly efficient chemical reduction of O2 through cytochrome c oxidase, mitochondria still generate significant levels of ROS (27). Cellular and mitochondrial physiological levels of ROS are reached when production and scavenging are balanced (28). Mitochondrial dysfunction is believed to play an important role in a variety of diseases including diabetes, obesity, dyslipidaemia, hypertension, arrhythmias, and sudden cardiac death (29–31).
In the setting of cardiovascular risk factors, namely hyperglycemia, mitochondrial ROS can be regarded as an upstream biochemical event responsible for the activation of pro-inflammatory pathways (i.e., NF-kB), protein kinase C as well as advanced glycation end products (AGEs) (32). An increasing body of evidence has contributed to unveil different sources of mitochondrial ROS in endothelial cells. Studies in isolated mitochondria have shown that superoxide anion formation at complexes I and III accounts for 0.1-2% of the total (33). In addition to complexes I and III, the nicotinamide adenine dinucleotide phosphate oxidase (NOX) 4—a ROS-generating enzyme involved in endothelial cell senescence, migration, angiogenesis, and adaptive responses to hypoxia—is highly expressed in vascular cells and has been localized to mitochondria (34). Moreover, the monoamine oxidase (MAO) family of enzymes—which is found in the outer mitochondrial membrane—generates hydrogen peroxide (H2O2) during catabolism of catecholamines and has been implicated in maladaptive cellular hypertrophy and apoptosis (35). MAO-A-induced ROS are involved in serotonin-induced vasoconstriction in vascular smooth muscle cells (36). Although endothelial cells are known to express MAO, its importance for endothelial function is poorly understood (37). The mitochondrial adaptor protein p66Shc was recently shown to be causally involved in mitochondrial ROS generation and cellular death. In conditions of cellular stress, p66Shc is phosphorylated at ser36 by protein kinase C beta2 (PKCβ2) and translocates to the mitochondria where it oxidizes cytochrome c, leading to accumulation of H2O2, PTP opening, and release of solutes and proapoptotic signals (38). The causal role of p66Shc in vascular disease is supported by the notion that its genetic deletion or gene silencing prevents age and hyperglycemia-induced endothelial dysfunction in mice (39–41). The prolyl-isomerase 1 (Pin1), which regulates p66Shc translocation to the mitochondria, has also shown to be causally implicated in the regulation of mitochondrial oxidative stress and integrity in experimental models of diabetes (42, 43). The mitochondrial ATP-sensitive potassium channel (mitoKATP) was also recently discovered as a potential source of mitochondrial ROS in cardiac myocytes (44). Although the exact mechanism of action remains elusive, mitoKATP seems to act as an uncoupling agent by reducing membrane potential and mitochondrial calcium. Pharmacological inhibition of mitoKATP was found to improve endothelial function and to prevent ischemia-induced cellular apoptosis (44). Several antioxidant enzymes play a pivotal role in maintaining redox balance in mitochondria. Manganese superoxide dismutase (MnSOD) represents one of the first line defense against accumulation of mitochondrial superoxide. MnSOD is located in the mitochondrial matrix and catalyzes the conversion of superoxide anion to hydrogen peroxide (45). Loss of MnSOD in mice leads to impaired endothelium-dependent vasodilation, suggesting its role in regulating vascular function. In addition, ApoE−/− MnSOD+/− mice display early mtDNA damage and accelerated atherosclerosis when compared to control animals (46). Levels of H2O2 are regulated by glutathione peroxidase-1, thioredoxin-2, peroxiridoxin-3, and glutaredoxin-2 (47). As noted, increased expression of these enzymes is signaled by AMPK and PGC-1α in response to H2O2 and other free radicals in endothelial cells (48). Studies in experimental models have shown that reduced expression of mitochondrial antioxidant enzymes can induce mitochondrial damage, endothelial dysfunction, and atherogenesis (45, 46). Conversely, overexpression of these proteins is protective against the development of vascular disease (49).
Although the role of mitochondrial ROS in vascular damage is well-established, only few studies have explored the specific contribution of mitochondria-derived ROS in the pathophysiology of endothelial dysfunction in humans. Mitochondrial ROS production and membrane hyperpolarization are significantly altered in visceral fat arteries and peripheral blood mononuclear cells isolated from patients with obesity and type 2 diabetes (50, 51). Furthermore, impaired endothelium-dependent vasodilation in freshly isolated arterioles from diabetic individuals is reversed by mild membrane depolarization or mitochondria-targeted antioxidants (50).
Recent evidence indicates that epigenetic changes, defined as plastic modifications of DNA/histone complexes, are heavily implicated in the regulation of mitochondrial and vascular function (52, 53). Studies conducted over the last few years have unmasked a complex intersection among environmental factors, mitochondrial metabolism, epigenetic signals and transcriptional programs (54, 55). Epigenetic changes acquired during the life time may derail the expression of genes involved in mitochondrial homeostasis (52). On the other hand, metabolic alterations occurring in mitochondria may affect the availability of substrates for chromatin-modifying enzymes, thus leading to maladaptive epigenetic signatures altering chromatin accessibility and, hence, gene transcription (Figure 2) (54). Indeed, the availability of some intermediate mitochondrial metabolites (ATP, AcCoA, NADH, α-ketoglutarate) has shown to foster different patterns of epigenetic modifications. For examples, iron, α-ketoglutarate (α-KG) and O2 are needed both for histone demethylation—catalysed by iron-containing jumonji-domain (jmjC) demethylases (56)—as well as for DNA demethylation of 5-methylcytosine—catalysed by the ten-eleven translocation family of dioxygenases (TET) (57). Therefore, mitochondrial sensitivity determined by environmental factors and lifestyle changes (sedentarism, physical activity, overnutrition, balanced nutrition) will favor, or prevent, the effects of metabolic disorders.
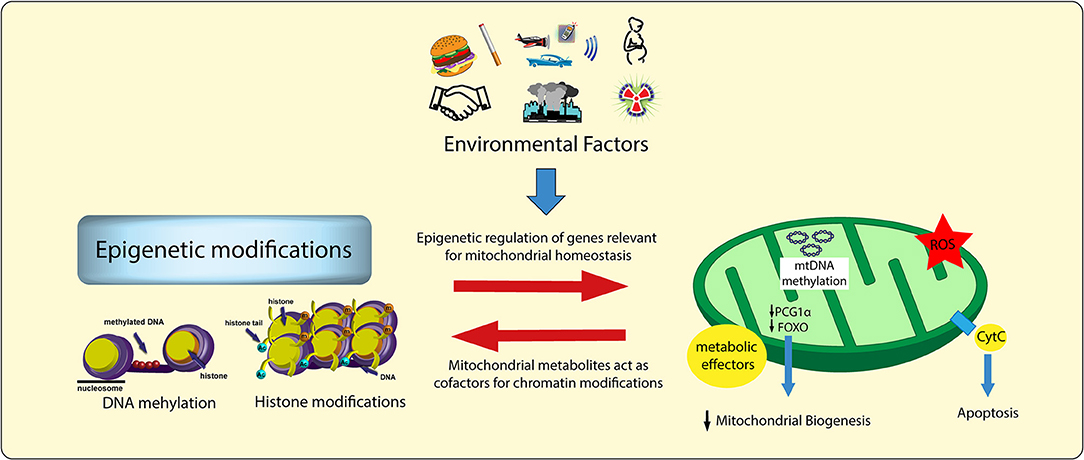
Figure 2. Environmental factors, chromatin modifications, and mitochondrial damage. Environmental factors lead to specific epigenetic signatures as well as to alterations of mitochondrial intermediate metabolites (i.e., acetyl-CoA, FAD+, NAD+). These two processes influence each other, thus leading to a vicious cycle responsible for adverse chromatin modifications, maladaptive transcriptional programs, and vascular dysfunction. ROS, reactive oxygen species.
Epigenetic mechanisms can be divided into three main categories: (i) chemical modifications of DNA (i.e., methylation); (ii) post-translational modifications of histone tails; (iii) regulation of gene expression by non-coding RNAs [i.e., microRNAs, long non-coding RNAs (lncRNAs)] (58). In the present review, we will focus on the modifications of DNA/histone complexes and their impact on mitochondrial integrity and functionality.
Methylation of DNA mainly takes place at the level of CpG regions of gene promoters through the attachment of methyl group (CH3) from S-adenosyl methionine (SAM) to the C5 position in the cytosine-paired-with-guanine (CpG) dinucleotide sequences (59). CpG sequences are generally located into promoter regions of genes, however, they can also be located within gene bodies (58). Promoter methylation is generally associated with transcriptional repression, while gene body methylation is associated with enhanced transcription (60). Promoter methylation hampers gene expression mainly via two mechanisms: (i) by fostering transcriptional silencing, or (ii) by preventing the recruitment of transcription factors (61). Specifically, methylated cytosines are recognized by DNA methyl-binding proteins (MBPs) that repress gene transcription by preventing the interaction of transcription factors with the promoter (62). Alternatively, DNA methylation may recruit specific proteins that may also favor the recruitment of enzymes catalysing histone posttranslational modifications (PTMs) with subsequent gene repression (63, 64).
DNA methylation is a relatively stable epigenetic signature, it can be tissue-specific and, most importantly, it can be transmitted to the offspring, a phenomenon known as “epigenetic inheritance” (65). Different families of enzymes, known as methyltransferases (DNMTs), are involved in the regulation of DNA methylation: DNMT1 is responsible for the maintenance of methylation patterns in the genome by replicating the hemi-methylated CpG sites (66), whereas Dnmt3a/b are considered de novo methyltransferases (67). Methylation of DNA is a dynamic and reversible process governed by methyl-writing and -erasing enzymes (58). DNA demethylation can be achieved by either passive or active mechanisms (58). Active DNA demethylation consists in the removal of the methyl group by breaking a carbon–carbon bond. DNA demethylation may follow two main pathways: the first is dependent on cytosine deamination (AID, APOBEC3G, FTO) while the second is dependent on the oxidation of methylated cytosines (68). This latter reaction is catalysed by members of the Ten-eleven translocation (TET) proteins family (TET1-3) that convert 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC) (69, 70). TET1 is mostly found in embryonic stem cells, whereas TET2 and TET3 are ubiquitously expressed. TET1- 3 proteins could further oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) that are recognized and excised by the thymine DNA glycosylase (TDG) via the base excision repair pathway (70, 71). By contrast, passive DNA demethylation is the result of DNMT1 inhibition during DNA replication (69).
DNA is packaged into repeating units called nucleosomes by wrapping around multimeric histone proteins. When nucleosomes are organized into tightly packed bundles (heterochromatin), the transcriptional machinery is hampered by a reduction of chromatin accessibility. Conversely, when chromatin is relaxed (euchromatin), DNA is more accessible to transcription factors, and gene transcription may occur (72). Histones are amenable to many posttranslational modifications (PTMs), which include methylation, acetylation, ubiquitination, phosphorylation, SUMOylation, GlcNAcylation, carbonylation, and ADP-ribosylation (73, 74). Of interest, these modifications may cluster in different patterns to regulate chromatin accessibility (59, 72, 75). Albeit the biological significance of many PTMs remains to be elucidated, considerable advances have been made in the understanding of lysine acetylation and methylation (74).
Histone acetylation, characterized by the addition of positively charged acetyl groups to amino acid residues at the level of histone tails, reduces the affinity of histones for DNA thus increasing chromatin accessibility (76). Acetylation occurs mainly on lysine residues on histones H3 and H4; this mark mainly associates with activation of transcription by enhancing chromatin accessibility (77). In this context, bromodomain and extra-terminal proteins recognize histone acetylation marks and initiate the assembly of the transcriptional machinery (78). By contrast, non-acetylated histones have been observed in transcriptionally silent genes where chromatin is compact (79). Acetylation is modulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs) which are involved in addition or removal of an acetyl group, respectively (80). This modification is driven by recognition and binding of transcription factors able to recruit one of a growing family of HATs, namely CBP/p300, MYST, and GNAT (59, 73). HATs catalyse the addition of two-carbon acetyl groups to lysine residues from acetyl-CoA thus leading to gene expression (81). On the other hand, removal of acetyl groups from histone residues by HDACs represses gene transcription (82, 83). Several HDACs have been reported in humans, and they are subdivided into four classes (Class I, IIa, IIb, III, and IV) (84, 85).
In contrast to lysine acetylation, which enhances gene expression, histone methylation may result in different chromatin states according to the methylated residue and the number of added methyl groups (79). Histone methylation is defined as the transfer of methyl group from S-adenosyl-L-methionine to lysine or arginine residues of histone proteins by histone methyltransferases (HMTs) (86). Histone methyltransferases (HMTs) have higher specificity as compared to HATs (87) and include several families of enzymes (EZH, SETD, PRDM, PRMT, METTL, and MLL) (88). Recent evidence indicates that a fine balance between histone methylation and demethylation plays a pivotal role in the regulation of chromatin accessibility.
Several lysine demethylases specific for diverse histone lysine residues have been identified (89). HDMs include members of UTX/Y, JARID1, JMJD, LSD, PHF, and FBXL enzyme families (88).
Interestingly, modifications of histones may reciprocally influence or eventually affect DNA methylation (74). In this regard, recent evidence suggests that DNA methylase (DNMTs), histone methyltransferase (HMTs), and histone acetyltransferase (HATs) are closely interconnected to regulate chromatin remodeling under specific stimuli (90). A well-described crosstalk between DNA methylation and histone H3K9 methylation, mediated by the heterochromatin protein 1 (HP1), represents a valid example of how histone modifications may facilitate the recruitment of enzymes (DNTM3a/b) involved in DNA methylation (91). Another example is methyl-CpG binding protein 2 (MECP2), which recruits the histone methyltransferase SUV39H1 only after binding methylated DNA (92, 93). Therefore, chromatin modifications may influence each other and can propagate.
Increasing evidence suggests that aberrant mitochondrial DNA (mtDNA) modification play an important role in disease development and progression (94). Since the vast majority of mitochondrial proteins are encoded in the nuclear genome, appropriate communication between the nuclear, cytoplasmic and mitochondrial compartments is essential for maintaining proper mitochondrial function. The mitochondrial genome consists of roughly 1,500 genes distributed across the maternal mtDNA and nuclear DNA (nDNA) (95). Human mtDNA is a 16.5-kb circular double-stranded DNA containing a heavy (H) and a light (L) strand located in the mitochondrial matrix (96, 97). mtDNA forms an mtDNA–protein complex, known as nucleoid, with a range of proteins including prohibitins, ATPase family AAA domain-containing protein 3 (ATAD3), mitochondrial transcription factor A (TFAM) and POLG (DNA polymerase gamma, catalytic subunit) (98, 99). In contrast to nDNA, human mtDNA is maternally inherited, is intronless, and lacks histones (100). It contains 37 genes encoding 13 subunit of the oxidative phosphorylation (OXPHOS) complexes I, III, IV, and V; two rRNAs; and 22 tRNAs (2). All other mitochondrial proteins, including those required for mtDNA replication and transcription, are encoded in the nucleus and translocated to the mitochondria using specialized import systems which often involve N-terminal mitochondrial targeting sequences (101).
Emerging evidence suggests that mtDNA may also be regulated at the epigenetic level in the form of mtDNA methylation (2). While nDNA methylation is a well-established feature, mtDNA methylation has been a matter of debate (94, 102). The prevailing opinion was that mtDNA cannot be methylated for two main reasons: (i) methylase cannot access mitochondria, and (ii) mtDNA is not complexed with histones (103). Only recently, mtDNA has been reported to contain 5-methylcytosine (5mC) as well as 5-hydroxymethylcytosine (5hmC) at CpG dinucleotides. In 2011, Shock et al. have identified a mitochondrially targeted DNMT1 transcript variant (mtDNMT1) that uses an upstream alternative translation start site leading to the inclusion of a mitochondrial targeting sequence (101). mtDNMT1 binds to the mitochondrial genome in a manner proportional to the density of CpG dinucleotides. Of note, cytosine methylation in mtDNA may play different role. Indeed, mtDNA methylation represses gene expression from the light-strand promoter. However, increased or no change in transcription of genes from the heavy-strand promoter raises the possibility of a different mode of action (104). This DNMT1 variant is upregulated by the hypoxia-responsive transcription factors peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1a) and nuclear respiratory factor 1 (NRF1) suggesting a regulatory role of mtDNMT1 during vascular oxidative stress (Figure 3) (101).
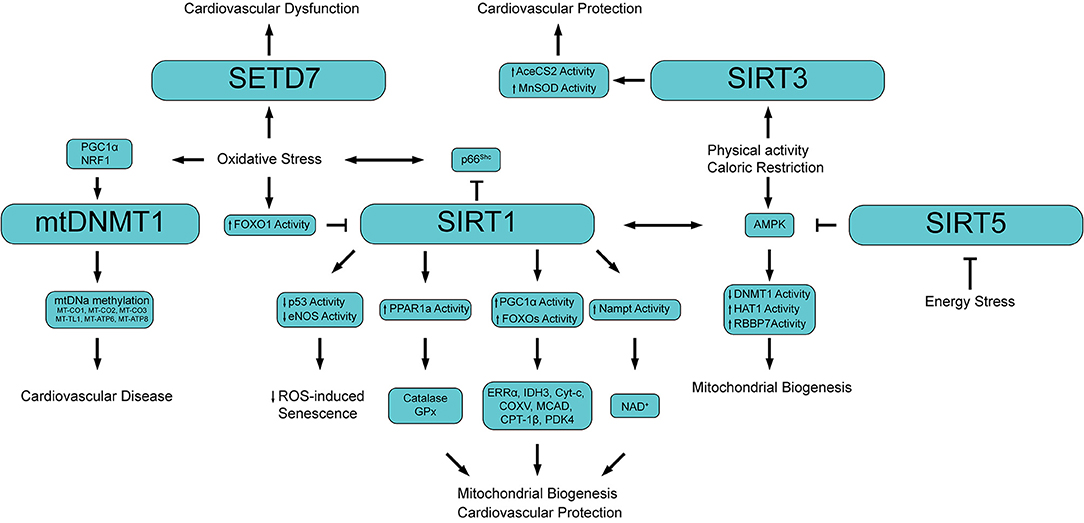
Figure 3. Schematic showing the main epigenetic networks regulating mitochondrial functionality and cardiovascular disease. PGC1α, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; NRF1, Nuclear respiratory factor 1; mtDNMT1, Mitochondrial DNA methyltransferase 1; MT-CO (1-3), Cytochrome c oxidase subunit (I, II, III); MT-TL1, Mitochondrially encoded tRNA-Leu 1; MT-ATP (6, 8) Mitochondrially encoded ATP synthase membrane (subunit 6, 8); FOXO1, Forkhead box O3; PPAR1a, Peroxisome proliferator-activated receptor 1 alpha; GPx, Glutathione peroxidase; ERRα, Estrogen-related receptor alpha; IDH3, isocitrate dehydrogenase 3; Cyt-c, Cytochrome c; COXV, Cytochrome c oxidase subunit 5; MCAD, Medium-chain acyl-CoA dehydrogenase; CPT-1β, Carnitine Palmitoyltransferase 1 beta; PDK4, Pyruvate dehydrogenase lipoamide kinase isozyme 4; Nampt, Nicotinamide phosphoribosyltransferase; NAD+, Nicotinamide adenine dinucleotide; AceCS2, acetyl-CoA synthetase 2; AMPK, 5′ adenosine monophosphate-activated protein kinase; HAT1, Histone acetyltransferase 1; RBBP7, Retinoblastoma binding protein 7.
Besides mtDNMT1, no other specific mitochondrially targeted isoforms of enzymes involved in DNA methylation or hydroxymethylation are known (100). Nevertheless, other enzymes, namely DNMT3A/B and ten–eleven translocation (TET) 1 and 2, have been detected in the mitochondrial protein fraction (105). Interestingly, the presence of these enzymes in the mitochondria seems to be tissue specific. Indeed, inside the mitochondria of ‘excitable tissues’ (heart, skeletal muscle, and neurons) only DNMT3A but not DNMT3b has been detected (106). Furthermore, epigenetic modifications of mtDNA can modulate the activity of nDNA, and vice versa (107). Under conditions of oxidative stress, such as exposure to hypoxia or ethanol, DNMT1 is upregulated and suppresses the expression of ND6 (101), while ND1 is upregulated. Although the significance of opposite ND1 and ND6 regulation is poorly understood, a proposed mechanism involves the interaction of MTERF1 (mitochondrial terminator factor 1) with 5-methylcytosine in the CpG dinucleotides and/or its interaction with mtDNA-bound mtDNMT1 (94).
An interesting study by Byun et al. showed a higher mtDNA methylation level in workers highly exposed to airborne pollutants compared to low airborne pollutant exposed subjects (108). In line with this finding, in a cohort of 81 individuals aged 18-91, methylation levels of the mitochondria gene 12S rRNA inversely correlated with age suggesting that mtDNA methylation may represent an epigenetic marker of ageing (109). In the retina of diabetic mice mtDNA methylation was found associated with mtDNA damage characterized by increased base mismatches and hypermethylated cytosines. Interestingly, inhibition of DNA methylation, or regulation of cytosine deamination, attenuated base mismatches at the D-loop thus preventing mitochondrial dysfunction and microvascular damage. In this study epigenetic signals of mtDNA were driven by oxidative stress as overexpression of Sod2 was able to prevent diabetes-induced D-loop hypermethylation and increase in base mismatches (110). Of clinical relevance, retinal microvasculature from human donors with diabetic retinopathy presented similar increase in D-loop methylation and decrease in mtDNA transcription (111). In another study, analysis of mtDNA methylation by bisulfite sequencing in senescent endothelial cells showed alteration in the methylation pattern of several genes regulating mitochondrial function and metabolism (112). Patients with cardiovascular disease display a significantly higher mtDNA methylation of genes encoding for cytochrome c oxidases (MT-CO1, MT-CO2, MT-CO3), tRNA leucine 1 (MT-TL1) and (1.67%, P = 0.0001) as well as genes involved in ATP synthesis (MT-ATP6 and MT-ATP8) (113). The latter study suggests that mtDNA methylation could serve as non-invasive and easy-to-obtain epigenetic biomarker and may be implicated in the etiology of CVD (Figure 3).
Growing evidence indicates that PTMs of histones, mainly at lysine and arginine residues, significantly affect chromatin accessibility thus enabling cell-specific transcriptional programs implicated in mitochondrial dysfunction and vascular disease (Figure 3). Sirtuins are class III histone deacetylases (HDACs), homologs of the yeast protein Silent Information Regulatory 2 (Sir2), a deacetylase involved in yeast metabolism and lifespan (104). The sirtuin family of deacetylases include seven enzymes differentially distributed throughout the cell: SIRT1 and SIRT2 which are mainly localized in both cytoplasmic and nuclear compartments; SIRT3 SIRT4, and SIRT5, which are localized in the mitochondria, and SIRT6 and SIRT7 which are located in the cell nucleus (29, 114, 115). The deacetylation reaction catalysed by sirtuins is NAD+-dependent, and leads to the formation of O-acetyl-ADP ribose (AADPR) which can be used as a donor group in ADP-ribosylation reactions (116). In term of activity, all the above-mentioned sirtuins display a deacetylase activity with the exception of SIRT4 which is mostly an ADP-ribosyl transferase, and SIRT6 which exhibits both activities (104).
Available evidence indicates that sirtuins act as pivotal regulators of life span and life-extending effects of calorie restriction (2). Among the different sirtuins, SIRT3 is particularly active in the mitochondria, where it is responsible for the deacetylation of the acetyl-CoA synthase enzyme (AceCS2) (117, 118). Under appropriate nutritional conditions, AceCS2 is completely inactivated upon acetylation at Lys-642, while it is rapidly reactivated by SIRT3 deacetylation (117). Deacetylation of AceCS2 by SIRT3 increases AceCS2 activity leading to the formation of O-acetyl-ADP-ribose and nicotinamide (118), important metabolites implicated in biosynthetic and regulatory purposes (119). In line with these studies, genetic deletion of SIRT3 in mice or gene downregulation as the result of high fat diet feeding, are associated with early metabolic abnormalities which are mainly the result of mitochondrial dysfunction (120). SIRT3 also regulates mitochondrial oxidative stress levels by deacetylation of the antioxidant enzyme MnSOD (121). Although not localized in the mitochondria, SIRT1 is a major regulator of mitochondrial function via deacetylation of PGC1α and FOXOs proteins (122). SIRT1-mediated activation of these target proteins leads to increased mitochondrial respiration and lipid oxidation through regulation of several genes (i.e., ERRα, IDH3, Cyt-c, COXV, MCAD, CPT-1β, and PDK4) required in energy-depleted cell (29, 123). SIRT1 is also critically involved in a dynamic cross-talk with AMPK, a key molecular effector involved in cellular metabolism. Activation of SIRT1/AMPK by physical activity or caloric restriction is associated with an increased usage of lipids as an energy source, mitochondrial biogenesis as well as with an increased expression of nicotinamide phosphoribosyl-transferase (Nampt), the rate-limiting enzyme in NAD+ bio-synthesis. The increase in Nampt activity leads to higher NAD+ production, which in turn activates SIRT1 (124).
Of note, activation of AMPK by SIRT1 seems to be particularly important for the phosphorylation of three main proteins involved in epigenetic remodeling: the DNA methyltransferase DNMT1, the histone acetyltransferase HAT1, and RBBP7, which inhibits DNMT1 and is a HAT1 coactivator (125). AMPK-mediated phosphorylation of these proteins triggered nucleosome remodeling thus favoring the transcription of nuclear-encoded genes involved in mitochondrial biogenesis and function (125). These results show that SIRT1-AMPK axis coordinates mitochondrial function with energy status through epigenetic regulation of nuclear gene expression. SIRT1 is also highly sensitive to the cellular redox state, and confers cardioprotection by counteracting oxidative stress through deacetylation of multiple cellular targets (126–128). In the human endothelium, SIRT1 antagonizes H2O2-induced premature senescence through its negative modulation of p53 by deacetylation of Lys-373, Lys-382, and Lys-320 (129). Conversely, endothelial SIRT1 overexpression reversed oxidative stress-induced premature senescence through activation of endothelial nitric oxide synthase (eNOS) (130). SIRT1 has also shown to deacetylate FOXO3 thus preventing cellular apoptosis via a mechanism involving the tumor suppressor p53 (131, 132). On the other hand, ROS-dependent acetylation of FOXO1 inhibits its transcriptional activity on SIRT1, catalase (CAT), and MnSOD target genes thus creating a detrimental vicious cycle driven by oxidative stress (133). This molecular circuitry is reinforced by the activation of the mitochondrial adaptor p66Shc which further amplifies ROS levels (134). Interestingly, SIRT1 controls mitochondrial oxidative stress by regulating the transcription of p66Shc (135–137). SIRT1-dependent deacetylation of histone 3 reduces chromatin accessibility on p66Shc promoter thus impeding transcription. By contrast, SIRT1 downregulation as the results of cardiovascular risk factors induces an open chromatin eventually leading to p66Shc expression, mitochondrial oxidative stress and endothelial dysfunction (138). It has also been shown that SIRT1 overexpression increases mitochondrial biogenesis and expression of antioxidant enzymes, namely catalase and glutathione peroxidase (GPx), via activation of the peroxisome proliferator-activated receptor coactivator (PPAR) 1-a activation (139).
SIRT5, a weak deacetylase with strong desuccinylase, demalonylase, and deglutarylase activity, has been also implicated in regulating different aspects of mitochondrial metabolism and cardiovascular function (140). SIRT5 downregulation was recently associated with mitochondrial dysfunction in endothelial progenitor cells of patients with arterial hypertension (141). Other studies reported that SIRT5 deficiency exert a protective role by suppressing mitochondrial ATP production and promoting AMPK activation in response to energy stress. Moreover, genetic deletion of SIRT5 protects against ischemic stroke via modulation of PI3K/Akt pathway (142).
Recent evidence suggests that in the diseased aorta containing atherosclerotic plaques and grafted arteriosclerosis, REF1/H3K9me3 pathway is suppressed thus leading to an increase in the mitochondrial translocation of the AIP1B isoform with subsequent generation of mitochondrial ROS and EC activation (143).
Mitochondrial-generated ROS have a major impact on DNA methylation. ROS can directly convert 5-methylcytosin (5mC) to 5-hydroxymethylcytosine (5hmC) which blocks the activity of DNMT1 leading to an improper methylation inheritance during mitosis and global hypomethylation (144). Moreover ROS can oxidize guanosine to 8-oxo-20-deoxyguanosine (8-oxodG) thus inhibiting methylation of adjacent cytosine and further contributing to global hypomethylation of DNA (145, 146). The formation of 8-oxodG in particular loci promotes the transcription of pro-inflammatory genes in response to TNF-α (147). Furthermore, 8-oxodG interacts with HIF1α thus affecting its ability to bind VEGF promoter with subsequent impairment of angiogenesis (148). In line with these observations, two recent meta-analyses showed that high levels of 8-oxodG are associated with atherosclerotic vascular disease and predict outcome (149, 150). High ROS levels also influence both repressive (H3K9me2/3 and H3K27me3) and active histone marks (H3K4me2/3) (151, 152).
Similarly to DNA methylation, histone methylation is dependent on SAM availability and is therefore reduced in the presence of high ROS levels (153, 154). In support of this hypothesis in a model of cardiac pressure overload the SET and MYND domain containing protein 1 (SMYD1) methyltransferase was significantly downregulated (155). On the other hand, several studies showed that hyperglycemia-induced oxidative stress increases the expression of the methyltransferase SETD7 and its epigenetic marker H3K4m eventually leading to enhanced transcription of inflammatory and oxidant genes, thus generating a vicious cycle (Figure 3) (156).
By serving as essential cofactors for most chromatin-modifying enzymes, important intermediates of cell metabolism and dietary intake allow the integration of metabolic information and transcriptional control (Figure 4). Fluctuating metabolite concentrations are therefore proposed to provide signalling cues for continual adjustment of gene expression by modulating the epigenome to influence chromatin dynamics. Additional biochemical evidence suggests that energy metabolite concentration could affect PTMs of the chromatin-modifying machinery itself, in turn regulating enzymatic activity, stability, and chromatin binding capacity associated with gene expression (54).
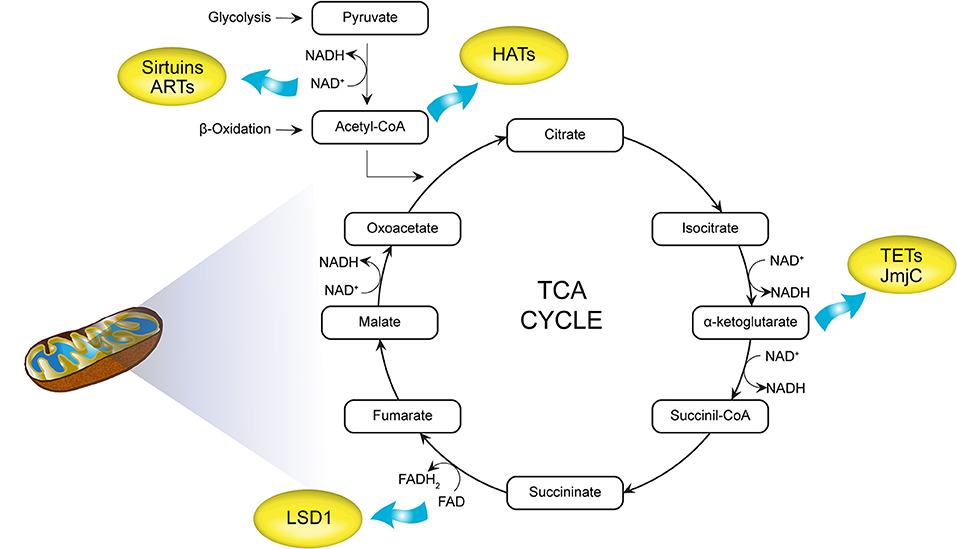
Figure 4. Intermediate mitochondrial metabolites as cofactors for chromatin modifications. acetyl-CoA generated by glycolysis and β-oxidation acts as a substrate for histone acetyltransferases (HATs). Nicotinamide adenine dinucleotide (NAD+) is required for histone deacetylases (HDACs; histone deacetylation) as well as ADP-ribosyltransferases (ARTs). α-Ketoglutarate and flavin adenine dinucleotide (FAD+) are cofactors for DNA (ten-eleven translocations, TETs) and histone demethylases [Jumonji C domain containing (JmjC), LSD1]. TCA, tricarboxylic acid cycle; HATs, histone acetyltransferases; NAD+, nicotinamide adenine dinucleotide; HDACs, histone deacetylases; ARTs, ADP-ribosyltransferases; FAD, flavin adenine dinucleotide; TETs, ten-eleven translocations.
NAD+ is an essential cofactor for reactions catalysed by the highly conserved SIRT HDAC family (2). Other NAD+ consuming enzymes such as ADP-ribosyltransferases have also been shown to covalently ADP-ribosylate core histones (157). PAR polymerases (PARPs) utilize NAD+ to catalyse poly(ADPribose) synthesis and are involved in the cellular stress response (158). Poly(ADP-ribose) polymerase-1 (PARP1), a major member of the PARP family, is a nuclear protein involved in chromatin remodeling and promotion of DNA repair (159). However, several studies report that in condition of oxidative stress PARP-1 also localizes to mitochondria (160–162). Mitochondrial PARP-1 is reported to actively participate in maintenance of functional integrity of the organelles (163) and to play a detrimental role when hyperactivated (160, 164). Furthermore, the potential role of PARP1 as a nuclear epigenetic regulator for the maintenance of mitochondrial DNA integrity has been suggested (159). Indeed, PARP-1 suppression reduces mtDNA integrity, as well as the expression of mitochondria-encoded respiratory complex subunits COX-1, COX-2, and ND-2 (164). Accordingly, PARP-1 localizes at promoters of nuclear genes encoding both the mtDNA repair proteins UNG1, MYH1, and APE1 and the mtDNA transcription factors TFB1M and TFB2M (164). Consistent with these findings, PARP-1 suppression impairs mitochondrial ATP production (164).
S-Adenosylmethionine (SAM) is produced by the condensation of methionine and ATP during the first of nine steps required for the conversion of methionine to succinyl-CoA, a predominantly cytoplasmic pathway that ends up in the mitochondria (29). It contains the active methyl-donor group utilized by most methyltransferase enzymes. It has been demonstrated that ROS can reduce SAM availability, thus limiting the activity of DNA and histone methyltransferases (145). This is achieved either by inhibiting methionine adenosyl-transferase and thus SAM synthesis or by inhibiting methionine synthase and thus methionine regeneration (56). Interestingly, long-term exposure to H2O2 decreased SAM levels leading to hypomethylation of the long interspersed nuclear element-1 (LINE-1) (165). LINE-1 hypomethylation as an indicator of global methylation status was found in blood from patients with ischaemic heart disease and stroke, and has been related to higher risk for these diseases (166).
Derived from the vitamin riboflavin (vitamin B2), mitochondrial-generated FAD functions as the prosthetic group for certain oxidation–reduction enzymes (2). For example, LSD1 demethylase is a FAD+-dependent enzyme capable of demethylating H3K4me1/2 and H3K9me1/2 (167). LSD1 activity is regulated by redox state and it is stimulated when FAD is oxidized (168). LSD1, in turn, regulates mitochondrial respiration and energy expenditure. Specifically, LSD1 binds directly to genes such as PGC1α, PDK4, FATP1, and adipose triacylglycerol lipase (ATGL), and represses their transcription associated with loss of H3K4 methylation (169).
The ketone body β-hydroxybutyrate (βOHB) modulates several signalling pathways with implications for metabolic disease and diabetes (170). Prolonged fasting, calorie restriction, strenuous exercise, or ketogenic diets are conditions associated with increases in serum concentrations β-OHB (171). Interestingly, βOHB is an endogenous inhibitor of many NAD+-independent HDACs (172). HDAC inhibition by βOHB might affect the pathogenesis of type 2 diabetes in at least two ways: through direct regulation of HDAC-dependent glucose metabolism, or by promoting resistance to oxidative stress (170). For examples, βOHB-mediated inhibition of HDAC1 and HDAC2 increases acetylation of histone H3K9 and H3K14 and establishes a permissive chromatin configuration for the expression of Foxo3 with subsequent transcription of its downstream antioxidant genes such as catalase and MnSOD (172). Similarly, βOHB may have similar effects on mitochondrial function, glucose homeostasis, and obesity through endogenous inhibition of HDAC3. The mechanism for these metabolic benefits of class I HDAC inhibition may be the upregulation of PGC1α in a variety of tissues (173, 174). Transcription of FGF21 is similarly upregulated via βOHB-mediated inhibition of HDAC3 which results in the activation of ketogenesis in obese mice (175). The microvascular and macrovascular complications of type 2 diabetes are thought to be due in part to increased oxidative stress brought on through several pathways including polyols, protein kinase C, hexosamine, and advanced glycosylation end products (176). In this context, the emerging role of βOHB in suppressing oxidative stress may be relevant for the management of diabetic complications. Other studies have previously suggested a role for both βOHB and HDAC inhibitors in the protection from oxidative or ischemic stress (170).
Connections between metabolic cofactors and enzymes associated with the removal of epigenetic methyl modifications are also emerging (54). The TET family of dioxygenases mediate the oxidation of 5mC. The potential for the TET family (TET1/2/3) to regulate diverse physiological functions including metabolic signalling requires the TCA cycle metabolite α-KG, and this activity is inhibited by 2-hydroxyglutarate (2HG) (2) (Figure 4). This means that oxygen deficiency and disturbances in mitochondrial metabolism could affect the activation of TET enzymes and thus control DNA methylation (177). Hearts of mice exposed to high-fat diet (HFD) showed reduced levels of αKG and this observation was paralleled by a compromised TET1 function. Accordingly, an exogenous source of αKG restored the DNA demethylation cycle, glucose uptake, and insulin response (178).
Jumonji C domain-containing histone demethylases are α-KG-dependent (177). Although studies are yet to determine the TET-metabolism connection, mutations in isocitrate dehydrogenase genes are associated with reduced α-KG and elevated 2HG levels leading to genome-wide changes in histone and DNA methylation patterns (54).
The Jumonji C domain (JmjC) containing lysine demethylases (KDM) are the largest group, which can be divided to six subgroups (KDM2-7) depending on their chromatin interacting domains and substrate specificity (179). The activation of these enzymes is also dependent on the presence of α-KG. Therefore, disturbances in Krebs cycle function can affect histone methylation and gene expression (177).
Acetyl-CoA generated from glucose and fatty acid metabolism feeds into the TCA cycle to contribute to cellular energy supply. Importantly, acetyl-CoA is the essential acetyl group donor to lysine acetylation reactions and both pharmacological and genetic interventions that modify cellular acetyl-CoA concentrations directly affect acetylated proteins including histones (180). Because histone acetylation is ubiquitously associated with open chromatin and gene expression, acetyl-CoA links intermediary carbon metabolism with chromatin dynamics and transcription (54).
Targeting epigenetic modifications is a highly promising approach to restore gene expression and to rescue or prevent mitochondrial insufficiency and vascular dysfunction. There are several examples of how specific interventions can be employed to modify the landscape of DNA/histone modifications in this setting.
Studies in knockout mice have shown that class I HDACs play a key role in regulating metabolism. Chronic treatment with butyrate, a broad HDAC inhibitor that is expected to phenocopy HDAC3 loss-of-function, prevents metabolic alterations in diet-induced obese as well as in aged mice, mainly by enhancing oxidative phosphorylation and beta-oxidation in mitochondria (181, 182). Butyrate treatment also improves mitochondrial biogenesis via epigenetic modulation of PGC-1α as well as induction of several microRNAs such as miR-133a-3p, miR-208b, and miR-499-5p, implicated in the regulation of mitochondrial potential and integrity (183). Similarly, the class I HDAC inhibitor SAHA, but not a class II HDAC inhibitor, increases the expression of PGC-1α thus leading to enhanced mitochondrial biogenesis, oxygen consumption in adipose tissue and skeletal muscle from mice with type 2 diabetes (174). These changes were associated with a significant improvement of insulin sensitivity, metabolic rate and oxidative metabolism (174). Moreover, treatment with SAHA was also found to reduce ischemia-reperfusion injury following myocardial infarction and to prevent apoptosis in cultured myocytes subjected to hypoxia/reoxygenation (184, 185).
Pharmacological modulation of sirtuins has also shown to impact on mitochondrial functionality and vascular function (186). Although primarily known as a nuclear protein, SIRT1-mediated deacetylation of PGC-1α has been extensively implicated in metabolic control and mitochondrial biogenesis, which was proposed to partially underline SIRT1 role in caloric restriction and impacts on longevity. Moreover, recent evidence suggests that modulation of SIRT1 activity may also affect the turnover of defective mitochondria by mitophagy (187). In line with these evidences, SIRT1 activation by resveratrol improves vascular function while attenuating dyslipidaemia and obesity-induced metabolic alterations in human subjects (188). SIRT1-dependent improvement of flow-mediated dilation can be partially explained by increased deacetylation of p66Shc promoter as well as posttranslational and transcriptional regulation of endothelial NO synthase (eNOS) (137, 189). Indeed, SIRT1 inhibition significantly increases p66Shc transcription, mitochondrial oxidative stress and organelle disruption. Whereas, in both the diabetic vasculature and myocardium activation of SIRT1 suppresses p66Shc signalling thus preventing the accumulation of H2O2 in mitochondria and cellular death (137, 138, 190). Pharmacological activation of SIRT3 by small molecules, namely 7-hydroxy-3-(4′-methoxyphenyl) coumarin (C12), also represents a promising approach to prevent mitochondrial ROS via deacetylation and activation of MnSOD (121).
Together with SIRT1, other epigenetic modulators participate to the transcriptional regulation of the mitochondrial adaptor p66Shc. Modulation of CpG DNA methylation by folates regulates p66Shc transcription (138). Consistently, a recent work found that homocysteine stimulates p66Shc transcription in human endothelial cells via specific CpG dinucleotides demethylation in the p66Shc promoter (191). Of note, p66Shc promoter CpG methylation was significantly reduced in peripheral blood leukocytes of patients with coronary artery disease and high plasma homocysteine levels, thus strengthening the relevance of p66Shc-related epigenetic changes in the context of cardiovascular disease (191). Moreover, metformin, a widely used antidiabetic drug, was found to modulate SIRT1-p66Shc signaling in experimental models of diabetes (138, 192, 193).
Inhibitors of histone acetyltransferases have also shown to revert mitochondrial oxidative stress. The dietary compound curcumin, an inhibitor of the histone acetyltransferase CBP/p300, has shown to rescue hyperglycemia-induced endothelial dysfunction by regulating the expression of several pro-oxidant and antioxidant enzymes involved in mitochondrial oxidative stress and mitochondrial biogenesis (194). Similarly, inhibition of another acetyltransferase, GCN5, prevents angiotensin II–mediated downregulation of catalase thus fostering accumulation of mitochondrial ROS (195).
In conclusion, evidence discussed so far strongly suggests that specific epigenetic signals are responsible for transcriptional changes leading to mitochondrial dysfunction and cardiovascular disease. In turn, the availability of mitochondrial intermediate metabolites controls the activation of chromatin modifying enzymes. The growing understanding of chromatin modifications and their impact on transcription, will open perspective for the development of personalized biomarkers and epigenetic therapies aimed at preventing mitochondrial dysfunction and cardiovascular disease.
SM, SA, FP, and SC drafted the manuscript and prepared the graphical illustrations. TL revised the manuscript and figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors FP.
FP is the recipient of a H.H. Sheikh Khalifa bin Hamad Al Thani Foundation Assistant Professorship at the Faculty of Medicine, University of Zurich. This work was supported by the Zürich Heart House, the Swiss Heart Foundation, Swiss Life Foundation, Kurt und Senta-Hermann Stiftung, the EMDO Stiftung and the Schweizerische Diabetes-Stiftung (to FP); the Holcim Foundation and the Swiss Heart Foundation (to SC).
1. Ogawa K, Miura T. Aphid polyphenisms: trans-generational developmental regulation through viviparity. Front Physiol. (2014) 5:1. doi: 10.3389/fphys.2014.00001
2. Aon MA, Cortassa S, Juhaszova M, Sollott SJ. Mitochondrial health, the epigenome and healthspan. Clin Sci. (2016) 130:1285–305. doi: 10.1042/CS20160002
3. Chandel NS. Mitochondria as signaling organelles. BMC Biol. (2014) 12:34. doi: 10.1186/1741-7007-12-34
4. Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. (2012) 16:9–17. doi: 10.1016/j.cmet.2012.06.001
5. Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. (2010) 10:12–31. doi: 10.1016/j.mito.2009.09.006
6. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. (2000) 192:1001–14. doi: 10.1084/jem.192.7.1001
7. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
8. Chen Y, Liu Y, Dorn GW II. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. (2011) 109:1327–31. doi: 10.1161/CIRCRESAHA.111.258723
9. Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. (2014) 16:495–501. doi: 10.1038/ncb2979
10. Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol. (2018) 19:93–108. doi: 10.1038/nrm.2017.129
11. Um JH, Yun J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. (2017) 50:299–307. doi: 10.5483/BMBRep.2017.50.6.056
12. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. (2003) 299:896–9. doi: 10.1126/science.1079368
13. Dominy JE Jr, Lee Y, Gerhart-Hines Z, Puigserver P. Nutrient-dependent regulation of PGC-1alpha's acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta. (2010) 1804:1676–83. doi: 10.1016/j.bbapap.2009.11.023
14. Patten IS, Arany Z. PGC-1 coactivators in the cardiovascular system. Trends Endocrinol Metab. (2012) 23:90–7. doi: 10.1016/j.tem.2011.09.007
15. Barth E, Stammler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. (1992) 24:669–81. doi: 10.1016/0022-2828(92)93381-S
16. Kadlec AO, Chabowski DS, Ait-Aissa K, Gutterman DD. Role of PGC-1alpha in vascular regulation: implications for atherosclerosis. Arterioscler Thromb Vasc Biol. (2016) 36:1467–74. doi: 10.1161/ATVBAHA.116.307123
17. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. (2013) 112:1171–1188. doi: 10.1161/CIRCRESAHA.111.300233
18. Craige SM, Kroller-Schon S, Li C, Kant S, Cai S, Chen K, et al. PGC-1alpha dictates endothelial function through regulation of eNOS expression. Sci Rep. (2016) 6:38210. doi: 10.1038/srep38210
19. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. (2011) 124:444–53. doi: 10.1161/CIRCULATIONAHA.110.014506
20. Haslip M, Dostanic I, Huang Y, Zhang Y, Russell KS, Jurczak MJ, et al. Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arterioscler Thromb Vasc Biol. (2015) 35:1166–78. doi: 10.1161/ATVBAHA.114.304865
21. Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J Cell Sci. (2010) 123:917–26. doi: 10.1242/jcs.059246
22. Mai S, Muster B, Bereiter-Hahn J, Jendrach M. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy. (2012) 8:47–62. doi: 10.4161/auto.8.1.18174
23. Wu NN, Zhang Y, Ren J. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid Med Cell Longev. (2019) 2019:9825061. doi: 10.1155/2019/9825061
24. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. (2005) 120:483–95. doi: 10.1016/j.cell.2005.02.001
25. Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. (1972) 20:145–7. doi: 10.1111/j.1532-5415.1972.tb00787.x
26. Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. (2007) 47:143–83. doi: 10.1146/annurev.pharmtox.47.120505.105122
27. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. (2003) 552:335–44. doi: 10.1113/jphysiol.2003.049478
28. Aon MA, Cortassa S, O'Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. (2010) 1797:865–77. doi: 10.1016/j.bbabio.2010.02.016
29. Kaelin WG Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. (2013) 153:56–69. doi: 10.1016/j.cell.2013.03.004
30. Akar FG, Aon MA, Tomaselli GF, O'Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. (2005) 115:3527–35. doi: 10.1172/JCI25371
31. Yang KC, Kyle JW, Makielski JC, Dudley SC Jr. Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res. (2015) 116:1937–55. doi: 10.1161/CIRCRESAHA.116.304691
32. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. (2000) 404:787–90. doi: 10.1038/35008121
33. Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal. (2011) 15:1517–30. doi: 10.1089/ars.2010.3642
34. Chen F, Haigh S, Barman S, Fulton DJ. From form to function: the role of Nox4 in the cardiovascular system. Front Physiol. (2012) 3:412. doi: 10.3389/fphys.2012.00412
35. Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, et al. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res. (2010) 106:193–202. doi: 10.1161/CIRCRESAHA.109.198366
36. Poon CC, Seto SW, Au AL, Zhang Q, Li RW, Lee WY, et al. Mitochondrial monoamine oxidase-A-mediated hydrogen peroxide generation enhances 5-hydroxytryptamine-induced contraction of rat basilar artery. Br J Pharmacol. (2010) 161:1086–98. doi: 10.1111/j.1476-5381.2010.00941.x
37. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
38. Cosentino F, Francia P, Camici GG, Pelicci PG, Luscher TF, Volpe M. Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arterioscler Thromb Vasc Biol. (2008) 28:622–8. doi: 10.1161/ATVBAHA.107.156059
39. Francia P, delli Gatti C, Bachschmid M, Martin-Padura I, Savoia C, Migliaccio E, et al. Deletion of p66Shc gene protects against age-related endothelial dysfunction. Circulation. (2004) 110:2889–95. doi: 10.1161/01.CIR.0000147731.24444.4D
40. Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, et al. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res. (2012) 111:278–289. doi: 10.1161/CIRCRESAHA.112.266593
41. Paneni F, Costantino S, Krankel N, Cosentino F, Luscher TF. Reprogramming ageing and longevity genes restores paracrine angiogenic properties of early outgrowth cells. Eur Heart J. (2016) 37:1733–7. doi: 10.1093/eurheartj/ehw073
42. Paneni F, Costantino S, Castello L, Battista R, Capretti G, Chiandotto S, et al. Targeting prolyl-isomerase Pin1 prevents mitochondrial oxidative stress and vascular dysfunction: insights in patients with diabetes. Eur Heart J. (2015) 36:817–28. doi: 10.1093/eurheartj/ehu179
43. Costantino S, Paneni F, Luscher TF, Cosentino F. Pin1 inhibitor Juglone prevents diabetic vascular dysfunction. Int J Cardiol. (2016) 203:702–7. doi: 10.1016/j.ijcard.2015.10.221
44. Queliconi BB, Wojtovich AP, Nadtochiy SM, Kowaltowski AJ, Brookes PS. Redox regulation of the mitochondrial K(ATP) channel in cardioprotection. Biochim Biophys Acta. (2011) 1813:1309–15. doi: 10.1016/j.bbamcr.2010.11.005
45. Brown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol. (2007) 27:1941–6. doi: 10.1161/ATVBAHA.107.146852
46. Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, et al. Mitochondrial integrity and function in atherogenesis. Circulation. (2002) 106:544–9. doi: 10.1161/01.CIR.0000023921.93743.89
47. Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation. (2003) 108:1912–6. doi: 10.1161/01.CIR.0000093660.86242.BB
48. Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. (2005) 66:562–73. doi: 10.1016/j.cardiores.2005.01.026
49. Widder JD, Fraccarollo D, Galuppo P, Hansen JM, Jones DP, Ertl G, et al. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension. (2009) 54:338–44. doi: 10.1161/HYPERTENSIONAHA.108.127928
50. Kizhakekuttu TJ, Wang J, Dharmashankar K, Ying R, Gutterman DD, Vita JA, et al. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol. (2012) 32:2531–9. doi: 10.1161/ATVBAHA.112.256024
51. Costantino S, Paneni F, Virdis A, Hussain S, Mohammed SA, Capretti G, et al. Interplay among H3K9-editing enzymes SUV39H1, JMJD2C and SRC-1 drives p66Shc transcription and vascular oxidative stress in obesity. Eur Heart J. (2019) 40:383–91. doi: 10.1093/eurheartj/ehx615
52. Kowluru RA. Mitochondrial stability in diabetic retinopathy: lessons learned from epigenetics. Diabetes. (2019) 68:241–7. doi: 10.2337/dbi18-0016
53. Costantino S, Libby P, Kishore R, Tardif JC, El-Osta A, Paneni F. Epigenetics and precision medicine in cardiovascular patients: from basic concepts to the clinical arena. Eur Heart J. (2018) 39:4150–8. doi: 10.1093/eurheartj/ehx568
54. Keating ST, El-Osta A. Epigenetics and metabolism. Circ Res. (2015) 116:715–36. doi: 10.1161/CIRCRESAHA.116.303936
55. Costantino S, Mohammed SA, Ambrosini S, Paneni F. Epigenetic processing in cardiometabolic disease. Atherosclerosis. (2019) 281:150–8. doi: 10.1016/j.atherosclerosis.2018.09.029
56. Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal. (2011) 15:551–89. doi: 10.1089/ars.2010.3492
57. Delatte B, Deplus R, Fuks F. Playing TETris with DNA modifications. EMBO J. (2014) 33:1198–211. doi: 10.15252/embj.201488290
58. Costantino S, Paneni F, Cosentino F. Targeting chromatin remodeling to prevent cardiovascular disease in diabetes. Curr Pharm Biotechnol. (2015) 16:531–43. doi: 10.2174/138920101606150407113644
59. Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. (2011) 123:2145–56. doi: 10.1161/CIRCULATIONAHA.110.956839
60. Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. (2010) 466:253–257. doi: 10.1038/nature09165
61. Costantino S, Ambrosini S, Paneni F. The epigenetic landscape in the cardiovascular complications of diabetes. J Endocrinol Invest. (2019) 42:505–11. doi: 10.1007/s40618-018-0956-3
62. Prasher D, Greenway SC, Singh RB. The impact of epigenetics on cardiovascular disease. Biochem Cell Biol. (2020) 98:12-22. doi: 10.1139/bcb-2019-0045
63. Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. (2007) 213:384–90. doi: 10.1002/jcp.21224
64. Matouk CC, Marsden PA. Epigenetic regulation of vascular endothelial gene expression. Circ Res. (2008) 102:873–87. doi: 10.1161/CIRCRESAHA.107.171025
65. Izquierdo AG, Crujeiras AB. Role of epigenomic mechanisms in the onset and management of insulin resistance. Rev Endocr Metab Disord. (2019) 20:89–102. doi: 10.1007/s11154-019-09485-0
66. Vilkaitis G, Suetake I, Klimasauskas S, Tajima S. Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J Biol Chem. (2005) 280:64–72. doi: 10.1074/jbc.M411126200
67. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. (1999) 99:247–57. doi: 10.1016/S0092-8674(00)81656-6
68. Aavik E, Babu M, Yla-Herttuala S. DNA methylation processes in atheosclerotic plaque. Atherosclerosis. (2019) 281:168–79. doi: 10.1016/j.atherosclerosis.2018.12.006
69. Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. (2010) 11:607–20. doi: 10.1038/nrm2950
70. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. (2013) 502:472–9. doi: 10.1038/nature12750
71. Franchini DM, Schmitz KM, Petersen-Mahrt SK. 5-Methylcytosine DNA demethylation: more than losing a methyl group. Annu Rev Genet. (2012) 46:419–41. doi: 10.1146/annurev-genet-110711-155451
72. Jenuwein T, Allis CD. Translating the histone code. Science. (2001) 293:1074–80. doi: 10.1126/science.1063127
73. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128:693–705. doi: 10.1016/j.cell.2007.02.005
74. Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. (2007) 128:669–81. doi: 10.1016/j.cell.2007.01.033
75. Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. (2005) 6:838–49. doi: 10.1038/nrm1761
76. Nicorescu I, Dallinga GM, de Winther MPJ, Stroes ESG, Bahjat M. Potential epigenetic therapeutics for atherosclerosis treatment. Atherosclerosis. (2019) 281:189–97. doi: 10.1016/j.atherosclerosis.2018.10.006
77. Gillette TG, Hill JA. Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ Res. (2015) 116:1245–53. doi: 10.1161/CIRCRESAHA.116.303630
78. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. (2014) 13:337–56. doi: 10.1038/nrd4286
79. Cooper ME, El-Osta A. Epigenetics: mechanisms and implications for diabetic complications. Circ Res. (2010) 107:1403–1413. doi: 10.1161/CIRCRESAHA.110.223552
80. Baccarelli A, Ghosh S. Environmental exposures, epigenetics and cardiovascular disease. Curr Opin Clin Nutr Metab Care. (2012) 15:323–9. doi: 10.1097/MCO.0b013e328354bf5c
81. Carrozza MJ, Utley RT, Workman JL, Cote J. The diverse functions of histone acetyltransferase complexes. Trends Genet. (2003) 19:321–9. doi: 10.1016/S0168-9525(03)00115-X
82. Lavebratt C, Almgren M, Ekstrom TJ. Epigenetic regulation in obesity. Int J Obes. (2012) 36:757–65. doi: 10.1038/ijo.2011.178
83. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. (2006) 5:769–84. doi: 10.1038/nrd2133
84. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. (2009) 10:32–42. doi: 10.1038/nrg2485
85. Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. (2003) 983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x
86. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. (2011) 21:381–95. doi: 10.1038/cr.2011.22
87. Bannister AJ, Kouzarides T. Reversing histone methylation. Nature. (2005) 436:1103–6. doi: 10.1038/nature04048
88. Stratton MS, Farina FM, Elia L. Epigenetics and vascular diseases. J Mol Cell Cardiol. (2019) 133:148–63. doi: 10.1016/j.yjmcc.2019.06.010
89. Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. (2006) 125:467–81. doi: 10.1016/j.cell.2006.03.028
90. Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. (2007) 128:707–19. doi: 10.1016/j.cell.2007.01.015
91. Eskeland R, Eberharter A, Imhof A. HP1 binding to chromatin methylated at H3K9 is enhanced by auxiliary factors. Mol Cell Biol. (2007) 27:453–65. doi: 10.1128/MCB.01576-06
92. Thambirajah AA, Ng MK, Frehlick LJ, Li A, Serpa JJ, Petrotchenko EV, et al. MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain. Nucleic Acids Res. (2012) 40:2884–97. doi: 10.1093/nar/gkr1066
93. Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. (2003) 31:2305–12. doi: 10.1093/nar/gkg332
94. Iacobazzi V, Castegna A, Infantino V, Andria G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab. (2013) 110:25–34. doi: 10.1016/j.ymgme.2013.07.012
95. Wallace DC, Fan W. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev. (2009) 23:1714–36. doi: 10.1101/gad.1784909
96. Yan C, Duanmu X, Zeng L, Liu B, Song Z. Mitochondrial DNA: distribution, mutations, and elimination. Cells. (2019) 8:379. doi: 10.3390/cells8040379
97. Birky CW Jr. Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc Natl Acad Sci USA. (1995) 92:11331–8. doi: 10.1073/pnas.92.25.11331
98. Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, et al. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. (2003) 31:1640–5. doi: 10.1093/nar/gkg251
99. Maniura-Weber K, Goffart S, Garstka HL, Montoya J, Wiesner RJ. Transient overexpression of mitochondrial transcription factor A (TFAM) is sufficient to stimulate mitochondrial DNA transcription, but not sufficient to increase mtDNA copy number in cultured cells. Nucleic Acids Res. (2004) 32:6015–27. doi: 10.1093/nar/gkh921
100. van der Wijst MG, Rots MG. Mitochondrial epigenetics: an overlooked layer of regulation? Trends Genet. (2015) 31:353–6. doi: 10.1016/j.tig.2015.03.009
101. Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci USA. (2011) 108:3630–5. doi: 10.1073/pnas.1012311108
102. Pollack Y, Kasir J, Shemer R, Metzger S, Szyf M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. (1984) 12:4811–24. doi: 10.1093/nar/12.12.4811
103. Satoh M, Kuroiwa T. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp Cell Res. (1991) 196:137–40. doi: 10.1016/0014-4827(91)90467-9
104. Cosentino C, Mostoslavsky R. Metabolism, longevity and epigenetics. Cell Mol Life Sci. (2013) 70:1525–41. doi: 10.1007/s00018-013-1295-3
105. Dzitoyeva S, Chen H, Manev H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol Aging. (2012) 33:2881–91. doi: 10.1016/j.neurobiolaging.2012.02.006
106. Wong M, Gertz B, Chestnut BA, Martin LJ. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front Cell Neurosci. (2013) 7:279. doi: 10.3389/fncel.2013.00279
107. Manev H, Dzitoyeva S. Progress in mitochondrial epigenetics. Biomol Concepts. (2013) 4:381–9. doi: 10.1515/bmc-2013-0005
108. Byun HM, Panni T, Motta V, Hou L, Nordio F, Apostoli P, et al. Effects of airborne pollutants on mitochondrial DNA methylation. Part Fibre Toxicol. (2013) 10:18. doi: 10.1186/1743-8977-10-18
109. Mawlood SK, Dennany L, Watson N, Dempster J, Pickard BS. Quantification of global mitochondrial DNA methylation levels and inverse correlation with age at two CpG sites. Aging. (2016) 8:636–41. doi: 10.18632/aging.100892
110. Mishra M, Kowluru RA. DNA methylation-a potential source of mitochondria DNA base mismatch in the development of diabetic retinopathy. Mol Neurobiol. (2019) 56:88–101. doi: 10.1007/s12035-018-1086-9
111. Mishra M, Kowluru RA. Epigenetic modification of mitochondrial DNA in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. (2015) 56:5133–42. doi: 10.1167/iovs.15-16937
112. Bianchessi V, Vinci MC, Nigro P, Rizzi V, Farina F, Capogrossi MC, et al. Methylation profiling by bisulfite sequencing analysis of the mtDNA non-coding region in replicative and senescent endothelial cells. Mitochondrion. (2016) 27:40–7. doi: 10.1016/j.mito.2016.02.004
113. Baccarelli AA, Byun HM. Platelet mitochondrial DNA methylation: a potential new marker of cardiovascular disease. Clin Epigenetics. (2015) 7:44. doi: 10.1186/s13148-015-0078-0
114. Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. (2010) 35:669–75. doi: 10.1016/j.tibs.2010.07.003
115. Guarente L. Franklin H. Epstein lecture: sirtuins, aging, and medicine. N Engl J Med. (2011) 364:2235–44. doi: 10.1056/NEJMra1100831
116. Sauve AA, Celic I, Avalos J, Deng H, Boeke JD, Schramm VL. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry. (2001) 40:15456–63. doi: 10.1021/bi011858j
117. Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA. (2006) 103:10230–5. doi: 10.1073/pnas.0604392103
118. Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA. (2006) 103:10224–10229. doi: 10.1073/pnas.0603968103
119. Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. (2006) 70:789–829. doi: 10.1128/MMBR.00040-05
120. Hirschey MD, Shimazu T, Huang JY, Schwer B, Verdin E. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb Symp Quant Biol. (2011) 76:267–77. doi: 10.1101/sqb.2011.76.010850
121. Lu J, Zhang H, Chen X, Zou Y, Li J, Wang L, et al. A small molecule activator of SIRT3 promotes deacetylation and activation of manganese superoxide dismutase. Free Radic Biol Med. (2017) 112:287–97. doi: 10.1016/j.freeradbiomed.2017.07.012
122. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. (2005) 434:113–8. doi: 10.1038/nature03354
123. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. (2006) 127:1109–22. doi: 10.1016/j.cell.2006.11.013
124. Posavec M, Timinszky G, Buschbeck M. Macro domains as metabolite sensors on chromatin. Cell Mol Life Sci. (2013) 70:1509–24. doi: 10.1007/s00018-013-1294-4
125. Marin TL, Gongol B, Zhang F, Martin M, Johnson DA, Xiao H, et al. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci Signal. (2017) 10. doi: 10.1126/scisignal.aaf7478
126. Nadtochiy SM, Redman E, Rahman I, Brookes PS. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc Res. (2011) 89:643–9. doi: 10.1093/cvr/cvq287
127. Nadtochiy SM, Yao H, McBurney MW, Gu W, Guarente L, Rahman I, et al. SIRT1-mediated acute cardioprotection. Am J Physiol Heart Circ Physiol. (2011) 301:H1506–12. doi: 10.1152/ajpheart.00587.2011
128. Vinciguerra M, Santini MP, Martinez C, Pazienza V, Claycomb WC, Giuliani A, et al. mIGF-1/JNK1/SirT1 signaling confers protection against oxidative stress in the heart. Aging Cell. (2012) 11:139–49. doi: 10.1111/j.1474-9726.2011.00766.x
129. Kao CL, Chen LK, Chang YL, Yung MC, Hsu CC, Chen YC, et al. Resveratrol protects human endothelium from H(2)O(2)-induced oxidative stress and senescence via SirT1 activation. J Atheroscler Thromb. (2010) 17:970–9. doi: 10.5551/jat.4333
130. Ota H, Eto M, Kano MR, Ogawa S, Iijima K, Akishita M, et al. Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler Thromb Vasc Biol. (2008) 28:1634–9. doi: 10.1161/ATVBAHA.108.164368
131. Huang K, Yan ZQ, Zhao D, Chen SG, Gao LZ, Zhang P, et al. SIRT1 and FOXO mediate contractile differentiation of vascular smooth muscle cells under cyclic stretch. Cell Physiol Biochem. (2015) 37:1817–29. doi: 10.1159/000438544
132. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. (2004) 303:2011–5. doi: 10.1126/science.1094637
133. D'Onofrio N, Servillo L, Balestrieri ML. SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal. (2018) 28:711–732. doi: 10.1089/ars.2017.7178
134. Carlomosti F, D'Agostino M, Beji S, Torcinaro A, Rizzi R, Zaccagnini G, et al. Oxidative stress-induced miR-200c disrupts the regulatory loop among SIRT1, FOXO1, and eNOS. Antioxid Redox Signal. (2017) 27:328–44. doi: 10.1089/ars.2016.6643
135. Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, et al. The p66Shc adaptor protein controls oxidative stress response and life span in mammals. Nature. (1999) 402:309–13. doi: 10.1038/46311
136. Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science. (2007) 315:659–63. doi: 10.1126/science.1135380
137. Zhou S, Chen HZ, Wan YZ, Zhang QJ, Wei YS, Huang S, et al. Repression of p66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res. (2011) 109:639–48. doi: 10.1161/CIRCRESAHA.111.243592
138. Paneni F, Volpe M, Luscher TF, Cosentino F. SIRT1, p66(Shc), and Set7/9 in vascular hyperglycemic memory: bringing all the strands together. Diabetes. (2013) 62:1800–7. doi: 10.2337/db12-1648
139. Tan M, Tang C, Zhang Y, Cheng Y, Cai L, Chen X, et al. SIRT1/PGC-1alpha signaling protects hepatocytes against mitochondrial oxidative stress induced by bile acids. Free Radic Res. (2015) 49:935–45. doi: 10.3109/10715762.2015.1016020
140. Tang X, Chen XF, Chen HZ, Liu DP. Mitochondrial Sirtuins in cardiometabolic diseases. Clin Sci. (2017) 131:2063–78. doi: 10.1042/CS20160685
141. Yu BB, Zhi H, Zhang XY, Liang JW, He J, Su C, et al. Mitochondrial dysfunction-mediated decline in angiogenic capacity of endothelial progenitor cells is associated with capillary rarefaction in patients with hypertension via downregulation of CXCR4/JAK2/SIRT5 signaling. EBioMedicine. (2019) 42:64–75. doi: 10.1016/j.ebiom.2019.03.031
142. Diaz-Canestro C, Merlini M, Bonetti NR, Liberale L, Wust P, Briand-Schumacher S, et al. Sirtuin 5 as a novel target to blunt blood-brain barrier damage induced by cerebral ischemia/reperfusion injury. Int J Cardiol. (2018) 260:148–55. doi: 10.1016/j.ijcard.2017.12.060
143. Li Z, Li L, Zhang H, Zhou HJ, Ji W, Min W. Short AIP1 (ASK1-Interacting Protein-1) isoform localizes to the mitochondria and promotes vascular dysfunction. Arterioscler Thromb Vasc Biol. (2020) 40:112–27. doi: 10.1161/ATVBAHA.119.312976
144. Madugundu GS, Cadet J, Wagner JR. Hydroxyl-radical-induced oxidation of 5-methylcytosine in isolated and cellular DNA. Nucleic Acids Res. (2014) 42:7450–60. doi: 10.1093/nar/gku334
145. Kietzmann T, Petry A, Shvetsova A, Gerhold JM, Gorlach A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br J Pharmacol. (2017) 174:1533–54. doi: 10.1111/bph.13792
146. Le DD, Fujimori DG. Protein and nucleic acid methylating enzymes: mechanisms and regulation. Curr Opin Chem Biol. (2012) 16:507–15. doi: 10.1016/j.cbpa.2012.09.014
147. Pan L, Zhu B, Hao W, Zeng X, Vlahopoulos SA, Hazra TK, et al. Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase-1-mediated epigenetic regulation of nuclear factor kappaB-driven gene expression. J Biol Chem. (2016) 291:25553–66. doi: 10.1074/jbc.M116.751453
148. Pastukh V, Roberts JT, Clark DW, Bardwell GC, Patel M, Al-Mehdi AB, et al. An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L1367–75. doi: 10.1152/ajplung.00236.2015
149. Kroese LJ, Scheffer PG. 8-hydroxy-2'-deoxyguanosine and cardiovascular disease: a systematic review. Curr Atheroscler Rep. (2014) 16:452. doi: 10.1007/s11883-014-0452-y
150. Di Minno A, Turnu L, Porro B, Squellerio I, Cavalca V, Tremoli E, et al. 8-Hydroxy-2-deoxyguanosine levels and cardiovascular disease: a systematic review and meta-analysis of the literature. Antioxid Redox Signal. (2016) 24:548–55. doi: 10.1089/ars.2015.6508
151. Chervona Y, Costa M. The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic Biol Med. (2012) 53:1041–7. doi: 10.1016/j.freeradbiomed.2012.07.020
152. Niu Y, DesMarais TL, Tong Z, Yao Y, Costa M. Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med. (2015) 82:22–8. doi: 10.1016/j.freeradbiomed.2015.01.028
153. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. (2015) 22:861–73. doi: 10.1016/j.cmet.2015.08.024
154. Chisholm NC, Henderson ML, Selvamani A, Park MJ, Dindot S, Miranda RC, et al. Histone methylation patterns in astrocytes are influenced by age following ischemia. Epigenetics. (2015) 10:142–52. doi: 10.1080/15592294.2014.1001219
155. Liu T, Wu C, Jain MR, Nagarajan N, Yan L, Dai H, et al. Master redox regulator Trx1 upregulates SMYD1 & modulates lysine methylation. Biochim Biophys Acta. (2015) 1854:1816–22. doi: 10.1016/j.bbapap.2015.09.006
156. Paneni F, Costantino S, Battista R, Castello L, Capretti G, Chiandotto S, et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ Cardiovasc Genet. (2015) 8:150–8. doi: 10.1161/CIRCGENETICS.114.000671
157. Messner S, Altmeyer M, Zhao H, Pozivil A, Roschitzki B, Gehrig P, et al. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. (2010) 38:6350–62. doi: 10.1093/nar/gkq463
158. Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. (2012) 26:417–32. doi: 10.1101/gad.183509.111
159. Szczesny B, Brunyanszki A, Olah G, Mitra S, Szabo C. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: implications for the regulation of mitochondrial function. Nucleic Acids Res. (2014) 42:13161–73. doi: 10.1093/nar/gku1089
160. Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. (2003) 278:18426–433. doi: 10.1074/jbc.M301295200
161. Ivana Scovassi A, Diederich M. Modulation of poly(ADP-ribosylation) in apoptotic cells. Biochem Pharmacol. (2004) 68:1041–7. doi: 10.1016/j.bcp.2004.04.023
162. Pankotai E, Lacza Z, Muranyi M, Szabo C. Intra-mitochondrial poly(ADP-ribosyl)ation: potential role for alpha-ketoglutarate dehydrogenase. Mitochondrion. (2009) 9:159–64. doi: 10.1016/j.mito.2009.01.013
163. Rossi MN, Carbone M, Mostocotto C, Mancone C, Tripodi M, Maione R, et al. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J Biol Chem. (2009) 284:31616–24. doi: 10.1074/jbc.M109.025882
164. Lapucci A, Pittelli M, Rapizzi E, Felici R, Moroni F, Chiarugi A. Poly(ADP-ribose) polymerase-1 is a nuclear epigenetic regulator of mitochondrial DNA repair and transcription. Mol Pharmacol. (2011) 79:932–40. doi: 10.1124/mol.110.070110
165. Kloypan C, Srisa-art M, Mutirangura A, Boonla C. LINE-1 hypomethylation induced by reactive oxygen species is mediated via depletion of S-adenosylmethionine. Cell Biochem Funct. (2015) 33:375–85. doi: 10.1002/cbf.3124
166. Baccarelli A, Wright R, Bollati V, Litonjua A, Zanobetti A, Tarantini L, et al. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology. (2010) 21:819–28. doi: 10.1097/EDE.0b013e3181f20457
167. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119:941–53. doi: 10.1016/j.cell.2004.12.012
168. Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol. (2012) 227:3169–77. doi: 10.1002/jcp.24054
169. Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S, et al. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat Commun. (2012) 3:758. doi: 10.1038/ncomms1755
170. Newman JC, Verdin E. beta-hydroxybutyrate: much more than a metabolite. Diabetes Res Clin Pract. (2014) 106:173–81. doi: 10.1016/j.diabres.2014.08.009
171. Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. (2013) 502:489–98. doi: 10.1038/nature12752
172. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. (2013) 339:211–4. doi: 10.1126/science.1227166
173. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. (2009) 58:1509–17. doi: 10.2337/db08-1637
174. Galmozzi A, Mitro N, Ferrari A, Gers E, Gilardi F, Godio C, et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes. (2013) 62:732–42. doi: 10.2337/db12-0548
175. Li H, Gao Z, Zhang J, Ye X, Xu A, Ye J, et al. Sodium butyrate stimulates expression of fibroblast growth factor 21 in liver by inhibition of histone deacetylase 3. Diabetes. (2012) 61:797–806. doi: 10.2337/db11-0846
176. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. (2010) 107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545
177. Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A. Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process. Cell Signal. (2014) 26:1598–603. doi: 10.1016/j.cellsig.2014.03.030
178. Spallotta F, Cencioni C, Atlante S, Garella D, Cocco M, Mori M, et al. Stable oxidative cytosine modifications accumulate in cardiac mesenchymal cells from type2 diabetes patients: rescue by alpha-ketoglutarate and TET-TDG functional reactivation. Circ Res. (2018) 122:31–46. doi: 10.1161/CIRCRESAHA.117.311300
179. Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. (2012) 48:491–507. doi: 10.1016/j.molcel.2012.11.006
180. Shi L, Tu BP. Protein acetylation as a means to regulate protein function in tune with metabolic state. Biochem Soc Trans. (2014) 42:1037–42. doi: 10.1042/BST20140135
181. Hong J, Jia Y, Pan S, Jia L, Li H, Han Z, et al. Butyrate alleviates high fat diet-induced obesity through activation of adiponectin-mediated pathway and stimulation of mitochondrial function in the skeletal muscle of mice. Oncotarget. (2016) 7:56071–82. doi: 10.18632/oncotarget.11267
182. Walsh ME, Bhattacharya A, Sataranatarajan K, Qaisar R, Sloane L, Rahman MM, et al. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell. (2015) 14:957–70. doi: 10.1111/acel.12387
183. Zhang Y, Yu B, Yu J, Zheng P, Huang Z, Luo Y, et al. Butyrate promotes slow-twitch myofiber formation and mitochondrial biogenesis in finishing pigs via inducing specific microRNAs and PGC-1alpha expression1. J Anim Sci. (2019) 97:3180–92. doi: 10.1093/jas/skz187
184. Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. (2014) 129:1139–51. doi: 10.1161/CIRCULATIONAHA.113.002416
185. Yang J, He J, Ismail M, Tweeten S, Zeng F, Gao L, et al. HDAC inhibition induces autophagy and mitochondrial biogenesis to maintain mitochondrial homeostasis during cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. (2019) 130:36–48. doi: 10.1016/j.yjmcc.2019.03.008
186. Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J. (2015) 36:3404–12. doi: 10.1093/eurheartj/ehv290
187. Ou X, Lee MR, Huang X, Messina-Graham S, Broxmeyer HE. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells. (2014) 32:1183–94. doi: 10.1002/stem.1641
188. Pollack RM, Crandall JP. Resveratrol: therapeutic potential for improving cardiometabolic health. Am J Hypertens. (2013) 26:1260–8. doi: 10.1093/ajh/hpt165
189. Costantino S, Paneni F, Battista R, Castello L, Capretti G, Chiandotto S, et al. Impact of glycemic variability on chromatin remodeling, oxidative stress, and endothelial dysfunction in patients with type 2 diabetes and with target HbA1c levels. Diabetes. (2017) 66:2472–82. doi: 10.2337/db17-0294
190. Costantino S, Paneni F, Mitchell K, Mohammed SA, Hussain S, Gkolfos C, et al. Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66(Shc). Int J Cardiol. (2018) 268:179–86. doi: 10.1016/j.ijcard.2018.04.082
191. Kim CS, Kim YR, Naqvi A, Kumar S, Hoffman TA, Jung SB, et al. Homocysteine promotes human endothelial cell dysfunction via site-specific epigenetic regulation of p66shc. Cardiovasc Res. (2011) 92:466–75. doi: 10.1093/cvr/cvr250
192. Arunachalam G, Samuel SM, Marei I, Ding H, Triggle CR. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br J Pharmacol. (2014) 171:523–35. doi: 10.1111/bph.12496
193. de Kreutzenberg SV, Ceolotto G, Cattelan A, Pagnin E, Mazzucato M, Garagnani P, et al. Metformin improves putative longevity effectors in peripheral mononuclear cells from subjects with prediabetes. A randomized controlled trial. Nutr Metab Cardiovasc Dis. (2015) 25:686–93. doi: 10.1016/j.numecd.2015.03.007
194. de Oliveira MR, Jardim FR, Setzer WN, Nabavi SM, Nabavi SF. Curcumin, mitochondrial biogenesis, and mitophagy: Exploring recent data and indicating future needs. Biotechnol Adv. (2016) 34:813–26. doi: 10.1016/j.biotechadv.2016.04.004
Keywords: epigenetics, mitochondria, vascular disease, oxidative stress, endothelial function
Citation: Mohammed SA, Ambrosini S, Lüscher T, Paneni F and Costantino S (2020) Epigenetic Control of Mitochondrial Function in the Vasculature. Front. Cardiovasc. Med. 7:28. doi: 10.3389/fcvm.2020.00028
Received: 11 November 2019; Accepted: 19 February 2020;
Published: 04 March 2020.
Edited by:
Sebastiano Sciarretta, Sapienza University of Rome, ItalyReviewed by:
Shiyou Chen, University of Missouri, United StatesCopyright © 2020 Mohammed, Ambrosini, Lüscher, Paneni and Costantino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah Costantino, c2FyYWguY29zdGFudGlub0B1emguY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.