 Denise Burtenshaw
Denise Burtenshaw Michael Kitching
Michael Kitching Eileen M. Redmond3
Eileen M. Redmond3 Paul A. Cahill
Paul A. Cahill- 1Vascular Biology & Therapeutics, School of Biotechnology, Dublin City University, Dublin, Ireland
- 2School of Chemistry, Dublin City University, Dublin, Ireland
- 3Department of Surgery, University of Rochester, Rochester, NY, United States
- 4Centre for Health Science, UHI Institute of Health Research and Innovation, Inverness, United Kingdom
Arteriosclerosis causes significant morbidity and mortality worldwide. Central to this process is the development of subclinical non-atherosclerotic intimal lesions before the appearance of pathologic intimal thickening and advanced atherosclerotic plaques. Intimal thickening is associated with several risk factors, including oxidative stress due to reactive oxygen species (ROS), inflammatory cytokines and lipid. The main ROS producing systems in-vivo are reduced nicotinamide dinucleotide phosphate (NADPH) oxidase (NOX). ROS effects are context specific. Exogenous ROS induces apoptosis and senescence, whereas intracellular ROS promotes stem cell differentiation, proliferation, and migration. Lineage tracing studies using murine models of subclinical atherosclerosis have revealed the contributory role of medial smooth muscle cells (SMCs), resident vascular stem cells, circulating bone-marrow progenitors and endothelial cells that undergo endothelial-mesenchymal-transition (EndMT). This review will address the putative physiological and patho-physiological roles of ROS in controlling vascular cell fate and ROS contribution to vascular regeneration and disease progression.
Introduction
Arteriosclerosis occurs when the arterial blood vessels that carry oxygen and nutrients from the heart to the rest of the body become thick and stiffen thereby restricting blood flow to vital organs (1). It is a common feature of aging while pulmonary hypertension, peripheral arterial disease (PAD), transplant arteriosclerosis and in-stent restenosis (ISR) following balloon angioplasty are all significant clinical outcomes peroxidation (2–5). Atherosclerosis is a specific type of arteriosclerosis and refers to the specific build-up of lipids, cholesterol and other substances in and on the artery wall forming a plaque which can further restrict blood flow (6). It is considered the main cause of cardiovascular disease and is characterized by the early development of subclinical atherosclerosis due to pathologic intimal thickening (PIT) within atherosclerotic-prone regions of the vasculature (7). Subclinical atherosclerosis is an early indicator of atherosclerotic burden and its reversal can prevent the progression to symptomatic cardiovascular disease (CVD) (1). For the purpose of this review, subclinical atherosclerosis refers to the early intimal thickening that occurs prior to the accumulation of lipid and early plaque formation (8).
Reactive oxygen species (ROS) are a class of highly reactive molecules derived from O2 metabolism (9). Members of the ROS family include: superoxide (O2−), alkoxyl radical (RO·), peroxyl radical (ROO·), hydroxyl radicals (OH·), peroxynitrate (ONOO−), hydrogen peroxide (H2O2), ozone (O3), and hypochlorus acid (HOCl). Physiological concentrations of ROS are important signaling molecules that maintain vascular homeostasis whereas excessive ROS production may result in oxidative stress leading to vascular disease progression. ROS maintain vascular cell homoeostasis by controlling the phenotype and fate of multiple cell types including endothelial cells (ECs), vascular smooth muscle cells (SMCs), adventitial cells, myeloid cells and resident stem/progenitor cells (10, 11).
Oxidative stress influences the onset and progression of subclinical atherosclerosis by inducing early endothelial cell activation, permeability changes to the endothelium, disruption of glycocalyx, activation of myeloid and progenitor stem cells leading to the eventual accumulation of vascular smooth muscle (SMCs)-like cells within the intima (12–14). This accumulation of cells promotes diffuse (DIT) and adaptive (AIT) intimal thickening and is considered an important nexus in the development of subclinical atherosclerosis. These cells may originate from (i) medial SMCs (15), (ii) resident vascular stem cells (16) (iii) circulating bone marrow-derived mesenchymal stem cells (17) and (iv) endothelial cells undergoing mesenchymal-stem-cell-transition (EndMT) (18). Concurrently, innate and adaptive immune cells that enter the vasculature may also participate in the pathology of hypertensive-induced arteriosclerosis by releasing several mediators including ROS that cause vascular damage leading to adaptive (AIT) and pathologic (PIT) intimal thickening (19). In this context, ROS affect resident myeloid cells that reside within intimal and/or adventitial layers of susceptible regions of arterial vessels that display low-grade inflammation (20–22). Upon exposure to hypercholesterolemia, these cells become laden with oxidized LDL and acquire foam cell morphology prior to the recruitment of monocytes that differentiate into macrophage foam cells (6).
Once thought to be nothing more than harmful by-products of cellular metabolism, it is now clear that low—moderate ROS levels contribute to cellular functions such as differentiation, migration, adhesion, senescence, growth and apoptosis (23). The main ROS producing systems in-vivo are reduced nicotinamide dinucleotide phosphate (NADPH) oxidase (NOX) (11), xanthine oxidase (XO) (24), the electron transport chain in the mitochondria (25), cytochrome P450 (26), lipoxygenases, heme oxygenase and cyclooxygenases (27), myeloperoxidase (28), monoamine oxidases (29) and uncoupled nitric oxide (NO) synthase (30). ROS can also be generated from exogenous sources such as UV light, air and water pollution, alcohol, tobacco smoke, transition and heavy metals, industrial solvents, pesticides, high temperature (31) (Figure 1). Table 1 lists the seven isoforms of NOX expressed in mammals. While, NOX represents the major source of vascular superoxide anion that generates oxidative stress (45), endothelial ROS is also generated in the mitochondria from the partial oxygen reduction to form superoxide and also participates in the activation of these cells following cholesterol loading (46). Similarly, macrophages produce elevated levels of mitochondrial ROS in a NOX-independent fashion (47).
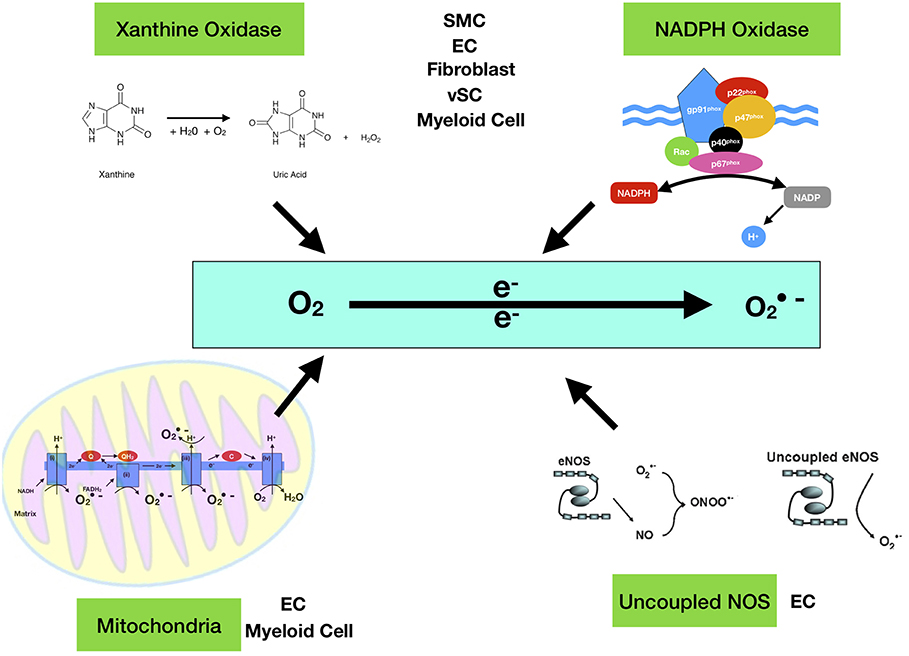
Figure 1. Enzymatic sources of superoxide anion (·O2−). The major enzymes responsible for ROS generation in the vasculature include mitochondria (mtROS), NAD(P)H oxidase, xanthine oxidase, and uncoupled NOS. NAD(P)H oxidase is a multi-subunit enzyme, comprising gp91phox (or its homologs, NOX1 and NOX4), p22phox, p47phox (or NOXO1), p67phox (or NOXA1), and p40phox. Smooth muscle cell (SMC), endothelial cell (EC), Myeloid Cell (monocytes and macrophages), vSC (vascular stem cell). The mitochondrial electron transport chain produces mtROS. Mitochondrial complexes I and II use electrons donated from NADH and FADH2 to reduce coenzyme Q during the process of oxidative phosphorylation (OXPHOS). Leakage of electrons at complex I and complex III from electron transport chains leads to partial reduction of oxygen to form superoxide [Quinol QH2, quinone Q and C cytochrome c].
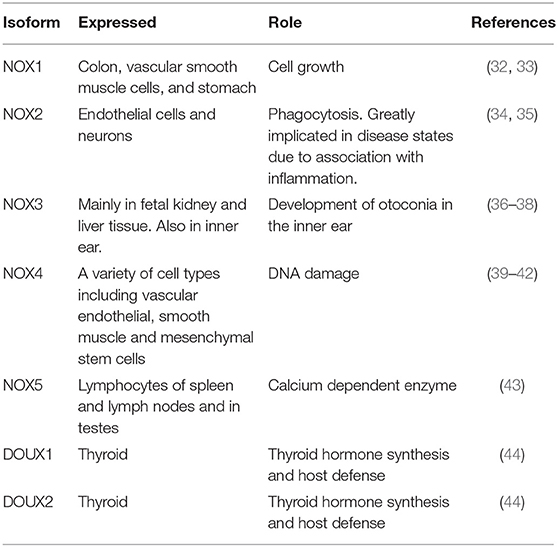
Table 1. Isoforms of NOX.
Initially O2− is formed from the reduction of molecular O2. O2− is the most pathologically relevant molecule due to its high chemical reactivity, therefore O2− requires rapid reduction to H2O2 by the enzyme superoxide dismutase (SOD) (48). H2O2 is thought to be the main ROS molecule involved in intracellular signaling. This reaction may also occur spontaneously in a process known as dismutation. O2− may react with H2O2 in the presence of iron (released from O2− oxidative damage to proteins containing FeS clusters) to generate damaging OH- radicals, or O2− may also react with NO to form ONOO− (31). ROS are important mediators and signal modifiers upon stimulation by growth factors (49, 50), cytokines (51), hypoxia (52), shear stress (53), and cyclic strain (54). In response, many important pathways are activated such as GPCR, Notch, Wnt-β-catenin, MAPK, JAK-STAT, NF-κB, and PI3K/AKT (55–60).
ROS acts as an intracellular signal through reversible oxidation of amino acid residues, most commonly cysteine (61). This induces a conformational change in the sensor protein and influences their function, stability, subcellular localization and protein-protein interaction. H2O2 is the most studied ROS mediator due to its stability and its ability to diffuse through the phospholipid bi-layer (62). H2O2 has been implicated in a variety of cellular processes (63), including proliferation and migration (64), differentiation (65), and apoptosis (66). O2− signaling is less understood due to its poor stability and the difficulty to specifically target O2− in vitro or in vivo (67). In spite of its low stability and poor diffusion, it can oxidize thiol groups of proteins in the immediate vicinity of where it was generated (68). O2− signaling has been associated with major epigenetic processes, including DNA methylation, histone methylation and histone acetylation (69). ROS also possess antimicrobial functions, important in phagocytosis and pathogen destruction (70).
Generation of ROS is tightly regulated by the ROS scavenging system, which are enzymes that neutralize ROS. These include SOD, catalase, heme-oxygenase-1 (HO-1), NADPH quinone reductase and, gamma-glutamylcysteine reductase (48). Oxidative stress is normally induced when the production of ROS overcomes the ROS scavenging system. This facilitates lipoprotein/phospholipid oxidation, protein denaturation, and DNA damage through free-radical-mediated chain reaction, primarily through the reduction of guanine residues to 8-oxoguanine (71). OH· radicals can also cause single/double strand breaks in DNA (71). The anti-oxidant defense response, primarily SOD, regulates ROS signaling by limiting the concentration of ROS to low or moderate levels, controlling the redox profile of the cell and ensure that ROS are localized close to their intended targets (70). SOD1 inhibition by tetrathiomolybdate increased intracellular O2− and H2O2 levels and attenuated growth factor mediated ERK1/2 signaling in endothelial and tumor cells (48). Glutathione peroxidase (GPx-1) has also an important anti-oxidant role in the generation of ROS. GPx-1 is inversely associated with CVD and important for maintenance of a normal level of GSH. It can also protect mitochondria against ROS-induced reoxygenation damage in vivo (72).
The overall consensus is that ROS production when not compensated for by scavenging endogenous antioxidants will lead to the rise of ROS beyond a “normal” or “physiological” threshold level. This results in a process termed “oxidative stress.” Intracellular ROS generation may be pathological or physiological (73). ROS is invariably generated from cellular metabolism or in response to various exogenous stimuli. While the main endogenous source of ROS is the electron transport chain of the mitochondria and cytosolic generation by NOX, other ROS sources are referred to as “professional” generators, capable of producing high levels of ROS in a spatial and temporal manner (74). NOX derived ROS has been implicated in cancer (75), diabetes (76), neurodegenerative disorders (77) and CVD (78).
Vascular Mitochondrial ROS (mtROS)
Mitochondria are unique in that they are not only a major source of ROS but are also particularly susceptible to oxidative damage by ROS. Consequently, mitochondria suffer oxidative damage with age that contributes to mitochondrial dysfunction (79). Under physiological conditions, mitochondrial metabolism results in the build-up of potentially damaging ROS which are neutralized by mitochondrial permeability transition pore (mPTP) openings that maintain healthy mitochondrial homeostasis. However, adaptive and maladaptive responses can occur that involve activation of mitochondrial channels such as mPTP and inner membrane anion channel (IMAC) resulting in intra- and intramitochondrial redox-environment changes leading to ROS release. Physiological levels of ROS produced in the mitochondria (mtROS) are critical components of downstream signaling pathways including those regulating immune responses and autophagy (59, 80).
Mitochondria have a four-layer structure, including outer mitochondrial membrane, intermembrane space, inner mitochondrial membrane, and matrix. Generation of mtROS occurs during the process of oxidative phosphorylation (OXPHOS) at the electron transport chain (ETC) located on the inner mitochondrial membrane. Five big protein complexes are involved in this process. These ETC complexes are named complex I (NADH dehydrogenase (ubiquinone), 45 protein subunits), complex II (succinate dehydrogenase, 4 protein subunits), complex III (ubiquinol-cytochrome c reductase, 10 protein subunits), complex IV (cytochrome c oxidase, 19 protein subunits), and complex V (ATP synthase, 19 protein subunits). Electrons donated from nicotine adenine dinucleotide (NADH) at complex I and flavin adenine dinucleotide (FADH2) at complex II pass through ETC and ultimately reduce O2 to water at complex IV. Positively charged protons (H+) are actively pumped from the mitochondrial matrix into the intermembrane space, resulting in the increased negative charges in the mitochondrial matrix and the upregulated positive charges in the intermembrane space, and thus creating a mitochondrial membrane potential (Δψm) across the inner mitochondrial membrane. This proton-motive force allows complex V - ATP synthase (ATP-ase) to generate ATP from adenosine diphosphate (ADP) and inorganic phosphate when protons re-enter the mitochondrial matrix through the complex V enzyme. However, the process of ETC is not perfect and leakage of electrons occurs at complex I and complex III resulting in partial reduction of oxygen to form superoxide (O2.−). It has been estimated that 0.2–2.0% of O2 consumed by mitochondria generates the superoxide (O2.−). There are three leakage events: complex I leaks O2.− toward the mitochondrial matrix, while complex III leaks O2.− toward both the intermembrane space and mitochondrial matrix (79) (Figure 1).
Overall, there are 11 sites of ROS production (superoxide and/or hydrogen peroxide) identified in mammalian mitochondria related to substrate metabolism, electron transport and oxidative phosphorylation (81). However, because mtROS and ATP production are both coupled to electron transport chain activity, it is unclear how mtROS is induced independently of ATP synthesis. Recent studies now suggest that mtROS is activated via unique calcium entry–mediated increase of proton leak and mitochondrial O2 reduction (46).
The regenerative cycle of mtROS formation and release is termed ROS-induced ROS release (RIRR). Reversible mPTP channel opening and associated ROS release constitutes an adaptive housekeeping function of potentially toxic levels of ROS (and Ca2+). At higher ROS levels, longer mPTP channel openings release a ROS burst leading to destruction of mitochondria, and if propagated from mitochondrion to mitochondrion, of the cell itself. The destructive function of RIRR serves a physiological role through removal of unwanted cells or damaged mitochondria. It may however also cause the pathological elimination of vital and essential mitochondria and cells. The adaptive release of sufficient ROS into the vicinity of mitochondria may also activate local pools of redox-sensitive enzymes involved in protective signaling pathways that limit ischemic damage to mitochondria and cells in that area. Maladaptive mPTP- or IMAC-related RIRR may also be playing a role in aging (79).
Collectively, both O2.− and H2O2 are considered the primary mtROS but have different fates. Given its electrophilic property and short half-life, O2.− undergoes radical-radical reaction with nitric oxide (NO) to form peroxynitrite (ONOO2.−) within mitochondria, a detrimental oxidant capable of induction of DNA damage, disruption of mitochondrial integrity, and irreversible modification of protein. In contrast, H2O2 is electrophobic and more stable and hence abundant within mitochondrion a (>100 times greater than that of O2.−) thereby rendering mitochondrial H2O2 an ideal signaling molecule in mammalian cells (63).
Importantly, ROS generation in the mitochondria appears to be an important aspect of ROS production for both endothelial cells (82) and intimal myeloid cells (primarily monocytes and macrophages) in atherosclerosis (83, 84).
Vascular NADPH Oxidase (NOX)
Vascular NADPH oxidases (NOXs) are ROS generating oxidases (85) Table 2. With the exception of NOX5, all NOX enzymes are heteroprotein transmembrane complexes with a core catalytic subunit and a number of regulatory subunits (NOX1, NOX2, NOX4, and NOX5 are expressed and functionally active in human vascular cells. In humans, NADPH oxidase had been thought to be a phagocyte specific enzyme (its catalytic unit: gp91phox) mediating bacterial killing by producing a burst of O2− (86). The p22phox, a membrane protein, forms a heterodimer with gp91phox, thereby stabilizing gp91phox and enhancing its O2−-producing activity. The ubiquitous expression of p22phox in non-phagocytic cells led to the identification of NOX1, a homolog of gp91phox, in non-phagocytic cells and facilitated the discovery other NOX proteins (45) (Figure 2).
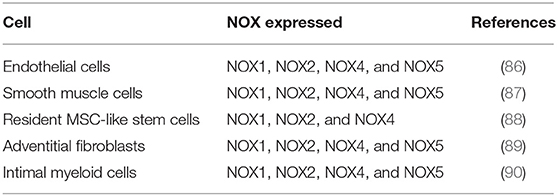
Table 2. Vascular NOX isoforms.
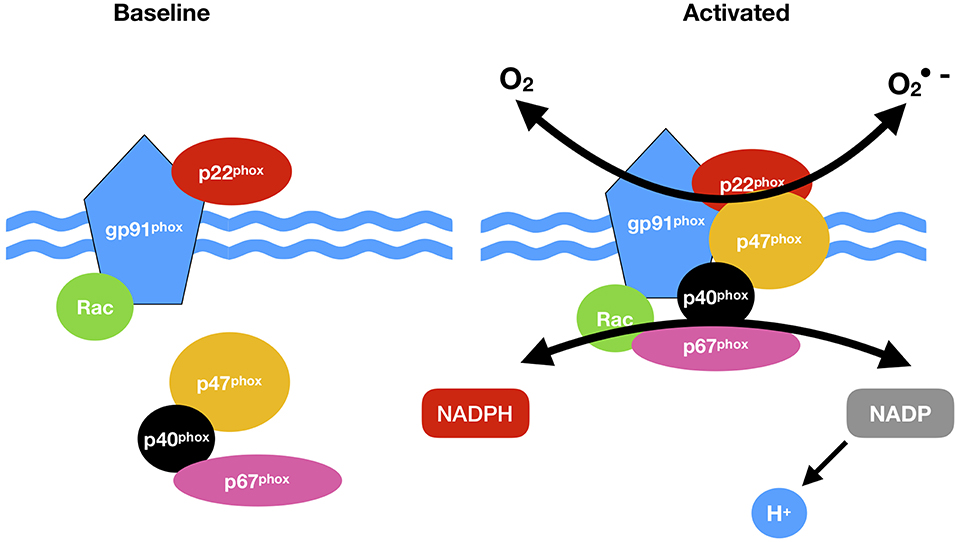
Figure 2. NADPH oxidase (NOX) activation. NOX comprises cytosolic (p47phox, p67 phox, p40 phox, and Rac) and membrane subunits (gp91 phox and p22 phox). During activation of NOX, cytosolic subunits comprise a multi-component enzyme and mi- grate to the plasma membrane to dock with the membrane subunits. This multi-subunit enzyme produces a superoxide anion (O2·).
NOX1, 2, 3, 4, and 5 along with Duox1 and 2 and are present in the plasma membrane, endoplasmic reticulum (NOX 2, 4, and 5), the mitochondria membrane and nuclear membrane (87). The classical NOX complex (NOX2) is comprised of the gp91-phox which is the main catalytic subunit that transfers NADPH electrons via FAD and the haem groups to O2 and constitutively forms a heterodimer with p22-phox on the membrane (91). Classical NOX is also comprised of three cytosolic subunits p47-phox, p67-phox and p40-phox along with the G-protein Rac (92) (Figure 2).
All NOX homologs have 6–7 transmembrane domains with two haem binding regions containing histidine residues and a NADPH binding region on the intracellular C-terminus to facilitate O2− production. The different isoforms of NOX contain homologs of the NOX2 gp91-phox subunit. Structural homology of the catalytic core is preserved within NOX1, NOX3, NOX4, NOX5, DUOX1, and DUOX2, however regulation, localization and function slightly vary across isoforms (93).
NOX1, 2, 3, and 5 mainly produce O2−, while NOX4, DUOX1, and DUOX2 generate mainly H2O2. NOX generates O2− by a complex reaction once NADPH binds to the cytosolic COOH terminus. Initially the electrons donated from NADPH are used to reduce FAAD to FADH. FADH is then used to reduce O2 on the other side of the membrane (93).
NOX is activated by phosphorylation though phagocytic particles, physiological or pathological cues such as hyperglycaemia, altered cellular hypoxia, and inflammation (76, 94, 95) Phosphorylation of p47-phox may be mediated by several serine kinases including protein kinase C isoforms, mitogen-activated protein kinases (MAPK), cyclic AMP dependent kinase, p21-activated kinases (PAK), PKB/AKT, protein kinase A (PKA), phosphatidylinositol-3-kinase (PI3K) and non-receptor associated protein kinases (e.g. JAK and SRC) (58, 96, 97).
NOX activators include cytokines (98), platelet derived growth factor (99), epidermal growth factor (50), TGF-β1 (100), mechanical forces such as pulsatile/oscillatory shear stress (101) cyclic stretch (102), hypoxia (52), and G protein coupled receptor agonists (55).
Protein-protein interactions among NOX and members of the thioredoxin family, and transient oscillations in intracellular concentration of various ions, may trigger the activation of NOX. Nuclear factor erythroid 2- related factor 2 (NrF2) is a negative regulator of NOX. Phosphorylation of p47-phox allows it to bind to a p40-phox-p67-phox complex (103)and facilitates the translocation of the trimer to the membrane where it binds to p22-phox thus assembling the active NOX complex (92).
It has recently been reported that NOX enzymes are present in extracellular vehicles (EVs) and microparticles (MPs) released from various cells, including endothelial cells (104, 105). EVs have been implicated in a number of pathological and physiological conditions such as cancer and atherosclerosis (106, 107). During septic shock, platelet derived exosomes may generate ROS through NOX-2, which forms ONOO- that induces endothelial apoptosis (106). NOX2 is present in circulating MPs from patients with hypercholesterolemia (108). However, as NOX2 activation requires the translocation of its cytosolic subunit p47 to the cytoplasmic membrane, it is important that p47 is also localized at the membrane surface or in proximity of the vesicle (85).
Subclinical Atherosclerosis
The arterial wall is comprised of the intima (the innermost layer of endothelial cells with some intimal myeloid cells surrounded by a basal lamina), the media (consists mostly of smooth muscle cells and some resident stem cells supported by the extracellular matrix) and the adventitia (the outer most layer containing a variety of cell types including fibroblasts, myeloid cells, macrophages, adipocytes and pericytes) (109) (Figure 3).
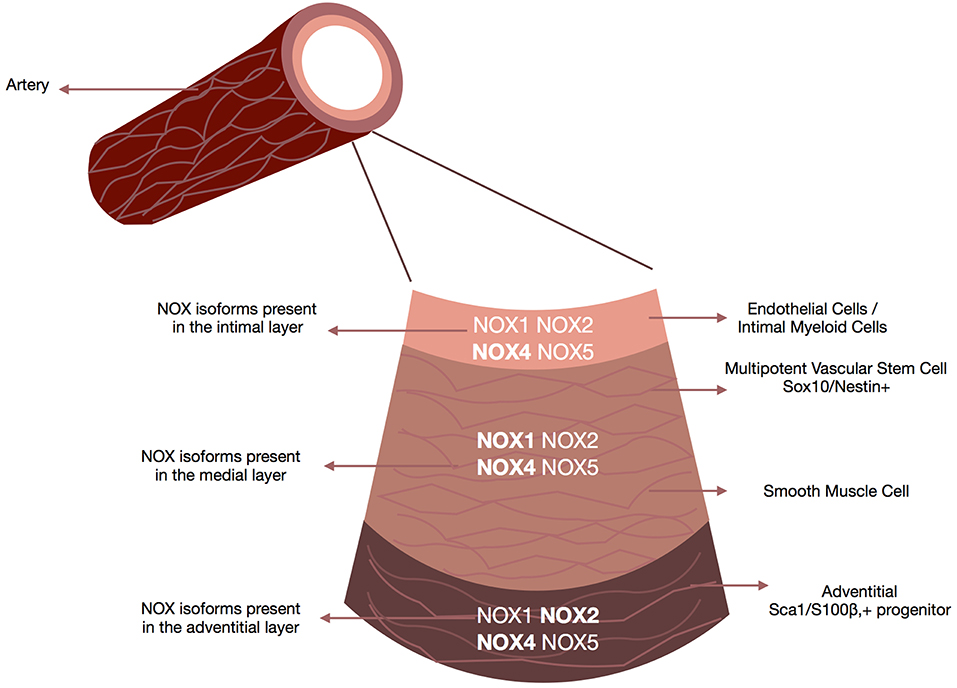
Figure 3. NOX enzymes present within the vascular walls. Schematic depicts the repertoire of NOX enzymes within all three layers of the vascular wall, the adventitia (i.e., fibroblasts, macrophages, and adventitial progenitor stem cells), the media (i.e., smooth muscle cells accounting for 90% of the vessel wall) and the intima (i.e., endothelial cells/smooth muscle cells). Although all isoforms are expressed at some levels within all three layers there are distinct NOX profiles associated with each layer. NOX4 is the predominant isoform in endothelial cells, NOX1 and NOX4 in smooth muscle cells, Nox4 in fibroblasts, and Nox2 and NOX4 in resident vascular stem cells.
Atherosclerosis is a chronic progressive inflammatory disease and the leading cause of death worldwide (6). Despite an extensive understanding of established/advanced lesion morphologies that lead to myocardial infarction or stroke due to thrombosis from acute plaque rupture or erosion, there still exists a superficial understanding of the initiation and progression of subclinical atherosclerosis. Central to this process is the development of non-atherosclerotic intimal lesions referred to as adaptive [AIT] or diffuse [DIT] intimal thickening, before the appearance of pathologic intimal thickening (PIT) leading to plaque formation in human vessels (7, 8, 110). Intimal thickening (AIT/DIT) leads to lipid deposition within the walls of the thickened artery and transpires before the build-up of foam cells which results in atheroma formation and restricted blood flow (1). Within the atheroma, there is also a significant accumulation of synthetic SMCs that are considered both protective when at the fibrous cap but also alternatively atherogenic if they become macrophage-like cells (6). Several studies have shown upregulation of NOX-based NAD(P)H oxidases during the progression of AIT following flow restriction due to carotid injury (111) and lipid diet (112). The importance of intimal thickening to subclinical atherosclerosis in advancing atherosclerosis has been clearly established in ApoE gene deficient mice fed on a western diet in combination with carotid artery ligation-induced injury (113, 114).
The origin of the SMC-like cells that contribute to DIT/AIT and make up the atheroma remains controversial. However, there are four proposed mechanisms based on recent lineage tracing analysis that include:
1) De-differentiation/reprogramming of a subset of medial vascular smooth muscle cells (15, 115)
2) Myogenic differentiation of a resident vascular stem cells (16, 116)
3) Myogenic differentiation of bone marrow-derived mesenchymal stem cells (17)
4) Endothelial-mesenchymal cell transition (EndMT) (18, 117)
Vascular Role of ROS
In the vascular bed, the main ROS of interest generated by NOX are H2O2 and O2− (11). H2O2 in low concentration is a vital signaling molecule under physiological conditions whereas O2− is associated with oxidative stress leading to pro-inflammatory and oxidative processes (Figure 3).
Cardiovascular risk factors such as hypercholesterolemia (118), hypertension (100), diabetes mellitus (76), and smoking (119) all increase ROS generation and decrease endothelial NO production. There is compelling evidence to support the role of ROS in intimal thickening leading to the progression of atherosclerosis in vivo using rodent models (10, 71, 111). Adaptive vascular lesions preferentially form within regions of disturbed blood flow leading to enhanced ROS and pathologic intimal thickening (8), and numerous human studies have demonstrated several NOX proteins including gp91phox and NOX4 contribute to increased intracellular oxidative stress in a cell-specific manner and thus may be involved in the genesis and progression of human coronary atherosclerotic disease (118). In vivo studies on the specific role of NOX homologs in vascular lesions have significantly advanced our understanding of the role of superoxide and increased NOX expression in injury models in mice (120), while antioxidant treatment with tempol or N-acetyl-cysteine protects against injury-induced lesion formation (121).
ROS in Vascular Endothelium
The vascular endothelium plays a critical role in vessel homeostasis by maintaining blood flow regulating blood flow, controlling macromolecule and fluid exchange with tissues and preventing leukocyte activation (122). NOX are in part localized to the plasma membrane, producing extracellular superoxide with a paracrine function but NOX 1, 2, and 4 are also localized in intracellular compartments with a perinuclear distribution (123). Sustained ROS levels contribute to endothelial dysfunction and activation of an inflammatory phenotype leading to the development of atherosclerosis (82). In vascular endothelial cells the main source of ROS is the electron leakage from the mitochondria (46).
Endothelial cell-dependent relaxation is primarily mediated by endothelial-derived hyperpolarizing factor (EDHF) and nitric oxide (NO), with H2O2 as the primary EDHF (39). NOX4 generates H2O2, which may react with NO and increase (124) or decrease (125) endothelial nitric oxide synthase (eNOS) expression and activity through a phosphoinositide 3-kinase-dependent and the inhibition of AP-1 activity, respectively. Flow, the preeminent stimulus for endothelial NO, also promotes the endothelial release of other factors that impact on vascular function, including activation of the lysosomal biogenesis transcription factor EB (TFEB) to decrease mTOR (mechanistic target of rapamycin) activity (80, 126). Indeed, ROS may induce autophagy by activating the major Ca2+ release channel on the lysosomal membrane through a TFEB pathway, facilitating the removal of damaged mitochondria and excess ROS (127). Moreover, athero-prone regions of the vasculature have enhanced TFEB levels linked with a reduction in H2O2, and superoxide (126).
Finally, proinflammatory lipids like lysophosphatidylcholines (LPC) are known to stimulate ROS formation in atherosclerosis, an effect that is attenuated by mitoTEMPO—a mitochondrial ROS scavenger. Therefore, an imbalance of redox-mediated signaling in endothelial cells may precipitate endothelial dysfunction that is a key event for the development of atherosclerosis (46).
NOX4 is also involved in endothelial progenitor stem cell proliferation, migration and cell survival after exposure to TNF-alpha (128). Superoxide (O2−) attenuates endothelial cell dependant relaxation, therefore SOD may play an important role to limit intracellular O2− concentration in endothelial cells by converting O2− to H2O2 (129).
ROS in Vascular Smooth Muscle Cells
The primary NOX isoforms in SMCs are NOX1 and NOX4 (130). Their localization and enzymatic activity differs in that NOX1 is primarily found in the plasma membrane, caveoli and endosomes, whereas NOX4 localizes to focal adhesions, the endoplasmic reticulum, and mitochondria. NOX1 interacts with multiple regulatory proteins to drive inducible O2−, while NOX4 is constitutively active and primarily generates H2O2 (131). NOX1 and NOX4 have highly specialized roles within smooth muscle cells (SMCs). Evidence suggests that NOX4 is involved in maintenance of SMC quiescence (132) while NOX1 has a role in modulating SMC function (33, 133). Most data to date has come from global transgenic murine studies that reveal the overall effect of NOX1 and NOX4 in normal and disease development (86). Ectopic expression of NOX1 in vascular smooth muscle promotes the production of ROS in response to ANG II and causes eNOS uncoupling and a decrease in nitric oxide bioavailability, resulting in impaired vasorelaxation (134). More recent studies have focused on SMC-specific transgenics that allow definition of the specific contribution of SMC NOX proteins to SMC function and phenotypic state, and to vascular disease development (86, 135). Using NoxO1 or p47phox gene deleted animals, ROS production stimulated by NoxO1 and p47phox limited endothelium-dependent relaxation and maintained blood pressure. However, NoxO1 and p47phox cannot substitute each other despite their similar effects on vascular function. Deletion of NoxO1 induced an anti-inflammatory phenotype, whereas p47phox deletion rather elicited a hyper-inflammatory response (136).
Activation of NOX1 contributes to matrix degradation, and the migration and proliferation of SMC (137). Protein kinase C-beta1-mediated phosphorylation of NOX1 is necessary for its interaction with the NOXA1 subunit and generation of superoxide (138). A peptide inhibitor of this process prevents SMC migration (139). Induction of SMC proliferation and hypertrophy leads to downregulation of NOX4 and upregulation of NOX1 expression, respectively. NOX4 downregulation leads to senescence of human vascular smooth muscle cells (140).
PDGF-BB-induced increases in NOX1 expression and H2O2 production promotes activation of c-Jun N-terminal kinase (JNK), cyclin D and extracellular signal-regulated kinase (ERK)1/2 signaling to enhance SMC migration and proliferation, respectively (141). On the other hand, Ang II-induced SMC hypertrophy is regulated by NOX1 activation of Ras, p38 mitogen kinase activated protein kinase (MAPK)/protein kinase B (Akt), and epidermal growth factor (EGF) receptor pathways (142). NOX1 mRNA expression is enhanced in phenotypic de-differentiated SMC (112). NOX1 deficient mice exhibit decreased proliferation and migration in response to PDGF-BB, whereas ectopic expression of NOX1 has the opposite effects (86).
In further agreement, ectopic SMC expression of human NOX1 facilitated enhanced Ang II-induced vascular O2− production, hypertension and vessel wall hypertrophy (143). Contradictory data from global NOX1 knockdown suggest that NOX1 deficiency may be protective after femoral wire-induced injury by attenuating neointima formation and cell proliferation (86). Indeed, vascular NOX1 levels are upregulated in carotid arteries following balloon injury. Utilizing a combination of genetic mouse models and cell culture studies, strong evidence has recently emerged that the NOX1 coactivator protein, NoxA1 also critically regulates SMC migration and phenotypic modulation in stenotic and atherosclerotic vascular remodeling (112, 144, 145).
In contrast to NOX1, NOX4 function is associated with SMC contractile proteins in vitro and maintenance of SMC in a quiescent contractile state (132). NOX4, as the primary isoform in SMCs is responsible for the baseline levels of ROS in maintaining the identity of differentiated SMCs (146). Indeed, NOX4 knockdown results in a decrease in SMC differentiation marker expression [smooth muscle myosin heavy chain 11 (SM-MHC), smooth muscle alpha actin (ACTA-2) and calponin 1 (CNN1)], while NOX4 overexpression increases their expression. NOX4 specifically may be required for maintenance of the contractile-type stress fibers in SMCs. TGF-β1 stimulates SMCs differentiation and specifically induces H2O2 generated by NOX4 via the SMAD signaling pathway (132). NOX4 mediates TGF-β1 induced SMC proliferation but not that by PDGF-BB or interferon gamma. NOX4 knockdown also results in decreased levels of serum response factor (SRF) required for CArG box dependent expression of SMC contractile proteins. NOX4 and CNN1 are both expressed within the neointima following balloon injury while ectopic SMC expression of a NOX4 dominant negative mutant reversed neointima formation following injury, in part by suppressing epoxide hydrolase 2 which inhibits SMC proliferation, migration and inflammation (147).
While not expressed in rodent vessels, NOX5 is present in human and porcine cells and is preferentially expressed in human coronary arteries and atherosclerotic lesions (43, 148). Putative regulators of NOX5 include interferon-gamma (IFNγ) which increases NOX5 production (43) while NOX5 knockdown impairs PDGF-BB mediated proliferation and ROS production (149)
Collectively, coordination of NOX1 and NOX4-dependent signaling facilitates de-differentiation and subsequent migration and proliferation of SMCs. Strategies such as those to inhibit NOX1 phosphorylation or NOX4 silencing may mitigate the development of cardiovascular disease.
ROS in Adventitial Cells
The adventitia used to be considered as an inert connective layer comprised of adventitial fibroblasts wrapped around the medial layer. However, accumulated data now suggests that the adventitia is a major site of immune and inflammatory cell trafficking that is facilitated by the vasa vasorum to maintain the medial layer and provide an important gateway for macrophage and leukocyte migration into the intima (150). It is also an important stem/progenitor cell niche ready to respond to arterial injury and thus acts as an essential regulator of vascular wall structure by contributing to the reorganization of the extracellular matrix (151). Adventitial cells express NOX1, NOX2, and NOX4 isoforms (152). NOX2, p22phox, p47phox, and p67phox are abundantly expressed in aortic vascular adventitia and in cultured adventitial fibroblasts in vitro (153). In contrast, NOX4 is weakly expressed in adventitial fibroblasts of human coronary arteries (118). NOX can be activated in adventitial cells by the similar vascular different stimuli including cytokines, hormones, metabolic factors and mechanical injury to stimulate the release of ROS (152). NOX1 and 4 are associated with hypoxic challenge in the adventitia (95). The primary role of NOX within the adventitia may be superoxide production and increased expression of adhesion molecules leading to chemotactic movement of leukocytes and their increased penetration into the vessel wall (142). Human coronary artery adventitial fibroblasts express NOX2 and NOX4 that produce superoxide in response to angiotensin II (Ang II) (118). This superoxide can be converted to H2O2 by extracellular SOD thus raising the possibility that H2O2 derived directly from NOX4 in the adventitial layer can also act as a paracrine mediator.
ROS in Intimal Myeloid Cells
Myeloid cells are present in the intima of the large arteries like the aorta, where vascular lesions and atherosclerosis plaques develop (154). NOX are primary sources of ROS in macrophages where a tumor necrosis factor–like weak inducer of apoptosis (TWEAK) fibroblast growth factor–inducible 14 (Fn14) TWEAK/Fn14 axis regulates NOX2-dependent ROS production (83). While it remains unclear whether endogenous NOX in macrophage has a direct impact on the progress of atherosclerosis, many studies have revealed a significant role of NOX-derived ROS in regulation of monocyte differentiation and macrophage functions (155). Recent evidence also indicates that human monocytes and macrophages express functionally active NOX5 (43) and that a NOX5-p22phox complex drives macrophage-dendritic differentiation (156).
Mitochondrial ROS is also an important source of ROS in macrophages and promotes MCP-1 production to promote monocyte infiltration and lesion inflammation (157). Another potential source of ROS in macrophages is the XO. XO inhibitors inhibit macrophage ROS formation, inflammatory cytokine release, and atherosclerosis (158). XO breaks down hypoxanthine and xanthine to uric acid and produces ROS, both of which may affect the function of macrophages. However, XO-dependent generation of ROS, rather than uric acid, mediates inflammatory cytokine production (159). Irrespective of origin, ROS can also significantly affect macrophage function whereby heme scavengers inhibit heme-mediated ROS production and ROS-mediated oxidative damage (160).
Pathological Role of ROS
Oxidative stress in arteriosclerosis results primarily from the activity of NOX enzymes (9) (Figure 4). However, their specific role in SMCs during the progression of subclinical arteriosclerosis remains unresolved. While global NOX1 deficient mice develop less neointimal thickening after wire-induced injury consistent with subdued SMC proliferation and migration rates in vitro and enhanced NOX1 expression in neointimal SMCs in vivo, SMC-specific ectopic expression of NOX1 failed to increase DIT/AIT (86). This enhanced NOX1 expression was associated with ERK1/2 (extracellular signal-regulated kinases 1/2) activation and enhanced MMP-9 (matrix metallopeptidase 9) (33). Gene expression network analysis of human arteriosclerotic vessels suggests the network hub gene glutathione peroxidase-1 (GPX1) is the most significantly downregulated following pathologic intimal thickening (161). Decreased GPX1 expression in atherosclerotic mice led to reductive stress via a time-dependent increase in glutathione suggesting that GPX1-dependent alterations in oxido-reductive stress promote vascular remodeling.
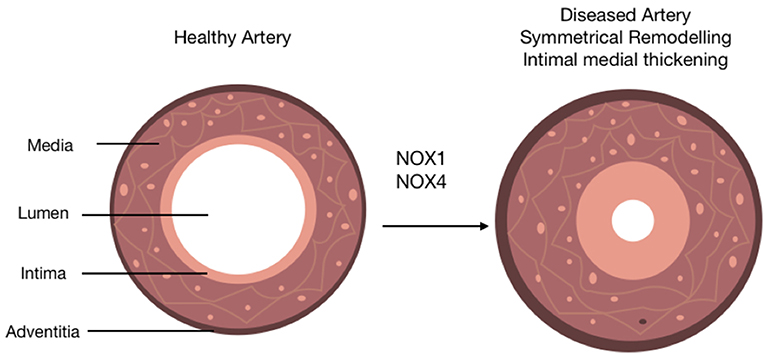
Figure 4. The role of NOX isoforms in vascular disease progression. Schematic represents the role of NOX 1/4 enzymes in the progression of arteriosclerosis. A healthy artery is depicted with three distinct layers, the outermost layer; the adventitia, the middle layer; the media and the innermost layer; the intima. The activity of NOX 1/4 enzymes and subsequent production of ROS leads to the progression of arteriosclerosis. This is characterized by the accumulation of neointimal SMC-like cells within the medial and intimal layers (intimal medial thickening), an induction of adventitial fibrosis represented by a slight increase out the adventitia, and a distinctive narrowing of the lumen subsequently resulting in restricted blood flow.
Endothelial NOX4 plays a critical role in the control of atherosclerosis where ROS is athero-protective via NOX4-dependent inhibition of inflammation and vascular remodeling (162). Ectopic expression of endothelial NOX4 in ApoE deficient mice reduced lesion formation, increased Treg numbers and decreased levels of effector T cells and chemokines (162). However, downregulation of NOX4 in human aortic ECs increased the expression of profibrotic CTGF (connective tissue growth factor), while decreasing endothelial H2O2 and reducing the levels of p-SMAD3 (phosphorylated mothers against decapentaplegic homolog 3) (40). NOX4 knockdown in vivo also leads to increased fibrillar collagens I and III in plaques, which is associated with elevated transforming growth factor-β expression and p-SMAD3 levels in diabetic lesions (135). The response of endothelial cells to endoplasmic reticulum (ER) stress during the progression of arteriosclerosis is governed by NOX4 and H2O2 (163). ER stress increases H2O2 in ER in a NOX4-dependent manner leading to oxidation of Ca2+-ATPase, elevated cytosolic calcium and RasGRF (Ras-specific guanine nucleotide releasing factor) activation. NOX generated ROS also impacts on XBP1 splicing (X-box-binding protein 1), a key protein that promotes EC apoptosis and atherosclerosis formation (164).
The presence of NOX4 in adventitial fibroblasts and the adventitial location of ROS production following AIT/DIT in murine models of vascular remodeling highlights their fundamental importance to vascular pathology and regeneration (165) (Figure 5). The functional significance of NOX4 in adventitial fibroblasts has led investigators to suggest an “outside in” process of vascular remodeling. p22phox protein and ROS production both increase within the adventitial layer of injured carotid arteries (111) while NOX4 overexpression stimulates migration and proliferation, as well as matrix gene expression, of adventitial fibroblasts (166). NOX4 also mediates TGF-β1 activation of fibroblasts, which promotes differentiation into a profibrotic myofibroblast phenotype and matrix production (167). Small molecule inhibitors of NOX4 reduce adventitial ROS generation and subsequent vascular remodeling (89). This “outside in” process is further supported by recent lineage tracing analysis using Gli-Cre-LoxP transgenic mice supporting a role for adventitial cells in contributing to DIT/AIT through hedgehog signaling pathways (151, 168) Notably, interaction of NOX4 with hedgehog has recently been demonstrated in gastric cancer cells (169). Overexpression of the hedgehog target gene, Gli1, inhibited the anti-mitogenic effect of NOX4 knockdown while concomitant overexpression of NOX4 increased Gli1 expression, an effect reversed by Gli1 depletion. Further, ROS generated by NOX4 was required for GLI1 expression, as shown by use of the ROS inhibitor, diphenylene iodonium (DPI).
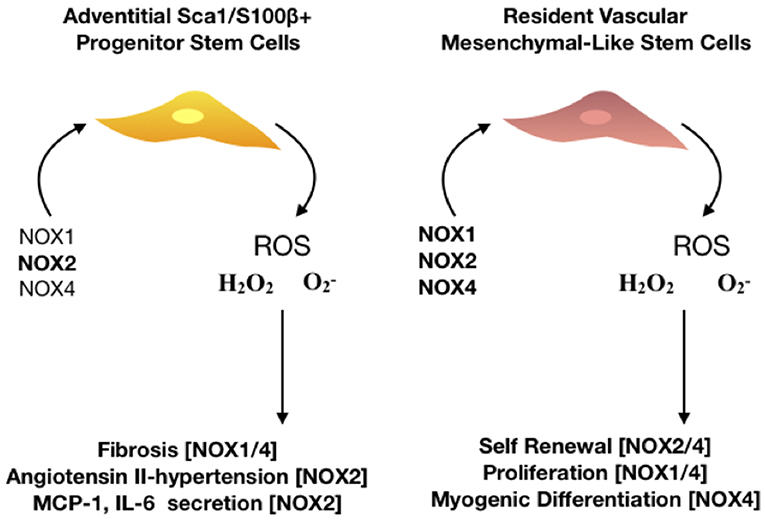
Figure 5. The role of NOX isoforms in vascular stem cell populations. Schematic represents the effect of NOX 1,2, and 4 enzymes on stem cell activity within the vasculature. There are two major resident stem cell populations that are effected by NOX enzymes, adventitial Sca1/S100β+ and resident vascular mesenchymal-like stem cells. NOX2 is predominantly associated with hypertensive vessels and promotes the secretion of monocyte chemoattractant protein (MCP-1) and interleukin 6 (IL-6) whilst NOX 1/4 are associated with hypoxic challenge and fibrosis in adventitial progenitor cells. Proliferation, self-renewal and differentiation of resident vascular stem cells is driven by NOX 1,2, and 4 as the production of ROS- mediates P13K/AKT dependent signaling whilst orchestrating a redox-mediated regulatory mechanisms of stem cell function of vascular repair.
In arteriosclerosis, innate and adaptive immune cells also enter the vasculature and participate in the pathology of AIT and PIT by releasing several mediators including ROS that cause vascular damage (170). Upon exposure to hypercholesterolemia, these cells get loaded with oxidized LDL and acquire foam cell morphology prior to the recruitment of monocytes that differentiate into macrophage foam cells. Atherosclerotic patient-derived monocytes/macrophages have exaggerated IL-6 and IL-1β levels, which was highly dependent on mitochondrial ROS but not NOX2 (171). Moreover, 8-oxoguanine glycosylase, a major DNA glycosylase responsible for removing mitochondrial oxidative stress–induced DNA damage, plays a protective role in atherosclerosis by preventing excessive inflammasome activation in macrophages, further supporting the critical role of macrophage mitochondrial oxidative stress in promoting atherosclerosis. While macrophages produce ROS through XO, there is still a lack of solid evidence demonstrating the role of macrophage XO in atherosclerosis (172).
ROS and Resident Vascular Stem Cells
It is widely accepted that the regulation of stem cell self-renewal and differentiation is crucial for tissue homeostasis, and in particular, vascular remodeling and fibrosis (173). Indeed several recent studies have highlighted the critical importance of the cellular oxidation-reduction (redox) state in modulating the balance between stem cell self-renewal and differentiation (41, 174). As a result, the study of ROS regulation in regenerative medicine has rapidly evolved to define the putative roles of oxidative stress in dictating the fate of multi-potent and pluripotent stem cells.
In adult vessels, resident vascular stem cells (rVSCs) are present in all three layers and are important in maintaining vessel homeostasis (16, 151, 175) These progenitor cells express various stem cell markers including Sca-1, cKit, CD34 and Flk1, S100β, Sox10, Sox17, and Nestin, are multi-potent and can differentiate into lineages of mesoderm and neuroectoderm origins including SMCs, osteoblasts, adipocytes and chrondrocytes (173). The influence of rVSC behavior on the development of subclinical atherosclerosis may be critical. Hence, a greater understanding of the regulatory mechanisms that control stem/progenitor cell expansion, migration, and differentiation is essential for targeted therapies. Accumulating evidence suggests that rVSCs are mobilized by local signal molecules in their microenvironment. Importantly, rVSCs are normally quiescent but can be activated in response to injury to participate variously in endothelial regeneration, adventitial fibrosis and/or neointimal SMC-like cell accumulation that drives subclinical arteriosclerosis and neointima formation (173). Cell fate mapping studies using transgenic mouse models have greatly extended our understanding of the fate of these cells and that of their progeny during pathologic vessel remodeling, and how they might be influenced by the redox state (71). While little is known about ROS control of resident vSCs, in general, these progenitors adopt a mesenchymal stem-like phenotype before they undergo myogenic differentiation to SMC-like cells and accumulate with the neointima during subclinical atherosclerosis (116). Vascular mesenchymal-like stem cells (MSCs) are multipotent stem cells that are defined by three main characteristics: plastic adherence, ability to naturally differentiate into a diverse set of tissues within the mesoderm lineage, and of self-renewal (173).
It is clear that NOX–derived ROS is a major regulator of MSC cell fate (41). There are conflicting reports on the antioxidant levels at baseline for MSCs and their subsequent resistance to oxidative stress (176, 177). This may be due to differences in the timing of MSC isolation, cultivation and exposure to oxidative insult as cellular senescence and cell age increase oxidative stress. Nevertheless, NOX1 and NOX4 derived ROS is hypothesized to be a redox messenger for rVSC derived MSC proliferation and differentiation (178). In general, low levels of ROS are associated with MSC maintenance and expansion in pluripotent embryonic stem cells (ESCs) and multipotent adult stem cells, whereas increased levels are associated with stem cell differentiation (174, 179). Pharmacological or genetic approaches to alter stem cell metabolism have been shown to directly influence stem cell activity since ROS generation in both embryonic and adult stem cells is mainly dependant on glycolysis (180). There is a large difference in energy metabolism and cellular redox status between pluripotent stem cells and terminally differentiated cells. As low levels of ROS are required in stem cells to maintain quiescence and self-renewal, it is likely that rVSCs and their MSC progenitors reside in a specialized microenvironment that is low in O2 (41).
Physiological low levels of ROS, either from exogenous H2O2 or hypoxia, play an important role in the regulation of MSC cell fate decision through activation of NOX-1 and NOX-4 (181, 182). In particular, ROS regulates cell expansion by (i) activation of miR-210 that triggers ERK1/2 and AKT activation in MSCs (ii) secretion of chemokines (e.g., CCL-2, CCL-4) through the activation of p38-mitogen-activated protein kinases (MAPK) pathway (183) and (ii) release vascular endothelial growth factor (VEGF) to promote angiogenesis (184). In contrast, high endogenous levels of ROS not only promote oxidative stress to disrupt adhesion through the down-regulation of key focal adhesion molecules including focal adhesion kinase (FAK), Src, and integrin expression (185), but also DNA damage (186) and a reduction in telomere length leading to MSC senescence (187). Oxidative stress also causes cell cycle arrest by inhibiting phosphorylated retinoblastoma (pRB) expression via a p38 MAPK/P16 pathway (188), disrupts mitochondrial cardiolipin-cytochrome c complexes and induces BAX-BAK dimerization to drive overall apoptosis and cell death (189). Finally, ROS may alter stem cell fate by influencing the epigenetic landscape via DNA methylation, post-translational histone modifications, ATP-dependent alterations to chromatin and non-coding RNA transcripts (69).
Myogenic Differentiation of Stem Cells- the Role of ROS
There is a direct correlation between NOX-derived ROS and modulation of multiple stem fates. Differentiation of mesenchymal stem cells is stimulated by differentiation factors such as TGF-β (190), epidermal growth factors (EGF) (191), wingless type MMTV integration site (wnt) proteins (192), fibroblast growth factor (FGF) (193). Many redox sensor proteins play a key role in altering stem cell fate, such as transcription factors NF-κB (194), forkhead box O (FOXO) (195), nuclear factor erythroid 2 (NRF2) (103) and the p53 (TRP53) tumor suppressor (196). Differentiation signals induce NOX4 in stem cells suggesting intracellular H2O2 may act as a non-specific intracellular differentiation signal and influence other activated pathways in promoting specific lineages (197).
MSCs undergo myogenic differentiation to SMC like-cells upon stimulation with TGF-β1, mechanical stress and sphingosylphosphorylcholine (SPC), and with co-cultivation with vascular endothelial cells (198). Little is known about how ROS contributes to myogenesis in rVSC but SPC promotes SMC differentiation in human MSCs, which is dependent on ROS activation of the DJ-1 pathway (199). ROS can also induce myogenic differentiation during the early stages in various stem cells when NOX4 is activated by TGF-β1 and/or PDGF-BB to generate ROS (H2O2 and O2−). NOX4-derived H2O2 up-regulates serum response factor (SRF) gene transcription and protein translation, which when phosphorylated binds CArG elements within the promoter-enhancer region of SMC-specific genes to regulate myogenic differentiation (132). NOX4-derived O2− also activates MAPK which increases SRF-mediated gene transcription activation to further drive differentiation (139, 200). At the later stage of myogenic differentiation, NOX4 is recruited to SMC myofilaments to maintain cells in a differentiated state (197). NOX4 also drives myogenic differentiation from mouse embryonic stem cells (ESCs) in-vitro, mediated by NRF-2 (200). Anti-oxidants such as selenium may also dictate stem cell differentiation, as they influence the cellular redox profile (201).
Endothelial to Mesenchymal Transition (EndMT)
Endothelial to mesenchymal transition (EndMT) is a process whereby an endothelial cell undergoes a series of molecular events that lead to a change in phenotype toward a mesenchymal cell (e.g., myofibroblast, smooth muscle cell) (202). EndMT plays a fundamental role during development, and recent evidence suggests that EndMT may be involved in cardiovascular diseases (CVDs), including atherosclerosis, pulmonary hypertension, valvular disease, and fibroelastosis. In particular, EndMT has been implicated in the progression of subclinical atherosclerosis as “transitioning” cells and is readily detected in human plaques (117). Oxidative stress is known to be involved in EndMT and subsequent vascular damage through TGF-β (203). Brain Arnt-like protein-1 (BMAL1) suppresses ROS production and a positive relationship exists between loss of BMAL1 expression and EndMT in atherosclerotic plaque vulnerability in human carotid plaques. In-vitro, BMAL1 inhibits oxidized low-density lipoprotein-induced intracellular ROS accumulation and subsequent EndMT in human aortic endothelial cells (204–206). Endothelial-specific NOX2 overexpression in transgenic mice enhances EndMT and has pronounced pro-fibrotic effects in the heart (207). Moreover, exposure of endothelial cells to H2O2 inhibits endothelial specific lineage genes and promotes EndMT due to TGF-β (208). Similarly, hypoxia promotes EndMT of human endothelial cells (117) by upregulating unregulated EndMT genes, SNAIL1 and SNAIL2 (209).
Antioxidants Effect on Atherosclerotic Disease
Extensive Cochrane meta-analysis of clinical studies suggests no significant benefit of anti-oxidants in CVD (210). Despite this, higher dietary intake and/or blood concentrations of vitamin C, carotenoids, and α-tocopherol (as markers of fruit and vegetable intake) were all associated with reduced risk of cardiovascular disease independent of anti-oxidant effects (211). Nevertheless, patient cohorts at high risk of cardiovascular disease exhibit a low plasma concentration of anti-oxidants such as β-carotene, α-tocopherol, and ascorbic acid (212). Epidemiological studies confirm an inverse relationship between plasma concentration of anti-oxidants and degree of atherosclerotic disease (213). EUK-207, an anti-oxidant therapy reduces endothelial P-selectin, von Willebrand factor A1-domain and platelet adhesion in mouse models of atherosclerosis concomitant with reduced plaque area and macrophage content. ROS scavenging may also affect stem cell differentiation, as complete scavenging with a flavoprotein inhibitor NAC completely inhibited human embryonic stem cell derived CD34+ differentiation (214). In general, anti-oxidants are beneficial to stem cell activities by [i] mitigating oxidative stress through neutralization of free radicals and increasing the expression of antioxidant enzymes and [ii] influencing the differentiation fate of precursor stem cells
Patient cohorts with high plasma concentrations of reductants including cryptoxanthin, lycopene and α-carotene have lower intimal thickening compared to subjects with a low concentration of these anti-oxidants (215), and an inverse correlation exits for α-carotene and β-carotene and subclinical atherosclerosis (216). Lycopene, which has the highest reducing capacity among the carotenoids (650 mV), significantly decreased plaques in transgenic mice while improving endothelial function (217). Moreover, there is an inverse correlation between the plasma concentration of the anti-oxidant Vitamin E and the development of cardiovascular diseases (210, 218). Another anti-oxidant, selenium (Se), is incorporated into proteins known as selenoproteins to reduce H2O2 and lipid/phospholipid hydro-peroxidases by Se-dependent glutathione peroxidases (GPXs), while low levels of GPx-1 (the primary selenoprotein in mammals) activity are associated with atherosclerosis and severity of disease (219). Finally, despite limited bioavailability and rapid degradation, dietary anthocyanins are antioxidants with potentially significant cardiovascular benefits (220).
Conclusions
Subclinical atherosclerosis is characterized by intimal thickening due to the accumulation of neointimal SMC-like cells derived from a heterogeneous population of parent cells including differentiated SMCs, resident vascular stem cells, bone-marrow derived MSCs and EndMT. The heterogeneity reflects the variable phenotypes and functions of these cells depending on the severity of the injury to the vessel wall. Indeed, many investigators are speculating that these phenotypes may represent the various different stages of resident stem cell mediated differentiation. Redox/ROS signaling through the activity of NOX and or mtROS controls the maintenance of these phenotypes and their contribution to intimal thickening and subclinical atherosclerosis.
While global and cell-specific knockdown studies have presented compelling evidence for the role of NOX isoforms in controlling cell fate during intimal thickening, similar studies that address the functional consequences of NOX knockdown on specific stem cell populations and intimal myeloid cells are required. Moreover, elucidation of the mechanisms dictating the migration, proliferation, and myogenic differentiation of resident vSCs vascular stem cells through NOX-dependent pathways will provide vital information for the development of more targeted therapies for treating subclinical atherosclerosis.
Author Contributions
DB and MK wrote the manuscript. DB and PC prepared the figures. MK prepared the tables. IM, PC, and ER reviewed and edited the manuscript.
Funding
This work was funded by the Health Research Board of Ireland and European Union's INTERREG VA Programme, managed by the Special EU Programmes Body (SEUPB).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Singh SS, Pilkerton CS, Shrader CD, Frisbee SJ. Subclinical atherosclerosis, cardiovascular health, and disease risk: Is there a case for the Cardiovascular Health Index in the primary prevention population? BMC Public Health. (2018) 18:677–83. doi: 10.1186/s12889-018-5263-6
2. Zanoli L, Lentini P, Briet M, Castellino P, House AA, London GM, et al. Arterial Stiffness in the Heart Disease of CKD. J Am Soc Nephrol. (2019) 30:ASN.2019020117. doi: 10.1681/ASN.2019020117
3. Mitra AK, Agrawal DK. In stent restenosis: bane of the stent era. J Clin Pathol. (2006) 59:232–9. doi: 10.1136/jcp.2005.025742
4. Russell PS, Chase CM, Sykes M, Ito H, Shaffer J, Colvin RB. Tolerance, mixed chimerism, and chronic transplant arteriopathy. J Immunol. (2001) 167:5731–40. doi: 10.4049/jimmunol.167.10.5731
5. Thenappan T, Chan SY, Weir EK. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am J Physiol Circ Physiol. (2018) 315:H1322–31. doi: 10.1152/ajpheart.00136.2018
6. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
7. Sakamoto A, Torii S, Jinnouchi H, Finn AV, Virmani R, Kolodgie FD. Pathologic intimal thickening: are we any closer to understand early transitional plaques that lead to symptomatic disease? Atherosclerosis. (2018) 274:227–9. doi: 10.1016/j.atherosclerosis.2018.04.033
8. Nakagawa K, Nakashima Y. Pathologic intimal thickening in human atherosclerosis is formed by extracellular accumulation of plasma-derived lipids and dispersion of intimal smooth muscle cells. Atherosclerosis. (2018) 274:235–42. doi: 10.1016/j.atherosclerosis.2018.03.039
9. Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem. (2015) 30:11–26. doi: 10.1007/s12291-014-0446-0
10. Singh U, Jialal I. Oxidative stress and atherosclerosis. Pathophysiology. (2006) 13:129–42. doi: 10.1016/j.pathophys.2006.05.002
11. Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH oxidases in vascular pathology. Antioxid Redox Signal. (2013) 20:2794–814. doi: 10.1089/ars.2013.5607
12. Elahi MM, Kong YX, Matata BM. Oxidative stress as a mediator of cardiovascular disease. Oxid Med Cell Longev. (2009) 2:259–69. doi: 10.4161/oxim.2.5.9441
13. Burtenshaw D, Hakimjavadi R, Redmond E, Cahill P. Nox, reactive oxygen species and regulation of vascular cell fate. Antioxidants. (2017) 6:90. doi: 10.3390/antiox6040090
14. Wang H, Li H, Hou Z, Pan L, Shen X, Li G. Role of oxidative stress in elevated blood pressure induced by high free fatty acids. Hypertens Res. (2009) 32:152–8. doi: 10.1038/hr.2008.35
15. Herring BP, Hoggatt AM, Burlak C, Offermanns S. Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury. Vasc Cell. (2014) 6:21. doi: 10.1186/2045-824X-6-21
16. Yuan F, Wang D, Xu K, Wang J, Zhang Z, Yang L, et al. Contribution of vascular cells to neointimal formation. PLoS ONE. (2017) 12:e0168914. doi: 10.1371/journal.pone.0168914
17. Han CL, Campbell GR, Campbell JH. Circulating bone marrow cells can contribute to neointimal formation. J Vasc Res. (2001) 38:113–19. doi: 10.1159/000051038
18. Cooley BC, Nevado J, Mellad J, Yang D, St. Hilaire C, Negro A, et al. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med. (2014) 6:227ra34. doi: 10.1126/scitranslmed.3006927
19. Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. (2018) 215:21–33. doi: 10.1084/jem.20171773
20. Shi W, Wang NJ, Shih DM, Sun VZ, Wang X, Lusis AJ. Determinants of atherosclerosis susceptibility in the C3H and C57BL/6 mouse model. Circ Res. (2012) 86:1078–84. doi: 10.1161/01.res.86.10.1078
21. Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. (2011) 31:1506–16. doi: 10.1161/ATVBAHA.110.221127
22. Cybulsky MI, Jongstra-Bilen J. Resident intimal dendritic cells and the initiation of atherosclerosis. Curr Opin Lipidol. (2010) 21:397–403. doi: 10.1097/MOL.0b013e32833ded96
23. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. (2014) 15:411–21. doi: 10.1038/nrm3801
24. Kelley EE. A new paradigm for XOR-catalyzed reactive species generation in the endothelium. Pharmacol Reports. (2015) 67:669–74. doi: 10.1016/j.pharep.2015.05.004
25. Dolinsky VW, Cole LK, Sparagna GC, Hatch GM. Cardiac mitochondrial energy metabolism in heart failure: role of cardiolipin and sirtuins. Biochim Biophys Acta. (2016) 1861:1544–54. doi: 10.1016/j.bbalip.2016.03.008
26. Hrycay EG, Bandiera SM. Involvement of Cytochrome P450 in reactive oxygen species formation and cancer. Adv Pharmacol. (2015) 74:35–84. doi: 10.1016/BS.APHA.2015.03.003
27. Cho KJ, Seo JM, Kim JH. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol Cells. (2011) 32:1–5. doi: 10.1007/s10059-011-1021-7
28. Parker H, Winterbourn CC. Reactive oxidants and myeloperoxidase and their involvement in neutrophil extracellular traps. Front Immunol. (2012) 3:424. doi: 10.3389/fimmu.2012.00424
29. Maggiorani D, Manzella N, Edmondson DE, Mattevi A, Parini A, Binda C, et al. Monoamine oxidases, oxidative stress, and altered mitochondrial dynamics in cardiac ageing. Oxid Med Cell Longev. (2017) 2017:1–8. doi: 10.1155/2017/3017947
30. Mitchell BM, Cook LG, Danchuk S, Puschett JB. Uncoupled endothelial nitric oxide synthase and oxidative stress in a rat model of pregnancy-induced hypertension. Am J Hypertens. (2007) 20:1297–304. doi: 10.1016/j.amjhyper.2007.08.007
31. Liochev SI, Fridovich I. Superoxide and iron: partners in crime. IUBMB Life. (1999) 48:157–61. doi: 10.1080/152165499307161
32. Juhasz A, Markel S, Gaur S, Liu H, Lu J, Jiang G, et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J Biol Chem. (2017) 292:7866–87. doi: 10.1074/jbc.M116.768283
33. Jagadeesha DK, Takapoo M, Banfi B, Bhalla RC, Miller FJ. Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovasc Res. (2012) 93:406–13. doi: 10.1093/cvr/cvr308
34. Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid Redox Signal. (2005) 7:308–17. doi: 10.1089/ars.2005.7.308
35. Cooney SJ, Bermudez-Sabogal SL, Byrnes KR. Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J Neuroinflammation. (2013) 10:917. doi: 10.1186/1742-2094-10-155
36. Bánfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. (2004) 279:46065–72. doi: 10.1074/jbc.M403046200
37. Gill PS, Wilcox CS. NADPH Oxidases in the Kidney. Antioxid Redox Signal. (2006) 8:1597–607. doi: 10.1089/ars.2006.8.1597
38. Liang S, Kisseleva T, Brenner DA. The role of NADPH Oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front Physiol. (2016) 7:17. doi: 10.3389/fphys.2016.00017
39. Muñoz M, Martínez MP, López-Oliva ME, Rodríguez C, Corbacho C, Carballido J, et al. Hydrogen peroxide derived from NADPH oxidase 4- and 2 contributes to the endothelium-dependent vasodilatation of intrarenal arteries. Redox Biol. (2018) 19:92–104. doi: 10.1016/j.redox.2018.08.004
40. Liu XH, Zhang QY, Pan LL, Liu SY, Xu P, Luo XL, et al. NADPH oxidase 4 contributes to connective tissue growth factor expression through Smad3-dependent signaling pathway. Free Radic Biol Med. (2016) 94:174–84. doi: 10.1016/j.freeradbiomed.2016.02.031
41. Atashi F, Modarressi A, Pepper MS. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: a review. Stem Cells Dev. (2015) 24:1150–63. doi: 10.1089/scd.2014.0484
42. Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caillou B, et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene. (2012) 31:1117–29. doi: 10.1038/onc.2011.327
43. Manea A, Manea SA, Gan AM, Constantin A, Fenyo IM, Raicu M, et al. Human monocytes and macrophages express NADPH oxidase 5; A potential source of reactive oxygen species in atherosclerosis. Biochem Biophys Res Commun. (2015) 461:172–9. doi: 10.1016/j.bbrc.2015.04.021
44. Ameziane-El-Hassani R, Morand S, Boucher JL, Frapart YM, Apostolou D, Agnandji D, et al. Dual oxidase-2 has an intrinsic Ca2+-dependent H 2O2-generating activity. J Biol Chem. (2005) 280:30046–54. doi: 10.1074/jbc.M500516200
45. Ago T, Kuroda J, Kamouchi M, Sadoshima J, Kitazono T. Pathophysiological roles of NADPH Oxidase/Nox family proteins in the vascular system. Circ J. (2011) 75:1791–800. doi: 10.1253/circj.CJ-11-0388
46. Li X, Fang P, Li Y, Kuo Y-M, Andrews AJ, Nanayakkara G, et al. Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arterioscler Thromb Vasc Biol. (2016) 36:1090–100. doi: 10.1161/ATVBAHA.115.306964
47. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167:457–470.e13. doi: 10.1016/j.cell.2016.08.064
48. Juarez JC, Manuia M, Burnett ME, Betancourt O, Boivin B, Shaw DE, et al. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc Natl Acad Sci USA. (2008) 105:7147–52. doi: 10.1073/pnas.0709451105
49. Bäumer AT, Ten Freyhaus H, Sauer H, Wartenberg M, Kappert K, Schnabel P, et al. Phosphatidylinositol 3-kinase-dependent membrane recruitment of Rac-1 and p47phox is critical for α-platelet-derived growth factor receptor-induced production of reactive oxygen species. J Biol Chem. (2008) 283:7864–76. doi: 10.1074/jbc.M704997200
50. Lien GS, Wu MS, Bien MY, Chen CH, Lin CH, Chen BC. Epidermal growth factor stimulates nuclear factor-κB activation and heme oxygenase-1 expression via c-Src, NADPH oxidase, PI3K, and Akt in human colon cancer cells. PLoS ONE. (2014) 9:e104891. doi: 10.1371/journal.pone.0104891
51. Weaver JR, Holman TR, Imai Y, Jadhav A, Kenyon V, Maloney DJ, et al. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol Cell Endocrinol. (2012) 358:88–95. doi: 10.1016/j.mce.2012.03.004
52. Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS, et al. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med. (2008) 45:1223–31. doi: 10.1016/j.freeradbiomed.2008.06.012
53. Chiu JJ, Wung BS, Shyy JYJ, Hsieh HJ, Wang DL. Reactive oxygen species are involved in shear stress-induced intercellular adhesion molecule-1 expression in endothelial cells. Arterioscler Thromb Vasc Biol. (1997) 17:3570–7. doi: 10.1161/01.ATV.17.12.3570
54. Cheng JJ, Wung BS, Chao YJ, Wang DL. Cyclic strain-induced reactive oxygen species involved in ICAM-1 gene induction in endothelial cells. Hypertension. (1998) 31:125–30. doi: 10.1161/01.HYP.31.1.125
55. Ushio-Fukai M. Vascular signaling through G protein-coupled receptors: new concepts. Curr Opin Nephrol Hypertens. (2009) 18:153–9. doi: 10.1097/MNH.0b013e3283252efe
56. Caliceti C, Nigro P, Rizzo P, Ferrari R. ROS, Notch, and Wnt signaling pathways: crosstalk between three major regulators of cardiovascular biology. Biomed Res Int. (2014) 2014:1–8. doi: 10.1155/2014/318714
57. Son Y, Kim S, Chung HT, Pae HO. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 528:27–48. doi: 10.1016/B978-0-12-405881-1.00002-1
59. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. (2011) 21:103–15. doi: 10.1038/cr.2010.178
60. Koundouros N, Poulogiannis G. Phosphoinositide 3-Kinase/Akt signaling and redox metabolism in cancer. Front Oncol. (2018) 8:160. doi: 10.3389/fonc.2018.00160
61. Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. (2003) 25:207–18. doi: 10.1007/s00726-003-0011-2
62. Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. (2014) 2:535–62. doi: 10.1016/j.redox.2014.02.006
63. Cai H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc Res. (2005) 68:26–36. doi: 10.1016/j.cardiores.2005.06.021
64. Polytarchou C, Hatziapostolou M, Papadimitriou E. Hydrogen peroxide stimulates proliferation and migration of human prostate cancer cells through activation of activator protein-1 and up-regulation of the heparin affin regulatory peptide gene. J Biol Chem. (2005) 280:40428–35. doi: 10.1074/jbc.M505120200
65. Lin C, Wang H. NADPH oxidase is involved in H 2 O 2 -induced differentiation of human promyelocytic leukaemia HL-60 cells. Cell Biol Int. (2012) 36:391–5. doi: 10.1042/cbi20110290
66. Saito Y, Nishio K, Ogawa Y, Kimata J, Kinumi T, Yoshida Y, et al. Turning point in apoptosis/necrosis induced by hydrogen peroxide. Free Radic Res. (2006) 40:619–30. doi: 10.1080/10715760600632552
67. Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, et al. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the American Heart Association. Circ Res. (2016) 119:e39–75. doi: 10.1161/RES.0000000000000110
68. Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. (2008) 45:549–61. doi: 10.1016/j.freeradbiomed.2008.05.004
69. Kietzmann T, Petry A, Shvetsova A, Gerhold JM, Görlach A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br J Pharmacol. (2017) 174:1533–54. doi: 10.1111/bph.13792
70. Kanzaki H, Wada S, Narimiya T, Yamaguchi Y, Katsumata Y, Itohiya K, et al. Pathways that regulate ROS scavenging enzymes, and their role in defense against tissue destruction in periodontitis. Front Physiol. (2017) 8:351. doi: 10.3389/fphys.2017.00351
71. Shah A, Gray K, Figg N, Finigan A, Starks L, Bennett M. Defective base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation. (2018) 138:1446–62. doi: 10.1161/CIRCULATIONAHA.117.033249
72. Thu VT, Kim HK, Ha SH, Yoo J-Y, Park WS, Kim N, et al. Glutathione peroxidase 1 protects mitochondria against hypoxia/reoxygenation damage in mouse hearts. Pflugers Arch. (2010) 460:55–68. doi: 10.1007/s00424-010-0811-7
73. Rhee SG. H2O2, a necessary evil for cell signaling. Science. (2006) 312:1882–3. doi: 10.1126/science.1130481
74. Aitken RJ. Reactive oxygen species as mediators of sperm capacitation and pathological damage. Mol Reprod Dev. (2017) 84:1039–52. doi: 10.1002/mrd.22871
75. Liou G-Y, Storz P. Reactive oxygen species in cancer. Free Radic Res. (2010) 44:479–96. doi: 10.3109/10715761003667554
76. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications review-Article. Cell Death Dis. (2018) 9:119. doi: 10.1038/s41419-017-0135-z
77. Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. (2018) 592:728–742. doi: 10.1002/1873-3468.12956
78. Ellulu MS, Patimah I, Khaza'ai H, Rahmat A, Abed Y, Ali F. Atherosclerotic cardiovascular disease: a review of initiators and protective factors. Inflammopharmacology. (2016) 24:1–10. doi: 10.1007/s10787-015-0255-y
79. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol Rev. (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
80. Zhang X, Yu L, Xu H. Lysosome calcium in ROS regulation of autophagy. Autophagy. (2016) 12:1954–5. doi: 10.1080/15548627.2016.1212787
81. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
82. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab. (2014) 25:452–63. doi: 10.1016/j.tem.2014.06.012
83. Madrigal-Matute J, Fernandez-Laso V, Sastre C, Llamas-Granda P, Egido J, Martin-Ventura JL, et al. TWEAK/Fn14 interaction promotes oxidative stress through NADPH oxidase activation in macrophages. Cardiovasc Res. (2015) 108:139–47. doi: 10.1093/cvr/cvv204
84. Jongstra-Bilen J, Haidari M, Zhu S-N, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. (2006) 203:2073–83. doi: 10.1084/jem.20060245
85. Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. (2008) 275:3249–77. doi: 10.1111/j.1742-4658.2008.06488.x
86. Rivera J, Sobey CG, Walduck AK, Drummond GR. Nox isoforms in vascular pathophysiology: insights from transgenic and knockout mouse models. Redox Rep. (2010) 15:50–63. doi: 10.1179/174329210x12650506623401
87. Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. (2012) 110:1364–90. doi: 10.1161/CIRCRESAHA.111.243972
88. Piccoli C, D'Aprile A, Ripoli M, Scrima R, Lecce L, Boffoli D, et al. Bone-marrow derived hematopoietic stem/progenitor cells express multiple isoforms of NADPH oxidase and produce constitutively reactive oxygen species. Biochem Biophys Res Commun. (2007) 353:965–72. doi: 10.1016/j.bbrc.2006.12.148
89. Barman SA, Chen F, Su Y, Dimitropoulou C, Wang Y, Catravas JD, et al. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler Thromb Vasc Biol. (2014) 34:1704–15. doi: 10.1161/ATVBAHA.114.303848
90. Reddy MM, Fernandes MS, Salgia R, Levine RL, Griffin JD, Sattler M. NADPH oxidases regulate cell growth and migration in myeloid cells transformed by oncogenic tyrosine kinases. Leukemia. (2011) 25:281–9. doi: 10.1038/leu.2010.263
91. Yu L, Quinn MT, Cross AR, Dinauer MC. Gp91phox is the heme binding subunit of the superoxide-generating NADPH oxidase. Proc Natl Acad Sci USA. (2002) 95:7993–8. doi: 10.1073/pnas.95.14.7993
92. Rastogi R, Geng X, Li F, Ding Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front Cell Neurosci. (2017) 10:301. doi: 10.3389/fncel.2016.00301
93. Magnani F, Mattevi A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr Opin Struct Biol. (2019) 59:91–7. doi: 10.1016/j.sbi.2019.03.001
94. Dupré-Crochet S, Erard M, Nüβe O. ROS production in phagocytes: why, when, and where? J Leukoc Biol. (2013) 94:657–70. doi: 10.1189/jlb.1012544
95. Li S, Tabar SS, Malec V, Eul BG, Klepetko W, Weissmann N, et al. NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid Redox Signal. (2008) 10:1687–98. doi: 10.1089/ars.2008.2035
96. Ago T, Kuribayashi F, Hiroaki H, Takeya R, Ito T, Kohda D, et al. Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc Natl Acad Sci USA. (2003) 100:4474–79. doi: 10.1073/pnas.0735712100
97. Fontayne A, Dang PMC, Gougerot-Pocidalo MA, El Benna J. Phosphorylation of p47phox sites by PKC α, βII, δ, and ζ: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. (2002) 41:7743–50. doi: 10.1021/bi011953s
98. Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun. (2017) 8:2247. doi: 10.1038/s41467-017-02325-2
99. Marumo T, Schini-Kerth VB, Fisslthaler B, Busse R. Platelet-derived growth factor–stimulated superoxide anion production modulates activation of transcription factor NF-κB and expression of monocyte chemoattractant protein 1 in human aortic smooth muscle cells. Circulation. (1997) 96:2361–7. doi: 10.1161/01.CIR.96.7.2361
100. Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, et al. Transforming growth factor-β1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Cell Mol Physiol. (2005) 290:L661–73. doi: 10.1152/ajplung.00269.2005
101. Hwang J, Ing MH, Salazar A, Lassègue B, Griendling K, Navab M, et al. Pulsatile versus oscillatory shear stress regulates nadph oxidase subunit expression: implication for native LDL oxidation. Circ Res. (2003) 93:1225–32. doi: 10.1161/01.RES.0000104087.29395.66
102. Liang X, Wang Z, Gao M, Wu S, Zhang J, Liu Q, et al. Cyclic stretch induced oxidative stress by mitochondrial and NADPH oxidase in retinal pigment epithelial cells. BMC Ophthalmol. (2019) 19:79. doi: 10.1186/s12886-019-1087-0
103. Murakami S, Motohashi H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic Biol Med. (2015) 88:168–78. doi: 10.1016/j.freeradbiomed.2015.06.030
104. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. (2013) 200:373–83. doi: 10.1083/jcb.201211138
105. Hervera A, De Virgiliis F, Palmisano I, Zhou L, Tantardini E, Kong G, et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat Cell Biol. (2018) 20:307–19. doi: 10.1038/s41556-018-0039-x
106. Gambim MH, de Oliveira do Carmo A, Marti L, Veríssimo-Filho S, Lopes LR, Janiszewski M. Platelet-derived exosomes induce endothelial cell apoptosis through peroxynitrite generation: experimental evidence for a novel mechanism of septic vascular dysfunction. Crit Care. (2007) 11:R107. doi: 10.1186/cc6133
107. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
108. Pignatelli P, Carnevale R, Cangemi R, Loffredo L, Sanguigni V, Stefanutti C, et al. Atorvastatin inhibits gp91phox circulating levels in patients with hypercholesterolemia. Arterioscler Thromb Vasc Biol. (2010) 30:360–7. doi: 10.1161/ATVBAHA.109.198622
109. Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. (2008) 111:5271–81. doi: 10.1182/blood-2008-01-078204
110. Finn AV, Kolodgie FD, Virmani R. Correlation between carotid intimal/medial thickness and atherosclerosis: a point of view from pathology. Arterioscler Thromb Vasc Biol. (2010) 30:177–81. doi: 10.1161/ATVBAHA.108.173609
111. Szöcs K, Lassègue B, Sorescu D, Hilenski LL, Valppu L, Couse TL, et al. Upregulation of Nox-Based NAD(P)H Oxidases in Restenosis After Carotid Injury. Arterioscler Thromb Vasc Biol. (2002) 22:21–7. doi: 10.1161/hq0102.102189
112. Vendrov AE, Sumida A, Canugovi C, Lozhkin A, Hayami T, Madamanchi NR, et al. NOXA1-dependent NADPH oxidase regulates redox signaling and phenotype of vascular smooth muscle cell during atherogenesis. Redox Biol. (2019) 21:101063. doi: 10.1016/j.redox.2018.11.021
113. Chang Z, Huangfu C, Grainger AT, Zhang J, Guo Q, Shi W. Accelerated atherogenesis in completely ligated common carotid artery of apolipoprotein E-deficient mice. Oncotarget. (2017) 8:110289–99. doi: 10.18632/oncotarget.22685
114. Nam D, Ni C-W, Rezvan A, Suo J, Budzyn K, Llanos A, et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Circ Physiol. (2009) 297:H1535–43. doi: 10.1152/ajpheart.00510.2009
115. Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD, et al. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res. (2016) 119:1313–23. doi: 10.1161/CIRCRESAHA.116.309799
116. Tang Z, Wang A, Yuan F, Yan Z, Liu B, Chu JS, et al. Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat Commun. (2012) 3:875. doi: 10.1038/ncomms1867
117. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, et al. Corrigendum: endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. (2017) 8:14710. doi: 10.1038/ncomms14710
118. Sorescu D, Weiss D, Lasségue B, Clempus RE, Szöcs K, Sorescu GP, et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. (2002) 105:1429–35. doi: 10.1161/01.CIR.0000012917.74432.66
119. Talukder MAH, Johnson WM, Varadharaj S, Lian J, Kearns PN, El-Mahdy MA, et al. Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. Am J Physiol Circ Physiol. (2010) 300:H388–96. doi: 10.1152/ajpheart.00868.2010
120. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. (2017) 120:713–35. doi: 10.1161/CIRCRESAHA.116.309326
121. Lu Y, Qin W, Shen T, Dou L, Man Y, Wang S, et al. The Antioxidant N-acetylcysteine promotes atherosclerotic plaque stabilization through suppression of RAGE, MMPs and NF-κB in ApoE-Deficient Mice. J Atheroscler Thromb. (2011) 18:998–1008. doi: 10.5551/jat.8870
122. Cahill PA, Redmond EM. Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis. (2016) 248:97–109. doi: 10.1016/j.atherosclerosis.2016.03.007
123. Li JM, Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem. (2002) 277:19952–60. doi: 10.1074/jbc.M110073200
124. Thomas SR, Chen K, Keaney JF. Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem. (2002) 277:6017–24. doi: 10.1074/jbc.M109107200
125. Kumar S, Sun X, Wiseman DA, Tian J, Umapathy NS, et al. Hydrogen peroxide decreases endothelial nitric oxide synthase promoter activity through the inhibition of Sp1 Activity. DNA Cell Biol. (2009) 28:119. doi: 10.1089/DNA.2008.0775
126. Lu H, Fan Y, Qiao C, Liang W, Hu W, Zhu T, et al. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci Signal. (2017) 10:eaah4214. doi: 10.1126/scisignal.aah4214
127. Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol. (2016) 32:255–78. doi: 10.1146/annurev-cellbio-111315-125407
128. Hakami NY, Ranjan AK, Hardikar AA, Dusting GJ, Peshavariya HM. Role of NADPH Oxidase-4 in human endothelial progenitor cells. Front Physiol. (2017) 8:150. doi: 10.3389/fphys.2017.00150
129. Dreher D, Junod AF. Differential effects of superoxide, hydrogen peroxide, and hydroxyl radical on intracellular calcium in human endothelial cells. J Cell Physiol. (1995) 162:147–53. doi: 10.1002/jcp.1041620118
130. Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2004) 24:677–83. doi: 10.1161/01.ATV.0000112024.13727.2c
131. Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HHHW, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. (2008) 45:1340–51. doi: 10.1016/j.freeradbiomed.2008.08.013
132. Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, et al. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. (2007) 27:42–8. doi: 10.1161/01.ATV.0000251500.94478.18
133. Yin W, Voit EO. Function and design of the Nox1 system in vascular smooth muscle cells. BMC Syst Biol. (2013) 7:20. doi: 10.1186/1752-0509-7-20
134. Dikalova AE, Góngora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Circ Physiol. (2010) 299:H673–9. doi: 10.1152/ajpheart.00242.2010
135. Gray SP, Di Marco E, Kennedy K, Chew P, Okabe J, El-Osta A, et al. Reactive oxygen species can provide atheroprotection via NOX4-dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol. (2016) 36:295–307. doi: 10.1161/ATVBAHA.115.307012
136. Rezende F, Moll F, Walter M, Helfinger V, Hahner F, Janetzko P, et al. The NADPH organizers NoxO1 and p47phox are both mediators of diabetes-induced vascular dysfunction in mice. Redox Biol. (2018) 15:12–21. doi: 10.1016/j.redox.2017.11.014
137. Xu S, Shriver AS, Jagadeesha DK, Chamseddine AH, Szócs K, Weintraub NL, et al. Increased expression of Nox1 in neointimal smooth muscle cells promotes activation of matrix metalloproteinase-9. J Vasc Res. (2012) 49:242–8. doi: 10.1159/000332958
138. Streeter J, Schickling BM, Jiang S, Stanic B, Thiel WH, Gakhar L, et al. Phosphorylation of Nox1 regulates association with NoxA1 activation domain. Circ Res. (2014) 115:911–8. doi: 10.1161/CIRCRESAHA.115.304267
139. Durgin BG, Straub AC. Redox control of vascular smooth muscle cell function and plasticity. Lab Investig. (2018) 98:1254–62. doi: 10.1038/s41374-018-0032-9
140. Przybylska D, Janiszewska D, Gozdzik A, Bielak-Zmijewska A, Sunderland P, Sikora E, et al. NOX4 downregulation leads to senescence of human vascular smooth muscle cells. Oncotarget. (2016) 7:66429–43. doi: 10.18632/oncotarget.12079
141. Lassègue B, Sorescu D, Szöcs K, Yin QQ, Akers M, Zhang Y, et al. Novel gp91phox homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. (2001) 88:888–94. doi: 10.1161/hh0901.090299
142. Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. (2003) 23:776–82. doi: 10.1161/01.ATV.0000066684.37829.16
143. Dikalova A, Clempus R, Lassègue B, Cheng G, McCoy J, Dikalov S, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. (2005) 112:2668–76. doi: 10.1161/CIRCULATIONAHA.105.538934
144. Niu XL, Madamanchi NR, Vendrov AE, Tchivilev I, Rojas M, Madamanchi C, et al. Nox activator 1: A potential target for modulation of vascular reactive oxygen species in atherosclerotic arteries. Circulation. (2010) 121:549–59. doi: 10.1161/CIRCULATIONAHA.109.908319
145. Orr AW, Woolard MD. Cardiovascular disease is obNOXious: new insights into NoxA1 in smooth muscle phenotype. Redox Biol. (2019) 22:101081. doi: 10.1016/j.redox.2018.101081
146. Deliri H, McNamara CA. Nox 4 regulation of vascular smooth muscle cell differentiation marker gene expression. Arterioscler Thromb Vasc Biol. (2007) 27:12–4. doi: 10.1161/01.ATV.0000254154.43871.50
147. Tong X, Khandelwal AR, Qin Z, Wu X, Chen L, Ago T, et al. Role of smooth muscle Nox4-based NADPH oxidase in neointimal hyperplasia. J Mol Cell Cardiol. (2015) 89:185–94. doi: 10.1016/j.yjmcc.2015.11.013
148. Montezano AC, Camargo LDL, Persson P, Rios FJ, Harvey AP, et al. NADPH Oxidase 5 Is a pro-contractile nox isoform and a point of cross-talk for calcium and redox signaling-implications in vascular function. J Am Heart Assoc. (2018) 7:9388. doi: 10.1161/JAHA.118.009388
149. Jay DB, Papaharalambus CA, Seidel-Rogol B, Dikalova AE, Lassègue B, Griendling KK. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic Biol Med. (2008) 45:329–35. doi: 10.1016/j.freeradbiomed.2008.04.024
150. Majesky MW, Dong XR, Hoglund V, Mahoney WM, Daum G. The adventitia: a dynamic interface containing resident progenitor cells. Arterioscler Thromb Vasc Biol. (2011) 31:1530–9. doi: 10.1161/ATVBAHA.110.221549
151. Kramann R, Goettsch C, Wongboonsin J, Iwata H, Schneider RK, Kuppe C, et al. Adventitial MSC-like cells are progenitors of vascular smooth muscle cells and drive vascular calcification in chronic kidney disease. Cell Stem Cell. (2016) 19:628–42. doi: 10.1016/j.stem.2016.08.001
152. Meijles DN, Pagano PJ. Recent advances in hypertension: nox and inflammation in the vascular adventitia. Hypertension. (2015) 67:14–9. doi: 10.1161/HYPERTENSIONAHA.115.03622
153. Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, Quinn MT. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc Natl Acad Sci USA. (1997) 94:14483–8. doi: 10.1073/pnas.94.26.14483
154. Roufaiel M, Gracey E, Siu A, Zhu SN, Lau A, Ibrahim H, et al. CCL19-CCR7-dependent reverse transendothelial migration of myeloid cells clears Chlamydia muridarum from the arterial intima. Nat Immunol. (2016) 17:1263–72. doi: 10.1038/ni.3564
155. Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu ZG. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. (2013) 23:898–914. doi: 10.1038/cr.2013.75
156. Marzaioli V, Hurtado-Nedelec M, Pintard C, Tlili A, Marie JC, Monteiro RC, et al. NOX5 and p22phox are 2 novel regulators of human monocytic differentiation into dendritic cells. Blood. (2017) 130:1734–45. doi: 10.1182/blood-2016-10-746347
157. Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-κB-mediated inflammation in macrophages. Circ Res. (2014) 114:421–33. doi: 10.1161/CIRCRESAHA.114.302153
158. Kushiyama A, Okubo H, Sakoda H, Kikuchi T, Fujishiro M, Sato H, et al. Xanthine oxidoreductase is involved in macrophage foam cell formation and atherosclerosis development. Arterioscler Thromb Vasc Biol. (2012) 32:291–8. doi: 10.1161/ATVBAHA.111.234559
159. Ives A, Nomura J, Martinon F, Roger T, LeRoy D, Miner JN, et al. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat Commun. (2015) 6:6555. doi: 10.1038/ncomms7555
160. Mehta NU, Grijalva V, Hama S, Wagner A, Navab M, Fogelman AM, et al. Apolipoprotein E-/- mice lacking hemopexin develop increased atherosclerosis via mechanisms that include oxidative stress and altered macrophage function. Arterioscler Thromb Vasc Biol. (2016) 36:1152–63. doi: 10.1161/ATVBAHA.115.306991
161. Ali ZA, De Perez VJ, Yuan K, Orcholski M, Pan S, Qi W, et al. Oxido-reductive regulation of vascular remodeling by receptor tyrosine kinase ROS1. J Clin Invest. (2014) 124:5159–74. doi: 10.1172/JCI77484
162. Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, et al. Endothelial NADPH oxidase 4 protects ApoE-/- mice from atherosclerotic lesions. Free Radic Biol Med. (2015) 89:1–7. doi: 10.1016/j.freeradbiomed.2015.07.004
163. Wu R-F, Ma Z, Liu Z, Terada LS. Nox4-Derived H2O2 mediates endoplasmic reticulum signaling through local ras activation. Mol Cell Biol. (2010) 30:3553–68. doi: 10.1128/mcb.01445-09
164. Martin D, Li Y, Yang J, Wang G, Margariti A, Jiang Z, et al. Unspliced X-box-binding protein 1 (XBP1) protects endothelial cells from oxidative stress through interaction with histone deacetylase 3. J Biol Chem. (2014) 289:30625–34. doi: 10.1074/jbc.M114.571984
165. Barman SA, Fulton D. Adventitial fibroblast Nox4 expression and ROS signaling in pulmonary arterial hypertension. Adv Exp Med Biol. (2017) 967:1–11. doi: 10.1007/978-3-319-63245-2_1
166. Haurani MJ, Pagano PJ. Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: bellwether for vascular disease? Cardiovasc Res. (2007) 75:679–89. doi: 10.1016/j.cardiores.2007.06.016
167. Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. (2011) 79:944–56. doi: 10.1038/ki.2010.516
168. Baker AH, Péault B. A Gli(1)ttering role for perivascular stem cells in blood vessel remodeling. Cell Stem Cell. (2016) 19:563–5. doi: 10.1016/j.stem.2016.10.011
169. Tang CT, Lin XL, Wu S, Liang Q, Yang L, Gao YJ, et al. NOX4-driven ROS formation regulates proliferation and apoptosis of gastric cancer cells through the GLI1 pathway. Cell Signal. (2018) 46:52–63. doi: 10.1016/j.cellsig.2018.02.007
170. Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. (2010) 106:383–90. doi: 10.1161/CIRCRESAHA.109.210781
171. Kigawa Y, Miyazaki T, Lei X-F, Kim-Kaneyama J-R, Miyazaki A. Functional heterogeneity of nadph oxidases in atherosclerotic and aneurysmal diseases. J Atheroscler Thromb. (2016) 24:1–13. doi: 10.5551/jat.33431
172. Tumurkhuu G, Shimada K, Dagvadorj J, Crother TR, Zhang W, Luthringer D, et al. Ogg1 -Dependent DNA repair regulates NLRP3 inflammasome and prevents atherosclerosis. Circ Res. (2016) 119:e76–90. doi: 10.1161/CIRCRESAHA.116.308362
173. Wang D, Li LAK, Dai T, Wang A, Li S. Adult stem cells in vascular remodeling. Theranostics. (2018) 8:815–29. doi: 10.7150/THNO.19577
174. Cieślar-Pobuda A, Yue J, Lee H-C, Skonieczna M, Wei Y-H. ROS and oxidative stress in stem cells. Oxid Med Cell Longev. (2017) 2017:1–2. doi: 10.1155/2017/5047168
175. Di Luca M, Fitzpatrick E, Liu W, Morrow D, Redmond EM, Cahill PA. Characterisation of resident multipotent vascular stem cells (MVSCS) from susceptible and non-susceptible arteriosclerotic regions of the mouse aorta. Heart. (2018) 104:A10. doi: 10.1136/heartjnl-2018-scf.25
176. Orciani M, Gorbi S, Benedetti M, Di Benedetto G, Mattioli-Belmonte M, Regoli F, et al. Oxidative stress defense in human-skin-derived mesenchymal stem cells versus human keratinocytes: different mechanisms of protection and cell selection. Free Radic Biol Med. (2010) 49:830–8. doi: 10.1016/j.freeradbiomed.2010.06.007
177. Valle-Prieto A, Conget PA. Human mesenchymal stem cells efficiently manage oxidative stress. Stem Cells Dev. (2010) 19:1885–93. doi: 10.1089/scd.2010.0093
178. Park SG, Kim JH, Xia Y, Sung J-H. Generation of reactive oxygen species in adipose-derived stem cells: friend or foe? Expert Opin Ther Targets. (2011) 15:1297–306. doi: 10.1517/14728222.2011.628315
179. Bigarella CL, Liang R, Ghaffari S. Stem cells and the impact of ROS signaling. Development. (2014) 141:4206–18. doi: 10.1242/dev.107086
180. Lee J, Cho YS, Jung H, Choi I. Pharmacological regulation of oxidative stress in stem cells. Oxid Med Cell Longev. (2018) 2018:1–13. doi: 10.1155/2018/4081890
181. Sela M, Tirza G, Ravid O, Volovitz I, Solodeev I, Friedman O, et al. NOX1-induced accumulation of reactive oxygen species in abdominal fat-derived mesenchymal stromal cells impinges on long-term proliferation. Cell Death Dis. (2015) 6:e1728–e1728. doi: 10.1038/cddis.2015.84
182. Tyurin-Kuzmin PA, Zhdanovskaya ND, Sukhova AA, Sagaradze GD, Albert EA, Ageeva LV, et al. Nox4 and duox1/2 mediate redox activation of mesenchymal cell migration by PDGF. PLoS ONE. (2016) 11:e0154157. doi: 10.1371/journal.pone.0154157
183. Kim JH, Park SG, Song SY, Kim JK, Sung JH. Reactive oxygen species-responsive miR-210 regulates proliferation and migration of adipose-derived stem cells via PTPN2. Cell Death Dis. (2013) 4:e588. doi: 10.1038/cddis.2013.117
184. De Barros S, Dehez S, Arnaud E, Barreau C, Cazavet A, Perez G, et al. Aging-related decrease of human ASC angiogenic potential is reversed by hypoxia preconditioning through ROS production. Mol Ther. (2013) 21:399–408. doi: 10.1038/mt.2012.213
185. Song H, Cha MJ, Song BW, Kim IK, Chang W, Lim S, et al. Reactive oxygen species inhibit adhesion of mesenchymal stem cells implanted into ischemic myocardium via interference of focal adhesion complex. Stem Cells. (2010) 28:555–63. doi: 10.1002/stem.302
186. Borodkina A, Shatrova A, Abushik P, Nikolsky N, Burova E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging. (2014) 6:481–95. doi: 10.18632/aging.100673
187. Guo Y-L, Chakraborty S, Rajan SS, Wang R, Huang F. Effects of oxidative stress on mouse embryonic stem cell proliferation, apoptosis, senescence, and self-renewal. Stem Cells Dev. (2010) 19:1321–31. doi: 10.1089/scd.2009.0313
188. Lee JS, Lee MO, Moon BH, Shim SH, Fornace AJ, Cha HJ. Senescent growth arrest in mesenchymal stem cells is bypassed by Wip1-mediated downregulation of intrinsic stress signaling pathways. Stem Cells. (2009) 27:1963–75. doi: 10.1002/stem.121
189. Wu C-C, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. (2013) 19:546–58. doi: 10.1089/ars.2012.4905
190. Ghosh D, McGrail DJ, Dawson MR. TGF-β1 Pretreatment improves the function of mesenchymal stem cells in the wound bed. Front Cell Dev Biol. (2017) 5:28. doi: 10.3389/fcell.2017.00028
191. Tamama K, Fan VH, Griffith LG, Blair HC, Wells A. Epidermal growth factor as a candidate for ex vivo expansion of bone marrow-derived mesenchymal stem cells. Stem Cells. (2005) 24:686–95. doi: 10.1634/stemcells.2005-0176
192. Gustafson B, Eliasson B, Smith U. Thiazolidinediones increase the wingless-type MMTV integration site family (WNT) inhibitor Dickkopf-1 in adipocytes: a link with osteogenesis. Diabetologia. (2010) 53:536–40. doi: 10.1007/s00125-009-1615-1
193. Tsutsumi S, Shimazu A, Miyazaki K, Pan H, Koike C, Yoshida E, et al. Retention of multilineage differentiation potential of mesenchymal cells during proliferation in response to FGF. Biochem Biophys Res Commun. (2001) 288:413–9. doi: 10.1006/bbrc.2001.5777
194. Cho HH, Shin KK, Kim YJ, Song JS, Kim JM, Bae YC, et al. NF-κB activation stimulates osteogenic differentiation of mesenchymal stem cells derived from human adipose tissue by increasing TAZ expression. J Cell Physiol. (2010) 223:168–77. doi: 10.1002/jcp.22024
195. Guérit D, Brondello J-M, Chuchana P, Philipot D, Toupet K, Bony C, et al. FOXO3A regulation by miRNA-29a controls chondrogenic differentiation of mesenchymal stem cells and cartilage formation. Stem Cells Dev. (2014) 23:1195–205. doi: 10.1089/scd.2013.0463
196. Kumar S, Sun X, Wedgwood S, Black SM. Hydrogen peroxide decreases endothelial nitric oxide synthase promoter activity through the inhibition of AP-1 activity. Am J Physiol Cell Mol Physiol. (2008) 295:L370–7. doi: 10.1152/ajplung.90205.2008
197. Xiao Q, Wang G, Luo Z, Xu Q. The mechanism of stem cell differentiation into smooth muscle cells. Thromb Haemost. (2010) 104:440–8. doi: 10.1160/TH09-11-0794
198. Jeon ES, Moon HJ, Lee MJ, Song HY, Kim YM, Bae YC, et al. Sphingosylphosphorylcholine induces differentiation of human mesenchymal stem cells into smooth-muscle-like cells through a TGF- -dependent mechanism. J Cell Sci. (2006) 119:4994–5005. doi: 10.1242/jcs.03281
199. Baek S, Lee KP, Jung SH, Cui L, Ko K, Kim B, et al. DJ-1 Regulates differentiation of human mesenchymal stem cells into smooth muscle-like cells in response to sphingosylphosphorylcholine. Proteomics. (2017) 17:1700208. doi: 10.1002/pmic.201700208
200. Xiao Q, Luo Z, Pepe AE, Margariti A, Zeng L, Xu Q. Embryonic stem cell differentiation into smooth muscle cells is mediated by Nox4-produced H 2 O 2. Am J Physiol Physiol. (2009) 296:C711–23. doi: 10.1152/ajpcell.00442.2008
201. Song SH, Kim K, Park JJ, Min KH, Suh W. Reactive oxygen species regulate the quiescence of CD34-positive cells derived from human embryonic stem cells. Cardiovasc Res. (2014) 103:147–55. doi: 10.1093/cvr/cvu106
202. Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, et al. Endothelial to mesenchymal transition in cardiovascular disease: JACC State-of-the-art review. J Am Coll Cardiol. (2019) 73:190–209. doi: 10.1016/j.jacc.2018.09.089
203. Thuan DTB, Zayed H, Eid AH, Abou-Saleh H, Nasrallah GK, Mangoni AA, et al. A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front Immunol. (2018) 9:1985. doi: 10.3389/fimmu.2018.01985
204. Anea CB, Cheng B, Sharma S, Kumar S, Caldwell RW, Yao L, et al. Increased superoxide and endothelial NO synthase uncoupling in blood vessels of Bmal1-knockout mice. Circ Res. (2012) 111:1157–65. doi: 10.1161/CIRCRESAHA.111.261750
205. Khapre RV, Kondratova AA, Susova O, Kondratov RV. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle. (2011) 10:4162–9. doi: 10.4161/cc.10.23.18381
206. Zhu M, Tang H, Tang X, Ma X, Guo D, Chen F. BMAL1 suppresses ROS-induced endothelial-to-mesenchymal transition and atherosclerosis plaque progression via BMP signaling. Am J Transl Res. (2018) 10:3150–61.
207. Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial- mesenchymal transition. J Am Coll Cardiol. (2014) 63:2734–41. doi: 10.1016/j.jacc.2014.02.572
208. Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGF -induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci. (2008) 121:3317–24. doi: 10.1242/jcs.028282
209. Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM. Snail Is a direct target of hypoxia-inducible factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem. (2015) 290:16553–664. doi: 10.1074/jbc.M115.636944
210. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Sao Paulo Med J. (2015) 133:164. doi: 10.1590/1516-3180.20151332T1
211. Leopold JA. Antioxidants and coronary artery disease: From pathophysiology to preventive therapy. Coron Artery Dis. (2015) 26:176–83. doi: 10.1097/MCA.0000000000000187
212. Leermakers ET, Darweesh SK, Baena CP, Moreira EM, Van Lent DM, Tielemans MJ, et al. The effects of lutein on cardiometabolic health across the life course: a systematic review and meta-analysis. Am J Clin Nutr. (2016) 103:481–94. doi: 10.3945/ajcn.115.120931
213. Atkinson T, Packwood W, Xie A, Liang S, Qi Y, Ruggeri Z, et al. Assessment of novel antioxidant therapy in atherosclerosis by contrast ultrasound molecular imaging. J Am Soc Echocardiogr. (2018) 31:1252–1259. doi: 10.1016/j.echo.2018.07.017
214. Shaban S, El-Husseny MWA, Abushouk AI, Salem AMA, Mamdouh M, Abdel-Daim MM. Effects of antioxidant supplements on the survival and differentiation of stem cells. Oxid Med Cell Longev. (2017) 2017:1–16. doi: 10.1155/2017/5032102
215. Karppi J, Kurl S, Laukkanen JA, Rissanen TH, Kauhanen J. Plasma carotenoids are related to intima - media thickness of the carotid artery wall in men from eastern Finland. J Intern Med. (2011) 270:478–85. doi: 10.1111/j.1365-2796.2011.02401.x
216. Gey KF, Puska P, Jordan P, Moser UK. Inverse correlation between plasma vitamin E and mortality from ischemic heart disease in cross-cultural epidemiology. Am J Clin Nutr. (1991) 53:326S−34. doi: 10.1093/ajcn/53.1.326S
217. D'Odorico A, Martines D, Kiechl S, Egger G, Oberhollenzer F, Bonvicini P, Sturniolo GC, et al. High plasma levels of α- and β-carotene are associated with a lower risk of atherosclerosis. Atherosclerosis. (2000) 153:231–9. doi: 10.1016/S0021-9150(00)00403-2
218. Gey KF, Puska P. Plasma Vitamins E and A inversely correlated to mortality from ischemic heart disease in cross-cultural epidemiology. Ann N Y Acad Sci. (1989) 570:268–82. doi: 10.1111/j.1749-6632.1989.tb14926.x
219. Espinola-Klein C, Rupprecht HJ, Bickel C, Schnabel R, Genth-Zotz S, Torzewski M, et al. Glutathione peroxidase-1 activity, atherosclerotic burden, and cardiovascular prognosis. Am J Cardiol. (2007) 99:808–12. doi: 10.1016/j.amjcard.2006.10.041
220. Goszcz K, Deakin SJ, Duthie GG, Stewart D, Megson IL. Bioavailable concentrations of delphinidin and its metabolite, gallic acid, induce antioxidant protection associated with increased intracellular glutathione in cultured endothelial cells. Oxid Med Cell Longev. (2017) 2017:1–17. doi: 10.1155/2017/9260701
Keywords: NOX, NAPDH oxidase, smooth muscle (physiology), endothelial cells, adventitial cells, stem cells, intimal thickening, arteriosclerosis
Citation: Burtenshaw D, Kitching M, Redmond EM, Megson IL and Cahill PA (2019) Reactive Oxygen Species (ROS), Intimal Thickening, and Subclinical Atherosclerotic Disease. Front. Cardiovasc. Med. 6:89. doi: 10.3389/fcvm.2019.00089
Received: 21 March 2019; Accepted: 14 June 2019;
Published: 02 August 2019.
Edited by:
Abdelali Agouni, Qatar University, QatarReviewed by:
Jenny Jongstra-Bilen, University Health Network (UHN), CanadaSimona Adriana Manea, Institute of Cellular Biology and Pathology (ICBP), Romania
Copyright © 2019 Burtenshaw, Kitching, Redmond, Megson and Cahill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul A. Cahill, cGF1bC5jYWhpbGwmI3gwMDA0MDtkY3UuaWU=
†These authors have contributed equally to this work