 Silje Madeleine Kalstø
Silje Madeleine Kalstø Joylene Elisabeth Siland
Joylene Elisabeth Siland Michiel Rienstra
Michiel Rienstra Ingrid E. Christophersen1,3*
Ingrid E. Christophersen1,3*- 1Department of Medical Research, Bærum Hospital, Vestre Viken Hospital Trust, Rud, Norway
- 2Department of Cardiology, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 3The Department of Medical Genetics, Oslo University Hospital, Oslo, Norway
Atrial fibrillation (AF) is the most common heart rhythm disorder worldwide and may have serious cardiovascular health consequences. AF is associated with increased risk of stroke, dementia, heart failure, and death. There are several known robust, clinical risk predictors for AF, such as male sex, increasing age, and hypertension; however, during the last couple of decades, a substantive genetic component has also been established. Over the last 10 years, the discovery of novel AF-related genetic variants has accelerated, increasing our understanding of mechanisms behind AF. Current studies are focusing on mapping the polygenic structure of AF, improving risk prediction, therapeutic development, and patient-specific management. Nevertheless, it is still difficult for clinicians to interpret the role of genetics in AF prediction and management. Here, we provide an overview of relevant topics within the genetics of AF and attempt to provide some guidance on how to interpret genetic advances and their implementation into clinical decision-making.
Introduction
Atrial fibrillation (AF) is the most common sustained heart rhythm disorder and is estimated to affect more than 30 million individuals worldwide (1). The lifetime risk of individuals older than 55 years of age is 37%, and AF is associated with an increased risk of stroke, dementia, heart failure, and death (2).
Many studies have investigated the risk factors for AF to improve prevention of the arrhythmia. The most important risk factors for AF are increasing age, male sex, hypertension, diabetes, obesity, myocardial infarction, heart failure, and stroke (3, 4); aggregation of risk factors further increases the risk for AF. However, the AF population is heterogeneous, and some individuals develop AF with no apparent risk factors. Despite all efforts, it remains difficult to predict AF based solely on clinical risk factors.
Ever since familial clustering of AF was demonstrated and the heritability of AF was established, large efforts have been made to identify the genetic causes of AF (2, 5, 6). With the advances of genetic sequencing and bioinformatic analyses, the field of genetic research has turned from candidate gene sequencing approaches to large-scale genome-wide analyses. Today, more than a hundred genetic loci have been associated with AF.
For clinicians, it remains difficult to interpret the genetic advances in the context of clinical risk factors, and translation of genetic discoveries to clinical decision-making remains unresolved. Here, we aim to provide clinicians with an overview of the relevant literature on the genetics of common AF using four directions earlier described (7): genetic mapping of AF, AF risk prediction, therapeutic development, and patient-specific management. We provide some guidance on the consequences of large-scale genetic studies for AF and whether genetic information is ready to be implemented into clinical practice—alternatively, what is needed to get there.
Familial Clustering of AF
One of the first descriptions of familial aggregation of AF was published in 1943, describing three brothers diagnosed with AF who had experienced irregular heart rate since childhood (8). More recently, several studies have described familial clustering of AF in larger populations (5, 6, 9–12). In 2004, Fox et al. showed that one-third of the individuals diagnosed with AF in the Framingham Heart Study had at least one parent also diagnosed with AF, providing evidence that parental AF increased the risk of AF in offspring in the general population (5). Short after this publication, the Icelandic nation-wide genealogic community-based data were used in an analysis of more than 5,000 individuals. An important finding was that the AF risk ratio declined exponentially when the proportion of identically shared alleles by descent diminished in relatives, confirming the importance of genetics in relation to AF risk. Similarly, a Danish twin study showed that having a co-twin with AF increased an individual's risk of AF and that the risk was doubled in monozygotic twins, compared to dizygotic twins (6). In 2010, the Framingham Heart Study confirmed that familial AF, particularly when premature in onset, slightly improved prediction of new-onset AF beyond clinical AF risk factors (13). Moreover, the risk of AF increased with an increasing number of affected first-degree relatives, and having an affected sibling conferred a similar magnitude of risk as parental AF. Together, the observations suggest that familial clustering of AF may be genetically caused.
Genetic Mapping of AF
Segregation Analyses and Candidate Gene Sequencing
Historically, segregation analyses and candidate gene sequencing were used to identify genotype–phenotype correlations, involving families or populations with high prevalence of AF. Segregation analysis is used to identify genetic markers co-segregating with the AF phenotype, utilizing the linkage phenomenon; because of genetic recombination, genetic markers that are located in close proximity are more likely to co-segregate through pedigrees than genetic markers located far apart. In 2003, a gain-of-function mutation in KCNQ1 was identified through segregation analysis, making it the first AF-associated gene (14). Candidate gene sequencing involves sequencing of candidate genes, based on our knowledge of their biological function, and thus an a priori hypothesis that increased or decreased function of the resulting proteins may lead to AF. Although many genetic variants, largely related to ion channels, have been associated with AF using segregation analysis and candidate gene sequencing, there are several limitations to these methods. The limited sample size in most of these studies and the low frequency of the genetic variants discovered have made them underpowered. The recent introduction of large reference databases of exomes and genomes has shown that many genetic variants proposed to be related to AF and other cardiac arrhythmia disorders through candidate gene sequencing are present at a much higher frequency in the population than what we would expect of a disease-causing variant (15–18).
Genome-Wide Association Studies on AF
The new millennium brought along the genome-wide association study (GWAS) approach, which was novel in its unbiased nature and its focus on common rather than rare genetic variants. One of the first GWAS was performed in 2002 by Ozaki et al. who identified a candidate locus associated with myocardial infarction (19). This achievement initiated an accelerated search for disease-associated loci through GWAS. The first large GWAS for AF, including 550 AF cases, was performed on Iceland in 2007 (20), identifying a genetic locus associated with AF on chromosome 4q25. The most significant variant was located in an intergenic region, with the nearest gene, PITX2, 150 kilobases (kb) away. Pitx2c is a homeodomain transcription factor involved in embryonic development, essential in the formation of the atria, the sinus node, and left-right heart asymmetry (20, 21). Figure 1 shows an overview of GWAS performed for AF to date. The association between the 4q25 locus and AF was replicated in two independent studies in 2009 (22, 23), and since then, PITX2 has continued to be the most significant genetic locus in AF GWAS. Gudbjartsson et al. expanded their GWAS to include 2,385 individuals with AF, and identified the ZFHX3 locus in 2009 (24). In the same period, Benjamin et al. showed a similar association between the ZFHX3 locus and AF in a European ancestry population (25). The ZFHX3 gene encodes a transcription factor enriched in cardiac tissue (21, 25), and there is evidence showing that interaction between ZFHX3 and Pitx2c or TBX5, both transcription factors active in embryogenic cardiogenesis, may increase the risk of AF significantly (26, 27).
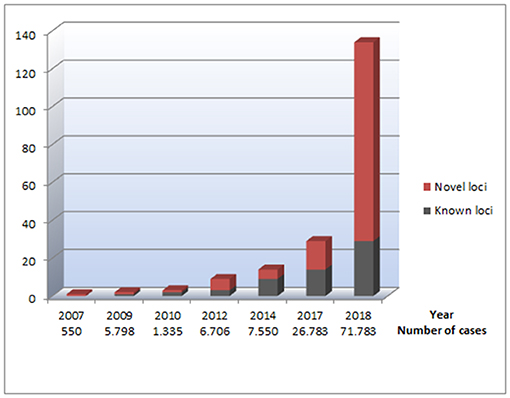
Figure 1. An overview of genetic loci identified through GWAS performed for AF. The figure illustrates the number of identified loci for AF, based on the data listed in Table 1. The x axis represents which years the different GWAS for AF was performed. Under each year, the total number of cases included in all GWAS for AF the current year is listed, illustrating the relationship between the increasing sample sizes and the increasing number of AF-associated loci identified. The Y axes represent the number of genetic loci associated with AF. The red parts of the columns represent genetic loci that have not previously been reported in relation to AF. The black parts of the columns represent previously reported genetic loci associated with AF, making the total of each column reflect the total number of AF-associated loci at a given time.
In 2010, the KCNN3 gene, encoding a calcium-gated potassium channel involved in atrial repolarization, was identified as the third AF-associated locus in a lone AF population by Ellinor et al. Two years later, the first GWAS meta-analysis from the international Atrial Fibrillation Genetics (AFGen) Consortium, including ~6,500 AF cases, revealed six new genetic loci for AF (28). The most significant novel association was found for the PRRX1 locus, which encodes a transcription factor highly expressed in the developing heart. Further studies on PRRX1 have shown that loss of PRRX1 expression results in shortening of the atrial action potential duration and may, thus, promote AF (29).
From 2007 to 2017, there was a steady increase in the identification of novel GWAS loci for AF, and a total of 14 new loci were identified in this period (20, 24, 25, 28, 30, 31) (Table 1).
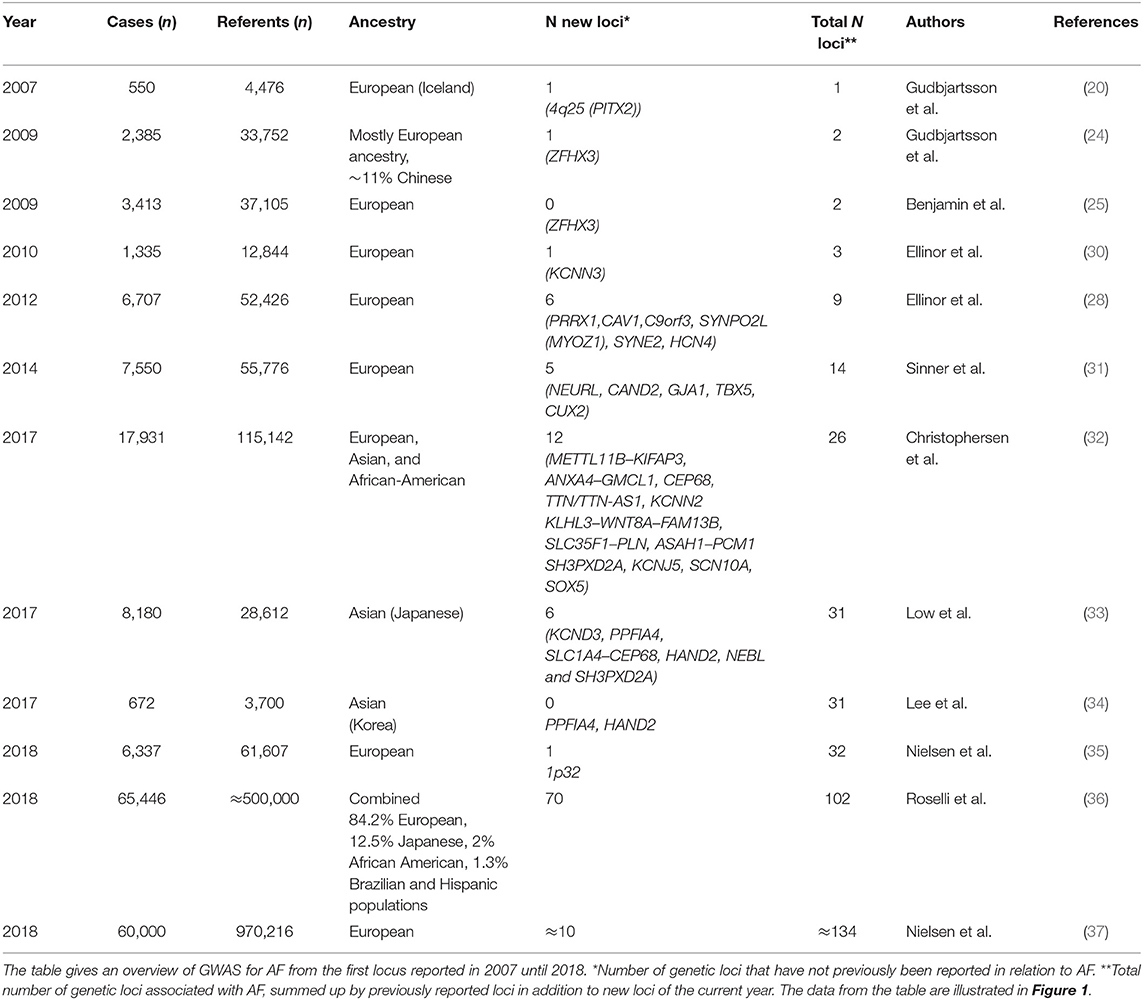
Table 1. Overview of GWAS for AF.
In 2017, Christophersen et al. identified 12 new loci associated with AF in a multiancestry GWAS meta-analysis from the AFGen Consortium, nearly doubling the number of AF risk loci. Their study included European, Asian, and African-American ancestry groups (32). An intriguing finding was the TTN locus, which encodes titin, the largest protein in the human body and an essential building block in the sarcomeres of striated muscle tissue. It is highly expressed in the human heart, in both atria and ventricles. Hence, titin dysfunction can alter the cardiomyocyte structure (38). A second biologically interesting discovery was the gene KCNN2, encoding the calcium-dependent potassium channel subunit SK2. SK2 forms a complex with SK3, which is encoded by the gene KCNN3 previously associated with AF in an early AF GWAS, as described above (30). SK2 blockers are under development as a new potential treatment against AF (39–42). GWAS in Korean and Japanese populations the same year identified six loci, of which the loci KCND3, PPFIA4, HAND2, and NEBL were specific to East-Asian AF patients (33, 34).
In 2018, a Norwegian GWAS including 6,337 cases and 61,607 referents (35) identified one novel risk locus on chromosome 1p32, with DMRTA and CDKN2C as suggested functional genes, potentially involved in structural remodeling of cardiac tissue as a substrate for AF.
Later in 2018, there was a major leap in the progression of AF GWAS when a large GWAS meta-analysis from the AFGen Consortium revealed 97 AF risk loci, of which 92 represented common variants and 70 were novel findings (36). The study included over half a million individuals of combined ancestry and approximately 65,000 AF cases. Another GWAS meta-analysis of the same magnitude was published the same year, including ~1 million individuals, of which ~60,000 were AF cases (37), identifying 111 AF-associated loci. A preliminary meta-analysis combining non-overlapping participants from these two largest GWAS performed, including ~93,000 AF cases, resulted in ~134 genetic loci associated with AF (36).
The new genetic discoveries provide optimism for future novel treatment options for AF, as many of the candidate genes identified may provide potential novel drug targets. Roselli et al. points out the SCN5A gene, encoding the most important sodium channel in heart tissue, and KCNH2 encoding a subunit of the potassium channel complex, as primary targets for antiarrhythmic channel blockers or inhibitors already in use (36). Of the 151 candidate genes identified, Nielsen et al. found potential drug interactions for 31 genes, describing a total of 475 potential drugs (37). Of the potential drugs identified, 78 are already known in relation to treatment for arrhythmias. For some of the drug target interactions, clinical trials may be in sight, and as mentioned above, the SK2 blocker seems to be a promising new treatment for AF.
It has been proven possible to carry out large AF GWAS meta-analyses in an order of magnitude that provides the statistical power needed to establish true genetic associations, through extensive international collaboration. So are we there yet, or do we need even bigger and better GWAS?
In order to approach an answer to this question, we can look to large GWAS made for other common complex diseases, such as major depressive disorder or schizophrenia. In 2018, Nishino et al. predicted the number of cases needed to detect significant genetic variants for these two psychiatric diseases (43). The numbers of cases needed to detect 1, 10, and 100 genetic variants by GWAS were calculated to be 7,000, 18,000, and 51,000 cases for schizophrenia, and 34,000, 61,000, and 118,000 cases for major depressive order, respectively. The authors pointed out that the number of genetic variants associated to major depressive disorder would rapidly increase, when 50,000 cases or more were included. In comparison, the largest AF GWAS thus far included a similar number of cases.
However, the power of a GWAS depends not only on the sample size, but equally important are the genetic coverage (genotyping chip and imputation panel density), the frequency of the genetic variants, the ancestry group, and the effect size (44–48). Nelson et al. have estimated the power of GWAS across a range of genotyping chips, ancestry groups, minor allele frequencies, and effect sizes, suggesting that we are close to having identified the most relevant common variants for AF in European ancestry groups. The pursuit of new common variants for AF will likely be more successful if we focus on non-European ancestry groups and on low-frequency variants with large effect size (46). Moreover, as discussed below, heritability estimates show that there is still a large proportion of missing heritability, which may partially be caused by unidentified common genetic variants.
Heritability of AF
The large number of common genetic variants now identified for AF has enabled estimation of the heritability of AF, using genetic data. In 2017, Weng et al. showed that genetic variation accounts for 22.1% of the total variance for AF (2), of which the 25 AF loci that were known at the time accounted for only 25%. The fraction of the heritability of AF explained by common genetic variants have increased to 42% based on the results of the largest GWAS thus far (36). As evident from the estimates of explained heritability, there is still a substantial proportion of “missing heritability.” Where the missing heritability is buried is debated. First, common genetic variants identified through GWAS are mostly located in intergenic or intronic regulatory regions. They may participate in disease development through regulatory mechanisms, thereby affecting the expression of multiple genes located in proximity and possibly further away. For example, the co-regulatory network involving TBX5 and PITX2 influences atrial development (36, 49). Gene–gene interactions can play a significant role in regulating cellular behavior and biological events. Two variants with small individual effects may have large interaction effects. Lin et al. showed that two genetic variants, rs7164883 at the HCN4 locus and rs4980345 at the SLC28A1 locus, were highly significantly associated with AF in a European ancestry discovery cohort from 15 studies. Moreover, eight additional gene–gene interactions were marginally significant, albeit unreplicated thus far (50).
Second, gene–environment interactions may contribute to the missing heritability. Some cross-trait associations have been found between genetic variants associated with AF risk factors, such as height, body mass index (BMI), and hypertension, and AF risk variants (36). These pleiotropic effects may also contribute to missing heritability. Last, unidentified common and rare genetic variants, including copy number variation, may partially explain the missing heritability of AF (51). The fast-developing high-throughput sequencing technology may play an important role in uncovering missing heritability for AF.
High-Throughput Sequencing in AF
During the last decades, genetic sequencing technology has evolved rapidly, and with that, the price for sequencing a whole genome has dropped dramatically (52). High-throughput sequencing describes a variety of techniques, including whole-exome and -genome sequencing, that enable high-resolution genetic analysis of all coding and non-coding regions of our genome. The advantage of high-throughput sequencing is the ability to analyze genetic variants with lower frequencies, which are more likely to have large effects on disease risk, as opposed to GWAS, which is mainly suitable for analysis of common variants that confer small effects on disease risk. Table 2 gives an overview of the main published high-throughput sequencing studies of AF. Figure 2 shows the main biological pathways implicated in AF pathophysiology by GWAS and high-throughput sequencing studies.
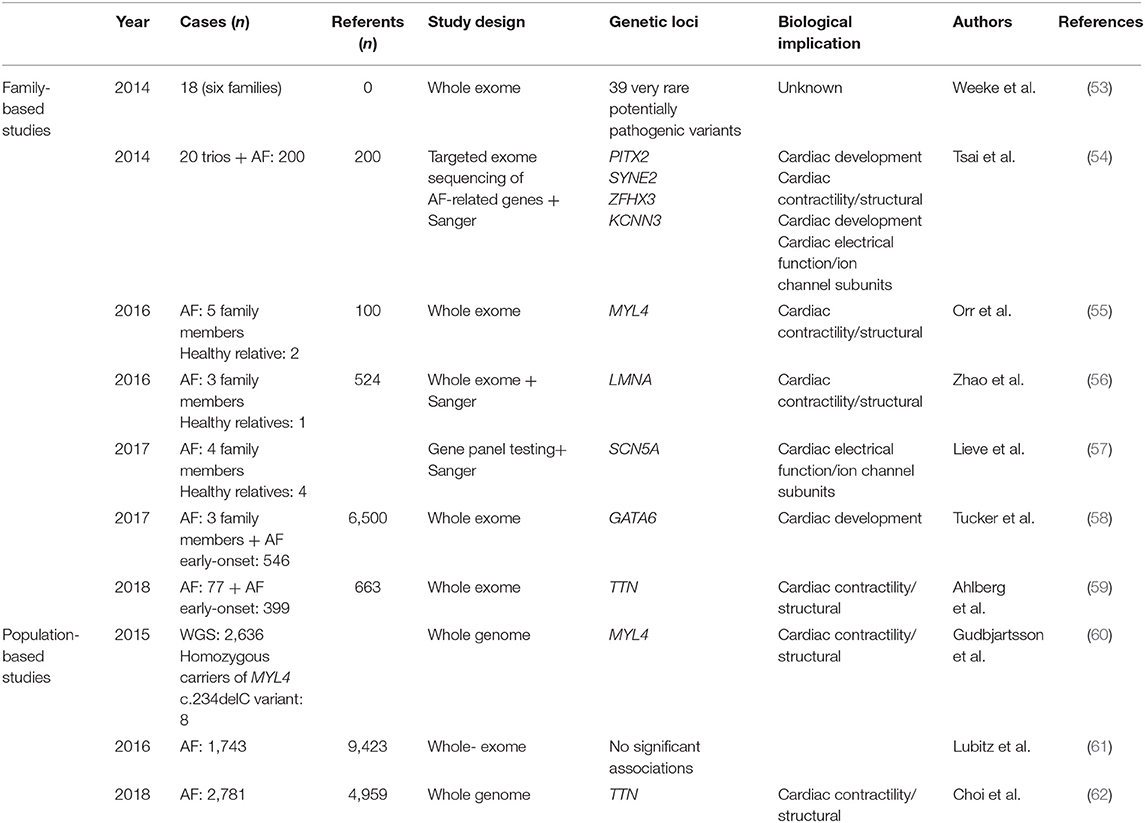
Table 2. Overview of high-throughput sequencing studies for AF, divided in family- and population-based studies.
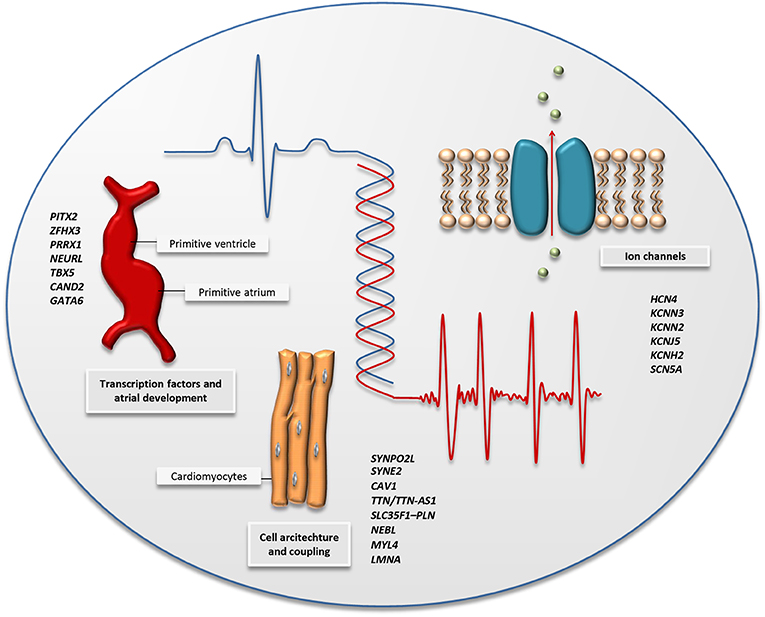
Figure 2. The figure illustrates the main biological pathways implicated by AF-associated variants identified by GWAS and high-throughput sequencing; cardiac transcription factors and embryonic cardiogenesis, the architecture of the cardiac cells, and ion channel function. A selection of genes associated with AF through GWAS and high-throughput sequencing is listed for each pathway and can also be found in Tables 1, 2.
High-Throughput Sequencing in Family Studies
One of the first reports of high-throughput sequencing in AF was performed in 2014, where 39 very rare variants [minor allele frequency (MAF) <0.04%] were identified through whole-exome sequencing in six families with aggregation of AF (53). The potentially pathogenic variants, with a range of 7 to 15 shared variants per family, underscored the complexity of the genetics of AF and suggested that non-coding regions may be more important in the search for causal genetic variants. However, the lack of sequencing of healthy family members leaves us uncertain whether the identified genetic variants truly segregate with the disease. The same year, five novel rare variants were identified performing high-throughput sequencing of nine AF-associated genes in 20 parent–offspring trios (54). One of the variants was located in the 5' untranslated region of the PITX2 gene, downregulating expression of Pitx2c in atrial myocytes. The remaining variants were exonic nonsense mutations in the genes SYNE2, ZFXH3, and KCNN3, and thus represent each of the three main pathological pathways for AF development suggested by prior GWASs (Figure 2). Whereas, mechanisms for AF development previously have been focused at cardiac electrical function, there are several genetic findings that now also point to structural changes in the atrium as a cause of the arrhythmia. Orr et al. highlighted one such association describing a family with aggregation of early-onset AF, cardiac conduction disease, and atrial myopathy (55). Through whole-exome sequencing, they identified a novel genetic variant in MYL4 shared by all five affected relatives. MYL4 is expressed in adult atria and embryonic muscle tissue, and it has been shown that loss-of-function variants in MYL4 can lead to atrial cardiomyopathy (63). Another genetic variant related to structural changes in the heart, a non-sense mutation in the LMNA gene (c.G1494A, p.Trp498Ter), was identified through whole-exome sequencing and Sanger sequencing in a four-generation AF family from northern China (56). LMNA encodes the nuclear membrane proteins Lamin A and Lamin C, and pathological variants are known to cause a broad variety of inherited diseases referred to as laminopathies, including myopathies in skeletal muscle with a dystrophy-like picture, cardiomyopathy, and conduction system disease (64–66).
In 2017, Lieve et al. performed a gene panel testing complemented with Sanger in a two-generation Dutch family, with a phenotype presenting with ventricular arrhythmias and early-onset AF (57). They found a gain-of-function variant in SCN5A, which encodes the alpha subunit of the main cardiac sodium channel, and with this, drew attention back to cardiac electrical function as a biological pathway for AF. The functional effect of the variant seemed to be increased channel availability and current duration. SCN5A has previously been shown to be one of the most important genes for overall atrial conduction (32). The same year, Tucker et al. identified a gain-of-function variant in GATA6 through whole-exome sequencing in two families with early-onset AF; one of the families also displayed atrioventricular septal defects (58). GATA6 encodes a cardiac transcription factor important to cardiac morphogenesis, and loss-of-function variants have previously been reported to be associated with congenital cardiac defects (67, 68). In addition, loss-of-function variants in GATA6 have been associated with lone AF through candidate gene studies (69, 70).
As described, the gene TTN has been associated with AF in two independent GWAS meta-analyses (32, 35), and results from recent high-throughput sequencing studies support this association (59, 62). Ahlberg et al. performed whole-exome sequencing in 24 families with aggregation of AF (59) and discovered that the frequency of titin-truncating variants (TTNtv) in cardiac transcripts was significantly higher in the individuals with AF compared to an unaffected referent group (16.7 vs. 0.5%, P = 1.76 × 10−6). The association was replicated in 399 individuals with early-onset AF (AF before age 40), showing an increased frequency of TTNtv in cases compared to referents (odds ratio [OR] = 36.8, 95%, confidence interval [CI] = 5.0–4692.5, P = 4.13 × 10−6). They further examined the effect of TTNtv on atrial development in a zebrafish model, which revealed disorganized sarcomeres and atrial fibrosis in adult heterozygous fish, and electrocardiogram (ECG) analysis showed prolongation of the PR interval, which is a known risk factor for AF (71).
High-Throughput Sequencing in Population-Based Studies
Large-scale high-throughput sequencing is merely in the initial phase when it comes to AF in the general population. In 2015, Gudbjartsson et al. performed large-scale whole-genome sequencing in 2,636 individuals on Iceland (60). They found a recessive frameshift mutation in MYL4 (c.234delC, MAF = 0.65%), increasing the risk for early-onset AF by 110-fold (recessive OR = 110.3, P = 5.2 × 10−10). Eight homozygous carriers of the MYL4 variant were identified. All eight carriers had been diagnosed with early-onset AF. The need for large sample sizes in order to have sufficient power to detect significant associations in high-throughput sequencing studies was demonstrated by Lubitz et al. who performed whole-exome sequencing in 1,734 individuals with AF and 9,423 referents, with no significant associations detected (61). Recently, Choi et al. reported results from whole-genome sequencing in almost 2,781 individuals with early-onset AF and 4,959 referents of European ancestry (62). They found an association between a rare (MAF = 0.1%) loss-of-function variant in TTN and early-onset AF and showed that the probability of being a carrier of the variant increased conversely with age at AF onset. As previously described, TTN encodes the large protein titin, which forms a crucial component in the sarcomeres of muscle tissue. An association between TTN variants and dilated cardiomyopathy (DCM) is well-established (72, 73), and patients with familial or early-onset DCM are routinely screened for variants in the gene (74). The recent discoveries indicate that TTNtv variants may contribute to AF without the presence of DCM (59, 62).
The population-based studies describing a role for TTN and MYL4 in the genetic background for AF are in line with the findings from the family-based studies by Orr and Ahlberg, described above. The results suggest that a proportion of AF-related genes or genetic regions may be involved in structural cardiac abnormalities, in addition to the well-known electrical abnormalities identified for several ion channel genes, and support the hypothesis that some types of AF may be considered to be atrial cardiomyopathies (75). The obvious fact that AF, as a heterogeneous disease, displays different causal mechanisms is important for differentiating treatment. In the future, we may routinely be identifying underlying causes of AF through genetic testing, before choice of treatment.
Mendelian Randomization Studies—Causal Effects of Genetic Variants
In a polygenic and heterogeneous disease, such as AF, it is difficult to prove causality. Causality can only be established when confounding factors are eliminated in the analyses. Residual confounding, reverse causation, and bias should be taken into consideration in any analyses of observational data (76, 77). Mendelian randomization is a statistical method that is currently used as a powerful control for reverse causation and confounding. In Mendelian randomization, genetic variants with known association to the exposure are used as instrumental variables to examine the causal effect of the exposure on the disease. The use of genetic variants of AF-related risk factors may clarify the complexity between AF and the environmental factors that interact with AF. A causal relation between BMI and incident AF has been suggested by observational data (4, 78), until recently confirmed by Chatterjee et al. using Mendelian randomization (79), supporting prevention of obesity as a therapeutic target to reduce incident AF. However, BMI covers weight and height but does not define the components of body mass, e.g., distinguish between fat mass and muscle mass. In an attempt to understand how body mass plays a causal role in AF, Tikkanen et al. evaluated the relation between fat mass and fat-free mass, and incident AF, using multivariate Mendelian randomization (80). During a follow-up period of 6.1 years, 10,852 incident AF cases occurred in the UK biobank population of 502,619 individuals (54% women, mean age = 56.5 ± 8.1 years). The multivariate Mendelian randomization showed that the effects of fat mass and fat-free mass were independently of each other associated with incident AF. Moreover, fat mass showed stronger causal associations in women, whereas fat-free mass displayed a similar association across sexes. Reductions in fat mass and fat-free mass likely follow different biological pathways, and both reduce AF risk independently.
Another anthropometric trait, height, is a well-known, independent risk factor for AF (81). More than 400 genetic loci have been associated with height (82–86), of which selected variants have also been associated with AF (87). Taller individuals have larger atria and more frequent premature atrial contractions. These are both strong predictors for AF and can influence the cardiac conduction system, reflected in PR interval and QRS duration on ECG (88–92). Kofler et al. investigated 2,149 individuals [54% women, median age = 37 (30–40) years], with a median height of 1.71 m, of the Genetic and Phenotypic Determinants of Blood Pressure and other Cardiovascular Risk Factors study (93). The Mendelian randomization showed a significant association between height and both PR interval and QRS duration. The authors propose that genetically determined body size has impact on the cardiac conduction system and thus may lead to increased risk of AF. However, the association may not be generalizable to all populations. The genetic variants associated with height discovered in data of the GIANT consortium and replication studies have been suggested to be biased by population stratification (94–97). The suggested polygenic adaptation of height, supported by average polygenic scores, increased from south to north across Europe, but in such steep line that is inconsistent with a neutral model evolution. Recently, new analyses were performed in the UK Biobank, in which the signals of selection were absent. Berg et al. highlights the challenge that comes with correcting for population stratification in GWAS of polygenic traits and distinguishing differences in polygenic scores between populations.
Hyperthyroidism increases the risk of AF, and hypothyroidism is associated with a reduced risk of AF. The increased risk or AF persists despite treatment against hyperthyroidism (98, 99). In a Mendelian randomization of 55,114 AF cases and nearly half a million referents, supporting evidence for a causal pituitary–thyroid–cardiac axis was found. Low thyrotropin and an increased ratio of triiodothyronine: free thyroxine was genetically associated with AF. However, due to the lack of genetic instruments, especially for triiodothyronine, and the lack of association with increased thyroxine, a link between a specific agent of the thyroid and AF can still not be addressed (100). Although Mendelian randomization seems a good tool for identifying causality, knowledge gaps and relatively small sample sizes can still prevent the discovery of true associations between AF risk factors and AF.
AF Risk Prediction
Genetic Risk Interacts With Lifestyle and Mediates Lifetime AF Risk
The incidence and prevalence of AF have increased over the past decades (101, 102), which may have influenced the increase in lifetime risk that we see in AF (37% among individuals aged >55 years) (103, 104), compared with prior estimates (men 23.8%, women 22.2% at age 55 years in the Rotterdam study; men 25.9%, women 23.2% at age 50 years in the Framingham Heart Study) (105, 106). The burden of clinical risk factors for AF contributes to the increase in lifetime risk. Advanced age is a well-known risk factor for AF, in addition to male sex, hypertension, obesity, smoking, alcohol intake, sleep apnea, diabetes, hyperthyroidism, previous myocardial infarction, and heart failure (107, 108). Clinical risk factors for AF are known to be strongly associated with arrhythmia, with large effect sizes. For instance, hypertension has been shown to increase risk of AF by 40% in women and 50% in men, diabetes increases the risk by 60% in women and 40% in men, whereas a 10-year increase in age doubles the risk for AF (109). As described above, most known genetic variants associated with AF have small effect sizes, increasing the risk for AF in the range 0.1–20.0%, except for the 4q25 locus, for which an effect size up to ~48% has been estimated (36). So how is the interplay between genetic and clinical risk factors for AF? Said et al. aimed to investigate the association of combined health behavior and genetic risk group in cardiovascular disease, including AF, in a cohort of nearly 340,000 individuals included from the UK Biobank (110). They showed that both high genetic risk and poor behavioral lifestyle are associated with increased risk of new onset of disease, yet there was no interaction effect observed between genetic risk profile and lifestyle. Participants were divided into groups according to lifestyle and genetic risk, where lifestyle was defined as ideal, poor, or intermediate, according to smoking, BMI, and physical activity. Genetic risk was calculated based on genetic variants associated with AF through GWASs. In the low genetic risk group, poor lifestyle increased the risk for new-onset AF more than 5-fold [hazard ratio (HR) 5.41, 95% CI 4.29–6.81, P < 0.001], compared to ideal lifestyle. Genetic composition and lifestyle had a logon additive effect on risk for disease, and the relative effect of poor lifestyle was comparable between genetic risk groups. Said et al. point to the fact that everyone benefits from lifestyle intervention, but due to the logon additive effect of genetic risk and lifestyle, high genetic risk may be a suitable selection criterium for intervention.
Polygenic Risk Scores and Risk of Incident AF
Since common genetic variants confer small effect of risk of AF, polygenic risk scores can be constructed, summing the weighted risk of each genetic variant. Such a score can be used to evaluate the overall association of all known genetic variants with a specific phenotype. However, unlike a biomarker that represents an underlying biological pathway, the underlying biological pathways of genetic variants included in a polygenic risk score are unknown.
Lubitz et al. (111) have evaluated the association between several polygenic risk scores and incident AF. Because of the unknown true significance of each genetic variant, they used several significance thresholds and built risk scores with varying numbers (from 11 to 719) of included genetic variants. They found that polygenic risk scores were associated with AF beyond established clinical risk factors, underscoring the important contribution of genetic variants in the identification of individuals at risk for AF. The polygenic nature, the ongoing discovery of genetic variants associated with AF, and the unknown true significance of each genetic variant in the development of AF have motivated ongoing adjustments to the polygenic risk scores (111, 112). In a recent paper led by Lubitz, polygenic and clinical risk scores were combined to estimate the lifetime risk of AF in the Framingham Heart Study (104). More than 5,000 individuals were analyzed, and the overall lifetime risk of AF was estimated to be 37.1% for individuals older than 55 years. Both the polygenic risk score, including approximately a thousand genetic variants, and the clinical risk score contributed to the lifetime risk of AF. In individuals with low clinical risk for AF, the lifetime risk was doubled when moving from low to high polygenic risk [22.3% (95 CI, 15.4–29.1%) vs. 43.6% (95% CI, 35.6–51.6)], and the effect was similar in individuals with high clinical risk. Studies on polygenic risk scores for AF show that an estimation of genetic predisposition to AF is feasible with GWAS data and that polygenic risk scores can be utilized in lifetime risk models; however, regardless of the inherited predisposition to AF, the arrhythmia develops at an older age when individuals have a low burden of clinical risk factors. Modifiable risk factor management as for hypertension, obesity, smoking, and obstructive sleep apnea is still important to reduce AF risk. Future studies should determine to what extent AF risk factors can compete with polygenic AF risk.
Polygenic AF and Risk of Stroke
Ten years ago, cardioembolic stroke was described to be associated with the two strongest AF risk loci (chromosome 4q25 and 16q22) (24, 113). In the prospective Malmö Diet and Cancer Study, incident ischemic stroke was associated with an AF genetic risk score containing 12 AF risk variants. From the 27,471 individuals, 2,160 individuals developed AF, and 1,495 developed stroke. The AF genetic risk score was associated with incident AF, but also with incident ischemic stroke. The individuals in the top AF genetic risk score quintile had a 2-fold increased risk of incident AF after adjusting for clinical risk factors, and 23% increased risk of ischemic stroke (114). Recently, in a separate analysis, Lubitz et al. described an association between increased polygenic risk for AF and stroke in both individuals with and without established AF (111). Moreover, genetic risk for AF was strongly associated with cardioembolic stroke, suggesting that an elevated polygenic risk score may serve as a surrogate for thromboembolism from AF.
The risk of stroke increases when AF is present (115, 116), but AF genetic risk was also associated with cardioembolic stroke in the absence of a diagnosis of AF. The authors hypothesize that AF genetic risk is a clinically relevant marker for subclinical or previously undiagnosed AF. If individuals with subclinical or undiagnosed AF can be identified using a polygenic risk score for AF, this may be used to prevent stroke. Thus, in the future, polygenic risk scores for AF may be clinically useful in primary and secondary prevention of AF-related stroke.
Polygenic AF and Risk of Heart Failure
Heart failure may cause and be caused by AF. In a 38-year follow-up in the Framingham Heart study, heart failure was the strongest predictor of AF (117). Increasing age, non-ischemic etiology of heart failure, and a New York Heart Association (NYHA) class greater than II has been associated with the coexistence of AF (118). In the last 10 years, five GWAS have been conducted for heart failure. In aggregate, six genetic loci and five candidate genes were identified in 10,468 cases and 33,029 referents (119–124). Although heart failure and AF are strongly associated, only one shared genetic locus has been discovered. The ubiquitin–proteasome system enzyme 3 (USP3) gene is one of the novel genetic AF loci recently identified. USP3 produces a protease that breaks down and modulates many intracellular proteins and eliminates damaged and misfolded proteins and regulates cellular processes as apoptosis. In cardiomyocytes, USP regulates voltage-gated membrane channels, such as sodium and potassium channels, and cell surface receptors. Cardiac signal transduction pathways are regulated by USP through regulating proapoptotic factors (125, 126). The onset and progression of hypertrophic cardiomyopathy are possibly linked to low concentrations or dysfunction of this protease (126, 127). The USP-regulated cardiac signal transduction pathways may play a mechanistic role in AF, but further studies are warranted.
The GWAS that have been reported for heart failure have had limited statistical power, compared to the half a million individuals that led to the discovery of 134 genetic variants for AF. The heterogeneity of heart failure complicates the identification of the genetic susceptibility and phenotype of heart failure. There is a large garden of variety that contributes to heart failure that can easily lead to different heart failure phenotypes. Future studies may use the polygenic risk score for AF to investigate shared genetic loci between AF and related phenotypes. The complex nature of AF may be elucidated by investigating these pleiotropic effects.
Therapeutic Development Based on Genetic Discoveries for AF
Rate and Rhythm Control
Data on genetic predisposition for successful rate and/or rhythm control is scarce. A study of Barret et al. investigated genetic susceptibility and rate control after intravenous treatment with diltiazem at the emergency department, using genetic variants previously associated with atrioventricular nodal conduction, resting heart rate, and AF (128). Separate risk scores were constructed for each of the three phenotypes, and the primary outcome was the maximum heart rate decrease within 4 h after diltiazem administration. The secondary outcome was a ventricular heart rate <110 bpm within 4 h of diltiazem administration, defined as rate control. However, in the 127 eligible individuals, there was no significant association between the polygenic risk score for AF and successful rate control. The relatively small sample size may render the study underpowered to reveal any significant associations.
One of the largest studies in the field was reported from Parvez et al. in the Vanderbilt AF registry, including 478 caucasian individuals with documented AF (32% women, age 63 ± 14 years), who used at least one antiarrhythmic drug. During a 12-month follow-up period, successful rhythm control (> 75% reduction in symptomatic AF burden, defined as frequency, duration and severity of symptoms, in a patient who remained on the same antiarrhythmic drug for a minimum of 6 months) was established in 399 (84%) individuals. A genetic variant at chromosome 4q25 independently predicted successful rhythm control. As described previously, the gene closest to the 4q25 locus is PITX2, and loss of the cardiac isoform pitx2c can lead to failure to suppress a default pathway for the sinus node development of the pulmonary myocardium or the cardiomyocytes in the left atrium (129). More studies at the intersection with genetic data and detailed information of AF treatment are needed before genetically tailored treatment will be part of the guidelines.
The predictive ability of the three most strongly associated AF susceptibility loci in relation to AF recurrence after successful electrical cardioversion has been evaluated in a cohort of 184 individuals in the Vanderbilt AF registry. One hundred sixty-two individuals underwent successful electrical cardioverson, of which 108 individuals had AF recurrence. A genetic variant at chromosome 4q25 (rs2200733) was significantly associated with AF recurrence after electrical cardioversion (130). The outcome of electrical cardioversion of AF may be genetically predisposed; however, more and well-powered studies are needed to establish true associations. Furthermore, the biological pathways of established associations should be investigated to elucidate the mechanisms of AF.
Cardiac Catheter Ablation
Another treatment option for AF is radiofrequency catheter ablation. The use of genetic variants to predict AF catheter ablation outcome may be a promising clinical tool for the selection of patients for this procedure in the future. In 2013, Shoemaker et al. investigated 311 individuals in the Vanderbilt AF registry, of which 238 underwent only one ablation and 73 underwent two or more (131). Recurrence of AF was defined as AF, atrial flutter, or atrial tachycardia lasting more than 30 s and occuring in the blanking period of 3 months postablation. Of the 378 catheter-based ablations, 200 (53%) AF/atrial flutter recurrences were observed. The AF risk allele, rs2200733, at the 4q25 locus predicted shorter recurrence-free time after AF ablation.
In 2015, Shoemaker et al. explored the three genetic loci related to PITX2, KCNN3, and ZFHX3 that have been consistently been strongly associated with AF (132). In a combined cohort of Vanderbilt University, Heart Center Leipzig, and Massachusetts General hospital, 991 individuals with first-time AF ablation were analyzed. AF recurrence rate, defined as AF, atrial flutter, or atrial tachycardia lasting more than 30 s and occuring after 3 months postablation, was 42% during 12 months of follow-up. However, only the genetic variant rs2200733 at 4q25 predicted increased risk of recurrence.
Park et al. performed several analyses in the Korean Yonsei AF ablation population. They found that the genetic variant rs2106216 at the ZFHX3 locus on chromosome 16q22 was independently associated with a good response after AF ablation, but no association was found with the PITX locus. So far, the findings of Park et al. have not been replicated. The difference in ethnicity, patient characteristics, and the consistenty in ablation technique and relatively small sample size may explain the different results with previous studies (131–133). Recently, a polygenic risk score for AF was constructed with four genetic variants, rs1448818, rs2200733, rs6843082, and rs6838973, from the PITX2 locus and one from the ZFHX3 locus, rs2106261, to predict AF recurrence after ablation in another Korean population (134) from the Seoul National University Hospital and Korea University Guro Hospital. During 23 months of follow-up in 746 individuals (74% men, mean age = 59 ± 11 years), 168 (22.5%) had AF recurrence after AF catheter ablation. Increased polygenic risk for AF was strongly associated with AF recurrence; Individuals carrying 7 to 10 risk alleles of the 5 AF risk variants (rs1448818, rs2200733, rs6843082, rs6838973, and rs2106261) had a 2.66-fold increased risk of recurrence compared to the lowest risk group, where individuals carried zero to three risk alleles.
Large, well-phenotyped populations are necessary to discover new genetic variants associated with outcomes of AF ablation, e.g., a large-scale AF ablation GWAS may expose more common genetic variants related to AF recurrence after ablation. However, even when associations of genetic variants with AF ablation are established, underlying biological pathways leading to AF recurrence after ablation are still unknown. In aggregate, the current literature does not support the use of polygenic risk scores to guide AF management.
Patient Specific Management—Where Do We Go From Here?
The complex biology of AF and AF heritability has been increasingly elucidated in the era of AF genetics research. Nevertheless, our knowledge of the biologic mechanisms underlying the genetic associations is still limited. The translation of genetic information into clinical practice is difficult and far from ready to be implemented. So where do we go from here (Figure 3)?
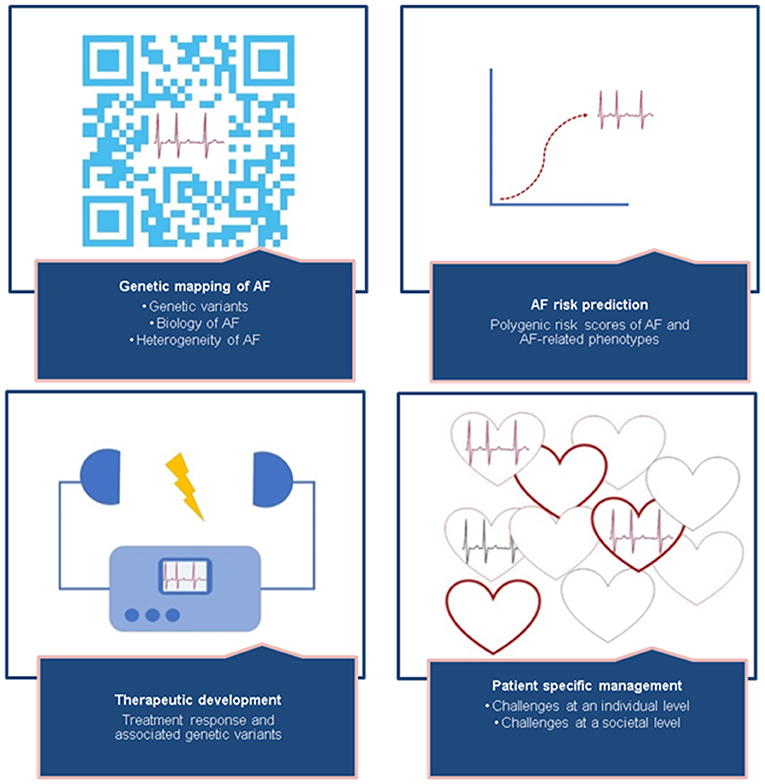
Figure 3. The missing heritability of AF can be revealed by focusing on mapping the polygenic structure of AF, improving risk prediction, therapeutic development, and patient-specific management. Details of suggested studies are described in Table 3. Future perspectives—translating AF genetics into clinical practice.
First, we have to unravel the missing heritability of AF. Although genetic mapping of AF is still ongoing, the discovery of novel genetic variants has made great leaps in the past decade, revealing 134 common genetic variants for AF. Undiscovered common genetic variants are still part of the missing heritability, and therefore, the search in AF GWAS studies should be continued. Efforts are also underway to pursue larger whole-genome sequencing studies, which may identify common and rare genetic variants for AF. Another important avenue is for more studies to focus on functional characterization of genetic variants, through expression quantitative trait loci (eQTL) analyses, proteomic, transcriptomic, metabolomic, cell and animal experiments, as well as further large-scale population-based studies revealing gene–gene and gene–environment interactions will expand the knowledge of the biology underlying AF.
We need to better understand the mechanisms underlying the complex pleiotropic relationship between AF and AF-related phenotypes. AF is known to be associated with multiple clinical risk factors, which may interact with or be the cause of AF. Well-powered Mendelian randomization analyses of AF and AF-related phenotypes, including endophenotypes such as ECG markers, may elucidate causal relations. Phenome-wide association studies (PheWAS) can be performed to identify associations between a genetic variant and multiple phenotypes, revealing novel genotype–phenotype associations.
Similarly to clinical AF risk prediction models, such as the CHARGE-AF risk score (135), we anticipate that polygenic risk scores for AF will be used in daily clinical practice in the future to guide prevention and treatment of AF. Polygenic risk scores may be used to reveal AF, but also AF-related phenotypes, like stroke, may attribute to AF mechanisms. Polygenic risk scores are a hot topic and have been increasingly performed in several fields of medical research. It is essential to view the clinical applicability of polygenic risk scores in perspective; unlike other clinical biomarkers, the underlying biological pathways of the genetic variants combined in a risk score are still largely unknown. There has been a high hope that discovering genetic causes of AF would also lead to the development of new drugs to treat the arrhythmia. It has been suggested that selecting drug targets that have a known genetic association with its related phenotype doubles the likelihood of success in developing new drugs (136). Although in the past decades the number of studies of genetic variants for AF increased extensively, underlying biological pathways are still unclear. The process has so far been challenging when it comes to development of new drugs for AF, mainly due to the fact that many novel genetic loci for AF are not directly “druggable” but require years in the wet-lab for their functional effects to be mapped out. Identifying statistical associations between genetic variants and AF without knowing neither (I) which specific parts of the genetic loci carry the causal effects nor (II) the underlying biological mechanisms carried by the causal variants will not automatically increase our success in establishing new therapeutic targets. Although there are a few new and promising drugs emerging that have been developed partly due to supporting genetic evidence (39–42), the yield of genetically identified drug targets has been disappointing. We need to identify the causal entities in the AF loci identified and disentangle their biological mechanisms in order to improve the development of new drugs and to understand how to improve AF interventions.
The complex and heterogeneous nature of AF demands patient-specific management. The current clinical classification of AF is not optimal, as it basically relies on the frequency and duration of AF episodes, and not underlying disease mechanisms, comorbidity, or daily function. Hence, the classification does not reflect the phenotypic and genotypic spectrum of AF, nor the fundamental difference between monogenic and polygenic AF forms. In association with disease, rare genetic variants tend to have a large effect and may represent monogenic disease, which has shown to be the case in several reports of familial AF (137). In families with known monogenic causes of AF, disease risk may be more easily predictable, depending on the penetrance, and it will potentially be possible to directly target the single gene or biological pathway involved in disease development. Nevertheless, individually targeted treatment of monogenic AF may be challenging due to the heterogenic nature of monogenic AF—almost no family carries the same genetic variant as the cause of their disease. Historically, monogenic variants leading to familial AF are involved in electrical abnormality. However, as we have discussed, more recent studies of familial AF have also revealed variants related to structural changes. In contrast, more common AF phenotypes are more likely to be multifactorial and polygenic, with multiple common genetic variants, as well as environmental factors, contributing to complex disease development, thus challenging both targeted treatment and estimation of disease risk. Although monogenic AF represents only a minority of AF cases, genetic information derived from this group provides valuable knowledge for interpreting data derived from GWAS and high-throughput sequencing and in the search for targeted treatment for more common forms of AF. Through increased insight into genetic causality and the various biological pathways implicated, it will open up for better classification of AF, thereby providing better opportunities for risk assessment and precision medicine for all types of AF. We have described a range of studies highlighting the possibility of individualized risk assessment, based on both genetic and clinical risk, and implicated the importance of addressing modifiable clinical risk factors, such as hypertension and obesity to prevent or delay the onset of AF. Potential challenges in clinical practice will be how to convey the concept of genetic risk to the individual patient and tackling the potential reverse effect on behavior when a patient gets insight into her or his own genetic risk. Another challenge can be the cost of implementing genetic screening of AF globally. However, potential reductions in the high lifetime risk of AF and costs of only partially effective treatments may contribute to a positive cost–benefit ratio of the introduction of genetic screening for AF. Overall, better understanding of the genetics of AF will hopefully provide better tools to prevent, detect, and treat AF, thus reducing the impact of the disease both at an individual level and in a public healthcare perspective (Table 3).

Table 3. Knowledge gaps in the genetics of AF.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
SK was supported by a Ph.D. grant from Vestre Viken Hospital Trust. IC was supported by a mobility grant from the Research Council of Norway (240149/F20).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. (2014) 129:837–47. doi: 10.1161/circulationaha.113.005119
2. Weng LC, Choi SH, Klarin D, Smith JG, Loh PR, Chaffin M, et al. Heritability of atrial fibrillation. Circ Cardiovasc Genet. (2017) 10:e001838. doi: 10.1161/circgenetics.117.001838
3. Camm AJ, Kirchhof P, Lip GY, Schotten U, Savelieva I, Ernst S, et al. Guidelines for the management of atrial fibrillation: the task force for the management of atrial fibrillation of the European Society of Cardiology (ESC). Eur Heart J. (2010) 31:2369–429. doi: 10.1093/eurheartj/ehq278
4. Vermond RA, Geelhoed B, Verweij N, Tieleman RG, Van der Harst P, Hillege HL, et al. Incidence of atrial fibrillation and relationship with cardiovascular events, heart failure, and mortality: a community-based study from the Netherlands. J Am Coll Cardiol. (2015) 66:1000–7. doi: 10.1016/j.jacc.2015.06.1314
5. Fox CS, Parise H, D'Agostino RB Sr., Lloyd-Jones DM, Vasan RS, Wang TJ, et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. (2004) 291:2851–5. doi: 10.1001/jama.291.23.2851
6. Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, et al. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. (2009) 2:378–83. doi: 10.1161/circep.108.786665
7. Bapat A, Anderson CD, Ellinor PT, Lubitz SA. Genomic basis of atrial fibrillation. Heart. (2018) 104:201–6. doi: 10.1136/heartjnl-2016-311027
8. Wolff L. Familial auricular fibrillation. N Engl J Med. (1943) 229:396–8. doi: 10.1056/nejm194309022291002
9. Darbar D, Herron KJ, Ballew JD, Jahangir A, Gersh BJ, Shen WK, et al. Familial atrial fibrillation is a genetically heterogeneous disorder. J Am Coll Cardiol. (2003) 41:2185–92. doi: 10.1016/s0735-1097(03)00465-0
10. Ellinor PT, Yoerger DM, Ruskin JN, MacRae CA. Familial aggregation in lone atrial fibrillation. Hum Genet. (2005) 118:179–84. doi: 10.1007/s00439-005-0034-8
11. Arnar DO, Thorvaldsson S, Manolio TA, Thorgeirsson G, Kristjansson K, Hakonarson H, et al. Familial aggregation of atrial fibrillation in Iceland. Eur Heart J. (2006) 27:708–12. doi: 10.1093/eurheartj/ehi727
12. Marcus GM, Smith LM, Vittinghoff E, Tseng ZH, Badhwar N, Lee BK, et al. A first-degree family history in lone atrial fibrillation patients. Heart Rhythm. (2008) 5:826–30. doi: 10.1016/j.hrthm.2008.02.016
13. Lubitz SA, Yin X, Fontes JD, Magnani JW, Rienstra M, Pai M, et al. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA. (2010) 304:2263–9. doi: 10.1001/jama.2010.1690
14. Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. (2003) 299(5604):251–4. doi: 10.1126/science.1077771
15. Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, et al. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur J Hum Genet. (2013) 21:918–28. doi: 10.1038/ejhg.2012.283
16. Risgaard B, Jabbari R, Refsgaard L, Holst AG, Haunso S, Sadjadieh A, et al. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet. (2013) 84:489–95. doi: 10.1111/cge.12126
17. Weeke P, Denny JC, Basterache L, Shaffer C, Bowton E, Ingram C, et al. Examining rare and low-frequency genetic variants previously associated with lone or familial forms of atrial fibrillation in an electronic medical record system: a cautionary note. Circ Cardiovasc Genet. (2015) 8:58–63. doi: 10.1161/circgenetics.114.000718
18. Christophersen IE, Ellinor PT. Genetics of atrial fibrillation: from families to genomes. J Hum Genet. (2016) 61:61–70. doi: 10.1038/jhg.2015.44
19. Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, Tsunoda T, et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet. (2002) 32:650–4. doi: 10.1038/ng1047
20. Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sigurdsson A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. (2007) 448:353–7. doi: 10.1038/nature06007
21. Paludan-Muller C, Svendsen JH, Olesen MS. The role of common genetic variants in atrial fibrillation. J Electrocardiol. (2016) 49:864–70. doi: 10.1016/j.jelectrocard.2016.08.012
22. Kaab S, Darbar D, van Noord C, Dupuis J, Pfeufer A, Newton-Cheh C, et al. Large scale replication and meta-analysis of variants on chromosome 4q25 associated with atrial fibrillation. Eur Heart J. (2009) 30:813–9. doi: 10.1093/eurheartj/ehn578
23. Shi L, Li C, Wang C, Xia Y, Wu G, Wang F, et al. Assessment of association of rs2200733 on chromosome 4q25 with atrial fibrillation and ischemic stroke in a Chinese Han population. Hum Genet. (2009) 126:843–9. doi: 10.1007/s00439-009-0737-3
24. Gudbjartsson DF, Holm H, Gretarsdottir S, Thorleifsson G, Walters GB, Thorgeirsson G, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. (2009) 41:876–8. doi: 10.1038/ng.417
25. Benjamin EJ, Rice KM, Arking DE, Pfeufer A, van Noord C, Smith AV, et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. (2009) 41:879–81. doi: 10.1038/ng.416
26. Huang Y, Wang C, Yao Y, Zuo X, Chen S, Xu C, et al. Molecular basis of gene–gene interaction: cyclic cross-regulation of gene expression and post-GWAS gene–gene interaction involved in atrial fibrillation. PLoS Genet. (2015) 11:e1005393. doi: 10.1371/journal.pgen.1005393
27. Mikhailov AT, Torrado M. Interplay between cardiac transcription factors and non-coding RNAs in predisposing to atrial fibrillation. J Mol Med (Berl). (2018) 96: 601–10. doi: 10.1007/s00109-018-1647-4
28. Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. (2012) 44:670–5. doi: 10.1038/ng.2261
29. Tucker NR, Dolmatova EV, Lin H, Cooper RR, Ye J, Hucker WJ, et al. Diminished PRRX1 expression is associated with increased risk of atrial fibrillation and shortening of the cardiac action potential. Circ Cardiovasc Genet. (2017) 10: e001902. doi: 10.1161/circgenetics.117.001902
30. Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. (2010) 42:240–4. doi: 10.1038/ng.537
31. Sinner MF, Tucker NR, Lunetta KL, Ozaki K, Smith JG, Trompet S, et al. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation. (2014) 130:1225–35. doi: 10.1161/circulationaha.114.009892
32. Christophersen IE, Rienstra M, Roselli C, Yin X, Geelhoed B, Barnard J, et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. (2017) 49:946–52. doi: 10.1038/ng.3843
33. Low SK, Takahashi A, Ebana Y, Ozaki K, Christophersen IE, Ellinor PT, et al. Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat Genet. (2017) 49:953–8. doi: 10.1038/ng.3842
34. Lee JY, Kim TH, Yang PS, Lim HE, Choi EK, Shim J, et al. Korean atrial fibrillation network genome-wide association study for early-onset atrial fibrillation identifies novel susceptibility loci. Eur Heart J. (2017) 38:2586–94. doi: 10.1093/eurheartj/ehx213
35. Nielsen JB, Fritsche LG, Zhou W, Teslovich TM, Holmen OL, Gustafsson S, et al. Genome-wide study of atrial fibrillation identifies seven risk loci and highlights biological pathways and regulatory elements involved in cardiac development. Am J Hum Genet. (2018) 102:103–15. doi: 10.1016/j.ajhg.2017.12.003
36. Roselli C, Chaffin MD, Weng LC, Aeschbacher S, Ahlberg G, Albert CM, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. (2018) 50:1225–33. doi: 10.1038/s41588-018-0133-9
37. Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. (2018) 50:1234–9. doi: 10.1038/s41588-018-0171-3
38. Peng J, Raddatz K, Molkentin JD, Wu Y, Labeit S, Granzier H, et al. Cardiac hypertrophy and reduced contractility in hearts deficient in the titin kinase region. Circulation. (2007) 115:743–51. doi: 10.1161/circulationaha.106.645499
39. Haugaard MM, Hesselkilde EZ, Pehrson S, Carstensen H, Flethoj M, Praestegaard KF, et al. Pharmacologic inhibition of small-conductance calcium-activated potassium (SK) channels by NS8593 reveals atrial antiarrhythmic potential in horses. Heart Rhythm. (2015) 12:825–35. doi: 10.1016/j.hrthm.2014.12.028
40. Diness JG, Skibsbye L, Simo-Vicens R, Santos JL, Lundegaard P, Citerni C, et al. Termination of vernakalant-resistant atrial fibrillation by inhibition of small-conductance Ca2+-activated K+ channels in pigs. Circ Arrhythm Electrophysiol. (2017) 10:e005125. doi: 10.1161/circep.117.005125
41. Simo-Vicens R, Kirchhoff JE, Dolce B, Abildgaard L, Speerschneider T, Sorensen US, et al. A new negative allosteric modulator, AP14145, for the study of small conductance calcium-activated potassium (KCa2) channels. Br J Pharmacol. (2017) 174:4396–408. doi: 10.1111/bph.14043
42. Simo-Vicens R, Sauter DRP, Grunnet M, Diness JG, Bentzen BH. Effect of antiarrhythmic drugs on small conductance calcium-activated potassium channels. Eur J Pharmacol. (2017) 803:118–23. doi: 10.1016/j.ejphar.2017.03.039
43. Nishino J, Ochi H, Kochi Y, Tsunoda T, Matsui S. Sample size for successful genome-wide association study of major depressive disorder. Front Genet. (2018) 9:227. doi: 10.3389/fgene.2018.00227
44. Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. (2012) 90:7–24. doi: 10.1016/j.ajhg.2011.11.029
45. Ball RD. Designing a GWAS: power, sample size, and data structure. Methods Mol Biol. (2013) 1019:37–98. doi: 10.1007/978-1-62703-447-0_3
46. Nelson SC, Doheny KF, Pugh EW, Romm JM, Ling H, Laurie CA, et al. Imputation-based genomic coverage assessments of current human genotyping arrays. G3 (Bethesda). (2013) 3:1795–807. doi: 10.1534/g3.113.007161
47. Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet. (2014) 15:335–46. doi: 10.1038/nrg3706
48. Wang Y, Li Y, Hao M, Liu X, Zhang M, Wang J, et al. Robust reference powered association test of genome-wide association studies. Front Genet. (2019) 10:319. doi: 10.3389/fgene.2019.00319
49. Nadadur RD, Broman MT, Boukens B, Mazurek SR, Yang X, van den Boogaard M, et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci Transl Med. (2016) 8:354ra115. doi: 10.1126/scitranslmed.aaf4891
50. Lin H, Mueller-Nurasyid M, Smith AV, Arking DE, Barnard J, Bartz TM, et al. Gene–gene interaction analyses for atrial fibrillation. Sci Rep. (2016) 6:35371. doi: 10.1038/srep35371
51. Parvez B, Darbar D. The “missing” link in atrial fibrillation heritability. J Electrocardiol. (2011) 44:641–4. doi: 10.1016/j.jelectrocard.2011.07.027
52. Lubitz SA, Ellinor PT. Next-generation sequencing for the diagnosis of cardiac arrhythmia syndromes. Heart Rhythm. (2015) 12:1062–70. doi: 10.1016/j.hrthm.2015.01.011
53. Weeke P, Muhammad R, Delaney JT, Shaffer C, Mosley JD, Blair M, et al. Whole-exome sequencing in familial atrial fibrillation. Eur Heart J. (2014) 35:2477–83. doi: 10.1093/eurheartj/ehu156
54. Tsai CT, Hsieh CS, Chang SN, Chuang EY, Juang JM, Lin LY, et al. Next-generation sequencing of nine atrial fibrillation candidate genes identified novel de novo mutations in patients with extreme trait of atrial fibrillation. J Med Genet. (2015) 52:28–36. doi: 10.1136/jmedgenet-2014-102618
55. Orr N, Arnaout R, Gula LJ, Spears DA, Leong-Sit P, Li Q, et al. A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun. (2016) 7:11303. doi: 10.1038/ncomms11303
56. Zhao J, Yao H, Li Z, Wang L, Liu G, Wang DW, et al. A novel nonsense mutation in LMNA gene identified by exome sequencing in an atrial fibrillation family. Eur J Med Genet. (2016) 59:396–400. doi: 10.1016/j.ejmg.2016.06.006
57. Lieve KV, Verkerk AO, Podliesna S, van der Werf C, Tanck MW, Hofman N, et al. Gain-of-function mutation in SCN5A causes ventricular arrhythmias and early onset atrial fibrillation. Int J Cardiol. (2017) 236:187–93. doi: 10.1016/j.ijcard.2017.01.113
58. Tucker NR, Mahida S, Ye J, Abraham EJ, Mina JA, Parsons VA, et al. Gain-of-function mutations in GATA6 lead to atrial fibrillation. Heart Rhythm. (2017) 14:284–91. doi: 10.1016/j.hrthm.2016.10.014
59. Ahlberg G, Refsgaard L, Lundegaard PR, Andreasen L, Ranthe MF, Linscheid N, et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun. (2018) 9:4316. doi: 10.1038/s41467-018-06618-y
60. Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet. (2015) 47:435–44. doi: 10.1038/ng.3247
61. Lubitz SA, Brody JA, Bihlmeyer NA, Roselli C, Weng LC, Christophersen IE, et al. Whole exome sequencing in atrial fibrillation. PLoS Genet. (2016) 12:e1006284. doi: 10.1371/journal.pgen.1006284
62. Choi SH, Weng LC, Roselli C, Lin H, Haggerty CM, Shoemaker MB, et al. Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA. (2018) 320:2354–64. doi: 10.1001/jama.2018.18179
63. Peng W, Li M, Li H, Tang K, Zhuang J, Zhang J, et al. Dysfunction of myosin light-chain 4 (MYL4) leads to heritable atrial cardiomyopathy with electrical, contractile, and structural components: evidence from genetically-engineered rats. J Am Heart Assoc. (2017) 6:e007030. doi: 10.1161/jaha.117.007030
64. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. (1999) 341:1715–24. doi: 10.1056/nejm199912023412302
65. Worman HJ. Nuclear lamins and laminopathies. J Pathol. (2012) 226:316–25. doi: 10.1002/path.2999
66. Marreddy Cheedipudi S, Matkovich SJ, Coarfa C, Hu X, Robertson MJ, Sweet ME, et al. Genomic reorganization of lamin-associated domains in cardiac myocytes is associated with differential gene expression and DNA methylation in human dilated cardiomyopathy. Circ Res. (2019) 124:1198–1213. doi: 10.1161/circresaha.118.314177
67. Maitra M, Koenig SN, Srivastava D, Garg V. Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatr Res. (2010) 68:281–5. doi: 10.1203/00006450-201011001-0054910.1203/PDR.0b013e3181ed17e4
68. Wang J, Luo XJ, Xin YF, Liu Y, Liu ZM, Wang Q, et al. Novel GATA6 mutations associated with congenital ventricular septal defect or tetralogy of fallot. DNA Cell Biol. (2012) 31:1610–7. doi: 10.1089/dna.2012.1814
69. Li J, Liu WD, Yang ZL, Yang YQ. Novel GATA6 loss-of-function mutation responsible for familial atrial fibrillation. Int J Mol Med. (2012) 30:783–90. doi: 10.3892/ijmm.2012.1068
70. Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu YJ, et al. GATA6 loss-of-function mutation in atrial fibrillation. Eur J Med Genet. (2012) 55:520–6. doi: 10.1016/j.ejmg.2012.06.007
71. Rasmussen PV, Nielsen JB, Skov MW, Pietersen A, Graff C, Lind B, et al. Electrocardiographic PR interval duration and cardiovascular risk: results from the Copenhagen ECG Study. Can J Cardiol. (2017) 33:674–81. doi: 10.1016/j.cjca.2017.02.015
72. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. (2012) 366:619–28. doi: 10.1056/NEJMoa1110186
73. Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. (2015) 7:270ra276. doi: 10.1126/scitranslmed.3010134
74. Peters S, Kumar S, Elliott P, Kalman JM, Fatkin D. Arrhythmic genotypes in familial dilated cardiomyopathy: implications for genetic testing and clinical management. Heart Lung Circ. (2019) 28:31–8. doi: 10.1016/j.hlc.2018.09.010
75. Nattel S. Close connections between contraction and rhythm: a new genetic cause of atrial fibrillation/cardiomyopathy and what it can teach us. Eur Heart J. (2017) 38:35–7. doi: 10.1093/eurheartj/ehw457
76. Smith GD, Ebrahim S. ‘Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. (2003) 32:1–22. doi: 10.1093/ije/dyg070
77. Burgess S, Labrecque JA. Mendelian randomization with a binary exposure variable: interpretation and presentation of causal estimates. Eur J Epidemiol. (2018) 33:947–52. doi: 10.1007/s10654-018-0424-6
78. Wang TJ, Parise H, Levy D, D'Agostino R.B. Sr., Wolf PA, Vasan RS, et al. Obesity and the risk of new-onset atrial fibrillation. JAMA. (2004) 292:2471–7. doi: 10.1001/jama.292.20.2471
79. Chatterjee NA, Giulianini F, Geelhoed B, Lunetta KL, Misialek JR, Niemeijer MN, et al. Genetic obesity and the risk of atrial fibrillation: causal estimates from Mendelian randomization. Circulation. (2017) 135:741–54. doi: 10.1161/circulationaha.116.024921
80. Tikkanen E, Gustafsson S, Knowles JW, Perez M, Burgess S, Ingelsson E. Body composition and atrial fibrillation: a Mendelian randomization study. Eur Heart J. (2019) 40:1277–82. doi: 10.1093/eurheartj/ehz003
81. Rosenberg MA, Patton KK, Sotoodehnia N, Karas MG, Kizer JR, Zimetbaum PJ, et al. The impact of height on the risk of atrial fibrillation: the Cardiovascular Health Study. Eur Heart J. (2012) 33:2709–17. doi: 10.1093/eurheartj/ehs301
82. Estrada K, Krawczak M, Schreiber S, van Duijn K, Stolk L, van Meurs JB, et al. A genome-wide association study of northwestern Europeans involves the C-type natriuretic peptide signaling pathway in the etiology of human height variation. Hum Mol Genet. (2009) 18:3516–24. doi: 10.1093/hmg/ddp296
83. Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. (2010) 42:565–9. doi: 10.1038/ng.608
84. Liu F, Hendriks AE, Ralf A, Boot AM, Benyi E, Savendahl L, et al. Common DNA variants predict tall stature in Europeans. Hum Genet. (2014) 133:587–97. doi: 10.1007/s00439-013-1394-0
85. Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. (2014) 46:1173–86. doi: 10.1038/ng.3097
86. Zhong K, Zhu G, Jing X, Hendriks AEJ, Drop SLS, Ikram MA, et al. Genome-wide compound heterozygote analysis highlights alleles associated with adult height in Europeans. Hum Genet. (2017) 136:1407–17. doi: 10.1007/s00439-017-1842-3
87. Rosenberg MA, Kaplan RC, Siscovick DS, Psaty BM, Heckbert SR, Newton-Cheh C, et al. Genetic variants related to height and risk of atrial fibrillation: the Cardiovascular Health Study. Am J Epidemiol. (2014) 180:215–22. doi: 10.1093/aje/kwu126
88. Abhayaratna WP, Seward JB, Appleton CP, Douglas PS, Oh JK, Tajik AJ, et al. Left atrial size: physiologic determinants and clinical applications. J Am Coll Cardiol. (2006) 47:2357–63. doi: 10.1016/j.jacc.2006.02.048
89. Rosengren A, Hauptman PJ, Lappas G, Olsson L, Wilhelmsen L, Swedberg K. Big men and atrial fibrillation: effects of body size and weight gain on risk of atrial fibrillation in men. Eur Heart J. (2009) 30:1113–20. doi: 10.1093/eurheartj/ehp076
90. Conen D, Adam M, Roche F, Barthelemy JC, Felber Dietrich D, Imboden M, et al. Premature atrial contractions in the general population: frequency and risk factors. Circulation. (2012) 126:2302–8. doi: 10.1161/circulationaha.112.112300
91. Dewland TA, Vittinghoff E, Mandyam MC, Heckbert SR, Siscovick DS, Stein PK, et al. Atrial ectopy as a predictor of incident atrial fibrillation: a cohort study. Ann Intern Med. (2013) 159:721–8. doi: 10.7326/0003-4819-159-11-201312030-00004
92. Chun KJ, Hwang JK, Park SJ, On YK, Kim JS, Park KM. Electrical PR interval variation predicts new occurrence of atrial fibrillation in patients with frequent premature atrial contractions. Medicine. (2016) 95:e3249. doi: 10.1097/md.0000000000003249
93. Kofler T, Theriault S, Bossard M, Aeschbacher S, Bernet S, Krisai P, et al. Relationships of measured and genetically determined height with the cardiac conduction system in healthy adults. Circ Arrhythm Electrophysiol. (2017) 10:e004735. doi: 10.1161/circep.116.004735
94. Zoledziewska M, Sidore C, Chiang CWK, Sanna S, Mulas A, Steri M, et al. Height-reducing variants and selection for short stature in Sardinia. Nat Genet. (2015) 47:1352–6. doi: 10.1038/ng.3403
95. Field Y, Boyle EA, Telis N, Gao Z, Gaulton KJ, Golan D, et al. Detection of human adaptation during the past 2000 years. Science. (2016) 354(6313):760–4. doi: 10.1126/science.aag0776
96. Tachmazidou I, Suveges D, Min JL, Ritchie GRS, Steinberg J, Walter K, et al. Whole-genome sequencing coupled to imputation discovers genetic signals for anthropometric traits. Am J Hum Genet. (2017) 100:865–84. doi: 10.1016/j.ajhg.2017.04.014
97. Berg JJ, Harpak A, Sinnott-Armstrong N, Joergensen AM, Mostafavi H, Field Y, et al. Reduced signal for polygenic adaptation of height in UK Biobank. Elife. (2019) 8:e39725. doi: 10.7554/eLife.39725
98. Selmer C, Olesen JB, Hansen ML, Lindhardsen J, Olsen AM, Madsen JC, et al. The spectrum of thyroid disease and risk of new onset atrial fibrillation: a large population cohort study. BMJ. (2012) 345:e7895. doi: 10.1136/bmj.e7895
99. Dekkers OM, Horvath-Puho E, Cannegieter SC, Vandenbroucke JP, Sorensen HT, Jorgensen JO. Acute cardiovascular events and all-cause mortality in patients with hyperthyroidism: a population-based cohort study. Eur J Endocrinol. (2017) 176:1–9. doi: 10.1530/eje-16-0576
100. Ellervik C, Roselli C, Christophersen IE, Alonso A, Pietzner M, Sitlani CM, et al. Assessment of the relationship between genetic determinants of thyroid function and atrial fibrillation: a Mendelian randomization study. JAMA Cardiol. (2019) 4:144–152. doi: 10.1001/jamacardio.2018.4635
101. Guo Y, Tian Y, Wang H, Si Q, Wang Y, Lip GYH. Prevalence, incidence, and lifetime risk of atrial fibrillation in China: new insights into the global burden of atrial fibrillation. Chest. (2015) 147:109–19. doi: 10.1378/chest.14-0321
102. Zulkifly H, Lip GYH, Lane DA. Epidemiology of atrial fibrillation. Int J Clin Pract. (2018) 72:e13070. doi: 10.1111/ijcp.13070
103. Staerk L, Wang B, Preis SR, Larson MG, Lubitz SA, Ellinor PT, et al. Lifetime risk of atrial fibrillation according to optimal, borderline, or elevated levels of risk factors: cohort study based on longitudinal data from the Framingham Heart Study. BMJ. (2018) 361:k1453. doi: 10.1136/bmj.k1453
104. Weng LC, Preis SR, Hulme OL, Larson MG, Choi SH, Wang B, et al. Genetic predisposition, clinical risk factor burden, and lifetime risk of atrial fibrillation. Circulation. (2018) 137:1027–38. doi: 10.1161/circulationaha.117.031431
105. Lloyd-Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation. (2004) 110:1042–6. doi: 10.1161/01.cir.0000140263.20897.42
106. Heeringa J, van der Kuip DA, Hofman A, Kors JA, van Herpen G, Stricker BH, et al. Prevalence, incidence and lifetime risk of atrial fibrillation: the Rotterdam study. Eur Heart J. (2006) 27:949–53. doi: 10.1093/eurheartj/ehi825
107. Schnabel RB, Yin X, Gona P, Larson MG, Beiser AS, McManus DD, et al. 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet. (2015) 386(9989):154–62. doi: 10.1016/s0140-6736(14)61774-8
108. Staerk L, Sherer JA, Ko D, Benjamin EJ, Helm RH. Atrial fibrillation: epidemiology, pathophysiology, and clinical outcomes. Circ Res. (2017) 120:1501–17. doi: 10.1161/circresaha.117.309732
109. Kannel WB, Wolf PA, Benjamin EJ, Levy D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol. (1998) 82:2N−9N.
110. Said MA, Verweij N, van der Harst P. Associations of combined genetic and lifestyle risks with incident cardiovascular disease and diabetes in the UK Biobank Study. JAMA Cardiol. (2018) 3:693–702. doi: 10.1001/jamacardio.2018.1717
111. Lubitz SA, Yin X, Lin HJ, Kolek M, Smith JG, Trompet S, et al. Genetic risk prediction of atrial fibrillation. Circulation. (2017) 135:1311–20. doi: 10.1161/circulationaha.116.024143
112. Ehret GB, Lamparter D, Hoggart CJ, Whittaker JC, Beckmann JS, Kutalik Z. A multi-SNP locus-association method reveals a substantial fraction of the missing heritability. Am J Hum Genet. (2012) 91:863–71. doi: 10.1016/j.ajhg.2012.09.013
113. Gretarsdottir S, Thorleifsson G, Manolescu A, Styrkarsdottir U, Helgadottir A, Gschwendtner A, et al. Risk variants for atrial fibrillation on chromosome 4q25 associate with ischemic stroke. Ann Neurol. (2008) 64:402–9. doi: 10.1002/ana.21480
114. Tada H, Shiffman D, Smith JG, Sjogren M, Lubitz SA, Ellinor PT, et al. Twelve-single nucleotide polymorphism genetic risk score identifies individuals at increased risk for future atrial fibrillation and stroke. Stroke. (2014) 45:2856–62. doi: 10.1161/strokeaha.114.006072
115. No_authors_listed. Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med. (1994) 154:1449–57.
116. Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. (2001) 285:2370–5. doi: 10.1001/jama.285.18.2370
117. Benjamin EJ, Levy D, Vaziri SM, D'Agostino RB, Belanger AJ, Wolf PA. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA. (1994) 271:840–4.
118. De Ferrari GM, Klersy C, Ferrero P, Fantoni C, Salerno-Uriarte D, Manca L, et al. Atrial fibrillation in heart failure patients: prevalence in daily practice and effect on the severity of symptoms. Data from the ALPHA study registry. Eur J Heart Fail. (2007) 9:502–9. doi: 10.1016/j.ejheart.2006.10.021
119. Larson MG, Atwood LD, Benjamin EJ, Cupples LA, D'Agostino RB Sr., Fox CS, et al. Framingham Heart Study 100K project: genome-wide associations for cardiovascular disease outcomes. BMC Med Genet. (2007) 8 (Suppl 1):S5. doi: 10.1186/1471-2350-8-s1-s5
120. Cappola TP, Li M, He J, Ky B, Gilmore J, Qu L, et al. Common variants in HSPB7 and FRMD4B associated with advanced heart failure. Circ Cardiovasc Genet. (2010) 3:147–54. doi: 10.1161/circgenetics.109.898395
121. Smith NL, Felix JF, Morrison AC, Demissie S, Glazer NL, Loehr LR, et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. (2010) 3:256–66. doi: 10.1161/circgenetics.109.895763
122. Villard E, Perret C, Gary F, Proust C, Dilanian G, Hengstenberg C, et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. (2011) 32:1065–76. doi: 10.1093/eurheartj/ehr105
123. Meder B, Ruhle F, Weis T, Homuth G, Keller A, Franke J, et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J. (2014) 35:1069–77. doi: 10.1093/eurheartj/eht251
124. van der Ende MY, Said MA, van Veldhuisen DJ, Verweij N, van der Harst P. Genome-wide studies of heart failure and endophenotypes: lessons learned and future directions. Cardiovasc Res. (2018) 114:1209–25. doi: 10.1093/cvr/cvy083
125. Willis MS, Patterson C. Into the heart: the emerging role of the ubiquitin-proteasome system. J Mol Cell Cardiol. (2006) 41:567–79. doi: 10.1016/j.yjmcc.2006.07.015
126. Wohlschlaeger J, Sixt SU, Stoeppler T, Schmitz KJ, Levkau B, Tsagakis K, et al. Ventricular unloading is associated with increased 20s proteasome protein expression in the myocardium. J Heart Lung Transplant. (2010) 29:125–32. doi: 10.1016/j.healun.2009.07.022
127. Rau CD, Lusis AJ, Wang Y. Genetics of common forms of heart failure: challenges and potential solutions. Curr Opin Cardiol. (2015) 30:222–7. doi: 10.1097/hco.0000000000000160
128. Barrett TW, Self WH, Darbar D, Jenkins CA, Wasserman BS, Kassim NA, et al. Association of atrial fibrillation risk alleles and response to acute rate control therapy. Am J Emerg Med. (2016) 34:735–40. doi: 10.1016/j.ajem.2016.01.034
129. Parvez B, Vaglio J, Rowan S, Muhammad R, Kucera G, Stubblefield T, et al. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J Am Coll Cardiol. (2012) 60:539–45. doi: 10.1016/j.jacc.2012.01.070
130. Parvez B, Shoemaker MB, Muhammad R, Richardson R, Jiang L, Blair MA, et al. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm. (2013) 10:849–55. doi: 10.1016/j.hrthm.2013.02.018
131. Benjamin Shoemaker M, Muhammad R, Parvez B, White BW, Streur M, Song Y, et al. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation. Heart Rhythm. (2013) 10:394–400. doi: 10.1016/j.hrthm.2012.11.012
132. Shoemaker MB, Bollmann A, Lubitz SA, Ueberham L, Saini H, Montgomery J, et al. Common genetic variants and response to atrial fibrillation ablation. Circ Arrhythm Electrophysiol. (2015) 8:296–302. doi: 10.1161/circep.114.001909
133. Husser D, Adams V, Piorkowski C, Hindricks G, Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol. (2010) 55:747–53. doi: 10.1016/j.jacc.2009.11.041
134. Choe WS, Kang JH, Choi EK, Shin SY, Lubitz SA, Ellinor PT, et al. A genetic risk score for atrial fibrillation predicts the response to catheter ablation. Korean Circ J. (2019) 49:338–49. doi: 10.4070/kcj.2018.0161
135. Alonso A, Krijthe BP, Aspelund T, Stepas KA, Pencina MJ, Moser CB, et al. Simple risk model predicts incidence of atrial fibrillation in a racially and geographically diverse population: the CHARGE-AF consortium. J Am Heart Assoc. (2013) 2:e000102. doi: 10.1161/jaha.112.000102
136. Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nature Genetics. (2015) 47:856. doi: 10.1038/ng.3314
Keywords: atrial fibrillation, genetics, genome-wide association studies (GWAS), heritability, precision medicine, personalized medicine, risk factors, whole genome sequencing
Citation: Kalstø SM, Siland JE, Rienstra M and Christophersen IE (2019) Atrial Fibrillation Genetics Update: Toward Clinical Implementation. Front. Cardiovasc. Med. 6:127. doi: 10.3389/fcvm.2019.00127
Received: 12 June 2019; Accepted: 12 August 2019;
Published: 06 September 2019.
Edited by:
Valeria Novelli, Agostino Gemelli University Polyclinic, ItalyReviewed by:
Ville-Petteri Makinen, South Australian Health and Medical Research Institute (SAHMRI), AustraliaSteven Clive Greenway, University of Calgary, Canada
Copyright © 2019 Kalstø, Siland, Rienstra and Christophersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ingrid E. Christophersen, uxchia@vestreviken.no
†These authors have contributed equally to this work