 Jenny E. Kanter
Jenny E. Kanter Cheng-Chieh Hsu
Cheng-Chieh Hsu Karin E. Bornfeldt
Karin E. Bornfeldt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 14 February 2020
Sec. Cardiovascular Metabolism
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.00010
This article is part of the Research Topic Diabetes Augmentation on Vascular Disease View all 8 articles
With the increasing prevalence of diabetes worldwide, vascular complications of diabetes are also on the rise. Diabetes results in an increased risk of macrovascular complications, with atherosclerotic cardiovascular disease (CVD) being the leading cause of death in adults with diabetes. The exact mechanisms for how diabetes promotes CVD risk are still unclear, although it is evident that monocytes and macrophages are key players in all stages of atherosclerosis both in the absence and presence of diabetes, and that phenotypes of these cells are altered by the diabetic environment. Evidence suggests that at least five pro-atherogenic mechanisms involving monocytes and macrophages contribute to the accelerated atherosclerotic lesion progression and hampered lesion regression associated with diabetes. These changes include (1) increased monocyte recruitment to lesions; (2) increased inflammatory activation; (3) altered macrophage lipid accumulation and metabolism; (4) increased macrophage cell death; and (5) reduced efferocytosis. Monocyte and macrophage phenotypes and mechanisms have been revealed mostly by different animal models of diabetes. The roles of specific changes in monocytes and macrophages in humans with diabetes remain largely unknown. There is an ongoing debate on whether the changes in monocytes and macrophages are caused by altered glucose levels, insulin deficiency or insulin resistance, lipid abnormalities, or combinations of these factors. Current research in humans and mouse models suggests that reduced clearance of triglyceride-rich lipoproteins and their remnants is one important mechanism whereby diabetes adversely affects macrophages and promotes atherosclerosis and CVD risk. Although monocytes and macrophages readily respond to the diabetic environment and can be seen as protagonists in diabetes-accelerated atherosclerosis, they are likely not instigators of the increased CVD risk.
To date, almost 10% of the US population has type 1 or type 2 diabetes mellitus (T1DM or T2DM), and the prevalence of diabetes continues to rise (1–4). The prevalence of diabetes worldwide is now approaching that of the US, with over 8% of people over the age of 18 having been diagnosed with diabetes. Diabetes is classically defined as hyperglycemia, such as fasting plasma glucose ≥7 mmol/L or glycated hemoglobin (HbA1c) ≥6.5% (5). Hyperglycemia develops largely as a result of impaired insulin production and/or insulin resistance, but dyslipidemia is often also present at varying degrees in patients with poor glycemic control. This is explained in part by insulin's non-glycemic effects, including its triglyceride-lowering actions (6). Thus, relative insulin deficiency or insulin resistance results in both elevated glucose levels and lipid levels.
Both forms of diabetes significantly increase the risk of atherosclerotic cardiovascular disease (CVD). Atherosclerotic lesion morphology appears to be similar in people with T1DM and T2DM based on post-mortem studies, and both forms of diabetes increase atherosclerosis disease burden, enlarge lesion necrotic cores and increase lesional macrophage content (based on CD68 immunoreactivity), as compared with lesions from subjects without diabetes (7).
Improved glycemic control using intensive insulin therapy reduces mortality and the risk of CVD in people with T1DM (8) even after the effect of the intense insulin therapy on glucose control has waned, and over time, a significant proportion of the beneficial effects (known as the legacy effect) of intensive insulin therapy is mediated by control of lipid risk factors (9). In T2DM, improved glucose control is often not associated with protection against incident CVD, at least not in subjects with established CVD (10, 11). However, in a recent study, participants with T2DM who had been randomly assigned to intensive glucose control for 5.6 years had a lower risk of CVD events (than those who received standard therapy) during the period in which the glycated hemoglobin curves were separated. There was no evidence of a legacy effect or a mortality benefit with intensive glucose control (12). It is not known whether the effects of intense glucose control on CVD risk are due to direct effects of glucose in the artery wall.
The observed dyslipidemia associated with diabetes is characterized, not primarily by elevated low-density lipoprotein (LDL) cholesterol levels, but rather by elevated triglyceride-rich lipoproteins (TRLs)—very low-density lipoproteins (VLDL), chylomicrons, and their remnant lipoprotein particles (RLPs)—and by lower high-density lipoprotein (HDL) cholesterol and smaller denser LDL particles. This lipid profile, commonly referred to as diabetic dyslipidemia (13), is most often seen in subjects with T2DM, less well-controlled T1DM, or in subjects with T1DM with additional traits of metabolic syndrome (3).
Furthermore, recent studies suggest that apolipoprotein C3 (APOC3), an apolipoprotein that prevents clearance of TRLs and their RLPs (14–18), predicts incident CVD in subjects with T1DM even when triglyceride levels are in the normal range or close to normal range (19, 20). The effect of APOC3 as a CVD risk factor was independent of glycemic control (HbA1c levels). Therefore, changes in lipids are likely to be critical in promoting CVD risk in both T1DM and T2DM. It is, however, possible that hyperglycemia acts in concert with the lipid changes. For example, it has been suggested that hyperglycemia, due to increased formation of advanced glycation end products, leads to cross-linking of extracellular matrix molecules and arterial stiffening (21, 22), which could, in turn, exacerbate the atherogenic process driven by other factors, such as lipids. Furthermore, when present, risk factors for CVD identified in the general population, such as smoking and elevated blood pressure, exacerbate CVD risk also in people with diabetes.
The metabolic hallmarks of diabetes discussed above affect multiple cell types in the artery wall, promoting pro-atherogenic processes and progression of atherosclerosis. In particular, diabetes results in changes in the myeloid cell compartment, which includes monocytes and macrophages. In this review, we will discuss diabetes-induced alterations in monocyte and macrophage phenotypes and how these alterations might contribute to the increased CVD burden in diabetes.
Monocytes and macrophages play essential roles in all stages of atherosclerosis (23), both in the presence and absence of diabetes (24). Initially, monocytes enter the subendothelial space in susceptible arteries in response to the retention of apolipoprotein B (APOB)-containing lipoproteins (25), which bind through positive charges in APOB to negatively charged extracellular matrix molecules, primarily glycosaminoglycans (26). The accumulation of monocytes in the artery wall and subsequent maturation of these cells to macrophages propagate a chronic, non-resolving low-grade arterial inflammation (27, 28). Macrophages also contribute to advanced lesions; macrophage death is largely responsible for formation and expansion of necrotic cores in advanced lesions (29). Lesions of atherosclerosis can regress in response to aggressive lipid lowering. Lesion regression is accompanied by a reduced abundance of lesional macrophages, rendering the lesion more fibrotic and likely more stable, and a pro-resolving “M2-like” macrophage phenotype (30, 31).
Early studies of the effects of diabetes on macrophages and other immune cells were pursued in part because of the interest in pancreatic immune cells contributing to diabetes development. Some of these studies suggested that diabetes suppresses macrophage functions, such as phagocytosis (32) and migration (33) in animal models of diabetes and in humans. Later studies indicated an increased activation of macrophages in diabetes (34). Different macrophage isolation methods and differences in macrophage populations might explain these discrepant findings.
Both mouse and human studies implicate macrophages as a key cell type in atherosclerosis associated with diabetes (35–37). Thus, autopsy and coronary atherectomy samples from humans have shown that lesions from people with diabetes are enriched in macrophages, as compared with specimens from patients without diabetes (38). Studies of mouse and porcine models of diabetes have revealed that arterial accumulation of glycosaminoglycans and accumulation of macrophages in early fatty streak lesions are accelerated by diabetes (39–42), consistent with an acceleration of monocyte recruitment and macrophage accumulation according to the response-to-retention hypothesis (43). Other mouse models have shown that diabetes hinders the regression of lesions through a process dependent on monocyte recruitment and skewing of the phenotype toward a more inflammatory lesional macrophage (36, 44–47). Diabetes also promotes progression of more advanced lesions in mouse and porcine models through mechanisms related to changes in macrophage abundance and phenotypes (19, 45, 46, 48–51). Thus, monocytes and macrophage are altered by the diabetic environment, and may be critical mediators of the proatherogenic effects of diabetes both in progressing and regressing lesions of atherosclerosis.
Elevated levels of circulating blood monocytes (monocytosis) can result in increased monocyte recruitment to lesions, thereby accelerating lesion progression, and can also impede atherosclerosis regression. Numerous animal studies support the idea that increased monocyte levels in circulation can result in increased monocyte recruitment to the artery wall (52–54). Hypercholesterolemia is a strong driver of hematopoiesis in mice, and the early myeloid precursors in bone marrow are especially sensitive to alterations in cholesterol homeostasis (55). Recently, Nagareddy et al. in the Goldberg, Fisher and Tall laboratories suggested that these bone marrow progenitor cells also respond to signals associated with hyperglycemia (44). Previous work from this group has shown that diabetes impairs atherosclerotic lesion regression in response to dramatic lipid-lowering (36). Nagareddy demonstrated that lowering blood glucose by using a sodium-glucose cotransporter 2 (SGLT2) inhibitor in mouse models of T1DM prevented diabetes-induced monocytosis, which in turn reduced monocyte recruitment to the artery wall and improved lesion regression in diabetic mice (44). Furthermore, this interesting study suggested that rather than glucose acting directly on the bone marrow hematopoietic progenitor cell compartment, neutrophil-derived S100A8/S100A9 stimulated myelopoiesis by activating the receptor for advanced glycation end products (RAGE) on bone marrow progenitor cells (44). S100A8 and S100A9 are damage-associated molecular pattern proteins released from neutrophils and monocytes (56). S100A8 and S100A9 form dimers and exhibit both intracellular and extracellular functions. Extracellularly, S100A8/S100A9 is believed to activate RAGE or the lipopolysaccharide (LPS) receptor toll-like receptor 4 (TLR4). Subsequent studies demonstrated that the increase in neutrophil S100A8/S100A9 release in diabetic mice also causes reticulated thrombocytosis by activating RAGE on hepatic Kupffer cells, resulting in increased interleukin-6 (IL-6) production and, through increased thrombopoietin production, thrombocytosis (57). This pathway was shown to contribute to diabetes-accelerated atherosclerosis. The stimulatory effect of diabetes on thrombocytosis was also prevented by SGLT2 inhibition, suggesting that SGLT2 inhibitors prevent both diabetes-induced monocytosis and thrombocytosis in these mouse models. The same group inferred a non-glucose mediated mechanism whereby obesity/T2DM could stimulate monocytosis (58). In the obese mouse models investigated, inflamed adipose tissue rather than neutrophils appears to be the primary source of S100A8/S100A9. S100A8/S100A9 prompts adipose tissue macrophages to release interleukin-1β (IL-1β) through a TLR4-dependent mechanism, which in turn stimulates myelopoiesis by acting on bone marrow progenitors (58).
Together, these studies demonstrate that monocytosis is associated with exacerbated atherosclerosis in diabetic mouse models and that the damage-associated molecular pattern molecules S100A8 and S100A9 play a critical role in this process. Similar mechanisms might be relevant in humans because plasma S100A8/S100A9 levels were shown to correlate with leukocyte counts and coronary artery disease in patients with T1DM (44) and to be increased in patients with T2DM (57). Furthermore, in a mouse model of T1DM, increased intraplaque hemorrhage in advanced lesions was shown to correlate with increased lesional S100A9 immunoreactivity (48). Very recently, diabetes-induced myelopoiesis, monocytosis, neutrophil production of S100A8/S100A9 and impaired lesion regression were found to be prevented by the increased levels of HDL in a human APOA1 transgenic mouse model (47), suggesting that diabetes-induced monocytosis and hampered lesion regression can be prevented by increasing cholesterol efflux from myeloid cells and their bone marrow progenitors.
However, diabetes is not consistently associated with monocytosis in mouse models, and monocytosis is not routinely shown to be present in people with diabetes. Furthermore, in a recent study, SGLT2 inhibition accelerated atherosclerosis regression in diabetic mice without altering circulating monocyte levels (59), demonstrating that SGLT2 inhibition does not act solely by reducing monocytosis in mouse models. In this study, SGLT2 inhibition prevented leukocyte adhesion to the artery wall as well as lesional macrophage proliferation, rather than affecting monocytosis. In mouse models, the ability of diabetes to induce monocytosis appears to be dependent on the diet. Thus, our studies have shown that hyperglycemic mouse models of T1DM fed a low-fat semi-purified diet do not exhibit monocytosis, yet show accelerated atherosclerosis (19, 60). In obese hyperlipidemic mouse models of T2DM or metabolic syndrome (pre-diabetes), on the other hand, monocytosis is consistently present and is likely to contribute to the accelerated atherosclerosis in these mice (61, 62). These findings are consistent with the observations that cholesterol promotes monocytosis by acting on bone marrow myeloid cell precursors (55) and that diabetes-induced myelopoiesis can be prevented by increasing cholesterol efflux through elevated HDL levels (47).
Together, these studies suggest that diabetes-induced monocytosis can contribute to, but is not necessary for, diabetes-accelerated atherosclerosis in mouse models. Thus, additional mechanisms are important in driving diabetes-accelerated atherosclerosis both in mouse models and in humans.
Monocytes enter peripheral tissues in response to tissue changes the body perceives as an injury, such as atherosclerosis. Once in tissues, these cells differentiate into macrophages. Macrophages are highly plastic cells that take on different phenotypes depending on their surrounding micro-environment. Their primary roles are to phagocytose pathogens and dead and dying cells, which contributes to healing of the perceived injury, and to produce chemokines and cytokines to call in additional leukocytes to aid in the protection of the organism when needed. Macrophages thereby contribute to both the pro-inflammatory and the pro-resolution phases of inflammation.
Many studies have demonstrated that diabetes causes monocytes to take on a more inflammatory phenotype, both in mouse models and in humans. For example, isolated CD14-positive human peripheral blood monocytes from subjects with T1DM or T2DM express and produce elevated levels of the pro-inflammatory cytokines IL-1β, tumor necrosis factor-α (TNF-α), and IL-6, as compared to monocytes isolated from control subjects (63–66). Likewise, blood monocytes from a mouse model of T1DM exhibit elevated levels of Tnfa mRNA (37). In addition to increased cytokine production under basal conditions, several groups have suggested that monocytes isolated from subjects with diabetes are “primed” to respond more robustly, as compared with monocytes isolated from controls, to inflammatory stimuli, such as LPS or interferon-γ. The increased expression of TLRs, such as TLR2 and TLR4, on the cell surface of monocytes from people with diabetes supports this hypothesis (63–65, 67). It is possible that the primed state of monocytes results in a heightened inflammatory activation once the monocytes encounter TLR ligands in the lesion (Figure 1).
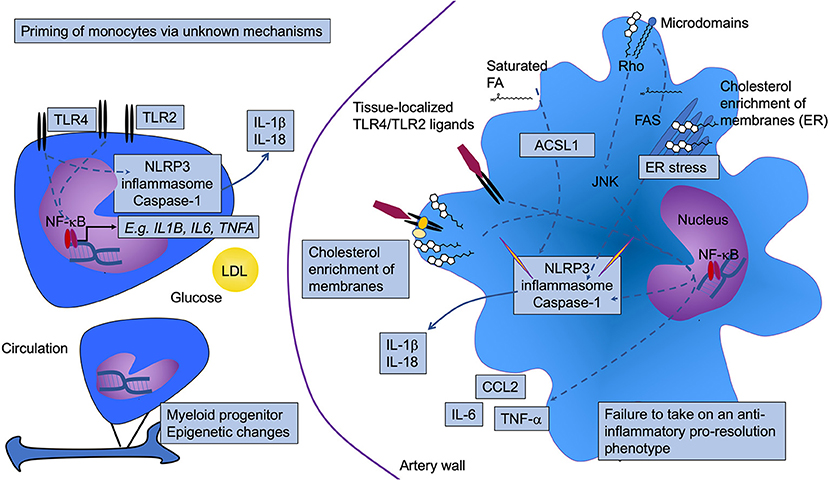
Figure 1. Diabetes is associated with increased monocyte and macrophage inflammatory status. Diabetes results in activation and priming of monocytes to take on a more inflammatory phenotype. Although the mechanism whereby the priming occurs is incompletely understood, glucose, low-density lipoprotein (LDL), and epigenetic modifications have been implicated. This phenotype includes increased gene expression of prototypical pro-inflammatory cytokines, such as IL-1β, TNF-α, and IL-6 (likely through activation of NF-κB), as well as increased expression of toll-like receptors (TLR) 2 and TLR4. Once the monocytes enter the artery wall and differentiate into macrophages, they also display a heightened inflammatory status. The elevated TLR2/4 levels may interact with tissue-localized TLR ligands. Alternatively, or in addition, the aberrant lipid loading observed in diabetes might result in changes in membrane cholesterol accumulation, resulting in increased proximity of TLRs with other proteins that enhance TLR signaling. Increased cholesterol and saturated fatty acids (FA), either through altered cellular membrane properties, as crystals, and/or as inducers of ER stress, can activate the NLRP3 inflammasome and caspase 1, thereby contributing to increased inflammation through release of IL-1β and IL-18. In human monocytes, TLR4 activation is sufficient for inflammasome and caspase 1 activation, as indicated by a direct arrow from TLR4 to the NLRP3 inflammasome and caspase 1. ACSL1 (long-chain acyl-CoA synthetase 1) is induced in inflammatory macrophages. This enzyme acts on free fatty acids to produce acyl-CoA species, which in the case of saturated acyl-CoAs have been shown to contribute to lipotoxicity and inflammasome activation. Fatty acid synthase (FAS) might also contribute by generating fatty acid substrates that are then incorporated into glycerolipids in the membrane where membrane microdomains regulate signaling via Rho activation of JNK. Pathways that have been shown to be altered by diabetes are highlighted by boxes. NLRP3, NOD-like receptor protein 3.
Post-mortem studies of atherosclerotic lesions from both subjects with T1DM and T2DM suggest that diabetes increases the abundance of CD68-positive lesion cells likely to be macrophages (7, 38). To date, there are no studies on macrophage function or phenotype in response to diabetes in human atherosclerotic lesions. However, isolated CD68-positive macrophages from chronic wounds suggest that diabetes results in a less anti-inflammatory and a more pro-inflammatory macrophage phenotype, characterized by reduced expression of CD206 (a pattern recognition receptor that can recognize microbial carbohydrates) and CD163 (a scavenger receptor for the hemoglobin-haptoglobin complex), which are often used as markers of the alternatively activated “M2” macrophage phenotype, and increased expression of the pro-inflammatory cytokine IL-12 upon ex vivo stimulation (68). Whether similar changes also occur in human lesional macrophages in response to diabetes is unknown.
Studies in obese hyperglycemic leptin receptor-deficient mice (a model of T2DM) imply that diabetes impairs the resolution of the inflammatory response during wound healing, with macrophages from diabetic mice continuing to produce pro-inflammatory markers even during the resolution phase (68, 69). At least in mice, similar processes transpire in atherosclerotic lesions in response to aggressive lipid-lowering, in which CD68-positive cells in diabetic mice fail to undergo a switch to a less inflammatory and more “M2-like” pro-resolution macrophage (36, 44). Under conditions of atherosclerotic lesion regression in response to aggressive lipid-lowering, a large proportion of the reduction in lesional macrophage accumulation could be attributed to reduced monocyte recruitment together with a stable rate of apoptosis of plaque macrophages [(70); Figure 1]. Interestingly, work from the Fisher laboratory demonstrated that lesion regression appears to require recruitment of the Ly6Chi monocyte population, and the conversion of these monocytes to “M2-like” alternatively activated macrophages within the lesion (30). This pathway appears to be impaired by diabetes. The mechanism whereby alternatively activated or pro-resolving macrophages contribute to lesion regression is still unclear. One possibility is that these macrophages are more effective in clearing dying or dead cells through efferocytosis than are inflammatory macrophages (71).
Interestingly, at least in mice, the changes observed in macrophages during wound healing are driven by epigenetic modifications that occur in the hematopoietic compartment (68, 69). The notion that diabetes results in lasting effects on the myeloid cell compartment are further highlighted by the fact that if blood monocytes are isolated from T2DM subjects or matched controls, and then differentiated into macrophages for 5 days in vitro, monocyte-derived macrophages from subjects with diabetes express elevated levels of numerous pro-inflammatory cytokines, including IL1B, CCL2, and IL6, as compared with monocyte-derived macrophages from controls (72). Using a similar model, it was shown that monocyte-derived macrophages from patients with T2DM exhibit increased expression of NLRP3 and inflammasome activation [(72); Figure 1].
The factors responsible for monocyte priming in diabetes are largely unknown. One study showed that the priming can be mimicked in vitro by a combination of LDL and high glucose and appears to be mediated by the redox-sensitive MAPK phosphatase MKP-1 (73). Furthermore, epigenetic changes have recently been shown to be responsible for a phenomenon called “trained immunity,” which allows innate immune cells to mount a response to reinfection (74). It has been suggested that diabetes might induce epigenetic changes in monocytes through trained immunity [(75); Figure 1], although research is needed to provide evidence for this hypothesis.
The recent Canakinumab Antiinflammatory Thrombosis Outcome Study (CANTOS) indicates a causative role for IL-1β in atherosclerotic cardiovascular disease in humans (76). Subjects who had already had a myocardial infarction and had elevated levels of high-sensitivity C-reactive protein were treated with a blocking antibody to IL-1β, and cardiovascular death, myocardial infarction, or stroke were evaluated. There was a significant reduction in the rate of CVD in those treated with the IL-1β blocking antibody, demonstrating for the first time a role for inflammation in CVD in humans in these high-risk subjects (76). Interestingly, subjects with and without diabetes within the trial had a similar reduction in major cardiovascular events (77). This might suggest that inflammation is not the culprit in the diabetes-acceleration of atherosclerosis. Alternatively, it might merely indicate that when comparing diabetes to other states of heightened inflammation (per inclusion criteria for this trial), there is no further role for inflammation associated with diabetes.
A very recent single-cell RNA-sequencing study describes the transcriptome of leukocytes from asymptomatic and symptomatic human carotid atherosclerotic lesions. Strikingly, the authors found that a TLR4 and IL-1β signaling signature was associated with macrophages from asymptomatic lesions, rather than symptomatic lesions (78). Instead, macrophages in symptomatic lesions were characterized by an alternatively activated signature, including IL-4-mediated signaling, suggesting a healing function. This finding indicates that macrophage phenotypes in symptomatic lesions are not geared to inflammation but rather to healing, whereas macrophages in lesions that have not yet caused symptoms harbor more active inflammation. These types of studies have not yet been done in people with diabetes.
In summary, there is evidence that diabetes promotes a more inflammatory monocyte and macrophage phenotype, and that the heightened inflammatory capacity of macrophages might be facilitated by priming of blood monocytes or potentially of myeloid progenitor cells by the diabetic environment (Figure 1). It is not known to what extent the increased inflammatory capacity of monocytes and macrophages directly contributes to atherosclerosis progression in diabetes.
Macrophages in lesions of atherosclerosis will engulf APOB-containing lipoprotein particles (LDL, VLDL, and RLPs), resulting in the formation of the hallmark cell type of atherosclerosis—foam cells. Large TRLs, such as chylomicrons derived from the gut after a meal, are believed to be too large to enter the artery wall without first being partly hydrolyzed to RLPs by lipoprotein lipase. While LDL needs to be modified, e.g., by oxidation, to be effectively ingested by macrophages, VLDLs and RLPs are readily taken up by macrophages without modification (79, 80). Early studies showed that VLDL isolated from patients with T2DM is taken up more effectively by mouse peritoneal macrophages, and results in increased lipid accumulation, as compared with VLDL from subjects without diabetes (81). In addition to macrophages, other cell types significantly contribute to foam cell formation in lesions, such as smooth muscle cells, which are known to transdifferentiate into macrophage-like cells after they become lipid-laden (82). Although the effect of diabetes on smooth muscle cell lipid loading and subsequent transdifferentiation to macrophage-like cells is unknown, it has been shown that macrophages become more loaded with cholesteryl esters in the presence of diabetes (19). This might be due in part to increased levels of RLPs in the interstitial fluid surrounding macrophages in the setting of diabetes [(19); Figure 2]. Diabetes has also been shown to lead to reduced expression of the cholesterol transport proteins ABCA1 and ABCG1 in macrophages (83–85), which would reduce cholesterol efflux and further increase their cellular cholesterol content. Furthermore, high glucose and reduced insulin signaling have been suggested to increase the expression of CD36, a lipid scavenger receptor, in macrophages and monocytes through post-transcriptional mechanisms (86, 87). Together, these observations demonstrate that macrophages carry a larger load of lipid in the presence of diabetes (Figure 2).
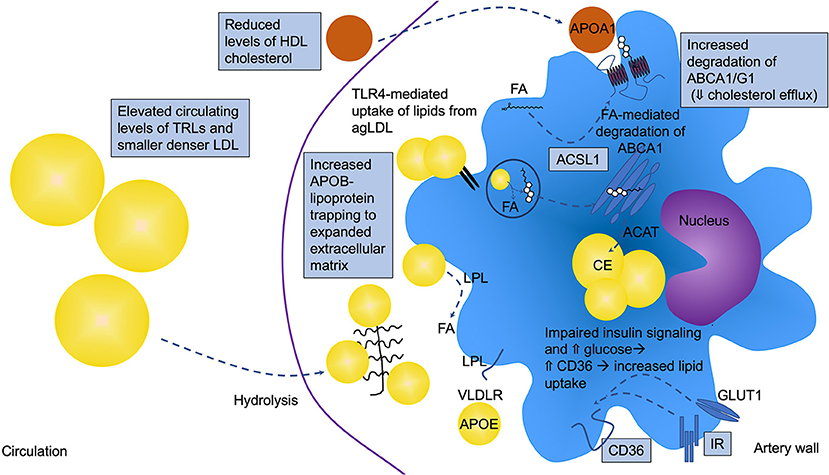
Figure 2. Altered macrophage lipid handling is critically involved in diabetes-accelerated atherosclerosis. Under diabetic conditions, macrophages are often lipid loaded and contain increased levels of cholesteryl ester (CE) in lipid droplets. T2DM and poorly controlled T1DM are associated with elevated circulating levels of triglyceride-rich lipoproteins (TRLs), which contribute to macrophage lipid loading, but the increased lipid loading can also be in part due to increased retention of atherogenic lipoproteins in the artery wall, such as LDL, VLDL, and remnant lipoprotein particles (RLPs). These APOB-containing lipoproteins can bind the expanded/altered extracellular matrix present in lesions in diabetes. Diabetes also alters the balance between uptake and efflux of lipids by modulating the levels of receptors and transporters involved in cholesterol and fatty acid uptake and efflux. CD36 and very low-density lipoprotein receptor (VLDLR), in addition to other receptors and mechanisms, mediate lipid uptake while ATP-binding cassette transporter A1 and G1 (ABCA1 and ABCG1) are critical mediators of cholesterol efflux to HDL and APOA1. ABCA1 and CD36 have been shown to be reduced and elevated, respectively, under diabetic conditions. Fatty acids have been demonstrated to mediate increased degradation of ABCA1 through a mechanism dependent on ACSL1. Impaired insulin signaling via the insulin receptor (IR) and elevated glucose can lead to increased CD36 levels. Furthermore, the elevated levels of TLR4 might result in increased uptake of lipids from aggregated LDL (agLDL), further contributing to the increased lipid loading. Lipoprotein lipase (LPL) aids in the hydrolysis of TRLs, generating free fatty acids (FA) that are easily taken up by cells. All of these pathways can be altered by the diabetic environment as indicated in the schematic by boxes, resulting in lipid accumulation and a pro-atherogenic macrophage phenotype.
It was long believed that the uptake of lipoproteins invariably results in an increased pro-inflammatory state of macrophages (88–90). However, recent studies indicate the opposite is true, at least under some circumstances, perhaps when the macrophage's ability to handle the increased lipid load is unharmed (91, 92). Accordingly, Kim et al. recently demonstrated that foamy macrophages isolated from atherosclerotic lesions had an attenuated inflammatory repertoire. In contrast, the non-foamy macrophages expressed elevated levels of pro-inflammatory markers and overlapped with the inflammatory macrophage cluster identified by single-cell RNA sequencing (92). Along the same lines, we recently demonstrated that macrophage lipid loading and their inflammatory capacity do not go hand-in-hand in the setting of diabetes (19). We observed, by using a mouse model of T1DM, that diabetes results in both heightened inflammation and increased cholesteryl ester loading in cells isolated from the peritoneal cavity. However, when the diabetic mice were treated with an antisense oligonucleotide (ASO) to APOC3 in vivo, the increased macrophage cholesteryl ester loading associated with diabetes was prevented while the heightened Il1b and Tnfa gene expression was unaffected (19). Moreover, diabetes-accelerated atherosclerosis closely associated with the prevention of macrophage cholesteryl ester accumulation and was dramatically reduced by the APOC3 ASO, suggesting that reduced lipid loading of macrophages might be more important for atherosclerosis than inflammatory changes in these cells in diabetes.
Macrophage lipid loading results in an impaired migratory capacity, which could contribute to trapping of the macrophage within the lesion [(93, 94); Figure 3]. Work from the Randolph laboratory suggests that macrophages within the atherosclerotic lesion have a limited migratory capacity (95) and that macrophages are stagnant cells with limited mobility once recruited to lesions, especially if the cells display a morphology consistent with lipid-laden cells. Smaller cells nearer the shoulder region and surface of the plaque exhibit increased movement; however, when monocytes were labeled at different time-points, there was no overlap in the macrophages being labeled, suggesting little movement of the macrophages once they were located deeper in the lesion. This important experiment also showed that monocytes accumulate in waves and implied that there is little phagocytosis of older macrophages by newly infiltrating cells (95). Lipid loading extends beyond macrophages; monocytes have been shown to accumulate lipid droplets, especially if they encounter TRLs, which in turn impair their migration (96). Our recent findings suggest that diabetes increases the ensnaring of pro-atherogenic particles containing APOB, APOC3, and APOE (indicating that they might be TRLs or RLPs derived from TRLs) in the artery wall and that this contributes to accelerated atherosclerosis (19). The extracellular matrix, which is responsible for retention of these lipoprotein particles in the artery wall, is expanded and altered under diabetic conditions (39, 42), and VLDL and LDL particles enriched in APOC3 bind better to matrix proteins (97), potentially indicating that under diabetic conditions, macrophages encounter more lipids within the artery wall (Figure 2).
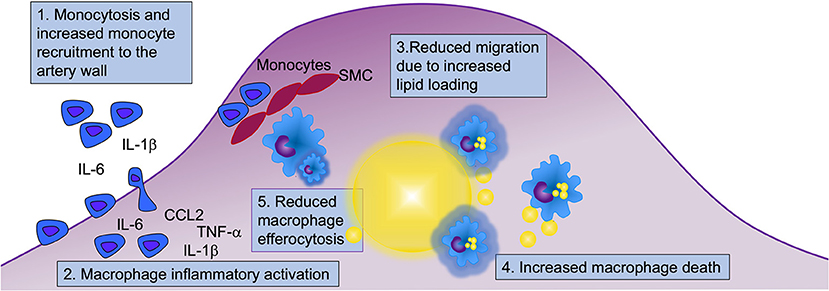
Figure 3. Myeloid cells contribute to diabetes-accelerated atherosclerosis and hindered lesion regression through at least five mechanisms. Diabetes alters many aspects of myeloid cell biology that can contribute to accelerated atherosclerosis and hampered lesion regression. Both monocytes and macrophages display an augmented inflammatory potential, and under certain conditions, diabetes results in elevated levels of monocytes in circulation, which when present contribute to increased recruitment of monocytes to the artery wall. Diabetes increases macrophage lipid loading (indicated by yellow lipid droplets in blue macrophages), which in turn might reduce their ability to migrate. Lipid overload may drive macrophage cell death, and together with reduced efferocytosis, this is likely to contribute to the acceleration of necrotic core formation and progression of atherosclerosis in diabetes. Five mechanisms involving monocytes and macrophages that are likely to contribute to diabetes-accelerated atherosclerosis are highlighted (numbered in boxes). SMC, smooth muscle cells.
There are, however, several circumstances in which lipid handling goes awry and lipids promote inflammatory activation of macrophages (Figure 1). One such mechanism is NLRP3 inflammasome activation by cholesterol crystals, which are present in lesions of atherosclerosis (98). A similar mechanism has been proposed for crystals generated by saturated fatty acids (99). The extent to which cholesterol- or saturated fatty acid crystals activate macrophages in vivo still needs to be investigated, as some have cautioned that the crystalline material may have been formed during sample processing due to the crystallization of lipids in refrigerated samples (100). The NLRP3 inflammasome is a multimeric complex that processes pro-IL-1β and pro-IL-18 into their mature forms. The NLRP3 inflammasome typically requires two signals—a priming step depending on TLR activation that induces the transcription of Nlrp3 and Casp1 (caspase 1)—and an activating step resulting in the assembly and activation of caspase 1 and subsequent processing of pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18 (101). The activation step can be achieved by crystalline materials, ion flux induced e.g., by ATP, and ER stressors. Interestingly, human monocytes, as opposed to human macrophages, release processed IL-1β after a one-time stimulation of TLRs (102). This was explained by the finding that these cells also release endogenous ATP (102), or that an alternative inflammasome pathway exists in these cells (103). The ATP released from newly recruited monocytes might therefore contribute to inflammasome activation in human lesional macrophages by providing ATP as an activating signal.
Many groups have reported that diabetes results in increased expression of genes that would be consistent with increased inflammation and with priming/and or activation of the NLRP3 inflammasome in both T1DM and T2DM (36, 37, 44, 58, 66, 104–109) (see the above section), but it is not yet clear what the diabetes signal that drives augmented NLRP3 activation might be. It is possible that increased intracellular cholesterol crystals, crystals formed by saturated fatty acids, or altered membrane levels of cholesterol or saturated fatty acids might explain this phenomenon. Aberrant intracellular cholesterol handling can also trigger the inflammasome via increased ER cholesterol accumulation [(100, 110, 111); Figure 1]. In diabetes, the aberrant cholesterol accumulation in membranes might be due to reduced expression of the cholesterol transport proteins ABCA1 and ABCG1 in macrophages (83–85). These transporters efflux cholesterol to APOA1 and HDL, thereby reducing cellular and membrane cholesterol levels (Figure 2). The ER membrane appears to serve as an important sensor of intracellular cholesterol homeostasis (112). Some, but not all studies, have found that deletion of the inflammasome reduces atherosclerosis under non-diabetic conditions (63, 98, 100, 113, 114). Further studies are needed to investigate the role of the NLRP3 inflammasome in macrophages in the setting of diabetes.
Based primarily on studies of macrophages deficient in ABCA1 and ABCG1, in which cholesterol loading is particularly high due to the defective cholesterol efflux, it has been shown that TLR4 signaling is enhanced by cellular cholesterol accumulation. The mechanism is believed to be due to increased activation of TLR4 due to increased proximity of TLR4 with its binding proteins MD2 and CD14 in lipid rafts [(115); Figure 1]. Furthermore, a recent study suggested a different role of TLR4 in relation to macrophage lipid loading. This study demonstrated that TLR4 is required for macrophage lipid uptake from aggregated LDL, by promoting the release of exosomal enzymes for degradation of lipids in these aggregates (116). Accordingly, TLR4-deficiency reduced lipid loading of macrophages through a pathway involving PI3K and Akt (Figure 2). Direct evidence for these pathways in macrophages in diabetes is missing, but the models would fit with the increased inflammatory activation and lipid loading of these cells in diabetes.
In addition, increased fatty acid load is likely to contribute to macrophage activation through several mechanisms. Recent studies have revealed that fatty acid synthesis through fatty acid synthase (FAS) is required for macrophage inflammatory activation (117). Loss of FAS altered membrane order and composition, impairing the retention of plasma membrane cholesterol and disrupting Rho GTPase trafficking, resulting in impaired cell adhesion, migration, and activation (117). Interestingly, the effect of FAS-deficiency could be reversed by the addition of cholesterol, suggesting that endogenous fatty acids are critical for correct membrane cholesterol localization (Figure 1).
Disruption of fatty acid storage in inert triacylglycerol is associated with saturated fatty acid-induced inflammatory changes in macrophages (118). However, lipid uptake via the VLDL receptor (VLDLR) results in pro-inflammatory effects in macrophages concomitant with both increased triacylglycerol and C16:0 ceramide levels (119). VLDLR binds APOE, rather than APOB, and acts in concert with LPL to promote uptake of lipids from APOE-containing VLDL, RLPs and chylomicrons (120). Furthermore, we and others have demonstrated that deletion of acyl-CoA synthetase 1 (ACSL1), an enzyme that esterifies fatty acids into their acyl-CoA derivatives, a step necessary for most downstream fatty acid metabolism, reduces inflammation and lipotoxicity in response to saturated fatty acids and diabetes (37, 121, 122). Moreover, high levels of unsaturated fatty acids (C18:1 and C18:2) induce degradation of ABCA1, reducing cholesterol efflux capacity through a mechanism mediated by ACSL1 in macrophages (123). Myeloid cell-targeted deletion of ACSL1 also prevented diabetes-accelerated atherosclerosis (37), demonstrating the important role of fatty acid metabolism in atherosclerosis in the setting of diabetes (Figures 1, 2).
Together, these studies indicate that aberrant lipid accumulation and handling in macrophages can cause, or is associated with, increased inflammation. Prevention of macrophage lipid loading in diabetic mice associates with prevention of accelerated atherosclerosis (19). Further studies are needed in order to clarify the effects of diabetes on macrophage cholesterol and fatty acid handling, the relationships among lipids and inflammation, and the relative impacts of these pathways on atherosclerosis.
Human imaging studies suggest that diabetes accelerates lesion progression. Thus, Won et al. used coronary computed tomography angiography to demonstrate that atherosclerotic lesion progression is faster in people with diabetes than in those without, despite having lower LDL cholesterol at baseline (124). In a similar study, Kim et al. demonstrated that plaque progression (necrotic core expansion), was accelerated in diabetes—again in the absence of elevated LDL cholesterol (125). These studies highlight the concept that diabetes accelerates atherosclerotic lesion progression even in settings in which LDL cholesterol levels are lower or similar as compared with the non-diabetic control cohort. In both the aforementioned studies, mean LDL cholesterol levels were ~100 mg/dL, which is in the normal range. Mouse studies of diabetes-accelerated atherosclerosis also support this concept (39).
As the atherosclerotic lesion progresses, a necrotic core is formed. Necrotic core expansion is thought to be due, at least in part, to the death and reduced clearance of dead and dying macrophages (Figure 3). Both human pathology studies and imaging studies have demonstrated that diabetes results in an expansion of lesional necrotic cores associated with lesion progression (126–129). Necrotic core expansion can also be observed in mouse models of T1DM (19) and of T2DM/metabolic syndrome (61, 130). It is unclear to what extent the expanded necrotic cores observed in diabetes are due to a selective modulation of the myeloid cell compartment, resulting in increased macrophage death or reduced clearance of dead and dying macrophages, or if the expanded necrotic core is a marker of a faster-progressing lesion, which might be due to increased recruitment and turnover of macrophages (Figure 3). Alternatively, all three scenarios could be occurring. For example, deletion of insulin receptors in myeloid cells results in increased susceptibility of macrophages to apoptosis in response to altered cholesterol metabolism, causing larger necrotic cores in mouse models [(131); Figure 3]. Thus, insufficient insulin signaling might contribute directly to increased macrophage death and necrotic core expansion. Furthermore, if there indeed is increased inflammasome activation in diabetes, this can result in pyroptosis, an inflammatory type of cell death linked to the NLRP3 inflammasome that has been proposed to play a role in atherosclerosis (132). Future research will reveal whether pyroptosis or other cell death pathways are increased in macrophages in diabetes and if such mechanisms contribute to diabetes-accelerated lesion progression.
The clearance of dying or dead cells by phagocytes is termed efferocytosis. Efferocytosis has been shown to be impaired in mouse models of T2DM, an effect ascribed to the increased membrane content of saturated fatty acids (130). Reduced efferocytosis can contribute to the expansion of necrotic cores and the progression of the atherosclerotic lesion [(133); Figure 3].
Efferocytosis requires metabolic reprogramming to increase glucose uptake and aerobic glycolysis (134). A prevailing hypothesis in the field of diabetes is that if a cell is bathed in excessive glucose, it ought to take up more glucose, especially if the cell expresses non-insulin dependent glucose transporters, such GLUT1. Macrophages express relatively high levels of GLUT1 and lower levels of the insulin-sensitive GLUT4. However, because GLUT1 exhibits a Km for glucose of 1–7 mM, it is generally believed to be nearly saturated at physiological ranges of glucose (135). Instead, increased glucose influx through GLUT1 might be a result of inflammatory activation of macrophages (136). To investigate if increased glucose flux into myeloid cells per se could mimic the effects of diabetes in non-diabetic mice, we overexpressed GLUT1 under control of the CD68 promoter in hematopoietic cells, which results in expression primarily in myeloid cells (136). Forcing macrophage to take up more glucose through GLUT1 overexpression did not induce inflammatory changes in primary macrophages ex vivo or accelerate atherosclerosis or alter necrotic core size (136). However, deletion of GLUT1 prevented normal inflammatory activation of these cells by LPS or LPS and interferon-γ (136, 137). Thus, GLUT1 is required for normal inflammatory activation of macrophages, consistent with previous studies (138), but increased glucose flux through GLUT1 is not sufficient to promote lesion progression in non-diabetic mice. However, if GLUT1 is deleted selectively in myeloid cells in non-diabetic mice, efferocytosis is reduced, and the necrotic core area is expanded, because of the reliance on glucose metabolism to carry out phagocytosis and efferocytosis (134, 137). Conversely, GLUT1 deletion from the entire hematopoietic compartment results in reduced myelopoiesis in response to dyslipidemia, reduced monocyte recruitment to the artery wall, and reduced atherosclerosis (139). Together, these three studies highlight the complexity of glucose metabolism within different closely related cell types. Increased glucose uptake through GLUT1 in myeloid cells does not appear to be sufficient for inflammatory activation or lesion progression, but GLUT1 is required for several functions of myeloid cells and their precursors.
The findings to date suggest that diabetes enhances necrotic core formation in lesions, perhaps because it promotes lesion progression to more advanced lesions, and possibly in part because it has direct effects on macrophage death and adverse effects on efferocytosis. The effects of diabetes on necrotic core expansion do not appear to be due to increased glucose uptake in macrophages, but rather to be dependent on a reduction in efferocytosis due to increased macrophage membrane saturation (130), and perhaps to increased macrophage death due to insufficient insulin signaling or other mechanisms. Lesions with large necrotic cores are believed to be responsible for a significant proportion of the clinical events of CVD, being less stable and more susceptible to fissuring or rupturing.
Since monocytes are circulating cells that enter all injured tissues, do some of the changes observed in lesional macrophages also occur in other tissues affected by diabetes complications? In addition to macrovascular disease, diabetes increases the risk of microvascular complications, such as diabetic retinopathy and diabetic kidney disease (DKD). Several lines of evidence suggest that the presence of microvascular disease dramatically increases the risk of macrovascular complications (140–142). Whether this is due to a shared underlying mechanism or if one complication accelerates the other is unknown. If diabetes influences the myeloid cell compartment and alters monocyte and macrophage function as it relates to atherogenesis, perhaps changes in monocytes and macrophages also contribute to other complications of diabetes, such as DKD. Such changes in monocytes and macrophages might, in part, explain why DKD and atherogenesis are tightly linked.
Do monocytes and macrophages play a causative role in DKD? Macrophages are known to accumulate in the glomerulus and renal interstitium in kidney disease (143–145). Monocyte depletion studies in preclinical models suggest that monocytes and macrophages play a direct pathological role in DKD, perhaps by affecting podocyte barrier function (146). Similarly, deletion or reduction of CCL2, a key chemokine involved in monocyte recruitment, reduces diabetic kidney disease in mice (147–149). More recently, two clinical trials targeting CCL2 have been completed with promising improvements of albuminuria in humans with T2DM and kidney disease (150, 151). Very recently, Niewczas et al. highlighted the importance of chronic inflammation in the progression of kidney disease in diabetes (152). A cluster of 17 kidney disease risk signature proteins that include TNF family proteins and several cytokines predicted the risk of end-stage renal disease, with a least some of these proteins being produced in the glomerulus, and some of them correlating with histopathological changes associated with diabetes, such as glomerulosclerosis (152). This network included the IL-1 receptor, which might suggest activation of the inflammasome pathway.
Lipids accumulate in glomeruli in DKD (153), consistent with the reduced ABCA1 expression in kidneys from diabetic mice (83). TNF-α induces cholesterol accumulation in cultured podocytes, which in turn exacerbates apoptosis in these cells (154), suggesting links among lipid accumulation, lipotoxicity, and inflammatory stimuli. Consistent with this notion, dyslipidemia associated with atherosclerosis augmented albuminuria in a mouse model of DKD (155). Interestingly, the pro-atherogenic dyslipidemia was associated with increased glomerular macrophage accumulation and a dramatic increase in renal cortex inflammation, suggesting that dyslipidemia contributes to DKD, perhaps via augmented macrophage activation analogous to that in the artery wall. Further research is needed to address the links among diabetes-induced macrophage phenotypic changes and DKD and other diabetes complications.
In 2008, the Food and Drug Administration recommended to include cardiovascular endpoints in all phase 2 and phase 3 clinical trials of new anti-diabetic therapies. As a result of this recommendation, new anti-diabetic therapeutics were revealed to have unexpected protective effects on cardiovascular outcomes. Glucagon-like peptide 1 (GLP-1) receptor agonists, which increase insulin secretion, and SGLT2 inhibitors, which lower blood glucose levels by preventing glucose reabsorption in the kidney, both were shown to reduce cardiovascular events in patients with T2DM (156–160). GLP-1 receptor agonists have multiple beneficial effects in several organs in humans, resulting in reduced risk of atherosclerotic CVD events and CVD death. Among the many beneficial effects of GLP-1 receptor activation are anti-inflammatory effects, direct effects in the heart, increased natriuresis and diuresis, reduced body weight, reduced coagulation, and a reduction in post-prandial lipids, as reviewed extensively elsewhere (161). SGLT2 inhibitors display the greatest beneficial effect on heart failure and progression of renal disease, and have only modest beneficial effects on reducing atherosclerotic CVD, and primarily are effective in patients with established atherosclerotic disease (162). Furthermore, SGLT2 inhibitors exert beneficial actions independent of glucose control, consistent with results from the recent DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial, which indicated beneficial effects of this SGLT2 inhibitor (dapagliflozin) in patients with heart failure and a reduced ejection fraction even when diabetes was not present (163). Suggested beneficial mechanisms not directly related to blood glucose-lowering effects include natriuresis and osmotic diuresis, reduced inflammation, reduced oxidative stress, reduced arterial stiffness, reductions in blood pressure and body weight, and renoprotective effects (164). According to the safety profiles, SGLT2 inhibitors do not appear to reduce monocyte counts in humans with T2DM, suggesting that the effects of SGLT2 inhibitors on monocytosis in diabetic mice might not be translated to humans. Other clinical trials have demonstrated beneficial effects on CVD outcomes of icosapent ethyl ester, a highly pure fish oil ester, in cohorts with and without T2DM (165, 166).
Do these drugs have direct effects on monocytes and macrophages and is it possible that some of the beneficial effects are due to alterations of lesion macrophages? SGLT2 inhibitors have been shown to prevent macrophage foam cell formation in diabetic mice concomitant with reduced atherosclerosis (46). The mechanism was suggested to be due to downregulation of the increased LOX1 and ACAT and upregulation of the decreased ABCA1 in macrophages from diabetic mice treated with the SGLT2 inhibitors. Overall however, current research suggests that the main therapeutic effects of SGLT2 inhibitors are unlikely to be mediated by effects in myeloid cells, at least in humans.
GLP-1 receptor agonists prevent incident atherosclerotic CVD in people with diabetes. Some of the effects might be mediated by direct effects on macrophages, although GLP-1 receptor agonists exert effects in a plethora of cell types, and macrophages are unlikely to be a major point of action. Nevertheless, exendin-4 (a GLP-1 receptor agonist) has been shown to reduce atherosclerosis and monocyte adhesion to aortas as well as inflammatory gene expression in macrophages in mice, likely by increasing cAMP levels in these cells (167).
In relation to the beneficial effects of icosapent ethyl ester on incident CVD, fish oils have been shown to have beneficial effects in macrophages, including reversal of the defective efferocytosis associated with diabetes (130). Although the mechanism of icosapent ethyl ester might be distinct from that of natural mixed fish oils, it is relevant that fish oils can be converted to bioactive lipid mediators involved in the resolution of inflammation (168).
More research is needed to understand the beneficial effects of these therapies in patients with diabetes. Such studies could also shed additional light on the mechanisms whereby diabetes increases CVD risk.
Diabetes creates an environment that fosters the initiation and progression of the atherosclerotic lesion as well as a hampered lesion regression through at least five likely mechanisms in monocytes and macrophages (Figure 3). First, diabetes increases monocyte recruitment to the lesion, especially in the setting of monocytosis. However, while monocytosis can exacerbate monocyte recruitment to lesions, it is not required for diabetes-accelerated atherosclerosis. Second, monocytes appear to be primed in circulation to have an increased inflammatory potential, and once in the artery wall, their boosted cytokine/chemokine production can contribute to increased recruitment of new monocytes and activation of macrophages and other lesional cells. Third, diabetes increases macrophage lipid loading, perhaps mainly due to trapping of TRLs and RLPs or modified LDL within the artery wall via increased retention of these lipoprotein particles through binding to glycosaminoglycans. Once loaded with lipids, macrophages struggle to migrate and egress out of the lesion, contributing to necrotic core expansion. Fourth, due to increased sensitivity to cell death induced by the elevated lipid environment and/or insufficient insulin signaling, and, fifth, reduced efferocytosis capacity, lesions progress faster under diabetic conditions than under non-diabetic conditions and develop expanded necrotic cores. Similar mechanisms are involved in mediating the detrimental effects of diabetes on lesion regression.
Many pieces of this puzzle remain to be found. Although monocytes and macrophages are present in all stages of atherosclerosis and their function is altered by diabetes, a major question is whether the myeloid cell compartment could be successfully targeted for prevention of diabetes-accelerated atherosclerosis and CVD. The CANTOS trial was the first trial to target inflammation and produce a protective effect on CVD. Such an approach might be feasible and successful in people with diabetes and heightened inflammation. Furthermore, based on the research available to date, new therapies that target apolipoproteins, such as APOC3, would be predicted to be effective in preventing CVD in patients with diabetes and elevated levels of APOC3. Other important questions for future research include the mechanisms whereby diabetes induces adverse effects in myeloid cells. For example, what exactly goes awry in myeloid cell lipid handling in the setting of diabetes, and what is the mechanism whereby lipids accumulate in these cells? What is the role of inflammatory changes vis-à-vis lipid over-accumulation? What changes are critical for necrotic core expansion and CVD events? Do different myeloid cell populations respond differently to the diabetic environment? Some of these important answers are likely to be revealed by new methodology, such as single-cell RNA-sequencing, proteomics, metabolomics and lipidomics.
JK and C-CH wrote the manuscript. KB wrote parts of the manuscript and edited the full manuscript.
Research in the authors' laboratories was supported by the American Diabetes Association grant 1-16-IBS-153 (JK), the National Institutes of Health grants DK076169 and DK115255 (P&F to JK), DP3DK108209, R01HL127694, R01HL126028, P01HL092969, R21AI135447 (KB), and the Diabetes Research Center at the University of Washington, P30DK017047.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Emerging Risk Factors C, Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. (2010) 375:2215–22. doi: 10.1016/S0140-6736(10)60484-9
2. Rawshani A, Rawshani A, Franzen S, Eliasson B, Svensson AM, Miftaraj M, et al. Mortality and cardiovascular disease in type 1 and type 2 diabetes. N Engl J Med. (2017) 376:1407–18. doi: 10.1056/NEJMoa1608664
3. Laakso M, Kuusisto J. Insulin resistance and hyperglycaemia in cardiovascular disease development. Nat Rev Endocrinol. (2014) 10:293–302. doi: 10.1038/nrendo.2014.29
4. Juutilainen A, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Type 2 diabetes as a “coronary heart disease equivalent”: an 18-year prospective population-based study in Finnish subjects. Diabetes Care. (2005) 28:2901–7. doi: 10.2337/diacare.28.12.2901
5. American Diabetes Association. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2019. Diabetes Care. (2019) 42(Suppl. 1):S13–28. doi: 10.2337/dc19-S002
6. Yki-Jarvinen H. Nonglycemic effects of insulin. Clin Cornerstone. (2003) 5(Suppl. 4):S6–12. doi: 10.1016/S1098-3597(03)90061-5
7. Burke AP, Kolodgie FD, Zieske A, Fowler DR, Weber DK, Varghese PJ, et al. Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler Thromb Vasc Biol. (2004) 24:1266–71. doi: 10.1161/01.ATV.0000131783.74034.97
8. Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. (2005) 353:2643–53. doi: 10.1056/NEJMoa052187
9. Bebu I, Braffett BH, Pop-Busui R, Orchard TJ, Nathan DM, Lachin JM, et al. The relationship of blood glucose with cardiovascular disease is mediated over time by traditional risk factors in type 1 diabetes: the DCCT/EDIC study. Diabetologia. (2017) 60:2084–91. doi: 10.1007/s00125-017-4374-4
10. Chait A, Bornfeldt KE. Diabetes and atherosclerosis: is there a role for hyperglycemia? J Lipid Res. (2009) 50(Suppl.):S335–9. doi: 10.1194/jlr.R800059-JLR200
11. Bornfeldt KE. Does elevated glucose promote atherosclerosis? Pros and cons. Circ Res. (2016) 119:190–3. doi: 10.1161/CIRCRESAHA.116.308873
12. Reaven PD, Emanuele NV, Wiitala WL, Bahn GD, Reda DJ, McCarren M, et al. Intensive glucose control in patients with type 2 diabetes - 15-year follow-up. N Engl J Med. (2019) 380:2215–24. doi: 10.1056/NEJMoa1806802
13. Xiao C, Dash S, Morgantini C, Hegele RA, Lewis GF. Pharmacological targeting of the atherogenic dyslipidemia complex: the next frontier in CVD prevention beyond lowering LDL cholesterol. Diabetes. (2016) 65:1767–78. doi: 10.2337/db16-0046
14. Khetarpal SA, Zeng X, Millar JS, Vitali C, Somasundara AVH, Zanoni P, et al. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med. (2017) 23:1086–94. doi: 10.1038/nm.4390
15. Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. (2008) 322:1702–5. doi: 10.1126/science.1161524
16. TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. (2014) 371:22–31. doi: 10.1056/NEJMoa1307095
17. Taskinen MR, Packard CJ, Boren J. Emerging evidence that ApoC-III inhibitors provide novel options to reduce the residual CVD. Curr Atheroscler Rep. (2019) 21:27. doi: 10.1007/s11883-019-0791-9
18. Sandesara PB, Virani SS, Fazio S, Shapiro MD. The forgotten lipids: triglycerides, remnant cholesterol, and atherosclerotic cardiovascular disease risk. Endocr Rev. (2019) 40:537–57. doi: 10.1210/er.2018-00184
19. Kanter JE, Shao B, Kramer F, Barnhart S, Shimizu-Albergine M, Vaisar T, et al. Increased apolipoprotein C3 drives cardiovascular risk in type 1 diabetes. J Clin Invest. (2019) 130:4165–79. doi: 10.1172/JCI127308
20. Basu A, Bebu I, Jenkins AJ, Stoner JA, Zhang Y, Klein RL, et al. Serum apolipoproteins and apolipoprotein-defined lipoprotein subclasses: a hypothesis-generating prospective study of cardiovascular events in T1D. J Lipid Res. (2019) 60:1432–9. doi: 10.1194/jlr.P090647
21. Conway BN, Aroda VR, Maynard JD, Matter N, Fernandez S, Ratner RE, et al. Skin intrinsic fluorescence is associated with coronary artery disease in individuals with long duration of type 1 diabetes. Diabetes Care. (2012) 35:2331–6. doi: 10.2337/dc12-0053
22. Senatus LM, Schmidt AM. The AGE-RAGE axis: implications for age-associated arterial diseases. Front Genet. (2017) 8:187. doi: 10.3389/fgene.2017.00187
23. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. (2016) 118:653–67. doi: 10.1161/CIRCRESAHA.115.306256
24. Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. (2011) 14:575–85. doi: 10.1016/j.cmet.2011.07.015
25. Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. (1995) 15:551–61. doi: 10.1161/01.ATV.15.5.551
26. Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. (2002) 417:750–4. doi: 10.1038/nature00804
27. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
28. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
29. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. (2011) 145:341–55. doi: 10.1016/j.cell.2011.04.005
30. Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest. (2017) 127:2904–15. doi: 10.1172/JCI75005
31. MacDougall ED, Kramer F, Polinsky P, Barnhart S, Askari B, Johansson F, et al. Aggressive very low-density lipoprotein (VLDL) and LDL lowering by gene transfer of the VLDL receptor combined with a low-fat diet regimen induces regression and reduces macrophage content in advanced atherosclerotic lesions in LDL receptor-deficient mice. Am J Pathol. (2006) 168:2064–73. doi: 10.2353/ajpath.2006.051009
32. Saiki O, Negoro S, Tsuyuguchi I, Yamamura Y. Depressed immunological defence mechanisms in mice with experimentally induced diabetes. Infect Immun. (1980) 28:127–31. doi: 10.1128/IAI.28.1.127-131.1980
33. Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol Med Microbiol. (1999) 26:259–65. doi: 10.1111/j.1574-695X.1999.tb01397.x
34. Cornell RP. Reticuloendothelial hyperphagocytosis occurs in streptozotocin-diabetic rats. Studies with colloidal carbon, albumin microaggregates, and soluble fibrin monomers. Diabetes. (1982) 31:110–8. doi: 10.2337/diabetes.31.2.110
35. Virmani R, Burke AP, Kolodgie F. Morphological characteristics of coronary atherosclerosis in diabetes mellitus. Can J Cardiol. (2006) (22 Suppl. B):81B−4. doi: 10.1016/S0828-282X(06)70991-6
36. Parathath S, Grauer L, Huang LS, Sanson M, Distel E, Goldberg IJ, et al. Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes. (2011) 60:1759–69. doi: 10.2337/db10-0778
37. Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc Natl Acad Sci USA. (2012) 109:E715–24. doi: 10.1073/pnas.1111600109
38. Moreno PR, Murcia AM, Palacios IF, Leon MN, Bernardi VH, Fuster V, et al. Coronary composition and macrophage infiltration in atherectomy specimens from patients with diabetes mellitus. Circulation. (2000) 102:2180–4. doi: 10.1161/01.CIR.102.18.2180
39. Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, et al. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. (2004) 114:659–68. doi: 10.1172/JCI200417867
40. Lamharzi N, Renard CB, Kramer F, Pennathur S, Heinecke JW, Chait A, et al. Hyperlipidemia in concert with hyperglycemia stimulates the proliferation of macrophages in atherosclerotic lesions: potential role of glucose-oxidized LDL. Diabetes. (2004) 53:3217–25. doi: 10.2337/diabetes.53.12.3217
41. Vedantham S, Noh H, Ananthakrishnan R, Son N, Hallam K, Hu Y, et al. Human aldose reductase expression accelerates atherosclerosis in diabetic apolipoprotein E-/- mice. Arterioscler Thromb Vasc Biol. (2011) 31:1805–13. doi: 10.1161/ATVBAHA.111.226902
42. McDonald TO, Gerrity RG, Jen C, Chen HJ, Wark K, Wight TN, et al. Diabetes and arterial extracellular matrix changes in a porcine model of atherosclerosis. J Histochem Cytochem. (2007) 55:1149–57. doi: 10.1369/jhc.7A7221.2007
43. Williams KJ, Tabas I. The response-to-retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol. (1998) 9:471–4. doi: 10.1097/00041433-199810000-00012
44. Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. (2013) 17:695–708. doi: 10.1016/j.cmet.2013.04.001
45. Kong L, Shen X, Lin L, Leitges M, Rosario R, Zou YS, et al. PKCβ promotes vascular inflammation and acceleration of atherosclerosis in diabetic ApoE null mice. Arterioscler Thromb Vasc Biol. (2013) 33:1779–87. doi: 10.1161/ATVBAHA.112.301113
46. Terasaki M, Hiromura M, Mori Y, Kohashi K, Nagashima M, Kushima H, et al. Amelioration of hyperglycemia with a sodium-glucose cotransporter 2 inhibitor prevents macrophage-driven atherosclerosis through macrophage foam cell formation suppression in type 1 and type 2 diabetic mice. PLoS ONE. (2015) 10:e0143396. doi: 10.1371/journal.pone.0143396
47. Barrett TJ, Distel E, Murphy AJ, Hu J, Garshick MS, Ogando Y, et al. Apolipoprotein AI) promotes atherosclerosis regression in diabetic mice by suppressing myelopoiesis and plaque inflammation. Circulation. (2019) 140:1170–84. doi: 10.1161/CIRCULATIONAHA.119.039476
48. Johansson F, Kramer F, Barnhart S, Kanter JE, Vaisar T, Merrill RD, et al. Type 1 diabetes promotes disruption of advanced atherosclerotic lesions in LDL receptor-deficient mice. Proc Natl Acad Sci USA. (2008) 105:2082–7. doi: 10.1073/pnas.0709958105
49. Candido R, Allen TJ, Lassila M, Cao Z, Thallas V, Cooper ME, et al. Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation. (2004) 109:1536–42. doi: 10.1161/01.CIR.0000124061.78478.94
50. Hayek T, Hussein K, Aviram M, Coleman R, Keidar S, Pavoltzky E, et al. Macrophage foam-cell formation in streptozotocin-induced diabetic mice: stimulatory effect of glucose. Atherosclerosis. (2005) 183:25–33. doi: 10.1016/j.atherosclerosis.2005.02.018
51. Gerrity RG, Natarajan R, Nadler JL, Kimsey T. Diabetes-induced accelerated atherosclerosis in swine. Diabetes. (2001) 50:1654–65. doi: 10.2337/diabetes.50.7.1654
52. Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res. (2014) 114:1757–71. doi: 10.1161/CIRCRESAHA.114.301174
53. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. (2007) 117:195–205. doi: 10.1172/JCI29950
54. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. (2007) 117:185–94. doi: 10.1172/JCI28549
55. Murphy AJ, Dragoljevic D, Tall AR. Cholesterol efflux pathways regulate myelopoiesis: a potential link to altered macrophage function in atherosclerosis. Front Immunol. (2014) 5:490. doi: 10.3389/fimmu.2014.00490
56. Averill MM, Kerkhoff C, Bornfeldt KE. S100A8 and S100A9 in cardiovascular biology and disease. Arterioscler Thromb Vasc Biol. (2012) 32:223–9. doi: 10.1161/ATVBAHA.111.236927
57. Kraakman MJ, Lee MK, Al-Sharea A, Dragoljevic D, Barrett TJ, Montenont E, et al. Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J Clin Invest. (2017) 127:2133–47. doi: 10.1172/JCI92450
58. Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. (2014) 19:821–35. doi: 10.1016/j.cmet.2014.03.029
59. Pennig J, Scherrer P, Gissler MC, Anto-Michel N, Hoppe N, Funer L, et al. Glucose lowering by SGLT2-inhibitor empagliflozin accelerates atherosclerosis regression in hyperglycemic STZ-diabetic mice. Sci Rep. (2019) 9:17937. doi: 10.1038/s41598-019-54224-9
60. Vallerie SN, Kramer F, Barnhart S, Kanter JE, Breyer RM, Andreasson KI, et al. Myeloid cell prostaglandin E2 receptor EP4 modulates cytokine production but not atherogenesis in a mouse model of type 1 diabetes. PLoS ONE. (2016) 11:e0158316. doi: 10.1371/journal.pone.0158316
61. Kanter JE, Kramer F, Barnhart S, Duggan JM, Shimizu-Albergine M, Kothari V, et al. A novel strategy to prevent advanced atherosclerosis and lower blood glucose in a mouse model of metabolic syndrome. Diabetes. (2018) 67:946–59. doi: 10.2337/db17-0744
62. Wall VZ, Barnhart S, Kanter JE, Kramer F, Shimizu-Albergine M, Adhikari N, et al. Smooth muscle glucose metabolism promotes monocyte recruitment and atherosclerosis in a mouse model of metabolic syndrome. JCI Insight. (2018) 3:96544. doi: 10.1172/jci.insight.96544
63. Bradshaw EM, Raddassi K, Elyaman W, Orban T, Gottlieb PA, Kent SC, et al. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J Immunol. (2009) 183:4432–9. doi: 10.4049/jimmunol.0900576
64. Giulietti A, van Etten E, Overbergh L, Stoffels K, Bouillon R, Mathieu C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile. 1,25-dihydroxyvitamin D(3) works as anti-inflammatory. Diabetes Res Clin Pract. (2007) 77:47–57. doi: 10.1016/j.diabres.2006.10.007
65. Devaraj S, Glaser N, Griffen S, Wang-Polagruto J, Miguelino E, Jialal I. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes. (2006) 55:774–9. doi: 10.2337/diabetes.55.03.06.db05-1417
66. Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. (2010) 33:861–8. doi: 10.2337/dc09-1799
67. Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab. (2008) 93:578–83. doi: 10.1210/jc.2007-2185
68. Gallagher KA, Joshi A, Carson WF, Schaller M, Allen R, Mukerjee S, et al. Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes. (2015) 64:1420–30. doi: 10.2337/db14-0872
69. Yan J, Tie G, Wang S, Tutto A, DeMarco N, Khair L, et al. Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat Commun. (2018) 9:33. doi: 10.1038/s41467-017-02425-z
70. Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J Clin Invest. (2011) 121:2025–36. doi: 10.1172/JCI43802
71. Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. (2019). doi: 10.1038/s41577-019-0240-6. [Epub ahead of print].
72. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. (2013) 62:194–204. doi: 10.2337/db12-0420
73. Kim HS, Ullevig SL, Zamora D, Lee CF, Asmis R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc Natl Acad Sci USA. (2012) 109:E2803–12. doi: 10.1073/pnas.1212596109
74. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
75. van Diepen JA, Thiem K, Stienstra R, Riksen NP, Tack CJ, Netea MG. Diabetes propels the risk for cardiovascular disease: sweet monocytes becoming aggressive? Cell Mol Life Sci. (2016) 73:4675–84. doi: 10.1007/s00018-016-2316-9
76. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
77. Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC, et al. Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J Am Coll Cardiol. (2018) 71:2392–401. doi: 10.1016/j.jacc.2018.03.002
78. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–158. doi: 10.1038/s41591-019-0590-4
79. Van Lenten BJ, Fogelman AM, Jackson RL, Shapiro S, Haberland ME, Edwards PA. Receptor-mediated uptake of remnant lipoproteins by cholesterol-loaded human monocyte-macrophages. J Biol Chem. (1985) 260:8783–8.
80. Whitman SC, Miller DB, Wolfe BM, Hegele RA, Huff MW. Uptake of type III hypertriglyceridemic VLDL by macrophages is enhanced by oxidation, especially after remnant formation. Arterioscler Thromb Vasc Biol. (1997) 17:1707–15. doi: 10.1161/01.ATV.17.9.1707
81. Kraemer FB, Chen YD, Lopez RD, Reaven GM. Effects of noninsulin-dependent diabetes mellitus on the uptake of very low density lipoproteins by thioglycolate-elicited mouse peritoneal macrophages. J Clin Endocrinol Metab. (1985) 61:335–42. doi: 10.1210/jcem-61-2-335
82. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
83. Tang C, Kanter JE, Bornfeldt KE, Leboeuf RC, Oram JF. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J Lipid Res. (2010) 51:1719–28. doi: 10.1194/jlr.M003525
84. Mauldin JP, Srinivasan S, Mulya A, Gebre A, Parks JS, Daugherty A, et al. Reduction in ABCG1 in type 2 diabetic mice increases macrophage foam cell formation. J Biol Chem. (2006) 281:21216–24. doi: 10.1074/jbc.M510952200
85. Mauldin JP, Nagelin MH, Wojcik AJ, Srinivasan S, Skaflen MD, Ayers CR, et al. Reduced expression of ATP-binding cassette transporter G1 increases cholesterol accumulation in macrophages of patients with type 2 diabetes mellitus. Circulation. (2008) 117:2785–92. doi: 10.1161/CIRCULATIONAHA.107.741314
86. Griffin E, Re A, Hamel N, Fu C, Bush H, McCaffrey T, et al. A link between diabetes and atherosclerosis: glucose regulates expression of CD36 at the level of translation. Nat Med. (2001) 7:840–6. doi: 10.1038/89969
87. Sampson MJ, Davies IR, Braschi S, Ivory K, Hughes DA. Increased expression of a scavenger receptor (CD36) in monocytes from subjects with type 2 diabetes. Atherosclerosis. (2003) 167:129–34. doi: 10.1016/S0021915002004215
88. Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. (2009) 50(Suppl.):S376–81. doi: 10.1194/jlr.R800087-JLR200
89. Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. (2005) 25:1213–9. doi: 10.1161/01.ATV.0000159891.73193.31
90. Miller YI, Shyy JY. Context-dependent role of oxidized lipids and lipoproteins in inflammation. Trends Endocrinol Metab. (2017) 28:143–52. doi: 10.1016/j.tem.2016.11.002
91. Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. (2012) 151:138–52. doi: 10.1016/j.cell.2012.06.054
92. Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. (2018) 123:1127–42. doi: 10.1161/CIRCRESAHA.118.312804
93. Pagler TA, Wang M, Mondal M, Murphy AJ, Westerterp M, Moore KJ, et al. Deletion of ABCA1 and ABCG1 impairs macrophage migration because of increased Rac1 signaling. Circ Res. (2011) 108:194–200. doi: 10.1161/CIRCRESAHA.110.228619
94. Nagao T, Qin C, Grosheva I, Maxfield FR, Pierini LM. Elevated cholesterol levels in the plasma membranes of macrophages inhibit migration by disrupting RhoA regulation. Arterioscler Thromb Vasc Biol. (2007) 27:1596–602. doi: 10.1161/ATVBAHA.107.145086
95. Williams JW, Martel C, Potteaux S, Esaulova E, Ingersoll MA, Elvington A, et al. Limited macrophage positional dynamics in progressing or regressing murine atherosclerotic plaques-brief report. Arterioscler Thromb Vasc Biol. (2018) 38:1702–10. doi: 10.1161/ATVBAHA.118.311319
96. Jackson WD, Weinrich TW, Woollard KJ. Very-low and low-density lipoproteins induce neutral lipid accumulation and impair migration in monocyte subsets. Sci Rep. (2016) 6:20038. doi: 10.1038/srep20038
97. Olin-Lewis K, Krauss RM, La Belle M, Blanche PJ, Barrett PH, Wight TN, et al. ApoC-III content of apoB-containing lipoproteins is associated with binding to the vascular proteoglycan biglycan. J Lipid Res. (2002) 43:1969–77. doi: 10.1194/jlr.M200322-JLR200
98. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
99. Karasawa T, Kawashima A, Usui-Kawanishi F, Watanabe S, Kimura H, Kamata R, et al. Saturated fatty acids undergo intracellular crystallization and activate the NLRP3 inflammasome in macrophages. Arterioscler Thromb Vasc Biol. (2018) 38:744–56. doi: 10.1161/ATVBAHA.117.310581
100. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation. (2018) 138:898–912. doi: 10.1161/CIRCULATIONAHA.117.032636
101. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
102. Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood. (2009) 113:2324–35. doi: 10.1182/blood-2008-03-146720
103. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
104. Devaraj S, Jialal I, Yun JM, Bremer A. Demonstration of increased toll-like receptor 2 and toll-like receptor 4 expression in monocytes of type 1 diabetes mellitus patients with microvascular complications. Metabolism. (2011) 60:256–9. doi: 10.1016/j.metabol.2010.01.005
105. Devaraj S, Cheung AT, Jialal I, Griffen SC, Nguyen D, Glaser N, et al. Evidence of increased inflammation and microcirculatory abnormalities in patients with type 1 diabetes and their role in microvascular complications. Diabetes. (2007) 56:2790–6. doi: 10.2337/db07-0784
106. Willecke F, Yuan C, Oka K, Chan L, Hu Y, Barnhart S, et al. Effects of high fat feeding and diabetes on regression of atherosclerosis induced by low-density lipoprotein receptor gene therapy in LDL receptor-deficient mice. PLoS ONE. (2015) 10:e0128996. doi: 10.1371/journal.pone.0128996
107. Zaharieva E, Kamenov Z, Velikova T, Tsakova A, El-Darawish Y, Okamura H. Interleukin-18 serum level is elevated in type 2 diabetes and latent autoimmune diabetes. Endocr Connect. (2018) 7:179–85. doi: 10.1530/EC-17-0273
108. Nakamura A, Shikata K, Hiramatsu M, Nakatou T, Kitamura T, Wada J, et al. Serum interleukin-18 levels are associated with nephropathy and atherosclerosis in Japanese patients with type 2 diabetes. Diabetes Care. (2005) 28:2890–5. doi: 10.2337/diacare.28.12.2890
109. Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tsutsumi Z, Takahashi S, et al. Elevated levels of interleukin-18 and tumor necrosis factor-α in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism. (2003) 52:605–8. doi: 10.1053/meta.2003.50096
110. Tall AR, Westerterp M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res. (2019) 60:721–7. doi: 10.1194/jlr.S091280
111. de la Roche M, Hamilton C, Mortensen R, Jeyaprakash AA, Ghosh S, Anand PK. Trafficking of cholesterol to the ER is required for NLRP3 inflammasome activation. J Cell Biol. (2018) 217:3560–76. doi: 10.1083/jcb.201709057
112. Fotakis P, Kothari V, Thomas DG, Westerterp M, Molusky MM, Altin E, et al. Anti-inflammatory effects of HDL (High-Density Lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. (2019) 39:e253–72. doi: 10.1161/ATVBAHA.119.313253
113. Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, et al. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. (2011) 2:e137. doi: 10.1038/cddis.2011.18
114. Zhuang T, Liu J, Chen X, Zhang L, Pi J, Sun H, et al. Endothelial Foxp1 suppresses atherosclerosis via modulation of Nlrp3 inflammasome activation. Circ Res. (2019) 125:590–605. doi: 10.1161/CIRCRESAHA.118.314402
115. Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol. (2010) 30:139–43. doi: 10.1161/ATVBAHA.108.179283
116. Singh RK, Haka AS, Asmal A, Barbosa-Lorenzi VC, Grosheva I, Chin HF, et al. TLR4 (Toll-Like Receptor 4)-dependent signaling drives extracellular catabolism of LDL (Low-Density Lipoprotein) aggregates. Arterioscler Thromb Vasc Biol. (2020) 40:86–102. doi: 10.1161/ATVBAHA.119.313200
117. Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. (2016) 539:294–8. doi: 10.1038/nature20117
118. Koliwad SK, Streeper RS, Monetti M, Cornelissen I, Chan L, Terayama K, et al. DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J Clin Invest. (2010) 120:756–67. doi: 10.1172/JCI36066
119. Shin KC, Hwang I, Choe SS, Park J, Ji Y, Kim JI, et al. Macrophage VLDLR mediates obesity-induced insulin resistance with adipose tissue inflammation. Nat Commun. (2017) 8:1087. doi: 10.1038/s41467-017-01232-w
120. Takahashi S, Sakai J, Fujino T, Miyamori I, Yamamoto TT. The very low density lipoprotein (VLDL) receptor–a peripheral lipoprotein receptor for remnant lipoproteins into fatty acid active tissues. Mol Cell Biochem. (2003) 248:121–7. doi: 10.1023/a:1024184201941
121. Kalugotla G, He L, Weber KJ, Daemen S, Reller A, Razani B, et al. Frontline Science: Acyl-CoA synthetase 1 exacerbates lipotoxic inflammasome activation in primary macrophages. J Leukoc Biol. (2019) 106:803–14. doi: 10.1002/JLB.3HI0219-045RR
122. Ruiz M, Bodhicharla R, Stahlman M, Svensk E, Busayavalasa K, Palmgren H, et al. Evolutionarily conserved long-chain Acyl-CoA synthetases regulate membrane composition and fluidity. Elife. (2019) 8:e47733. doi: 10.7554/eLife.47733.sa2
123. Kanter JE, Tang C, Oram JF, Bornfeldt KE. Acyl-CoA synthetase 1 is required for oleate and linoleate mediated inhibition of cholesterol efflux through ATP-binding cassette transporter A1 in macrophages. Biochim Biophys Acta. (2012) 1821:358–64. doi: 10.1016/j.bbalip.2011.10.008
124. Won KB, Lee SE, Lee BK, Park HB, Heo R, Rizvi A, et al. Longitudinal assessment of coronary plaque volume change related to glycemic status using serial coronary computed tomography angiography: A PARADIGM (Progression of AtheRosclerotic PlAque DetermIned by Computed TomoGraphic Angiography Imaging) substudy. J Cardiovasc Comput Tomogr. (2019) 13:142–7. doi: 10.1016/j.jcct.2018.12.002
125. Kim U, Leipsic JA, Sellers SL, Shao M, Blanke P, Hadamitzky M, et al. Natural history of diabetic coronary atherosclerosis by quantitative measurement of serial coronary computed tomographic angiography: results of the PARADIGM Study. JACC Cardiovasc Imaging. (2018) 11:1461–71. doi: 10.1016/j.jcmg.2018.04.009
126. Yahagi K, Kolodgie FD, Lutter C, Mori H, Romero ME, Finn AV, et al. Pathology of human coronary and carotid artery atherosclerosis and vascular calcification in diabetes mellitus. Arterioscler Thromb Vasc Biol. (2017) 37:191–204. doi: 10.1161/ATVBAHA.116.306256
127. Marso SP, Mercado N, Maehara A, Weisz G, Mintz GS, McPherson J, et al. Plaque composition and clinical outcomes in acute coronary syndrome patients with metabolic syndrome or diabetes. JACC Cardiovasc Imaging. (2012) 5(Suppl. 3):S42–52. doi: 10.1016/j.jcmg.2012.01.008
128. Gao X, Song J, Watase H, Hippe DS, Zhao X, Canton G, et al. Differences in carotid plaques between symptomatic patients with and without diabetes mellitus. Arterioscler Thromb Vasc Biol. (2019) 39:1234–9. doi: 10.1161/ATVBAHA.118.312092
129. Sun B, Li X, Liu X, Ge X, Lu Q, Zhao X, et al. Association between carotid plaque characteristics and acute cerebral infarction determined by MRI in patients with type 2 diabetes mellitus. Cardiovasc Diabetol. (2017) 16:111. doi: 10.1186/s12933-017-0592-9
130. Li S, Sun Y, Liang CP, Thorp EB, Han S, Jehle AW, et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ Res. (2009) 105:1072–82. doi: 10.1161/CIRCRESAHA.109.199570
131. Han S, Liang CP, DeVries-Seimon T, Ranalletta M, Welch CL, Collins-Fletcher K, et al. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. (2006) 3:257–66. doi: 10.1016/j.cmet.2006.02.008
132. Rayner KJ. Cell death in the vessel wall: the good, the bad, the ugly. Arterioscler Thromb Vasc Biol. (2017) 37:e75–81. doi: 10.1161/ATVBAHA.117.309229
133. Doran AC, Ozcan L, Cai B, Zheng Z, Fredman G, Rymond CC, et al. CAMKIIγ suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J Clin Invest. (2017) 127:4075–89. doi: 10.1172/JCI94735
134. Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L, et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature. (2018) 563:714–8. doi: 10.1038/s41586-018-0735-5
135. Manolescu AR, Witkowska K, Kinnaird A, Cessford T, Cheeseman C. Facilitated hexose transporters: new perspectives on form and function. Physiology. (2007) 22:234–40. doi: 10.1152/physiol.00011.2007
136. Nishizawa T, Kanter JE, Kramer F, Barnhart S, Shen X, Vivekanandan-Giri A, et al. Testing the role of myeloid cell glucose flux in inflammation and atherosclerosis. Cell Rep. (2014) 7:356–65. doi: 10.1016/j.celrep.2014.03.028
137. Freemerman AJ, Zhao L, Pingili AK, Teng B, Cozzo AJ, Fuller AM, et al. Myeloid Slc2a1-deficient murine model revealed macrophage activation and metabolic phenotype are fueled by GLUT1. J Immunol. (2019) 202:1265–86. doi: 10.4049/jimmunol.1800002
138. Gautier EL, Westerterp M, Bhagwat N, Cremers S, Shih A, Abdel-Wahab O, et al. HDL and Glut1 inhibition reverse a hypermetabolic state in mouse models of myeloproliferative disorders. J Exp Med. (2013) 210:339–53. doi: 10.1084/jem.20121357
139. Sarrazy V, Viaud M, Westerterp M, Ivanov S, Giorgetti-Peraldi S, Guinamard R, et al. Disruption of Glut1 in hematopoietic stem cells prevents myelopoiesis and enhanced glucose flux in atheromatous plaques of ApoE(-/-) mice. Circ Res. (2016) 118:1062–77. doi: 10.1161/CIRCRESAHA.115.307599
140. Sabanayagam C, Chee ML, Banu R, Cheng CY, Lim SC, Tai ES, et al. Association of diabetic retinopathy and diabetic kidney disease with all-cause and cardiovascular mortality in a multiethnic Asian population. JAMA Netw Open. (2019) 2:e191540. doi: 10.1001/jamanetworkopen.2019.1540
141. Pongrac Barlovic D, Harjutsalo V, Gordin D, Kallio M, Forsblom C, King G, et al. The association of severe diabetic retinopathy with cardiovascular outcomes in long-standing type 1 diabetes: a longitudinal follow-up. Diabetes Care. (2018) 41:2487–94. doi: 10.2337/dc18-0476
142. Rawshani A, Rawshani A, Franzen S, Sattar N, Eliasson B, Svensson AM, et al. Risk factors, mortality, and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. (2018) 379:633–44. doi: 10.1056/NEJMoa1800256
143. Haller H, Bertram A, Nadrowitz F, Menne J. Monocyte chemoattractant protein-1 and the kidney. Curr Opin Nephrol Hypertens. (2016) 25:42–9. doi: 10.1097/MNH.0000000000000186
144. Klessens CQF, Zandbergen M, Wolterbeek R, Bruijn JA, Rabelink TJ, Bajema IM, et al. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol Dial Transplant. (2017) 32:1322–9. doi: 10.1093/ndt/gfw260
145. Nguyen D, Ping F, Mu W, Hill P, Atkins RC, Chadban SJ. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology. (2006) 11:226–31. doi: 10.1111/j.1440-1797.2006.00576.x
146. You H, Gao T, Cooper TK, Brian Reeves W, Awad AS. Macrophages directly mediate diabetic renal injury. Am J Physiol Renal Physiol. (2013) 305:F1719–27. doi: 10.1152/ajprenal.00141.2013
147. Ninichuk V, Clauss S, Kulkarni O, Schmid H, Segerer S, Radomska E, et al. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36–3'PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am J Pathol. (2008) 172:628–37. doi: 10.2353/ajpath.2008.070601
148. Sullivan T, Miao Z, Dairaghi DJ, Krasinski A, Wang Y, Zhao BN, et al. CCR2 antagonist CCX140-B provides renal and glycemic benefits in diabetic transgenic human CCR2 knockin mice. Am J Physiol Renal Physiol. (2013) 305:F1288–97. doi: 10.1152/ajprenal.00316.2013
149. Seok SJ, Lee ES, Kim GT, Hyun M, Lee JH, Chen S, et al. Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol Dial Transplant. (2013) 28:1700–10. doi: 10.1093/ndt/gfs555
150. de Zeeuw D, Bekker P, Henkel E, Hasslacher C, Gouni-Berthold I, Mehling H, et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol. (2015) 3:687–96. doi: 10.1016/S2213-8587(15)00261-2
151. Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, et al. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant. (2017) 32:307–15. doi: 10.1093/ndt/gfv459
152. Niewczas MA, Pavkov ME, Skupien J, Smiles A, Md Dom ZI, Wilson JM, et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat Med. (2019) 25:805–13. doi: 10.1038/s41591-019-0415-5
153. Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res. (2014) 55:561–72. doi: 10.1194/jlr.P040501
154. Pedigo CE, Ducasa GM, Leclercq F, Sloan A, Mitrofanova A, Hashmi T, et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J Clin Invest. (2016) 126:3336–50. doi: 10.1172/JCI85939
155. Bornfeldt KE, Kramer F, Batorsky A, Choi J, Hudkins KL, Tontonoz P, et al. A novel type 2 diabetes mouse model of combined diabetic kidney disease and atherosclerosis. Am J Pathol. (2018) 188:343–52. doi: 10.1016/j.ajpath.2017.10.012
156. Schmidt AM. Diabetes mellitus and cardiovascular disease. Arterioscler Thromb Vasc Biol. (2019) 39:558–68. doi: 10.1161/ATVBAHA.119.310961
157. Abdul-Ghani M, DeFronzo RA, Del Prato S, Chilton R, Singh R, Ryder REJ. Cardiovascular disease and type 2 diabetes: has the dawn of a new era arrived? Diabetes Care. (2017) 40:813–20. doi: 10.2337/dc16-2736
158. Cavender MA, Norhammar A, Birkeland KI, Jorgensen ME, Wilding JP, Khunti K, et al. SGLT-2 inhibitors and cardiovascular risk: an analysis of CVD-REAL. J Am Coll Cardiol. (2018) 71:2497–506. doi: 10.1016/j.jacc.2018.01.085
159. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
160. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
161. Drucker DJ. The cardiovascular biology of glucagon-like peptide-1. Cell Metab. (2016) 24:15–30. doi: 10.1016/j.cmet.2016.06.009
162. Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. (2019) 393:31–9. doi: 10.1016/S0140-6736(18)32590-X
163. McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. (2019) 381:1995–2008. doi: 10.1056/NEJMoa1911303
164. Verma S. Potential mechanisms of sodium-glucose co-transporter 2 inhibitor-related cardiovascular benefits. Am J Cardiol. (2019) 124(Suppl. 1):S36–44. doi: 10.1016/j.amjcard.2019.10.028
165. Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. (2007) 369:1090–8. doi: 10.1016/S0140-6736(07)60527-3
166. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent Ethyl for hypertriglyceridemia. N Engl J Med. (2019) 380:11–22. doi: 10.1056/NEJMoa1812792
167. Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, et al. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes. (2010) 59:1030–7. doi: 10.2337/db09-1694
Keywords: apolipoprotein C3, atherosclerosis, diabetes, cytokines, efferocytosis, monocytosis, necrotic core
Citation: Kanter JE, Hsu C-C and Bornfeldt KE (2020) Monocytes and Macrophages as Protagonists in Vascular Complications of Diabetes. Front. Cardiovasc. Med. 7:10. doi: 10.3389/fcvm.2020.00010
Received: 05 November 2019; Accepted: 27 January 2020;
Published: 14 February 2020.
Edited by:
Godfrey Getz, University of Chicago, United StatesReviewed by:
Naoki Ishimori, Hokkaido University Hospital, JapanCopyright © 2020 Kanter, Hsu and Bornfeldt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin E. Bornfeldt, Ym9ybmZAdXcuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.