 Anais Audebrand
Anais Audebrand Laurent Désaubry
Laurent Désaubry Canan G. Nebigil
Canan G. Nebigil
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 24 January 2020
Sec. Cardio-Oncology
Volume 6 - 2019 | https://doi.org/10.3389/fcvm.2019.00194
This article is part of the Research Topic Emerging Challenges of Cardiovascular and Metabolic Dysfunctions in Cardio-oncology: From Bench to Bedside View all 11 articles
Novel anticancer medicines, including targeted therapies and immune checkpoint inhibitors, have greatly improved the management of cancers. However, both conventional and new anticancer treatments induce cardiac adverse effects, which remain a critical issue in clinic. Cardiotoxicity induced by anti-cancer treatments compromise vasospastic and thromboembolic ischemia, dysrhythmia, hypertension, myocarditis, and cardiac dysfunction that can result in heart failure. Importantly, none of the strategies to prevent cardiotoxicity from anticancer therapies is completely safe and satisfactory. Certain clinically used cardioprotective drugs can even contribute to cancer induction. Since G protein coupled receptors (GPCRs) are target of forty percent of clinically used drugs, here we discuss the newly identified cardioprotective agents that bind GPCRs of adrenalin, adenosine, melatonin, ghrelin, galanin, apelin, prokineticin and cannabidiol. We hope to provoke further drug development studies considering these GPCRs as potential targets to be translated to treatment of human heart failure induced by anticancer drugs.
New anticancer treatments have improved overall mortality (1). However, most of the anticancer drugs display a wide array of cardiovascular toxicities, leading to interruption of cancer therapies and maladaptive remodeling in hearts, affecting the short- and long-term quality of life (2–4). Oxidative stress and inflammation are inter-reliant processes involved in cardiovascular diseases and cancers (5, 6), along with apoptosis (7, 8) and necrosis (9). Tissue resident and circulating inflammatory cells (such as macrophages, mast cells, neutrophils, and monocytes) can also release both reactive oxygen species (ROS) and reactive nitrogen species (RNS) to induce an oxidative stress (6). Due to negligible detoxification capacity, the heart is particularly susceptible to ROS and RNS injury (10). Thus, high levels of ROS and RNS can debilitate cardiac cellular signaling pathways and can augment the gene expression of proinflammatory (11) and antioxidant defenses as the major cause for necrosis and apoptosis.
Classic chemotherapeutics particularly anthracyclines are the prototype of drugs causing cardiotoxicity (12). They can induce acute cardiotoxicity, including reversible hypotension, pericarditis and transient electrocardiographic abnormalities (changes in the ST-T waves, QT prolongation), and vasodilatation (13). However, after completion of cumulative dose regimens, anthracyclines promote irreversible cardiomyopathy (classified as type (1) cardiotoxicity), leading to heart failure (HF) (13, 14). Doxorubicin (DOX), the most frequently used anthracyclines can cause irreversible type 1 cardiotoxicity via accumulation of ROS and RNS (15, 16). They also target Topoisomerase IIβ (Top IIβ) in cardiomyocytes to induce DNA damage and apoptosis. Recently, the anthracycline mediated cardiotoxicity has been reviewed by Nebigil (17).
Targeted therapies also provoke some degree of cardiotoxicity. Targeting key tyrosine kinases (TKs) with TK antibodies and inhibitors has a remarkable achievement in cancer management. However, they also induce cardiotoxicity, because they block pathways that also regulate myocardial function (18). This cardiotoxicity is often reversible, and thus classified as type 2 cardiotoxicity (19, 20). It results in ultrastructural changes in cardiomyocytes, with reversible cardiac dysfunctions such as elevated blood pressure, thromboembolism, pericardial thickening, and arrhythmia (21). Type 1 and 2 forms of cardiotoxicity can overlap, when the classic and targeted therapeutics used together or subsequently. For example, in patient treated with anthracyclines earlier, trastuzumab, a monoclonal antibody anti-HER-2 can cause irreversible cardiac damage and left ventricular (LV) dysfunction (18, 22, 23). On the other hand, 27%, of patients who received both anthracycline and trastuzumab encountered cardiac dysfunction, while this rate was of 2-16% for patients treated with anthracyclines alone (24).
Recent studies have demonstrated that patients treated with immune checkpoint inhibitors (25) also develop myocarditis due to immune-related adverse events (6, 26). The therapeutic mechanisms of inhibitors mostly rely on blocking either the cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) or programmed cell death protein-1 (PD-1) pathways, while activating the host's immune system against cancer (27). CTLA-4 and PD-1 act as immune response inhibitors (6, 28). They suppress the T-cell response in order to prevent autoimmunity and maintain T-cell tolerance. Cardiac immune-related adverse events appear more frequently in patients treated with CTLA-4 antagonists compared with PD-1 inhibitors (29) and the myocarditis risk increases with combination therapy, leading to discontinuation in approximately 50% of patients (30, 31) probably due to targeting PD-1 and CTLA-4 in cardiomyocytes as well.
There are several cardioprotective therapeutics that have been used against anticancer-mediated cardiotoxicity. Their properties are summarized in Table 1.
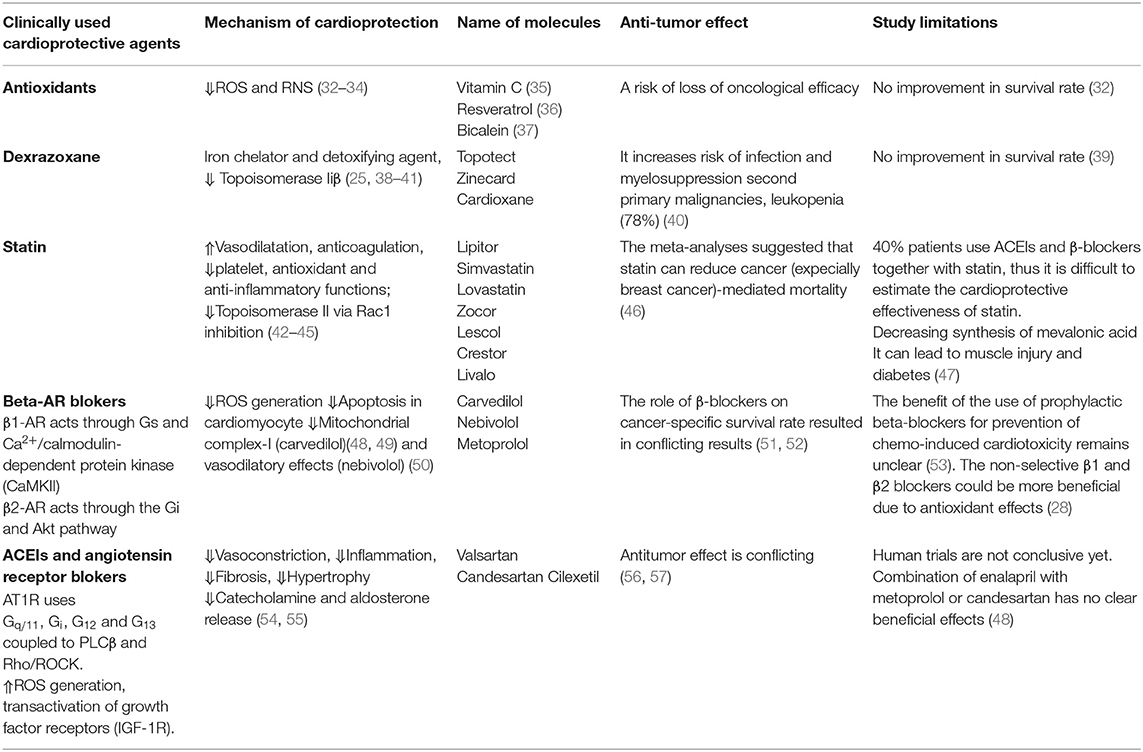
Table 1. Prophylactic cardioprotective agents.
Beneficial effects of antioxidants on LV remodeling and amelioration of contractility have been demonstrated in many experimental models of HF. For example, vitamin C effectively mitigates DOX-induced oxidative stress and apoptosis in rats (35). Resveratrol, a polyphenolic compound has also both prophylactic and therapeutic benefits in reversing DOX induced apoptosis and fibrosis in rat myocardium (36). Baicalein, a bioflavonoid can alleviate cardiotoxicity in mice (37). However, elimination of ROS and RNS by antioxidant drugs may be detrimental and even impair physiological cellular functions (58). There is also a risk of loss of oncological efficacy, because of the overlapping mechanisms with cardioprotective effects. Nevertheless, in clinic these approaches did not significantly improve survival rate and they may even increase mortality if they do not have other pharmacological properties (32, 59).
Dexrazoxane is an iron chelator and detoxifying agent that can prevent anthracycline-associated cardiotoxicity. It also acts on Topoisomerase IIβ to promote cardioprotective effects. Dexrazoxane is the only Food and Drug Administration (FDA) and the European Medicines Agency (EMA) approved cardioprotective drug to against chemotherapeutics-mediated HF (38, 60). However, its use in children and adolescent were forbidden by EMA in 2011, because it increases risk of infection, myelosuppression and second primary malignancies. These restrictions by EMA have been partially altered based on the new findings in 2018 (39). Only use of dexrazoxane was allowed in patients who have received a cumulative DOX at the dose of 300 mg/m (2) and are continuing with this medicine. Although dexrazoxane is a valuable option to prevent cardiotoxicity, it induces a severe leukopenia in 78% of cancer patients (40). Use of dexrazoxane is not recommended with non-anthracycline chemotherapy regimens.
Statins are used to lower low-density lipoprotein (LDL) and cholesterol amount in the blood on patients suffering to arterosclerosis (61). The mechanism involved in this action is due to inhibition of HMG-CoA reductase, which is involved the biosynthesis of cholesterol. Statins also display significant vasodilatation, platelet inhibition, anti-inflammatory, and antioxidant effects due to their pleiotropic effects (62, 63). Statin (atorvastatin) could be effective in maintenance of LV ejection fraction (LVEF) in patients treated with anthracycline (42). Moreover, it could limit oxidative stress and vascular inflammation (64) and activate autophagy (43) to promote cardioprotective effects against dasatinib. Statins also inhibits Top IIβ mediated DNA damage via Rac1 inhibition. Recent meta-analyses suggest that statins are at least equally potent as dexrazoxane in the prevention of anthracycline-induced cardiotoxicity (65). Calvillo-Argüelles and colleagues have found that in HER2+ breast cancer patients treated with trastuzumab with or without anthracycline, the concomitant statin use was associated with a lower risk of cardiotoxicity (44). Although, several studies on the influence of statin therapy on development of cancer risk resulted in conflicting results, the recent meta-analyses suggested that statin can reduce cancer-mediated mortality (46). However, there are some studies show that statin induces myopathies that may be due to decreased synthesis of mevalonic acid, leading to decreased energy generation and muscle injury. Another side effect associated with statin usage is new-onset diabetes (47). Many of the beneficial effects of a statin is due to inhibition of heterotrimeric G proteins, including Ras and Rho or Rac1 signaling (45). Thus, the specific Rho and Rac inhibitors may be more preferable targets for future chemo-preventive strategies.
As seven transmembrane (7TM) domain proteins, G protein-coupled receptors (GPCRs) represent the largest family of cell surface proteins (66). GPCRs regulate many physiological processes in every tissue, making the GPCR superfamily a major target for therapeutic intervention (67). The binding of agonists to GPCRs not only initiates the “classical,” signaling cascades through heterotrimeric G proteins (composed of the three subunits, Gα, Gβ, and Gγ). It can also activate G-protein-independent pathways involving β-arrestin (68, 69). Indeed, β-arrestins are identified as scaffolding proteins for MAP kinases and serine/threonine kinases cascades (70). The discovery that some GPCRs prefer to activate G-protein- or arrestin-mediated pathways has given rise to efforts to produce signal biased drugs (71). The drug discovery efforts aim to produce “biased” and/or allosteric ligands with less adverse effects without compromising their efficacy (72). In cardiovascular system, GPCRs can lead to hypertrophy, apoptosis, contraction, and cardiomyocytes survival. Some of the GPCR targeted therapeutics are used in clinic for treatment of heart failure and cardiotoxicity (Table 1).
β-adrenergic receptors (β-ARs) play a crucial role in cardiovascular regulation. It exists 3 types of β-ARs: β1, β2 and β3. Cardiac adrenergic receptor corresponding to β1-ARs whereas β2-ARs are localized on blood vessels. β1-ARs, are coupled to the Gαs and activate adenylyl cyclase to exert a positive inotropic, chronotropic and dromotropic effects in the heart. Indeed, β1-ARs increase heart rate, cardiac contractility and myocardial oxygen demand, thus promoting myocardial ischemia in patients with coronary heart disease. More importantly, persistent β1-ARs induce myocyte apoptosis and hypertrophy by activating CaMKII. On the opposite, persistent β2-ARs activation protects myocardium through a Gαi-mediated pathway, and activating PI3K, and Akt kinase probably via small G proteins (73). Administration of β2-AR agonist and β1-AR antagonist seems to be better than β2-AR antagonist in HF prevention. Interestingly, β3-AR is activated by catecholamines at higher concentration than those required to activate β1-AR and β2-AR (73). Thus, β3-AR plays an important protective role in the cardiovascular system during sympathetic over-stimulation.
It exists three mains β-AR blockers. The first generation of β-blockers, such as propranolol, inhibits both β1 and β2-ARs. The second generation of β-blockers (metoprolol) are cardioselective (β1-ARs).
The third generation of β-blockers (carvedilol and nebivolol) are vasodilators that not only inhibit β1 and α1-adrenoreceptors, but they also activate β3-adrenergic receptors (74). Carvedilol also reduces ROS generation and apoptosis in cardiomyocyte (49). Nebivolol has a vasodilatory effect mediated by nitric oxide release and avoid vasoconstriction to decrease blood pressure in hypertensive patients (50). Two clinical studies showed that carvedilol prevent cardiotoxicity in female patients diagnosed with breast cancer (75, 76). This cardioprotective effects has been attributed to its antioxidant and anti-apoptotic properties rather than its β-AR blocking activity, because carvedilol inhibits mitochondrial complex-I that promotes cardiotoxicity (77). This cardioprotective effect of carvedilol is superior than metoprolol and atenolol for preventing cardiomyocytes against DOX-induced apoptosis (78). In contrast, Avila and his colleague showed that carvedilol has no impact on the LVEF reduction induced by anthracycline in breast cancer patients (53). The recent meta-analyses on cancer patients have demonstrated that the use of β-blockers is not associated with cancer prognosis (51). Indeed, several studies on the influence of β-blockers on cancer-specific survival rate resulted in conflicting results (51, 52). The beneficial effects of non-selective β1 and β2 blockers could be due to their antioxidant effects (28).
Renin-angiotensin-aldosterone (RAAS) system regulates the cardiac and renal functions. Ang-II interacts with two GPCRs: AT-1R and AT-2R that are associated with opposite functions (79). However, most of the effects of renin-angiotensin system (RAS) are mediated by AT-1R, which promotes vasoconstriction, inflammation, fibrosis, hypertrophy, and releasing of catecholamine and aldosterone. AT-2 is implicated to vasodilatations, inhibition on cell growth, apoptosis, and bradykinin releasing. Increasing of Ang-II also stimulates sympathetic system and the production of aldosterone, leading to LV hypertrophy (80). Reduction of excessive Ang-II and aldosterone decrease cardiovascular morbidity and mortality. Indeed, AT-1R blockers ACE inhibitors are of paramount importance in treatment of cardiovascular diseases, including hypertension (54).
Several clinical trials indicate that Angiotensin-II receptor blockers (ARB) alleviate anthracycline cardiotoxicity (55), however, prospective trials are still needed for further validation. The expression of AngII and AT-1R have been found in many cell types of the tumor microenvironment (56). Thus, the RAS may alter remodeling of the tumor microenvironment and the immuno-suppressive milieu, thereby affecting tumor growth. In contrast, meta-analysis derived from the results of a group of trials demonstrated that ARB may promote the occurrences of new tumors (especially lung cancer) (57). These findings warrant further investigation.
The cardioprotective effects of combined ACEIs/ARBs and β-blockers have been evaluated during anthracycline, trastuzumab, or sequential chemotherapy. The combination of carvedilol and enalapril has been shown to preserve the LV function in adult patients treated with anthracyclines (81). However, other trials with combination of enalapril with metoprolol (82) or candesartan with metoprolol (83), ended up with disappointing results. Indeed, Guglin and his colleague recently demonstrated that both lisinopril and carvedilol do not prevent the cardiotoxicity of trastuzumab monotherapy in breast cancer patients (48). However, both drugs significantly alleviated the cardiotoxicity of anthracycline and trastuzumab sequential therapy. Although, ARBs, ACEIs, and β-blockers are necessary for treatment of HF, long-term studies are essential to validate whether ARBs have cardioprotective effects against the chronic or late-onset types of cardiotoxicities induced by cancer treatments.
We discus here newly identified GPCR agonists that exhibit cardioprotective effects against anti-cancer drugs in in vitro and in vivo preclinical models (Figure 1 and Table 2).
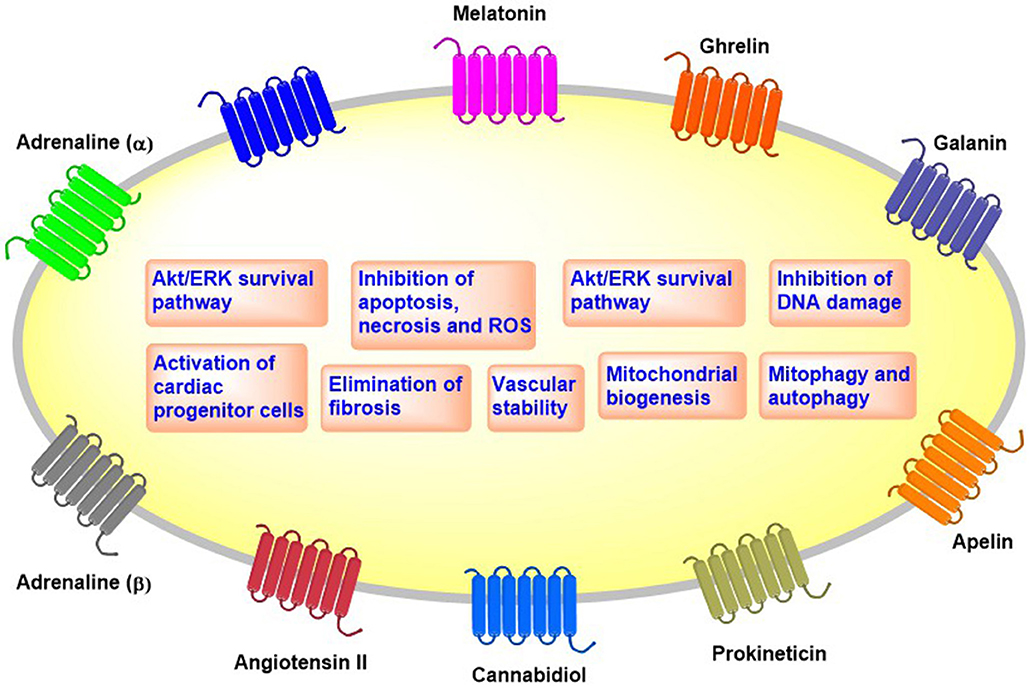
Figure 1. Overview of the cellular effects of cardioprotective GPCRs.
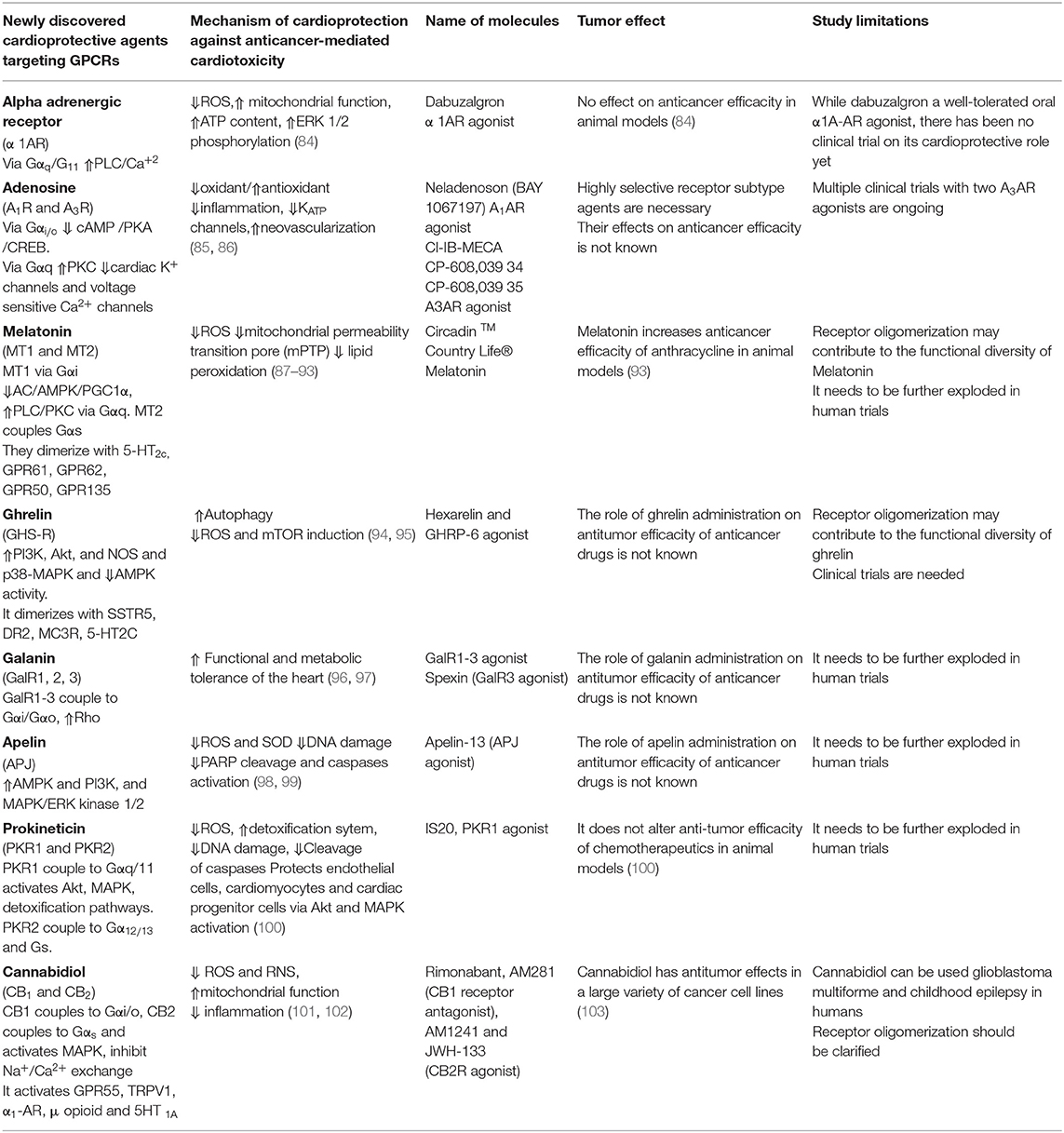
Table 2. Newly discovered cardioprotective agents targeting GPCRs.
Both the adrenergic receptors alpha 1 (α-AR1) and alpha 2 (α-AR2) bind catecholamines (epinephrine and norepinephrine). The α-AR1 couples to Gαq type, resulting in activation of phospholipase C, increasing Inositol trisphosphate (IP3) and diacylglycerol (DAG), and ultimately increasing the intracellular Ca2+ levels, leading to smooth muscle contraction and glycogenolysis (104). Cardiac α1-ARs activate phospholipase C and MAPK to promote ischemic preconditioning (105), cardiac hypertrophy (106)and cardiac cell survival (107). The knockout of α1A/α1B-adrenoceptor in mice develops small hearts (108) and aggravates the pressure overload–induced HF. In support of this study a large-scale clinical trial showed that doxazosin, an inhibitor of α-AR1 signaling, increases HF in hypertension patients (109). The α2-AR acts via Gαi/o to an inhibit adenylyl cyclase, decreasing the available cAMP (110). It also decreases neurotransmitter release and central vasodilation.
Dabuzalgron is a selective α1AR agonist that has been clinically examined against urinary incontinence (111). Recent study in mice showed that dabuzalgron displayed a strong cardioprotection against DOX-induced cardiotoxicity (84). It reduces ROS production and fibrosis, enhances contractile function, and preserves myocardial ATP content via regulating mitochondrial function, in DOX-treated mice. Cardioprotective signaling pathways of α1-AR is not limited to activation of MAPK1/2 pathways (84), it also activates pro-survival pathways such as A kinase anchoring protein-Lbc (AKAP-Lbc) and its anchored protein kinase D1 (PKD1) in cardiotoxicity mice models (112). Future studies should determine whether dabuzalgron can be used to treat chemotherapeutics-mediated HF in cancer patients.
Adenosine is a naturally occurring nucleoside formed by the degradation of ATP. Extracellular adenosine concentrations rise in response to hypoxia and other stress (113). However, chronic adenosine elevation can increase inflammation, cytokine release, and induces brain dopamine depletion, fibrosis and kidney damage (114). The adenosine receptors A1R, A2AR, A2BR, and A3R can sense an imbalance of demand and supply of oxygen and nutrients (115). Adenosine exerts a significant cardioprotective effect during cardiac ischemia by activation of the A1R and A3R (86, 116). However, full A1R agonists have promote several cardiovascular adverse effects due to its off-target activation as well as desensitization of A1R, leading to tachyphylaxis (117). In contrast, a selective partial agonist for A1AR improves cardiac function without promoting atrioventricular blocks, bradycardia, or unfavorable effect on blood pressure (118, 119).
A selective A3R agonist (Cl-IB-ME) mitigates bradycardia, elevated serum creatine kinase levels and cardiac histopathological changes in DOX-treated mice. Cardioprotective effect of Cl-IB-ME involves the inhibition of ROS production and inflammation induced by DOX in vivo (85). A3AR activation also prevents perioperative myocardial ischemic injury (120), protects ischemic cardiomyocytes by preconditioning (121), and induces ischemic tolerance that is dependent on KATP channels (122). This cardioprotective effects A3R agonists were absence in A3AR deficient mouse cardiomyocytes, showing an A3AR-mediated effect. On the opposite to A1AR, A3AR is expressed at very low levels in adult ventricular cardiomyocytes. The efficacy of two A3AR agonists is currently examined in multiple clinical trials (123).
Melatonin is a pineal gland hormone synthesized from the amino acid tryptophan and is secreted into both the bloodstream and cerebrospinal fluid. It regulates circadian, seasonal, and transgenerational time cycles. Melatonin acts through 2 GPCRs, MT1, and MT2 that are linked to Gαi/Gαo or Gαq/Gα11 to induce anti-adrenergic effects (124). These melatonin receptors are ubiquitously present in central and peripheral organs, including the cardiovascular system. Melatonin regulates blood pressure and heart rate either normalizing the circadian rhythm of blood pressure and ameliorating nocturnal hypertension, or directly acting on heart and blood vessels (125). They also regulate the renin-angiotensin system (126) and mitochondrial function (127).
Melatonin inhibits necrosis and apoptosis, and improves DOX-mediated cardiac dysfunction without compromising the antitumor effect of DOX in mice (87) and rats (88). The mechanism involved in cardioprotective effect against DOX-cardiotoxicity has been attributed to its antioxidant effect (89) and suppression of lipid peroxidation (90). Recent studies showed that melatonin activates AMPK, PGC1α (91), and sirtuins (92) to attenuate acute DOX-cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Indeed, high doses of melatonin are essential to reach adequate subcellular concentrations to exert these cardioprotective effects (128).
Ramelteon, is a dual MT1 and MT2 melatonin receptor agonist used for insomnia that displays a strong cardioprotective effect in the models of ischemic HF induced by the coronary artery ligation (129), chronic intermittent hypoxia-induced HF (130), and isoproterenol-induced myocardial infarction (131, 132). Unfortunately, the effect of ramelteon in anticancer-mediated cardiotoxicity has not been studied yet. Melatonin can also enhance antitumor effects of anthracycline in animal model (93). Thus, the combined treatment of anthracyclines and melatonin needs to be further explored in cancer patients.
Ghrelin is a growth hormone-releasing and orexigenic peptide that acts through growth hormone secretagogue receptor (GHS-R) in the brain. However, expression of GHS-R in cardiovascular system is controversial. Ghrelin regulates energy balance, body weight maintenance, and metabolism (133). Roles of ghrelin in protecting heart function and reducing mortality after myocardial infarction are partly due to its role on the cardiac vagal afferent nerve terminals (inhibition of cardiac sympathetic and activation of cardiac parasympathetic nerve activity) (134). Ghrelin significantly decreased blood pressure and heart rate in healthy human (135) and prevents the arrhythmia in the mice model of myocardial infarction (136).
Ghrelin significantly improves LV functions and attenuates fibrosis (137) and development of cachexia (138) in rat HF model. Ghrelin inhibits the DOX -induced cardiotoxicity in mice hearts and cardiomyocytes by blocking AMPK activity and activating the p38-MAPK pathway, which suppresses excessive autophagy (94). A ghrelin-containing salmon extract given per os was found to alleviate the cardiotoxicity of DOX in mice, mimicking cardioprotective effect of synthetic ghrelin (95). Cardioprotective effect of ghrelin can also be due to its angiogenic properties in ischemic tissue (139–141). Ghrelin via GHS-R ameliorates impaired angiogenesis by increasing VEGF levels in the ischemic hearts of diabetic rats (140) and in a rat myocardial infarction model (142). Despite the potent synthetic agonist of GHS-R, RM-131 plays an anticatabolic effect in chronic HF models of rat (143), its role in anti-cancer drug mediated cardiotoxicity has not been studied yet.
Galanin is a neuropeptide present in the nervous system and some organs (144) that uses 3 kinds of GPCRs called GalR1, GalR2 and GalR3 that are all expressed in the cardiovascular system (145). The elevated sympathetic activity during cardiac failure stimulates the release of galanin. This neuropeptide is a one of the sympathetic co-transmitters together with ATP and neuropeptide Y (NPY), in addition to norepinephrine. Galanin released by sympathetic nerves may diminish vagal neurotransmission (146). Indeed, galanin via GalR1 inhibits vagal bradycardia (147). In accord with this study, GalR1 inhibitor, M40 improves cardiac function and attenuate remodeling after myocardial infarction in rats (148). In contrast, an peptide agonist of galanin receptors and the full-length galanin reduce infarct size and the cardiac damage markers in ischemia and reperfusion rat model (96). Indeed, the natural N fragments of Galanin that have more affinity to GalR2 than GalR1 and GalR3 (145) limit acute myocardial infarction in rats in vivo (149). Moreover, natural galanin and GalR2 agonist have shown to increase cell viability by suppressing caspase-3 and 9 activity against hypoxic insults in other cells (97).
The GalR1-3 agonist [RAla14, His15]-galanin (2-15) exhibits cardioprotective properties against DOX-mediated cardiac injury in rats. Coadministration of this agonist with DOX has prevented the increase in plasma CK-MB activity and improved the parameters of cardiac function and caused weight gain. The obtained results demonstrate the ability of a novel agonist of galanin receptors GalR1-3 to attenuate DOX-induced cardiotoxicity (150). To conclude, galanin peptides via GalR1-3 alleviate the cardiac dysfunctions induced by DOX. The role of GalR1-3 agonist on anti-tumor effect of DOX in cancer mice model needs to be studied.
Apelin is an endogenous peptide that acts trough the APJ receptor that is 54% identical with AngII receptor. However, angiotensin II does not bind to APJ (151). Mature apelin, apelin-36, and its shorter forms (apelin-17, -12, and -13) result from the cleavage of pre-pro-apelin. Apelin itself can also be cleaved in vitro by the angiotensin-converting enzyme 2 (ACE2) (152). Apelin has a positive inotropic effect in vitro (153) and is involved in lowering arterial blood pressure (154), inducing arterial vasodilation (155), and improving cardiac output (156). It protects the heart against ischemia/reperfusion-mediated injury and promotes angiogenesis (157).
Moreover, in APJ knockout mice exhibited more severe heart injury, including impaired contractility functions and survival rate after DOX treatments as compare to wild type mice receiving DOX (98). On the other hand, apelin protects H9c2 cardiomyocytes overexpressing APJ against DOX-mediated cell death. These findings all together have suggested that the suppression of APJ expression can worsen DOX-induced cardiotoxicity. Impairment of the endogenous apelin-APJ system may partially depress the protective signaling in DOX-treated hearts (98). Apelin-13 pretreatment attenuates cisplatin-induced cardiotoxicity by inhibiting apoptosis in cardiomyocytes via activation of MAPKs and PI3K/Akt signaling in vitro and in vivo in mice heart (99). The mechanism of cardioprotection in vivo involves an attenuation of the ROS and superoxide anion accumulation, inhibition of DNA damage, and suppression of PARP and caspases as well as an improvement in angiogenesis.
Importantly, high levels of apelin and APJ have been found in several cancer types that may be connected with obesity. For example, increase levels of Apelin-12 in colon cancer patients with obesity (158), or elevated levels of apelin-36 in endometrial and breast cancer patients with obesity (159–161) have been found. The role of AJP agonist on anti-tumor effect of anti-cancer agents in cancer mice model needs to be studied. Thus, promoting APJ signaling in heart may represent an interesting strategy to alleviate the cardiotoxicity of anticancer treatments.
Prokineticins are peptides found in milk and macrophages (162). These peptides are called prokineticin because of their first identified biological activity was a prokinetic effect on smooth muscle cells of the gastrointestinal tract (163). Prokineticins exist as two isoforms, PROK1 and PROK2 that are expressed in all mammalian tissues (164). They are angiogenic factors (165) and induce mitogenic and survival pathway in lymphocytes and hematopoietic stem cells (166), neuronal cells (167, 168), cardiomyocytes (169), and endothelial cells (170). PROK1 and PROK2 exert their biological activity on prokineticin receptors 1 and 2 (PKR1 and PKR2) (171).
We have showed that PROK2/PKR1 can induce angiogenesis, while PROK2/PKR2 signaling promotes endothelial cell fenestration and disorganization (170). In cardiomyocytes PKR1 signaling activates Gα11/Akt pathway to reduce cardiomyocyte death (169), while PKR2 signaling induces hypertrophic cardiomyopathy (172). Indeed, PKR1 gene therapy promotes resistance to ischemia, protects heart against myocardial infarction, and ameliorates heart structure and function (169). Overexpression of PKR1 in transgenic mice hearts promotes neovascularization, suggesting a novel myocardial-epicardial interaction that is involved in differentiation of epicardial progenitor cells (EPDCs) in to vasculogenic cells type by a paracrine PROK2/PKR1 signaling (173).
PKR1 signaling controls epithelial mesenchymal transformation (EMT) during heart (174) and kidney development (175). PKR1 controls fate of tcf21+ fibroblast (176) and Wt1+ epicardial cells (174). PKR1 epigenetically controls stemness and differentiation of these cells, unraveling a new neovasculogenic pathway vs. adipogenesis (177). PKR1 inhibits adipogenesis and reduce adipocyte accumulation under high fat diet regime of mice (178, 179). PKR1 controls trans-endothelial insulin uptake, preadipocyte proliferation and adipogenesis (180). Lack of PKR1 in mice induces developmental defect in heart and kidney and in adult stage insulin resistance and obesity (181, 182).
In 2015, Gasser et al. discovered the first PKR1 agonists called IS20 (183). This agonist prevents the formation of cardiac lesions and ameliorates the cardiac function and survival after myocardial infarction in mice. IS20 inhibits DOX-mediated cardiotoxicity in cultured cardiac cells including cardiomyocytes, endothelial and progenitor cell as well as in mice models of acute and chronic cardiotoxicity. Importantly, these small molecules did not alter cytotoxic effect of DOX in cancer cells and in vivo cancer cell line- derived xenograft mice model (100). This study also described how classic chemotherapeutics, anthracyclines affect cardiac cells in dose-and time-dependent manner and how they impair NFR2 defense mechanism. These results indicate that PKR1 is a target for development of cardioprotective drugs.
Cannabidiol is the most abundant non-psychoactive, derived cannabinoid (184). In the low nanomolar range, cannabidiol act as an antagonist of cannabinoid 1 receptor (CB1R) and cannabinoid 2 receptor (CB2R), while it has agonist/inverse agonist actions at micromolar concentrations (185, 186). Cannabidiol activate TRPV1 channel and several GPCRs, including the orphan receptor GPR55, the putative Abn-CBD receptor, α1-adrenoreceptors, 5HT1A receptors and μ opioid receptors (187). Several studies showed cardioprotective effects of cannabidiol in animal models of myocardial ischemic reperfusion injury (188), and myocardial infarction (189). It also ameliorates cardiac functions in diabetic cardiomyopathy (186).
Cannabidiol protects hearts against DOX-induced cardiac injury, in rats (101) and in mice (102). It improves cardiac dysfunction by (i) attenuating ROS /RNS accumulation, (ii) preserving mitochondrial function and biogenesis, (iii) promoting cell survival, and (v) decreasing myocardial inflammation. The involvement of CB1 and CB2 signaling were not clarified in these studies. Recent data has shown that CB1R and CB2R receptors have opposite effects. Indeed CB1R antagonists and CB2R agonists both protect the heart against clozapine-toxicity (190). Thus, CB1R antagonist reduces DOX-induced cardiotoxicity and decreased cortical cerebral infarction (191). By contrast, two CB2R agonists JWH-133, AM 1241 alleviate quetiapine cardiotoxicity (192). Moreover, cannabidiol by itself display cytotoxicity in many cancer cell lines, and anti-tumor effects in cancer mice models (103), suggesting that cannabidiol may have a synergistic effect with antineoplastic drugs in the use of cardioprotective agents. In fact, the cannabinoid HU-331 has been shown to be more potent and less cardiotoxic than DOX (193). Indeed, Insys Therapeutics has obtained FDA orphan drug designation for Cannabidiol for the treatment of multiform glioblastoma and childhood epilepsy.
Cardiotoxicity induced by anti-cancer therapy may occur when the anticancer agent targets a common signaling pathway that are essential to maintain the functions of both cardiac and cancer cells. It can also involve off-target effects due to non-selective actions of anti-cancer agents. The choice of the cardioprotective therapeutic approach relies on the delicate balance between the efficiency of anti-neoplastic drugs and the management of cardiovascular complication.
Cardioprotective utility of GPCR ligands will require validation of preferentially expression of these GPCRs in both cancer and cardiac cells, and identification of their signaling (e.g., G-protein- or arrestin-mediated pathways) and functional roles (Figure 2A). Whether these cardioprotective ligands interfere with the anti-tumor effect of the chemotherapeutics should be studied as well. The human inducible pluripotent stem cell derived cardiomyocytes (hiPSC-CMs), iPSC-CM-derived 3D cultures and organoids provide human-based model systems to explore the molecular mechanisms of cardiotoxicity and cardioprotection (194). They may also serve as a platform for personalized medicine. Thus, GPCR ligand efficacy can be optimized and their side-effects can be examined in hiPSC-CMs and organoids.
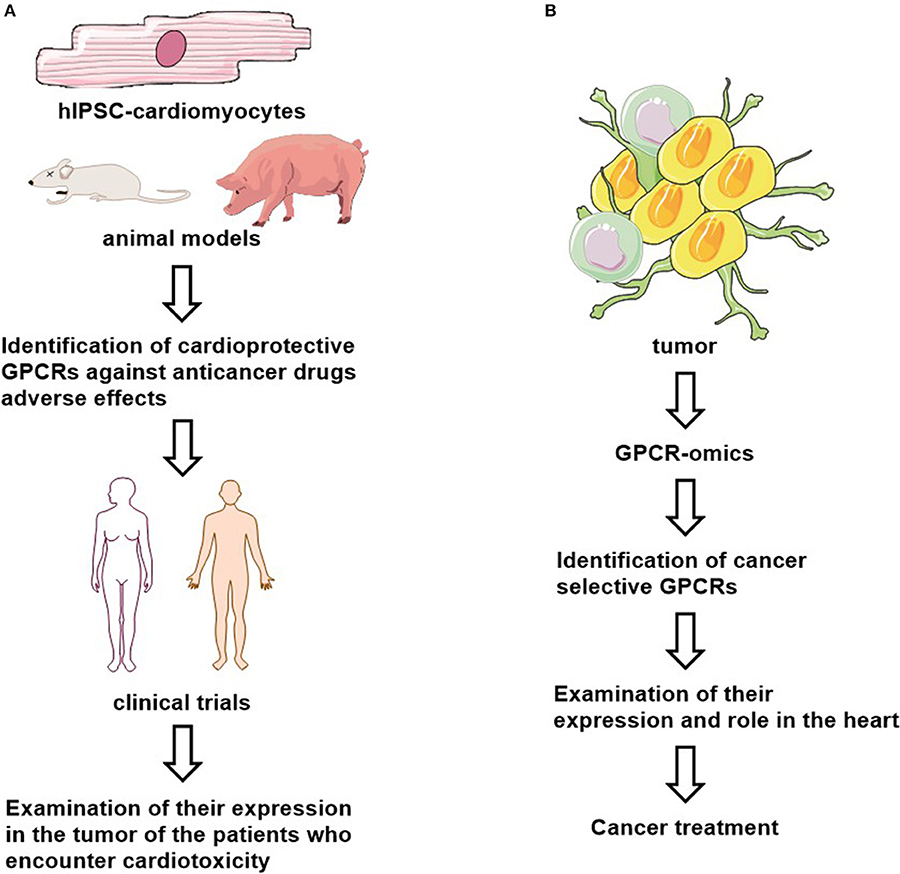
Figure 2. Proposed cardioprotective (A) and anti-cancer (B) drug studies.
In addition, most of the data regarding the efficacy of cardioprotective GPCR-ligands against cancer therapy mediated-cardiotoxicity have been obtained from small animal models of cardiotoxicity and cancer cell-derived xenograft mice models. Therefore, further studies in bigger animals are necessary to examine their efficacy and adverse effects before these findings can be translated to a human study.
Interestingly, certain cancer cell types may retain a GPCR expression pattern via serving novel biomarkers and/or as valuable therapeutic targets. For example, GPR161 is functionally expressed in breast cancer (195) and GPRC5A in pancreatic cancer (196) and GPR68 in the tumor microenvironment (197). However, both CD97 and GPR56 are highly express in multiple cancer types and in normal tissues (198). Moreover, many mutated GPCRs such as GPR110, GPR112, GPR125, GPR126, GPR98, and GPR110 have been found in certain cancers (199). These findings suggest that different types of cancers may be characterized by a specific onco-GPCR-ome (67). It could be interesting to examine if there is a “GPCR signature” in heart as well. In precision medicine, selectively targeting GPCRs in specific cancers can lead to a novel class of anti-cancer drugs with less adverse cardiac effects, after defining their expression and their role in heart (Figure 2B).
AA, LD, and CN participated in writing and drawings of the manuscript.
The publication was made possible by grants from European ERA-NET, ERA-CVD-JC2016, French government managed by Agence Nationale de la Recherche (ANR-16-ECVD-0005-01) Centre National de la Recherche Scientifique (CNRS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
GPCR, G protein-coupled receptor; DOX, Doxorubicin; HF, Heart failure; ROS, Reactive oxygen species; RNS, Reactive nitrogen species; Top Iiβ, Topoisomerase Iiβ; LVEF, LV ejection fraction; HER2, Hergulin2; CTLA-4, T-lymphocyte associated antigen-4; PD-1, Programmed cell death protein-1; LDL, low-density lipoprotein; HMG-CoA, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase; PI3K, Phosphoinositide 3-kinases; MAPK, Mitogen-activated protein kinases; β-ARs, β-adrenergic receptors; ? -ARs???-adrenergic receptors; CaMKII, Calmodulin-dependent protein kinase II; Ang-II, Angiotensin II; AT-1R and AT-2R, Angiotensin receptors; RAS, Renin-angiotensin system; ARB, Angiotensin-II receptor blockers; IP3, Inositol trisphosphate; DAG, Diacylglycerol; PKD1, Anchored protein kinase D1; ATP, Adenosine-triphosphate; A1R, A2AR, A2BR and A3R, Adenosine receptors; MT1 and MT2, Melatonin receptors; mPTP, Mitochondrial permeability transition pore; GHS-R, Ghrelin receptor, growth hormone secretagogue receptor; VEGF, vascular endothelial growth factor; GalR1, GalR2 and GalR3, Galanin receptors; APJ, Apelin receptor; ACE2, Angiotensin-converting enzyme 2; PARP, Poly(ADP-ribose) polymerase; PROK1 and PROK2, Prokineticins 1 and 2; PKR1 and PKR2, Prokineticin receptors; hiPSC-CMs, Inducible pluripotent stem cell derived cardiomyocytes.
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. (2011) 61:69–90. doi: 10.3322/caac.20107
2. Moslehi JJ, Deininger M. Tyrosine kinase inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol. (2015) 33:4210–8. doi: 10.1200/JCO.2015.62.4718
3. Mercurio V, Pirozzi F, Lazzarini E, Marone G, Rizzo P, Agnetti G, et al. Models of Heart Failure Based on the Cardiotoxicity of Anticancer Drugs. J Card Fail. (2016) 22:449–58. doi: 10.1016/j.cardfail.2016.04.008
4. Armenian SH, Lacchetti C, Lenihan D. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: american society of clinical oncology clinical practice guideline summary. J Oncol Pract. (2017) 13:270–5. doi: 10.1200/JOP.2016.018770
5. Galdiero MR, Varricchi G, Marone G. The immune network in thyroid cancer. Oncoimmunology. (2016) 5:e1168556. doi: 10.1080/2162402X.2016.1168556
6. Varricchi G, Galdiero MR, Tocchetti CG. Cardiac toxicity of immune checkpoint inhibitors: cardio-oncology meets immunology. Circulation. (2017) 136:1989–92. doi: 10.1161/CIRCULATIONAHA.117.029626
7. Priya LB, Baskaran R, Huang CY, Padma VV. Neferine ameliorates cardiomyoblast apoptosis induced by doxorubicin: possible role in modulating NADPH oxidase/ROS-mediated NFkappaB redox signaling cascade. Sci Rep. (2017) 7:12283. doi: 10.1038/s41598-017-12060-9
8. Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci Rep. (2017) 7:44735. doi: 10.1038/srep44735
9. Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal D, Fernyhough P, et al. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci USA. (2014) 111:E5537–44. doi: 10.1073/pnas.1414665111
10. Minotti G, Salvatorelli E, Menna P. Pharmacological foundations of cardio-oncology. J Pharmacol Exp Ther. (2010) 334:2–8. doi: 10.1124/jpet.110.165860
11. Biswas SK. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxid Med Cell Longev. (2016) 2016:5698931. doi: 10.1155/2016/5698931
12. Ghigo A, Li M, Hirsch E. New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim Biophys Acta. (2016) 1863:1916–25. doi: 10.1016/j.bbamcr.2016.01.021
13. Licata S, Saponiero A, Mordente A, Minotti G. Doxorubicin metabolism and toxicity in human myocardium: role of cytoplasmic deglycosidation and carbonyl reduction. Chem Res Toxicol. (2000) 13:414–20. doi: 10.1021/tx000013q
14. Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. (2007) 49:330–52. doi: 10.1016/j.pcad.2006.10.002
15. Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat Rev Cardiol. (2010) 7:564–75. doi: 10.1038/nrcardio.2010.121
16. Scott JM, Khakoo A, Mackey JR, Haykowsky MJ, Douglas PS, Jones LW. Modulation of anthracycline-induced cardiotoxicity by aerobic exercise in breast cancer current evidence and underlying mechanisms. Circulation. (2011) 124:642–50. doi: 10.1161/CIRCULATIONAHA.111.021774
17. Nebigil CG, Désaubry L. Updates in anthracycline-mediated cardiotoxicity. Front Pharmacol. (2018) 9:1262. doi: 10.3389/fphar.2018.01262
18. Suter TM, Ewer MS. Cancer drugs and the heart: importance and management. Eur Heart J. (2013) 34:1102–11. doi: 10.1093/eurheartj/ehs181
19. Ewer MS, Lippman SM. Type II chemotherapy-related cardiac dysfunction: time to recognize a new entity. J Clin Oncol. (2005) 23:2900–2. doi: 10.1200/JCO.2005.05.827
20. Ewer MS, Vooletich MT, Durand JB, Woods ML, Davis JR, Valero V, et al. Reversibility of trastuzumab-related cardiotoxicity: new insights based on clinical course and response to medical treatment. J Clin Oncol. (2005) 23:7820–6. doi: 10.1200/JCO.2005.13.300
21. Chen ZI, Ai DI. Cardiotoxicity associated with targeted cancer therapies. Mol Clin Oncol. (2016) 4:675–81. doi: 10.3892/mco.2016.800
22. Timolati F, Ott D, Pentassuglia L, Giraud MN, Perriard JC, Suter TM, et al. Neuregulin-1 beta attenuates doxorubicin-induced alterations of excitation-contraction coupling and reduces oxidative stress in adult rat cardiomyocytes. J Mol Cell Cardiol. (2006) 41:845–54. doi: 10.1016/j.yjmcc.2006.08.002
23. Zamorano JL, Lancellotti P, Rodriguez Munoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur J Heart Fail. (2017) 19:9–42. doi: 10.1002/ejhf.654
24. Bengala C, Zamagni C, Pedrazzoli P, Matteucci P, Ballestrero A, Da Prada G, et al. Cardiac toxicity of trastuzumab in metastatic breast cancer patients previously treated with high-dose chemotherapy: a retrospective study. Br J Cancer. (2006) 94:1016–20. doi: 10.1038/sj.bjc.6603060
25. Swain SM, Vici P. The current and future role of dexrazoxane as a cardioprotectant in anthracycline treatment: expert panel review. J Cancer Res Clin Oncol. (2004) 130:1–7. doi: 10.1007/s00432-003-0498-7
26. Altan M, Toki MI, Gettinger SN, Carvajal-Hausdorf DE, Zugazagoitia J, Sinard JH, et al. Immune checkpoint inhibitor-associated pericarditis. J Thorac Oncol. (2019) 14:1102–8. doi: 10.1016/j.jtho.2019.02.026
27. Tajiri K, Ieda M. Cardiac complications in immune checkpoint inhibition therapy. Front Cardiovasc Med. (2019) 6:3. doi: 10.3389/fcvm.2019.00003
28. Varricchi G, Galdiero MR, Marone G, Criscuolo G, Triassi M, Bonaduce D, et al. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open. (2017) 2:e000247. doi: 10.1136/esmoopen-2017-000247
29. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. (2012) 24:207–12. doi: 10.1016/j.coi.2011.12.009
30. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. (2016) 375:1749–55. doi: 10.1056/NEJMoa1609214
31. Tawbi HA, Bolejack V, Burgess M, Schuetze S. PD-1 inhibition in sarcoma still needs investigation - Authors' reply. Lancet Oncol. (2018) 19:e7. doi: 10.1016/S1470-2045(17)30922-1
32. Arcaro A, Pirozzi F, Angelini A, Chimenti C, Crotti L, Giordano C, et al. Novel perspectives in redox biology and pathophysiology of failing myocytes: modulation of the intramyocardial redox milieu for therapeutic interventions-a review article from the working group of cardiac cell biology, italian society of cardiology. Oxid Med Cell Longev. (2016) 2016:6353469. doi: 10.1155/2016/6353469
33. Aon MA, Cortassa S, O'Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. (2010) 1797:865–77. doi: 10.1016/j.bbabio.2010.02.016
34. Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol. (2014) 73:26–33. doi: 10.1016/j.yjmcc.2014.03.011
35. Akolkar G, da Silva Dias D, Ayyappan P, Bagchi AK, Jassal DS, Salemi VMC, et al. Vitamin C mitigates oxidative/nitrosative stress and inflammation in doxorubicin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol. (2017) 313:H795–809. doi: 10.1152/ajpheart.00253.2017
36. Shoukry HS, Ammar HI, Rashed LA, Zikri MB, Shamaa AA, Abou Elfadl SG, et al. Prophylactic supplementation of resveratrol is more effective than its therapeutic use against doxorubicin induced cardiotoxicity. PLoS ONE. (2017) 12:e0181535. doi: 10.1371/journal.pone.0181535
37. Sahu BD, Kumar JM, Kuncha M, Borkar RM, Srinivas R, Sistla R. Baicalein alleviates doxorubicin-induced cardiotoxicity via suppression of myocardial oxidative stress and apoptosis in mice. Life Sci. (2016) 144:8–18. doi: 10.1016/j.lfs.2015.11.018
38. Deng S, Yan T, Jendrny C, Nemecek A, Vincetic M, Godtel-Armbrust U, et al. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC Cancer. (2014) 14:842. doi: 10.1186/1471-2407-14-842
39. Reichardt P, Tabone MD, Mora J, Morland B, Jones RL. Risk-benefit of dexrazoxane for preventing anthracycline-related cardiotoxicity: re-evaluating the European labeling. Future Oncol. (2018) 14:2663–76. doi: 10.2217/fon-2018-0210
40. Cvetkovic RS, Scott LJ. Dexrazoxane : a review of its use for cardioprotection during anthracycline chemotherapy. Drugs. (2005) 65:1005–24. doi: 10.2165/00003495-200565070-00008
41. Shaikh F, Dupuis LL, Alexander S, Gupta A, Mertens L, Nathan PC. Cardioprotection and second malignant neoplasms associated with dexrazoxane in children receiving anthracycline chemotherapy: a systematic review and meta-analysis. J Natl Cancer Inst. (2016) 108:djv357. doi: 10.1093/jnci/djv357
42. Acar Z, Kale A, Turgut M, Demircan S, Durna K, Demir S, et al. Efficiency of atorvastatin in the protection of anthracycline-induced cardiomyopathy. J Am Coll Cardiol. (2011) 58:988–9. doi: 10.1016/j.jacc.2011.05.025
43. Andres AM, Hernandez G, Lee P, Huang C, Ratliff EP, Sin J, et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal. (2014) 21:1960–73. doi: 10.1089/ars.2013.5416
44. Calvillo-Arguelles O, Abdel-Qadir H, Michalowska M, Billia F, Suntheralingam S, Amir E, et al. Cardioprotective effect of statins in patients with her2-positive breast cancer receiving trastuzumab therapy. Can J Cardiol. (2019) 35:153–9. doi: 10.1016/j.cjca.2018.11.028
45. Henninger C, Fritz G. Statins in anthracycline-induced cardiotoxicity: Rac and Rho, and the heartbreakers. Cell Death Dis. (2017) 8:e2564. doi: 10.1038/cddis.2016.418
46. Zhong S, Zhang X, Chen L, Ma T, Tang J, Zhao J. Statin use and mortality in cancer patients: Systematic review and meta-analysis of observational studies. Cancer Treat Rev. (2015) 41:554–67. doi: 10.1016/j.ctrv.2015.04.005
47. Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. (2010) 375:735–42. doi: 10.1016/S0140-6736(09)61965-6.
48. Guglin M, Krischer J, Tamura R, Fink A, Bello-Matricaria L, McCaskill-Stevens W, et al. Randomized trial of lisinopril versus carvedilol to prevent trastuzumab cardiotoxicity in patients with breast cancer. J Am Coll Cardiol. (2019) 73:2859–68. doi: 10.1016/j.jacc.2019.03.495
49. Spallarossa P, Garibaldi S, Altieri P, Fabbi P, Manca V, Nasti S, et al. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J Mol Cell Cardiol. (2004) 37:837–46. doi: 10.1016/j.yjmcc.2004.05.024
50. Mason RP, Kalinowski L, Jacob RF, Jacoby AM, Malinski T. Nebivolol reduces nitroxidative stress and restores nitric oxide bioavailability in endothelium of black Americans. Circulation. (2005) 112:3795–801. doi: 10.1161/CIRCULATIONAHA.105.556233
51. Na Z, Qiao X, Hao X, Fan L, Xiao Y, Shao Y, et al. The effects of beta-blocker use on cancer prognosis: a meta-analysis based on 319,006 patients. Onco Targets Ther. (2018) 11:4913–44. doi: 10.2147/OTT.S167422
52. Weberpals J, Jansen L, Carr PR, Hoffmeister M, Brenner H. Beta blockers and cancer prognosis - The role of immortal time bias: a systematic review and meta-analysis. Cancer Treat Rev. (2016) 47:1–11. doi: 10.1016/j.ctrv.2016.04.004
53. Avila MS, Ayub-Ferreira SM, de Barros Wanderley MR Jr, das Dores Cruz F, Goncalves Brandao SM, Rigaud VOC, et al. Carvedilol for prevention of chemotherapy-related cardiotoxicity: the CECCY trial. J Am Coll Cardiol. (2018) 71:2281–90. doi: 10.1016/j.jacc.2018.02.049
54. Ferrario CM, Mullick AE. Renin angiotensin aldosterone inhibition in the treatment of cardiovascular disease. Pharmacol Res. (2017) 125:57–71. doi: 10.1016/j.phrs.2017.05.020
55. Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci Transl Med. (2017) 9:eaan5616. doi: 10.1126/scitranslmed.aan5616
56. Cortez-Retamozo V, Etzrodt M, Newton A, Ryan R, Pucci F, Sio SW, et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity. (2013) 38:296–308. doi: 10.1016/j.immuni.2012.10.015
57. Sipahi I, Debanne SM, Rowland DY, Simon DI, Fang JC. Angiotensin-receptor blockade and risk of cancer: meta-analysis of randomised controlled trials. Lancet Oncol. (2010) 11:627–36. doi: 10.1016/S1470-2045(10)70106-6
58. Cortassa S, O'Rourke B, Aon MA. Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim Biophys Acta. (2014) 1837:287–95. doi: 10.1016/j.bbabio.2013.11.007
59. Kirk JA, Paolocci N. New redox-related arrows in the arsenal of cardiac disease treatment. Antioxid Redox Signal. (2014) 21:1945–8. doi: 10.1089/ars.2014.6124
60. van Dalen EC, Caron HN, Dickinson HO, Kremer LC. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst Rev. (2005) 2005:Cd003917. doi: 10.1002/14651858.CD003917.pub2
61. Reddy R, Chahoud G, Mehta JL. Modulation of cardiovascular remodeling with statins: fact or fiction? Curr Vasc Pharmacol. (2005) 3:69–79. doi: 10.2174/1570161052773915
62. Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. (2005) 45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748
63. Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. (2017) 120:229–43. doi: 10.1161/CIRCRESAHA.116.308537
65. Kalam K, Marwick TH. Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: a systematic review and meta-analysis. Eur J Cancer. (2013) 49:2900–9. doi: 10.1016/j.ejca.2013.04.030
66. Lefkowitz RJ. Seven transmembrane receptors: something old, something new. Acta Physiol. (2007) 190:9–19. doi: 10.1111/j.1365-201X.2007.01693.x
67. Insel PA, Sriram K, Wiley SZ, Wilderman A, Katakia T, McCann T, et al. GPCRomics: GPCR expression in cancer cells and tumors identifies new, potential biomarkers and therapeutic targets. Front Pharmacol. (2018) 9:431. doi: 10.3389/fphar.2018.00431
68. Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA. (2003) 100:11406–11. doi: 10.1073/pnas.1936664100
69. Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. (2005) 308:512–7. doi: 10.1126/science.1109237
70. Zhou XE, Melcher K, Xu HE. Understanding the GPCR biased signaling through G protein and arrestin complex structures. Curr Opin Struct Biol. (2017) 45:150–9. doi: 10.1016/j.sbi.2017.05.004
71. Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. (2011) 36:457–69. doi: 10.1016/j.tibs.2011.06.003
72. Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. (2000) 275:17201–10. doi: 10.1074/jbc.M910348199
73. Zheng M, Zhu W, Han Q, Xiao RP. Emerging concepts and therapeutic implications of beta-adrenergic receptor subtype signaling. Pharmacol Ther. (2005) 108:257–68. doi: 10.1016/j.pharmthera.2005.04.006
74. do Vale GT, Ceron CS, Gonzaga NA, Simplicio JA, Padovan JC. Three generations of beta-blockers: history, class differences and clinical applicability. Curr Hypertens Rev. (2019) 15:22–31. doi: 10.2174/1573402114666180918102735
75. Nabati M, Janbabai G, Baghyari S, Esmaili K, Yazdani J. Cardioprotective effects of carvedilol in inhibiting doxorubicin-induced cardiotoxicity. J Cardiovasc Pharmacol. (2017) 69:279–85. doi: 10.1097/FJC.0000000000000470
76. Tashakori Beheshti A, Mostafavi Toroghi H, Hosseini G, Zarifian A, Homaei Shandiz F, Fazlinezhad A. Carvedilol administration can prevent doxorubicin-induced cardiotoxicity: a double-blind randomized trial. Cardiology. (2016) 134:47–53. doi: 10.1159/000442722
77. Oliveira PJ, Goncalves L, Monteiro P, Providencia LA, Moreno AJ. Are the antioxidant properties of carvedilol important for the protection of cardiac mitochondria? Curr Vasc Pharmacol. (2005) 3:147–158. doi: 10.2174/1570161053586903
78. Kalay N, Basar E, Ozdogru I, Er O, Cetinkaya Y, Dogan A, et al. Protective effects of carvedilol against anthracycline-induced cardiomyopathy. J Am Coll Cardiol. (2006) 48:2258–62. doi: 10.1016/j.jacc.2006.07.052
79. Porrello ER, Delbridge LM. Cardiomyocyte autophagy is regulated by angiotensin II type 1 and type 2 receptors. Autophagy. (2009) 5:1215–6. doi: 10.4161/auto.5.8.10153
80. Turu G, Balla A, Hunyady L. The role of beta-arrestin proteins in organization of signaling and regulation of the AT1 angiotensin receptor. Front Endocrinol. (2019) 10:519. doi: 10.3389/fendo.2019.00519
81. Bosch X, Rovira M, Sitges M, Domenech A, Ortiz-Perez JT, de Caralt TM, et al. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: the OVERCOME trial (preventiOn of left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of Malignant hEmopathies). J Am Coll Cardiol. (2013) 61:2355–62. doi: 10.1016/j.jacc.2013.02.072
82. Georgakopoulos P, Roussou P, Matsakas E, Karavidas A, Anagnostopoulos N, Marinakis T, et al. Cardioprotective effect of metoprolol and enalapril in doxorubicin-treated lymphoma patients: a prospective, parallel-group, randomized, controlled study with 36-month follow-up. Am J Hematol. (2010) 85:894–6. doi: 10.1002/ajh.21840
83. Gulati G, Heck SL, Ree AH, Hoffmann P, Schulz-Menger J, Fagerland MW, et al. Prevention of cardiac dysfunction during adjuvant breast cancer therapy (PRADA): a 2 x 2 factorial, randomized, placebo-controlled, double-blind clinical trial of candesartan and metoprolol. Eur Heart J. (2016) 37:1671–80. doi: 10.1093/eurheartj/ehw022
84. Beak J, Huang W, Parker JS, Hicks ST, Patterson C, Simpson PC, et al. An oral selective Alpha-1A adrenergic receptor agonist prevents doxorubicin cardiotoxicity. JACC Basic Transl Sci. (2017) 2:39–53. doi: 10.1016/j.jacbts.2016.10.006
85. Galal A, El-Bakly WM, Al Haleem EN, El-Demerdash E. Selective A3 adenosine receptor agonist protects against doxorubicin-induced cardiotoxicity. Cancer Chemother Pharmacol. (2016) 77:309–22. doi: 10.1007/s00280-015-2937-y
86. Shneyvays V, Mamedova L, Zinman T, Jacobson K, Shainberg A. Activation of A(3)adenosine receptor protects against doxorubicin-induced cardiotoxicity. J Mol Cell Cardiol. (2001) 33:1249–61. doi: 10.1006/jmcc.2001.1387
87. Liu X, Chen Z, Chua CC, Ma YS, Youngberg GA, Hamdy R, et al. Melatonin as an effective protector against doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol. (2002) 283:H254–63. doi: 10.1152/ajpheart.01023.2001
88. Aygun H, Gul SS. Cardioprotective effect of melatonin and agomelatine on doxorubicin-induced cardiotoxicity in a rat model: an electrocardiographic, scintigraphic and biochemical study. Bratisl Lek Listy. (2019) 120:249–55. doi: 10.4149/BLL_2019_045
89. Xu MF, Ho S, Qian ZM, Tang PL. Melatonin protects against cardiac toxicity of doxorubicin in rat. J Pineal Res. (2001) 31:301–7. doi: 10.1034/j.1600-079X.2001.310403.x
90. Balli E, Mete UO, Tuli A, Tap O, Kaya M. Effect of melatonin on the cardiotoxicity of doxorubicin. Histol Histopathol. (2004) 19:1101–8. doi: 10.14670/HH-19.1101.
91. Liu D, Ma Z, Di S, Yang Y, Yang J, Xu L, et al. AMPK/PGC1alpha activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic Biol Med. (2018) 129:59–72. doi: 10.1016/j.freeradbiomed.2018.08.032
92. Govender J, Loos B, Marais E, Engelbrecht AM. Melatonin improves cardiac and mitochondrial function during doxorubicin-induced cardiotoxicity: A possible role for peroxisome proliferator-activated receptor gamma coactivator 1-alpha and sirtuin activity? Toxicol Appl Pharmacol. (2018) 358:86–101. doi: 10.1016/j.taap.2018.06.031
93. Kim C, Kim N, Joo H, Youm JB, Park WS, Cuong DV, et al. Modulation by melatonin of the cardiotoxic and antitumor activities of adriamycin. J Cardiovasc Pharmacol. (2005) 46:200–10. doi: 10.1097/01.fjc.0000171750.97822.a2
94. Wang X, Wang XL, Chen HL, Wu D, Chen JX, Wang XX, et al. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem Pharmacol. (2014) 88:334–50. doi: 10.1016/j.bcp.2014.01.040
95. Kihara M, Kaiya H, Win ZP, Kitajima Y, Nishikawa M. Protective effect of dietary ghrelin-containing salmon stomach extract on mortality and cardiotoxicity in doxorubicin-induced mouse model of heart failure. J Food Sci. (2016) 81:H2858–65. doi: 10.1111/1750-3841.13526
96. Studneva I, Serebryakova L, Veselova O, Pal'keeva M, Molokoedov A, Ovchinnikov M, et al. Galanin receptors activation modulates myocardial metabolic and antioxidant responses to ischaemia/reperfusion stress. Clin Exp Pharmacol Physiol. (2019) 46:1174–82. doi: 10.1111/1440-1681.13164
97. Sun J, Xu S, Li H, Li L, Xu ZD. Galanin protects rat cortical astrocyte from oxidative stress: involvement of GalR2 and pERK1/2 signal pathway. Med Inflamm. (2019) 2019:2716028. doi: 10.1155/2019/2716028
98. Hamada J, Baasanjav A, Ono N, Murata K, Kako K, Ishida J, et al. Possible involvement of downregulation of the apelin-APJ system in doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol. (2015) 308:H931–41. doi: 10.1152/ajpheart.00703.2013
99. Zhang P, Yi LH, Meng GY, Zhang HY, Sun HH, Cui LQ. Apelin-13 attenuates cisplatin-induced cardiotoxicity through inhibition of ROS-mediated DNA damage and regulation of MAPKs and AKT pathways. Free Radic Res. (2017) 51:449–59. doi: 10.1080/10715762.2017.1313414
100. Gasser AYWC, Audebrand YW, Daglayan A, Charavin A, Escoubet M, Karpov B, et al. Prokineticin receptor-1 signaling inhibits dose- and time-dependent anthracycline-induced cardiovascular toxicity via myocardial and vascular protection. JACC Cardio Oncol. (2019) 1:84–102. doi: 10.1016/j.jaccao.2019.06.003
101. Fouad AA, Albuali WH, Al-Mulhim AS, Jresat I. Cardioprotective effect of cannabidiol in rats exposed to doxorubicin toxicity. Environ Toxicol Pharmacol. (2013) 36:347–57. doi: 10.1016/j.etap.2013.04.018
102. Hao E, Mukhopadhyay P, Cao Z, Erdelyi K, Holovac E, Liaudet L, et al. Cannabidiol protects against doxorubicin-induced cardiomyopathy by modulating mitochondrial function and biogenesis. Mol Med. (2015) 21:38–45. doi: 10.2119/molmed.2014.00261
103. Massi P, Solinas M, Cinquina V, Parolaro D. Cannabidiol as potential anticancer drug. Br J Clin Pharmacol. (2013) 75:303–12. doi: 10.1111/j.1365-2125.2012.04298.x
104. Biazi GR, Frasson IG, Miksza DR, de Morais H, de Fatima Silva F, Bertolini GL, et al. Decreased hepatic response to glucagon, adrenergic agonists, and cAMP in glycogenolysis, gluconeogenesis, and glycolysis in tumor-bearing rats. J Cell Biochem. (2018) 119:7300–9. doi: 10.1002/jcb.27027
105. Kudej RK, Shen YT, Peppas AP, Huang CH, Chen W, Yan L, et al. Obligatory role of cardiac nerves and alpha1-adrenergic receptors for the second window of ischemic preconditioning in conscious pigs. Circ Res. (2006) 99:1270–6. doi: 10.1161/01.RES.0000251282.79411.44
106. Sabri A, Pak E, Alcott SA, Wilson BA, Steinberg SF. Coupling function of endogenous alpha(1)- and beta-adrenergic receptors in mouse cardiomyocytes. Circ Res. (2000) 86:1047–53. doi: 10.1161/01.RES.86.10.1047
107. Wright CD, Chen Q, Baye NL, Huang Y, Healy CL, Kasinathan S, et al. Nuclear alpha1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ Res. (2008) 103:992–1000. doi: 10.1161/CIRCRESAHA.108.176024
108. O'Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, et al. The alpha(1A/C)- and alpha(1B)-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J Clin Invest. (2003) 111:1783–91. doi: 10.1172/JCI200316100
109. O'Connell TD, Block RC, Huang SP, Shearer GC. omega3-Polyunsaturated fatty acids for heart failure: Effects of dose on efficacy and novel signaling through free fatty acid receptor 4. J Mol Cell Cardiol. (2017) 103:74–92. doi: 10.1016/j.yjmcc.2016.12.003
110. Durkee CA, Covelo A, Lines J, Kofuji P, Aguilar J, Araque A. Gi/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia. (2019) 67:1076–93. doi: 10.1002/glia.23589
111. Musselman DM, Ford AP, Gennevois DJ, Harbison ML, Laurent AL, Mokatrin AS, et al. A randomized crossover study to evaluate Ro 115-1240, a selective alpha1A/1L-adrenoceptor partial agonist in women with stress urinary incontinence. BJU Int. (2004) 93:78–83. doi: 10.1111/j.1464-410X.2004.04560.x
112. Caso S, Maric D, Arambasic M, Cotecchia S, Diviani D. AKAP-Lbc mediates protection against doxorubicin-induced cardiomyocyte toxicity. Biochim Biophys Acta Mol Cell Res. (2017) 1864:2336–46. doi: 10.1016/j.bbamcr.2017.09.007
113. Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets–what are the challenges? Nat Rev Drug Discov. (2013) 12:265–86. doi: 10.1038/nrd3955
114. Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K. Pathological overproduction: the bad side of adenosine. Br J Pharmacol. (2017) 174:1945–60. doi: 10.1111/bph.13763
115. Sobrevia L, Fredholm BB. Adenosine – from molecular mechanisms to pathophysiology. Mol Aspects Med. (2017) 55:1–3. doi: 10.1016/j.mam.2017.06.003
116. McIntosh VJ, Lasley RD. Adenosine receptor-mediated cardioprotection: are all 4 subtypes required or redundant? J Cardiovasc Pharmacol Ther. (2012) 17:21–33. doi: 10.1177/1074248410396877
117. Deb PK. Therapeutic potentials of adenosine receptors: the state of the art. Curr Pharm Des. (2019) 25:2789–91. doi: 10.2174/138161282526191007143942
118. Meibom D, Albrecht-Kupper B, Diedrichs N, Hubsch W, Kast R, Kramer T, et al. Neladenoson bialanate hydrochloride: a prodrug of a partial adenosine A1 receptor agonist for the chronic treatment of heart diseases. ChemMedChem. (2017) 12:728–37. doi: 10.1002/cmdc.201700151
119. Voors AA, Dungen HD, Senni M, Nodari S, Agostoni P, Ponikowski P, et al. Safety and tolerability of neladenoson bialanate, a novel oral partial adenosine A1 receptor agonist, in patients with chronic heart failure. J Clin Pharmacol. (2017) 57:440–51. doi: 10.1002/jcph.828
120. DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, et al. 3'-Aminoadenosine-5'-uronamides: discovery of the first highly selective agonist at the human adenosine A3 receptor. J Med Chem. (2003) 46:353–5. doi: 10.1021/jm0255724
121. Lasley RD. Adenosine receptor-mediated cardioprotection-current limitations and future directions. Front Pharmacol. (2018) 9:310. doi: 10.3389/fphar.2018.00310
122. Wan TC, Tampo A, Kwok WM, Auchampach JA. Ability of CP-532,903 to protect mouse hearts from ischemia/reperfusion injury is dependent on expression of A3 adenosine receptors in cardiomyoyctes. Biochem Pharmacol. (2019) 163:21–31. doi: 10.1016/j.bcp.2019.01.022
123. Jacobson KA, Tosh DK, Jain S, Gao ZG. Historical and current adenosine receptor agonists in preclinical and clinical development. Front Cell Neurosci. (2019) 13:124. doi: 10.3389/fncel.2019.00124
124. Baltatu OC, Senar S, Campos LA, Cipolla-Neto J. Cardioprotective melatonin: translating from proof-of-concept studies to therapeutic use. Int J Mol Sci. (2019) 20:4342. doi: 10.3390/ijms20184342
125. Stroethoff M, Christoph I, Behmenburg F, Raupach A, Bunte S, Senpolat S, et al. Melatonin receptor agonist ramelteon reduces ischemia-reperfusion injury through activation of mitochondrial potassium channels. J Cardiovasc Pharmacol. (2018) 72:106–11. doi: 10.1097/FJC.0000000000000600
126. Campos LA, Cipolla-Neto J, Amaral FG, Michelini LC, Bader M, Baltatu OC. The Angiotensin-melatonin axis. Int J Hypertens. (2013) 2013:521783. doi: 10.1155/2013/521783
127. Baltatu OC, Amaral FG, Campos LA, Cipolla-Neto J. Melatonin, mitochondria and hypertension. Cell Mol Life Sci. (2017) 74:3955–64. doi: 10.1007/s00018-017-2613-y
128. Govender J, Loos B, Marais E, Engelbrecht AM. Mitochondrial catastrophe during doxorubicin-induced cardiotoxicity: a review of the protective role of melatonin. J Pineal Res. (2014) 57:367–80. doi: 10.1111/jpi.12176
129. Pei H, Du J, Song X, He L, Zhang Y, Li X, et al. Melatonin prevents adverse myocardial infarction remodeling via Notch1/Mfn2 pathway. Free Radic Biol Med. (2016) 97:408–17. doi: 10.1016/j.freeradbiomed.2016.06.015
130. Yeung HM, Hung MW, Lau CF, Fung ML. Cardioprotective effects of melatonin against myocardial injuries induced by chronic intermittent hypoxia in rats. J Pineal Res. (2015) 58:12–25. doi: 10.1111/jpi.12190
131. Mukherjee D, Roy SG, Bandyopadhyay A, Chattopadhyay A, Basu A, Mitra E, et al. Melatonin protects against isoproterenol-induced myocardial injury in the rat: antioxidative mechanisms. J Pineal Res. (2010) 48:251–62. doi: 10.1111/j.1600-079X.2010.00749.x
132. Patel V, Upaganlawar A, Zalawadia R, Balaraman R. Cardioprotective effect of melatonin against isoproterenol induced myocardial infarction in rats: a biochemical, electrocardiographic and histoarchitectural evaluation. Eur J Pharmacol. (2010) 644:160–8. doi: 10.1016/j.ejphar.2010.06.065
133. Tokudome T, Otani K, Miyazato M, Kangawa K. Ghrelin and the heart. Peptides. (2019) 111:42–6. doi: 10.1016/j.peptides.2018.05.006
134. Soeki T, Kishimoto I, Schwenke DO, Tokudome T, Horio T, Yoshida M, et al. Ghrelin suppresses cardiac sympathetic activity and prevents early left ventricular remodeling in rats with myocardial infarction. Am J Physiol Heart Circ Physiol. (2008) 294:H426–32. doi: 10.1152/ajpheart.00643.2007
135. Zhang CJ, Bidlingmaier M, Altaye M, Page LC, D'Alessio D, Tschop MH, et al. Acute administration of acyl, but not desacyl ghrelin, decreases blood pressure in healthy humans. Eur J Endocrinol. (2017) 176:123–32. doi: 10.1530/EJE-16-0789
136. Soeki T, Niki T, Uematsu E, Bando S, Matsuura T, Kusunose K, et al. Ghrelin protects the heart against ischemia-induced arrhythmias by preserving connexin-43 protein. Heart Vessels. (2013) 28:795–801. doi: 10.1007/s00380-013-0333-2
137. Yang C, Liu J, Liu K, Du B, Shi K, Ding M, et al. Ghrelin suppresses cardiac fibrosis of post-myocardial infarction heart failure rats by adjusting the activin A-follistatin imbalance. Peptides. (2018) 99:27–35. doi: 10.1016/j.peptides.2017.10.018
138. Nagaya N, Uematsu M, Kojima M, Ikeda Y, Yoshihara F, Shimizu W, et al. Chronic administration of ghrelin improves left ventricular dysfunction and attenuates development of cardiac cachexia in rats with heart failure. Circulation. (2001) 104:1430–5. doi: 10.1161/hc3601.095575
139. Katare R, Rawal S, Munasinghe PE, Tsuchimochi H, Inagaki T, Fujii Y, et al. Ghrelin promotes functional angiogenesis in a mouse model of critical limb ischemia through activation of proangiogenic MicroRNAs. Endocrinology. (2016) 157:432–45. doi: 10.1210/en.2015-1799
140. Wang L, Chen Q, Li G, Ke D. Ghrelin ameliorates impaired angiogenesis of ischemic myocardium through GHSR1a-mediated AMPK/eNOS signal pathway in diabetic rats. Peptides. (2015) 73:77–87. doi: 10.1016/j.peptides.2015.09.004
141. Yuan MJ, Wang T, Kong B, Wang X, Huang CX, Wang D. GHSR-1a is a novel pro-angiogenic and anti-remodeling target in rats after myocardial infarction. Eur J Pharmacol. (2016) 788:218–25. doi: 10.1016/j.ejphar.2016.06.032
142. Yuan MJ, He H, Hu HY, Li Q, Hong J, Huang CX. Myocardial angiogenesis after chronic ghrelin treatment in a rat myocardial infarction model. Regul Pept. (2012) 179:39–42. doi: 10.1016/j.regpep.2012.08.013
143. Lenk K, Palus S, Schur R, Datta R, Dong J, Culler MD, et al. Effect of ghrelin and its analogues, BIM-28131 and BIM-28125, on the expression of myostatin in a rat heart failure model. J Cachexia Sarcopenia Muscle. (2013) 4:63–9. doi: 10.1007/s13539-012-0085-3
144. Tatemoto K, Rokaeus A, Jornvall H, McDonald TJ, Mutt V. Galanin - a novel biologically active peptide from porcine intestine. FEBS Lett. (1983) 164:124–8. doi: 10.1016/0014-5793(83)80033-7
145. Webling KE, Runesson J, Bartfai T, Langel U. Galanin receptors and ligands. Front Endocrinol. (2012) 3:146. doi: 10.3389/fendo.2012.00146
146. Potter EK, Smith-White MA. Galanin modulates cholinergic neurotransmission in the heart. Neuropeptides. (2005) 39:345–8. doi: 10.1016/j.npep.2004.12.006
147. Herring N, Cranley J, Lokale MN, Li D, Shanks J, Alston EN, et al. The cardiac sympathetic co-transmitter galanin reduces acetylcholine release and vagal bradycardia: implications for neural control of cardiac excitability. J Mol Cell Cardiol. (2012) 52:667–76. doi: 10.1016/j.yjmcc.2011.11.016
148. Chen A, Li M, Song L, Zhang Y, Luo Z, Zhang W, et al. Effects of the galanin receptor antagonist M40 on cardiac function and remodeling in rats with heart failure. Cardiovasc Ther. (2015) 33:288–93. doi: 10.1111/1755-5922.12144
149. Serebryakova LI, Studneva IM, Ovchinnikov MV, Veselova OM, Molokoedov AS, Arzamastsev EV, et al. [Cardiometabolic efficacy and toxicological evaluation of a pharmacological galanin receptor agonist]. Biomed Khim. (2019) 65:231–8. doi: 10.18097/PBMC20196503231
150. Studneva IM, Palkeeva ME, Veselova OM, Molokoedov AS, Lubimov RO, Ovchinnikov MV, et al. [Protective action of a modified fragment of galanine in rats with doxorubicin-induced heart failure]. Biomed Khim. (2019) 65:51–6. doi: 10.18097/PBMC20196501051
151. Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y, et al. Characterization of apelin, the ligand for the APJ receptor. J Neurochem. (2000) 74:34–41. doi: 10.1046/j.1471-4159.2000.0740034.x
152. Sato T, Suzuki T, Watanabe H, Kadowaki A, Fukamizu A, Liu PP, et al. Apelin is a positive regulator of ACE2 in failing hearts. J Clin Invest. (2013) 123:5203–11. doi: 10.1172/JCI69608
153. Szokodi I, Tavi P, Foldes G, Voutilainen-Myllyla S, Ilves M, Tokola H, et al. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res. (2002) 91:434–40. doi: 10.1161/01.RES.0000033522.37861.69
154. Tatemoto K, Takayama K, Zou MX, Kumaki I, Zhang W, Kumano K, et al. The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regul Pept. (2001) 99:87–92. doi: 10.1016/S0167-0115(01)00236-1
155. Japp AG, Cruden NL, Amer DA, Li VK, Goudie EB, Johnston NR, et al. Vascular effects of apelin in vivo in man. J Am Coll Cardiol. (2008) 52:908–13. doi: 10.1016/j.jacc.2008.06.013
156. Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, et al. Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure. Circulation. (2010) 121:1818–27. doi: 10.1161/CIRCULATIONAHA.109.911339
157. Mughal A, O'Rourke ST. Vascular effects of apelin: mechanisms and therapeutic potential. Pharmacol Ther. (2018) 190:139–47. doi: 10.1016/j.pharmthera.2018.05.013
158. Zuurbier L, Rahman A, Cordes M, Scheick J, Wong TJ, Rustenburg F, et al. Apelin: a putative novel predictive biomarker for bevacizumab response in colorectal cancer. Oncotarget. (2017) 8:42949–61. doi: 10.18632/oncotarget.17306
159. Altinkaya SO, Nergiz S, Kucuk M, Yuksel H. Apelin levels are higher in obese patients with endometrial cancer. J Obstet Gynaecol Res. (2015) 41:294–300. doi: 10.1111/jog.12503
160. Cabia B, Andrade S, Carreira MC, Casanueva FF, Crujeiras AB. A role for novel adipose tissue-secreted factors in obesity-related carcinogenesis. Obes Rev. (2016) 17:361–76. doi: 10.1111/obr.12377
161. Uribesalgo I, Hoffmann D, Zhang Y, Kavirayani A, Lazovic J, Berta J, et al. Apelin inhibition prevents resistance and metastasis associated with anti-angiogenic therapy. EMBO Mol Med. (2019) 11:e9266. doi: 10.15252/emmm.201809266
162. Nebigil CG. Prokineticin receptors in cardiovascular function: foe or friend? Trends Cardiovasc Med. (2009) 19:55–60. doi: 10.1016/j.tcm.2009.04.007
163. Li M, Bullock CM, Knauer DJ, Ehlert FJ, Zhou QY. Identification of two prokineticin cDNAs: recombinant proteins potently contract gastrointestinal smooth muscle. Mol Pharmacol. (2001) 59:692–8. doi: 10.1124/mol.59.4.692
164. Ngan ES, Tam PK. Prokineticin-signaling pathway. Int J Biochem Cell Biol. (2008) 40:1679–84. doi: 10.1016/j.biocel.2008.03.010
165. LeCouter J, Lin R, Tejada M, Frantz G, Peale F, Hillan KJ, et al. The endocrine-gland-derived VEGF homologue Bv8 promotes angiogenesis in the testis: localization of Bv8 receptors to endothelial cells. Proc Natl Acad Sci USA. (2003) 100:2685–90. doi: 10.1073/pnas.0337667100
166. LeCouter J, Zlot C, Tejada M, Peale F, Ferrara N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc Natl Acad Sci USA. (2004) 101:16813–18. doi: 10.1073/pnas.0407697101
167. Landucci E, Lattanzi R, Gerace E, Scartabelli T, Balboni G, Negri L, et al. Prokineticins are neuroprotective in models of cerebral ischemia and ischemic tolerance in vitro. Neuropharmacology. (2016) 108:39–48. doi: 10.1016/j.neuropharm.2016.04.043
168. Gordon R, Neal ML, Luo J, Langley MR, Harischandra DS, Panicker N, et al. Prokineticin-2 upregulation during neuronal injury mediates a compensatory protective response against dopaminergic neuronal degeneration. Nat Commun. (2016) 7:12932. doi: 10.1038/ncomms12932
169. Urayama K, Guilini C, Messaddeq N, Hu K, Steenman M, Kurose H, et al. The prokineticin receptor-1 (GPR73) promotes cardiomyocyte survival and angiogenesis. Faseb j. (2007) 21:2980–93. doi: 10.1096/fj.07-8116com
170. Guilini C, Urayama K, Turkeri G, Dedeoglu DB, Kurose H, Messaddeq N, et al. Divergent roles of prokineticin receptors in the endothelial cells: angiogenesis and fenestration. Am J Physiol Heart Circ Physiol. (2010) 298:H844–52. doi: 10.1152/ajpheart.00898.2009
171. Kaser A, Winklmayr M, Lepperdinger G, Kreil G. The AVIT protein family. Secreted cysteine-rich vertebrate proteins with diverse functions. EMBO Rep. (2003) 4:469–73. doi: 10.1038/sj.embor.embor830
172. Urayama K, Dedeoglu DB, Guilini C, Frantz S, Ertl G, Messaddeq N, et al. Transgenic myocardial overexpression of prokineticin receptor-2 (GPR73b) induces hypertrophy and capillary vessel leakage. Cardiovasc Res. (2009) 81:28–37. doi: 10.1093/cvr/cvn251
173. Urayama K, Guilini C, Turkeri G, Takir S, Kurose H, Messaddeq N, et al. Prokineticin receptor-1 induces neovascularization and epicardial-derived progenitor cell differentiation. Arterioscl Thromb Vasc Biol. (2008) 28:841–9. doi: 10.1161/ATVBAHA.108.162404
174. Arora H, Boulberdaa M, Qureshi R, Bitirim V, Gasser A, Messaddeq N, et al. Prokineticin receptor-1 signaling promotes Epicardial to Mesenchymal Transition during heart development. Sci Rep. (2016) 6:25541. doi: 10.1038/srep25541
175. Arora H, Boulberdaa M, Qureshi R, Bitirim V, Messadeq N, Dolle P, et al. Prokineticin receptor 1 is required for mesenchymal-epithelial transition in kidney development. FASEB J. (2016) 30:2733–40. doi: 10.1096/fj.201600181R
176. Qureshi R, Kindo M, Arora H, Boulberdaa M, Steenman M, Nebigil CG. Prokineticin receptor-1-dependent paracrine and autocrine pathways control cardiac tcf21(+) fibroblast progenitor cell transformation into adipocytes and vascular cells. Sci Rep. (2017) 7:12804. doi: 10.1038/s41598-017-13198-2
177. Qureshi R, Kindo M, Boulberdaa M, von Hunolstein JJ, Steenman M, Nebigil CG. A prokineticin-driven epigenetic switch regulates human epicardial cell stemness and fate. Stem Cells. (2018) 36:1589–602. doi: 10.1002/stem.2866
178. Szatkowski C, Vallet J, Dormishian M, Messaddeq N, Valet P, Boulberdaa M, et al. Prokineticin receptor 1 as a novel suppressor of preadipocyte proliferation and differentiation to control obesity. PLoS ONE. (2013) 8:e81175. doi: 10.1371/journal.pone.0081175
179. Von Hunolstein JJ, Nebigil CG. Can prokineticin prevent obesity and insulin resistance? Curr Opin Endocrinol Diabet Obesity. (2015) 22:367–73. doi: 10.1097/MED.0000000000000185
180. Dormishian M, Turkeri G, Urayama K, Nguyen TL, Boulberdaa M, Messaddeq N, et al. Prokineticin receptor-1 is a new regulator of endothelial insulin uptake and capillary formation to control insulin sensitivity and cardiovascular and kidney functions. J Am Heart Assoc. (2013) 2:e000411. doi: 10.1161/JAHA.113.000411
181. Boulberdaa M, Turkeri G, Urayama K, Dormishian M, Szatkowski C, Zimmer L, et al. Genetic inactivation of prokineticin receptor-1 leads to heart and kidney disorders. Arterioscl Thromb Vasc Biol. (2011) 31:842–50. doi: 10.1161/ATVBAHA.110.222323
182. Boulberdaa M, Urayama K, Nebigil CG. Prokineticin receptor 1 (PKR1) signalling in cardiovascular and kidney functions. Cardiovasc Res. (2011) 92:191–8. doi: 10.1093/cvr/cvr228
183. Gasser A, Brogi S, Urayama K, Nishi T, Kurose H, Tafi A, et al. Discovery and cardioprotective effects of the first non-Peptide agonists of the G protein-coupled prokineticin receptor-1. PLoS ONE. (2015) 10:e0121027. doi: 10.1371/journal.pone.0121027
184. Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. (2006) 58:389–462. doi: 10.1124/pr.58.3.2
185. Stanley CP, Hind WH, O'Sullivan SE. Is the cardiovascular system a therapeutic target for cannabidiol? Br J Clin Pharmacol. (2013) 75:313–22. doi: 10.1111/j.1365-2125.2012.04351.x
186. Rajesh M, Mukhopadhyay P, Hasko G, Liaudet L, Mackie K, Pacher P. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br J Pharmacol. (2010) 160:688–700. doi: 10.1111/j.1476-5381.2010.00712.x
187. Pertwee RG. Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond. Addict Biol. (2008) 13:147–59. doi: 10.1111/j.1369-1600.2008.00108.x
188. Durst R, Danenberg H, Gallily R, Mechoulam R, Meir K, Grad E, et al. Cannabidiol, a nonpsychoactive Cannabis constituent, protects against myocardial ischemic reperfusion injury. Am J Physiol Heart Circ Physiol. (2007) 293:H3602–7. doi: 10.1152/ajpheart.00098.2007
189. Walsh SK, Hepburn CY, Kane KA, Wainwright CL. Acute administration of cannabidiol in vivo suppresses ischaemia-induced cardiac arrhythmias and reduces infarct size when given at reperfusion. Br J Pharmacol. (2010) 160:1234–42. doi: 10.1111/j.1476-5381.2010.00755.x
190. Williams DW, Askew LC, Jones E, Clements JE. CCR2 signaling selectively regulates IFN-alpha: role of beta-arrestin 2 in IFNAR1 internalization. J Immunol. (2019) 202:105–18. doi: 10.4049/jimmunol.1800598
191. Lim SY, Davidson SM, Yellon DM, Smith CC. The cannabinoid CB1 receptor antagonist, rimonabant, protects against acute myocardial infarction. Basic Res Cardiol. (2009) 104:781–92. doi: 10.1007/s00395-009-0034-2
192. Li X, Peng Z, Zhou Y, Wang J, Lin X, Dong X, et al. Quetiapine induces myocardial necroptotic cell death through bidirectional regulation of cannabinoid receptors. Toxicol Lett. (2019) 313:77–90. doi: 10.1016/j.toxlet.2019.06.005
193. Kogan NM, Schlesinger M, Peters M, Marincheva G, Beeri R, Mechoulam R. A cannabinoid anticancer quinone, HU-331, is more potent and less cardiotoxic than doxorubicin: a comparative in vivo study. J Pharmacol Exp Ther. (2007) 322:646–53. doi: 10.1124/jpet.107.120865
194. Nair P, Prado M, Perea-Gil I, Karakikes I. Concise review: precision matchmaking: induced pluripotent stem cells meet cardio-oncology. Stem Cells Transl Med. (2019) 8:758–67. doi: 10.1002/sctm.18-0279
195. Feigin ME, Xue B, Hammell MC, Muthuswamy SK. G-protein-coupled receptor GPR161 is overexpressed in breast cancer and is a promoter of cell proliferation and invasion. Proc Natl Acad Sci USA. (2014) 111:4191–6. doi: 10.1073/pnas.1320239111
196. Zhou H, Telonis AG, Jing Y, Xia NL, Biederman L, Jimbo M, et al. GPRC5A is a potential oncogene in pancreatic ductal adenocarcinoma cells that is upregulated by gemcitabine with help from HuR. Cell Death Dis. (2016) 7:e2294. doi: 10.1038/cddis.2016.169
197. Wiley SZ, Sriram K, Liang W, Chang SE, French R, McCann T, et al. GPR68, a proton-sensing GPCR, mediates interaction of cancer-associated fibroblasts and cancer cells. FASEB J. (2018) 32:1170–83. doi: 10.1096/fj.201700834R
198. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. (2015) 347:1260419. doi: 10.1126/science.1260419
Keywords: GPCRs, cardiotoxicity, melatonin, ghrelin, galanin, apelin, prokineticin, cannabidiol
Citation: Audebrand A, Désaubry L and Nebigil CG (2020) Targeting GPCRs Against Cardiotoxicity Induced by Anticancer Treatments. Front. Cardiovasc. Med. 6:194. doi: 10.3389/fcvm.2019.00194
Received: 13 November 2019; Accepted: 23 December 2019;
Published: 24 January 2020.
Edited by:
Carlo Gabriele Tocchetti, University of Naples Federico II, ItalyReviewed by:
Claudia Penna, University of Turin, ItalyCopyright © 2020 Audebrand, Désaubry and Nebigil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Canan G. Nebigil, bmViaWdpbEB1bmlzdHJhLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.