 David Ngai
David Ngai Marsel Lino
Marsel Lino Michelle P. Bendeck
Michelle P. Bendeck- 1Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
- 2Ted Rogers Centre for Heart Research, University of Toronto, Toronto, ON, Canada
- 3Department of Medicine, University of Toronto, Toronto, ON, Canada
Vascular calcification is a complex pathological process occurring in patients with atherosclerosis, type 2 diabetes, and chronic kidney disease. The extracellular matrix, via matricrine-receptor signaling plays important roles in the pathogenesis of calcification. Calcification is mediated by osteochondrocytic-like cells that arise from transdifferentiating vascular smooth muscle cells. Recent advances in our understanding of the plasticity of vascular smooth muscle cell and other cells of mesenchymal origin have furthered our understanding of how these cells transdifferentiate into osteochondrocytic-like cells in response to environmental cues. In the present review, we examine the role of the extracellular matrix in the regulation of cell behavior and differentiation in the context of vascular calcification. In pathological calcification, the extracellular matrix not only provides a scaffold for mineral deposition, but also acts as an active signaling entity. In recent years, extracellular matrix components have been shown to influence cellular signaling through matrix receptors such as the discoidin domain receptor family, integrins, and elastin receptors, all of which can modulate osteochondrocytic differentiation and calcification. Changes in extracellular matrix stiffness and composition are detected by these receptors which in turn modulate downstream signaling pathways and cytoskeletal dynamics, which are critical to osteogenic differentiation. This review will focus on recent literature that highlights the role of cell-matrix interactions and how they influence cellular behavior, and osteochondrocytic transdifferentiation in the pathogenesis of cardiovascular calcification.
Introduction
Vascular calcification is a pathology characterized by ectopic calcification of the vessel wall of muscular or elastic arteries. Vascular calcification is often observed in atherosclerosis, type 2 diabetes (T2D), chronic kidney disease, and aging, and contributes to increased cardiovascular morbidity and mortality independent of other known risk factors (1–3). Vascular calcification increases the risk of myocardial infarction and heart failure and is the leading cause of the death in patients with chronic kidney disease (4, 5). In addition, patients with high calcium deposition that also have peripheral artery disease have approximately a 25% increase in the risk of limb amputation (6). Despite the widespread incidence of vascular calcification, effective, and targeted therapies are still lacking.
Previously thought to be a passive process of crystal deposition on the surrounding extracellular matrix (ECM), it is now appreciated that vascular calcification pathogenesis is an active process involving the transdifferentiation of vascular smooth muscle cells (VSMCs) into osteochondrocytic cells (7–9). The nucleation and propagation of calcium phosphate crystals in the vessel wall is mediated by matrix vesicles secreted by osteochondrocytic cells deposited on components of the ECM such as collagens and elastin (10). Furthermore, previous studies have found many of the transcriptional and regulatory signaling pathways involved in normal bone formation are also present in calcifying vessels (11–14). Some examples include paracrine signaling molecules like bone morphogenetic protein 2 (BMP-2) (15), the master osteogenic transcription factor runt-related transcription factor 2 (Runx2) (14), expression of pro-calcification enzymes such as alkaline phosphatase (16), and loss of calcification inhibitors such as pyrophosphate (17), matrix GLA protein (18), and fetuin-A (19).
Much research on vascular calcification has highlighted the impact of systemic factors such as inflammation, lipids, glucose, and phosphate on VSMC transdifferentiation. In addition to these factors, it has become evident that the local ECM tissue microenvironment not only acts as a scaffold for cells and hydroxyapatite deposition, but it also plays an important role as a signaling entity, modulator of inflammation, and of cell phenotype. This has been observed in VSMC transdifferentiation to osteochondrocytic cells as well as in other cell types such as mesenchymal stem cells. Matrix binding receptors such as the integrins and discoidin domain receptors (DDRs) have been implicated in the regulation of osteogenic programs. Modulation of cell phenotype by ECM stiffness sensed through matrix binding receptors has also been recognized to be important in osteogenic differentiation. Furthermore, indirect effects of the ECM on vascular calcification may be induced by creating changes in systemic factors or overall metabolism.
In this article, we review the current state of knowledge on the ECM and its role in osteogenesis and vascular calcification at a physiological level, and from a molecular and cellular standpoint. We review mechanisms of ectopic calcification and osteogenic differentiation to provide context for further discussion of matricrine regulation of calcification. We then review ECM proteins and receptors which have been implicated in calcification, in particular DDRs, integrins, the elastin receptor complex (ERC), and receptor for advanced glycation end products (RAGE). Next is a discussion of two important matrix localized calcification inhibitors, osteopontin and matrix Gla proteins (MGP). Finally, compelling evidence for an integrated mechanosensitive matricrine signaling axis involving receptors coupled to the cytoskeleton is discussed. A better understanding of the pathobiology will help to identify potential targets for more effective therapeutics or treatment options.
Ectopic Calcification
Ectopic calcification is characterized by the pathological deposition of calcium phosphate crystals within the extracellular matrix of soft tissues, including the vasculature (20). Vascular calcification is a form of ectopic mineralization and can be classified as one of two main types: (1) atherosclerotic (intimal) calcification, and (2) medial calcification (21). As suggested by the name, intimal calcification occurs within atherosclerotic plaques, while medial calcification occurs within the medial layer of the vasculature. The cellular mechanisms driving both forms of vascular calcification are similar and share features in common with osteogenic programs (22), however remain distinct in their pathology. Risk factors associated with vascular calcification include chronic kidney disease (CKD) (5, 21, 23), T2D (24, 25), inflammation (26), and age to name a few (27, 28). Medial calcification is more common in patients suffering from T2D and CKD and is thought to be initiated by the nucleation of calcium phosphate crystals in matrix vesicles or on elastin and is exacerbated by metabolic factors such as hyperglycemia and insulin resistance (8). Intimal calcification on the other hand is more closely associated with atherosclerosis and is mediated by local inflammation (8). Both medial and intimal calcification involve the transdifferentiation of VSMCs into osteochondrocytes via mechanisms that are currently being investigated (11, 29–31).
Osteogenic differentiation and bone mineralization are highly regulated processes involving multiple interacting factors including intracellular signaling cascades, secreted factors, extracellular inhibitors of mineralization, and cell-matrix interactions. Several signaling molecules such as fibroblast growth factors (FGFs), transforming growth factors (TGFs), and bone morphogenetic proteins (BMPs) play a role in osteogenic differentiation. The most widely studied are the BMPs, particularly BMP-2. BMPs signal through BMP receptors, transducing downstream signaling through SMAD1/5/8, which in concert with SMAD4 upregulates the expression of the master osteogenic transcription factor, Runx2. Runx2 commits mesenchymal progenitors to the osteoblast lineage and drives osteochondrocytic transdifferentiation and calcification of VSMCs (14, 32). In addition, Runx2 regulates the expression of osteoblast-related genes such as osteocalcin, osterix, and type I collagen (33, 34). Runx2 activity is subject to regulation by phosphorylation events mediated by kinases such as Akt (12, 35), ERK1/2 (36), p38 (37), JNK (36), GSK3β (37), and CDK1 (in response to glucose) (38). Phosphorylation of Runx2 can be activating or inhibitory. Moreover, Runx2 activity is also regulated by nuclear translocation, DNA-binding capacity and interaction with transcriptional co-factors, as well as protection from degradation by binding partners such as Cbfb (39).
Following osteogenic differentiation, calcification can progress by the nucleation and propagation of hydroxyapatite crystals in the ECM. This is mediated by the secretion of matrix vesicles by osteoblasts or osteochondrocytic cells in normal bone formation and pathological calcification, respectively (40, 41). Matrix vesicles are membrane-bound particles of approximately 100 nm in diameter and are a driving force for the initiation and propagation of mineralization. The ECM is an important site for matrix vesicle deposition and scaffolding for biomineralization (41, 42). Secreted matrix vesicles can become deposited and cluster on collagens in the ECM (40, 41). Collagen density has been shown to be negatively correlated to matrix vesicle clustering and biomineralization both in in vitro and in vivo models of vascular calcification (41).
There are examples of diseases that demonstrate the importance of the ECM in the progression of calcification. Osteogenesis imperfecta is an inherited disease involving a mutation in the α1 or α2 chains of type I collagen leading to reduced deposition of normal collagen fibrils and production of structurally abnormal collagens (43). Improper mineralization of hydroxyapatite crystals on the collagen scaffolding leads to the development of fragile and brittle bones (44). Polymorphisms in the Sp1 binding site of the Col1α1 gene are associated with osteoporosis, a disease resulting in reduced bone mineral density and increased risk of bone fracture (45). This polymorphism increases binding of the Sp1 transcription factor to the Col1α1 gene promoter, causing an increase from the normal 2:1 ratio of Col1α1 to Col1α2 mRNA and protein produced by osteoblasts (46). This may be a causal mechanism for the reduced bone quality and bone mass in osteoporotic patients with this polymorphism.
Extracellular Matrix
The ECM is important in the regulation of cellular phenotype, homeostasis, and development in addition to providing physical support and organization of cells into tissues and organs (47). Collagens are the main component of the ECM and are made up of α-chains that assume a triple-helical conformation with a repeating Gly-X-Y amino acid motif where X and Y constitute any amino acid (47). Fibrillar collagens are composed of three α-chains, although more than 40 α-chains have been discovered in humans (48). Collagens can form supramolecular structures and can be classified as fibrillar, fibrillar-associated collagens with interrupted triple-helix (FACIT), membrane-associated collagens with interrupted triple-helix (MACIT), long chain, short chain, filamentous, or basement membrane comprised solely of type IV collagen. Although collagens are its main constituent, the ECM also consists of elastin, proteoglycans, lecticans, laminin, and fibronectin (FN). Like collagens, they have unique tertiary structures and contribute to the organization and complexity of the ECM and are reviewed in greater detail elsewhere (47).
The ECM is often referred to as the “matrisome” and consists of over 300 components that have been compiled and reviewed elsewhere (49). The relationship between the ECM and the cells residing within it is a reciprocal one, as matrix binding receptors sense the biochemical and physical makeup of the ECM and transduce signals to the cell which can in turn contribute to ECM remodeling. The major classes of ECM receptors (integrins, DDRs, and ERC) are discussed in further detail in this review.
Another important component of the “matrisome” is matrix bound proteins, such as growth factors that have important functions in the regulation of cell growth, plasticity, and metabolism. ECM components can bind to and sequester growth factors, storing them in a “solid phase” until their release is enabled (50). For example, FN and vitronectin contain known hepatocyte growth factor (HGF) binding sites, and endothelial cell migration was augmented by HGF complexed to FN or vitronectin (51). Similarly, vascular endothelial growth factor (VEGF) was shown to bind tenascin-X (52), and FN (53). FN fragments containing the VEGF binding domain as well as the α5β1 integrin binding domain were required for the maximal induction of endothelial cell migration and proliferation (53). In some cases, components of the ECM are required for ligand presentation and binding to its receptor. For instance, fibroblast growth factor (FGF) is known to bind to heparin sulfate, a requirement for FGF binding to the FGF receptor (54). Similarly, TGF-β is sequestered in the matrix, and TGF-β ligands are presented by integral membrane proteoglycans (55). In addition to acting as a growth factor reservoir, some ECM components contain growth factor-like domains that activate growth factor receptors directly. Two examples of this are laminins and tenascin, both known to contain EGF domains (56, 57). A hallmark of chondrogenic tissues is type II collagen, which is secreted into the ECM as two splice variants, type IIA and type IIB (58). Chondrocytes secrete mainly type IIB collagen (58), while type IIA collagen is secreted by epithelial and mesenchymal stem cells (59). Type IIA collagen was shown to bind to BMP-2 and TGF-β1, which are important to chondrogenesis and endochondral bone formation (58).
The composition of the ECM is highly dynamic and is regulated by a balance of matrix deposition and degradation by matrix proteases such as the matrix metalloproteinases (MMPs). A total of 23 MMPs have been identified in humans and can collectively degrade all ECM proteins (60). MMPs are secreted as zymogens that require enzymatic cleavage for activation. Endogenous inhibitors of MMPs, tissue inhibitors of metalloproteinases (TIMPs) 1–4, are present to prevent excessive cleavage of matrix components. TIMPs 1, 2, and 4 exist soluble in the extracellular milieu whereas TIMP3 is bound to the ECM (61). Dysregulation of matrix turnover resulting in excessive or insufficient matrix degradation can result in pathologies such as tissue fibrosis and contribute to the production of bioactive signaling molecules signaling through ECM receptors (62, 63). Degradation of matrix components can also result in altered release of growth factors sequestered in the ECM, for example TGF-β (64). Matrix turnover by MMPs is prominent in diseases associated with vascular calcification such as atherosclerosis and T2D, and thus release of pro-osteogenic growth factors may contribute to the pathogenesis of vascular calcification.
Direct signaling through ECM receptors by the matrisome also play an important role in the maintenance of cellular phenotype and function and will be the main focus of this review. Specifically, we will review work on the DDRs, the integrins, the ERC, and the RAGE, and their functions in osteogenesis and vascular calcification. A summary of these receptors, their ligands, and pro-calcific signaling and functions is provided in Table 1.
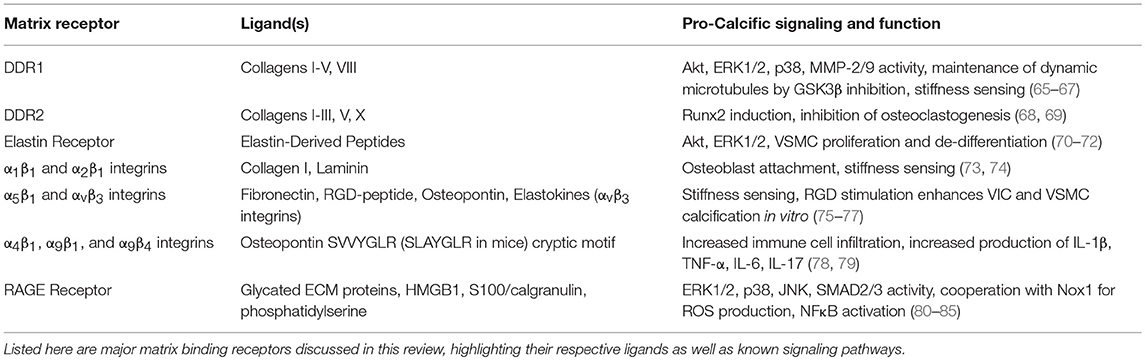
Table 1. Summary of matrix-binding receptors, their respective ligands, and their potential functions in promoting calcification.
Discoidin Domain Receptors
There are two DDRs, DDR1 and DDR2, which are collagen binding receptor tyrosine kinases. DDRs have been implicated in cellular processes regulating migration, adhesion, proliferation, as well as in the pathogenesis of fibrosis (86, 87), cancer (88, 89), atherosclerosis (90–92), and vascular calcification (16, 65). The DDRs are activated upon binding to native triple-helical collagens, undergoing autophosphorylation of the cytoplasmic domain which leads to downstream signaling (93). Compared to other receptor tyrosine kinases, the DDRs have delayed phosphorylation kinetics (93). For instance, the epidermal growth factor receptor (EGFR) and the fibroblast growth factor receptor (FGFR) are maximally phosphorylated in a period of seconds to minutes after ligand binding, and are then negatively regulated (94). In contrast, type-I collagen mediated DDR1 phosphorylation peaks at 90 min and is sustained for a period of 18 h (93). In cells grown in suspension, however, DDR1 phosphorylation was accelerated (95), demonstrating a context and adhesion dependent effect on DDR1 phosphorylation and function. Once activated, DDR1 can bind to signaling molecules that include PI3K subunits p85 and p110, STAT-1a/b,−3, and−5b, as well as guanine exchange factors PLC-γ1 and Vav1/2 (96). DDR1 has also been shown to activate P38 (97), ERK1/2 (98), and PI3K/Akt signaling pathways that are important in regulating cellular functions related to proliferation, metabolism and cell differentiation (65, 99).
Recent work from our laboratory has demonstrated that DDR1 promotes vascular calcification in atherosclerosis and diabetes (16, 65). Ahmad et al. studied Ldlr−/− mice fed a high-fat diet to stimulate the development of atherosclerotic plaques. DDR1 deficiency resulted in marked reductions in vascular calcification of the atherosclerotic plaques (16). VSMCs harvested from Ddr1−/− mice exhibited decreased alkaline phosphatase activity and matrix calcification in in vitro calcification assays. We next fed a diabetogenic diet to Ldlr−/− mice to induce diabetes and atherosclerosis (65). We found that DDR1 deletion decreased vascular calcification and abolished Runx2 nuclear localization in vivo. Cell culture experiments revealed that DDR1 signals via PI3K/Akt and P38 to activate Runx2 leading to VSMC transdifferentiation to an osteochondrocyte-like phenotype. Moreover, we showed that microtubules were required for the translocation of Runx2 to the nucleus, and microtubules were disrupted in DDR1 deficient VSMCs. In contrast to our recent work, a previous study reported that matrix calcification was significantly upregulated in DDR1 deficient VSMCs (100). A probable reason for this discrepancy is the use of different calcification media with a high concentration of β-glycerophosphate compared to our high glucose and phosphate media, because the former media causes cell death and passive calcification which likely obscures late stage phenotypic differences between the cells.
DDR2 has been implicated in chondrogenesis and in cartilage and bone remodeling (68, 69, 101–103). DDR2 is expressed in fibrocartilage within the temporomandibular joint, and deletion of DDR2 resulted in delayed condyle mineralization (104). DDR2 is also involved in bone remodeling. DDR2 overexpression inhibited osteoclast differentiation, and silencing DDR2 enhanced osteoclast differentiation, demonstrating that DDR2 is an important inhibitor of osteoclastogenesis (68). High expression of DDR2 has also been detected in synovial fibroblasts from patients with rheumatoid arthritis. DDR2 overexpression resulted in increased MMP-13 expression dependent on Runx2 and AP1 binding to the MMP-13 promoter (105). Taken together these studies highlight important functions for both DDRs in the regulation of osteogenesis, and specifically for DDR1 in regulating vascular calcification.
Integrins
Integrins are a family of heterodimeric ECM receptors formed by the dimerization of an α and a β subunit. In humans, 24 distinct heterodimers have been identified with different combinations of 18 α subunits and 8 β subunits (106, 107). Each heterodimer has distinct binding affinities and sequence recognition capabilities to ECM proteins. Upon ligand binding, integrins undergo a conformational change allowing for interaction with downstream intracellular signaling mediators such as focal adhesion kinase (FAK), Rho GTPases, and paxillin. In addition to conformational changes resulting from ligand binding, physical forces can induce structural changes to integrins. Integrins have been implicated in a number of cellular processes such as adhesion, migration, proliferation, and cellular differentiation.
Integrins have a role in osteogenic differentiation and both physiological and pathological calcification. During bone formation, osteoblasts and chondrocytes deposit ECM proteins such as osteopontin and type I collagen which activate integrins. The integrin-ECM interaction guides the progression of bone growth and mineralization. Of the integrins, β1 and β3 integrins have been studied more extensively. Stimulation of the FN receptor, α5β1 integrin, in adipose stem cells suppressed osteogenic differentiation (108). Transgenic mice expressing dominant-negative β1 integrin in osteoblasts had impaired bone formation, due to impaired adhesion of osteoblasts to the ECM (73). In addition to bone development, integrins have been shown to influence osteochondrocytic transdifferentiation and pathological calcification. Different matrix proteins elicit different effects on in vitro VSMC calcification and administration with α5-integrin blocking antibodies attenuated FN-mediated enhancement of calcification (75). This is in contrast to observations stated previously on the role of α5β1 integrin in adipose stem cells and osteogenesis, suggesting a cell type-specific role of α5β1 integrin in modulating osteogenesis. Work done on valve interstitial cell (VIC) calcification showed that stimulation with the β3-integrin ligand Arg-Gly-Asp (RGD) peptide, a motif found in FN, promoted osteochondrocytic differentiation and calcified nodule formation (76). This highlights the importance of integrin signaling to respond to cues from the ECM to modulate osteogenesis and calcification.
Elastin Receptor Complex
Elastin is a matrix component of large elastic and muscular arteries and is composed of polymerized tropoelastin monomers laid over a scaffold of fibrillin microfibrils, bound to elastin binding, and crosslinking proteins. Primarily synthesized and deposited during fetal development, elastin expression and synthesis of elastin fibers drops off significantly postnatally becoming undetectable by adulthood. Under physiological conditions, turnover of elastin is minimal and the protein has a half-life of several decades (109). In aging or pathological states such as atherosclerosis, elastin fiber integrity is compromised due to degradation by proteases such as MMP-2,−9, and−12 (110, 111), neutrophil elastases (112), and cysteine proteases (113). The proteolysis of elastin leads to the release of bioactive elastin-derived peptides called elastokines. Elastokines signal through cell surface receptors on smooth muscle cells to mediate a wide variety of activities. The elastin receptor complex (ERC) was the first receptor identified and remains the best studied (114), though αvβ3 and αvβ5 integrins (115, 116) as well as galectin-3 (117) can also respond to elastokines.
The ERC is a trimeric protein complex composed of two membrane-bound subunits, protective protein/cathepsin A (PPCA) and neuraminidase-1 (Neu-1), and the elastin-binding subunit, called elastin binding protein (EBP) (118). EBP is responsible for ligand binding through recognizing peptide motifs and Neu-1 is crucial for signal transduction (119, 120). The elastin receptor-activating elastokines include the VGVAPG repeat peptide and GXXPG-containing peptides, where X is any hydrophobic amino acid (119, 121, 122). Negative regulation of elastin receptor signaling is achieved through allosteric inhibition by galactose or lactose, which inhibits EBP binding to elastokines (123). Elastin receptor activation induces multiple intracellular events including activation of tyrosine kinases such as FAK and c-Src converging on activation of ERK1/2 and Akt (70–72).
Proteolytic degradation of elastin and signaling by elastokines and tropoelastin has been implicated in the progression of vascular calcification. Initial observations were made correlating the expression of MMP-2 and MMP-9 with regions of calcified elastin and osteogenic differentiation of VSMCs in vivo (124). More recent studies have shown that elastin breaks colocalize with regions of calcification in the aortas of Marfan syndrome patients (125). Treatment of VSMCs with elastokines promoted osteogenic differentiation and calcification in vitro and this was further enhanced with concomitant TGF-β treatment (126), or culture in high-glucose or high-phosphate media (127, 128).
Studies in mice with chemical or genetic inhibition of proteases have demonstrated that elastin degradation contributes to calcification. One of the earliest studies to directly demonstrate the effect of elastolysis on vascular calcification was done using a rat model of abdominal aortic aneurysm induced by low dose CaCl2 treatment (129). This protocol induced elastin breaks by increasing MMP-2 and MMP-9 activity, and tissues were harvested prior to the development of inflammatory response and alterations in vessel morphology due to abnormal hemodynamics. CaCl2 treatment caused calcification of the elastic fibers where breaks had been induced (129). Furthermore, MMP-2/MMP-9 deficiency or treatment with AlCl3, an inhibitor of MMP-2/9, attenuated degradation of elastic fibers and calcification (129). In other studies, the MMP inhibitor doxycycline was used to inhibit elastase activity in vitamin D3 or warfarin-induced rat models of vascular calcification and it was shown that this significantly inhibited vascular calcification (130–132). Nephrectomy is a commonly used model of chronic kidney disease which results in vascular calcification. Knockout of the elastolytic protease, cathepsin S (catS), in nephrectomised ApoE−/− mice attenuates elastin degradation and vascular and valvular calcification (133). Analysis of the blood biochemistry of ApoE−/−; CatS−/− mice following nephrectomy revealed reduced Cystatin C (marker for dysfunctional glomerular filtration), increased cholesterol, and similar phosphate and calcium concentrations compared to ApoE−/−; CatS+/+ mice following nephrectomy (133). Furthermore, histological analysis of carotid artery plaques from these two groups of mice revealed similar macrophage content as assessed by Mac-3 staining (133). Administration of cathepsin S to VSMCs treated with elastin peptides in culture enhanced in vitro calcification (133). TGF-β is normally sequestered in the ECM by the matrix-bound large latent complex (LLC) until released by matrix degrading proteases during matrix remodeling. The latent TGF-β binding proteins (LTBPs) are key components of the LLC and tether to elastin scaffold proteins and elastin crosslinking proteins such as fibrillin-1 and fibulin-5 (134, 135). Given these findings, proteolytic degradation of elastin and release of TGF-β may be a contributing mechanism to the development of vascular calcification. TGF-β is a known mediator of fibrosis and has been shown to stimulate VSMC calcification. Interestingly, calcifying vessels in mice lacking MGP had enhanced expression of elastin and abnormal elastin structure in the medial layer compared to non-calcifying wild-type mice (136). Furthermore, elastin haploinsufficiency (Eln+/−) on the MGP-null background reduced spontaneous vascular calcification (136). These results suggest that the quantity of elastin produced influences calcification, possibly through the scaffolding role of elastin for hydroxyapatite deposition or availability of elastin for the generation of elastokines which influence osteochondrocytic transdifferentiation.
Receptor for Advanced Glycation End Products (RAGE)
Vascular calcification is a major complication in T2D. One of the prominent hallmarks of T2D is hyperglycemia. Excessive circulating glucose can result in non-enzymatic glycation of proteins, altering their structural and functional properties. Glycation is a chemical modification by which a sugar moiety covalently modifies amino acid residues on proteins. Given the relatively long half-life and length of ECM proteins such as collagens, they are one of the major protein groups that are glycated in diabetes (137). Glycation of collagens can lead to structural deficits resulting in impairment of collagen cross-linking (138), increased matrix and tissue stiffness (139), inhibition of matrix turnover (140), defective cell-matrix interactions (141), and reduced sliding of collagen fibrils (142, 143). In addition to structural changes, glycated proteins can serve as ligands for the multi-ligand RAGE (144). RAGE are cell surface receptors found on most immune cells as well as on other cell types such as VSMCs (145–148). Aside from advanced glycation end products (AGEs), other ligands have been identified for RAGE such as the secreted proteins S-100/calgranulin (80) and high mobility group box 1 (HMGB1) (149), as well as phosphatidylserine (150). Ligands like S100/calgranulin family members can act through RAGE to promote the inflammatory NFκB pathway as well as ROS signaling through cooperation with NADPH oxidase-1 (Nox1) upon ligand binding (80, 81, 151). Downstream kinase mediators such as ERK1/2, p38, and JNK as well as TGF-β pathways are also activated by RAGE stimulation (82–85). A soluble RAGE is secreted by cells to act as a decoy receptor to attenuate RAGE signaling (149).
Recent studies have identified an important role for membrane RAGE and Nox1 signaling in VSMC osteochondrocytic differentiation and vascular calcification (81, 152). RAGE is upregulated in calcified tissues in rat models of diabetic aortic medial calcification as well as aortic valve calcification in ApoE−/− mice (153, 154). NFκB can increase the expression of RAGE, thus providing a positive feedback mechanism for this pathway (155, 156). In the assessment of human diabetic patients following foot amputation, it was found that levels of circulating AGEs and expression of RAGE in the anterior tibial artery wall was positively correlated with the extent of calcification (157). Circulating levels of the inhibitory soluble RAGE were reduced in patients with calcific aortic valve stenosis and with vascular calcification (158, 159). Ex vivo stimulation of diabetic rat femoral arteries with the RAGE ligand N-methylpyridinium enhanced calcification (160). Treatment of human VSMCs with AGEs from the serum of diabetic patients upregulated the expression of alkaline phosphatase, Runx2, and other osteochondrocytic proteins (161). On the other hand, RAGE-blocking antibodies, p38 inhibition, and Nox inhibition prevented calcification and VSMC osteochondrocytic transdifferentiation (81, 83). In vivo studies in ApoE−/− mice fed a high cholesterol diet showed that RAGE deficiency attenuated valvular calcification, and this was associated with reduced ER stress and inflammation without changes in lipid profile (154).
Osteopontin
Osteopontin is a matrix protein secreted by osteoblasts, macrophages, smooth muscle cells, and chondrocytes. This protein upon its secretion can integrate into the matrix through its negatively charged amino acid residues. Furthermore, it possesses a RGD sequence allowing for its recognition by integrin heterodimers including the αvβ3 integrin. Osteopontin also contains a distinct binding site for CD44 on immune cells which promotes cell adhesion and migration. Osteopontin contains a cleavage site for thrombin, which upon cleavage reveals a cryptic site for recognition and signal transduction by alternative integrin heterodimers α4β1, α9β1, and α9β4 (162–164). Recognition of the cryptic site by integrins enhances immune cell migration and promotes inflammation (78, 79). Bone marrow derived macrophages likely recognize osteopontin primarily through the cryptic site since flow cytometry has identified that ~95% are positive for α4 and α9 integrins, but only ~5% for αv integrins (165).
Osteopontin has a strong affinity for calcium allowing it to interact with hydroxyapatite (166). It has been identified to have a role in inhibiting biomineralization and negatively regulating calcium crystal formation both in physiological bone formation (167) and pathological ectopic calcification (168). Osteopontin also has roles modulating the formation and function of bone remodeling cells such as osteoclasts. Osteopontin stabilizes osteoclasts on the bone as it facilitates resorption and promotes the survival of osteoclasts (169–171). Osteopontin is upregulated in calcified vessels and possesses an inhibitory role in vascular calcification (172). Furthermore, circulating osteopontin has been identified as a biomarker for vascular calcification in diabetic patients (173). Knockout of osteopontin in spontaneously calcifying MGP-deficient mice or mice fed a high-phosphate diet accelerated the development of vascular calcification and death (174, 175). In vitro calcification of cultured VSMCs was found to be exacerbated by genetic deletion of osteopontin and prevented by reintroducing the osteopontin gene by retroviral transfection (176). Furthermore, loss of osteopontin promotes apoptosis of VSMCs, which may contribute to enhanced calcification (177). These studies implicate a beneficial effect of osteopontin in slowing the progression of vascular calcification. Interestingly, human peripheral monocytes and macrophages from hypertensive patients with vascular calcification had reduced potential to form osteoclasts following osteopontin stimulation (178). This indicates that osteopontin has a suppressive role for the turnover of hydroxyapatite in this context.
In addition to its effects on biomineralization, osteopontin has numerous roles in regulating smooth muscle cell phenotype and inflammation. Osteopontin promotes smooth muscle cell migration (179) and phenotypic switching by downregulating contractile markers and increasing proliferation (180, 181). Osteopontin is expressed widely by immune cells such as macrophages and is upregulated during the process of inflammation and wound healing (182, 183). It can facilitate differentiation of monocytes and allow adhesion and migration of immune cells through the engagement of adhesion receptors including integrins and CD44 (184–186). The osteopontin promoter contains pro-inflammatory response elements such as AP-1-binding sites and NFκB binding sites (187, 188). Stimulation of macrophages with LPS and pro-inflammatory cytokines such as IL-1β induces the expression of osteopontin (189). Osteopontin knockout in ApoE−/− mice attenuated macrophage infiltration into plaques and MMP-2/9 activity, resulting in a reduction in atherogenesis (190). Stimulation of monocytes/macrophages from hypertensive patients with vascular calcification with recombinant osteopontin reduced inflammatory cytokine secretion and increased anti-inflammatory cytokine production (178). However, osteopontin had no effect on monocytes/macrophages from hypertensive patients without vascular calcification (178). This finding of an anti-inflammatory effect contrasts with the findings stated previously. However, it may suggest a context-dependent response by monocytes/macrophages to osteopontin and through the influence of other factors in the calcifying environment.
Matrix Gla Protein (MGP)
MGP is a vitamin K-dependent, secreted matrix protein expressed by all tissues and multiple cell types including VSMCs, macrophages, and osteoblasts. MGP was first isolated in bone and was found to accumulate significantly in the ECM of bone compared to non-calcifying tissues such as the kidney, lungs, and heart (191). The mRNA expression of MGP in osteoblasts, however, was lower than that of the non-calcifying tissues (191). MGP was implicated as an inhibitor of vascular calcification following a study showing that its deletion in mice led to spontaneous vascular calcification and death within 2 months from arterial rupture (192). MGP possesses anti-calcific effects through inhibition of hydroxyapatite mineral formation (193) and BMP signaling (194, 195). Examining the expression pattern of MGP in human vessels, normal vessels exhibit a gradient with high levels of MGP on the luminal side that decreases gradually toward the medial layer (196). VSMCs normally express MGP and high levels of MGP can be found in the fibrous cap of atherosclerotic lesions (196). A negative correlation was found with areas of low or absent MGP expression in the vessel with calcification and Runx2 expression for both intimal and medial calcification (196). MGP expression is downregulated in calcified atherosclerotic human arteries compared to non-calcified vessels (196). In vitro calcification studies done with bovine VSMCs found reduced MGP mRNA expression in calcifying cells and had an inverse correlation with the extent of calcification (197). Inhibition of calcification with bisphosphonates restored the expression of MGP mRNA in VSMCs cultured in calcifying media, suggesting that the process of calcification precedes the downregulation of MGP (197). Some conflicting evidence was found, however, showing MGP mRNA was upregulated in calcifying human VSMCs in vitro (198). This is likely a result of species-specific differences or time-dependent fluctuations in the expression of MGP. For MGP activity, the protein must be gamma-carboxylated in a vitamin K-dependent manner (199). The anti-coagulation medication, warfarin, is known to be a vitamin K antagonist. A previous study has found that 2 weeks of warfarin treatment in young rats induced significant focal vascular calcification (200). Isolated VSMCs treated with warfarin have reduced carboxylated MGP and increased in vitro calcification (201). This was shown to be vitamin K-dependent as the reintroduction of vitamin K to warfarin-treated rats could inhibit calcification (201, 202).
Matrix Stiffness Sensing
The stiffness of the matrix is measured by how much force is necessary to deform a substrate. Stiffness is often measured as an elastic modulus in kilopascals (kPa). The elastic modulus is defined as the ratio of the stress, which is the force of deformation divided by the area to which it is applied, to the strain, which is the ratio describing the relative deformation compared to the object's original state. The composition, density, and structural integrity of the extracellular matrix play a significant role in determining the stiffness of a tissue. Increased density of collagens such as type I collagen and the glycoprotein FN enhance stiffness whereas glycosaminoglycans such as hyaluronic acid permit greater compliance of the ECM. Increased matrix degradation by MMPs reduces matrix stiffness, whereas increased crosslinking of matrix proteins by lysyl oxidase (LOX) and glycation of matrix proteins enhance matrix stiffness.
In addition to biochemical and chemical mediators, cells experience a variety of physical cues that influence their behavior, phenotype, and differentiation. By a process called mechanotransduction, cells can convert physical cues or forces into biochemical signals affecting cellular responses. The stiffness of the matrix is detected by matrix-binding cell surface receptors including the integrin family of receptors and DDRs. The mechanism by which this occurs is illustrated schematically in Figure 1. Matrix receptors sense the stiffness of the ECM by binding and tugging on the ECM via their extracellular domains and interacting with signaling molecules and the cytoskeleton via their cytosolic domains. Resistance or stiffness of the ECM triggers the reorganization of the cytoskeleton, polymerization of actin monomers (G-actin) into filamentous actin (F-actin) and bundling of F-actin into contractile units known as stress fibers. Cells contract in response to external forces, with myosin generating tensional forces on the actin fibers increasing the stiffness of the cytoskeleton (203, 204). Intact microtubules are also necessary for cells to respond to externally applied forces (204). Tension forces generated by actin stress fibers deform the nuclear envelope through their interaction with the LINC (linkers of the nucleoskeleton to the cytoskeleton) complexes (205). Increased tension on the nuclear envelope promotes the expression and reduces degradation of an abundant structural protein in the nuclear envelope, Lamin A (206). This increases the tensile strength of the nuclear envelope when cytoskeletal tension is high.
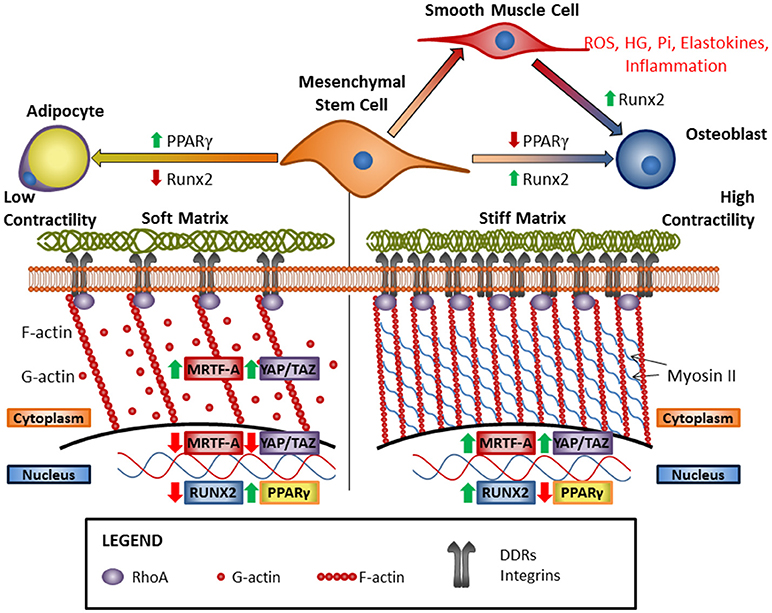
Figure 1. Increasing matrix stiffness drives osteogenic differentiation via cooperative ECM receptor signaling and modulation of cytoskeletal dynamics. Classically, matrix stiffness is known to be sensed by integrins, but recent research has shown that DDR1 can also act as a mechanosensory receptor and is an important mediator of vascular calcification. Vascular calcification occurs as VSMCs transdifferentiate into osteoblast-like cells and can be promoted by RUNX2, high-glucose (HG), inorganic phosphate (Pi), elastokines, and inflammation. Increased matrix stiffness leads to osteogenic differentiation of mesenchymal stem cells and the cytoskeleton plays an integral role in this process. Stress fiber formation due to increased RhoA activity results in increased nuclear internalization of fibrogenic/myogenic transcription co-factors MRTF-A and YAP/TAZ, and osteogenic transcription factor Runx2, and the concomitant inhibition of adipogenic transcription factor PPARγ. Conversely, reduced ECM stiffness leads to reductions in ECM receptor activation and stress fiber formation, leading to nuclear exclusion of MRTF-A and YAP/TAZ, and reduced Runx2 activity. The resulting increase in PPARγ activity results in adipogenic differentiation.
Since collagens and FN are abundant in the ECM, it is not surprising that integrins binding these molecules play important roles in mechanotransduction. Numerous studies have demonstrated the role of type I collagen-binding α1β1 and α2β1 integrin heterodimers (74, 207, 208), and FN-binding α5β1 and αvβ3 integrin heterodimers (77, 209) in mechanotransduction for a variety of cell types including mesenchymal stem cells, fibroblasts, and HEK293 cells. In more recent years, Ghosh et al. showed that DDR1 was a mechanotransducer in adipose-derived stem cells (ASCs) (66). In ASCs, increased compliance of the substrate and reduced cell contractility promoted aromatase and estrogen expression. Knocking down DDR1 with siRNAs attenuated aromatase expression on a soft substrate without influencing stress fiber formation, suggesting that DDR1 helps cells sense matrix compliance (66). Coelho et al. have shown that DDR1 activation is enhanced on type I collagen-coated stiff substrates compared to soft substrates, and that interaction of DDR1 with non-muscle myosin IIA triggers cell contraction and reorganization of the fibrillar collagen ECM (210). Furthermore, in vivo studies of rat dermal wound healing found increased DDR1 expression and activation in myofibroblasts when mechanical force was applied to the wound by splinting (210). DDR2 expression has been shown to increase with increasing matrix stiffness (211) and DDR2 has been implicated in cell shape changes in response to changes in matrix stiffness (212). However, evidence is still lacking to show whether or not this receptor is a mediator of mechanosensing.
Matrix Stiffness and Osteogenesis
A seminal paper published by Engler and colleagues showed that mesenchymal stem cell fate was determined by substrate stiffness (32). Osteogenesis was favored and promoted on a type I collagen-coated substrate with a stiffness of 34 kPa, which is within the range of stiffnesses of collagenous pre-calcified bone (25–40 kPa). Furthermore, pharmacological inhibition of non-muscle myosin IIa by blebbistatin blocked differentiation of mesenchymal stem cells. This highlights the importance of stiffness sensing in osteogenic cell differentiation.
Three transcription factors are responsive to changes in matrix stiffness, and they play roles in osteoblast differentiation, smooth muscle differentiation, and vascular smooth muscle calcification. These are the Yes-associated protein (YAP) and its related protein Tafazzin (TAZ) (213), and Myocardin related transcription factor-A (MRTF-A) (214). All three depend on actin polymerization for the regulation of cytoplasmic and nuclear localization and therefore activity (215–217) (illustrated schematically in Figure 1). MRTF-A is sequestered in the cytoplasm by binding to G-actin but is released to translocate to the nucleus upon actin polymerization to F-actin in cells under mechanical stress (215). In a similar mechanism, YAP/TAZ binds to angiomotin (AMOT) in the cytoplasm until G-actin polymerizes to F-actin, which binds AMOT and allows YAP/TAZ to translocate to the nucleus (218). Actin polymerization also inhibits the Hippo pathway, which is the canonical pathway that mediates YAP/TAZ phosphorylation and prevents its nuclear localization (216). In addition, mechanical stress can induce deformation of the nucleus allowing YAP to be transported into the nucleus through nuclear pores (219). In mechanotransduction, actin polymerization is able to regulate YAP/TAZ nuclear localization independently of canonical Hippo pathway signaling (217). This establishes YAP/TAZ as a mechanosensitive transcriptional co-factor, although further studies are necessary to understand the mechanisms at work.
In mesenchymal stem cells, TAZ forms an activating complex with Runx2 to promote osteogenesis and calcification while suppressing peroxisome proliferator-activated receptor-γ (PPARγ) -mediated adipogenesis (33, 220). YAP/TAZ can influence vascular calcification by enhancing phenotypic switching of VSMCs, or by promoting the inflammatory response. YAP blocks serum response factor (SRF) transcriptional activity to facilitate the conversion of VSMCs from the contractile to the synthetic phenotype, a process which precedes vascular calcification (221, 222). In addition, YAP/TAZ is upregulated in endothelial cells exposed to pro-atherogenic oscillatory blood flow and stimulates the inflammatory response which exacerbates vascular calcification (223, 224).
MRTF-A is an important transcriptional co-factor for SRF, the master regulator of VSMC specific gene expression (225). MRTF-A has also been implicated in osteogenic differentiation of mesenchymal stem cells, and in the transdifferentiation of heart VICs and VSMCs during pathological calcification. MRTF-A KO mice have defects in osteogenesis (226). In human aortic valve fibrosis and calcification, there is upregulation of MRTF-A and smooth muscle α-actin expression in calcified regions, concurrent with differentiation from the VIC to the myofibroblast phenotype (227). In VSMCs grown in normal media, MRTF-A acts downstream of BMPs to maintain the differentiated smooth muscle phenotype (228). However, under calcifying conditions, Runx2 interferes with MRTF-A/SRF to downregulate VSMC-specific genes and upregulate osteochondrocytic genes (229). Since both MRTF-A and YAP/TAZ are regulated by stiffness and actin dynamics, there is extensive crosstalk between the pathways which can be cooperative or mutually inhibitory (230–232). Furthermore, the net response from their signaling is modulated by activation of TGF-β-Smad3 (230). Excessive matrix turnover in vascular pathologies may result in increased release of the pro-fibrotic TGF-β, thus increasing matrix stiffness and cooperative signaling with MRTF-A or YAP/TAZ in VSMCs to enhance osteochondrocytic differentiation.
Cytoskeleton and Nucleoskeleton in Osteogenesis: Roles of Tubulin and Lamin A
The actin and microtubule cytoskeletons play important roles in mechanotransduction (233), transport of vesicles and proteins (234), and are involved in mediating osteogenesis (235–238). Microtubules can participate in signal cascades by affecting the distribution and compartmentalization of signaling molecules and transcription factors. Recently, we have identified DDR1 as an important mediator of vascular calcification, acting in part to maintain an intact microtubule cytoskeleton which allows Runx2 translocation to the nucleus (65). Studies in tumor cells have demonstrated that Runx2 binds to α-tubulin via its amino terminus, and that these interactions with tubulin are necessary for the nuclear export of Runx2 and sequestration of the transcription factor in the cytoplasm in these cells (239). Interestingly, a microtubule-associated protein, doublecortin-like and CAM kinase-like 1 (DCAMKL1), has been identified as a regulator of osteogenesis. DCAMKL1 blunted osteogenesis by inhibiting Runx2 and genetic disruption of DCAMKL1 in mice resulted in increased bone mass (240). These studies support the notion that microtubules affect osteogenesis by regulating the intracellular localization of Runx2, however the precise mechanisms are complex and vary between different cell types.
Lamin A is a mechanosensitive nuclear envelope protein, a link between the cytoplasmic and nuclear cytoskeleton, and can form complexes with transcription factors. Lamin A is an important reservoir and regulator of transcription factors and co-factors involved in cell fate specification for cells in the mesenchymal lineage, and likely acts by several overlapping mechanisms (241). Lamin A regulates YAP and SRF transcriptional activity by promoting YAP nuclear localization and regulating nuclear actin dynamics to promote SRF activity (206). Lamin A also binds to and promotes Runx2 activity and calcification in VSMCs, osteoblasts, and mesenchymal stem cells, facilitating nuclear transport of Runx2 and enhancing its DNA-binding and transcriptional activity (242, 243). Lamin A is upregulated in rat VSMCs cultured in high calcium and phosphate calcifying media (242). The unprocessed Lamin A precursor, prelamin A, accumulates in calcifying and senescent VSMCs in vitro and promotes the osteogenic transcriptional program (244). Increased accumulation of prelamin A in calcifying vascular cells was also observed in vivo in child dialysis patients with medial calcification (244).
Conclusion
Our understanding of the pathology of vascular calcification has advanced considerably, and it is now understood to be an active process driven by VSMC transdifferentiation into osteochondrocyte-like cells. There has been increased interest in understanding how cell-matrix interactions influence cellular responses during vascular calcification. The ECM is constantly changing in normal physiology and in pathology, and its role extends beyond that of a scaffold to that of a signaling mediator with important effects on the transcriptional regulation of cell phenotype. Further investigation into matricrine regulation of cell phenotypes will lead to the development of novel therapeutics to prevent or reverse vascular calcification.
Author Contributions
MB, DN, and ML conceived of the ideas and outline for the manuscript. DN and ML wrote the first drafts, and MB made suggestions for improvements and additions, and edited the manuscript.
Funding
Funded by the Canadian Institutes for Health Research grant MOP133592 to MB. Canadian Institutes for Health Research funded the researchers for related projects, and the publication costs of this manuscript. DN was funded by scholarships from the Canadian Institutes of Health Research and the Banting and Best Diabetes Centre. ML was funded by scholarships from the Banting and Best Diabetes Centre and Heart & Stroke Richard Lewar Centre of Excellence.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Budoff MJ, Nasir K, Katz R, Takasu J, Carr JJ, Wong ND, et al. Thoracic aortic calcification and coronary heart disease events: the multi-ethnic study of atherosclerosis (MESA). Atherosclerosis (2011) 215:196–202. doi: 10.1016/j.atherosclerosis.2010.11.017
2. Wilson PW, Kauppila LI, O'Donnell CJ, Kiel DP, Hannan M, Polak JM, et al. Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation (2001) 103:1529–34. doi: 10.1161/01.CIR.103.11.1529
3. Santos RD, Rumberger JA, Budoff MJ, Shaw LJ, Orakzai SH, Berman D, et al. Thoracic aorta calcification detected by electron beam tomography predicts all-cause mortality. Atherosclerosis (2010) 209:131–5. doi: 10.1016/j.atherosclerosis.2009.08.025
4. Chen J, Budoff MJ, Reilly MP, Yang W, Rosas SE, Rahman M, et al. Coronary artery calcification and risk of cardiovascular disease and death among patients with chronic kidney disease. JAMA Cardiol. (2017) 2:635–43. doi: 10.1001/jamacardio.2017.0363
5. Mizobuchi M, Towler D, Slatopolsky E. Vascular calcification: the killer of patients with chronic kidney disease. J Am Soc Nephrol. (2009) 20:1453–64. doi: 10.1681/ASN.2008070692
6. Huang CL, Wu IH, Wu YW, Hwang JJ, Wang SS, Chen WJ, et al. Association of lower extremity arterial calcification with amputation and mortality in patients with symptomatic peripheral artery disease. PLoS ONE (2014) 9:e90201. doi: 10.1371/journal.pone.0090201
7. Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. (2004) 24:1161–70. doi: 10.1161/01.ATV.0000133194.94939.42
8. Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. (2014) 34:715–23. doi: 10.1161/ATVBAHA.113.302070
9. Giachelli CM. The emerging role of phosphate in vascular calcification. Kidney Int. (2009) 75:890–7. doi: 10.1038/ki.2008.644
10. Shao JS, Cai J, Towler DA. Molecular mechanisms of vascular calcification: lessons learned from the aorta. Arterioscler Thromb Vasc Biol. (2006) 26:1423–30. doi: 10.1161/01.ATV.0000220441.42041.20
11. Andrade MC, Carmo LS, Farias-Silva E, Liberman M. Msx2 is required for vascular smooth muscle cells osteoblastic differentiation but not calcification in insulin-resistant ob/ob mice. Atherosclerosis (2017) 265:14–21. doi: 10.1016/j.atherosclerosis.2017.07.028
12. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. (2008) 283:15319–27. doi: 10.1074/jbc.M800021200
13. Cheng SL, Shao JS, Halstead LR, Distelhorst K, Sierra O, Towler DA. Activation of vascular smooth muscle parathyroid hormone receptor inhibits Wnt/beta-catenin signaling and aortic fibrosis in diabetic arteriosclerosis. Circ Res. (2010) 107:271–82. doi: 10.1161/CIRCRESAHA.110.219899
14. Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, et al. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. (2012) 111:543–52. doi: 10.1161/CIRCRESAHA.112.267237
15. Chen NX, Duan D, O'Neill KD, Moe SM. High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol Dial Transplant. (2006) 21:3435–42. doi: 10.1093/ndt/gfl429
16. Ahmad PJ, Trcka D, Xue S, Franco C, Speer MY, Giachelli CM, et al. Discoidin domain receptor-1 deficiency attenuates atherosclerotic calcification and smooth muscle cell-mediated mineralization. Am J Pathol. (2009) 175:2686–96. doi: 10.2353/ajpath.2009.080734
17. Lomashvili KA. Phosphate-induced vascular calcification: role of pyrophosphate and osteopontin. J Am Soc Nephrol. (2004) 15:1392–401. doi: 10.1097/01.Asn.0000128955.83129.9c
18. Jono S, Ikari Y, Vermeer C, Dissel P, Hasegawa K, Shioi A, et al. Matrix Gla protein is associated with coronary artery calcification as assessed by electron-beam computed tomography. Thromb Haemost. (2004) 91:790–4. doi: 10.1160/TH03-08-0572
19. Westenfeld R, Schafer C, Kruger T, Haarmann C, Schurgers LJ, Reutelingsperger C, et al. Fetuin-A protects against atherosclerotic calcification in CKD. J Am Soc Nephrol. (2009) 20:1264–74. doi: 10.1681/ASN.2008060572
20. Giachelli CM. Ectopic calcification: gathering hard facts about soft tissue mineralization. Am J Pathol. (1999) 154:671–5. doi: 10.1016/S0002-9440(10)65313-8
21. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. (2011) 109:697–711. doi: 10.1161/CIRCRESAHA.110.234914
22. Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. (2010) 7:528–36. doi: 10.1038/nrcardio.2010.115
23. Amann K. Media calcification and intima calcification are distinct entities in chronic kidney disease. Clin J Am Soc Nephrol. (2008) 3:1599–605. doi: 10.2215/CJN.02120508
24. Chen NX, Moe SM. Arterial calcification in diabetes. Curr Diab Rep. (2003) 3:28–32. doi: 10.1007/s11892-003-0049-2
25. Heath JM, Sun Y, Yuan K, Bradley WE, Litovsky S, Dell'Italia LJ, et al. Activation of AKT by O-linked N-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ Res. (2014) 114:1094–102. doi: 10.1161/CIRCRESAHA.114.302968
26. Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, et al. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr-/- mice. Arterioscler Thromb Vasc Biol. (2007) 27:2589–96. doi: 10.1161/ATVBAHA.107.153668
27. Chuang ML, Gona P, Oyama-Manabe N, Manders ES, Salton CJ, Hoffmann U, et al. Risk factor differences in calcified and noncalcified aortic plaque: the Framingham Heart Study. Arterioscler Thromb Vasc Biol. (2014) 34:1580–6. doi: 10.1161/ATVBAHA.114.303600
28. Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation (2010) 121:505–11. doi: 10.1161/CIRCULATIONAHA.109.886655
29. Alam MU, Kirton JP, Wilkinson FL, Towers E, Sinha S, Rouhi M, et al. Calcification is associated with loss of functional calcium-sensing receptor in vascular smooth muscle cells. Cardiovasc Res. (2009) 81:260–8. doi: 10.1093/cvr/cvn279
30. Chen TC, Lin CT, Chien SJ, Chang SF, Chen CN. Regulation of calcification in human aortic smooth muscle cells infected with high-glucose-treated Porphyromonas gingivalis. J Cell Physiol. (2018) 233:4759–69. doi: 10.1002/jcp.26268
31. Olesen P, Nguyen K, Wogensen L, Ledet T, Rasmussen LM. Calcification of human vascular smooth muscle cells: associations with osteoprotegerin expression and acceleration by high-dose insulin. Am J Physiol Heart Circ Physiol. (2007) 292:H1058–64. doi: 10.1152/ajpheart.00047.2006
32. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell (2006) 126:677–89. doi: 10.1016/j.cell.2006.06.044
33. Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science (2005) 309:1074–8. doi: 10.1126/science.1110955
34. Schroeder TM, Jensen ED, Westendorf JJ. Runx2: a master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Res C Embryo Today (2005) 75:213–25. doi: 10.1002/bdrc.20043
35. Choi YH, Kim YJ, Jeong HM, Jin YH, Yeo CY, Lee KY. Akt enhances Runx2 protein stability by regulating Smurf2 function during osteoblast differentiation. FEBS J. (2014) 281:3656–66. doi: 10.1111/febs.12887
36. Huang YF, Lin JJ, Lin CH, Su Y, Hung SC. c-Jun N-terminal kinase 1 negatively regulates osteoblastic differentiation induced by BMP2 via phosphorylation of Runx2 at Ser104. J Bone Miner Res. (2012) 27:1093–105. doi: 10.1002/jbmr.1548
37. Kugimiya F, Kawaguchi H, Ohba S, Kawamura N, Hirata M, Chikuda H, et al. GSK-3beta controls osteogenesis through regulating Runx2 activity. PLoS ONE (2007) 2:e837. doi: 10.1371/journal.pone.0000837
38. Pierce AD, Anglin IE, Vitolo MI, Mochin MT, Underwood KF, Goldblum SE, et al. Glucose-activated RUNX2 phosphorylation promotes endothelial cell proliferation and an angiogenic phenotype. J Cell Biochem. (2012) 113:282–92. doi: 10.1002/jcb.23354
39. Komori T. Requisite roles of Runx2 and Cbfb in skeletal development. J Bone Miner Metab. (2003) 21:193–7. doi: 10.1007/s00774-002-0408-0
40. Bernard GW, Daniel CP. An electron microscopy study of initial intramembranous osteogenesis. Am J Anat. (1969) 125:271–90. doi: 10.1002/aja.1001250303
41. Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. (2016) 15:335–43. doi: 10.1038/nmat4519
42. Starcher BC, Urry DW. Elastin coacervate as a matrix for calcification. Biochem Biophys Res Commun. (1973) 53:210–6. doi: 10.1016/0006-291X(73)91421-6
43. Forlino A, Marini JC. Osteogenesis imperfecta. Lancet (2016) 387:1657–71. doi: 10.1016/s0140-6736(15)00728-x
44. Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet (2004) 363:1377–85. doi: 10.1016/S0140-6736(04)16051-0
45. Xie P, Liu B, Zhang L, Chen R, Yang B, Dong J, et al. Association of COL1A1 polymorphisms with osteoporosis: a meta-analysis of clinical studies. Int J Clin Exp Med. (2015) 8:14764–81.
46. Mann V, Hobson EE, Li B, Stewart TL, Grant SF, Robins SP, et al. A COL1A1 Sp1 binding site polymorphism predisposes to osteoporotic fracture by affecting bone density and quality. J Clin Invest. (2001) 107:899–907. doi: 10.1172/JCI10347
47. Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol. (2014) 15:771–85. doi: 10.1038/nrm3902
48. Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol. (2011) 3:a004978. doi: 10.1101/cshperspect.a004978
49. Hynes RO, Naba A. Overview of the matrisome–an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol. (2012) 4:a004903. doi: 10.1101/cshperspect.a004903
50. Hynes RO. The extracellular matrix: not just pretty fibrils. Science (2009) 326:1216–9. doi: 10.1126/science.1176009
51. Rahman S, Patel Y, Murray J, Patel KV, Sumathipala R, Sobel M, et al. Novel hepatocyte growth factor (HGF) binding domains on fibronectin and vitronectin coordinate a distinct and amplified Met-integrin induced signalling pathway in endothelial cells. BMC Cell Biol. (2005) 6:8. doi: 10.1186/1471-2121-6-8
52. Ishitsuka T, Ikuta T, Ariga H, Matsumoto K. Serum tenascin-X strongly binds to vascular endothelial growth factor. Biol Pharm Bull. (2009) 32:1004–11. doi: 10.1248/bpb.32.1004
53. Wijelath ES, Rahman S, Namekata M, Murray J, Nishimura T, Mostafavi-Pour Z, et al. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ Res. (2006) 99:853–60. doi: 10.1161/01.RES.0000246849.17887.66
54. Mohammadi M, Olsen SK, Goetz R. A protein canyon in the FGF-FGF receptor dimer selects from an a la carte menu of heparan sulfate motifs. Curr Opin Struct Biol. (2005) 15:506–16. doi: 10.1016/j.sbi.2005.09.002
55. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell (2003) 113:685–700. doi: 10.1016/S0092-8674(03)00432-X
56. Panayotou G, End P, Aumailley M, Timpl R, Engel J. Domains of laminin with growth-factor activity. Cell (1989) 56:93–101. doi: 10.1016/0092-8674(89)90987-2
57. Schenk S, Hintermann E, Bilban M, Koshikawa N, Hojilla C, Khokha R, et al. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J Cell Biol. (2003) 161:197–209. doi: 10.1083/jcb.200208145
58. Zhu Y, Oganesian A, Keene DR, Sandell LJ. Type IIA procollagen containing the cysteine-rich amino propeptide is deposited in the extracellular matrix of prechondrogenic tissue and binds to TGF-beta1 and BMP-2. J Cell Biol. (1999) 144:1069–80. doi: 10.1083/jcb.144.5.1069
59. Sandell LJ, Morris N, Robbins JR, Goldring MB. Alternatively spliced type II procollagen mRNAs define distinct populations of cells during vertebral development: differential expression of the amino-propeptide. J Cell Biol. (1991) 114:1307–19. doi: 10.1083/jcb.114.6.1307
60. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. (2014) 15:786–801. doi: 10.1038/nrm3904
61. Leco KJ KR, Pavloff N, Hawkes SP, Edwards DR. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is an extracellular matrix-associated protein with a distinctive pattern of expression in mouse cells and tissues. J Biol Chem. (1994) 269:9352–60.
62. Decaris ML, Gatmaitan M, FlorCruz S, Luo F, Li K, Holmes WE, et al. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol Cell Proteomics (2014) 13:1741–52. doi: 10.1074/mcp.M113.037267
63. Ricard-Blum S, Vallet SD. Fragments generated upon extracellular matrix remodeling: biological regulators and potential drugs. Matrix Biol. (2017) S0945-053X(17)30277-9. doi: 10.1016/j.matbio.2017.11.005
64. Jones JA, Barbour JR, Stroud RE, Bouges S, Stephens SL, Spinale FG, et al. Altered transforming growth factor-beta signaling in a murine model of thoracic aortic aneurysm. J Vasc Res. (2008) 45:457–68. doi: 10.1159/000127437
65. Lino M, Wan MH, Rocca AS, Ngai D, Shobeiri N, Hou G, et al. Diabetic vascular calcification mediated by the collagen receptor discoidin domain receptor 1 via the Phosphoinositide 3-Kinase/Akt/Runt-related transcription factor 2 signaling axis. Arterioscler Thromb Vasc Biol. (2018) 38:1878–89. doi: 10.1161/ATVBAHA.118.311238
66. Ghosh S, Ashcraft K, Jahid MJ, April C, Ghajar CM, Ruan J, et al. Regulation of adipose oestrogen output by mechanical stress. Nat Commun. (2013) 4:1821. doi: 10.1038/ncomms2794
67. Hou G, Vogel W, Bendeck MP. The discoidin domain receptor tyrosine kinase DDR1 in arterial wound repair. J Clin Invest. (2001) 107:727–35. doi: 10.1172/JCI10720
68. Zhang Y, Su J, Wu S, Teng Y, Yin Z, Guo Y, et al. DDR2 (discoidin domain receptor 2) suppresses osteoclastogenesis and is a potential therapeutic target in osteoporosis. Sci Signal. (2015) 8:ra31. doi: 10.1126/scisignal.2005835
69. Zhang Y, Su J, Yu J, Bu X, Ren T, Liu X, et al. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res. (2011) 26:604–17. doi: 10.1002/jbmr.225
70. Duca L, Lambert E, Debret R, Rothhut B, Blanchevoye C, Delacoux F, et al. Elastin peptides activate extracellular signal-regulated kinase 1/2 via a Ras-independent mechanism requiring both p110gamma/Raf-1 and protein kinase A/B-Raf signaling in human skin fibroblasts. Mol Pharmacol. (2005) 67:1315–24. doi: 10.1124/mol.104.002725
71. Mochizuki S, Brassart B, Hinek A. Signaling pathways transduced through the elastin receptor facilitate proliferation of arterial smooth muscle cells. J Biol Chem. (2002) 277:44854–63. doi: 10.1074/jbc.M205630200
72. Fahem A, Robinet A, Cauchard JH, Duca L, Soula-Rothhut M, Rothhut B, et al. Elastokine-mediated up-regulation of MT1-MMP is triggered by nitric oxide in endothelial cells. Int J Biochem Cell Biol. (2008) 40:1581–96. doi: 10.1016/j.biocel.2007.11.022
73. Zimmerman D, Jin F, Leboy P, Hardy S, Damsky C. Impaired bone formation in transgenic mice resulting from altered integrin function in osteoblasts. Dev Biol. (2000) 220:2–15. doi: 10.1006/dbio.2000.9633
74. Shih YR, Tseng KF, Lai HY, Lin CH, Lee OK. Matrix stiffness regulation of integrin-mediated mechanotransduction during osteogenic differentiation of human mesenchymal stem cells. J Bone Miner Res. (2011) 26:730–8. doi: 10.1002/jbmr.278
75. Watson KE, Parhami F, Shin V, Demer LL. Fibronectin and collagen I matrixes promote calcification of vascular cells in vitro, whereas collagen IV matrix is inhibitory. Arterioscler Thromb Vasc Biol. (1998) 18:1964–71. doi: 10.1161/01.Atv.18.12.1964
76. Gu X, Masters KS. Regulation of valvular interstitial cell calcification by adhesive peptide sequences. J Biomed Mater Res A (2010) 93:1620–30. doi: 10.1002/jbm.a.32660
77. Balcioglu HE, van Hoorn H, Donato DM, Schmidt T, Danen EH. The integrin expression profile modulates orientation and dynamics of force transmission at cell-matrix adhesions. J Cell Sci. (2015) 128:1316–26. doi: 10.1242/jcs.156950
78. Boggio E, Dianzani C, Gigliotti CL, Soluri MF, Clemente N, Cappellano G, et al. Thrombin cleavage of osteopontin modulates its activities in human cells in vitro and mouse experimental autoimmune encephalomyelitis in vivo. J Immunol Res. (2016) 2016:9345495. doi: 10.1155/2016/9345495
79. Yamamoto N, Sakai F, Kon S, Morimoto J, Kimura C, Yamazaki H, et al. Essential role of the cryptic epitope SLAYGLR within osteopontin in a murine model of rheumatoid arthritis. J Clin Invest. (2003) 112:181–8. doi: 10.1172/jci17778
80. Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell (1999) 97:889–901. doi: 10.1016/S0092-8674(00)80801-6
81. Hofmann Bowman MA, Gawdzik J, Bukhari U, Husain AN, Toth PT, Kim G, et al. S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein E-null mice by activating an osteogenic gene regulatory program. Arterioscler Thromb Vasc Biol. (2011) 31:337–44. doi: 10.1161/ATVBAHA.110.217745
82. Ishihara K, Tsutsumi K, Kawane S, Nakajima M, Kasaoka T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. (2003) 550:107–13. doi: 10.1016/s0014-5793(03)00846-9
83. Tanikawa T, Okada Y, Tanikawa R, Tanaka Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J Vasc Res. (2009) 46:572–80. doi: 10.1159/000226225
84. Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest. (2008) 118:183–94. doi: 10.1172/JCI32703
85. Li JH, Huang XR, Zhu HJ, Oldfield M, Cooper M, Truong LD, et al. Advanced glycation end products activate Smad signaling via TGF-b-dependent and -independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. (2003) 36:1–10. doi: 10.1111/j.1523-1755.2004.66004.x
86. Coelho NM, McCulloch CA. Mechanical signaling through the discoidin domain receptor 1 plays a central role in tissue fibrosis. Cell Adh Migr. (2018) 26:1–15. doi: 10.1080/19336918.2018.1448353
87. Gross O, Girgert R, Beirowski B, Kretzler M, Kang HG, Kruegel J, et al. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. (2010) 29:346–56. doi: 10.1016/j.matbio.2010.03.002
88. Aguilera KY, Huang H, Du W, Hagopian MM, Wang Z, Hinz S, et al. Inhibition of Discoidin domain receptor 1 reduces collagen-mediated tumorigenicity in pancreatic ductal adenocarcinoma. Mol Cancer Ther. (2017) 16:2473–85. doi: 10.1158/1535-7163.MCT-16-0834
89. Ambrogio C, Darbo E, Lee SW, Santamaria D. A putative role for Discoidin Domain Receptor 1 in cancer chemoresistance. Cell Adh Migr. (2018) 12:1–4. doi: 10.1080/19336918.2018.1445954
90. Franco C, Ahmad PJ, Hou G, Wong E, Bendeck MP. Increased cell and matrix accumulation during atherogenesis in mice with vessel wall-specific deletion of discoidin domain receptor 1. Circ Res. (2010) 106:1775–83. doi: 10.1161/CIRCRESAHA.109.213637
91. Franco C, Britto K, Wong E, Hou G, Zhu SN, Chen M, et al. Discoidin domain receptor 1 on bone marrow-derived cells promotes macrophage accumulation during atherogenesis. Circ Res. (2009) 105:1141–8. doi: 10.1161/CIRCRESAHA.109.207357
92. Franco C, Hou G, Ahmad PJ, Fu EY, Koh L, Vogel WF, et al. Discoidin domain receptor 1 (ddr1) deletion decreases atherosclerosis by accelerating matrix accumulation and reducing inflammation in low-density lipoprotein receptor-deficient mice. Circ Res. (2008) 102:1202–11. doi: 10.1161/CIRCRESAHA.107.170662
93. Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell (1997) 1:13–23. doi: 10.1016/S1097-2765(00)80003-9
94. Dengjel J, Akimov V, Olsen JV, Bunkenborg J, Mann M, Blagoev B, et al. Quantitative proteomic assessment of very early cellular signaling events. Nat Biotechnol. (2007) 25:566–8. doi: 10.1038/nbt1301
95. L'Hote C G, Thomas PH, Ganesan TS. Functional analysis of discoidin domain receptor 1: effect of adhesion on DDR1 phosphorylation. FASEB J. (2002) 16:234–6. doi: 10.1096/fj.01-0414fje
96. Lemeer S, Bluwstein A, Wu Z, Leberfinger J, Muller K, Kramer K, et al. Phosphotyrosine mediated protein interactions of the discoidin domain receptor 1. J Proteomics (2012) 75:3465–77. doi: 10.1016/j.jprot.2011.10.007
97. Ge C, Yang Q, Zhao G, Yu H, Kirkwood KL, Franceschi RT. Interactions between extracellular signal-regulated kinase 1/2 and p38 MAP kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res. (2012) 27:538–51. doi: 10.1002/jbmr.561
98. Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, et al. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J Biol Chem. (2009) 284:32533–43. doi: 10.1074/jbc.M109.040980
99. Dejmek J, Dib K, Jonsson M, Andersson T. Wnt-5a and G-protein signaling are required for collagen-induced DDR1 receptor activation and normal mammary cell adhesion. Int J Cancer (2003) 103:344–51. doi: 10.1002/ijc.10752
100. Krohn JB, Hutcheson JD, Martinez-Martinez E, Irvin WS, Bouten CV, Bertazzo S, et al. Discoidin domain receptor-1 regulates calcific extracellular vesicle release in vascular smooth muscle cell fibrocalcific response via transforming growth factor-beta signaling. Arterioscler Thromb Vasc Biol. (2016) 36:525–33. doi: 10.1161/ATVBAHA.115.307009
101. Salazar A, Polur I, Servais JM, Li Y, Xu L. Delayed progression of condylar cartilage degeneration, by reduction of the discoidin domain receptor 2, in the temporomandibular joints of osteoarthritic mouse models. J Oral Pathol Med. (2014) 43:317–21. doi: 10.1111/jop.12137
102. Xu L, Servais J, Polur I, Kim D, Lee PL, Chung K, et al. Attenuation of osteoarthritis progression by reduction of discoidin domain receptor 2 in mice. Arthritis Rheum. (2010) 62:2736–44. doi: 10.1002/art.27582
103. Zhang S, Zhong Y, Li R, Wang W, Zeng L, Wang Z, et al. Experimental chondrocyte hypertrophy is promoted by the activation of discoidin domain receptor 2. Mol Med Rep. (2014) 10:1543–8. doi: 10.3892/mmr.2014.2340
104. Ge C, Mohamed F, Binrayes A, Kapila S, Franceschi RT. Selective role of discoidin domain receptor 2 in murine temporomandibular joint development and aging. J Dent Res. (2018) 97:321–8. doi: 10.1177/0022034517738190
105. Su J, Yu J, Ren T, Zhang W, Zhang Y, Liu X, et al. Discoidin domain receptor 2 is associated with the increased expression of matrix metalloproteinase-13 in synovial fibroblasts of rheumatoid arthritis. Mol Cell Biochem. (2009) 330:141–52. doi: 10.1007/s11010-009-0127-0
106. Kapp TG, Rechenmacher F, Neubauer S, Maltsev OV, Cavalcanti-Adam EA, Zarka R, et al. A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins. Sci Rep. (2017) 7:39805. doi: 10.1038/srep39805
107. Shimaoka M, Springer TA. Therapeutic antagonists and conformational regulation of integrin function. Nat Rev Drug Discov. (2003) 2:703–16. doi: 10.1038/nrd1174
108. Di Maggio N, Martella E, Frismantiene A, Resink TJ, Schreiner S, Lucarelli E, et al. Extracellular matrix and alpha5beta1 integrin signaling control the maintenance of bone formation capacity by human adipose-derived stromal cells. Sci Rep. (2017) 7:44398. doi: 10.1038/srep44398
109. Robert L, Robert AM, Fulop T. Rapid increase in human life expectancy: will it soon be limited by the aging of elastin? Biogerontology (2008) 9:119–33. doi: 10.1007/s10522-007-9122-6
110. Ishii T, Asuwa N. Collagen and elastin degradation by matrix metalloproteinases and tissue inhibitors of matrix metalloproteinase in aortic dissection. Hum Pathol. (2000) 31:640–6. doi: 10.1053/hupa.2000.7642
111. Liu SL, Bae YH, Yu C, Monslow J, Hawthorne EA, Castagnino P, et al. Matrix metalloproteinase-12 is an essential mediator of acute and chronic arterial stiffening. Sci Rep. (2015) 5:17189. doi: 10.1038/srep17189
112. Dollery CM, Owen CA, Sukhova GK, Krettek A, Shapiro SD, Libby P. Neutrophil elastase in human atherosclerotic plaques: production by macrophages. Circulation (2003) 107:2829–36. doi: 10.1161/01.CIR.0000072792.65250.4A
113. Smith ER, Tomlinson LA, Ford ML, McMahon LP, Rajkumar C, Holt SG. Elastin degradation is associated with progressive aortic stiffening and all-cause mortality in predialysis chronic kidney disease. Hypertension (2012) 59:973–8. doi: 10.1161/HYPERTENSIONAHA.111.187807
114. Wrenn DS, Hinek A, Mecham RP. Kinetics of receptor-mediated binding of tropoelastin to ligament fibroblasts. J Biol Chem. (1988) 263:2280–4.
115. Rodgers UR, Weiss AS. Integrin alpha v beta 3 binds a unique non-RGD site near the C-terminus of human tropoelastin. Biochimie (2004) 86:173–8. doi: 10.1016/j.biochi.2004.03.002
116. Lee P, Bax DV, Bilek MM, Weiss AS. A novel cell adhesion region in tropoelastin mediates attachment to integrin alphaVbeta5. J Biol Chem. (2014) 289:1467–77. doi: 10.1074/jbc.M113.518381
117. Pocza P, Suli-Vargha H, Darvas Z, Falus A. Locally generated VGVAPG and VAPG elastin-derived peptides amplify melanoma invasion via the galectin-3 receptor. Int J Cancer (2008) 122:1972–80. doi: 10.1002/ijc.23296
118. Hinek A, Pshezhetsky AV, von Itzstein M, Starcher B. Lysosomal sialidase (neuraminidase-1) is targeted to the cell surface in a multiprotein complex that facilitates elastic fiber assembly. J Biol Chem. (2006) 281:3698–710. doi: 10.1074/jbc.M508736200
119. Mecham R, Hinek A, Entwistle R, Wrenn DS, Griffin GL, Senior RM. Elastin binds to a multifunctional 67-kilodalton peripheral membrane protein. Biochemistry (1989) 28:3716–22. doi: 10.1021/bi00435a014
120. Duca L, Blanchevoye C, Cantarelli B, Ghoneim C, Dedieu S, Delacoux F, et al. The elastin receptor complex transduces signals through the catalytic activity of its Neu-1 subunit. J Biol Chem. (2007) 282:12484–91. doi: 10.1074/jbc.M609505200
121. Brassart B, Fuchs P, Huet E, Alix AJ, Wallach J, Tamburro AM, et al. Conformational dependence of collagenase (matrix metalloproteinase-1) up-regulation by elastin peptides in cultured fibroblasts. J Biol Chem. (2001) 276:5222–7. doi: 10.1074/jbc.M003642200
122. Heinz A, Jung MC, Jahreis G, Rusciani A, Duca L, Debelle L, et al. The action of neutrophil serine proteases on elastin and its precursor. Biochimie (2012) 94:192–202. doi: 10.1016/j.biochi.2011.10.006
123. Hinek A, Wrenn D, Mecham R, Barondes S. The elastin receptor: a galactoside-binding protein. Science (1988) 239:1539–41. doi: 10.1126/science.2832941
124. Vyavahare N, Jones PL, Tallapragada S, Levy RJ. Inhibition of matrix metalloproteinase activity attenuates Tenascin-C production and calcification of implanted purified elastin in rats. Am J Pathol. (2000) 157:885–93. doi: 10.1016/s0002-9440(10)64602-0
125. Wanga S, Hibender S, Ridwan Y, van Roomen C, Vos M, van der Made I, et al. Aortic microcalcification is associated with elastin fragmentation in Marfan syndrome. J Pathol. (2017) 243:294–306. doi: 10.1002/path.4949
126. Simionescu A, Philips K, Vyavahare N. Elastin-derived peptides and TGF-beta1 induce osteogenic responses in smooth muscle cells. Biochem Biophys Res Commun. (2005) 334:524–32. doi: 10.1016/j.bbrc.2005.06.119
127. Sinha A, Vyavahare NR. High-glucose levels and elastin degradation products accelerate osteogenesis in vascular smooth muscle cells. Diab Vasc Dis Res. (2013) 10:410–9. doi: 10.1177/1479164113485101
128. Pai A, Leaf EM, El-Abbadi M, Giachelli CM. Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am J Pathol. (2011) 178:764–73. doi: 10.1016/j.ajpath.2010.10.006
129. Basalyga DM, Simionescu DT, Xiong W, Baxter BT, Starcher BC, Vyavahare NR. Elastin degradation and calcification in an abdominal aorta injury model: role of matrix metalloproteinases. Circulation (2004) 110:3480–7. doi: 10.1161/01.CIR.0000148367.08413.E9
130. Qin X, Corriere MA, Matrisian LM, Guzman RJ. Matrix metalloproteinase inhibition attenuates aortic calcification. Arterioscler Thromb Vasc Biol. (2006) 26:1510–6. doi: 10.1161/01.ATV.0000225807.76419.a7
131. Bouvet C, Moreau S, Blanchette J, de Blois D, Moreau P. Sequential activation of matrix metalloproteinase 9 and transforming growth factor beta in arterial elastocalcinosis. Arterioscler Thromb Vasc Biol. (2008) 28:856–62. doi: 10.1161/ATVBAHA.107.153056
132. Hosaka N, Mizobuchi M, Ogata H, Kumata C, Kondo F, Koiwa F, et al. Elastin degradation accelerates phosphate-induced mineralization of vascular smooth muscle cells. Calcif Tissue Int. (2009) 85:523–9. doi: 10.1007/s00223-009-9297-8
133. Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, et al. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation (2009) 119:1785–94. doi: 10.1161/CIRCULATIONAHA.108.827972
134. Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. (2003) 278:2750–7. doi: 10.1074/jbc.M209256200
135. Noda K, Dabovic B, Takagi K, Inoue T, Horiguchi M, Hirai M, et al. Latent TGF-beta binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proc Natl Acad Sci USA. (2013) 110:2852–7. doi: 10.1073/pnas.1215779110
136. Khavandgar Z, Roman H, Li J, Lee S, Vali H, Brinckmann J, et al. Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J Bone Miner Res. (2014) 29:327–37. doi: 10.1002/jbmr.2039
137. Kasper M, Funk RHW. Age-related changes in cells and tissues due to advanced glycation end products (AGEs). Arch Gerontol Geriatr (2001) 32:233–43. doi: 10.1016/S0167-4943(01)00103-0
138. Khosravi R, Sodek KL, Faibish M, Trackman PC. Collagen advanced glycation inhibits its Discoidin Domain Receptor 2 (DDR2)-mediated induction of lysyl oxidase in osteoblasts. Bone (2014) 58:33–41. doi: 10.1016/j.bone.2013.10.001
139. Mason BN, Starchenko A, Williams RM, Bonassar LJ, Reinhart-King CA. Tuning three-dimensional collagen matrix stiffness independently of collagen concentration modulates endothelial cell behavior. Acta Biomater. (2013) 9:4635–44. doi: 10.1016/j.actbio.2012.08.007
140. Chong SA, Lee W, Arora PD, Laschinger C, Young EW, Simmons CA, et al. Methylglyoxal inhibits the binding step of collagen phagocytosis. J Biol Chem. (2007) 282:8510–20. doi: 10.1074/jbc.M609859200
141. Paul RG, Bailey AJ. The effect of advanced glycation end-product formation upon cell-matrix interactions. Int J Biochem Cell Biol. (1999) 31:653–60. doi: 10.1016/S1357-2725(99)00023-0
142. Gautieri A, Passini FS, Silvan U, Guizar-Sicairos M, Carimati G, Volpi P, et al. Advanced glycation end-products: mechanics of aged collagen from molecule to tissue. Matrix Biol. (2017) 59:95–108. doi: 10.1016/j.matbio.2016.09.001
143. Fessel G, Li Y, Diederich V, Guizar-Sicairos M, Schneider P, Sell DR, et al. Advanced glycation end-products reduce collagen molecular sliding to affect collagen fibril damage mechanisms but not stiffness. PLoS ONE (2014) 9:e110948. doi: 10.1371/journal.pone.0110948
144. Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YCE, et al. Cloning and expression of a cell surface receptor for advanced glycation end products of proteins. J Biol Chem. (1992) 267:14998–5004.
145. Collison KS, Parhar RS, Saleh SS, Meyer BF, Kwaasi AA, Hammami MM, et al. RAGE-mediated neutrophil dysfunction is evoked by advanced glycation end products (AGEs). J Leukocyte Biol. (2002) 71:433–44. doi: 10.1189/jlb.71.3.433
146. Gebhardt C, Riehl A, Durchdewald M, Nemeth J, Furstenberger G, Muller-Decker K, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. (2008) 205:275–85. doi: 10.1084/jem.20070679
147. Chen Y, Akirav EM, Chen W, Henegariu O, Moser B, Desai D, et al. RAGE ligation affects T cell activation and controls T cell differentiation. J Immunol. (2008) 181:4272–8. doi: 10.4049/jimmunol.181.6.4272
148. Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. (1993) 143:1699–712.
149. Zhang F, Su X, Huang G, Xin XF, Cao EH, Shi Y, et al. sRAGE alleviates neutrophilic asthma by blocking HMGB1/RAGE signalling in airway dendritic cells. Sci Rep. (2017) 7:14268. doi: 10.1038/s41598-017-14667-4
150. He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. (2011) 12:358–64. doi: 10.1038/embor.2011.28
151. Borchi E, Bargelli V, Guidotti V, Berti A, Stefani M, Nediani C, et al. Mild exposure of RIN-5F beta-cells to human islet amyloid polypeptide aggregates upregulates antioxidant enzymes via NADPH oxidase-RAGE: an hormetic stimulus. Redox Biol. (2013) 2:114–22. doi: 10.1016/j.redox.2013.12.005
152. Ren X, Shao H, Wei Q, Sun Z, Liu N. Advanced glycation end-products enhance calcification in vascular smooth muscle cells. J Int Med Res. (2009) 37:847–54. doi: 10.1177/147323000903700329
153. Wei Q, Ren X, Jiang Y, Jin H, Liu N, Li J. Advanced glycation end products accelerate rat vascular calcification through RAGE/oxidative stress. BMC Cardiovasc Disord. (2013) 13:13. doi: 10.1186/1471-2261-13-13
154. Wang B, Cai Z, Liu B, Liu Z, Zhou X, Dong N, et al. RAGE deficiency alleviates aortic valve calcification in ApoE(-/-) mice via the inhibition of endoplasmic reticulum stress. Biochim Biophys Acta (2017) 1863:781–92. doi: 10.1016/j.bbadis.2016.12.012
155. Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem. (1997) 272:16498–506. doi: 10.1074/jbc.272.26.16498
156. Chuong C, Katz J, Pauley KM, Bulosan M, Cha S. RAGE expression and NF-kappaB activation attenuated by extracellular domain of RAGE in human salivary gland cell line. J Cell Physiol. (2009) 221:430–4. doi: 10.1002/jcp.21873
157. Wang Z, Li L, Du R, Yan J, Liu N, Yuan W, et al. CML/RAGE signal induces calcification cascade in diabetes. Diabetol Metab Syndr. (2016) 8:83. doi: 10.1186/s13098-016-0196-7
158. Basta G, Corciu AI, Vianello A, Del Turco S, Foffa I, Navarra T, et al. Circulating soluble receptor for advanced glycation end-product levels are decreased in patients with calcific aortic valve stenosis. Atherosclerosis (2010) 210:614–8. doi: 10.1016/j.atherosclerosis.2009.12.029
159. Kim HS, Chung W, Kim AJ, Ro H, Chang JH, Lee HH, et al. Circulating levels of soluble receptor for advanced glycation end product are inversely associated with vascular calcification in patients on haemodialysis independent of S100A12 (EN-RAGE) levels. Nephrology (2013) 18:777–82. doi: 10.1111/nep.12166
160. Brodeur MR, Bouvet C, Bouchard S, Moreau S, Leblond J, Deblois D, et al. Reduction of advanced-glycation end products levels and inhibition of RAGE signaling decreases rat vascular calcification induced by diabetes. PLoS ONE (2014) 9:e85922. doi: 10.1371/journal.pone.0085922
161. Suga T, Iso T, Shimizu T, Tanaka T, Yamagishi S, Takeuchi M, et al. Activation of receptor for advanced glycation end products induces osteogenic differentiation of vascular smooth muscle cells. J Atheroscler Thromb. (2011) 18:670–83. doi: 10.5551/jat.7120
162. Green PM, Ludbrook SB, Miller DD, Horgan CMT, Barry ST. Structural elements of the osteopontin SVVYGLR motif important for the interaction with a4 integrins. FEBS Lett. (2001) 503:75–9. doi: 10.1016/S0014-5793(01)02690-4
163. Yokosaki Y, Matsuura N, Sasaki T, Murakami I, Schneider H, Higashiyama S, et al. The integrin a9b1 binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J Biol Chem. (1999) 274:36328–34. doi: 10.1074/jbc.274.51.36328
164. Ito K, Kon S, Nakayama Y, Kurotaki D, Saito Y, Kanayama M, et al. The differential amino acid requirement within osteopontin in alpha4 and alpha9 integrin-mediated cell binding and migration. Matrix Biol. (2009) 28:11–9. doi: 10.1016/j.matbio.2008.10.002
165. Lund SA, Wilson CL, Raines EW, Tang J, Giachelli CM, Scatena M. Osteopontin mediates macrophage chemotaxis via alpha4 and alpha9 integrins and survival via the alpha4 integrin. J Cell Biochem. (2013) 114:1194–202. doi: 10.1002/jcb.24462
166. Klaning E, Christensen B, Sorensen ES, Vorup-Jensen T, Jensen JK. Osteopontin binds multiple calcium ions with high affinity and independently of phosphorylation status. Bone (2014) 66:90–5. doi: 10.1016/j.bone.2014.05.020
167. Yuan Q, Jiang Y, Zhao X, Sato T, Densmore M, Schuler C, et al. Increased osteopontin contributes to inhibition of bone mineralization in FGF23-deficient mice. J Bone Miner Res. (2014) 29:693–704. doi: 10.1002/jbmr.2079
168. Steitz SA, Speer MY, McKee MD, Liaw L, Almeida M, Yang H, et al. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol. (2002) 161:2035–46. doi: 10.1016/S0002-9440(10)64482-3
169. Chellaiah MA, Kizer N, Biswas R, Alvarez U, Strauss-Schoenberger J, Rifas L, et al. Osteopontin deficiency produces osteoclast dysfunction due to reduced CD44 surface expression. Mol Biol Cell. (2003) 14:173–89. doi: 10.1091/mbc.e02-06-0354
170. Asou Y, Rittling SR, Yoshitake H, Tsuji K, Shinomiya K, Nifuji A, et al. Osteopontin facilitates angiogenesis, accumulation of osteoclasts, and resorption in ectopic bone. Endocrinology (2001) 142:1325–32. doi: 10.1210/endo.142.3.8006
171. Tanabe N, Wheal BD, Kwon J, Chen HH, Shugg RP, Sims SM, et al. Osteopontin signals through calcium and nuclear factor of activated T cells (NFAT) in osteoclasts: a novel RGD-dependent pathway promoting cell survival. J Biol Chem. (2011) 286:39871–81. doi: 10.1074/jbc.M111.295048
172. Ahmed S, Oneill KD, Hood AF, Evan AP, Moe SM. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am J Kidney Dis. (2001) 37:1267–76. doi: 10.1053/ajkd.2001.24533
173. Berezin AE, Kremzer AA. Circulating osteopontin as a marker of early coronary vascular calcification in type two diabetes mellitus patients with known asymptomatic coronary artery disease. Atherosclerosis (2013) 229:475–81. doi: 10.1016/j.atherosclerosis.2013.06.003
174. Speer MY, McKee MD, Guldberg RE, Liaw L, Yang H-Y, Tung E, et al. Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein–deficient mice. J Exp Med. (2002) 196:1047–55. doi: 10.1084/jem.20020911
175. Paloian NJ, Leaf EM, Giachelli CM. Osteopontin protects against high phosphate-induced nephrocalcinosis and vascular calcification. Kidney Int. (2016) 89:1027–36. doi: 10.1016/j.kint.2015.12.046
176. Speer MY, Chien YC, Quan M, Yang HY, Vali H, McKee MD, et al. Smooth muscle cells deficient in osteopontin have enhanced susceptibility to calcification in vitro. Cardiovasc Res. (2005) 66:324–33. doi: 10.1016/j.cardiores.2005.01.023
177. Weintraub AS, Schnapp LN, Lin XJ, Taubman MB. Osteopontin deficiency in rat vascular smooth muscle cells is associated with an inability to adhere to collagen and increased apoptosis. Lab Invest. (2000) 80:1603–15. doi: 10.1038/labinvest.3780171
178. Ge Q, Ruan CC, Ma Y, Tang XF, Wu QH, Wang JG, et al. Osteopontin regulates macrophage activation and osteoclast formation in hypertensive patients with vascular calcification. Sci Rep. (2017) 7:40253. doi: 10.1038/srep40253
179. Yue TL, McKenna PJ, Ohlstein EH, Farach-Carson MC, Butler WT, Johanson K, et al. Osteopontin-stimulated vascular smooth muscle cell migration is mediated by b3 integrin. Exp Cell Res. (1994) 214:459–64. doi: 10.1006/excr.1994.1282
180. Gao H, Steffen MC, Ramos KS. Osteopontin regulates alpha-smooth muscle actin and calponin in vascular smooth muscle cells. Cell Biol Int. (2012) 36:155–61. doi: 10.1042/CBI20100240
181. Parrish AR, Ramos KS. Differential processing of osteopontin characterizes the proliferative vascular smooth muscle cell phenotype induced by allylamine. J Cell Biochem. (1997) 65:267–75.
182. Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. (2007) 117:2877–88. doi: 10.1172/JCI31986
183. Liaw L, Birk DE, Ballas CB, Whitsitt JS, Davidson JM, Hogan BLM. Altered wound healing in mice lacking a functional osteopontin gene (ssp1). J Clin Invest. (1998) 101:1468–78. doi: 10.1172/JCI1122
184. Giachelli CM, Lombardi D, Johnson RJ, Murry CE, Almeida M. Evidence for a role of osteopontin in macrophage infiltration in response to pathological stimuli in vivo. Am J Pathol. (1998) 152:353–8.
185. Crawford HC, Matrisian LM, Liaw L. Distinct roles of osteopontin in host defense activity and tumor survival during squamous cell carcinoma progression in vivo. Cancer Res. (1998) 58:5206–15.
186. Weber GF, Zawaideh S, Hikita S, Kumar VA, Cantor H, Ashkar S. Phosphorylation-dependent interaction of osteopontin with its receptors regulates macrophage migration and activation. J Leukoc Biol (2002) 72:752–61. doi: 10.1189/jlb.72.4.752
187. Guo X, Zhang YP, Mitchell DA, Denhardt DT, Chambers AF. Identification of a ras-activated enhancer in the mouse osteopontin promoter and its interaction with a putative ETS-related transcription factor whose activity correlates with the metastatic potential of the cell. Mol Cell Biol. (1995) 15:476–87. doi: 10.1128/MCB.15.1.476
188. Zhao W, Wang L, Zhang M, Wang P, Zhang L, Yuan C, et al. NF-kappaB- and AP-1-mediated DNA looping regulates osteopontin transcription in endotoxin-stimulated murine macrophages. J Immunol. (2011) 186:3173–9. doi: 10.4049/jimmunol.1003626
189. Shimodaira T, Matsuda K, Uchibori T, Sugano M, Uehara T, Honda T. Upregulation of osteopontin expression via the interaction of macrophages and fibroblasts under IL-1b stimulation. Cytokine (2018) 110:63–9. doi: 10.1016/j.cyto.2018.04.025
190. Bruemmer D, Collins AR, Noh G, Wang W, Territo M, Arias-Magallona S, et al. Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. (2003) 112:1318–31. doi: 10.1172/JCI18141
191. Fraser JD PP. Lung, heart, and kidney express high levels of mRNA for the vitamin K-dependent matrix Gla protein. J Biol Chem. (1988) 263:11033–6.
192. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature (1997) 386:78–81. doi: 10.1038/386078a0
193. O'Young J, Liao Y, Xiao Y, Jalkanen J, Lajoie G, Karttunen M, et al. Matrix Gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J Am Chem Soc. (2011) 133:18406–12. doi: 10.1021/ja207628k
194. Malhotra R, Burke MF, Martyn T, Shakartzi HR, Thayer TE, O'Rourke C, et al. Inhibition of bone morphogenetic protein signal transduction prevents the medial vascular calcification associated with matrix Gla protein deficiency. PLoS ONE (2015) 10:e0117098. doi: 10.1371/journal.pone.0117098
195. Xue W, Wallin R, Olmsted-Davis EA, Borras T. Matrix GLA protein function in human trabecular meshwork cells: inhibition of BMP2-induced calcification process. Invest Ophthalmol Vis Sci. (2006) 47:997–1007. doi: 10.1167/iovs.05-1106
196. Engelse MA, Neele JM, Bronckers AL, Pannekoek H, de Vries CJ. Vascular calcification: expression patterns of the osteoblast-specific gene core binding factor alpha-1 and the protective factor matrix gla protein in human atherogenesis. Cardiovasc Res. (2001) 52:281–9.
197. Mori K, Shioi A, Jono S, Nishizawa Y, Morii H. Expression of matrix Gla protein (MGP) in an in vitro model of vascular calcification. FEBS Lett. (1998) 433:19–22. doi: 10.1016/S0014-5793(98)00870-9
198. Proudfoot D, Skepper JN, Shanahan CM, Weissberg PL. Calcification of human vascular cells in vitro is correlated with high levels of matrix Gla protein and low levels of osteopontin expression. Arterioscler Thromb Vasc Biol. (1998) 18:379–88.
199. Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J Cell Biol. (2004) 165:625–30. doi: 10.1083/jcb.200402046
200. Price PA, Faus SA, Williamson MK. Warfarin-induced artery calcification is accelerated by growth and vitamin D. Arterioscler Thromb Vasc Biol. (2000) 20:317–27. doi: 10.1161/01.ATV.20.2.317
201. Schurgers LJ, Spronk HM, Skepper JN, Hackeng TM, Shanahan CM, Vermeer C, et al. Post-translational modifications regulate matrix Gla protein function: importance for inhibition of vascular smooth muscle cell calcification. J Thromb Haemost. (2007) 5:2503–11. doi: 10.1111/j.1538-7836.2007.02758.x
202. Schurgers LJ, Spronk HM, Soute BA, Schiffers PM, DeMey JG, Vermeer C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood (2007) 109:2823–31. doi: 10.1182/blood-2006-07-035345
203. Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin–cytoskeleton linkages. Cell (1997) 88:39–48. doi: 10.1016/s0092-8674(00)81856-5
204. Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell-surface and through the cytoskeleton. Science (1993) 260:1124–7. doi: 10.1126/science.7684161
205. Lovett DB, Shekhar N, Nickerson JA, Roux KJ, Lele TP. Modulation of nuclear shape by substrate rigidity. Cell Mol Bioeng. (2013) 6:230–8. doi: 10.1007/s12195-013-0270-2
206. Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science (2013) 341:1240104. doi: 10.1126/science.1240104
207. Yeh YC, Ling JY, Chen WC, Lin HH, Tang MJ. Mechanotransduction of matrix stiffness in regulation of focal adhesion size and number: reciprocal regulation of caveolin-1 and beta1 integrin. Sci Rep. (2017) 7:15008. doi: 10.1038/s41598-017-14932-6
208. Jiang L, Sun Z, Chen X, Li J, Xu Y, Zu Y, et al. Cells sensing mechanical cues: stiffness influences the lifetime of cell-extracellular matrix interactions by affecting the loading rate. ACS Nano (2016) 10:207–17. doi: 10.1021/acsnano.5b03157
209. Jiang G, Huang AH, Cai Y, Tanase M, Sheetz MP. Rigidity sensing at the leading edge through alphavbeta3 integrins and RPTPalpha. Biophys J. (2006) 90:1804–9. doi: 10.1529/biophysj.105.072462
210. Coelho NM, Arora PD, van Putten S, Boo S, Petrovic P, Lin AX, et al. Discoidin domain receptor 1 mediates myosin-dependent collagen contraction. Cell Rep. (2017) 18:1774–90. doi: 10.1016/j.celrep.2017.01.061
211. Kim D, You E, Jeong J, Ko P, Kim JW, Rhee S. DDR2 controls the epithelial-mesenchymal-transition-related gene expression via c-Myb acetylation upon matrix stiffening. Sci Rep. (2017) 7:6847. doi: 10.1038/s41598-017-07126-7
212. Prager-Khoutorsky M, Lichtenstein A, Krishnan R, Rajendran K, Mayo A, Kam Z, et al. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat Cell Biol. (2011) 13:1457–65. doi: 10.1038/ncb2370
213. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature (2011) 474:179–83. doi: 10.1038/nature10137
214. Zhao XH, Laschinger C, Arora P, Szaszi K, Kapus A, McCulloch CA. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci. (2007) 120:1801–9. doi: 10.1242/jcs.001586
215. Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell (2003) 113:329–42. doi: 10.1016/S0092-8674(03)00278-2
216. Reddy P, Deguchi M, Cheng Y, Hsueh AJ. Actin cytoskeleton regulates Hippo signaling. PLoS ONE (2013) 8:e73763. doi: 10.1371/journal.pone.0073763
217. Das A, Fischer RS, Pan D, Waterman CM. YAP Nuclear localization in the absence of cell-cell contact is mediated by a filamentous actin-dependent, myosin II- and phospho-YAP-independent pathway during extracellular matrix mechanosensing. J Biol Chem. (2016) 291:6096–110. doi: 10.1074/jbc.M115.708313
218. Mana-Capelli S, Paramasivam M, Dutta S, McCollum D. Angiomotins link F-actin architecture to Hippo pathway signaling. Mol Biol Cell. (2014) 25:1676–85. doi: 10.1091/mbc.E13-11-0701
219. Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell (2017) 171:1397–410 e14. doi: 10.1016/j.cell.2017.10.008
220. Matsumoto Y, La Rose J, Kent OA, Wagner MJ, Narimatsu M, Levy AD, et al. Reciprocal stabilization of ABL and TAZ regulates osteoblastogenesis through transcription factor RUNX2. J Clin Invest. (2016) 126:4482–96. doi: 10.1172/JCI87802
221. Xie C, Guo Y, Zhu T, Zhang J, Ma PX, Chen YE. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem. (2012) 287:14598–605. doi: 10.1074/jbc.M111.329268
222. Feng X, Liu P, Zhou X, Li MT, Li FL, Wang Z, et al. Thromboxane A2 activates YAP/TAZ protein to induce vascular smooth muscle cell proliferation and migration. J Biol Chem. (2016) 291:18947–58. doi: 10.1074/jbc.M116.739722
223. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA. (2016) 113:11525–30. doi: 10.1073/pnas.1613121113
224. Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature (2016) 540:579–82. doi: 10.1038/nature20602
225. Wang Z, Wang DZ, Teg Pipes CG, Olson EN. Myocardin is a master regulator of smooth muscle cell gene expression. PNAS (2003) 100:7129–34. doi: 10.1073/pnas.1232341100
226. Bian H, Lin JZ, Li C, Farmer SR. Myocardin-related transcription factor A (MRTFA) regulates the fate of bone marrow mesenchymal stem cells and its absence in mice leads to osteopenia. Mol Metab. (2016) 5:970–9. doi: 10.1016/j.molmet.2016.08.012
227. Latif N, Sarathchandra P, Chester AH, Yacoub MH. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur Heart J. (2015) 36:1335–45. doi: 10.1093/eurheartj/eht547
228. Lagna G, Ku MM, Nguyen PH, Neuman NA, Davis BN, Hata A. Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors. J Biol Chem. (2007) 282:37244–55. doi: 10.1074/jbc.M708137200
229. Tanaka T, Sato H, Doi H, Yoshida CA, Shimizu T, Matsui H, et al. Runx2 represses myocardin-mediated differentiation and facilitates osteogenic conversion of vascular smooth muscle cells. Mol Cell Biol. (2008) 28:1147–60. doi: 10.1128/MCB.01771-07
230. Speight P, Kofler M, Szaszi K, Kapus A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFbeta-regulated Smad3. Nat Commun. (2016) 7:11642. doi: 10.1038/ncomms11642
231. Kim T, Hwang D, Lee D, Kim JH, Kim SY, Lim DS. MRTF potentiates TEAD-YAP transcriptional activity causing metastasis. EMBO J. (2017) 36:520–35. doi: 10.15252/embj.201695137
232. Yu O, Miyamoto S, Brown JH. MRTF-A and YAP exert dual control in GPCR and RhoA-mediated transcriptional regulation and cell proliferation. Mol Cell Biol. (2015) 30:15. doi: 10.1128/mcb.00772-15
233. Chang H, Knothe Tate ML. Structure-function relationships in the stem cell's mechanical world B: emergent anisotropy of the cytoskeleton correlates to volume and shape changing stress exposure. Mol Cell Biomech. (2011) 8:297–318. doi: 10.3970/mcb.2011.008.297
234. Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature (2010) 463:485–92. doi: 10.1038/nature08908
235. Arnsdorf EJ, Tummala P, Kwon RY, Jacobs CR. Mechanically induced osteogenic differentiation–the role of RhoA, ROCKII and cytoskeletal dynamics. J Cell Sci. (2009) 122:546–53. doi: 10.1242/jcs.036293
236. Geissler S, Textor M, Kuhnisch J, Konnig D, Klein O, Ode A, et al. Functional comparison of chronological and in vitro aging: differential role of the cytoskeleton and mitochondria in mesenchymal stromal cells. PLoS ONE (2012) 7:e52700. doi: 10.1371/journal.pone.0052700
237. Lee K, Kim H, Jeong D. Microtubule stabilization attenuates vascular calcification through the inhibition of osteogenic signaling and matrix vesicle release. Biochem Biophys Res Commun. (2014) 451:436–41. doi: 10.1016/j.bbrc.2014.08.007
238. Lee K, Kim H, Jeong D. Protein kinase C regulates vascular calcification via cytoskeleton reorganization and osteogenic signaling. Biochem Biophys Res Commun. (2014) 453:793–7. doi: 10.1016/j.bbrc.2014.10.026
239. Pockwinse SM, Rajgopal A, Young DW, Mujeeb KA, Nickerson J, Javed A, et al. Microtubule-dependent nuclear-cytoplasmic shuttling of Runx2. J Cell Physiol. (2006) 206:354–62. doi: 10.1002/jcp.20469
240. Zou W, Greenblatt MB, Brady N, Lotinun S, Zhai B, de Rivera H, et al. The microtubule-associated protein DCAMKL1 regulates osteoblast function via repression of Runx2. J Exp Med. (2013) 210:1793–806. doi: 10.1084/jem.20111790
241. Andres V, Gonzalez JM. Role of A-type lamins in signaling, transcription, and chromatin organization. J Cell Biol. (2009) 187:945–57. doi: 10.1083/jcb.200904124
242. Quiros-Gonzalez I, Roman-Garcia P, Alonso-Montes C, Barrio-Vazquez S, Carrillo-Lopez N, Naves-Diaz M, et al. Lamin A is involved in the development of vascular calcification induced by chronic kidney failure and phosphorus load. Bone (2016) 84:160–8. doi: 10.1016/j.bone.2016.01.005
243. Akter R, Rivas D, Geneau G, Drissi H, Duque G. Effect of lamin A/C knockdown on osteoblast differentiation and function. J Bone Miner Res. (2009) 24:283–93. doi: 10.1359/jbmr.081010
Keywords: vascular calcification, extracellular matrix, mechanotransduction, collagen, integrin, discoidin domain receptor, cytoskeleton, osteogenesis
Citation: Ngai D, Lino M and Bendeck MP (2018) Cell-Matrix Interactions and Matricrine Signaling in the Pathogenesis of Vascular Calcification. Front. Cardiovasc. Med. 5:174. doi: 10.3389/fcvm.2018.00174
Received: 10 October 2018; Accepted: 21 November 2018;
Published: 07 December 2018.
Edited by:
Yabing Chen, University of Alabama at Birmingham, United StatesReviewed by:
Sasha Anna Singh, Harvard Medical School, United StatesAdriana Georgescu, Institute of Cellular Biology and Pathology (ICBP), Romania
Copyright © 2018 Ngai, Lino and Bendeck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michelle P. Bendeck, bWljaGVsbGUuYmVuZGVja0B1dG9yb250by5jYQ==