 Veronica Dusi
Veronica Dusi Gaetano Maria De Ferrari
Gaetano Maria De Ferrari Luigi Pugliese
Luigi Pugliese Peter J. Schwartz
Peter J. Schwartz- 1Department of Molecular Medicine, Section of Cardiology, University of Pavia, Pavia, Italy
- 2Cardiac Intensive Care Unit, Arrhythmia and Electrophysiology and Experimental Cardiology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
- 3Unit of General Surgery 2, Department of Surgery, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
- 4Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Istituto Auxologico Italiano, IRCCS, Milan, Italy
Left cardiac sympathetic denervation (LCSD) is a surgical antiadrenergic intervention with a strong antiarrhythmic effect, supported by preclinical as well as clinical data. The mechanism of action of LCSD in structurally normal hearts with increased arrhythmic susceptibility (such as those of patients with channelopathies) is not limited to the antagonism of acute catecholamines release in the heart. LCSD also conveys a strong anti-fibrillatory action that was first demonstrated over 40 years ago and provides the rationale for its use in almost any cardiac condition at increased risk of ventricular fibrillation. The molecular mechanisms involved in the final antiarrhythmic effect of LCSD turned out to be much broader than anticipated. Beside the vagotonic effect at different levels of the neuraxis, other new mechanisms have been recently proposed, such as the antagonism of neuronal remodeling, the antagonism of neuropeptide Y effects, and the correction of neuronal nitric oxide synthase (nNOS) imbalance. The beneficial effects of LCSD have never been associated with a detectable deterioration of cardiac performance. Finally, patients express a high degree of satisfaction with the procedure. In this review, we focus on the rationale, results and our personal approach to LCSD in patients with channelopathies such as long QT syndrome and catecholaminergic polymorphic ventricular tachycardia.
Introduction
The management of patients at risk of life-threatening arrhythmias is challenging, more now than ever. On one hand, our capability to identify the subjects at higher risk of sudden cardiac death (SCD) is still limited (1). On the other, the widespread availability of implantable cardioverter defibrillators (ICDs) is a double edge sword. Not only because of the risk of side effects but also because in peculiar settings ICDs may even become pro-arrhythmic. Additionally, recurrent ICD shocks have a dramatic impact on the quality of life. These drawbacks are particularly evident in young patients with inherited arrhythmogenic disorders. The management of these subjects is further complicated by the unlikely feasibility of randomized clinical trials in this setting, which may give the wrong perception of lack of strong evidence for a specific treatment. Left cardiac sympathetic denervation (LCSD) is an extremely effective but still underutilized anti-adrenergic therapy. LCSD has a strong physiological rationale, combined with consistent preclinical results, and clinical data from well-conducted multicenter registries.
In this review we will first summarize the history and the antiarrhythmic rationale for LCSD, including well-established antiarrhythmic mechanisms as well as potential new mechanisms. Then, we will present the clinical results of LCSD in Long QT Syndrome (LQTS) and Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT), including both secondary and primary prevention. Finally, we will provide our approach for LCSD use in LQTS and CPVT.
Organization of Cardiac Sympathetic Nervous System in Humans
The two opposite branches of cardiac autonomic nervous system (ANS), namely the sympathetic and the parasympathetic nervous system, share a common embryological origin from the neuronal crest (2). The sympathetic cardiac ANS follows typical patterns in most people, although variants are seen (3, 4). It is constituted by the mediastinal cardiac plexus, the paravertebral sympathetic ganglia, the dorsal root ganglia (DRG), the spinal cord, and the brain stem. Cardiac sympathetic afferent fibers provide beat-to-beat information centrally as their sensory endings are mechanoreceptors (5). The extracardiac afferent stations, containing pseudounipolar nerve cells, are the DRG from C7 to T4 spinal cord level. Of note, cardiac sympathetic afferent fibers travel across the paravertebral sympathetic ganglia (without having synapsis) before reaching the DRG. Efferent sympathetic preganglionic neurons have their soma in the intermediolateral column of spinal cord and synapses on postganglionic neurons located in the lower cervical and upper thoracic paravertebral ganglia. The lowest cervical ganglion (C8) and the highest thoracic ganglion (T1) are generally fused bilaterally to constitute the left and the right stellate ganglia (also referred to as cervicothoracic ganglia). In <3% of human sympathetic chains, the second thoracic ganglion (T2) is fused as well, constituting a trilobal (C8-T1-T2) stellate ganglion (3). The stellate ganglia convey a consistent amount of cardiac sympathetic postganglionic fibers. The remaining is provided by T2–4 paravertebral ganglia. Figure 1 summarizes cardiac nervous system organization in humans.
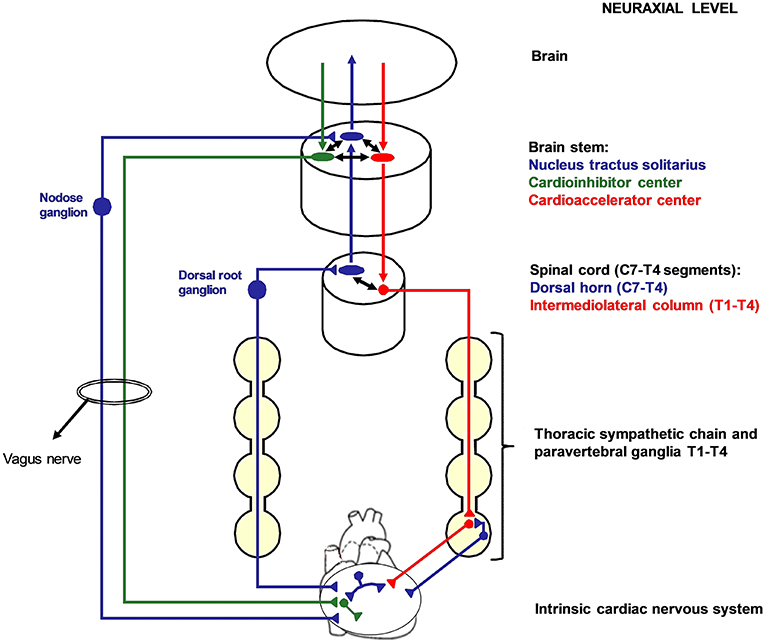
Figure 1. Cardiac nervous system organization in humans. Blue: afferent nervous system with its ganglia: nodose ganglia and C7-T4 dorsal root ganglia (DRG). Green: parasympathetic efferent nervous system. Red: sympathetic efferent nervous system. All the afferent and efferent structures outside the central nervous systems are bilateral, although mostly represented as unilateral for simplicity. Cardiac afferent fibers traveling across the paravertebral sympathetic ganglia (usually referred to as cardiac sympathetic afferent fibers) directly reach the DRG without having synapsis before. These fibers mediate cardio-cardiac sympathoexcitatory spinal reflexes that significantly increase the sympathetic output to the heart. Left cardiac sympathetic denervation (LCSD) consists in the removal of the left thoracic sympathetic chain and paravertebral ganglia from T1 to T4. Since ipsilateral DRG are spared by LCSD, a left afferent reinnervation from the DRG to the heart is theoretically possible with time. On the other hand, the left efferent sympathetic system from T1 to T4 is interrupted at a preganglionic level; therefore, no ipsilateral efferent sympathetic reinnervation is possible after LCSD.
Historical Prospective
In 1899, (6) Francois-Frank was the first to suggest that the removal of cervicothoracic sympathetic nervous system could prevent angina pectoris episodes. The first intervention was performed in 1916 by Jonnesco (7). He removed the left stellate ganglion (LSG) in a patient suffering incapacitating angina associated with cardiac arrhythmias, with effective and long-lasting suppression of both conditions. This pioneering intervention was strongly criticized due to the potential detrimental effects of depriving patients of the warning signal represented by pain. Moreover, the consequences of left stellectomy on coronary flow were still unclear. In 1929, Leriche and Fontaine (8) demonstrated that the sympathetic nerves exert a vasoconstrictive effect on the coronary arteries and not a vasodilator one, as previously thought. Subsequently, several clinical studies were performed in both Europe and the USA, confirming that left stellectomy was able to prevent anginal attacks (9), and to improve exercise tolerance (10). Concerning the optimal extension of the procedure, cervico-thoracic denervation (removal of the stellate ganglion and T2–T4 thoracic ganglia) proved to be the most effective. Finally, in the 60s, despite its clear efficacy, left cardiac sympathetic denervation (LCSD) was progressively abandoned for the treatment of angina due to the widespread usage of surgical coronary artery bypass graft and β-adrenergic-receptor blockers (11).
Except for some case reports (12, 13) the antiarrhythmic potential of cardiac sympathetic denervation in humans remained largely unexplored until the 70s. In 1971, Moss and McDonald (14) were the first to report LCSD in a LQTS patient with recurrent syncopal episodes. The rationale was based on a canine study (15) showing a consistent QT interval prolongation after either right stellate ganglionectomy or LSG stimulation. The patient underwent removal of the sympathetic ganglia from C7 to T2, including the entire LSG. Besides the suppression of arrhythmias, a persistent QT interval reduction was noticed. Subsequently, other groups tried to reproduce the beneficial effects on QT interval through the reversible percutaneous block of the LSG, with inconsistent results (16). Of note, at that time the appearance of Horner syndrome was considered as a good marker of the effective blockade of cardiac nerves. On the contrary, as pointed out already in 1975 (17), the Horner syndrome simply indicates an effective blockade of the sympathetic fibers traveling in the upper part of the stellate ganglion and innervating the eye. It does not necessarily indicate the block of the sympathetic fibers reaching the heart. Moreover, unlike in dogs and cats, in humans cardiac sympathetic innervation is not entirely provided by the stellate ganglia.
A better understanding of the rationale for LCSD in LQTS originated from the work by Schwartz and associates. Schwartz started from the observation in his first LQTS patient that sympathetic activation was triggering macroscopic T wave alternans, and he then reproduced in cats both QT prolongation and T-wave alternans by electrical stimulation of the LSG (18). On this basis, the young patient was treated with LCSD (remaining asymptomatic more than 40 years after) and sympathetic imbalance with left-sided dominance was proposed as the pathophysiological mechanisms of LQTS (17, 19). This concept prompted a large series of experimental studies investigating the consequences of unilateral (right or left) cardiac sympathetic denervation (20–22).
Antiarrhythmic Rationale and Mechanisms of Action of Cardiac Sympathetic Denervation
Antiadrenergic Effects
In 1976, Schwartz et al. showed in anesthetized dogs (20) that ischemia-related arrhythmias were increased by right stellate ganglion block and decreased by LSG block. In a conscious canine model (21), still considered as the most-clinically relevant experimental model of SCD (dogs with a healed myocardial infarction (MI) exposed to a brief coronary artery occlusion while exercising on a treadmill) left stellectomy confirmed its protective effect. The antagonism of ischemia-induced sympathetic activation (23) as well as the quantitative dominance of the left sided sympathetic nerves over the right (22) were the first antiarrhythmic mechanisms proposed for the protection associated with LSG block or removal. Next came the demonstration that VF threshold, a reliable and quantitative marker of cardiac electrical stability, was lower after unilateral right stellectomy and much higher after left stellectomy (24). These animal data provide a solid rationale for LCSD which goes far beyond LQTS and ischemia-related arrhythmias and could extend to every cardiac condition characterized by an increased susceptibility to VF. A major mechanism contributing to the protection is the net decrease in norepinephrine (NE) released in the left ventricle during sympathetic neural activation. Of note, the neural release of NE is an extremely inhomogeneous phenomenon (25–27). Indeed, sympathetic nerve stimulation rather than circulating norepinephrine, modulates T-peak to T-end interval (an ECG marker of dispersion of repolarization) by increasing global dispersion of repolarization (25, 28). In turn, a spatially inhomogeneous ventricular repolarization is a very well-defined pro-arrhythmic marker, both for scar related arrhythmias (29) and for functional reentrant arrhythmias such as polymorphic ventricular tachycardia and VF (30). The temporal dispersion of ventricular repolarization is important as well and, together with spatial dispersion, may lead to T wave alternans, an ECG marker of high electrical instability, both in case of macro- (18) and of microvolt alternans (31). Besides acting on the arrhythmic substrate, NE, like epinephrine, also modulates the trigger. Not only does it enhance automaticity in pacemaker cells in both the atria and the ventricles (32), but it also increases triggered activity including both early (EAD) (33) and delayed (DAD) afterdepolarizations (34, 35). Finally, LCSD has α-adrenergic-receptor blocking properties. Indeed, similarly to the effect of α-adrenergic-receptor blockade, and opposite to that of β-Blockade, LCSD increases myocardial reactive hyperemia, an index of the capability of the coronary bed to dilate (36). In addition to providing the basis for the antianginal effect, this could contribute to the antiarrhythmic efficacy (37).
Vagotonic Effect
Animal studies clearly showed that LCSD is accompanied by a reflex increase in cardiac parasympathetic (vagal) efferent activity (38). In fact, LCSD interrupts the majority of centrally projecting cardiac sympathetic afferents, which have an inhibitory effect on the vagal outflow directed to the heart. In turn, experimental (39, 40) and clinical data (41, 42) showed that blunted vagal tone and reflexes can favor life-threatening arrhythmias; conversely, these arrhythmias can be counteracted in animals by direct vagal stimulation (43) or pharmacological activation (44). So far, the experience with direct vagal stimulation in humans is limited to heart failure patients (45–48). Accordingly, the vagotonic effect of LCSD is particularly relevant from an anti-arrhythmic point of view in conditions characterized by a chronic and progressive increase in sympathetic tone and a parallel decrease in central parasympathetic drive, such as myocardial infarction (MI) and heart failure (49). Some of these concepts are at the basis of an ongoing clinical trial which examines the potential benefit associated with LCSD in patients with advanced heart failure (50).
Other Antiarrhythmic Mechanisms
Antagonism of Neuronal Remodeling
In 2000, Cao et al. (51) demonstrated in dogs that an increased intra-cardiac sympathetic nerve regeneration (nerve sprouting), obtained by infusing nerve growth factor to the LSG, was associated with a greater susceptibility to spontaneous ventricular arrhythmias. Of note, an intracardiac neuronal remodeling including both denervation and nerve sprouting (52) may occur after any kind of myocardial injury (53). Several animal studies consistently showed the high arrhythmic susceptibility of the denervated myocardium (54, 55). Similarly, in patients with cardiomyopathy and an ejection fraction ≤35%, the degree of cardiac sympathetic denervation quantified either by cardiac iodine-123 metaiodobenzylguanidine (123I- MIBG) imaging (56) or by positron emission tomography with 11 C-meta-hydroxyephedrine (11C-HED PET) (57) was significantly associated with ventricular arrhythmic risk. The process of neuronal remodeling is not limited to the heart, involving also extracardiac structures such as the sympathetic thoracic ganglia and the DRG (58). Myocardial infarction in animal models, independently of the site, is associated with an increase in nerve density, neuronal size, and neuropeptide Y expression in both the left and right stellate ganglia (59, 60). The same remodeling was described in humans. In 2012, Ajijola et al. (61) reported a significant neuronal enlargement and an increased synaptic density in the LSG of patients with refractory ventricular arrhythmias and structural heart disease undergoing LCSD. A few years later the same group further enriched the description of the sympathetic ganglia in patients with cardiomyopathy and refractory ventricular arrhythmias undergoing cardiac sympathetic denervation (62) showing the presence of a remarkable inflammatory cells infiltration (CD3+ T cells and neutrophils), combined with neurochemical remodeling, oxidative stress, and satellite glial cell activation. Of note, among the 16 patients studied (mean 45 ± 15 years), 5 had no macroscopically clear myocardial scar at pre-operatory multimodal imaging. Almost no signs of local inflammation or neuronal remodeling were observed in the stellate ganglia used as controls, obtained from 8 organ donors (mean 28 ± 8 years) with normal hearts deceased either for traumatic reasons or by natural causes.
These findings raise the intriguing question about the potential primary role of sympathetic ganglia inflammation in triggering adrenergic related ventricular arrhythmias in structurally normal hearts. Rizzo et al. (63) found mild but distinct inflammatory infiltrates composed of CD3+ and CD8+ T cells and macrophages in the LSG of 12 LQTS/ CPVT patients (mean 23 ± 17 years). They were all heavily symptomatic patients who received LCSD in secondary prevention. The authors specifically searched for neurotropic viruses as a potential trigger for the immune cell infiltration, with negative findings. They proposed that T-cell–mediated cytotoxicity toward ganglion cells may prompt an increase in sympathetic efferent activity toward the heart, therefore acting as a trigger and/or an enhancer of electrical instability in patients already predisposed to arrhythmias, as it occurs in LQTS and CPVT patients. Of note, as pointed out by Moss et al. (64) in the editorial comments of the paper, all patients had either recurrent syncopal episodes or many ICD shocks before the ganglionectomy, although the time frame between the last events and LCSD was not provided by the authors. Syncopal events are associated with transient generalized hypoperfusion, while ICD shocks can damage the myocardium and the neuronal fibers (65). Therefore, the mild auto immune mediated ganglionic remodeling observed by Rizzo et al. could be the consequence rather than the cause of the arrhythmic episodes. Moreover, the stellate ganglia used as controls, obtained from 10 accidently deceased patients (mean 35 ± 18 years), showed signs of inflammatory activity with the same immunohistological pattern, albeit to a lesser extent. Finally, no specific data supporting an increased sympathetic neuronal activity, such as increased neuronal size, increased synaptic density or a neurochemical shift in adrenergic phenotype were provided, as opposed to the neuronal hypertrophy and adrenergic shift demonstrated by Ajijola et al. (61, 62) in the stellate ganglia of patients with cardiomyopathy (even without overt scar) and intractable ventricular arrhythmias.
When interpreting these results, it's important to remember that cardiac sympathetic ganglia are not routinely evaluated by pathologist in the postmortem examination. Therefore, histological findings from these tissues among sudden arrhythmic death victims are lacking. On the other hand, a direct and non-invasive anatomopathological assessment of cardiac sympathetic ganglia in living patients is challenging both with labeled positron emission tomography tracers and with magnetic resonance. Indirect information about ongoing extracardiac neuronal remodeling processes can be obtained through cardiac 123I-MIBG or 11C-HED PET images, which are by the way unable to distinguish between anatomical (related to a reduced fiber density) rather than purely functional neuronal fibers abnormalities. As a matter of fact, an abnormal 123I-MIBG cardiac scintigraphy as compared with healthy controls was reported in LQTS (66, 67) patients as well as in patients with idiopathic ventricular tachycardia and fibrillation (68).
Overall, the intriguing question about the potential pro arrhythmic role of sympathetic ganglia inflammatory processes in channelopathies is still largely unsolved and should be properly assessed by larger studies. Nevertheless, cardiac 123I-MIBG data seem to support the presence of primary sympathetic nervous system abnormalities in these patients.
Antagonism of Neuropeptide Y
Neurotramitters other than NE released by sympathetic efferent fibers are an area of intense research. Co-release mainly occurs during high-level neuronal stimulation (69). The most studied sympathetic co-transmitter is neuropeptide Y (NPY) that has a long biological half-life and can be measured in peripheral blood (70). NYP was shown to inhibit acetylcholine (ACh) release from cardiac vagal postganglionic nerves (71–74) through Y2 receptors activation (75). NPY may also act on Y1 receptors on ventricular cardiomyocytes, affecting their electrophysiological properties. Optical mapping experiments in rats showed that NPY steepens the action potential duration restitution curve (76). Moreover, in Langendorff-perfused rat hearts with intact innervation only the combination of Y1 receptor antagonist with metoprolol was able to fully prevent the fall in VF threshold produced by prolonged high-frequency stellate stimulation (76). Finally, NPY is also a potent vasoconstrictor (77). In man, several studies already reported that plasmatic NPY levels rise following acute coronary syndromes (78) and in heart failure, showing a positive correlation with severity of heart failure and 1 year mortality (79, 80).
Correction of nNOS Imbalance
An additional neurotransmitter, which has recently gained attention, is neuronal nitric oxide (nNO). Neuronal nitric oxide synthase (nNOS), together with its adaptor protein (CAPON, codified by the gene NOS1AP, nitric oxide synthase 1 adaptor protein), is located in both intrinsic cardiac vagal neurons and postganglionic sympathetic neurons of the stellate ganglia. It acts locally as an intrinsic neuromodulator i.e., it is not released in the synaptic space but it acts in the synaptic cleft via stimulation of soluble guanylate cyclase, to generate cGMP. In turn, this prompts opposite effects in parasympathetic and sympathetic neurons. In parasympathetic neurons it leads to an increased release of Ach (81, 82), while in sympathetic neurons it causes a reduction in NE release (83, 84). Animal studies using viral vectors showed that an increase in nNOS may reverse impaired vagal (85) and exaggerated sympathetic drive (86, 87) in the spontaneously hypertensive rat. Moreover, in guinea pig overexpression of nNOS increased acetylcholine release and was associated with a trend of improved survival following MI (88). Interestingly, genetic studies not only consistently correlated genetic variation in NOS1AP with QT-interval duration in the general population (89–92), but also demonstrated their association with the risk for sudden death in general population (93) and the risk of drug-induced QT prolongation and ventricular arrhythmia (94). Additionally, NOS1AP was proved to be a genetic modifier in LQTS, both in a founder LQT1 population (95) and in a non-selected LQTS population including different genotypes (96). Of note, NOS1AP gene is also expressed at the cardiac level, and CAPON overexpression in isolated guinea pig myocytes causes attenuation of L-type calcium current, a slight increase in rapid delayed rectifier current (IKr), and a shortening of action potential (97). So far, an increase in L-type calcium current (which is also enhanced by sympathetic activation) due to CAPON under expression has been advocated as the main mechanism responsible for NOS1AP genetic variant impact on QT interval duration and arrhythmias susceptibility. Nevertheless, it is intriguing to speculate that in LQTS patients (as well as in the general population), even in absence of overt inflammatory changes within the stellate ganglia, CAPON under expression (on genetic bases) may lead to an increased NE release during sympathetic activation and therefore an increased arrhythmic risk. Of note, the disruption in CAPON expression in LQTS could also be the functional result of a mild ganglionitis rather than the cause of it, potentially contributing to explain the pro arrhythmic impact of the mild auto immune mediated ganglionitis described by Rizzo et al. (63).
Additional Effects of Cardiac Sympathetic Denervation on the Heart
Catecholamines, besides the arrhythmogenic potential, physiologically modulate nearly all cardiac functions, including inotropy, chronotropy, dromotropy, and lusitropy. Therefore, before systematically proposing LCSD in man, several experimental studies were performed in order to exclude any potential detrimental effect on the heart. In conscious dogs with a healed MI performing a submaximal exercise stress test, left ventricular contractility (assessed by dP/dt max) was not affected by left stellectomy (36). Moreover, LCSD did not reduce resting heart rate (HR) or chronotropic competence during effort. On the contrary, HR increase during exercise was slightly (6%) greater after LCSD. This apparently paradoxical effect was thought to be related to a controlateral reflex increase in right stellate ganglion activity. In fact, due to the asymmetric distributions of sympathetic cardiac nerves, the sinus node is under a predominant right-sided sympathetic control (98). In the same animal model (36) the maximal increase in HR during exercise was, respectively, 19 and 26% lower as compared to baseline (intact innervation) after bilateral and right only stellectomy. Finally, albeit no specific data about AV conduction were provided, the mean maximal HR reached (around 250 bpm) during effort after left stellectomy strongly argues against a significant impact of left stellectomy on dromotropy during sinus rhythm and in physiological conditions of sympathetic activation. This finding was in agreement with previous studies which showed that sympathetic innervation to the atria and the AV node is provided by both right and left sympathetic chain (99). Accordingly, recent data from patients with paroxysmal atrial fibrillation (AF) show equivalent electrophysiological effects of right and left stellate ganglion block (SGB) on both atria: unilateral temporary SGB with lidocaine slightly prolongs atrial effective refractory period and consistently reduces AF inducibility and AF episodes duration (100).
Finally, a last concern was that LCSD could lead to post-denervation supersensitivity, a pro-arrhythmic condition characterized by increased sensitivity of the left ventricle to catecholamines after complete denervation. From a theoretical point of view this possibility appeared unlikely, because right-sided sympathetic nerves (preserved after LCSD) are known to contribute to left ventricular innervation (101, 102). Animal studies confirmed that catecholamine stores in the myocardium were not completely depleted after LCSD (103, 104). Moreover, unilateral left stellectomy did not increase either dP/dt max or the incidence of ventricular arrhythmias in response to intravenous norepinephrine (105). Of note, LCSD is a preganglionic denervation; therefore no ipsilateral sympathetic efferent reinnervation is possible.
LCSD in Channelopathies
LCSD in Long QT Syndrome: Reported Results
The first large-scale evaluation of LCSD efficacy in LQTS was published in 1991 (106). Among the 85 reported patients, 99% were symptomatic before surgery, including 60% who suffered at least one aborted cardiac arrest (ACA). After LCSD, symptomatic patients decreased from 99 to 45% (P < 0.0001), and the mean number of cardiac events/patient dropped from 22 to 1. Of note, there were no ICDs. Therefore, this report truly reflects the impact of LCSD on SCD: it occurred in 8% of this high-risk group during 6 years of mean follow-up. The largest series of LQTS patients undergoing LCSD was reported in 2004 (107). As in the previous study, 99% of the patients were symptomatic before surgery, including 48% with a previous ACA and 75% with recurrent syncope despite maximum-dose β-Blockers. The majority were female (69%), the median age at surgery was 17 years and the mean QTc was 543 ± 65 ms. The average follow-up periods pre and post-LCSD were 5 and 8 years, respectively. After LCSD, 46% of the patients remained asymptomatic, syncope occurred in 31%, ACA in 16%, and SCD in 7%. Mean yearly number of cardiac events/patient dropped by 91% (P < 0.001). Among the 5 patients with a preoperative ICD the median number shocks/patient decreased from 25 to 0. Of note, 51 patients (35%) were genotyped, including 18 LQT1, 15 LQT2, 8 LQT3 and 9 patients with Jervell and Lange-Nielsen syndrome (JLN). As expected, LCSD appeared to be more effective in LQT1 than in LQT2. Despite the very limited numbers, patients with LQT3 and JLN did not seem to have a worse outcome compared with LQT1 patients. Finally, after LCSD a clinically significant mean reduction of QTc interval (39 ms) was noticed. Neither a preoperative QTc value ≥500 ms nor a change <40 ms were associated with a higher risk of recurrences. On the other hand, the persistence of a QTc ≥ 500 ms within 6 months from surgery appeared to carry a significantly higher risk of future events.
Subsequently, a large program of LCSD in LQTS was started by Ackerman at the Mayo Clinic, with equally positive results (108). In 2013, he reported a specific analysis on predictors of recurrences after LCSD in LQTS (109). They studied 52 consecutive LQTS patients undergoing LCSD between 2005 and 2010 at Mayo Clinic (23 LQT1, 9 LTQ2, 4 LQT3, 9 carrying multiple mutations, 3 JLN, and 4 genotype negative). All the procedures were performed using the minimally invasive, video-assisted thoracoscopic technique (VATS), and the sympathetic chain was removed from T1 to T4. Mean age at surgery was 10 years, 54% were female and mean QTc pre LCSD was 528 ± 74 ms. Most of them (61%) had LCSD as primary prevention because of either high-risk conditions or β-Blocker intolerance. This is a significant difference with the two previously reported populations and reflects the growing confidence in the benefit of the procedure. Overall, 12 subjects suffered cardiac events after LCSD (mean follow-up 3.6 years). Among them, only 5 (10%) had no discernible reduction of the arrhythmic episodes (true non-responders). These 5 high risk patients, all heavily symptomatic before LCSD, included 3 LQT3 patients and 2 LQT1 patients with multiple mutations. All of them had a very early onset of the disease (4 at birth, one in the first year of life) with QTc values above 600 ms. On the contrary, none of the 12 patients who received LCSD for β-Blocker intolerance experienced events during follow up.
In the following years, other centers all over the word started to perform LCSD and to report their results, overall confirming the positive post-procedural outcomes (110–112). The majority were small case series, yet in 2015 Waddell-Smith et al. (113) reported about 40 LQTS patients treated with thoracoscopic LCSD in New Zealand. LCSD related side effects and the quality of life after LCSD were the main topics analyzed. Most patients were female (70%) and LQT1 (57%), 11 were LQT2, 1 LQT3 and 5 had a negative genetic test. Half of the patients were completely asymptomatic before the procedure, and only 2 (5%) had surgery because of recurrences on β- Blockers. The two main indications for LCSD were β-Blocker intolerance or contraindication (35% of the patients) and β-Blocker non-adherence (25%). Interestingly, 10% of the patients specifically requested the procedure to their cardiologists either to increase their sense of protection or because of their desire to perform high level sports. These data confirm the diffusion and the increase in confidence in the procedure. During a median follow up of 2.5 years only 2 patients (5%), including 1 JLN, had arrhythmic events (syncopal episodes). All patients reported high levels of postoperative satisfaction. Table 1 summarizes indications and results of the largest case series reported of LCSD in LQTS with at least 1 year of follow up.
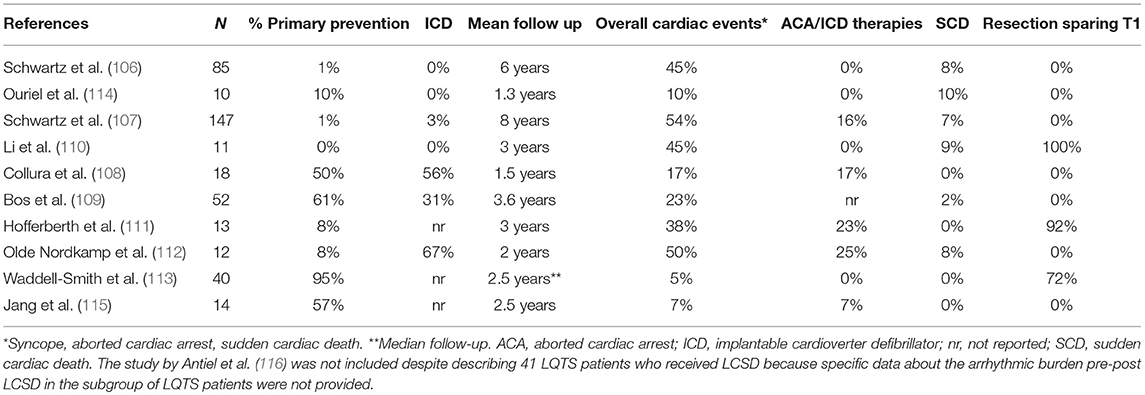
Table 1. Largest case series reported of LCSD in LQTS (at least 10 patients with at least 1 year of follow up).
LCSD in Long QT Syndrome: Our Approach
LCSD is now a mainstay in the management of LQTS patients (117, 118). Most experts agree that whenever ICD shocks occur in LQTS patients on optimized medical therapy, LCSD should be offered. We believe that, considering the high impact of LCSD on quality of life in this setting, the procedure should be undertaken without delay after the first breakthrough ICD intervention. ICD recurrences can be very detrimental and may lead to depression and even to suicidal attempts, particularly in these adolescents already predisposed to both anxiety and depression because of the underlying disease (119, 120). Moreover, the acute proarrhythmic potential of ICD shocks due to pain perception, fear and subsequent increase in the sympathetic drive on the heart should never be neglected, as will be discussed in detail for CPVT patients. The management of subjects with a first syncopal episode occurring despite maximum tolerated dose β-Blocker therapy is more challenging. As a referral group with a long-standing experience in the treatment of LQTS patients, we advise caution before directly implanting an ICD in these cases. Instead, a careful clinical evaluation is needed. Due to its high efficacy and optimal tolerability, we believe that LCSD should be offered first, clearly explaining to the patients and their families that the procedure is not an alternative to ICD implantation (that may always be considered in a later stage) and that the overall risk of life-threatening events after LCSD is low, unless the patient shows characteristics of high risk. At the same time, the life-spanning risk of complications and psychological consequences related to ICD implantation in these young patients is high (121) and should be properly acknowledged during patient and family counseling. Overall, a proper patient-physician communication in this setting requires to offer LCSD as therapeutic option even if the center is not performing the procedure as an inside facility. Ignorance and/or omission may carry medicolegal implications for the physician (122). On the other hand, in the case of markers of high risk such as onset of the symptoms in the first year of life and/or the persistence of QTc values exceeding 550 ms after LCSD, an ICD could be considered immediately after LCSD. Another difficult issue is the management of patients who never suffered arrhythmic episodes on therapy (and even before) but with either high risk LQTS phenotype or β-blocker intolerance, which represents the so-called primary prevention. In these cases LCSD should be offered before ICD implantation, with the clear intention to serve as bridge to an ICD in the most severe cases. Of course, additional pharmacological strategies such as mexiletine, already proposed in 1995 (123) and now widely used (124–126) should be offered as well, according to the genotype and the specific mutation. Finally, an additional indication for LCSD in LQTS is β-Blocker non-compliance. Generally, patients and their families managed in referral centers are well-instructed about the importance of strictly adhering to the prescribed medical therapy. Nevertheless, young subjects, particularly adolescents, are challenging to manage and may refuse therapy. Since β-Blocker non-compliance is a very well-defined risk factor for arrhythmic events in LQTS (127), if suspected and not modifiable, this condition should prompt to consider LCSD as additional protective measure. Concerning the indication to right cardiac sympathetic denervation (RCSD) in LQTS, we reserve it for patients not responding to LCSD. We discourage RCSD or a direct bilateral cardiac sympathetic denervation (BCSD) in patients not carrying an ICD (or pacemaker) due to the potential pro-arrhythmic effect of the induced (and largely unpredictable) bradycardia, particularly in LQT2 and LQT3 patients.
LCSD in Catecholaminergic Polymorphic Ventricular Tachycardia
The efficacy of LCSD in CPVT is not surprising from a pathophysiological point of view. Indeed, the disease is characterized by an intrinsic increase in the sensitivity of the heart to catecholamines due to mutations affecting the diastolic release of calcium from the sarcoplasmic reticulum. The first case series (3 patients) describing the long-lasting efficacy of LCSD in high risk CVPT was published in 2008 (128). We subsequently reported in 2015 the largest case series of LCSD in CPVT (129). It was a multicentric, international study involving 63 CPVT patients (71% RyR2 positive, 8% CASQ2 positive) who underwent LCSD between 1988 and 2014 at 11 centers worldwide. The majority (n = 54, 86%) had the procedure in secondary prevention, 97% were on β-Blockers, 24% on flecainide. The median post-LCSD follow-up was 37 months. In the 9 asymptomatic patients there were no cardiac events during follow-up. Among the 54 patients with prior major cardiac events either on (n = 38) or off (n = 16) optimal medical therapy, 13 (24%) had at least 1 recurrence, but only 1 patient died suddenly (after having been switched from nadolol to metoprolol). Specifically, the percentage of patients with cardiac events despite optimal medical therapy (n = 38) was reduced from 100 to 32% (P < 0.001) after LCSD, and among 29 patients with a pre-surgical ICD, the rate of shocks dropped by 93% from 3.6 to 0.6 per person per year (P < 0.001). Among the 13 patients with cardiac events after LCSD, only 5 (8%) had no reduction in the number of events as compared to before LCSD (true non-responders). Importantly, the only predictor of response was the extension of LCSD: 71% of the 7 patients with incomplete LCSD had recurrences as compared to 17% of those with a complete LCSD (P < 0.01). Among the 38 most severe patients, 100% of those with incomplete LCSD had recurrences (Figure 2). The most common reason for not performing a complete denervation was to reduce the risk of Horner syndrome. This is not justified since the incidence of permanent Horner syndrome when removing only the lower part of the stellate ganglion (T1) is extremely low (<2%). On the other hand, the antiarrhythmic protection when T1 is spared seems to be significantly lower, in agreement with pre-clinical data (130). In a subsequent exploratory sub analysis of the same population we focused on the 38 patients with an ICD (131). Our preliminary data suggest a reduction in supraventricular arrhythmias (SVA) leading to inappropriate ICD shocks after LCSD. Of course, this observation needs to be confirmed in a larger group of CPVT patients, but it seems very plausible from a pathophysiological point of view. Atrial arrhythmias (both atrial tachycardia and AF) in CPVT are typically triggered by catecholamines in the setting of structurally normal atria. Moreover, experimental animal models suggest that LCSD may increase the threshold for atrial arrhythmias onset and maintenance and reduce ventricular rate during atrial fibrillation (132–134).
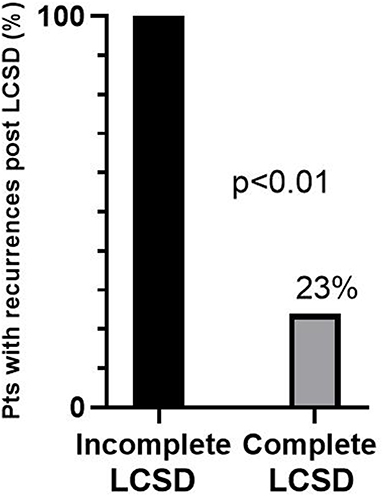
Figure 2. Percentages of recurrences after left cardiac sympathetic denervation (LCSD) in 38 CPVT patients who had previously suffered major cardiac events despite optimal medical therapy. The majority received a complete LCSD (n = 33), the remaining (n = 5) an incomplete LCSD. Modified from De Ferrari et al. (129) with permission.
Subsequently, a multicentric pediatric registry including 18 CPVT patients undergoing LCSD confirmed our results, showing no recurrences of ventricular arrhythmias in 89% of the subjects (135).
LCSD in Catecholaminergic Polymorphic Ventricular Tachycardia: our Approach
LCSD is now an established therapy also for CPVT (117, 118). Our recommendations for LCSD in CPVT are similar to those already discussed for LQTS (first ICD shock or syncope on optimized medical therapy, β-Blockers intolerance or non-compliance), bearing in mind that the decision to implant an ICD in CPVT patients must be considered even more carefully than in LQTS. Indeed, due to the exquisite sensitiveness to catecholamine of their hearts, combined with a generally good hemodynamic tolerability of both rapid polymorphic VT and bidirectional tachycardia (which usually precede VF), CPVT patients are at high risk of electrical storms. This happens because the pain and the fear of the first ICD shock, which generally occurs in a condition of preserved consciousness, elicit a massive neural release of catecholamines, starting a vicious circle. As a matter of fact, in our registry of LCSD in CPVT (and therefore in an already selected subgroup of high-risk patients) we found that 36% of the patients who received an ICD before LCSD suffered at least one electrical storm or end of treatment condition (136). On the contrary, among the 26 pts with no ICD before LCSD, excluding two who had an electrical storm as first manifestation of the disease, none had such episodes on medical therapy. In agreement with this concept, sporadic cases of death in CPVT patients because of ongoing ventricular arrhythmias and exhaustion of ICD shocks have been reported for over 10 years (137–139). Very recently, the largest CPVT meta-analysis ever published (140) including 503 patients with an ICD (median age 15 years) reported a 1.4% mortality rate during follow-up, driven by 4 deaths due to electrical storms. The high incidence of both electrical storms (19.6%) and inappropriate shocks (20.8%) in trans venous ICD recipients is in full agreement with our data (129), as well as the disquieting rate of ICD-related complications (32.4%). Only 3 ICD patients had a subcutaneous ICD (S-ICD); 2 of them received inappropriate shocks due T-wave oversensing. Of note, the mortality rate among the 412 patients treated without ICD was similar to those with an ICD (2%).
Finally, beyond being potentially pro-arrhythmic and often not necessary, ICD shocks in CPVT patients may also be ineffective. Indeed, rapid polymorphic VT or bidirectional VT episodes may be not only self-limiting with the interruption of the stressor (such as physical activity) without the need for shock, but could also be less susceptible to cardioversion compared to VF episodes. Miyake et al. (141) demonstrated that among 10 CPVT patients who received a total of 75 appropriate shocks, only 57% of the shocks were successful in primary termination of the arrhythmias. The underlying rhythm in all successful ICD shocks at first attempt was VF, while no episode of polymorphic VT or bidirectional VT was successfully treated at the first attempt. Subsequently, Roses-Noguer F et al. (142) found an even lower success rate of the first appropriate ICD shock in CPVT (32%), confirming the ineffectiveness on triggered arrhythmias as compared to VF. Moreover, also antitachycardia pacing therapies (ATPs), as expected, proved to be ineffective in CPVT.
For all the above mentioned reasons, the management of high risk CPVT patients is particularly challenging. An optimized antiadrenergic therapy based on the clinical phenotype should always be the main therapeutic goal, whether or not the patient is implanted with an ICD (or is a candidate to). Indeed, in complete agreement with the pathophysiology of the disease, β-Blockers (143) and LCSD (129) are the only therapeutic interventions with a proven efficacy on SCD, aborted cardiac arrest and ICD shocks. Flecainide, despite promising in vitro (144) and in vivo (135, 144–147) data mainly showing its efficacy on effort induced arrhythmias, still lacks a validation on hard clinical end points. Nevertheless, a first pharmacological attempt with flecainide in association to β-adrenergic blockade seems reasonable in β-Blocker non-responders, particularly if the patient has already been implanted with an ICD. Finally, as for LQTS patients, a careful ICD programming with a single VF zone, long detection times and no ATPs, is crucial in CPVT patients.
Conclusions
LCSD was proposed over one century ago for the treatment of angina pectoris. The antiarrhythmic potential of the technique, albeit evident since the first procedure by Jonnesco in 1916, took long to be fully appreciated (148). For many years the studies on LCSD were considered with skepticism, especially because there seemed to be just one group to support it. Finally, clinical data from well-conducted multicenter registries largely confirmed the preclinical findings, showing that LCSD is an effective treatment for drug-refractory ventricular arrhythmias in both LQTS and CPVT and LCSD is now recommended in recent guidelines (117, 118). Not surprisingly, considered the mechanism of action, the efficacy and potential indication of LCSD in channelopathies goes far beyond secondary prevention, potentially including many still asymptomatic patients with high-risk features for SCD despite optimized medical therapy. Regardless of this consistent body of evidence, LCSD is still an underutilized resource, as opposed to the often abused use of ICD in the same group of patients. From the technical point of view, the advantages of the thoracoscopic approach are such that it is difficult to see much room for different surgical approaches that might carry greater risks (149). LCSD can not only improve quality of life but also prevent fatal events that may still occur in patients with ICD due to the vicious circle of catecholamine-induced and maintained electrical storms.
Author Contributions
VD and LP wrote a general manuscript draft. GD and PS supervised the process and edited the manuscript to its final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has received funding from the European Union's Horizon 2020 research and innovation programme under acronym ESCAPE-NET, registered under grant agreement No 733381. The authors are grateful to Pinuccia De Tomasi for expert editorial support.
References
1. Wellens HJJ, Schwartz PJ, Lindemans FW, Buxton AE, Goldberger JJ, Hohnloser SH, et al. Risk stratification for sudden cardiac death: current status and challenges for the future. Eur Heart J. (2014) 35:1642–51. doi: 10.1093/eurheartj/ehu176
2. Vincent JW, Rubart M, Firulli AB. Ontogeny of cardiac sympathetic innervation and its implications for cardiac disease. Pediatr Cardiol. (2012) 33:923–8. doi: 10.1007/s00246-012-0248-1
3. Kawashima T. The autonomic nervous system of the human heart with special reference to its origin, course, and peripheral distribution. Anat Embryol. (2005) 209:425–38. doi: 10.1007/s00429-005-0462-1
4. Janes RD, Brandys JC, Hopkins DA, Johnstone DE, Murphy DA, Armour JA. Anatomy of human extrinsic cardiac nerves and ganglia. Am J Cardiol. (1986) 57:299–309. doi: 10.1016/0002-9149(86)90908-2
5. Malliani A, Recordati G, Schwartz PJ. Nervous activity of afferent cardiac sympathetic fibres with atrial and ventricular endings. J Physiol. (1973) 229:457–69. doi: 10.1113/jphysiol.1973.sp010147
6. Francois-Frank CA. Signification physiologique de la resection du sympathique dans la maladie de Basedow, l'epilepsie, l'idiotic et le glaucoma. Bull Acad Méd Paris. (1989) 41:565–94.
7. Jonnesco T. Traitement chirurgical de l'angine de poitrine par la résection du sympathique cervico-thoracique [French]. Presse Méd. (1921) 20:221–30.
8. Leriche R, Fontaine R. Rôle du ganglion étoile gauche dans le déterminisme de la crise de l'angine de poitrine [French]. C. R. Acad Sci. (1929) 188:279–80.
9. Burnett CF, Evans JA. Follow-up report on resection of the anginal pathway in thirty-three patients. JAMA. (1956) 162:709–12. doi: 10.1001/jama.1956.02970250009003
10. Cox WV. Influence of stellate ganglion block on angina pectoris and the post-exercise ECG. Am J Med. (1956) 252:289–95.
11. Schwartz PJ. The rationale and the role of left stellectomy for the prevention of malignant arrhythmias. Ann N Y Acad Sci. (1984) 427:199–221. doi: 10.1111/j.1749-6632.1984.tb20785.x
12. Estes EH, Izlar HR. Recurrent ventricular tachycardia: a case successfully treated by bilateral cardiac sympathectomy. Am J Med. (1961) 31:493–7. doi: 10.1016/0002-9343(61)90132-2
13. Zipes DP, Festoff B, Schaal SF, Cox C, Sealy WC, Wallace AG. Treatment of ventricular arrhythmia by permanent atrial pacemaker and cardiac sympathectomy. Ann Intern Med. (1968) 68:591–7. doi: 10.7326/0003-4819-68-3-591
14. Moss AJ, McDonald J. Unilateral cervicothoracic sympathetic ganglionectomy for the treatment of long QT interval syndrome. N Engl J Med. (1971) 285:903–4. doi: 10.1056/NEJM197110142851607
15. Yanowitz F, Preston JB, Abildskov JA. Functional distribution of right and left stellate innervation to the ventricles: production of neurogenic electrocardiographic changes by unilateral alteration of sympathetic tone. Circ Res. (1966) 18:416–28. doi: 10.1161/01.RES.18.4.416
16. Schwartz PJ. Cardiac sympathetic denervation to prevent life-threatening arrhythmias. Nat Rev Cardiol. (2014) 11:346–53. doi: 10.1038/nrcardio.2014.19
17. Schwartz PJ, Periti M, Malliani A. The long QT syndrome. Am Heart J. (1975) 89:378–90. doi: 10.1016/0002-8703(75)90089-7
18. Schwartz PJ, Malliani A. Electrical alternation of the T wave: clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long QT syndrome. Am Heart J. (1975) 89:45–50. doi: 10.1016/0002-8703(75)90008-3
19. Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J. (1985) 109:399–411. doi: 10.1016/0002-8703(85)90626-X
20. Schwartz P J, Stone HL, Brown AM. Effects of unilateral stellate ganglion blockade on the arrhythmias associated with coronary occlusion. Am. Heart J. (1976) 92:589–99. doi: 10.1016/S0002-8703(76)80078-6
21. Schwartz PJ, Billman GE, Stone HL. Autonomic mechanisms in ventricular fibrillation induced by myocardial ischemia during exercise in dogs with healed myocardial infarction: an experimental preparation for sudden cardiac death. Circulation. (1984) 69:790–800. doi: 10.1161/01.CIR.69.4.790
22. Schwartz PJ. Sympathetic imbalance and cardiac arrythmias. In: Randall WC, editor. Nervous Control of Cardiovascular Function. New York, NY; Oxford: Oxford University Press (1984). p. 225–52.
23. Malliani A, Schwartz PJ, Zanchetti A. A sympathetic reflex elicited by experimental coronary occlusion. Am J Physiol. (1969) 217:703–9. doi: 10.1152/ajplegacy.1969.217.3.703
24. Schwartz PJ, Snebold NG, Brown AM. Effects of unilateral cardiac sympathetic denervation on the ventricular fibrillation threshold. Am. J. Cardiol. (1976) 37:1034–40. doi: 10.1016/0002-9149(76)90420-3
25. Han J, Moe GK. Nonuniform recovery of excitability in ventricular muscle. Circ Res. (1964) 14:44–60. doi: 10.1161/01.RES.14.1.44
26. Opthof T, Misier AR, Coronel R, Vermeulen JT, Verberne HJ, Frank RG, et al. Dispersion of refractoriness in canine ventricular myocardium. Effects of sympathetic stimulation. Circ Res. (1991) 68:1204–15. doi: 10.1161/01.RES.68.5.1204
27. Ramirez RJ, Ajijola OA, Zhou W, Holmström B, Lüning H, Laks MM, et al. A new electrocardiographic marker for sympathetic nerve stimulation: modulation of repolarization by stimulation of stellate ganglia. J Electrocardiol. (2011) 44:694–9. doi: 10.1016/j.jelectrocard.2011.07.030
28. Yagishita D, Chui RW, Yamakawa K, Rajendran PS, Ajijola OA, Nakamura K, et al. Sympathetic nerve stimulation, not circulating norepinephrine, modulates T-peak to T-end interval by increasing global dispersion of repolarization. Circ Arrhythm Electrophysiol. (2015) 8:174–85. doi: 10.1161/CIRCEP.114.002195
29. Vaseghi M, Lux RL, Mahajan A, Shivkumar K. Sympathetic stimulation increases dispersion of repolarization in humans with myocardial infarction. Am J Physiol Heart Circ Physiol. (2012) 302:H1838–46. doi: 10.1152/ajpheart.01106.2011
30. Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol. (2007) 293:H2024–38. doi: 10.1152/ajpheart.00355.2007
31. Nearing BD, Oesterle SN, Verrier RL. Quantification of ischaemia induced vulnerability by precordial T-wave alternans analysis in dog and human. Cardiovasc Res. (1994) 28:1440–9. doi: 10.1093/cvr/28.9.1440
32. Zipes DP, Barber MJ, Takahashi N, Gilmour RF Jr. Influence of the autonomic nervous system on the genesis of cardiac arrhythmias. Pacing Clin Electrophysiol. (1983) 6:1210–20. doi: 10.1111/j.1540-8159.1983.tb04459.x
33. Ben-David J, Zipes DP. Differential response to right and left ansae subclaviae stimulation of early afterdepolarizations and ventricular tachycardia induced by cesium in dogs. Circulation. (1988) 78:1241–50. doi: 10.1161/01.CIR.78.5.1241
34. Charpentier F, Drouin E, Gauthier C, LeMarec H. Early after/depolarizations and triggered activity: mechanisms and autonomic regulation. Fundam Clin Pharmacol. (1993) 7:39–49. doi: 10.1111/j.1472-8206.1993.tb00216.x
35. Priori SG, Mantica M, Schwartz PJ. Delayed after depolarizations elicited in vivo by left stellate ganglion stimulation. Circulation. (1988) 78:178–85. doi: 10.1161/01.CIR.78.1.178
36. Schwartz PJ, Stone HL. Effects of unilateral stellectomy upon cardiac performance during exercise in dogs. Circ Res. (1979) 44:637–45. doi: 10.1161/01.RES.44.5.637
37. Schwartz PJ, Vanoli E, Zaza A, Zuanetti G. The effect of antiarrhythmic drugs on life-threatening arrhythmias induced by the interaction between acute myocardial ischemia and sympathetic hyperactivity. Am. Heart J. (1985) 109:937–48. doi: 10.1016/0002-8703(85)90233-9
38. Cerati D, Schwartz PJ. Single cardiac vagal fibers activity, acute myocardial ischemia, and risk for sudden death. Circ Res. (1991) 69:1389–1401. doi: 10.1161/01.RES.69.5.1389
39. Billman GE, Schwartz PJ, Stone HL. Baroreceptor reflex control of heart rate: a predictor of sudden cardiac death. Circulation. (1982) 66:874–80. doi: 10.1161/01.CIR.66.4.874
40. Schwartz PJ, Vanoli E, Stramba-Badiale M, De Ferrari GM, Billman GE, Foreman RD. Autonomic mechanisms and sudden death: new insights from analysis of baroreceptor reflexes in conscious dogs with and without a myocardial infarction. Circulation. (1998) 78:969–79. doi: 10.1161/01.CIR.78.4.969
41. La Rovere MT, Bigger JT Jr, Marcus FI, Mortara A, Schwartz PJ. Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. Lancet. (1998) 351:478–84. doi: 10.1016/S0140-6736(97)11144-8
42. La Rovere MT, Pinna GD, Hohnloser SH, Marcus FI, Mortara A, Nohara R, et al. Baroreflex sensitivity and heart rate variability in the identification of patients at risk for life-threatening arrhythmias: implications for clinical trials. Circulation. (2001) 103:2072–7. doi: 10.1161/01.CIR.103.16.2072
43. Vanoli E, De Ferrari GM, Stramba-Badiale M, Hull SS Jr, Foreman RD, Schwartz PJ. Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ Res. (1991) 68:1471–81. doi: 10.1161/01.RES.68.5.1471
44. De Ferrari GM, Vanoli E, Curcuruto P, Tommasini G, Schwartz PJ. Prevention of life-threatening arrhythmias by pharmacologic stimulation of the muscarinic receptors with oxotremorine. Am Heart J. (1992) 124:883–90. doi: 10.1016/0002-8703(92)90968-2
45. De Ferrari GM, Crijns HJ, Borggrefe M, Milasinovic G, Smid J, Zabel M, et al. Chronic vagus nerve stimulation: a new and promising therapeutic approach for chronic heart failure. Eur Heart J. (2011) 32:847–55. doi: 10.1093/eurheartj/ehq391
46. Zannad F, De Ferrari GM, Tuinenburg AE, Wright D, Brugada J, Butter C, et al. Chronic vagal stimulation for the treatment of low ejection fraction heart failure: results of the NEural Cardiac TherApy foR Heart Failure (NECTAR-HF) randomized controlled trial. Eur Heart J. (2015) 36:425–33. doi: 10.1093/eurheartj/ehu345
47. Gold MD, Van Veldhuisen DJ, Hauptman PJ, Borggrefe M, Kubo SH, Lieberman RA, et al. Vagus nerve stimulation for the treatment of heart failure The INOVATE-HF Trial. J Am Coll Cardiol. (2016) 68:149–58. doi: 10.1016/j.jacc.2016.03.525
48. De Ferrari GM, Dusi V. Vagus nerve stimulation for the treatment of heart failure. G Ital Cardiol. (2015) 16:147–54. doi: 10.1714/1820.19822
49. Schwartz PJ, De Ferrari GM. Sympathetic-parasympathetic interaction in health and disease: abnormalities and relevance in heart failure. Heart Fail Rev. (2011) 16:101–7. doi: 10.1007/s10741-010-9179-1
50. Chin A, Ntsekhe M, Viljoen C, Rossouw J, Pennel T, Schwartz PJ. Rationale and design of a prospective study to assess the effect of left cardiac sympathetic denervation in chronic heart failure. Int J Cardiol. (2017) 248:227–31. doi: 10.1016/j.ijcard.2017.08.012
51. Cao JM, Chen LS, KenKnight BH, Ohara T, Lee MH, Tsai J, et al. Nerve sprouting and sudden cardiac death. Circ Res. (2000) 86:816–21. doi: 10.1161/01.RES.86.7.816
52. Oh YS, Jong AY, Kim DT, Li H, Wang C, Zemljic-Harpf A, et al. Spatial distribution of nerve sprouting after myocardial infarction in mice. Heart Rhythm. (2006) 3:728–36. doi: 10.1016/j.hrthm.2006.02.005
53. Fu SY, Gordon T. The cellular and molecular basis of peripheral nerve regeneration. Mol Neurobiol. (1997) 14:67–116. doi: 10.1007/BF02740621
54. Inoue H, Zipes DP. Time course of denervation of efferent sympathetic and vagal nerves after occlusion of the coronary artery in the canine heart. Circ Res. (1988) 62:1111–20. doi: 10.1161/01.RES.62.6.1111
55. Inoue H, Zipes DP. Results of sympathetic denervation in the canine heart: supersensitivity that may be arrhythmogenic. Circulation. (1987) 75:877–87. doi: 10.1161/01.CIR.75.4.877
56. Jacobson AF, Senior R, Cerqueira MD, Wong ND, Thomas GS, Lopez VA, et al. Myocardial iodine-123 meta-iodobenzylguanidine imaging and cardiac events in heart failure. Results of the prospective ADMIRE-HF (AdreView Myocardial Imaging for Risk Evaluation in Heart Failure) study. J Am Coll Cardiol. (2010) 55:2212–21. doi: 10.1016/j.jacc.2010.01.014
57. Fallavollita JA, Heavey BM, Luisi AJ Jr, Michalek SM, Baldwa S, Mashtare TL Jr, et al. Regional myocardial sympathetic denervation predicts the risk of sudden cardiac arrest in ischemic cardiomyopathy. J Am Coll Cardiol. (2014) 63:141–9. doi: 10.1016/j.jacc.2013.07.096
58. Nakamura K, Ajijola OA, Aliotta E, Armour JA, Ardell JL, Shivkumar K. Pathological effects of chronic myocardial infarction on peripheral neurons mediating cardiac neurotransmission. Auton Neurosci. (2016) 197:34–40. doi: 10.1016/j.autneu.2016.05.001
59. Nguyen BL, Li H, Fishbein MC, Lin SF, Gaudio C, Chen PS, et al. Acute myocardial infarction induces bilateral stellate ganglia neural re modeling in rabbits. Cardiovasc Pathol. (2012) 21:143–8. doi: 10.1016/j.carpath.2011.08.001
60. Ajijola OA, Yagishita D, Reddy NK, Yamakawa K, Vaseghi M, Downs AM, et al. Remodeling of stellate ganglion neurons after spatially targeted myocardial infarction: neuropeptide and morphologic changes. Heart Rhythm. (2015) 12:1027–35. doi: 10.1016/j.hrthm.2015.01.045
61. Ajijola OA, Wisco JJ, Lambert HW, Mahajan A, Stark E, Fishbein MC, et al. Extracardiac neural remodeling in humans with cardiomyopathy. Circ Arrhythm Electrophysiol. (2012) 5:1010–116. doi: 10.1161/CIRCEP.112.972836
62. Ajijola OA, Hoover DB, Simerly TM, Brown TC, Yanagawa J, Biniwale RM, et al. Inflammation, oxidative stress, and glial cell activation characterize stellate ganglia from humans with electrical storm. JCI Insight. (2017) 2:e94715. doi: 10.1172/jci.insight.94715
63. Rizzo S, Basso C, Troost D, Aronica E, Frigo AC, Driessen AH, et al. T-cell-mediated inflammatory activity in the stellate ganglia of patients with ion-channel disease and severe ventricular arrhythmias. Circ Arrhythm Electrophysiol. (2014) 7:224–9. doi: 10.1161/CIRCEP.113.001184
64. Moss AJ, Ryan DH, Yeaney GA. Ganglionitis and genetic cardiac arrhythmias: more questions than answers. Circ Arrhythm Electrophysiol. (2014) 7:190–1. doi: 10.1161/CIRCEP.114.001589
65. Epstein AE, Kay GN, Plumb VJ, Dailey SM, Anderson PG. Gross and microscopic pathological changes associated with non thoracotomy implantable defibrillator leads. Circulation. (1998) 98:1517–24. doi: 10.1161/01.CIR.98.15.1517
66. Müller KD, Jakob H, Neuzner J, Grebe SF, Schlepper M, Pitschner HF.123I-metaiodobenzylguanidine scintigraphy in the detection of irregular regional sympathetic innervation in long QT syndrome. Eur Heart J. (1993) 14:316–25.
67. Kies P, Paul M, Gerss J, Stegger L, Mönnig G, Schober O, et al. Impaired cardiac sympathetic innervation in symptomatic patients with long QT syndrome. Eur J Nucl Med Mol Imaging. (2011) 38:1899–907. doi: 10.1007/s00259-011-1852-7
68. Schäfers M, Wichter T, Lerch H, Matheja P, Kuwert T, Schäfers K, et al. Cardiac 123I-MIBG uptake in idiopathic ventricular tachycardia and fibrillation. J Nucl Med. (1999) 40:1–5.
69. Burnstock G. Do some nerve cells release more than one transmitter? Neuroscience. (1976) 1:239–48. doi: 10.1016/0306-4522(76)90054-3
70. Burnstock G. Autonomic neurotransmission: 60 years since Sir Henry Dale. Annu Rev Pharmacol Toxicol. (2009) 49:1–30. doi: 10.1146/annurev.pharmtox.052808.102215
71. Potter EK, McCloskey DI. Peripheral inhibition of cardiac vagal action by sympathetic adrenergic stimulation. Proc Aust Soc Clin Exp Pharmacol. (1982) 13:99.
72. Potter E. Presynaptic inhibition of cardiac vagal postganglionic nerves by neuropeptide Y. Neurosci Lett. (1987) 83:101–6. doi: 10.1016/0304-3940(87)90223-0
73. Smith-White MA, Hardy TA, Brock JA, Potter EK. Effects of a selective neuropeptide Y Y2 receptor antagonist, BIIE0246, on Y2 receptors at peripheral neuroeffector junctions. Br J Pharmacol. (2001) 132:861–8. doi: 10.1038/sj.bjp.0703879
74. Smith-White MA, Herzog H, Potter EK. Role of neuropeptide Y Y(2) receptors in modulation of cardiac parasympathetic neurotransmission. Regul Pept. (2002) 103:105–11. doi: 10.1016/S0167-0115(01)00368-8
75. Herring N, Lokale MN, Danson EJ, Heaton DA, Paterson DJ. Neuropeptide Y reduces acetylcholine release and vagal bradycardia via a Y2 receptor-mediated, protein kinase C-dependent pathway. J Mol Cell Cardiol. (2008) 44:477–85. doi: 10.1016/j.yjmcc.2007.10.001
76. Herring N. Autonomic control of the heart: going beyond the classical neurotransmitters. Exp Physiol. (2015) 100:354–8. doi: 10.1113/expphysiol.2014.080184
77. Edvinsson L, Copeland JR, Emson PC, McCulloch J, Uddman R. Nerve fibers containing neuropeptide Y in the cerebrovascular bed: immunocytochemistry, radioimmunoassay, and vasomotor effects. J Cereb Blood Flow Metab. (1987) 7:45–57. doi: 10.1038/jcbfm.1987.7
78. Cuculi F, Herring N, De Caterina AR, Banning AP, Prendergast BD, Forfar JC, et al. Relationship of plasma neuropeptide Y with angiographic, electrocardiographic and coronary physiology indices of reperfusion during ST elevation myocardial infarction. Heart. (2013) 99:1198–203. doi: 10.1136/heartjnl-2012-303443
79. Hulting J, Sollevi A, Ullman B, Franco-Cereceda A, Lundberg JM. Plasma neuropeptide Y on admission to a coronary care unit: raised levels in patients with left heart failure. Cardiovasc Res. (1990) 24:102–8. doi: 10.1093/cvr/24.2.102
80. Ullman B, Hulting J, Lundberg JM. Prognostic value of plasma neuropeptide-Y in coronary care unit patients with and without acute myocardial infarction. Eur Heart J. (1994) 15:454–61. doi: 10.1093/oxfordjournals.eurheartj.a060526
81. Herring N, Golding S, Paterson DJ. Pre-synaptic NO-cGMP pathway modulates vagal control of heart rate in isolated adult guinea pig atria. J Mol Cell Cardiol. (2000) 32:1795–804. doi: 10.1006/jmcc.2000.1214
82. Herring N, Paterson DJ. Nitric oxide-cGMP pathway facilitates acetylcholine release and bradycardia during vagal nerve stimulation in the guinea-pig in vitro. J Physiol. (2001) 535:507–18. doi: 10.1111/j.1469-7793.2001.00507.x
83. Schwarz P, Diem R, Dun NJ, Forstermann U. Endogenous and exogenous nitric oxide inhibits norepinephrine release from rat heart sympathetic nerves. Circ Res. (1995) 77:841–8. doi: 10.1161/01.RES.77.4.841
84. Wang L, Henrich M, Buckler KJ, McMenamin M, Mee CJ, Sattelle DB, et al. Neuronal nitric oxide synthase gene transfer decreases [Ca2+]i in cardiac sympathetic neurons. J Mol Cell Cardiol. (2007) 43:717–25. doi: 10.1016/j.yjmcc.2007.09.005
85. Heaton DA, Li D, Almond SC, Dawson TA, Wang L, Channon KM, et al. Gene transfer of neuronal nitric oxide synthase into intracardiac ganglia reverses vagal impairment in hypertensive rats. Hypertension. (2007) 49:380–8. doi: 10.1161/01.HYP.0000255792.97033.f7
86. Li D, Wang L, Lee CW, Dawson TA, Paterson DJ. Noradrenergic cell specific gene transfer with neuronal nitric oxide synthase reduces cardiac sympathetic neurotransmission in hypertensive rats. Hypertension. (2007) 50:69–74. doi: 10.1161/HYPERTENSIONAHA.107.088591
87. Li D, Nikiforova N, Lu CJ, Wannop K, McMenamin M, Lee CW, et al. Targeted neuronal nitric oxide synthase transgene delivery into stellate neurons reverses impaired intracellular calcium transients in prehypertensive rats. Hypertension. (2013) 61:202–7. doi: 10.1161/HYPERTENSIONAHA.111.00105
88. Dawson TA, Li D, Woodward T, Barber Z, Wang L, Paterson DJ. Cardiac cholinergic NO-cGMP signaling following acute myocardial infarction and nNOS gene transfer. Am J Physiol Heart Circ Physiol. (2008) 295:H990–8. doi: 10.1152/ajpheart.00492.2008
89. Arking DE, Pfeufer A, Post W, Kao WH, Newton-Cheh C, Ikeda M, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. (2006) 38:644–51. doi: 10.1038/ng1790
90. Eijgelsheim M, Aarnoudse AL, Rivadeneira F, Kors JA, Witteman JC, Hofman A, et al. Identification of a common variant at the NOS1AP locus strongly associated to QT-interval duration. Hum Mol Genet. (2009) 18:347–57. doi: 10.1093/hmg/ddn341
91. Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. (2009) 41:399–406. doi: 10.1038/ng.364
92. Pfeufer A, Sanna S, Arking DE, Müller M, Gateva V, Fuchsberger C, et al. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. (2009) 41:407–14. doi: 10.1038/ng.362
93. Kao WH, Arking DE, Post W, Rea TD, Sotoodehnia N, Prineas RJ, et al. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community-based populations. Circulation. (2009) 119:940–51. doi: 10.1161/CIRCULATIONAHA.108.791723
94. Jamshidi Y, Nolte IM, Dalageorgou C, Zheng D, Johnson T, Bastiaenen R, et al. Common variation in the NOS1AP Gene is associated with drug-induced QT prolongation and ventricular arrhythmia. J Am Coll Cardiol. (2012) 60:841–50. doi: 10.1016/j.jacc.2012.03.031
95. Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, et al. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. (2009) 120:1657–63. doi: 10.1161/CIRCULATIONAHA.109.879643
96. Tomás M, Napolitano C, De Giuli L, Bloise R, Subirana I, Malovini A, et al. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol. (2010) 55:2745–52. doi: 10.1016/j.jacc.2009.12.065
97. Chang KC, Barth AS, Sasano T, Kizana E, Kashiwakura Y, Zhang Y, et al. CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc Natl Acad Sci USA. (2008) 105:4477–82. doi: 10.1073/pnas.0709118105
98. Randall WC, Rohse WG. The augmentor action of the sympathetic cardiac nerves. Circ. Res. (1956) 4:470–5. doi: 10.1161/01.RES.4.4.470
99. Geis WP, Kaye MP, Randall WC. Major autonomic pathways to the atria and S-A and A-V nodes in the canine heart. Am J Physiol. (1973) 224:202–8. doi: 10.1152/ajplegacy.1973.224.1.202
100. Leftheriotis D, Flevari P, Kossyvakis C, Katsaras D, Batistaki C, Arvaniti C, et al. Acute effects of unilateral temporary stellate ganglion block on human atrial electrophysiological properties and atrial fibrillation inducibility. Heart Rhythm. (2016) 13:2111–7. doi: 10.1016/j.hrthm.2016.06.025
101. Randall WC, Armour JA, Geis WP, Lippincott DB. Regional cardiac distribution of the sympathetic nerves. Fed Proc. (1972) 31:1199–208.
102. Kralios FA, Martin L, Burgess MJ, Millar K. Local ventricular repolarization changes due to sympathetic nerve-branch stimulation. Am J Physiol. (1975) 228:1621–6. doi: 10.1152/ajplegacy.1975.228.5.1621
103. Goodall M, Kirshner N. Effect of cervico-thoracic ganglionectomy on the adrenaline and noradrenaline content in the mammalian heart. J. Clin lnvest. (1956) 35:649–56.
104. Hertting G, Schlefthaler TH. The effect of stellate ganglion excision on the catecholamine content and the uptake of 3H-norepinephrine in the heart of the cat. lnt J Neuropharmacol. (1964) 3:65–9.
105. Schwartz PJ, Stone HL. Left stellectomy and denervation supersensitivity in conscious dogs. Am J Cardiol. (1982) 49:1185–90. doi: 10.1016/0002-9149(82)90043-1
106. Schwartz PJ, Locati EH, Moss AJ, Crampton RS, Trazzi R, Ruberti U. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome: a worldwide report. Circulation. (1991) 84:503–11. doi: 10.1161/01.CIR.84.2.503
107. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long QT syndrome. Circulation. (2004) 109:1826–33. doi: 10.1161/01.CIR.0000125523.14403.1E
108. Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. (2009) 6:752–9. doi: 10.1016/j.hrthm.2009.03.024
109. Bos JM, Bos KM, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation in Long QT Syndrome analysis of therapeutic nonresponders. Circ Arrhythm Electrophysiol. (2013) 6:705–11. doi: 10.1161/CIRCEP.113.000102
110. Li J, Liu Y, Yang F, Jiang G, Li C, Hu D, et al. Video-Assisted thoracoscopic left cardiac sympathetic denervation: a reliable minimally invasive approach for congenital long-QT syndrome. Ann Thorac Surg. (2008) 86:1955–9. doi: 10.1016/j.athoracsur.2008.07.100
111. Hofferberth SC, Cecchin F, Loberman D, Fynn-Thompson F. Left thoracoscopic sympathectomy for cardiac denervation in patients with life-threatening ventricular arrhythmias. J Thorac Cardiovasc Surg. (2014) 147:404–11. doi: 10.1016/j.jtcvs.2013.07.064
112. Olde Nordkamp LRA, Driessen AHG, Odero A, Blom NA, Koolbergen DR, Schwartz PJ, et al. Left cardiac sympathetic denervation in the Netherlands for the treatment of inherited arrhythmia syndromes. Neth Heart J. (2014) 22:160–6. doi: 10.1007/s12471-014-0523-2
113. Waddell-Smith KE, Ertresvaag KN, Li J, Chaudhuri K, Crawford JR, Hamill JK, et al. Physical and psychological consequences of left cardiac sympathetic denervation in Long-QT syndrome and catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. (2015) 8:1151–8. doi: 10.1161/CIRCEP.115.003159
114. Ouriel K, Moss A. Long QT syndrome: an indication for cervicothoracic sympathectomy. Cardiovasc Surg. (1995) 3:475–8. doi: 10.1016/0967-2109(95)94444-2
115. Jang S, Cho Y, Kim N, Kim C, Sohn J, Roh J, et al. Video assisted thorascopic left cardiac sympathetic denervation in patients with hereditary ventricular arrhythmias. Pacing Clin Electrophysiol. (2017) 40:232–41. doi: 10.1111/pace.13008
116. Antiel R, Bos M, Joyce D, Owen Moir HC, Ackerman M. Quality of life after videoscopic left cardiac sympathetic denervation in patients with potentially life threatening cardiac channelopathies/cardiomyopathies. Heart Rhythm. (2016) 13:62–9. doi: 10.1016/j.hrthm.2015.09.001
117. Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the european society of cardiology (ESC). Eur Heart J. (2015) 36:2793–867. doi: 10.1093/eurheartj/ehv316
118. Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American college of cardiology/American heart association task force on clinical practice guidelines and the heart rhythm society. J Am Coll Cardiol. (2018) 72:e91–e220. doi: 10.1016/j.jacc.2017.10.054
119. Hamang A, Eide GE, Rokne B, Nordin K, Bjorvatn C, Øyen N. Predictors of heart-focused anxiety in patients undergoing genetic investigation and counseling of long QT syndrome or hypertrophic cardiomyopathy: a one year follow-up. J Genet Couns. (2012) 21:72–84. doi: 10.1007/s10897-011-9393-6
120. Hintsa T, Keltikangas-Järvinen L, Puttonen S, Ravaja N, Toivonen L, Kontula K, et al. Depressive symptoms in the congenital long QT syndrome. Ann Med. (2009) 41:516–21. doi: 10.1080/07853890903037254
121. Schwartz PJ, Spazzolini C, Priori SG, Crotti L, Vicentini A, Landolina M, et al. Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them? Data from the European Long- QT Syndrome Implantable Cardioverter-Defibrillator (LQTS ICD) Registry. Circulation. (2010) 122:1272–82. doi: 10.1161/CIRCULATIONAHA.110.950147
122. Schwartz PJ. Efficacy of left cardiac sympathetic denervation has an unforeseen side effect: medicolegal complications. Heart Rhythm. (2010) 7:1330–2. doi: 10.1016/j.hrthm.2010.04.038
123. Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantù F, Towbin JA, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. (1995) 92:3381–6. doi: 10.1161/01.CIR.92.12.3381
124. Schwartz OJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD: true or false? Heart Rhythm. (2009) 6:113–20. doi: 10.1016/j.hrthm.2008.10.017
125. Mazzanti A, Maragna R, Faragli A, Monteforte N, Bloise R, Memmi M, et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with Long QT Syndrome type 3. J Am Coll Cardiol. (2016) 67:1053–8. doi: 10.1016/j.jacc.2015.12.033
126. Bos JM, Crotti L, Rohatgi RK, Schwartz PJ, Ackerman MJ. Mexiletine shortens the QT interval in patients with either type 1 or type 2 Long QT. Heart Rhythm. (2018) 15 (Suppl.):S450.
127. Vincent GM, Schwartz PJ, Denjoy I, Swan H, Bithell C, Spazzolini C, et al. High Efficacy of beta-blockers in long-QT syndrome type 1. Contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. (2009) 119:215–21. doi: 10.1161/CIRCULATIONAHA.108.772533
128. Wilde AA, Bhuiyan ZA, Crotti L, Facchini M, De Ferrari GM, Paul T, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. (2008) 358:2024–9. doi: 10.1056/NEJMoa0708006
129. De Ferrari GM, Dusi V, Spazzolini C, Bos JM, Abrams DJ, Berul CI, et al. Clinical management of catecholaminergic polymorphic ventricular tachycardia: the role of left cardiac sympathetic denervation. Circulation. (2015) 131:2185–93. doi: 10.1161/CIRCULATIONAHA.115.015731
130. Buckley U, Yamakawa K, Takamiya T, Armour JA, Shivkumar K, Ardell JL. Targeted stellate decentralization: implications for sympathetic control of ventricular electrophysiology Heart Rhythm. (2016) 13:282–8. doi: 10.1016/j.hrthm.2015.08.022
131. Dusi V, De Ferrari GM, Bos JM, Moir CR, Shkolnikova M, Abrams DJ, et al. Supraventricular arrhythmias and implantable cardioverter defibrillator shocks in catecholaminergic polymorphic ventricular tachycardia. Eur Heart J. (2016) 37:83
132. Ogawa M, Tan AY, Song J, Kobayashi K, Fishbein MC, Lin SF, et al. Cryoablation of stellate ganglia and atrial arrhythmia in ambulatory dogs with pacing-induced heart failure. Heart Rhythm. (2009) 6:1772–9. doi: 10.1016/j.hrthm.2009.08.011
133. Chinda K, Tsai WC, Chan YH, Lin AY, Patel J, Zhao Y, et al. Intermittent left cervical vagal nerve stimulation damages the stellate ganglia and reduces the ventricular rate during sustained atrial fibrillation in ambulatory dogs. Heart Rhythm. (2016) 13:771–80. doi: 10.1016/j.hrthm.2015.11.031
134. Ye Zhao, Yuan Y, Tsai WU, Jiang Z, Zhi-peng Tian, Shen C, et al. Antiarrhythmic effects of stimulating the left dorsal branch of the thoracic nerve in a canine model of paroxysmal atrial tachyarrhythmias. Heart Rhythm. (2018) 15:1242–51. doi: 10.1016/j.hrthm.2018.04.009
135. Roston TM, Vinocur JM, Maginot KR, Mohammed S, Salerno JC, Etheridge SP, et al. Catecholaminergic polymorphic ventricular tachycardia in children: analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ Arrhythm Electrophysiol. (2015) 8:633–42. doi: 10.1161/CIRCEP.114.002217
136. Dusi V, De Ferrari GM, Bos JM, Moir CR, Wilde AAM, Shkolnikova M, et al. Electrical storms in catecholaminergic polymorphic ventricular tachycardia: findings from the international registry of left cardiac sympathetic denervation. Eur Heart J. (2016) 37:215.
137. Mohamed U, Gollob MH, Gow RM, Krahn AD. Sudden cardiac death despite an implantable cardioverter-defibrillator in a young female with catecholaminergic ventricular tachycardia. Heart Rhythm. (2006) 3:1486–9. doi: 10.1016/j.hrthm.2006.08.018
138. Palanca V, Quesada A, Trigo A, Jimenez J. [Arrhythmic storm induced by AICD discharge in a patient with catecholaminergic polymorphic ventricular tachycardia]. Rev Esp Cardiol. (2006) 59:1079–80. doi: 10.1157/13093988
139. Pizzale S, Gollob MH, Gow R, Birnie DH. Sudden death in a young man with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. J Cardiovasc Electrophysiol. (2008) 19:1319–21. doi: 10.1111/j.1540-8167.2008.01211.x
140. Roston TM, Jones K, Hawkins NM, Bos JM, Schwartz PJ, Perry F, et al. Implantable cardioverter-defibrillator use in catecholaminergic polymorphic ventricular tachycardia: a systematic review. Heart Rhythm. (2018) 12:1791–9. doi: 10.1016/j.hrthm.2018.06.046
141. Miyake CY, Webster G, Czosek RJ, Kantoch MJ, Dubin AM, Avasarala K, et al. Efficacy of implantable cardioverter defibrillators in young patients with catecholaminergic polymorphic ventricular tachycardia. Success depends on substrate. Circ Arrhythm Electrophysiol. (2013) 6:579–87. doi: 10.1161/CIRCEP.113.000170
142. Roses-Noguer F, Jarman JW, Clague JR, Till J. Outcomes of defibrillator therapy in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. (2014) 11:58–66. doi: 10.1016/j.hrthm.2013.10.027
143. Hayashi M, Denjoy I, Extramiana F, Maltret A, Buisson NR, Lupoglazoff JM, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. (2009) 119:2426–34. doi: 10.1161/CIRCULATIONAHA.108.829267
144. Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. (2009) 15:380–3. doi: 10.1038/nm.1942
145. Van der Werf C, Kannankeril PJ, Sacher F, Krahn AD, Viskin S, Leenhardt A, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. (2011) 57:2244–54. doi: 10.1016/j.jacc.2011.01.026
146. Watanabe H, van der Werf C, Roses-Noguer F, Adler A, Sumitomo N, Veltmann C, et al. Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. (2013) 10:542–7. doi: 10.1016/j.hrthm.2012.12.035
147. Wangüemert Pérez F, Hernández Afonso JS, Groba Marco MDV, Caballero Dorta E, Álvarez Acosta L, Campuzano Larrea O, et al. Flecainide reduces ventricular arrhythmias in patients with genotype RyR2-positive catecholaminergic polymorphic ventricular tachycardia. Rev Esp Cardiol. (2018) 71:185–91. doi: 10.1016/j.recesp.2017.04.014
148. Schwartz PJ, De Ferrari GM, Pugliese L. Cardiac sympathetic denervation 100 years later: Jonnesco would have never believed it. Int J Cardiol. (2017) 15:237:25–8. doi: 10.1016/j.ijcard.2017.03.020
Keywords: sudden cardiac death, cardiac sympathetic denervation, long QT syndrome, catecholaminergic polymorphic ventricular tachycardia, cardiac autonomic nervous system
Citation: Dusi V, De Ferrari GM, Pugliese L and Schwartz PJ (2019) Cardiac Sympathetic Denervation in Channelopathies. Front. Cardiovasc. Med. 6:27. doi: 10.3389/fcvm.2019.00027
Received: 11 December 2018; Accepted: 01 March 2019;
Published: 26 March 2019.
Edited by:
Marina Cerrone, New York University, United StatesReviewed by:
Richard Hauer, ICIN Netherlands Heart Institute (KNAW), NetherlandsElizabeth S. Kaufman, MetroHealth, United States
Copyright © 2019 Dusi, De Ferrari, Pugliese and Schwartz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter J. Schwartz, cGV0ZXIuc2Nod2FydHpAdW5pcHYuaXQ=; cC5zY2h3YXJ0ekBhdXhvbG9naWNvLml0