 Yasuhiro Maejima
Yasuhiro Maejima
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 08 January 2020
Sec. Cardiovascular Metabolism
Volume 6 - 2019 | https://doi.org/10.3389/fcvm.2019.00186
This article is part of the Research Topic Mitochondrial Dysfunction and Cardiovascular Diseases View all 10 articles
Three cardiovascular outcome trials of sodium glucose cotransporter 2 (SGLT2) inhibitors, including the EMPA-REG OUTCOME trial, CANVAS Program, and DECLARE TIMI 58 trial, revealed that SGLT2 inhibitors were superior to a matching placebo in reducing cardiovascular events, including mortality and hospitalization for heart failure, in patients with type 2 diabetes. However, the detailed mechanism underlying the beneficial effects that SGLT2 inhibitors exert on cardiovascular diseases remains to be elucidated. We herein review the latest findings of the salutary mechanisms of SGLT2 inhibitors in cardiomyocytes, especially focusing on their mitochondrial function-mediated beneficial effects. The administration of SGLT2 inhibitors leads to the elevation of plasma levels of ketone bodies, which are an efficient energy source in the failing heart, by promoting oxidation of the mitochondrial coenzyme Q couple and enhancing the free energy of cytosolic ATP hydrolysis. SGLT2 inhibitors also promote sodium metabolism-mediated cardioprotective effects. These compounds could reduce the intracellular sodium overload to improve mitochondrial energetics and oxidative defense in the heart through binding with NHE and/or SMIT1. Furthermore, SGLT2 inhibitors could modulate mitochondrial dynamics by regulating the fusion and fission of mitochondria. Together with ongoing large-scale clinical trials to evaluate the efficacy of SGLT2 inhibitors in patients with heart failure, intensive investigations regarding the mechanism through which SGLT2 inhibitors promote the restoration in cases of heart failure would lead to the establishment of these drugs as potent anti-heart failure drugs.
Sodium glucose cotransporter (SGLT) is a channel protein that imports glucose into the intracellular space together with sodium ions (Na+) using the gradient of the Na+ concentration between inside and outside of the cells (Figure 1A) (1). SGLTs are expressed in limited organs, including the brain, small intestine, and renal tubule of mammals. Phlorizin, a phloretin that connects with glucose via glucoside bonding, is a natural compound derived from the bark of the apple tree root (Figure 1B). The administration of phlorizin leads to renal glycosuria, as this compound can inhibit SGLT1/2 located on the renal tubule, which results in the alleviation of hyperglycemia by discharging glucose to urine (Figure 1A) (2, 3). However, the intake of phlorizin causes severe diarrhea because this compound also inhibits small intestinal SGLT1, thereby suppressing the reabsorption of glucose together with water in the intestinal tract. To overcome this weakness of phlorizin, intensive analyses were conducted to investigate the molecular structures of both phlorizin and the SGLT receptor. Based on these analyses, highly selective SGLT2 inhibitors were developed as a novel type of anti-diabetes drug (Figure 1B) (4). In recent years, several cardiovascular outcome studies to test the safety of glucose-lowering drugs have demonstrated that SGLT2 inhibitors have a potential protective effect against cardiovascular events that is comparable to existing anti-heart failure drugs. However, it remains unknown how SGLT2 inhibitors exert such beneficial effects in patients with cardiovascular diseases. One of the major reasons why this has not been elucidated is that SGLT2 is not expressed in cardiomyocytes (5). Thus, it is largely believed that SGLT2 inhibitors play a protective role via the modulation of the internal environment outside of the myocardium (6). On the other hand, several investigators have shown that SGLT2 inhibitors directly manifest protective effects in the heart (6). In both cases, it is assumed that SGLT2 inhibitors exert their protective effects by restoring the mitochondrial function in cardiomyocytes. We herein review the current understanding on how SGLT2 inhibitors mitigate cardiac dysfunction through mitochondrial protection-mediated mechanisms.
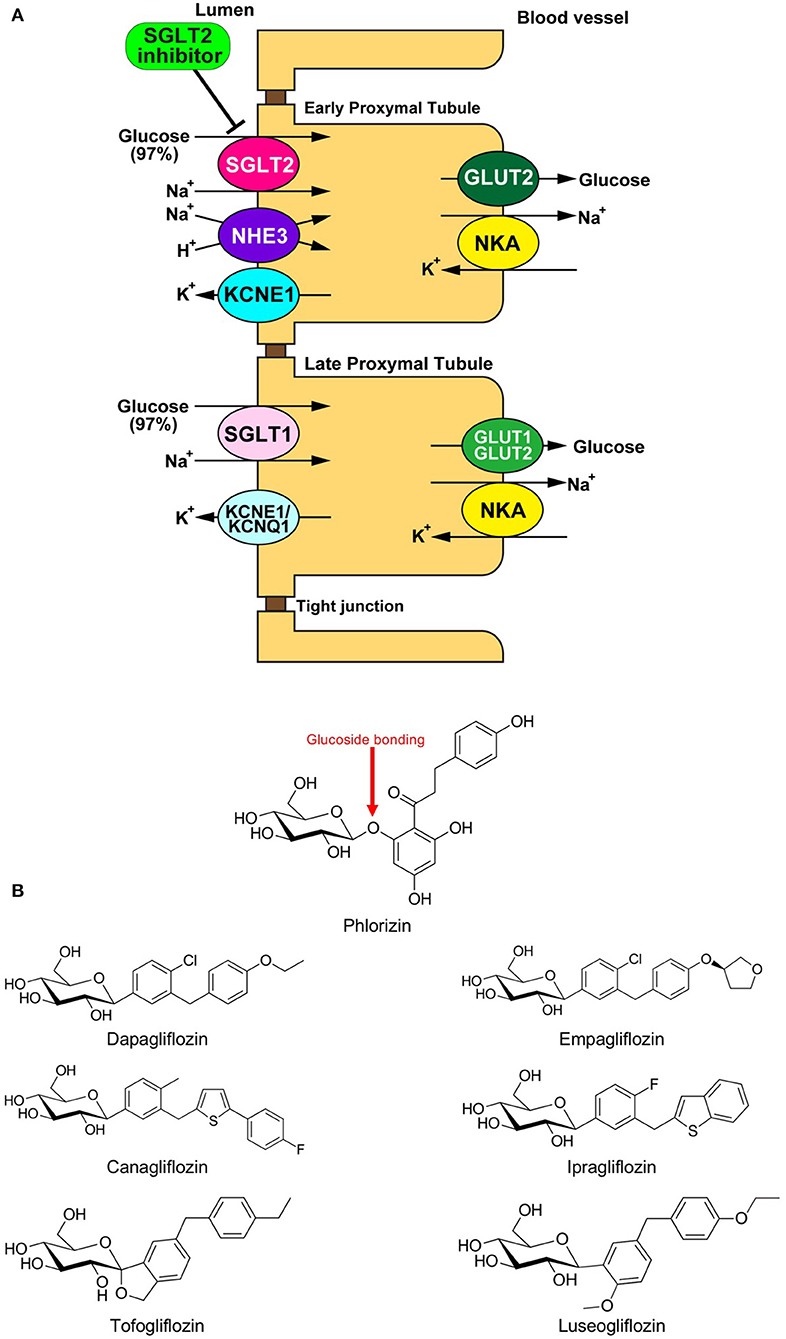
Figure 1. (A) Physiology of glucose reabsorption in the renal proximal tubules and the target of SGLT2 inhibitors. GLUT, glucose transporter; KCNE1, potassium voltage-gated channel Isk-related family member 1; KCNQ1, potassium voltage-gated channel KQT-like subfamily member 1; NHE, Na+/H+ exchanger; NKA, Na+/K+ ATPase; SGLT, sodium-dependent glucose transporter. (B) Chemical structural formulas of Phlorizin and SGLT2 inhibitors (Dapagliflozin, Empagliflozin, Canagliflozin, Ipragliflozin, Tofogliflozin, and Luseogliflozin).
The EMPA-REG OUTCOME trial, a cardiovascular outcome trial (CVOT) of the SGLT2 inhibitor empagliflozin, demonstrated that empagliflozin was superior to a matching placebo in reducing cardiovascular events, including mortality and hospitalization for heart failure in patients with type 2 diabetes and established cardiovascular diseases (7, 8) (Table 1). The CANVAS Program, which consists of the CANVAS study and CANVAS-R, CVOTs assessed the cardiovascular safety and efficacy of the SGLT2 inhibitor canagliflozin in patients with type 2 diabetes and established cardiovascular disease, and also revealed that canagliflozin reduced the risk of a composite outcome of major adverse cardiovascular events in comparison to a matching placebo (9) (Table 1). Furthermore, the DECLARE TIMI 58 trial demonstrated that the SGLT2 inhibitor dapagliflozin reduced the risk of cardiovascular death or hospitalization for heart failure in comparison to a matching placebo in patients with type 2 diabetes and either a high cardiovascular risk or established atherosclerotic cardiovascular disease (10) (Table 1). As most patients in these trials did not have a diagnosis of heart failure at the time of study entry, the merit of treatment with an SGLT2 inhibitor largely reflected the prevention of heart failure development (11). Furthermore, the fact that reduction in the risk of hospitalization for heart failure emerged early after randomization raised the possibility that the mechanisms of the SGLT2 inhibitor-mediated cardiovascular benefits differ from those of existing glucose-lowering therapies that exert their effects independently of glycemic control. Indeed, a series of preclinical investigations demonstrated the effectiveness of SGLT2 inhibitors in animal models of non-diabetic heart failure. Byrne et al. revealed that the administration of empagliflozin alleviated left ventricular systolic dysfunction in non-diabetic mice subjected to pressure overload both in vivo and ex vivo (12). Andreadou et al. and Yurista et al. demonstrated that the administration of empagliflozin reduced the infarcted area of the myocardium, thereby improving the cardiac function in experimental non-diabetic myocardial infarction models (13, 14). In this background, randomized clinical trials were designed to explore the effects of SGLT2 inhibitors in patients with established heart failure with or without diabetes. Recently, the DAPA-HF trial demonstrated the significant advantage of dapagliflozin in reducing major adverse outcomes, such as unexpected hospitalization due to the exacerbation of heart failure, in patients with established heart failure with a reduced ejection fraction (HFrEF) (15). However, for SGLT2 inhibitors to be safely used for the treatment of non-diabetic heart failure, it is essential to elucidate their mechanism of action in detail. Thus far, a number of hypothesized mechanisms have proposed to explain the benefits of SGLT2 inhibitors in heart failure (6). Some investigators suggested that SGLT2 inhibitor-mediated natriuresis reduces the plasma volume or interstitial fluid, thereby favorably influencing ventricular remodeling by reducing the cardiac volume (16). Other investigators suggested that SGLT2 inhibitors alleviate heart failure through the suppression of sympathetic nervous activity, as evidenced by the reduction in arterial blood pressure without an increase in heart rate (7, 17). Still others hypothesized that SGLT2 inhibitors enhance the synthesis of erythropoietin by restoring the activity of “neural crest-derived” fibroblasts surrounding the renal proximal tubules, which, in turn, increases the delivery of oxygen to the failing myocardium (18). Thus, the targets through which SGLT2 inhibitors exert their protective effects against heart failure are mainly located outside of the heart. However, some investigations regarding this issue demonstrated that SGLT2 inhibitors have the potential to directly protect cardiomyocytes. Most such investigations have argued that SGLT2 inhibitors directly alleviate cardiac dysfunction through the modulation of mitochondria-associated mechanisms, including ketone body metabolism, sodium metabolism, and mitochondrial dynamics.
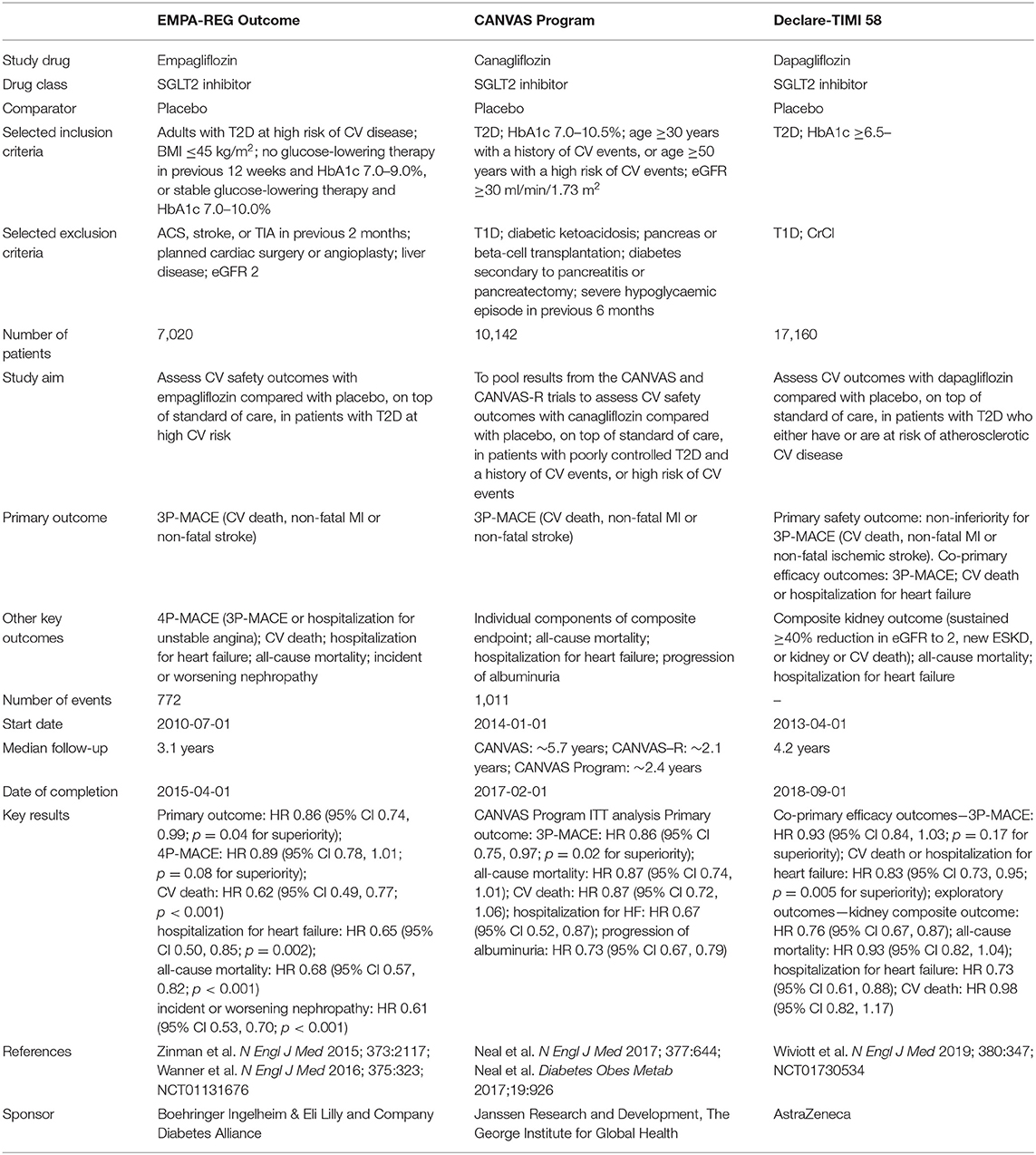
Table 1. Summary of cardiovascular outcome trials with SGLT2 inhibitors.
The inhibition of SGLT2 induces glucosuria, which thereby lowers plasma glucose levels, resulting in a reduction in the insulin level and an increase in the glucagon level during the fasting state. Such hormonal changes facilitate lipolysis in adipose tissue, and—at the same time—promote the conversion of carbohydrate to fat in whole-body substrate utilization. Thus, the administration of SGLT2 inhibitors could elevate ketone body levels in humans (Figure 2) (19). Ketone bodies, which are composed of acetoacetate (AcAc), β-hydroxybutyrate (βOHB), and acetone, are exclusively generated in the liver when the supply of glucose is impaired due to either a reduction of exogenous influx or deterioration of insulin signaling, or when the amount of free fatty acids (FFAs) is excessive due to the hyperactivation of lipolysis (20). During such situations, fatty acid β-oxidation is upregulated, thereby increasing the NADH/NAD+ ratio, which in turn promotes the conversion of AcAc to βOHB in the mitochondria of the liver (Figure 2). FFAs, a major source of ketone bodies, are taken up into hepatocytes, and β-oxidation transforms FFAs into acetyl-CoA and acetoacetyol-CoA (AcAc-CoA). 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 (HMGCS2), a rate-limiting mitochondrial enzyme, catalyzes the condensation of acetyl-CoA and AcAc-CoA to generate 3-hydroxy-3-methtylglutaryl-CoA (HMG-CoA) (21). Subsequently, 3-hydroxy-3-methylglutaryl-coenzyme A lyase (HMGCL) sequentially cleaves HMG-CoA into acetyl-CoA and AcAc (22, 23). Then, D-β-hydroxybutyrate dehydrogenase (BDH1) converts AcAc to βOHB, a more stable form of ketone body. In addition, both the kidneys and intestines play a critical role in maintaining ketone body homeostasis by regulating the ketone-reabsorptive capacity through sodium-dependent monocarboxylate transporter (SMCT) 1 and 2. Ketone bodies diffuse into the circulation and are used as an energy source in various organs (24). In the mitochondria of the heart, ketone bodies are rapidly converted to acetyl CoA through catalyzation with several enzymes, such as βOHB dehydrogenase (BDH1), succinyl-CoA:3-oxoacid-CoA transferase (SCOT), and mitochondrial acetyl-CoA acetyltransferase 1 (25).
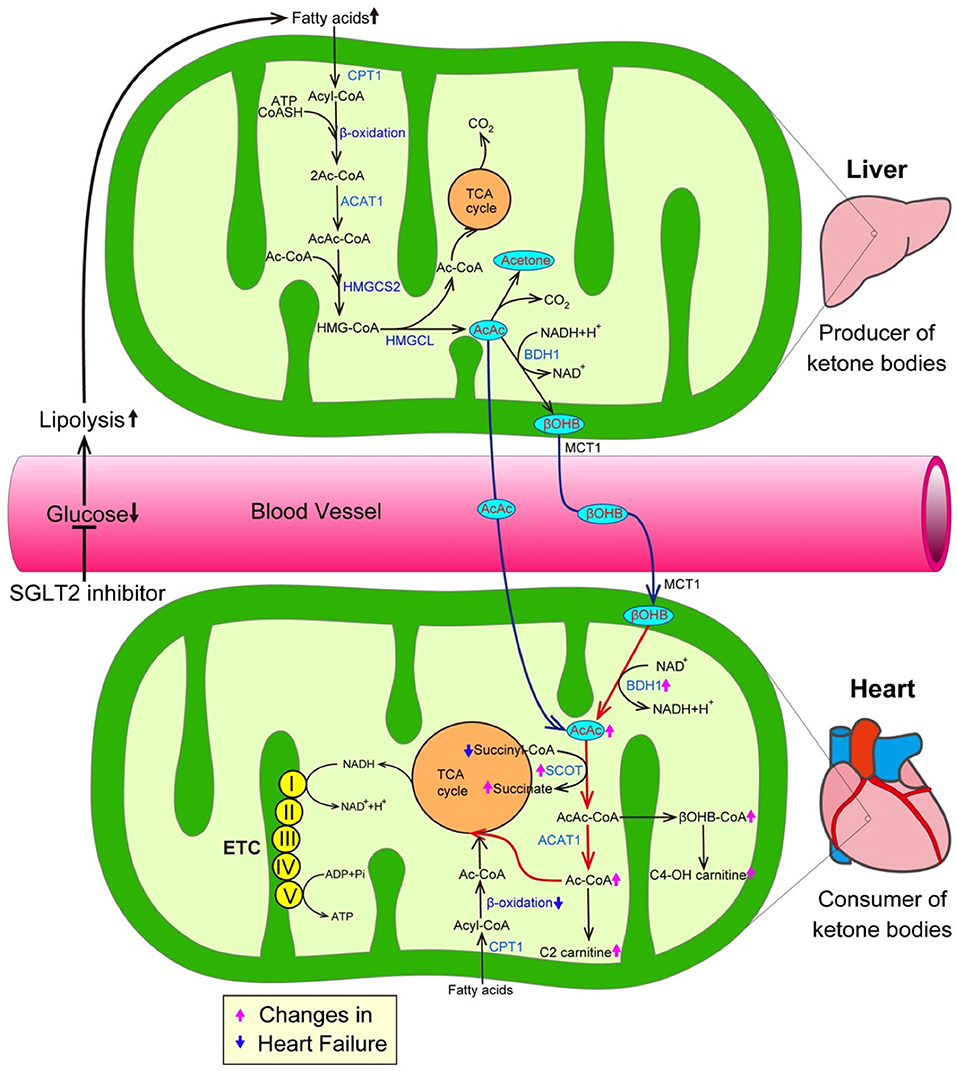
Figure 2. SGLT2 inhibitors increase the amount of ketone bodies, thereby promoting cardioprotective effects. The inhibition of SGLT2 reduces plasma glucose levels, thereby promoting lipolysis in adipose tissue, which in turn enhances the generation of ketone bodies. On the other hand, a growing body of evidence suggests that ketone bodies are favorable substrates in energy production because the conversion of ketone bodies to acetyl-CoA is much easier in comparison to the conversion of FFAs and glucose to acetyl-CoA. Furthermore, transcriptional level changes of ketone oxidation-related genes would be associated with the substrate shift to ketone bodies in the failing heart. Both pink and blue arrows show the changes in heart failure. AcAc CoA, Acetoacetyl CoA; ACAT1, Acetyl-CoA acetyltransferase; ADP, Adenosine diphosphate; ATP, Adenosine triphosphate; BDH1, Mitochondrial β-hydroxybutyrate dehydrogenase; βOHB, β-hydroxybutyrate; βOHB CoA, β-hydroxybutyryl CoA; C2-carnitine, Acetylcarnitine; C4-OH carnitine, Hydroxybutyrylcarnitine; CPT1, Carnitine palmitoyltransferase 1; ETC, Electron transport chain; HMGCL, 3-hydroxy-3-methylglutaryl-coenzyme A lyase; HMGCS2, 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2; and SCOT, Succinyl-CoA:3-oxoacid-CoA transferase.
The mammalian heart requires a vast amount of energy to maintain a normal contractile function and intracellular energy storage is limited. Thus, cardiomyocytes must generate an enormous amount of adenosine triphosphate (ATP) via the oxidation of carbon fuel. Under normal conditions, the predominant energy source of cardiomyocytes is FFAs, which provide 60% of the myocardial ATP demand by β-oxidation (26). The remaining 40% of the myocardial ATP demand is provided by carbohydrate oxidation, including glycolysis. The proportions of the energy sources of cardiomyocytes dynamically changes according to conditions such as exercise, feeding and starvation. When the mitochondrial oxidative metabolism balance of cardiomyocytes is seriously damaged due to various stresses including hypoxia and pressure overload, the major origin of ATP shifts from β-oxidation-mediated FA degradation to carbohydrate oxidation-mediated glucose catabolism. Such metabolic adaptation during hypoxia is reasonable because the glycolysis pathway can work, even under anaerobic conditions. However, as the efficiency of ATP production in glycolysis is significantly lower than that in mitochondrial oxidative metabolism, more efficient energy sources are required in the failing heart, in which the oxygen supply is impaired for an extended period of time (27). From this perspective, ketone bodies are a favorable substrate for energy production because the conversion of ketone bodies to acetyl-CoA is much easier in comparison to the conversion of FFAs and glucose (Figure 2) (28, 29). More importantly, ketone bodies can lead to the more efficient oxidation of the mitochondrial coenzyme Q couple and enhance the free energy of cytosolic ATP hydrolysis. Furthermore, changes at the transcriptional level of ketone oxidation-related genes would be associated with the substrate shift to ketone bodies in the failing heart. Indeed, previous investigations revealed that ketone metabolism is increased accompanied with the decrease of fatty acid oxidation in failing heart, as evidenced by the elevation of the levels of BDH1 and ketone body-derived materials, such as hydroxybutyryl-carnitine (C4OH-carnitine), βOHB-CoA and acetyl-carnitine (C2-carnitine) (28, 29) (Figure 2). In addition, a number of studies have demonstrated that the intake of a ketogenic diet extends longevity and the health span (30). Shimazu et al. revealed part of its mechanism. Treatment with βOHB inhibits histone deacetylase, thereby promoting FoxO3A and MT2 activity, which, in turn, markedly reduce oxidative stress and extend the life span in mice (31). Furthermore, ketone bodies possess anti-inflammatory activity (32). Youm et al. demonstrated that ketone bodies play an anti-inflammatory role by inhibiting the activity of the NOD-like receptor pyrin domain containing protein 3 (NLRP3) inflammasomes in animal models (33).
Thus, the elevation of ketone levels by SGLT2 inhibition might have a beneficial effect in patients with heart failure through multiple mechanisms.
As the inhibition of SGLT2 induces natriuresis as well as glycosuria because SGLT2 cotransports glucose with sodium, SGLT2 blockade could alter intracellular sodium homeostasis. Sodium plays an important role in mitochondrial redox regulation and excitation-contraction coupling in cardiomyocytes (Figure 3) (34, 35). Indeed, to produce energy in the form of ATP, cardiomyocytes primarily depend on the mitochondrial oxidative phosphorylation system (OXPHOS). Nicotinamide adenine dinucleotide (NADH), a reducing equivalent that is produced from the tricarboxylic acid (TCA) cycle, donates its electron to complexes I, III and IV of the electron transport chain (ETC), thereby promoting the translocation of H+ to the mitochondrial intermembrane space. The reduced form of flavin adenine dinucleotide (FADH2) also participates in the ETC reaction by donating its electron to complex II. The concentration gradient of H+ translocation generated by this reaction drives the conversion from ADP to ATP at the F1/F0-ATP synthase. The increase of ADP caused by the increased energy demand enhances the production of ATP at the F1/F0-ATP synthase, thereby promoting the oxidization of NADH to NAD+. Concurrently, the increase of cytosolic Ca2+ transients by β-adrenergic stimulation promotes the uptake of mitochondrial Ca2+ through the mitochondrial Ca2+ uniporter (MCU) (36). Then, Ca2+ activates the dehydrogenases of the TCA cycle to promote the regeneration of NADH (37). Thus, OXPHOS acts in concert with the TCA cycle to preserve constant ratios of ATP/ADP and NADH/NAD+ (38). In addition, nicotinamide adenine dinucleotide phosphate (NADPH) which is produced from NADH and TCA cycle products such as malate and isocitrate, plays a critical role in maintaining oxidative defense by donating electrons to reduced glutathione, thioredoxin, and glutaredoxin pools. Thus, the mitochondrial Ca2+ uptake is crucial for preserving the mitochondrial antioxidative capacity as well as for matching the energy supply to the demand (39). Ca2+ handling in cardiomyocytes is closely coordinated with Na+ handling through the activity of the sarcolemmal Na+/Ca2+ exchanger (NCX) and the mitochondrial Na+/Ca2+ exchanger (NCLX). The cardiac NCX entirely bails out Ca2+ to the extracellular space under physiological conditions. However, NCX sets out to import Ca2+ to the cytosol in the early phase of the action potential, depending on the membrane potential and the Na+ and Ca2+ transmembrane gradients (40). The cardiac NCLX is mainly responsible for the extrusion of Ca2+ from mitochondria. However, as the kinetics of NCLX are slower in comparison to the uptake of Ca2+ via the MCU, it is susceptible to the accumulation of Ca2+ in mitochondria after increasing the rate and amplitude of cytosolic Ca2+ transients.
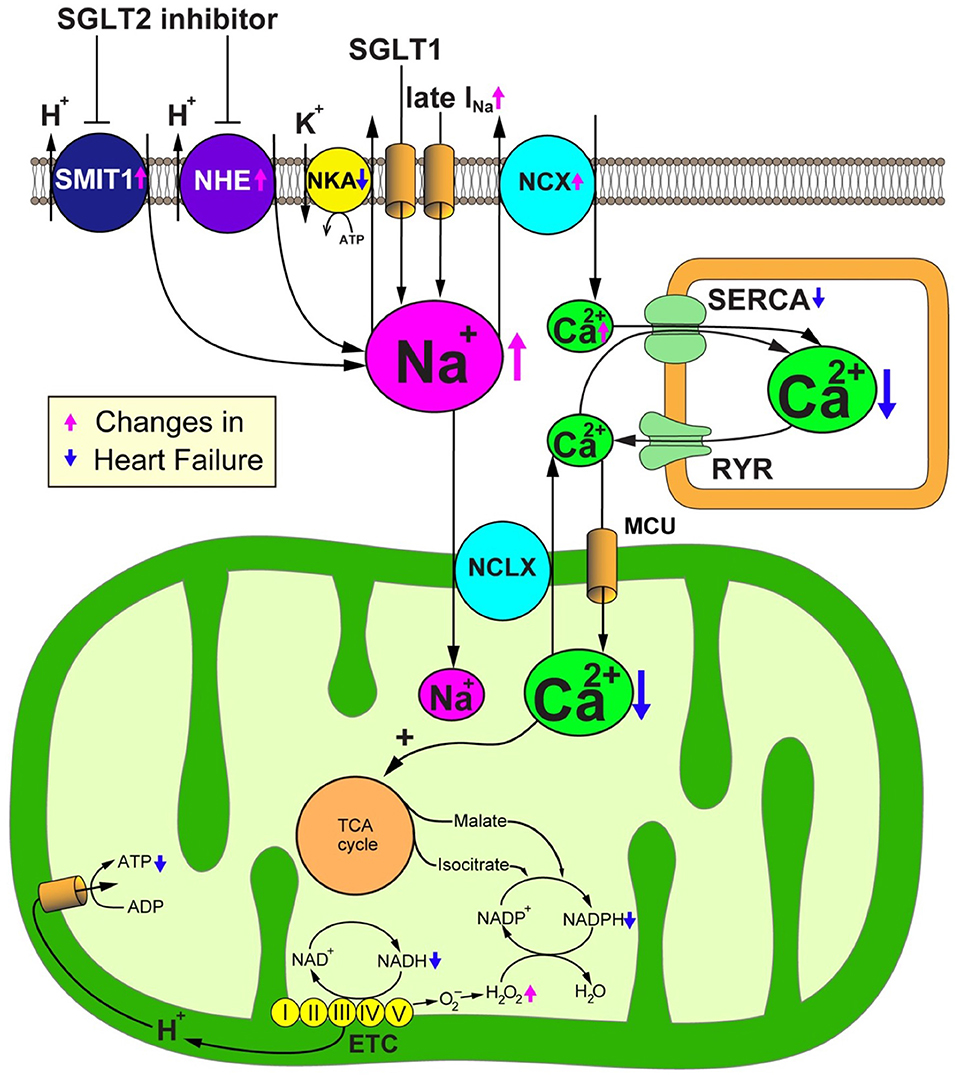
Figure 3. SGLT2 inhibitors promote sodium metabolism-mediated cardioprotective effects. Failing cardiomyocytes show elevated intracellular Na+ concentrations due to (1) increased Na+ influx via the late Na+ current (INa), (2) enhanced sarcolemmal Na+/H+ exchanger (NHE) activity, (3) reduced Na+/K+ ATPase (NKA) activity, and in the case of the diabetic heart, (4) the increased expression and activity of Na+-glucose cotransporter 1 (SGLT1). Intracellular overload of Na+ promotes Ca2+ efflux from mitochondria through the mitochondrial Na+/Ca2+ exchanger (NCLX). The reduction of the Ca2+ concentration in the mitochondrial matrix deteriorates the Ca2+-induced upregulation of TCA cycle dehydrogenases in response to workload transition, thereby disturbing the regeneration of reducing equivalents that are essential for preserving the antioxidative capacity and matching the energy supply to the energy demand. SGLT2 inhibitors would have a salutary role in failing cardiomyocytes through their alleviation of Na+ and Ca2+ handling through NHE inhibition. ADP, adenosine diphosphate; ATP, adenosine triphosphate; ETC, electron transport chain; MCU, mitochondrial Ca2+ uniporter; NAD+/NADH, nicotine amide dinucleotide oxidized/reduced; NCX, sarcolemmal Na+/Ca2+ exchanger; NKA, Na+/K+ ATPase; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+ ATPase.
Heart failure is closely associated with the impairment of both Ca2+ and Na+ handling in cardiomyocytes. Indeed, the amplitude and velocity of cytosolic Ca2+ transients are decreased in failing cardiomyocytes. Furthermore, the elevation of diastolic cytosolic Ca2+ ([Ca2+]c) and Na+ concentrations ([Na+]c) is observed in failing cardiomyocytes (41, 42). The impairment of Ca2+ handling is due to the decrease of the Ca2+ uptake by the sarco/endoplasmic reticulum Ca2+- ATPase (SERCA) and the leak of Ca2+ from the sarcoplasmic reticulum (SR) via ryanodine receptors (43, 44). The increase in the expression and activity of the NCX promotes the export of Ca2+ into the extracellular space, and thereby also reduces the Ca2+ load of the SR (45). Furthermore, the reduction of the release of Ca2+ from the SR results in the impairment of the mitochondrial Ca2+ uptake and steady-state Ca2+ concentration ([Ca2+]m) (46). On the other hand, excessive influx of Ca2+ into the mitochondria is detrimental to cardiomyocytes. The elevation of [Ca2+]m triggers depolarization of mitochondrial inner membrane potential, generation of reactive oxygen species (ROS), and opening the mitochondrial permeability transition pore (47, 48), thereby promoting the release of pro-apoptotic proteins, such as cytochrome c, into the cytosol (49).
Increasing lines of evidence suggest that [Na+]c is significantly elevated in failing cardiomyocytes as a result of (1) increased Na+ influx via the late Na+ current (INa) (41), (2) enhancement of sarcolemmal Na+/H+ exchanger (NHE) activity (50), (3) reduction of Na+/K+ ATPase (NKA) activity (51), and—in the case of diabetic heart—(4) the increased expression and activity of the Na+-glucose cotransporter 1 (SGLT1) (52) (Figure 2). Generally, the increase of [Na+]c should trigger positive effects on cytosolic Ca2+ handling because intracellular Na+ overload prevents the NCX from exporting Ca2+ during the diastolic phase and promotes the reverse-mode function of the NCX during the action potential—thereby enhancing additional trans-sarcolemmal Ca2+ influx to achieve the elevation of Ca2+ in the SR and increase the amplitude of cytosolic Ca2+ transients. However, from a metabolic point of view, the elevation of [Na+]c results in detrimental effects, especially in mitochondria. As Ca2+ is pumped out of mitochondria to the cytosol by an NCLX, the elevation of [Na+]c enhances the driving force for mitochondrial Ca2+ efflux. The decrease of [Ca2+]m suppresses the Ca2+-induced upregulation of dehydrogenases in the TCA cycle, thereby attenuating the production of both NADH and NADPH (46). The decreased production of NADH causes ATP depletion. The reduction of the amount of NADPH causes the impairment of mitochondrial antioxidative defense because the donation of electrons from NADPH is indispensable for antioxidative enzymes, such as peroxiredoxin, glutathione peroxidase, and glutaredoxin. Thus, the elevation of [Na+]c enhances oxidative stress, thereby aggravating the vulnerability of the heart to arrhythmias and neurohormonal hyperactivation. Furthermore, the increase of [Na+]c eventually causes the emission of mitochondrial ROS, which results in the further deterioration of the intracellular Na+ overload (35). Based on these facts, reducing the intracellular Na+ overload to improve mitochondrial energetics and oxidative defense could be a promising therapeutic strategy for heart failure (Figure 3).
With regard to the beneficial effects of SGLT2 inhibitors on heart failure, it was initially considered that SGLT2 inhibitors have no direct effect on cardiomyocytes because SGLT2 is not expressed in the heart in either healthy subjects or under pathological conditions (5). However, a recent investigation demonstrated that empagliflozin reduced [Na+]c and [Ca2+]c in isolated cardiomyocytes (53). According to this report, empagliflozin directly reduced myocardial [Na+]c and [Ca2+]c and elevated [Ca2+]m by suppressing myocardial NHE flux, independently of glucose transport. Habibi et al. demonstrated that the administration of empagliflozin mitigates diastolic dysfunction in db/db mice (54). The author of the present study found that empagliflozin suppresses the expression of serum- and glucocorticoid-inducible kinase 1 (SGK1) in the myocardium. As SGK1 activity may modulate NHE activity through Akt-mediated signaling, these results suggest that empagliflozin could restore myocardial [Na+]c in a sustained manner (55). Examinations using 23Na+ magnetic resonance imaging revealed that the tissue Na+ content in diabetic patients was markedly reduced by treatment with dapagliflozin (56). An in silico docking study demonstrated that three SGLT2 inhibitors, empagliflozin, dapagliflozin, and canagliflozin, showed high binding affinity with the extracellular Na+-binding site of NHE (57). In this study, the authors confirmed—by in vitro experiments—that empagliflozin, dapagliflozin and canagliflozin directly inhibit the cardiac NHE flux and reduce [Na+]c.
The expression of NHE is upregulated in the failing heart, possibly through the acidification of the intracellular environment due to increased conversion of pyruvate to lactate (58). Similarly, the NHE activity of cardiomyocytes of the animal models of type 2 diabetes and the suppression of [Na+]c by NHE inhibition with cariproride was found to be cardioprotective (59, 60). Specifically, cariproride significantly suppressed the elevation of [Na+]c at the end of ischemia and inhibited ventricular arrhythmia during reperfusion in a db/db mouse model of ischemia/reperfusion (59). In the Goto–Kakizaki rat model of type 2 diabetes, which does not develop hypertension, obesity or hyperlipidemia, the NHE activity of cardiomyocytes is markedly upregulated, which results in an increase in [Na+]c. In this model, the intracellular Na+ overload was closely associated with the Akt-mediated progression of left ventricular hypertrophy. Consistently, the administration of cariproride significantly suppressed both [Na+]c and Akt activation, resulting in the attenuation of cardiac hypertrophy (60).
There are seven SGLT isoforms (SGLT1 to 6 and sodium-myoinositol cotransporter 1, SMIT1). Among these, only SGLT1 and SMIT1 are expressed in the mammalian heart. The overexpression of SMIT1 activates NOX2, increases ROS, and exacerbates glucotoxicity in cardiomyocytes. Consistently, the deletion of SMIT1 prevented hyperglycemia-induced NOX2 activation (61). Thus far, the physiological role of SMIT1 in the heart remains unknown, as the deletion of SMIT1 does not alter the cardiac phenotype. Interestingly, however, SMIT1 is hardly associated with the glucose uptake in the heart, regardless of any glycemic conditions. Thus, SMIT1-mediated NOX2 activation would modulate glucose sensitization, which could trigger ionic signaling ([Na+]c and [Ca2+]c via the NCX) into cells in association with the changes in the extracellular glucose concentration. Concomitantly, intracellular signaling via protein kinase C (PKC)-β, a calcium-dependent serine/threonine kinase, could be the link to ionic changes downstream of SMIT1. The IC50 of empagliflozin and canagliflozin for SMIT1 are estimated to be 8.3 and 5.6 μM, respectively (62, 63). Indeed, empagliflozin is even able to reduce [Na+]c in the absence of glucose (53).
Mitochondria continuously fuse and divide in highly regulated manners to maintain their functions, which include metabolism, energy production, intracellular signaling, and the regulation of apoptosis. The enhancement of mitochondrial fusion would allow for the making up of “healthy” mitochondria, resulting in the normalization of the overall mitochondrial function. In response to various stresses, mitochondria undergo stress-induced mitochondrial hyperfusion (64), which thereby enhances ATP production, which—in turn—plays a pro-survival role. On the other hand, damaged mitochondria must be removed to preserve mitochondrial homeostasis. To this end, mitochondrial fission could be enhanced to more easily remove dysfunctional mitochondria via mitochondria-selective autophagy, termed mitophagy (65). Several key regulators are required for the operation of such mitochondrial dynamics. Mitochondrial fusion is regulated by mitofusin1 (Mfn1), mitofusin2 (Mfn2), and Opa1 (Figure 4) (66). On the other hand, mitochondrial fission is regulated by the recruitment of Dynamin-related protein 1 (Drp1) to specific sites on the outer mitochondrial membrane in coordination with mitochondrial fission 1 (Fis1) and mitochondrial fission factor (Mff) (Figure 4) (67).
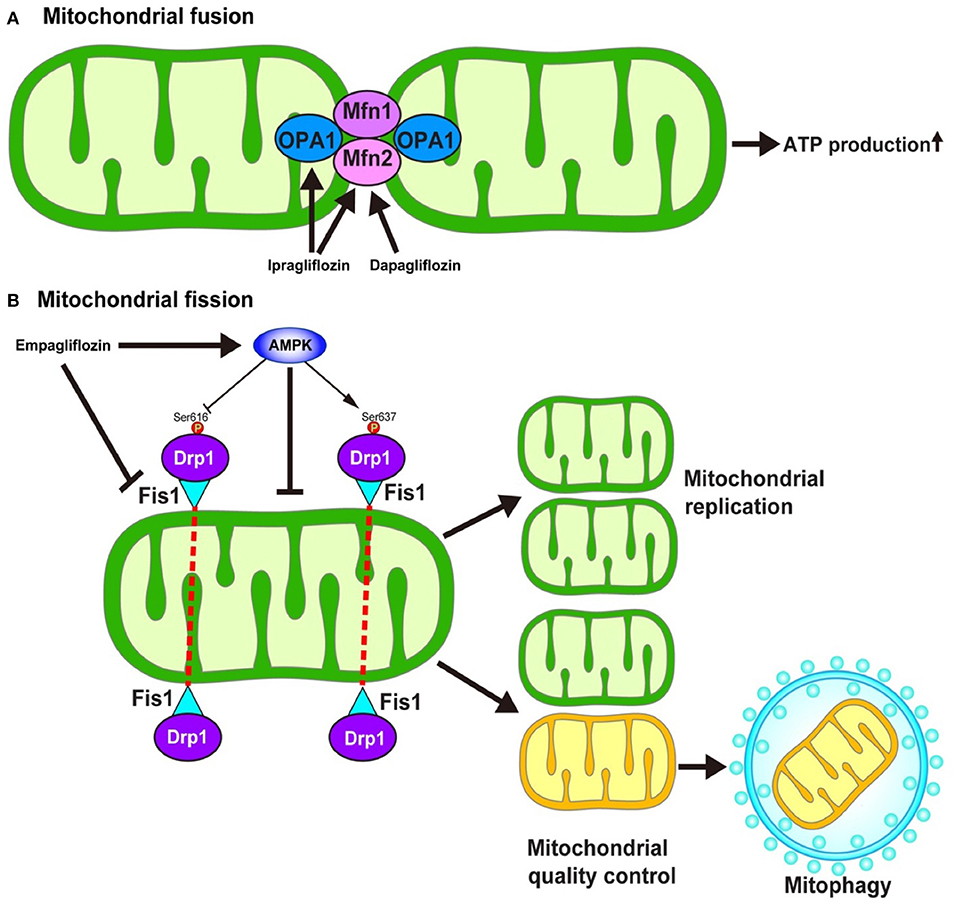
Figure 4. Hypothesized mechanism of the modulation of mitochondrial dynamics by SGLT2 inhibitors. The inhibition of SGLT2 might be associated with the mitochondrial dynamics through the regulation of (A) mitochondrial fusion and (B) mitochondrial fission. However, the detailed mechanism as to how SGLT2 inhibitors modulate the regulators of mitochondrial dynamics is largely unknown. AMPK, AMP-activated protein kinase; Drp1, Dynamin-related protein 1; Fis1, Mitochondrial fission 1 protein; Mfn, Mitofusin; Ser, Serine.
The impairment of mitochondrial fusion via the downregulation of Mfn1 and Mfn2 aggravates cardiac dysfunction both at baseline and in response to stress (68). On the other hand, the inhibition of mitochondrial fission by the pharmacological suppression of Drp1 with Mdivi-1 reduces the size of infarcts that develop in response to ischemia/reperfusion (I/R) (69). In contrast, the inhibition of mitochondrial fission by genetic modulation, such as the knockdown of Fis1 mRNA or the expression of dominant-negative mutation in Drp1, inhibits mitophagy which results in metabolic dysfunction in INS1 cells (70), suggesting that mitochondrial fission has a two-sided nature with respect to cell survival in the myocardium.
Increasing lines of evidence suggest that SGLT2 inhibitors may modulate mitochondrial dynamics. Ipragliflozin alleviates the mitochondrial dysfunction induced by a high-fat diet by restoring the levels of Opa1 and Mfn2 to normal values in vivo without reducing body weight or blood glucose levels in rat models (71). Similarly, dapagliflozin normalizes the Mfn1/Mfn2 ratio in the rat model of metabolic syndrome, thereby suppressing prolonged ventricular repolarization (72). Empagliflozin restores the AMP/ATP ratio, thereby activating adenosine monophosphate (AMP)-activated protein kinase (AMPK) (73). The activation of AMPK causes an increase in Drp1S637 phosphorylation and a decrease in Drp1S616 phosphorylation, which results in the suppression of mitochondrial fission. Another study demonstrated that empagliflozin normalized the size and number of mitochondria in the OLETF diabetic rat heart and that the diabetes-induced excessive reduction in mitochondrial size after MI was inhibited by empagliflozin via the suppression of Fis1 upregulation and following ROS production, which results in the reduction of the MI size (74).
Thus, inhibition of SGLT2 is closely associated with the mitochondrial dynamics through the regulation of fusion and fission of mitochondria. Although several hypotheses have been proposed (71, 74, 75), the detailed molecular mechanism through which mitochondrial fusion and fission are modulated by the administration of SGLT2 inhibitors is largely unknown. Furthermore, it remains to be determined whether the effect of SGLT2 inhibitors on AMPK activity, one of the key molecules in the regulation of mitochondrial fission, is a class effect or a drug-specific effect. Indeed, Mancini et al. reported that canagliflozin, but not dapagliflozin or empagliflozin, could enhance AMPK activity both in human umbilical vein endothelial cells and human arterial endothelial cells (76). In addition, the precise roles of mitochondrial fission and fusion in the development of heart failure remain to be determined.
We reviewed the proposed cardioprotective effect of SGLT2 inhibitors, which is mediated through the improvement of the mitochondrial function by (1) increasing ketone body usage, (2) the mitigation of sodium metabolism, and (3) the modulation mitochondrial dynamics. However, many questions remain to be solved to validate these hypotheses. Indeed, it remains controversial whether SGLT2 inhibitors could be directly involved in the protective effects of cardiomyocytes, which do not express SGLT2. In particular, regarding the regulation of mitochondrial dynamics, previous studies merely observed the change in the expression levels of factors that regulate the mitochondrial dynamics (e.g., Mfn1 or Drp1) in response to the administration of SGLT2 inhibitors. Thus, the molecular mechanism through which these compounds modulate mitochondrial fusion and fission remains to be elucidated. Regarding the association with ketone body metabolism, it is necessary to determine whether the favorable effects induced by the increase in ketone bodies would be limited in the alteration of the mitochondrial energy metabolism. Furthermore, the possibility that these drugs could regulate different target molecule(s) other than SGLT2 (i.e., have off-target effects) should be examined. Indeed, the hypothesis that SGLT2 inhibitors regulate sodium metabolism is based on the fact that SGLT2 inhibitors possess the potential to inhibit both NHE and SMIT1.
As stated above, the DAPA-HF trial demonstrated that dapagliflozin plays a protective role in patients with established HFrEF, regardless of the presence of diabetes (15). Currently, the EMPEROR-Reduced trial [NCT03057977] to evaluate the efficacy of empagliflozin vs. placebo on top of guideline-directed medical therapy in HFrEF patients with or without diabetes is ongoing (77). If empagliflozin is proven to be beneficial in patients with HFrEF based on the results of this trial, it would provide more robust evidence of the beneficial effect of SGLT2 inhibitors on heart failure. At the same time, two randomized clinical trials are evaluating the effects of SGLT2 inhibitors in patients with established heart failure with a preserved ejection fraction (HFpEF), regardless the presence of diabetes. One is the EMPEROR-Preserved trial [NCT03057951] with empagliflozin (78), and the other one is the DELIVER trial [NCT03619213] with dapagliflozin. Several preclinical studies proposed the mechanism how SGLT2 inhibitor alleviates cardiac diastolic dysfunction, a major cause of HFpEF. For example, Juni et al. demonstrated that Empagliflozin suppresses TNF-α-induced mitochondrial and cytoplasmic ROS accumulation, thereby restoring cardiac microvascular endothelial cell-derived NO delivery, which in turn leads to reinstatement of cardiac relaxation and contraction (79). There are great expectations regarding the result of these clinical trials because, at the time of writing, no drugs have been demonstrated to be effective for the treatment of HFpEF (80).
As is the case with the positive effects, unfavorable aspects of SGLT2 inhibitor administration for the heart failure patients should be considered. Increasing lines of evidence suggest that sarcopenia is one of the major risk factors for morbidity and mortality of heart failure. Past clinical observations demonstrated that the skeletal muscle mass reduction is observed in a given number of patients with diabetes who were treated with SGLT2 inhibitors. Also, the decreased exercise capacity, one of the major causes of sarcopenia which is the consequence of mitochondrial dysfunction in skeletal muscles, is an independent predictor of the poor prognosis of patients with heart failure (81). Thus, basically, the patients who are susceptible to sarcopenia should not be prescribed SGLT2 inhibitors. On the other hand, a recent investigation demonstrated the intriguing result that Empagliflozin restored decreased exercise endurance capacity by alleviating skeletal muscle fatty acid oxidation in an animal heart failure model (82). In any case, we should carefully determine which kind of patients are optimal for the treatment with SGLT2 inhibitors.
Taken together, unremitting efforts to elucidate the molecular mechanism through which the administration of SGLT2 inhibitors alleviates heart failure, as well as clinical studies of these compounds for non-diabetic heart failure could shift their classification from merely anti-diabetic drugs to potent anti-heart failure drugs.
The author confirms being the sole contributor of this work and has approved it for publication.
This work was supported in part by a JSPS KAKENHI Grant-in-Aid for Scientific Research (C) (17K09570), the Smoking Research Foundation, and Bristol-Myers Squibb Research Grant 2018.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author would like to thank Dr. Shun Nakagama, Dr. Yuka Shiheido-Watanabe, Dr. Noriko Tamura, Dr. Tetsuo Sasano, and Ms. Noriko Tamura, for their excellent contributions. Also, the author thanks Brian Quinn (Japan Medical Communication) for critical reading.
1. Pajor AM, Wright EM. Cloning and functional expression of a mammalian Na+/nucleoside cotransporter. A member of the SGLT family. J Biol Chem. (1992) 267:3557–60.
2. Diedrich DF. Competitive inhibition of intestinal glucose transport by phlorizin analogs. Arch Biochem Biophys. (1966) 117:248–56. doi: 10.1016/0003-9861(66)90409-7
3. Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to PHARMACOLOGY 2013/14: transporters. Br J Pharmacol. (2013) 170:1706–96. doi: 10.1111/bph.12450
4. Chao EC, Henry RR. SGLT2 inhibition–a novel strategy for diabetes treatment. Nat Rev Drug Discov. (2010) 9:551–9. doi: 10.1038/nrd3180
5. Chen J, Williams S, Ho S, Loraine H, Hagan D, Whaley JM, et al. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. (2010) 1:57–92. doi: 10.1007/s13300-010-0006-4
6. Chin KL, Ofori-Asenso R, Hopper I, von Lueder TG, Reid CM, Zoungas S, et al. Potential mechanisms underlying the cardiovascular benefits of sodium glucose cotransporter 2 inhibitors: a systematic review of data from preclinical studies. Cardiovasc Res. (2019) 115:266–76. doi: 10.1093/cvr/cvy295
7. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
8. Fitchett D, Zinman B, Wanner C, Lachin JM, Hantel S, Salsali A, et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME® trial. Eur Heart J. (2016) 37:1526–34. doi: 10.1093/eurheartj/ehv728
9. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
10. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2019) 380:347–57. doi: 10.1056/NEJMoa1812389
11. Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. (2019) 393:31–9. doi: 10.1016/S0140-6736(18)32590-X
12. Byrne NJ, Parajuli N, Levasseur JL, Boisvenue J, Beker DL, Masson G, et al. Empagliflozin prevents worsening of cardiac function in an experimental model of pressure overload-induced heart failure. JACC Basic Transl Sci. (2017) 2:347–54. doi: 10.1016/j.jacbts.2017.07.003
13. Andreadou I, Efentakis P, Balafas E, Togliatto G, Davos CH, Varela A, et al. Empagliflozin limits myocardial infarction in vivo and cell death in vitro: role of STAT3, mitochondria, and redox aspects. Front Physiol. (2017) 8:1077. doi: 10.3389/fphys.2017.01077
14. Yurista SR, Sillje HHW, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, et al. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. (2019) 21:862–73. doi: 10.1002/ejhf.1473
15. McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. (2019) 381:1995–2008. doi: 10.1056/NEJMoa1911303
16. Dekkers CCJ, Sjostrom CD, Greasley PJ, Cain V, Boulton DW, and Heerspink HJL. Effects of the sodium-glucose co-transporter-2 inhibitor dapagliflozin on estimated plasma volume in patients with type 2 diabetes. Diabetes Obes Metab. (2019) 21:2667–73. doi: 10.1111/dom.13855
17. Blood Pressure Lowering Treatment Trialists' Collaboration. Blood pressure-lowering treatment based on cardiovascular risk: a meta-analysis of individual patient data. Lancet. (2014) 384:591–8. doi: 10.1016/S0140-6736(14)61212-5
18. Sano M, Takei M, Shiraishi Y, Suzuki Y. Increased Hematocrit During sodium-glucose cotransporter 2 inhibitor therapy indicates recovery of tubulointerstitial function in diabetic kidneys. J Clin Med Res. (2016) 8:844–7. doi: 10.14740/jocmr2760w
19. Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME Trial: a “thrifty substrate” hypothesis. Diabetes Care. (2016) 39:1108–14. doi: 10.2337/dc16-0330
20. Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. (2017) 25:262–84. doi: 10.1016/j.cmet.2016.12.022
21. Qiu H, Novikov A, Vallon V. Ketosis and diabetic ketoacidosis in response to SGLT2 inhibitors: basic mechanisms and therapeutic perspectives. Diabetes Metab Res Rev. (2017) 33:e2886. doi: 10.1002/dmrr.2886
22. Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J. (1967) 103:514–27. doi: 10.1042/bj1030514
23. Krebs HA, Wallace PG, Hems R, Freedland RA. Rates of ketone-body formation in the perfused rat liver. Biochem J. (1969) 112:595–600. doi: 10.1042/bj1120595
24. Ganapathy V, Thangaraju M, Gopal E, Martin PM, Itagaki S, Miyauchi S, et al. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. Aaps J. (2008) 10:193–9. doi: 10.1208/s12248-008-9022-y
25. Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME Study? A unifying hypothesis. Diabetes Care. (2016) 39:1115–22. doi: 10.2337/dc16-0542
26. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
27. Neubauer S, The failing heart–an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
28. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The failing heart relies on ketone bodies as a fuel. Circulation. (2016) 133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
29. Bedi KC. Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. (2016) 133:706–16. doi: 10.1161/CIRCULATIONAHA.115.017545
30. Johnson AA, Stolzing A. The role of lipid metabolism in aging, lifespan regulation, and age-related disease. Aging Cell. (2019) 18: e13048. doi: 10.1111/acel.13048
31. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. (2013) 339:211–4. doi: 10.1126/science.1227166
32. Ruskin DN, Kawamura M, Masino SA. Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS One. (2009) 4:e8349. doi: 10.1371/journal.pone.0008349
33. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. (2015) 21:263–9. doi: 10.1038/nm.3804
34. Clancy CE, Chen-Izu Y, Bers DM, Belardinelli L, Boyden PA, Csernoch L, et al. Deranged sodium to sudden death. J Physiol. (2015) 593:1331–45. doi: 10.1113/jphysiol.2014.281204
35. Bay J, Kohlhaas M, Maack C. Intracellular Na(+) and cardiac metabolism. J Mol Cell Cardiol. (2013) 61:20–7. doi: 10.1016/j.yjmcc.2013.05.010
36. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. (2006) 99:172–82. doi: 10.1161/01.RES.0000232546.92777.05
37. Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res. (2018) 122:1460–78. doi: 10.1161/CIRCRESAHA.118.310082
38. Maack C, O'Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. (2007) 102:369–92. doi: 10.1007/s00395-007-0666-z
39. Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, et al. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. (2010) 121:1606–13. doi: 10.1161/CIRCULATIONAHA.109.914911
40. Weber CR, Piacentino V III, Houser SR, Bers DM. Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation. (2003) 108:2224–9. doi: 10.1161/01.CIR.0000095274.72486.94
41. Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. (2002) 105:2543–8. doi: 10.1161/01.CIR.0000016701.85760.97
42. Lambert R, Srodulski S, Peng X, Margulies KB, Despa F, Despa S. Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-glucose cotransport. J Am Heart Assoc. (2015) 4:e002183. doi: 10.1161/JAHA.115.002183
43. O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. (1999) 84:562–70. doi: 10.1161/01.RES.84.5.562
44. Hobai IA, Maack C, O'Rourke B, Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. (2004) 95: 292–9. doi: 10.1161/01.RES.0000136817.28691.2d
45. Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, et al. Gene expression of the cardiac Na(+)-Ca2+ exchanger in end-stage human heart failure. Circ Res. (1994) 75:443–53. doi: 10.1161/01.RES.75.3.443
46. Kohlhaas M, and Maack C, Adverse bioenergetic consequences of Na+-Ca2+ exchanger-mediated Ca2+ influx in cardiac myocytes. Circulation. (2010) 122:2273–80. doi: 10.1161/CIRCULATIONAHA.110.968057
47. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. (2004) 287:C817–33. doi: 10.1152/ajpcell.00139.2004
48. Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. (2002) 84:153–66. doi: 10.1016/S0300-9084(02)01375-5
49. Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. (2004) 95:957–70. doi: 10.1161/01.RES.0000148632.35500.d9
50. Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. [Na+]i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc Res. (2003) 57:986–95. doi: 10.1016/S0008-6363(02)00848-9
51. Swift F, Birkeland JA, Tovsrud N, Enger UH, Aronsen JM, Louch WE, et al. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc Res. (2008) 78:71–8. doi: 10.1093/cvr/cvn013
52. Banerjee SK, McGaffin KR, Pastor-Soler NM, Ahmad F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc Res. (2009) 84:111–8. doi: 10.1093/cvr/cvp190
53. Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, et al. Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia. (2017) 60:568–73. doi: 10.1007/s00125-016-4134-x
54. Habibi J, Aroor AR, Sowers JR, Jia G, Hayden MR, Garro M, et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol. (2017) 16:9. doi: 10.1186/s12933-016-0489-z
55. Voelkl J, Lin Y, Alesutan I, Ahmed MS, Pasham V, Mia S, et al. Sgk1 sensitivity of Na(+)/H(+) exchanger activity and cardiac remodeling following pressure overload. Basic Res Cardiol. (2012) 107:236. doi: 10.1007/s00395-011-0236-2
56. Karg MV, Bosch A, Kannenkeril D, Striepe K, Ott C, Schneider MP, et al. SGLT-2-inhibition with dapagliflozin reduces tissue sodium content: a randomised controlled trial. Cardiovasc Diabetol. (2018) 17:5. doi: 10.1186/s12933-017-0654-z
57. Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia. (2018) 61:722–6. doi: 10.1007/s00125-017-4509-7
58. Takewaki S, Kuro-o M, Hiroi Y, Yamazaki T, Noguchi T, Miyagishi A, et al. Activation of Na(+)-H+ antiporter (NHE-1) gene expression during growth, hypertrophy and proliferation of the rabbit cardiovascular system. J Mol Cell Cardiol. (1995) 27:729–42. doi: 10.1016/S0022-2828(08)80063-6
59. Anzawa R, Bernard M, Tamareille S, Baetz D, Confort-Gouny S, Gascard JP, et al. Intracellular sodium increase and susceptibility to ischaemia in hearts from type 2 diabetic db/db mice. Diabetologia. (2006) 49:598–606. doi: 10.1007/s00125-005-0091-5
60. Darmellah A, Baetz D, Prunier F, Tamareille S, Rucker-Martin C, Feuvray D. Enhanced activity of the myocardial Na+/H+ exchanger contributes to left ventricular hypertrophy in the Goto-Kakizaki rat model of type 2 diabetes: critical role of Akt. Diabetologia. (2007) 50:1335–44. doi: 10.1007/s00125-007-0628-x
61. Van Steenbergen A, Balteau M, Ginion A, Ferte L, Battault S, Ravenstein CM, et al. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci Rep. (2017) 7:41166. doi: 10.1038/srep41166
62. Suzuki M, Honda K, Fukazawa M, Ozawa K, Hagita H, Kawai T, et al. Tofogliflozin, a potent and highly specific sodium/glucose cotransporter 2 inhibitor, improves glycemic control in diabetic rats and mice. J Pharmacol Exp Ther. (2012) 341:692–701. doi: 10.1124/jpet.112.191593
63. Scheen AJ. Evaluating SGLT2 inhibitors for type 2 diabetes: pharmacokinetic and toxicological considerations. Expert Opin Drug Metab Toxicol. (2014) 10:647–63. doi: 10.1517/17425255.2014.873788
64. Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. (2009) 28:1589–600. doi: 10.1038/emboj.2009.89
65. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. (2018) 80:1–26. doi: 10.1146/annurev-physiol-021317-121427
66. Maejima Y, Chen Y, Isobe M, Gustafsson AB, Kitsis RN, Sadoshima J. Recent progress in research on molecular mechanisms of autophagy in the heart. Am J Physiol Heart Circ Physiol. (2015) 308:H259–68. doi: 10.1152/ajpheart.00711.2014
67. Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, et al. Drp1-Dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. (2016) 133:1249–63. doi: 10.1161/CIRCULATIONAHA.115.020502
68. Chen Y, Liu Y, Dorn GW II. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. (2011) 109:1327–31. doi: 10.1161/CIRCRESAHA.111.258723
69. Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. (2010) 121:2012–22. doi: 10.1161/CIRCULATIONAHA.109.906610
70. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. (2008) 27:433–46. doi: 10.1038/sj.emboj.7601963
71. Takagi S, Li J, Takagaki Y, Kitada M, Nitta K, Takasu T, et al. Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J Diabetes Invest. (2018) 9:1025–32. doi: 10.1111/jdi.12802
72. Durak A, Olgar Y, Degirmenci S, Akkus E, Tuncay E, Turan B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc Diabetol. (2018) 17:144. doi: 10.1186/s12933-018-0790-0
73. Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. (2018) 15:335–46. doi: 10.1016/j.redox.2017.12.019
74. Mizuno M, Kuno A, Yano T, Miki T, Oshima H, Sato T, et al. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol Rep. (2018) 6:e13741. doi: 10.14814/phy2.13741
75. Esterline RL, Vaag A, Oscarsson J, Vora J. MECHANISMS IN ENDOCRINOLOGY: SGLT2 inhibitors: clinical benefits by restoration of normal diurnal metabolism? Eur J Endocrinol. (2018) 178:R113–25. doi: 10.1530/EJE-17-0832
76. Mancini SJ, Boyd D, Katwan OJ, Strembitska A, Almabrouk TA, Kennedy S, et al. Canagliflozin inhibits interleukin-1β-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP-activated protein kinase-dependent and -independent mechanisms. Sci Rep. (2018) 8:5276. doi: 10.1038/s41598-018-23420-4
77. Packer M, Butler J, Filippatos GS, Jamal W, Salsali A, Schnee J, et al. Evaluation of the effect of sodium-glucose co-transporter 2 inhibition with empagliflozin on morbidity and mortality of patients with chronic heart failure and a reduced ejection fraction: rationale for and design of the EMPEROR-reduced trial. Eur J Heart Fail. (2019) 21:1270–8. doi: 10.1002/ejhf.1536
78. Anker SD, Butler J, Filippatos GS, Jamal W, Salsali A, Schnee J, et al. Evaluation of the effects of sodium-glucose co-transporter 2 inhibition with empagliflozin on morbidity and mortality in patients with chronic heart failure and a preserved ejection fraction: rationale for and design of the EMPEROR-preserved trial. Eur J Heart Fail. (2019) 21:1279–87. doi: 10.1002/ejhf.1596
79. Juni RP, Kuster DWD, Goebel M, Helmes M, Musters RJP, van der Velden J, et al. Cardiac microvascular endothelial enhancement of cardiomyocyte function is impaired by inflammation and restored by empagliflozin. JACC Basic Transl Sci. (2019) 4:575–91. doi: 10.1016/j.jacbts.2019.04.003
80. Bonsu KO, Arunmanakul P, Chaiyakunapruk N. Pharmacological treatments for heart failure with preserved ejection fraction-a systematic review and indirect comparison. Heart Fail Rev. (2018) 23:147–56. doi: 10.1007/s10741-018-9679-y
81. Kinugawa S, Takada S, Matsushima S, Okita K, Tsutsui H. Skeletal muscle abnormalities in heart failure. Int Heart J. (2015) 56:475–84. doi: 10.1536/ihj.15-108
Keywords: SGLT2, mitochondria, ketone body, NHE, fusion, fission
Citation: Maejima Y (2020) SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 6:186. doi: 10.3389/fcvm.2019.00186
Received: 03 November 2019; Accepted: 10 December 2019;
Published: 08 January 2020.
Edited by:
Junichi Sadoshima, University of Medicine and Dentistry of New Jersey, United StatesReviewed by:
Junco Shibayama Warren, The University of Utah, United StatesCopyright © 2020 Maejima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yasuhiro Maejima, eW1hZWppLmN2bUB0bWQuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.