
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 17 December 2019
Sec. Structural Interventional Cardiology
Volume 6 - 2019 | https://doi.org/10.3389/fcvm.2019.00182
This article is part of the Research TopicCurrent Issues in Managing and Treating Thoracic Aortic DiseasesView all articles
 Shohreh Maleki1
Shohreh Maleki1 Flore-Anne Poujade1
Flore-Anne Poujade1 Otto Bergman1Jesper R. Gådin1Nancy Simon1
Otto Bergman1Jesper R. Gådin1Nancy Simon1 Karin Lång1
Karin Lång1 Anders Franco-Cereceda2Simon C. Body3
Anders Franco-Cereceda2Simon C. Body3 Hanna M. Björck1
Hanna M. Björck1 Per Eriksson1*
Per Eriksson1*Thoracic aortic aneurysm (TAA) is the progressive enlargement of the aorta due to destructive changes in the connective tissue of the aortic wall. Aneurysm development is silent and often first manifested by the drastic events of aortic dissection or rupture. As yet, therapeutic agents that halt or reverse the process of aortic wall deterioration are absent, and the only available therapeutic recommendation is elective prophylactic surgical intervention. Being born with a bicuspid instead of the normal tricuspid aortic valve (TAV) is a major risk factor for developing aneurysm in the ascending aorta later in life. Although the pathophysiology of the increased aneurysm susceptibility is not known, recent studies are suggestive of a transformation of aortic endothelium into a more mesenchymal state i.e., an endothelial-to-mesenchymal transition in these individuals. This process involves the loss of endothelial cell features, resulting in junction instability and enhanced vascular permeability of the ascending aorta that may lay the ground for increased aneurysm susceptibility. This finding differentiates and further emphasizes the specific characteristics of aneurysm development in individuals with a bicuspid aortic valve (BAV). This review discusses the possibility of a developmental fate shared between the aortic endothelium and aortic valves. It further speculates about the impact of aortic endothelium phenotypic shift on aneurysm development in individuals with a BAV and revisits previous studies in the light of the new findings.
Thoracic aortic aneurysm (TAA) is a potentially deadly disease associated with progressive expansion and degeneration of the aorta. One of the highest risk factors for developing TAA is the possession of a bicuspid aortic valve (BAV) instead of the normal tricuspid aortic valve (TAV). BAV is the most common congenital cardiac disorder, more frequent in males and Caucasians and has a prevalence of 0.5–2% in the human population (1). Importantly, individuals with a BAV have an 80 times increased risk of developing aortic aneurysm compared to the general population (2). BAV is a complex disease with unknown etiology for the higher aneurysm susceptibility, and the importance of inheritance vs. exposure of ascending aortas (AscA) to non-physiological hemodynamics is currently debated.
The inheritance of BAV in human has been intensively studied and is beyond the scope of this review. Briefly, several genes i.e., NOTCH1, ACTA2, GATA4/5, NKX2.5, and SMAD6 have been characterized in association with familial non-syndromic BAV (1, 3–5). As yet, the high prevalence of sporadic BAV is not compatible with the few characterized genes for familial inheritance and this area of research is still open for new findings. Regarding the influence of shear stress, the last decade has witnessed a major breakthrough in studying the non-physiological hemodynamics caused by a BAV and its possible impact on AscA pathogenesis. Numerous original research and review articles have been allocated to this subject to which the interested readers can refer (6–12). With increasing data obtained on non-physiological hemodynamic of BAV patients, the common consensus emerging is that both genetics and hemodynamics contribute to aortopathy in BAV.
We and others have shown that ascending aortic aneurysm has different etiologies in patients with BAV and TAV [e.g., (13, 14)]. A deeper insight into ongoing molecular processes in the AscA prior to and after aneurysm manifestation is a prerequisite for understanding and preventing aortic degeneration. Moreover, discovering the inheritance of BAV aorthopathy, i.e., the set of genetic and/or epigenetic alterations that leads to AscA aneurysm coupled to a BAV, requires detailed cellular and molecular knowledge of interactions between different embryonic progenitors that act at the common window of space and time to determine the fate of aortic valve and AscA simultaneously.
Two recently published articles by us and others, showed an alteration of intimal endothelium in aneurysmal (15) and non-aneurysmal (16) BAV AscA to a more mesenchymal phenotype and discussed the possible contribution of the phenomenon endothelial mesenchymal transition (EndMT) to the development of aneurysm in these patients. These, and a number of other relevant observations, open up a new avenue in the field of aneurysm. As is highlighted in the title, this review will concentrate only on possible mechanisms of induction and cellular/molecular impact of the EndMT process on the higher susceptibility to develop aneurysm in individuals with BAV. The second objective is to explore if induction of this process in the intima and, as we have observed and will discuss later in this review, most probably also in the media, would clarify better the differences in onset and extent of disease manifestation and pathological changes induced by aneurysms in AscAs of humans with a BAV. Hence, throughout this review we use the term EndMT/EMT (epithelial mesenchymal transition) to describe the result obtained from intima-media of AscA and EndMT when observation is limited to the endothelial layer. We hope this review will widen the scope and add new dimensions and perspectives to the field of aneurysm research.
To explore the possible connection between the formation of a BAV and altered endothelial function in AscA, we should first consider the developmental context within which the fate of semilunar valves (aortic and pulmonic valves) and ascending aortic endothelium is determined. This requires a short review of the cardiac development and formation of cardiac cushion or primordia of aortic valves from endocardium. In the coming sections, we summarize a set of experiments done in transgenic models that have aided us to gain a clearer picture of the inter-connection between embryogenesis of aortic valves and the AscA.
In the human embryo, the linear heart tube forms by differentiation of cardiomyocytes within the primitive cardiac mesoderm, termed the cardiac crescent, during the third week of embryonic development (17). The heart tube is composed of the inner lining/endocardium and an outer layer/myocardium, separated by extracellular matrix known as cardiac jelly. Later during gestation, the cardiac tube loops and elongates by the addition of myocardium and mesenchymal tissues lying outside the early heart; the second heart field (SHF) progenitors and migrating cardiac neural crest cells (NCC). During looping, the first manifestation of cardiac valve formation appears as the expansion and swelling of the cardiac jelly in the atrioventricular canal, and somewhat later in the outflow tract (OFT), form the cardiac cushions via EndMT. Development and remodeling of semilunar valves and OFT septation into the AscA and the pulmonary trunk is accomplished by concerted interaction of OFT cushion with NCC and SHF progenitors with the net effect of complete separation of the systemic and pulmonary circulation (14, 18–21).
The pioneering set of experiments performed by Jonathan Epstein's group (19, 22–24), provided a framework for explaining the relationship between the morphogenesis of aortic valves and AscA. According to this model, the development of the aortic valve and primitive AscA is spatiotemporally related and involves a coordinated signal exchange between endocardium, SHF and NCC progenitors. Impaired function of each cardiac progenitor influences the embryonic development of the other, as well as the endocardia EndMT/EMT and cushion formation, resulting in the formation of a BAV or other aortic valves anomalies. One example of such interaction was shown recently by formation of a BAV as a consequence of deletion of an endothelial specific gene NOS3 in mice, manifesting impaired distribution of NCC and SHF within the aortic valve leaflets (21).
The model was further backed by experiments demonstrating that proper formation and/or septation of the OFT into AscA and pulmonary trunk was also tightly coupled to signals generated from endocardial cushion and cardiac progenitors and abolition of function in any of the involved compartment, SHF (25, 26), NCC (27–29), or endocardium (30, 31) resulted in defective aortic valve morphogenesis and/or septation of OFT. Nonetheless, to our knowledge, among all studies cited above, only three (23, 25, 28) extended their observations to the pathological changes of the AscA in connection to valve anomalies, without any particular reference to the state of aortic endothelium.
Epithelial mesenchymal transition (EMT) is one of the most studied biological processes due to its fundamental role in the onset of cancer metastasis. EMT program induces epithelial cells to lose their adhesion, polarity and cell-cell junctions, restructure their cytoskeleton and become more invasive and motile i.e., acquire “mesenchymal” state (see Figure 1). However, EMT is also essential for normal physiological processes such as embryogenesis or wound healing, but may be aberrantly reactivated in pathological conditions (32, 33). EndMT, a specific form of EMT originally discovered in endocardium during cardiac development (34), shares many similarities with EMT and is activated during similar biological processes and diseases (35–37). EndMT/EMT can also be induced in response to certain environmental changes such as oxidative stress, inflammation, or hemodynamic changes (38). Several signaling pathways with major roles in embryogenesis such as, NOTCH, TGFβ, WNT, FGF, EGF, are also regulators of the EndMT/EMT program (33, 39). Although several transcription factors monitor EndMT/EMT, five of them, e.g., ZEB1/ZEB2, SNAI1/SNAI2, and TWIST are considered to be the key factors for activation of EndMT/EMT (40, 41).
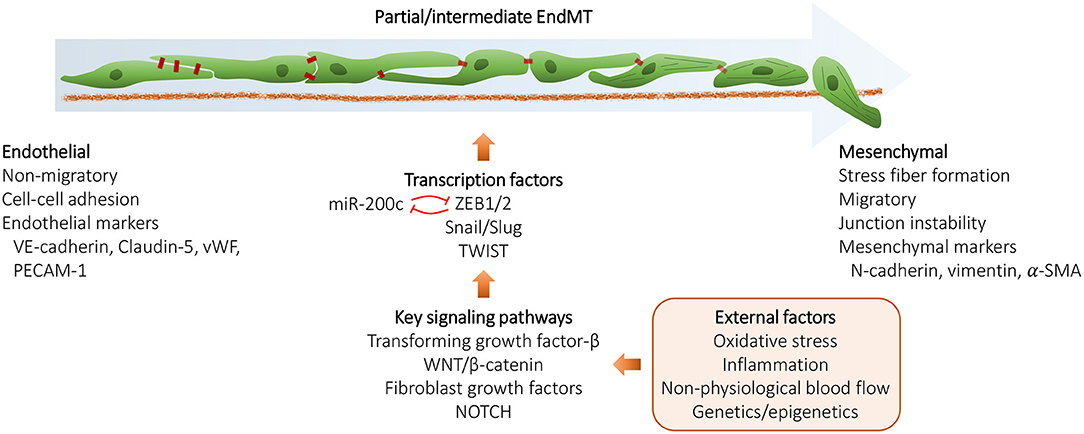
Figure 1. Schematic representation of the endothelial to mesenchymal transition (EndMT) process. During EndMT, endothelial cells lose endothelial cell features and acquire mesenchymal cell markers, resulting in junction instability, enhanced vascular permeability, and potentially cellular senescence. EndMT can be triggered by various external factors and involves signaling pathways, such as TGF-β, WNT/β-catenin, FGFs and NOTCH that converge and induce the expression of EndMT transcription factors ZEB1/2, Snail, Slug, and TWIST. EndMT/EMT may also be regulated by a number of microRNAs, the key microRNA family being miR-200 family that acts by suppressing ZEB1 and ZEB2 mRNA expression by a negative feedback loop.
Disruption of cell junctions is central to EndMT/EMT and that is achieved partly by transcription regulation of the junction proteins and partly by modification, turnover and degradation of junction proteins via endocytosis (42, 43). An early event in EndMT/EMT is the regulation of Cadherin superfamily expression, for instance downregulation of E-cadherin/VE-cadherin (CDH1/CDH5) in EMT and EndMT respectively, and upregulation of N-cadherin also known as mesenchymal cadherin (CDH2) (44, 45). Depending on the biological context and the outcome, EndMT/EMT is divided into three subtypes. Type I, which is activated during embryogenesis producing mesenchymal cells, Type II, occurring during tissue repair producing fibroblasts, and Type III, producing tumor cells activated during the metastatic propagation of cancers (33, 46) (Table 1).

Table 1. Occurrence and outcome of the different EndMT/EMT subtypes.
Like many other biological processes, EndMT/EMT is regulated by a number of microRNAs (47). One of the key microRNA families governing this process is miR-200 family that acts by suppression of ZEB1 and ZEB2 mRNA expression by a negative feedback loop, thereby coordinating EndMT/EMT with Cadherin expression (48, 49).
In spite of ample evidence presented for the role of endothelium in regulating the development, remodeling and functional integrity of vascular smooth muscle cells (VSMCs) (50–52), the aortic media has been in the center of attention in the aneurysm community. However, in late years, the involvement of the intima in aneurysm development has been brought into focus (53–55), which may be particularly interesting in the case of patients with BAV considering the numerous patient-based studies demonstrating exposure of non-physiological hemodynamic forces to the BAV AscA. One of the first experimental indications of endothelium involvement in aneurysm was the pioneering experiment with the Angiotensin II (ANGII)–infusion mice model where cell specific deletion of ANGII receptor, AT1a, in endothelial cells (EC) could attenuate ascending aortic aneurysm (56). Interestingly, the EC-specific transgenes showed structural reorganization of aortic media emblematic of aneurysm, arguing that the signals initiated in endothelium could induce aneurysm and accompanying pathological changes in the SMC. In a review article published 2013 (57), we discussed the intimal EC response to shear stress and the mechanisms by which this response could be relayed to and induce the pathological changes observed in media of AscAs in BAV aneurysmal patients. Furthermore, a recent and excellent review discusses the role of the endothelium and the potential mechanisms by which it influences the media layer in relation to the development of BAV-associated TAA, but with no focus on the EndMT/EMT process (58).
The crucial role of cells with endothelial/endocardium origin in the morphogenesis of aortic valve and OFT have been established by numerous experimental studies in transgenic animals (59–65) (Table 2).
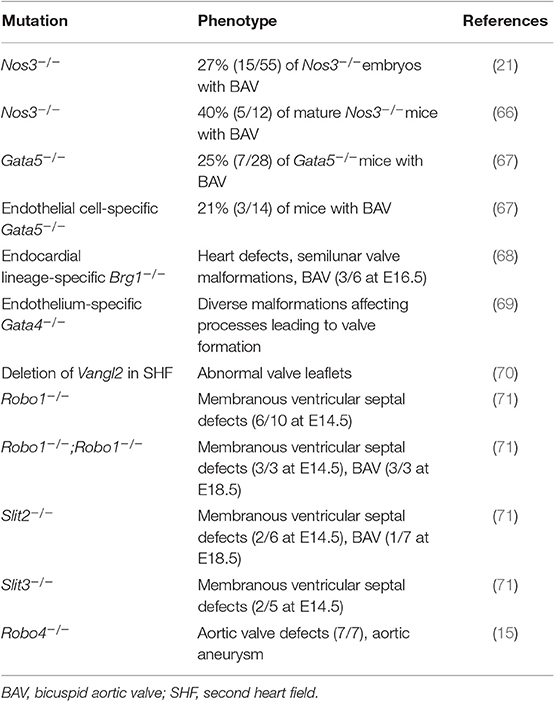
Table 2. Selection of transgenic mice studies analyzing the effect of certain mutations on aortic valve formation and integrity.
Several animal models with either a mutated endothelial specific gene or EC specific gene mutations were shown to develop progenies with BAVs. Mice deleted for endothelial specific nitric oxide synthase (eNOS the product of NOS3 gene) gave rise to 40% progeny with a BAV (66). Laforest et al. (67) were the first to propose that aberrant EC migration and differentiation was associated with the formation of a BAV in GATA5−/−mice mutants with 25% BAV progeny. EC-specific GATA5 deletion also resulted in mice progeny with a BAV, and GATA5−/− offspring with BAV had significantly lower expression of endothelial specific markers, CDH5 and TIE2, as well as decreased expression of eNOS in their left ventricles and OFT in comparison to the wildtype, indicative of altered endothelial function reminiscent of ROBO4 mutations in human BAV (see below). Unfortunately, neither of these studies addressed the extent or existence of concomitant aorthopathy or the status of AscA intima. Nonetheless, EC-specificity of NOS3 together with the fact that GATA5 is restricted mostly to the endocardium, disappears at mid gestation stage, and is required for early differentiation of cardiac progenitors into endothelial/endocardial cells (72) hints to a connection between BAV phenotype and disturbed endothelial function.
The association between functional integrity of AscA endothelium, inheritance of a BAV, and development of aneurysm has now been experimentally demonstrated in a newly published report (15). In this study, the association of heterozygous mutation of ROBO4, a gene important for vascular integrity by regulating endothelial barrier function (73), with non-syndromic familial inheritance of human BAV was established. Histological examination of sections from AscA of a patient with heterozygote mutation of ROBO4 and AscA aneurysm compared to control individual with matching age and sex demonstrated decreased intimal expression of ROBO4, increased fibro-proliferative phenotype of intima and sub-intimal region, and disrupted endothelial barrier function as judged by albumin staining. Treatment of human aortic ECs with siRNA against ROBO4 or expression of a mutant variant in the presence of inhibited endogenous ROBO4 resulted in loss of endothelial barrier function accompanied by downregulation of EC-adherence junction cadherin, CDH5, and tight junction component TJP1 at mRNA and protein levels and transformation of these cells to a mesenchymal state. Moreover, a direct involvement of SLIT-ROBO signaling in the formation of cardiac cushion and inheritance of BAV has been shown (71), and expression of ROBO4 in the endothelium of the aortic valve and proximal AscA was shown to persist throughout the mice postnatal life (15). Loss of function mutations of ROBO4 in mice revealed thickened aortic valves with or without BAV and in some cases, AscA aneurysm. These animals showed low penetrance and male predominance characteristic of human BAV (15). These observations nicely tied the inheritance of BAV and aneurysm of AscA to EC breakdown of barrier function and acquisition of mesenchymal state.
Changes in endothelial barrier function is most probably only limited to the proximal AscA in BAV patients and should have resulted from the impaired interaction between cardiac progenitors at the point where the common fate of aortic valve and AscA is determined. In line with that, using lineage tracing of specific markers of SHF mesenchyme showed that these progenitors could give rise to both SMC and endothelium of OFT (31, 64, 74) and the SHF specific markers could be traced both in the endocardium and endothelium of the developing AscA (75, 76), implying close developmental ties between these tissues in early embryogenesis. Indeed, it is thus likely that the impaired interaction also alters the VSMC population in the BAV aorta, rendering it more sensitive to TAA development.
During the development of aortic valves and OFT, there is a defined boundary for mesenchyme produced by endocardium, NCC and SHF progenitors (77). A defective mesenchyme production by each could be compensated for by the extension of other compartments into the segment that was not their normal niche. This change of mesenchymal boundaries and compensation by others has been shown to give rise to abnormal formation of aortic valves and septation of OFT (21, 31, 78, 79) as well as progenies with BAV (21, 68). Normally, the AscA is populated by a mosaic of SMCs arising from SHF mesoderm and migrating cardiac NCC, and recently, it was demonstrated in mice that NCC stemmed SMCs reside in areas close to intima and SHF generated SMC lie closer to the adventitia (76, 80). One possibility is thus that the impaired signal exchange between cardiac progenitors during development of aortic valves and OFT affects the distal OFT in such a way that a higher proportion of cardiac NCC populate the AscAs in BAV, filling some of the territory normally occupied by SMC of SHF origin.
One line of evidence supporting this hypothesis is the link between cancer cells and embryonic neural cells. Inhibition of a few chromatin modification enzymes in several cancer cell lines resulted in the loss of malignant features and differentiation to neuron-like cells. Further, a major part of mesenchymal marker genes activated during cancer promotion were only expressed in embryonic neural cells, including NCC, and not in other types of embryonic cells suggestive of a common regulatory network between tumorigenesis and neural development (81). If aortic media has a higher content of SMC with NCC origin in BAV, the appearance of EMT signals and cancer-related metabolic pathways among differentially regulated pathways between BAV and TAV is expected. In addition, the SMCs developed from NCC are more “immature” and proliferative and less contractile. As the elastin deposition in arteries takes place during fetal development, NCC-originated SMC may have different elastic properties. Whether or not a diversion from normal proportions of SMC could change the composition of collagen and elastic fibers is not known. All these factors can contribute to increased vulnerability of a cardinal vessel with high pressure function such as aorta.
Although it is almost impossible to disentangle the genetic from hemodynamic causes, distortion of endothelial-related functions in aneurysmal tissue of BAV patients has been described in several patient-based studies. For example, circulating endothelial progenitor cells as a marker of EC repair efficiency were significantly lower in BAV compared to TAV patients (82) as well as being lower in BAV patients with aortic regurgitation or stenosis compared to functional BAVs (83). In a study of male subjects of comparable age, systemic endothelial dysfunction was reported in BAV patients with proximal aortic dilation compared to non-dilated individuals with a BAV (84). Using multivariate analysis comparing BAV and TAV patients with dilated and non-dilated AscA, BAV morphology turned out to be the main predictor of increased circulating PECAM+ endothelial-specific microparticles, independent of the type of cusp fusion or disease (31). Notably, circulating PECAM+ microparticles were significantly decreased in patients who underwent aortic valve surgery, establishing endothelial damage in BAV individuals probably due to exposure to non-physiological blood flow, although the possibility of an inherited defect synergizing hemodynamic factors cannot be ruled out. Several laboratories that focused on eNOS content in the AscA reported a differential expression of eNOS between BAV and TAV aneurysmal patients, both at transcriptional and translational levels (85–88).
In a recent study, we provided cytological evidence for intimal instability and induction of EndMT-like process in non-dilated AscA of BAV patients due to downregulation and enhanced protein turnover of VE-Cadherin (CDH5) in addition to decreased expression of endothelial specific Claudin-5 (CLDN5). Moreover, mRNA expression of N-cadherin (CDH2) increased in dilated AscA of BAV patients compared to dilated AscA of TAV patients (16). Further, we showed that alteration in cadherin expression was accompanied by formation of pseudopodia and stress fibers in endothelium of non-dilated BAV, which is a second key hallmark of transition to a mesenchymal state.
A pertinent issue to raise here is if the prenatal, as well as lifelong exposure of BAV AscA to non-physiological flow, could contribute to the induction of EndMT/EMT in AscA of adult individuals. Using different set-ups, the influence of shear stress on the induction of EndMT in EC have been studied (29, 89–92). During embryonic development, subjecting the OFT to increased hemodynamic load via banding was shown to enhance the EndMT of the cardiac cushion in chicken embryos (93, 94). Also, changes in blood flow by banding of thoracic aorta resulted in enhanced EndMT/EMT in regions exposed to disturbed flow in mice (29). Indeed, DNA methylation studies performed by us further supported the notion that EndMT/EMT induction may be partly due to the exposure of the AscA to disturbed flow. Also, we further observed that the methylation signature in non-dilated BAV aorta was significantly associated with a methylation profile associated with oscillatory flow. Further, several key EMT transcription factors, such as ZEB1, SNAI2 and TWIST1 became hypomethylated in EC subjected to oscillatory flow indicating their increased activity. In addition, BAV and TAV primary ECs showed a different response to perturbed flow, with substantially fewer genes changing their expression in BAV ECs, indicating an impaired flow-response of BAV ECs.
In prenatal life, the exposure of cardiovascular system to blood flow starts with the onset of the first heartbeat and the early valve primordia has been shown to perform functions that are equivalent to the mature valves of adult heart (95, 96). For instance, ablation of NCC in quail embryos resulted in malformation of the OFT endocardial cushion and valves with consequent disturbed hemodynamic in OFT (31). Hence, impaired EndMT during cushion formation causing the formation of abnormal semilunar valves, would simultaneously subject the primitive aorta to non-physiological hemodynamic stresses at early stages of cardiogenesis. As hemodynamic factors function hand in hand and in parallel to genetic factors from early stages of OFT morphogenesis, it is difficult to separate the relative importance of each one for the induction of EndMT/EMT in AscA.
Proper cell-cell communication between different arterial layers is fundamental for vascular function and integrity. Intimal shear stress and intimal/medial strain will be propagated to other vascular layer, influencing structure and function. In response to shear stress, EC-SMC communication can influence SMC phenotype and proliferation (47, 50, 97–102) via mediators such as microRNAs (31, 81, 103), gap junction (104), or activation of certain signaling pathways through ligand receptor interaction (105–108). Another possible route of cell-cell communication is the inclusion of extra vesicular bodies by endocytosis that can transfer molecular characteristics between different cell types (109). Thus, a variety of molecular messengers are capable of transferring the EndMT/EMT induced in one section to other vascular compartments.
We have previously performed comparative studies on BAV and TAV aortic intima-media specimen, from non-dilated or dilated aortas, to investigate differences at genomic, proteomic and epigenomic levels. First, by combining large-scale proteomic pathway analysis on differentially-expressed proteins in non-dilated aortas, we showed enrichment of genes belonging to EMT, protein degradation and trafficking, cell junction dynamics, apoptosis, cell cycle and cancer-related biological processes (16). Second, to identify possible regulatory microRNAs (miRs) underlying the observed protein signature, we combined proteomic data with an in-silico network analysis approach (110). This procedure identified the miR-200 family, known to be important regulators of EndMT/EMT activity (48), as a key modulator of the ongoing biological process that differs between non-dilated BAV and TAV aorta. (48). Lastly, DNA methylation studies further showed enrichment of EMT genes in non-dilated AscA of BAV patients (111). Similarly, analysis of intima-media in dilated AscA of BAV and TAV patients identified EMT as the top GO term and several key transcription factors for EMT, including ZEB1, SNAI2, and TWIST2, were hypomethylated in dilated BAV aorta.
Collectively, these results, at the levels of mRNA expression, proteomic, DNA methylation, and microRNA regulation displayed an “EMT” signature in the aortic intima-media, and a major consideration would be what EndMT/EMT-like processes could mean for the state of medial SMC. One possible explanation is the different proliferative capacity of aortic SMC between BAV and TAV. Several upregulated proteins in non-dilated aorta of BAV compared to TAV patients included proteins associated with increased cell proliferation and invasion. One clear example was Yes-Associated Protein 1 (YAP1) that has been shown to regulate division and differentiation of VSMC from cardiovascular progenitors (112, 113), particularly in NCC-derived SMC (114). Furthermore, we documented significantly higher expression of Ki67 protein in SMC nuclei of non-dilated AscA of BAV patients (16). This result is in line with reported “immaturity” of SMC in dilated and non-dilated AscAs of BAV patients (14, 115). Compatible with that, an epigenetic study of dilated AscA of BAV patients, found a strong and significant non-CpG hypomethylation in aortic media that was interpreted as high proliferative SMC in this region (116).
Repair deficiency may be due to genetically impaired production or recruitment of stem/progenitor cells, and/or inefficient induction of repair-promoting signaling pathways. In non-regenerating adult tissues such as the aorta, the existence of mechanisms that can instigate the terminally differentiated VSMCs to resume proliferative cycle is vital for vascular repair. In the past decade, reservoirs of different stem/progenitor cells that can migrate and prime the vascular repair have been discovered (117) and the importance of functional stem/progenitor cells, their number, and the maintained proliferative capacity in vessel homeostasis have been discussed. Several different sources of VSMC progenitors or stem cells that can dedifferentiate and participate in repair and remodeling in physiological or disease situations have been identified (118, 119). These cells either reside within the adult VSMC tissues or reach the damaged VSMC via circulation or migration from the neighboring tissues (117–119). Adventitia, particularly in the aorta, has long been recognized as a main niche for vascular progenitors. The source of progenitors within adventitia is situated in the border area between the outer media and inner adventitia within a region highly rich in sonic hedgehog (SHH) signaling (119, 120). Nonetheless, in several arteries including the aorta, a subset of adventitial stem cells produced by fully differentiated residential VSMC has been reported to migrate into the adventitia to enrich the adventitial pool of vascular stem cells (58). One possibility for the higher susceptibility of AscAs to aneurysm in individuals with a BAV is an impaired function of the adventitia. Unfortunately, adventitia in BAV has also been neglected by aneurysm researchers. Studying adventitia, particularly the SHH rich region may be relevant for high susceptibility to aneurysm in BAV patients.
Searching for markers of SMC immaturity, Roostalu et al. (119) identified CD146/MCAM that was expressed by a small sub-population of SMC in adult descending aortas at sub-intimal regions and around aortic branch points and bifurcations, which remained immature also in adult tissue. Interestingly, the expression and activation of YAP1 protein, an inhibitor of VSMC differentiation (112) and the regulator of CD146 transcription (121), was also higher in these regions. This SMC sub-population was shown to proliferate and perform arterial repair in the case of minor injuries while severe injuries were repaired by cells migrating from adventitia. Whether or not such a subset of VSMC exists in human vs. mouse aorta and is populating the branching points of coronary arteries or arteries stemming from AscA or aortic arch is not clear. Nonetheless, we see an upregulation of YAP1 protein in non-dilated AscA of BAV patients (16) and gene expression analyses (13) revealed an increase of CD146/MCAM mRNA in dilated vs. non-dilated BAV AscA raising the possibility of increased immaturity in VSMC in BAV resulted from dilation. A systematic study of the role and regional distribution of CD146 expression and its relationship to VSMC “immaturity” of AscA in BAV may further clarify underlying mechanism of aortic dilation in BAV aortas.
One of the prime consequences of high proliferation of SMC and repair deficiency is premature aging. In line with this, Grewal et al. proposed that susceptibility to aneurysm in BAV was due to the SMC immaturity while in TAV was due to inflammation and enhanced aging (14, 115). This interpretation was questioned by Forte and Della Corte who proposed “premature aging” instead of immaturity being the cause of aortopathy in BAV (122). In support of premature aging, significantly shorter telomere length and lower wound healing capacity of aneurysmal SMC isolated from BAV as compared to control donors and TAV was reported (123). The two interpretations may not be mutually exclusive and induction of EndMT/EMT in BAV aorta can reconcile and encompass both proposals.
Aging and senescence are not equivalent and while aging organisms accumulate senescent cells, senescence can function as a response to a variety of stress situations unrelated to aging. Telomere shortening was originally believed to be the major cause of cellular senescence. However, several other inducers of senescence independent of telomere shortening have lately been identified, including hypoxia, oncogene-induced senescence, exposure to UV or gamma radiation, loss of tumor suppressing genes and mitochondrial dysfunction (124–127). Cellular senescence induced by telomere shortening is an intrinsic part of cell cycle check point causing permanent growth arrest and endowing the organism with tumor suppressive activity. Relevantly, cellular senescence and EndMT/EMT have both been considered as biological mechanisms guiding cancer progression and metastasis and recent findings marks the discovery of a cross talk between the two processes (128, 129). An example of such a crosstalk in non-cancerous cells is the induction of EndMT described for aging human aortic EC (130). Moreover, activation of all key transcription factors of the EndMT/EMT process has been reported in senescence (128, 129). ZEB1 has particularly been shown to be the link between cellular senescence and EMT (50). Thus, the immaturity in BAV can turn into premature senescence with all accompanying consequences. Our collective data on differentially regulated pathways between BAV and TAV i.e., hypoxia, oncogene-related pathways such as WNT or MYC, and UV response may well be due to premature senescence (16, 110, 111), giving support to the possibility of EndMT/EMT conversion to senescence and aortic degeneration in BAV. Compatible with shorter telomeres and senescence-induced activation of cell cycle checkpoint, we also observed significantly increased protein expression of the cyclin-dependent kinase inhibitor P27 (the product of CDKN1B gene) in non-dilated BAV AscA (16). Exposure to disturbed flow is a factor that can turn immaturity to premature aging. Indeed, disturbed flow was shown to stimulate senescence in ECs in mouse models and cell culture by a P53/P21-dependent mechanism (131).
The underlying molecular mechanisms for the BAV-associated aneurysm susceptibility remain to be elucidated. BAV patients differ in the extent and onset of medial degeneration and some individuals with a BAV may never develop aneurysm. The notion of disturbed signal exchange between cardiac progenitors causing distorted mesenchymal boundaries in aortic walls has backing in transgene studies and provides a molecular framework to explain some of these ambiguities. An extensive population of less differentiated, less contractile SMCs with NCC origin in the sub-intimal area of the aorta, the dysfunctional intima, in addition to constant exposure to non-physiological blood flow, subject the walls to a high risk. The degree of aortic susceptibility may depend on the location of the boundary and the magnitude of NCC contribution. Several different genes mentioned above, and many more yet to be discovered, can influence the boundary determination in OFT that can explain the difficulties of identifying responsible factors in the inheritance of non-familial BAVs.
Another level of complexity that may influence the disease outcome is the EndMT/EMT program itself. Recently, the process of EndMT/EMT has been revisited and it is currently defined as a way to provide more “plasticity” to the tissue. To perform that task, many EndMT/EMT transcription factors also interact with or recruit epigenetic modifiers in addition to their direct role in the regulation of transcription (41, 132). Moreover, the notion of EndMT/EMT being a transition between two alternative states of “epithelial” and “mesenchymal” have been challenged and most studies point to the tissues remaining in an intermediate state harboring both epithelial and mesenchymal features (40, 41). This state is termed partial or intermediate EndMT/EMT or “metastable” and can even become a final state of the tissues in some disease cases (40, 41). In the case of cancer, the existence of some pre-cancerous stem cells maintaining some, but not all, of the genetic features of cancer cells, that can be complemented and produce a fully transformed cell upon epigenetic changes have been reported (132). To extrapolate these ideas to aneurysm, the non-dilated aorta in BAV may be in a “metastable” state of EndMT/EMT or using cancer terminology, in a pre-cancerous state that may stay stable or be shifted further to a more mesenchymal state and aortic dilation. This is supported by the more aggravated EMT signature in dilation of BAV reported by us. In addition, the extent of the shift could depend on a spectrum of events such as cellular context, different pathways and/or different transcription factors initiating the transition, degree of epigenetic modifications, environmental cues, individual genetic background, and a wide range of other factors that may influence the outcome of aortic diseases.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the Swedish Research Council [2016-01761] and the Swedish Heart-Lung Foundation [20180451]. This study also received a private donation from Fredrik Lundberg. Fredrik Lundberg had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Prakash SK, Bosse Y, Muehlschlegel JD, Michelena HI, Limongelli G, Della Corte A, et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J Am College Cardiol. (2014) 64:832–9. doi: 10.1016/j.jacc.2014.04.073
2. Michelena HI, Khanna AD, Mahoney D, Margaryan E, Topilsky Y, Suri RM, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA. (2011) 306:1104–12. doi: 10.1001/jama.2011.1286
3. Laforest B, Nemer M. Genetic insights into bicuspid aortic valve formation. Cardiol Res Pract. (2012) 2012:180297. doi: 10.1155/2012/180297
4. Andreassi MG, Della Corte A. Genetics of bicuspid aortic valve aortopathy. Curr Opin Cardiol. (2016) 31:585–92. doi: 10.1097/HCO.0000000000000328
5. Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic bases of bicuspid aortic valve: the contribution of traditional and high-throughput sequencing approaches on research and diagnosis. Front Physiol. (2017) 8:612. doi: 10.3389/fphys.2017.00612
6. Girdauskas E, Disha K, Borger MA, Kuntze T. Relation of bicuspid aortic valve morphology to the dilatation pattern of the proximal aorta: focus on the transvalvular flow. Cardiol Res Pract. (2012) 2012:478259. doi: 10.1155/2012/478259
7. Hope MD, Sigovan M, Wrenn SJ, Saloner D, Dyverfeldt P. MRI hemodynamic markers of progressive bicuspid aortic valve-related aortic disease. J Magn Reson Imaging. (2014) 40:140–5. doi: 10.1002/jmri.24362
8. Spinale FG, Bolger AF. Fate versus flow: wall shear stress in the aortopathy associated with bicuspid aortic valves. J Am College Cardiol. (2015) 66:901–4. doi: 10.1016/j.jacc.2015.07.002
9. Guzzardi DG, Barker AJ, van Ooij P, Malaisrie SC, Puthumana JJ, Belke DD, et al. Valve-related hemodynamics mediate human bicuspid aortopathy: insights from wall shear stress mapping. J Am College Cardiol. (2015) 66:892–900. doi: 10.1016/j.jacc.2015.06.1310
10. Burris NS, Hope MD. Bicuspid valve-related aortic disease: flow assessment with conventional phase-contrast MRI. Acad Radiol. (2015) 22:690–6. doi: 10.1016/j.acra.2015.01.010
11. Fedak PW, Barker AJ, Verma S. Year in review: bicuspid aortopathy. Curr Opin Cardiol. (2016) 31:132–8. doi: 10.1097/HCO.0000000000000258
12. Bollache E, Guzzardi DG, Sattari S, Olsen KE, Di Martino ES, Malaisrie SC, et al. Aortic valve-mediated wall shear stress is heterogeneous and predicts regional aortic elastic fiber thinning in bicuspid aortic valve-associated aortopathy. J Thorac Cardiovasc Surg. (2018) 156:2112–20 e2. doi: 10.1016/j.jtcvs.2018.05.095
13. Folkersen L, Wagsater D, Paloschi V, Jackson V, Petrini J, Kurtovic S, et al. Unraveling the divergent gene expression profiles in bicuspid and tricuspid aortic valve patients with thoracic aortic dilatation - the ASAP study. Mol Med. (2011) 17:1365–73. doi: 10.2119/molmed.2011.00286
14. Grewal N, DeRuiter MC, Jongbloed MR, Goumans MJ, Klautz RJ, Poelmann RE, et al. Normal and abnormal development of the aortic wall and valve: correlation with clinical entities. Netherlands Heart J. (2014) 22:363–9. doi: 10.1007/s12471-014-0576-2
15. Gould RA, Aziz H, Woods CE, Seman-Senderos MA, Sparks E, Preuss C, et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat Genet. (2019) 51:42–50. doi: 10.1038/s41588-018-0265-y
16. Maleki S, Kjellqvist S, Paloschi V, Magne J, Branca RM, Du L, et al. Mesenchymal state of intimal cells may explain higher propensity to ascending aortic aneurysm in bicuspid aortic valves. Sci Rep. (2016) 6:35712. doi: 10.1038/srep35712
17. Moorman A, Webb S, Brown NA, Lamers W, Anderson RH. Development of the heart: (1) formation of the cardiac chambers and arterial trunks. Heart. (2003) 89:806–14. doi: 10.1136/heart.89.7.806
18. Epstein JA. Franklin H. Epstein Lecture. Cardiac development and implications for heart disease. N Engl J Med. (2010) 363:1638–47. doi: 10.1056/NEJMra1003941
19. Rentschler S, Jain R, Epstein JA. Tissue-tissue interactions during morphogenesis of the outflow tract. Pediatr Cardiol. (2010) 31:408–13. doi: 10.1007/s00246-009-9611-2
20. Keyte AL, Alonzo-Johnsen M, Hutson MR. Evolutionary and developmental origins of the cardiac neural crest: building a divided outflow tract. Birth Defects Res C Embryo Today. (2014) 102:309–23. doi: 10.1002/bdrc.21076
21. Peterson JC, Chughtai M, Wisse LJ, Gittenberger-de Groot AC, Feng Q, Goumans MTH, et al. Bicuspid aortic valve formation: Nos3 mutation leads to abnormal lineage patterning of neural crest cells and the second heart field. Dis Model Mech. (2018) 11:dmm034637. doi: 10.1242/dmm.034637
22. High FA, Jain R, Stoller JZ, Antonucci NB, Lu MM, Loomes KM, et al. Murine Jagged1/Notch signaling in the second heart field orchestrates Fgf8 expression and tissue-tissue interactions during outflow tract development. J Clin Invest. (2009) 119:1986–96. doi: 10.1172/JCI38922
23. Jain R, Engleka KA, Rentschler SL, Manderfield LJ, Li L, Yuan L, et al. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J Clin Invest. (2011) 121:422–30. doi: 10.1172/JCI44244
24. de la Pompa JL, Epstein JA. Coordinating tissue interactions: notch signaling in cardiac development and disease. Devel Cell. (2012) 22:244–54. doi: 10.1016/j.devcel.2012.01.014
25. Lewandowski SL, Janardhan HP, Trivedi CM. Histone deacetylase 3 coordinates deacetylase-independent epigenetic silencing of transforming growth factor-β1 (TGF-β1) to orchestrate second heart field development. J Biol Chem. (2015) 290:27067–89. doi: 10.1074/jbc.M115.684753
26. Leung C, Liu Y, Lu X, Kim M, Drysdale TA, Feng Q. Rac1 signaling is required for anterior second heart field cellular organization and cardiac outflow tract development. J Am Heart Assoc. (2015) 5:e002508. doi: 10.1161/JAHA.115.002508
27. Bradshaw L, Chaudhry B, Hildreth V, Webb S, Henderson DJ. Dual role for neural crest cells during outflow tract septation in the neural crest-deficient mutant Splotch(2H). J Anat. (2009) 214:245–57. doi: 10.1111/j.1469-7580.2008.01028.x
28. Phillips HM, Mahendran P, Singh E, Anderson RH, Chaudhry B, Henderson DJ. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc Res. (2013) 99:452–60. doi: 10.1093/cvr/cvt132
29. Moonen JR, Lee ES, Schmidt M, Maleszewska M, Koerts JA, Brouwer LA, et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc Res. (2015) 108:377–86. doi: 10.1093/cvr/cvv175
30. Milgrom-Hoffman M, Michailovici I, Ferrara N, Zelzer E, Tzahor E. Endothelial cells regulate neural crest and second heart field morphogenesis. Biol Open. (2014) 3:679–88. doi: 10.1242/bio.20148078
31. Alegret JM, Martinez-Micaelo N, Aragones G, Beltran-Debon R. Circulating endothelial microparticles are elevated in bicuspid aortic valve disease and related to aortic dilation. Int J Cardiol. (2016) 217:35–41. doi: 10.1016/j.ijcard.2016.04.184
32. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
33. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
34. Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. (1995) 77:1–6. doi: 10.1161/01.RES.77.1.1
35. Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. (2008) 99:1375–9. doi: 10.1038/sj.bjc.6604662
36. Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation. (2012) 125:1795–808. doi: 10.1161/CIRCULATIONAHA.111.040352
37. Sanchez-Duffhues G, Garcia de Vinuesa A, Ten Dijke P. Endothelial-to-mesenchymal transition in cardiovascular diseases: developmental signaling pathways gone awry. Dev Dyn. (2018) 247:492–508. doi: 10.1002/dvdy.24589
38. Muylaert DE, de Jong OG, Slaats GG, Nieuweboer FE, Fledderus JO, Goumans MJ, et al. Environmental influences on endothelial to mesenchymal transition in developing implanted cardiovascular tissue-engineered grafts. Tissue Eng Part B Rev. (2015) 22:58–67. doi: 10.1089/ten.teb.2015.0167
39. Zheng H, Kang Y. Multilayer control of the EMT master regulators. Oncogene. (2014) 33:1755–63. doi: 10.1038/onc.2013.128
40. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. (2016) 166:21–45. doi: 10.1016/j.cell.2016.06.028
41. Skrypek N, Goossens S, De Smedt E, Vandamme N, Berx G. Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet. (2017) 33:943–59. doi: 10.1016/j.tig.2017.08.004
42. Le Bras GF, Taubenslag KJ, Andl CD. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adh Migr. (2012) 6:365–73. doi: 10.4161/cam.21326
43. Voutsadakis IA. Ubiquitination and the Ubiquitin-Proteasome System as regulators of transcription and transcription factors in epithelial mesenchymal transition of cancer. Tumour Biol. (2012) 33:897–910. doi: 10.1007/s13277-012-0355-x
44. Gheldof A, Berx G. Cadherins and epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci. (2013) 116:317–36. doi: 10.1016/B978-0-12-394311-8.00014-5
45. van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer. (2014) 14:121–34. doi: 10.1038/nrc3647
46. Stone RC, Pastar I, Ojeh N, Chen V, Liu S, Garzon KI, et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. (2016) 365:495–506. doi: 10.1007/s00441-016-2464-0
47. Diaz-Lopez A, Moreno-Bueno G, Cano A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manage Res. (2014) 6:205–16. doi: 10.2147/CMAR.S38156
48. Hill L, Browne G, Tulchinsky E. ZEB/miR-200 feedback loop: at the crossroads of signal transduction in cancer. Int J Cancer. (2013) 132:745–54. doi: 10.1002/ijc.27708
49. Hilmarsdottir B, Briem E, Bergthorsson JT, Magnusson MK, Gudjonsson T. Functional role of the microRNA-200 family in breast morphogenesis and neoplasia. Genes. (2014) 5:804–20. doi: 10.3390/genes5030804
50. High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci USA. (2008) 105:1955–9. doi: 10.1073/pnas.0709663105
51. Xia Y, Bhattacharyya A, Roszell EE, Sandig M, Mequanint K. The role of endothelial cell-bound Jagged1 in Notch3-induced human coronary artery smooth muscle cell differentiation. Biomaterials. (2012) 33:2462–72. doi: 10.1016/j.biomaterials.2011.12.001
52. Scheppke L, Murphy EA, Zarpellon A, Hofmann JJ, Merkulova A, Shields DJ, et al. Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood. (2012) 119:2149–58. doi: 10.1182/blood-2011-04-348706
53. Ali OA, Chapman M, Nguyen TH, Chirkov YY, Heresztyn T, Mundisugih J, et al. Interactions between inflammatory activation and endothelial dysfunction selectively modulate valve disease progression in patients with bicuspid aortic valve. Heart. (2014) 100:800–5. doi: 10.1136/heartjnl-2014-305509
54. Malashicheva A, Kostina D, Kostina A, Irtyuga O, Voronkina I, Smagina L, et al. Phenotypic and functional changes of endothelial and smooth muscle cells in thoracic aortic aneurysms. Int J Vasc Med. (2016) 2016:3107879. doi: 10.1155/2016/3107879
55. Forte A, Bancone C, Cobellis G, Buonocore M, Santarpino G, Fischlein TJM, et al. A possible early biomarker for bicuspid aortopathy: circulating transforming growth factor β-1 to Soluble endoglin ratio. Circ Res. (2017) 120:1800–11. doi: 10.1161/CIRCRESAHA.117.310833
56. Rateri DL, Moorleghen JJ, Balakrishnan A, Owens AP 3rd, Howatt DA, Subramanian V, et al. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor-/- mice. Circ Res. (2011) 108:574–81. doi: 10.1161/CIRCRESAHA.110.222844
57. Maleki S, Bjorck HM, Paloschi V, Kjellqvist S, Folkersen L, Jackson V, et al. Aneurysm development in patients with Bicuspid Aortic Valve (BAV): possible connection to repair deficiency? Aorta. (2013) 1:13–22. doi: 10.12945/j.aorta.2013.12.011
58. Majesky MW, Horita H, Ostriker A, Lu S, Regan JN, Bagchi A, et al. Differentiated smooth muscle cells generate a subpopulation of resident vascular progenitor cells in the adventitia regulated by Klf4. Circ Res. (2017) 120:296–311. doi: 10.1161/CIRCRESAHA.116.309322
59. Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. (2001) 230:230–42. doi: 10.1006/dbio.2000.0106
60. de Lange FJ, Moorman AF, Anderson RH, Manner J, Soufan AT, de Gier-de Vries C, et al. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. (2004) 95:645–54. doi: 10.1161/01.RES.0000141429.13560.cb
61. Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn. (2004) 230:239–50. doi: 10.1002/dvdy.20051
62. DeLaughter DM, Saint-Jean L, Baldwin HS, Barnett JV. What chick and mouse models have taught us about the role of the endocardium in congenital heart disease. Birth Defects Res A Clin Mol Teratol. (2011) 91:511–25. doi: 10.1002/bdra.20809
63. Zhang T, Liu J, Zhang J, Thekkethottiyil EB, Macatee TL, Ismat FA, et al. Jun is required in Isl1-expressing progenitor cells for cardiovascular development. PLoS ONE. (2013) 8:e57032. doi: 10.1371/journal.pone.0057032
64. Puceat M. Embryological origin of the endocardium and derived valve progenitor cells: from developmental biology to stem cell-based valve repair. Biochim Biophys Acta. (2013) 1833:917–22. doi: 10.1016/j.bbamcr.2012.09.013
65. Luxan G, D'Amato G, MacGrogan D, de la Pompa JL. Endocardial notch signaling in cardiac development and disease. Circ Res. (2016) 118:e1–8. doi: 10.1161/CIRCRESAHA.115.305350
66. Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation. (2000) 101:2345–8. doi: 10.1161/01.CIR.101.20.2345
67. Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest. (2011) 121:2876–87. doi: 10.1172/JCI44555
68. Akerberg BN, Sarangam ML, Stankunas K. Endocardial Brg1 disruption illustrates the developmental origins of semilunar valve disease. Dev Biol. (2015) 407:158–72. doi: 10.1016/j.ydbio.2015.06.015
69. Rivera-Feliciano J, Lee KH, Kong SW, Rajagopal S, Ma Q, Springer Z, et al. Development of heart valves requires Gata4 expression in endothelial-derived cells. Development. (2006) 133:3607–18. doi: 10.1242/dev.02519
70. Eley L, Alqahtani AM, MacGrogan D, Richardson RV, Murphy L, Salguero-Jimenez A, et al. A novel source of arterial valve cells linked to bicuspid aortic valve without raphe in mice. Elife. (2018) 7:e34110. doi: 10.7554/eLife.34110
71. Mommersteeg MT, Yeh ML, Parnavelas JG, Andrews WD. Disrupted Slit-Robo signalling results in membranous ventricular septum defects and bicuspid aortic valves. Cardiovasc Res. (2015) 106:55–66. doi: 10.1093/cvr/cvv040
72. Nemer G, Nemer M. Cooperative interaction between GATA5 and NF-ATc regulates endothelial-endocardial differentiation of cardiogenic cells. Development. (2002) 129:4045–55.
73. Jones CA, London NR, Chen H, Park KW, Sauvaget D, Stockton RA, et al. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat Med. (2008) 14:448–53. doi: 10.1038/nm1742
74. Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, et al. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. (2006) 127:1151–65. doi: 10.1016/j.cell.2006.10.029
75. Keenan ID, Rhee HJ, Chaudhry B, Henderson DJ. Origin of non-cardiac endothelial cells from an Isl1+ lineage. FEBS Lett. (2012) 586:1790–4. doi: 10.1016/j.febslet.2012.05.014
76. Harmon AW, Nakano A. Nkx2–5 lineage tracing visualizes the distribution of second heart field-derived aortic smooth muscle. Genesis. (2013) 51:862–9. doi: 10.1002/dvg.22721
77. Wu B, Wang Y, Xiao F, Butcher JT, Yutzey KE, Zhou B. Developmental mechanisms of aortic valve malformation and disease. Annu Rev Physiol. (2017) 79:21–41. doi: 10.1146/annurev-physiol-022516-034001
78. Wu B, Wang Y, Lui W, Langworthy M, Tompkins KL, Hatzopoulos AK, et al. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ Res. (2011) 109:183–92. doi: 10.1161/CIRCRESAHA.111.245035
79. El Robrini N, Etchevers HC, Ryckebusch L, Faure E, Eudes N, Niederreither K, et al. Cardiac outflow morphogenesis depends on effects of retinoic acid signaling on multiple cell lineages. Dev Dyn. (2016) 245:388–401. doi: 10.1002/dvdy.24357
80. Sawada H, Rateri DL, Moorleghen JJ, Majesky MW, Daugherty A. Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta-brief report. Arterioscler Thromb Vasc Biol. (2017) 37:1722–6. doi: 10.1161/ATVBAHA.117.309599
81. Wang L, Qiu P, Jiao J, Hirai H, Xiong W, Zhang J, et al. Yes-associated protein inhibits transcription of myocardin and attenuates differentiation of vascular smooth muscle cell from cardiovascular progenitor cell lineage. Stem Cells. (2017) 35:351–61. doi: 10.1002/stem.2484
82. Balistreri CR, Crapanzano F, Schirone L, Allegra A, Pisano C, Ruvolo G, et al. Deregulation of Notch1 pathway and circulating endothelial progenitor cell (EPC) number in patients with bicuspid aortic valve with and without ascending aorta aneurysm. Sci Rep. (2018) 8:13834. doi: 10.1038/s41598-018-32170-2
83. Vaturi M, Perl L, Leshem-Lev D, Dadush O, Bental T, Shapira Y, et al. Circulating endothelial progenitor cells in patients with dysfunctional versus normally functioning congenitally bicuspid aortic valves. Am J Cardiol. (2011) 108:272–6. doi: 10.1016/j.amjcard.2011.03.039
84. Tzemos N, Lyseggen E, Silversides C, Jamorski M, Tong JH, Harvey P, et al. Endothelial function, carotid-femoral stiffness, and plasma matrix metalloproteinase-2 in men with bicuspid aortic valve and dilated aorta. J Am College Cardiol. (2010) 55:660–8. doi: 10.1016/j.jacc.2009.08.080
85. Aicher D, Urbich C, Zeiher A, Dimmeler S, Schafers HJ. Endothelial nitric oxide synthase in bicuspid aortic valve disease. Ann Thorac Surg. (2007) 83:1290–4. doi: 10.1016/j.athoracsur.2006.11.086
86. Mohamed SA, Radtke A, Saraei R, Bullerdiek J, Sorani H, Nimzyk R, et al. Locally different endothelial nitric oxide synthase protein levels in ascending aortic aneurysms of bicuspid and tricuspid aortic valve. Cardiol Res Pract. (2012) 2012:165957. doi: 10.1155/2012/165957
87. Henn D, Perttunen H, Gauer S, Schmied W, Porras C, Such M, et al. GATA5 and endothelial nitric oxide synthase expression in the ascending aorta is related to aortic size and valve morphology. Ann Thorac Surg. (2014) 97:2019–25. doi: 10.1016/j.athoracsur.2014.02.050
88. Kotlarczyk MP, Billaud M, Green BR, Hill JC, Shiva S, Kelley EE, et al. Regional disruptions in endothelial nitric oxide pathway associated with bicuspid aortic valve. Ann Thorac Surg. (2016) 102:1274–81. doi: 10.1016/j.athoracsur.2016.04.001
89. Egorova AD, Khedoe PP, Goumans MJ, Yoder BK, Nauli SM, ten Dijke P, et al. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ Res. (2011) 108:1093–101. doi: 10.1161/CIRCRESAHA.110.231860
90. Mahler GJ, Frendl CM, Cao Q, Butcher JT. Effects of shear stress pattern and magnitude on mesenchymal transformation and invasion of aortic valve endothelial cells. Biotechnol Bioeng. (2014) 111:2326–37. doi: 10.1002/bit.25291
91. Mina SG, Huang P, Murray BT, Mahler GJ. The role of shear stress and altered tissue properties on endothelial to mesenchymal transformation and tumor-endothelial cell interaction. Biomicrofluidics. (2017) 11:044104. doi: 10.1063/1.4991738
92. Mahmoud MM, Serbanovic-Canic J, Feng S, Souilhol C, Xing R, Hsiao S, et al. Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci Rep. (2017) 7:3375. doi: 10.1038/s41598-017-03532-z
93. Menon V, Eberth JF, Goodwin RL, Potts JD. Altered hemodynamics in the embryonic heart affects outflow valve development. J Cardiovasc Dev Dis. (2015) 2:108–24. doi: 10.3390/jcdd2020108
94. Midgett M, Lopez CS, David L, Maloyan A, Rugonyi S. Increased hemodynamic load in early embryonic stages alters endocardial to mesenchymal transition. Front Physiol. (2017) 8:56. doi: 10.3389/fphys.2017.00056
95. Butcher JT, McQuinn TC, Sedmera D, Turner D, Markwald RR. Transitions in early embryonic atrioventricular valvular function correspond with changes in cushion biomechanics that are predictable by tissue composition. Circ Res. (2007) 100:1503–11. doi: 10.1161/CIRCRESAHA.107.148684
96. Nomura-Kitabayashi A, Phoon CK, Kishigami S, Rosenthal J, Yamauchi Y, Abe K, et al. Outflow tract cushions perform a critical valve-like function in the early embryonic heart requiring BMPRIA-mediated signaling in cardiac neural crest. Am J Physiol Heart Circ Physiol. (2009) 297:H1617–28. doi: 10.1152/ajpheart.00304.2009
97. Chiu JJ, Chen LJ, Chen CN, Lee PL, Lee CI. A model for studying the effect of shear stress on interactions between vascular endothelial cells and smooth muscle cells. J Biomech. (2004) 37:531–9. doi: 10.1016/j.jbiomech.2003.08.012
98. Wang HQ, Huang LX, Qu MJ, Yan ZQ, Liu B, Shen BR, et al. Shear stress protects against endothelial regulation of vascular smooth muscle cell migration in a coculture system. Endothelium. (2006) 13:171–80. doi: 10.1080/10623320600760282
99. Mack PJ, Zhang Y, Chung S, Vickerman V, Kamm RD, Garcia-Cardena G. Biomechanical regulation of endothelium-dependent events critical for adaptive remodeling. J Biol Chem. (2009) 284:8412–20. doi: 10.1074/jbc.M804524200
100. Tsai MC, Chen L, Zhou J, Tang Z, Hsu TF, Wang Y, et al. Shear stress induces synthetic-to-contractile phenotypic modulation in smooth muscle cells via peroxisome proliferator-activated receptor alpha/delta activations by prostacyclin released by sheared endothelial cells. Circ Res. (2009) 105:471–80. doi: 10.1161/CIRCRESAHA.109.193656
101. Sakamoto N, Kiuchi T, Sato M. Development of an endothelial-smooth muscle cell coculture model using phenotype-controlled smooth muscle cells. Ann Biomed Eng. (2011) 39:2750–8. doi: 10.1007/s10439-011-0372-8
102. Wang L, Han Y, Shen Y, Yan ZQ, Zhang P, Yao QP, et al. Endothelial insulin-like growth factor-1 modulates proliferation and phenotype of smooth muscle cells induced by low shear stress. Ann Biomed Eng. (2014) 42:776–86. doi: 10.1007/s10439-013-0957-5
103. Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. (2012) 14:249–56. doi: 10.1038/ncb2441
104. Pfenniger A, Wong C, Sutter E, Cuhlmann S, Dunoyer-Geindre S, Mach F, et al. Shear stress modulates the expression of the atheroprotective protein Cx37 in endothelial cells. J Mol Cell Cardiol. (2012) 53:299–309. doi: 10.1016/j.yjmcc.2012.05.011
105. Lamboley M, Pittet P, Koenigsberger M, Sauser R, Beny JL, Meister JJ. Evidence for signaling via gap junctions from smooth muscle to endothelial cells in rat mesenteric arteries: possible implication of a second messenger. Cell Calcium. (2005) 37:311–20. doi: 10.1016/j.ceca.2004.11.004
106. Schmidt VJ, Wolfle SE, Boettcher M, de Wit C. Gap junctions synchronize vascular tone within the microcirculation. Pharmacol Rep. (2008) 60:68–74.
107. Palumbo R, Gaetano C, Antonini A, Pompilio G, Bracco E, Ronnstrand L, et al. Different effects of high and low shear stress on platelet-derived growth factor isoform release by endothelial cells: consequences for smooth muscle cell migration. Arterioscler Thromb Vasc Biol. (2002) 22:405–11. doi: 10.1161/hq0302.104528
108. Qi YX, Jiang J, Jiang XH, Wang XD, Ji SY, Han Y, et al. PDGF-BB and TGF-{beta}1 on cross-talk between endothelial and smooth muscle cells in vascular remodeling induced by low shear stress. Proc Natl Acad Sci USA. (2011) 108:1908–13. doi: 10.1073/pnas.1019219108
109. Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. (2014) 4:3. doi: 10.3402/jev.v3.24641
110. Maleki S, Cottrill KA, Poujade FA, Bhattachariya A, Bergman O, Gadin JR, et al. The mir-200 family regulates key pathogenic events in ascending aortas of individuals with bicuspid aortic valves. J Intern Med. (2019) 285:102–14. doi: 10.1111/joim.12833
111. Bjorck HM, Du L, Pulignani S, Paloschi V, Lundstromer K, Kostina AS, et al. Altered DNA methylation indicates an oscillatory flow mediated epithelial-to-mesenchymal transition signature in ascending aorta of patients with bicuspid aortic valve. Sci Rep. (2018) 8:2777. doi: 10.1038/s41598-018-20642-4
112. Jiang J, Wang K, Chen Y, Chen H, Nice EC, Huang C. Redox regulation in tumor cell epithelial-mesenchymal transition: molecular basis and therapeutic strategy. Signal Transduct Target Ther. (2017) 2:17036. doi: 10.1038/sigtrans.2017.36
113. Xie C, Guo Y, Zhu T, Zhang J, Ma PX, Chen YE. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem. (2012) 287:14598–605. doi: 10.1074/jbc.M111.329268
114. Manderfield LJ, Aghajanian H, Engleka KA, Lim LY, Liu F, Jain R, et al. Hippo signaling is required for Notch-dependent smooth muscle differentiation of neural crest. Development. (2015) 142:2962–71. doi: 10.1242/dev.125807
115. Grewal N, Franken R, Mulder BJ, Goumans MJ, Lindeman JH, Jongbloed MR, et al. Histopathology of aortic complications in bicuspid aortic valve versus Marfan syndrome: relevance for therapy? Heart Vessels. (2016) 31:795–806. doi: 10.1007/s00380-015-0703-z
116. Jones JA. Oxidative stress in bicuspid aortic valve-related aortopathy: Hand-me-downs and yoga pants. J Thorac Cardiovasc Surg. (2017) 154:1764–5. doi: 10.1016/j.jtcvs.2017.06.025
117. Psaltis PJ, Simari RD. Vascular wall progenitor cells in health and disease. Circ Res. (2015) 116:1392–412. doi: 10.1161/CIRCRESAHA.116.305368
118. Majesky MW, Dong XR, Regan JN, Hoglund VJ. Vascular smooth muscle progenitor cells: building and repairing blood vessels. Circ Res. (2011) 108:365–77. doi: 10.1161/CIRCRESAHA.110.223800
119. Roostalu U, Aldeiri B, Albertini A, Humphreys N, Simonsen-Jackson M, Wong JKF, et al. Distinct cellular mechanisms underlie smooth muscle turnover in vascular development and repair. Circ Res. (2018) 122:267–81. doi: 10.1161/CIRCRESAHA.117.312111
120. Majesky MW, Dong XR, Hoglund V, Daum G, Mahoney WM Jr. The adventitia: a progenitor cell niche for the vessel wall. Cells Tissues Organs. (2012) 195(1–2):73–81. doi: 10.1159/000331413
121. Wang G, Jacquet L, Karamariti E, Xu Q. Origin and differentiation of vascular smooth muscle cells. J Physiol. (2015) 593:3013–30. doi: 10.1113/JP270033
122. Forte A, Della Corte A. The aortic wall with bicuspid aortic valve: immature or prematurely aging? J Thorac Cardiovasc Surg. (2014) 148:2439–40. doi: 10.1016/j.jtcvs.2014.07.015
123. Blunder S, Messner B, Aschacher T, Zeller I, Turkcan A, Wiedemann D, et al. Characteristics of TAV- and BAV-associated thoracic aortic aneurysms–smooth muscle cell biology, expression profiling, and histological analyses. Atherosclerosis. (2012) 220:355–61. doi: 10.1016/j.atherosclerosis.2011.11.035
124. Di Mitri D, Alimonti A. Non-cell-autonomous regulation of cellular senescence in cancer. Trends Cell Biol. (2016) 26:215–26. doi: 10.1016/j.tcb.2015.10.005
125. Triana-Martinez F, Pedraza-Vazquez G, Maciel-Baron LA, Konigsberg M. Reflections on the role of senescence during development and aging. Arch Biochem Biophys. (2016) 598:40–9. doi: 10.1016/j.abb.2016.04.004
126. Gonzalez LC, Ghadaouia S, Martinez A, Rodier F. Premature aging/senescence in cancer cells facing therapy: good or bad? Biogerontology. (2016) 17:71–87. doi: 10.1007/s10522-015-9593-9
127. Schosserer M, Grillari J, Breitenbach M. The Dual role of cellular senescence in developing tumors and their response to cancer therapy. Front Oncol. (2017) 7:278. doi: 10.3389/fonc.2017.00278
128. Smit MA, Peeper DS. Epithelial-mesenchymal transition and senescence: two cancer-related processes are crossing paths. Aging. (2010) 2:735–41. doi: 10.18632/aging.100209
129. Kishi S, Bayliss PE, Hanai J. A prospective epigenetic paradigm between cellular senescence and epithelial-mesenchymal transition in organismal development and aging. Transl Res. (2015) 165:241–9. doi: 10.1016/j.trsl.2014.05.007
130. Fleenor BS, Marshall KD, Rippe C, Seals DR. Replicative aging induces endothelial to mesenchymal transition in human aortic endothelial cells: potential role of inflammation. J Vasc Res. (2012) 49:59–64. doi: 10.1159/000329681
131. Warboys CM, de Luca A, Amini N, Luong L, Duckles H, Hsiao S, et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol. (2014) 34:985–95. doi: 10.1161/ATVBAHA.114.303415
Keywords: bicuspid aortc valve, aneurysm, endothelial to mesenchymal transition (EndMT), ascending aorta, endothelial cell (EC)
Citation: Maleki S, Poujade F-A, Bergman O, Gådin JR, Simon N, Lång K, Franco-Cereceda A, Body SC, Björck HM and Eriksson P (2019) Endothelial/Epithelial Mesenchymal Transition in Ascending Aortas of Patients With Bicuspid Aortic Valve. Front. Cardiovasc. Med. 6:182. doi: 10.3389/fcvm.2019.00182
Received: 26 March 2019; Accepted: 21 November 2019;
Published: 17 December 2019.
Edited by:
Emanuela Branchetti, University of Pennsylvania, United StatesReviewed by:
Cristina Aurigemma, Agostino Gemelli University Polyclinic, ItalyCopyright © 2019 Maleki, Poujade, Bergman, Gådin, Simon, Lång, Franco-Cereceda, Body, Björck and Eriksson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Per Eriksson, cGVyLmVyaWtzc29uQGtpLnNl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.