 Lesca M. Holdt
Lesca M. Holdt Daniel Teupser*
Daniel Teupser*- Institute of Laboratory Medicine, University Hospital, LMU Munich, Munich, Germany
Ever since the first genome-wide association studies (GWAS) on coronary artery disease (CAD), the Chr9p21 risk locus has emerged as a top signal in GWAS of atherosclerotic cardiovascular disease, including stroke and peripheral artery disease. The CAD risk SNPs on Chr9p21 lie within a stretch of 58 kilobases of non-protein-coding DNA, containing the gene body of the long noncoding RNA (lncRNA) antisense non coding RNA in the INK4 locus (ANRIL). How risk is affected by the Chr9p21 locus in molecular detail is a matter of ongoing research. Here we will review recent advances in the understanding that ANRIL serves as a key risk effector molecule of atherogenesis at the locus. One focus of this review is the shift in understanding that genetic variation at Chr9p21 not only affects the abundance of ANRIL, and in some cases expression of the adjacent CDKN2A/B tumor suppressors, but also impacts ANRIL splicing, such that 3′-5′-linked circular noncoding ANRIL RNA species are produced. We describe how the balance of linear and circular ANRIL RNA, determined by the Chr9p21 genotype, regulates molecular pathways and cellular functions involved in atherogenesis. We end with an outlook on how manipulating circular ANRIL abundance may be exploited for therapeutic purposes.
Introduction
Since publication of the first genome-wide association studies (GWAS) of coronary artery disease (CAD) in 2007, Chr9p21 has emerged as the most significant risk locus associated with this frequent disease (1–4). The region contains a number of strongly interlinked SNPs within a stretch of 58 kilobases (kb) of non-protein-coding DNA. Later, the same haplotype block has been associated with other endpoints of atherosclerosis, such as stroke (5–11), peripheral artery disease (12–14), and also with different types of aneurysms (2, 8, 15, 16). Due to the availability of large study cohorts and the better resolution of genetic recombination in this region, it has now become clear that associations with other phenotypes at Chr9p21 fall in distinct haplotype blocks not overlapping with the CAD block (Figure 1A). Closely nearby, and proximal to the CAD locus, GWAS found associations with cancer, such as melanoma, glioma, basal cell carcinoma, and acute lymphoblastic leukemia [see (40) for review], and also with glaucoma, and diverse proliferative or inflammatory diseases, such as endometriosis of the reproductive tract (41), periodontitis (42), and platelet reactivity (43). The region located distally to the CAD region contains a distinct haplotype block associated with type 2 diabetes (44, 45).
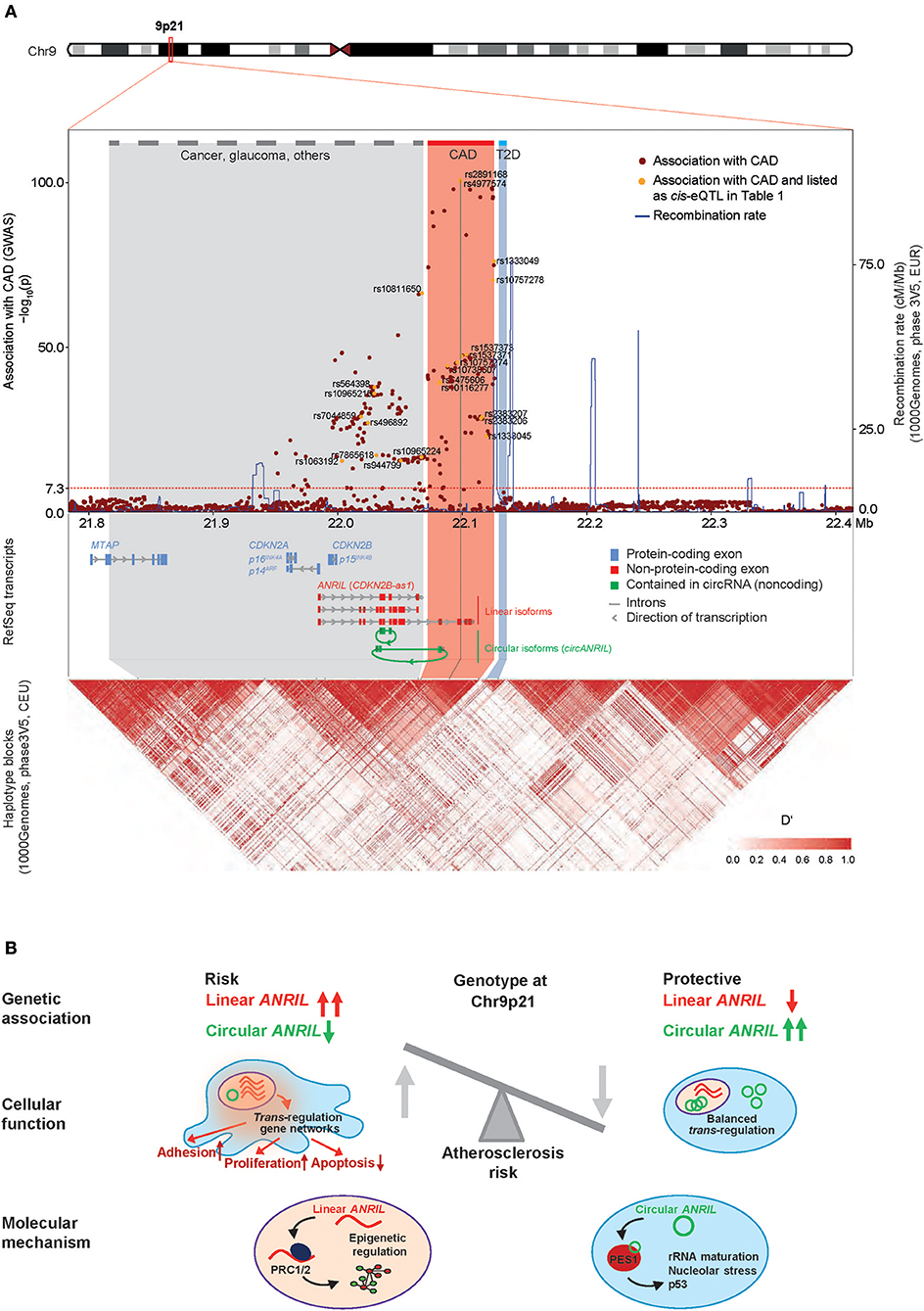
Figure 1. ANRIL and the CAD risk locus at Chr9p21. (A) Genomic mapping of SNPs identified in a region ± 300 kb around the top-ranking sentinel SNP rs4977574 based on data of the most recent large CAD GWAS (17). Chromosome ideogram and zoom-in onto RefSeq transcripts for ANRIL, CDKN2A, CDKN2B, and MTAP (top), regional association plot of CAD risk alleles and graph of recombination rate in the locus (middle), scaled linkage disequilibrium heatmap (D′) as derived from the 1000Genomes Project dataset (Phase3V5, CEU) (bottom). The threshold for significance of GWAS hits is indicated as horizontal dotted line (p < 5E-8). Dots for SNPs described in Table 1 are marked in yellow. The suspected core CAD risk region, corresponding to the distal region of ANRIL, has been defined experimentally through multiple CAD GWAS and is highlighted in red. The physical genomic map and the haplotype map are connected by oblique lines. Note that not all RNA transcripts and isoforms are depicted, and that type 2 diabetes (T2D, highlighted blue) and cancer risk regions (highlighted gray) are shown in simplified forms. (B) Model how the genotype at Chr9p21 controls the balance of linear and circular ANRIL RNA expression and potential molecular mechanisms of the different ANRIL isoforms. Linear ANRIL upregulation regulates gene expression in trans and pro-adhesive, pro-proliferative, anti-apoptotic cell functions. High levels of circANRIL inhibit over-proliferation of vascular cells by controlling rRNA maturation through impairing PES1 function in the PeBoW complex.
In the last 10 years, GWAS have been successfully used to increase the number of genetic loci implicated in CAD risk inheritance. The number of CAD risk loci in the genome rose from 56 by 2013 (24, 46–52) to 80 by 2015 (53–56), to 243 by 2017 (17). Concerning the Chr9p21 locus in these studies, the association rose steadily from p = 5.40 × 10−23 (rs4977575) (57), over p = 4.68−101 (rs4977574) (17) to p = 8.8 × 10−223 (rs4977574) (58). In populations of European descent, the allele frequency is very high (0.48), leading to the situation that approximately one-fourth of people are homozygous for the CAD risk alleles. CAD risk SNPs on Chr9p21 have recurrently been shown to have one of the top-ranking effect sizes [allele-specific odds ratio (OR) for CAD > 1.3] (3, 24). Despite the extent of effects, the Chr9p21 risk is independent of classically known CAD risk determinants, such as dyslipidemia, diabetes mellitus, age, and sex.
The Chr9p21 region contains at least 5 genes, which are, in part, tightly clustered and overlapping. These include the 3.8 kb long ANRIL non-coding RNA, and the tumor suppressors cyclin dependent kinase inhibitor CDKN2A/p16INK4A, CDKN2A/p14ARF, CDKN2B/p15INK4B, and methylthioadenosine phosphorylase (MTAP). ANRIL overlaps in antisense the full length of the p15 gene body, while sharing a bidirectional promoter with CDKN2A. Hence, it was also termed CDKN2B antisense RNA (CDKN2B-AS1). Only recently, the picture got even more complex: Advances in high-throughput sequencing and adaptions in bioinformatics mapping of RNA reads to reference genomes have revealed that thousands of genes in our genome produce not only mature linear RNA but also 3′-5′ covalently linked circular RNAs (circRNAs) (59). So far, two studies have shown that a number of circular ANRIL (circANRIL) isoforms exist, comprised of different exons, whereby a downstream exon is fused to an upstream exon by the enzymatic activity of the spliceosome in a reaction termed “backsplicing” [see (60, 61) for review]. Circularizing exons in ANRIL stemmed mostly from middle parts of the lncRNA (Figure 1A), which are in part also shared by the linear ANRIL isoforms. CircANRIL was found not only in many different cell lines, but also in many primary cell types, including vascular smooth muscle cells (VSMCs) and macrophages, as well as in heart and vascular tissue (22, 36).
A major focus in exploring how risk is effected by Chr9p21 has been on whether genetic variation affected expression of genes at the locus in cis (Figure 1A) or whether it elicited gene expression changes in trans. Top CAD-associated SNPs lie within the distal parts of long linear ANRIL isoforms (Figure 1A) and several studies have shown that they co-localize with sequences marked by chromatin modifications, RNA polymerase II transcription patterns and DNA motifs characteristic of bona-fide transcriptional enhancers (19, 35, 62–65). Using expression quantitative trait locus (eQTL) analyses in patient samples, several groups have by now investigated if the risk alleles at the locus were associated with the expression of specific target genes in cis (cis-eQTLs). Whereas studies investigating ANRIL expression have mostly used quantitative PCRs (qPCRs) targeting different exons from the lncRNA, expression of p14, p15, p16, or MTAP has either been investigated using genome-wide expression arrays or isoform-specific qPCRs. Here, we focus on studies investigating eQTLs in atherosclerosis cohorts but do not cover studies related to other phenotypes, such as cancer, which are reviewed elsewhere (66).
Cis-eQTLs at Chr9p21
ANRIL expression at Chr9p21 is complex and at least 20 linear isoforms as well as multiple circular isoforms have been reported [www.ensembl.org, (22, 36, 39)]. In principal, linear and circular isoforms can be distinguished by the fact that the latter derive from a backsplice event, where splicing of a downstream exon (e.g., exon 7) to an upstream exon (e.g., to exon 5) can be detected. Backsplicing of ex7-5 was the most common event observed in our own study in peripheral blood monocytes (36). Concordantly, Burd and colleagues have reported dominant backsplice isoforms spanning ex14-4 in peripheral blood T lymphocytes (22). In both studies, exon 1 and exons 17-20 were not contained in circularized ANRIL (Table 1). Thus, for classification reasons, results from studies targeting these exons will be referred to as proximal linear isoforms (containing the first ANRIL exons) and long linear isoforms (containing the distal exons 17-20) (Table 1). Since both linear and circular ANRIL may contain exons from the middle portion of the lncRNA (e.g., exons 4-16), a clear distinction as to whether linear or circular isoforms were investigated cannot be made in cases where these exons were targeted by qPCRs which were non-specific for backsplice junctions (Table 1).
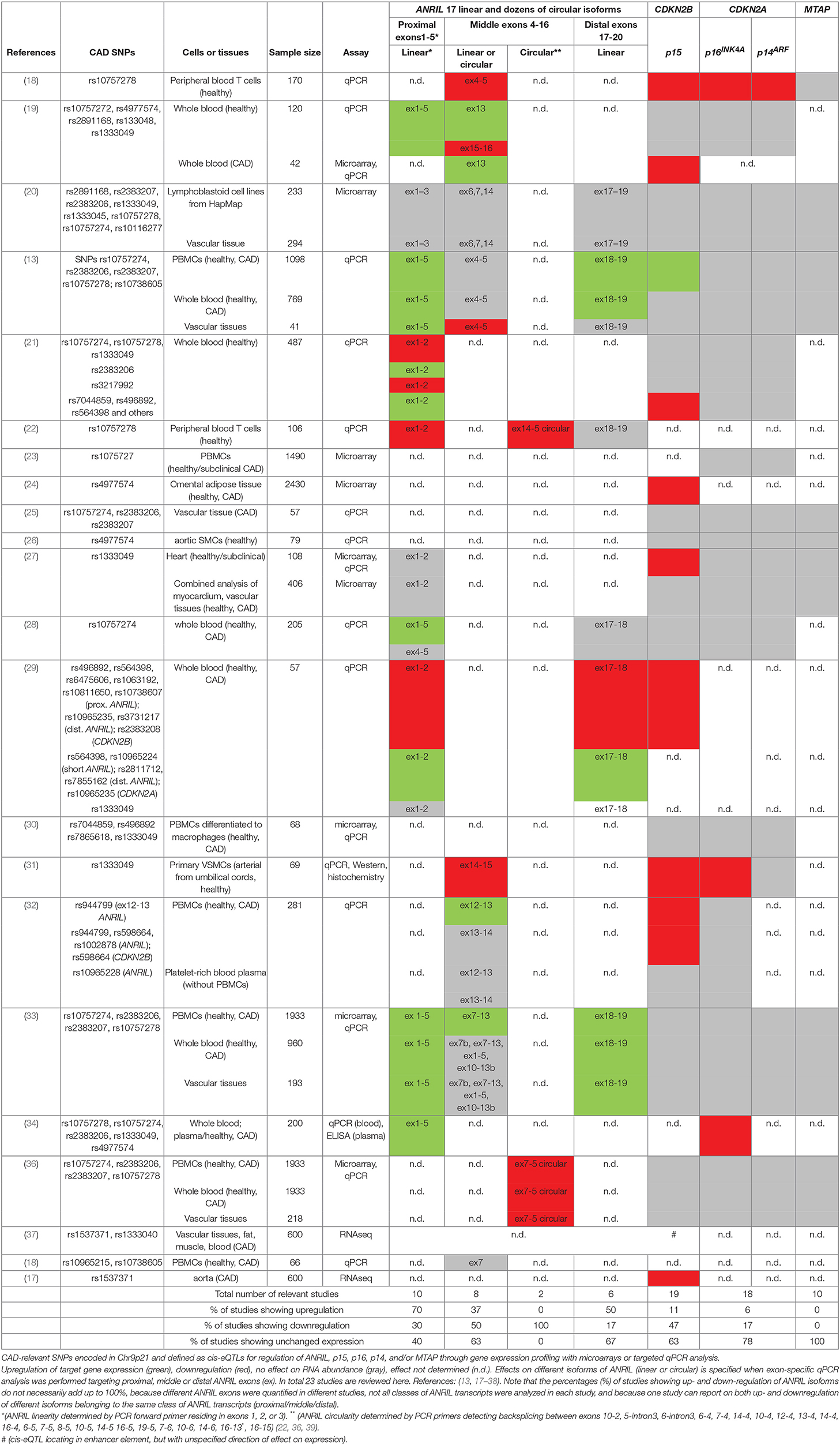
Table 1. Chr9p21 cis-eQTLs in patients.
As one of the first studies on Chr9p21, Jarinova et al. have shown that ANRIL expression was induced by the CAD risk SNP rs1333049 in peripheral blood monocytes (PBMCs). No significant effects on CDKN2A or on CDKN2B were recorded in that study (19). Over the years, comparable quantifications of these genes followed in whole blood, peripheral blood T lymphocytes, lymphoblastoid cells lines, aortic smooth muscle cells (SMCs) and in different tissue samples that are known to have a role in atherosclerosis. For example, vascular tissues such as carotid atherosclerotic plaque samples, samples from aorta, mammary artery, and from the heart ventricles have been analyzed, but also tissues like subcutaneous or omental fat have been used (Table 1). Of the 23 cis-eQTL studies conducted in the Chr9p21 CAD region to date, 16 investigated different isoforms of ANRIL, out of which 10 used assays targeting proximal ANRIL exons, 8 used assays targeting the middle region, 6 used assays targeting downstream linear ANRIL exons, and two investigated backsplices contained in circANRIL (Table 1). Complicating a clear-cut interpretation, in the different studies, different risk genotypes were used to indicate risk haplotypes. The expression of CDKN2A and of CDKN2B was investigated in 18 studies and MTAP in 10 studies (Table 1).
Overall, 80% of the studies investigating ANRIL expression found an association with the Chr9p21 genotype. Here, a trend toward higher expression of the proximal and distal exons contained in linear ANRIL in patients carrying the CAD-risk allele was observed (7 of 10 and 3 of 6 studies). In contrast, circular ANRIL was downregulated in the two published studies in patients carrying the Chr9p21 risk haplotype. No clear tendency was observed when assays targeting the middle region of ANRIL were used (Table 1). This is likely explained by the fact that these assays target both, linear and circular, ANRIL isoforms, which seem to be inversely regulated. With respect to the tumor suppressor genes contained at the Chr9p21 locus, 78 and 67% of the studies failed to find an association of CDKN2A and CDKN2B with Chr9p21, respectively. When reporting an association, specifically CDKN2B was down-regulated in the majority of studies (94%), yet its expression was not always anticorrelating with ANRIL expression (19, 21, 22, 29). MTAP expression was not associated with the Chr9p21 genotype in any of the published studies. Overall, the picture emerges that circular ANRIL and CDKN2B tend to be down-regulated in patients carrying the risk allele, whereas linear ANRIL isoforms tend to be inversely regulated (Figure 1B). It is currently unclear, why expression of p15 or of p14 and p16 were in many cases positively correlated with ANRIL (19, 21, 22, 27, 29, 32, 34, 65). Also, MTAP, which was not associated with Chr9p21 (Table 1), was in some conditions anticorrelating to ANRIL, but not in all cases or contexts (20, 34, 67). SNPs in ANRIL can hypothetically affect enhancers in both directions, either by disrupting transcription factor binding sites in open chromatin (68) or by increasing enhancer activity through yet unknown primary effects (24, 65).
In summary, many studies document cis-eQTLs for ANRIL or, separately, for CDKN2B (35). Throughout, from the existing data, it can be concluded that these effects are cell-type specific and combinatorial. Of note, many studies have investigated only very small cohorts and those, simultaneously testing both ANRIL and CDKN2B in larger cohorts (>1000 samples) identified much stronger effects of Chr9p21 on ANRIL than on CDKN2B (13, 33, 36). This observation might be explained by the haplotype block structure of the region, where effects of CAD lead SNPs are located within ANRIL but bleed through due to linkage disequilibrium, resulting in more subtle concomitant effects on CDKN2B expression. Another possibility is that the Chr9p21 genotype impacts transcription enhancers at the locus which contact and activate gene promoters affecting CAD. The consequences of such contacts would not be expected to be captured through traditional non-allelic RNA expression analysis. In fact, when allelic expression control through 3D-enhancer looping was specifically measured in a separate study in human coronary aortic SMCs (64), physical contacts of CAD variant-containing enhancers in the locus and the promoters of CDKN2A, CDKN2B, and ANRIL were corroborated.
Taken together, these data suggest that genetic variation within the core 9p21 CAD region relates to differential expression not only of ANRIL, but in specific cells or conditions, also of the CDKN2A/B tumor suppressors encoded in the locus. While either of these factors could potentially increase cell proliferation, or lead to unscheduled senescence, or elicit out of context inflammatory signaling, as far as based on work with cells in vitro, no study in humans or in mouse models has been able to decisively implicate a downstream effector pathway in vivo.
Trans-eQTLs at Chr9p21 and Molecular Functions of ANRIL in Transcriptional Regulation
As opposed to cis effects, two eQTL studies have so far detected modest and tissue-selective differential expression of dozens of genes associated with Chr9p21 genotype with genome-wide significance (19, 27). Affected genes were from a broad range of classes (AVPR2, PEAK1, FBLN1, KALRN, DAZL, STAU2, HLA-DQA1, BTNL8, PLEKHA6, TDGF1) in whole blood (19) and different, non-overlapping gene sets linked to tissue wounding, cell migration and inflammatory response, when analyzing heart tissue, plaques, aortas, and arteries (27).
Other, and in part, larger studies in vascular tissue (20), peripheral blood mononuclear cells (PBMC, n = 2280) (33) and in blood monocytes (n = 1490) (23) reported no significant expression association.
Though not directly comparable, another study showed that in macrophages cultured in vitro under stress-bearing IFNγ and LPS stimulation, the CAD risk genotype led to differential up- and downregulation of target genes outside the Chr9p21 locus and yet distinct from the previously mentioned studies (IL1B, IL12B, CASP5, CCL8, MT1A, MT1E, MUCL1, TNIP3, VCAN, ENPP2, NDP, CD163) (30). Also ANRIL knockdown in cultured cell lines (69–72) and overexpression of linear ANRIL affected the expression of non-overlapping gene sets in the genome in trans (33, 36).
How ANRIL exerts trans-regulation is not known, and despite a study that showed a physical interaction of ANRIL with promoters of target genes (33), this role is likely not a classical function as enhancer RNA [eRNA (73)], because it involved both up-and down-regulated genes, and was suggested to involve sequence homology (33). In the case of ANRIL, trans-regulation of target genes was ascribed to an ALU motif in both ANRIL and the target gene promoters (33). Similarly, an independent study found that ANRIL did not only silence its targets, but unexpectedly also upregulated target genes: For example, proinflammatory interleukins IL6/8 were found to be co-stimulated by ANRIL and YY1, a transcription-regulating factor that bound to the ANRIL RNA, especially in the context of TNFα/NFκB signaling (70). Therefore, opposite to what could have been expected from the reported physical interaction of ANRIL with proteins from the repressive Polycomb group complexes (74), ANRIL might be an activator, at least for some trans-regulated genes (33, 70) (see chapter 4 for details). Whether circANRIL, beyond regulating rRNA maturation, is involved in primary transcriptional control, alone or via impacting linear ANRIL's function, is not known (36). Nevertheless, it is interesting to note that circANRIL isoforms linked to CAD are produced from exons located in the middle of the ANRIL gene (22, 36), and as such do not include the ALU motif, which is important for gene trans-regulation by linear ANRIL and is located more distally in the gene (33). Thus, variation in ANRIL RNA at the molecular level (linear vs. circular) might impose a fundamental alteration in ANRIL effector function, while not offering any explanation per se on how linear ANRIL regulates genes, as scaffold for promoter-activating complexes, or as decoy/inhibitor of repressive chromatin-modifying complexes. Conservatively speaking, it seems possible that Chr9p21 CAD risk genotypes affects genomic expression both in cis and in trans, and linear ANRIL RNA may be one, but not the sole, important effector molecule for how the Chr9p21 locus transduces such effects (Figure 1B).
Correlation of Chr9p21 Genes With Atherosclerosis Severity in Humans and Mouse Models
Another piece of evidence for a functional role of ANRIL in determining CAD risk stems from correlation analysis with disease features in patient cohorts. Aside of the genetic association, ANRIL levels were often increased in CAD patients, and not only in atherosclerotic plaque tissue, but also in circulating PBMCs or whole blood. Here, linear ANRIL levels were positively correlated with the severity of atherosclerosis (13, 29, 75) whereas circANRIL was anticorrelated (36) (Figure 1B). Thus, while the genotype of Chr9p21 determines the production of atherogenic (linear) over antiatherogenic ANRIL RNA species (circular), CAD and peripheral artery disease-dependent changes may additionally feed into ANRIL regulation. For CDKN2B, two studies reported a correlation of the expression with atherosclerosis severity (34, 76), where the direction of the correlation (downregulation in plaques) was consistent from what could be expected from the association results. But another study reported increased p16INK4A levels to positively correlate with inflammation markers in plaques instead of anticorrelation (25). Together, results from association as well as correlation analyses have etablished ANRIL lncRNA as prime candidate at the Chr9p21 locus.
Molecular Function of ANRIL and CDKN2A/B in Atherogenesis
ANRIL belongs to the group of long non-coding RNAs and as such has been suggested to act as a molecular scaffold of chromatin-modifying complexes that control gene expression through modifying histone tails. Specifically, ANRIL was found to physically interact with the CBX7 protein inside the PRC1 Polycomb complex, one of the major gene repression complexes in cells (74). Knockdown of members of this Polycomb group complex led to increased expression of the CDKN2A and CDKN2B tumor suppressors in the Chr9p21 locus. Also, ongoing RNA polymerase II transcription was important for the association of the Polycomb proteins with the locus, indicative of the importance of RNA for recruitment. It was concluded that ANRIL's function may be, at least in part, to repress the CDKN2A and CDKN2B tumor suppressors. As a consequence increased ANRIL levels are thought to promote overproliferation and to be incompatible with senescence onset, a major function of CDKN2A/B. As described in chapter 2, other work has shown that recruitment of the Polycomb complexes may account also for how ANRIL regulates genes in trans on a genome-wide level: Overexpression of linear ANRIL isoforms in cultured cells was found to promote pro-atherogenic cell functions, such as proliferation and reduced apoptosis, and to trigger the differential expression of hundreds of genes, in this case without affecting CDKN2A/B suppressors. Results from that study therefore questioned whether ANRIL regulated these tumor suppressor genes in cis at all (36, 77).
How does circular ANRIL, whose abundances is reduced in CAD patients, fit into this model? Both in human peripheral blood T-lymphocytes, as well as in PBMCs, whole blood and endatherectomy plaque tissue, circANRIL isoforms were found to be downregulated in samples from CAD patients carrying the Chr9p21 risk allele (22, 36). In an initial model, it was suggested that the production of circANRIL from central ANRIL exons would shorten the linear ANRIL lncRNA and, thereby, impaired linear ANRIL's function in epigenetic control of target genes (22). In a second study, a more primary role was found for circANRIL that was, furthermore, independent of linear ANRIL (36). Here, circANRIL was found to be 10-fold more abundant than linear ANRIL. Mass-spectrometric analysis of proteins interacting with circANRIL showed that it bound to PES1 protein, a member of the evolutionarily conserved PeBoW complex. This complex is essential for proper rRNA-processing, that is the excision of RNA spacer elements from pre-ribosomal rRNA precursors. CircANRIL inhibited the activity of the PeBoW complex, as deduced from the accumulation of unsufficiently processed (and non-functional) 26S and 32S pre-rRNA intermediates when circANRIL was overexpressed (36). A deficit in rRNA maturation caused nucleolar stress and p53 activation, culminating in inhibition of cell proliferation and in an increase in apoptosis. Notably, the observed functions of circANRIL were inverse to that of linear ANRIL and, as shown by genomic knockout of linear ANRIL exons, independent from the presence of these lncRNA isoforms. Thus, experimental evidence from expression analysis in vivo and from genetic experiments both indicated that circANRIL was anti-atherogenic. Together, linear ANRIL confers overproliferation, and circular ANRIL protects from overproliferation, suggesting that the genotype of Chr9p21 is important to determine the balance of linear and circular ANRIL levels in SMCs and macrophages, and that a dominance of linear ANRIL in this ratio, even when small, over decades skews for CAD (36) (Figure 1B).
Whether suppressing linear ANRIL or boosting circularization is sufficient to protect from atherosclerotic cues in vivo is matter of ongoing research. The fact that ANRIL RNA is not conserved beyond primates complicates the functional in vivo analysis of the Chr9p21 locus. So far, insight on how CAD is controlled by Chr9p21 through genetic modeling in mouse mutants is fragmented. The genetic elements of Chr9p21 and their relative positioning are overall syntenically conserved in mouse chromosome 4. So far, only one study has investigated, if deletion of a 70 kb long portion of mouse Chr4 corresponding to the CAD haplotype block in humans had an effect on atherosclerosis in vivo (78). This region contains a multi-exon lncRNA, AK148321, which is, however, likely not corresponding to human ANRIL. Mutant mice (78) developed tumors, reminiscent of tumorigenesis associated with mutation in the Chr9p21 region. But despite some metabolic changes in the mutant mice and enhanced platelet activation, no significant change in atherosclerotic fatty lesion formation was observed (78), putting in question the validity of this mouse model for studying ANRIL-driven atherogenesis. On the other hand, the mutants did develop more vascular aneurysms (79), supporting that some aspects of CAD were indeed contained in the noncoding mouse sequence.
Overall, the picture is not yet fully clear. While the genetic data from mice support the importance of individual noncoding genetic elements and of some of the protein-coding tumor suppressors for regulation of atherosclerosis and other CAD entities, whether the lncRNA encoded in the locus regulates CAD mechanistically via epigenetically regulating the neighboring tumor suppressors in cis has not been determined. Nevertheless, mouse genetics remains an interesting research avenue to explore some aspects of Chr9p21 biology, at least relating to aneurysm, cancer, and glaucoma formation.
Summary
Starting from a GWAS signal for CAD in a “gene desert” on Chr9p21 in 2007, research in the last decade has firmly established this region as strongest genetic factor of human atherosclerosis and has contributed to a better understanding of the underlying pathophysiology. The picture has emerged that one of the major routes how this locus controls atherosclerosis risk is through regulating the expression of the lncRNA ANRIL in cis, where the risk allele leads to high levels of linear ANRIL but decreases circular ANRIL expression. Linear ANRIL has been established as molecular scaffold guiding epigenetic protein complexes and promoting pro-atherogentic cells functions. On the contrary, circularization shifts ANRIL's function toward controlling ribosomal RNA processing and controlling protein translation thereby promoting athero-protection (Figure 1B). The molecular mechanisms of how the ratio of linear and circular ANRIL is controlled by the genotype at the locus are currently not resolved and it will be important to determine which gene regulatory elements within the ANRIL gene are disturbed by causal CAD risk SNPs. Experimentally exploring details of the molecular effector mechanisms for linear ANRIL and for circular ANRIL will be paramount, but this task will not be trivial because linear and circular ANRIL isoforms always co-exist and in part share the same sequence. Not last, more nuanced relations between Chr9p21 genotype and gene expression output can be expected to be found in the future if, for example, analyses were to take into account cell type-specific and context (stress, inflammation, senescence)-specific effects, aspect that whole tissue expression profiling is currently missing. Additionally, although it is early days, measuring the levels of circANRIL/linear ANRIL, might offer a prognostic value and help improve CAD risk stratification or allow to better monitor treatment response or disease recurrence.Yet, since circANRIL levels are reduced in plaque tissue, and since circANRIL has been found to be anti-atherogenic with or without co-existing linear ANRIL, increasing circANRIL abundance in patients could also be of therapeutic relevance. Expressing circANRIL levels in the cells of the vasculature in CAD disease models might, therefore, be a promising next step to exploit the accumulated knowledge on the Chr9p21 CAD risk locus.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Bernd Northoff for bioinformatics analyses and his aid in preparing the Figure. This work was in part funded by the German Research Foundation (DFG) as part of the Collaborative Research Center CRC1123 Atherosclerosis–Mechanisms and Networks of Novel Therapeutic Targets (project B1) and by the Leducq-foundation CADgenomics.
References
1. McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science (2007) 316:1488–91. doi: 10.1126/science.1142447
2. Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science (2007) 316:1491–3. doi: 10.1126/science.1142842
3. Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. (2007) 357:443–53. doi: 10.1056/NEJMoa072366
4. Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature (2007) 447:661–78. doi: 10.1038/nature05911
5. Larson MG, Atwood LD, Benjamin EJ, Cupples LA, D'Agostino RBSr, Fox CS, et al. Framingham Heart Study 100K project: genome-wide associations for cardiovascular disease outcomes. BMC Med Genet. (2007) 8 Suppl 1:S5. doi: 10.1186/1471-2350-8-S1-S5
6. Matarin M, Brown WM, Singleton A, Hardy JA, Meschia JF, Investigators I. Whole genome analyses suggest ischemic stroke and heart disease share an association with polymorphisms on chromosome 9p21. Stroke (2008) 39:1586–9. doi: 10.1161/STROKEAHA.107.502963
7. Wahlstrand B, Orho-Melander M, Delling L, Kjeldsen S, Narkiewicz K, Almgren P, et al. The myocardial infarction associated CDKN2A/CDKN2B locus on chromosome 9p21 is associated with stroke independently of coronary events in patients with hypertension. J Hypertens. (2009) 27:769–73. doi: 10.1097/HJH.0b013e328326f7eb
8. Helgadottir A, Thorleifsson G, Magnusson KP, Gretarsdottir S, Steinthorsdottir V, Manolescu A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. (2008) 40:217–24. doi: 10.1038/ng.72
9. Gschwendtner A, Bevan S, Cole JW, Plourde A, Matarin M, Ross-Adams H, et al. Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Ann Neurol. (2009) 65:531–9. doi: 10.1002/ana.21590
10. International Stroke Genetics C, Wellcome Trust Case Control C, Bellenguez C, Bevan S, Gschwendtner A, Spencer CC, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet. (2012) 44:328–33. doi: 10.1038/ng.1081
11. Dichgans M, Malik R, Konig IR, Rosand J, Clarke R, Gretarsdottir S, et al. Shared genetic susceptibility to ischemic stroke and coronary artery disease: a genome-wide analysis of common variants. Stroke (2014) 45:24–36. doi: 10.1161/STROKEAHA.113.002707
12. Cluett C, McDermott MM, Guralnik J, Ferrucci L, Bandinelli S, Miljkovic I, et al. The 9p21 myocardial infarction risk allele increases risk of peripheral artery disease in older people. Circ Cardiovasc Genet. (2009) 2:347–53. doi: 10.1161/CIRCGENETICS.108.825935
13. Holdt LM, Beutner F, Scholz M, Gielen S, Gabel G, Bergert H, et al. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol. (2010) 30:620–7. doi: 10.1161/ATVBAHA.109.196832
14. Murabito JM, White CC, Kavousi M, Sun YV, Feitosa MF, Nambi V, et al. Association between chromosome 9p21 variants and the ankle-brachial index identified by a meta-analysis of 21 genome-wide association studies. Circ Cardiovasc Genet. (2012) 5:100–12. doi: 10.1161/CIRCGENETICS.111.961292
15. Bilguvar K, Yasuno K, Niemela M, Ruigrok YM, von Und Zu Fraunberg M, van Duijn CM, et al. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet. (2008) 40:1472–7. doi: 10.1038/ng.240
16. Yasuno K, Bilguvar K, Bijlenga P, Low SK, Krischek B, Auburger G, et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet. (2010) 42:420–5. doi: 10.1038/ng.563
17. Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. (2017) 49:1385–91. doi: 10.1038/ng.3913
18. Cheng J, Cai MY, Chen YN, Li ZC, Tang SS, Yang XL, et al. Variants in ANRIL gene correlated with its expression contribute to myocardial infarction risk. Oncotarget (2017) 8:12607–19. doi: 10.18632/oncotarget.14721
19. Jarinova O, Stewart AF, Roberts R, Wells G, Lau P, Naing T, et al. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. (2009) 29:1671–7. doi: 10.1161/ATVBAHA.109.189522
20. Folkersen L, Kyriakou T, Goel A, Peden J, Malarstig A, Paulsson-Berne G, et al. Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS ONE (2009) 4:e7677. doi: 10.1371/journal.pone.0007677
21. Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet. (2010) 6:e1000899. doi: 10.1371/journal.pgen.1000899
22. Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. (2010) 6:e1001233. doi: 10.1371/journal.pgen.1001233
23. Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, et al. Genetics and beyond–the transcriptome of human monocytes and disease susceptibility. PLoS ONE (2010) 5:e10693. doi: 10.1371/journal.pone.0010693
24. Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. (2011) 43:333–8. doi: 10.1038/ng.784
25. Holdt LM, Sass K, Gabel G, Bergert H, Thiery J, Teupser D. Expression of Chr9p21 genes CDKN2B (p15(INK4b)), CDKN2A (p16(INK4a), p14(ARF)) and MTAP in human atherosclerotic plaque. Atherosclerosis (2011) 214:264–70. doi: 10.1016/j.atherosclerosis.2010.06.029
26. Kim JB, Deluna A, Mungrue IN, Vu C, Pouldar D, Civelek M, et al. Effect of 9p21.3 coronary artery disease locus neighboring genes on atherosclerosis in mice. Circulation (2012) 126:1896–906. doi: 10.1161/CIRCULATIONAHA.111.064881
27. Pilbrow AP, Folkersen L, Pearson JF, Brown CM, McNoe L, Wang NM, et al. The chromosome 9p21.3 coronary heart disease risk allele is associated with altered gene expression in normal heart and vascular tissues. PLoS ONE (2012) 7:e39574. doi: 10.1371/journal.pone.0039574
28. Zhuang J, Peng W, Li H, Wang W, Wei Y, Li W, et al. Methylation of p15INK4b and expression of ANRIL on chromosome 9p21 are associated with coronary artery disease. PLoS ONE (2012) 7:e47193. doi: 10.1371/journal.pone.0047193
29. Congrains A, Kamide K, Oguro R, Yasuda O, Miyata K, Yamamoto E, et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis (2012) 220:449–55. doi: 10.1016/j.atherosclerosis.2011.11.017
30. Zollbrecht C, Grassl M, Fenk S, Hocherl R, Hubauer U, Reinhard W, et al. Expression pattern in human macrophages dependent on 9p21.3 coronary artery disease risk locus. Atherosclerosis (2013) 227:244–9. doi: 10.1016/j.atherosclerosis.2012.12.030
31. Motterle A, Pu X, Wood H, Xiao Q, Gor S, Ng FL, et al. Functional analyses of coronary artery disease associated variation on chromosome 9p21 in vascular smooth muscle cells. Hum Mol Genet. (2012) 21:4021–9. doi: 10.1093/hmg/dds224
32. Johnson AD, Hwang SJ, Voorman A, Morrison A, Peloso GM, Hsu YH, et al. Resequencing and clinical associations of the 9p21.3 region: a comprehensive investigation in the Framingham heart study. Circulation (2013) 127:799–810. doi: 10.1161/CIRCULATIONAHA.112.111559
33. Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K, et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. (2013) 9:e1003588. doi: 10.1371/journal.pgen.1003588
34. Shanker J, Arvind P, Jambunathan S, Nair J, Kakkar V. Genetic analysis of the 9p21.3 CAD risk locus in Asian Indians. Thromb Haemost. (2014) 111:960–9. doi: 10.1160/TH13-08-0706
35. Miller CL, Pjanic M, Wang T, Nguyen T, Cohain A, Lee JD, et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat Commun. (2016) 7:12092. doi: 10.1038/ncomms12092
36. Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. (2016) 7:12429. doi: 10.1038/ncomms12429
37. Franzen O, Ermel R, Cohain A, Akers NK, Di Narzo A, Talukdar HA, et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science (2016) 353:827–30. doi: 10.1126/science.aad6970
38. Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Mohlke KL, et al. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS ONE (2009) 4:e5027. doi: 10.1371/journal.pone.0005027
39. Sarkar D, Oghabian A, Bodiyabadu PK, Joseph WR, Leung EY, Finlay GJ, et al. Multiple isoforms of ANRIL in melanoma cells: structural complexity suggests variations in processing. Int J Mol Sci. 18:1378. doi: 10.3390/ijms18071378
40. Jeck WR, Siebold AP, Sharpless NE. Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell (2012) 11:727–31. doi: 10.1111/j.1474-9726.2012.00871.x
41. Nakaoka H, Gurumurthy A, Hayano T, Ahmadloo S, Omer WH, Yoshihara K, et al. Allelic imbalance in regulation of ANRIL through chromatin interaction at 9p21 endometriosis risk locus. PLoS Genet. (2016) 12:e1005893. doi: 10.1371/journal.pgen.1005893
42. Schaefer AS, Richter GM, Groessner-Schreiber B, Noack B, Nothnagel M, El Mokhtari NE, et al. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet. (2009) 5:e1000378. doi: 10.1371/journal.pgen.1000378
43. Musunuru K, Post WS, Herzog W, Shen H, O'Connell JR, McArdle PF, et al. Association of single nucleotide polymorphisms on chromosome 9p21.3 with platelet reactivity: a potential mechanism for increased vascular disease. Circ Cardiovasc Genet. (2010) 3:445–53. doi: 10.1161/CIRCGENETICS.109.923508
44. Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science (2007) 316:1341–5. doi: 10.1126/science.1142382
45. Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Mit LU, Novartis Institutes of BioMedical R, Saxena R, Voight BF, Lyssenko V, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science (2007) 316:1331–6. doi: 10.1126/science.1142358
46. Erdmann J, Grosshennig A, Braund PS, Konig IR, Hengstenberg C, Hall AS, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. (2009) 41:280–2. doi: 10.1038/ng.307
47. Myocardial Infarction Genetics C, Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. (2009) 41:334–41. doi: 10.1038/ng.327
48. Tregouet DA, Konig IR, Erdmann J, Munteanu A, Braund PS, Hall AS, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. (2009) 41:283–5. doi: 10.1038/ng.314
49. Consortium IKC. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. (2011) 7:e1002260. doi: 10.1371/journal.pgen.1002260
50. Wang Z, Jacobs KB, Yeager M, Hutchinson A, Sampson J, Chatterjee N, et al. Improved imputation of common and uncommon SNPs with a new reference set. Nat Genet. (2011) 44:6–7. doi: 10.1038/ng.1044
51. Consortium CAD, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. (2013) 45:25–33. doi: 10.1038/ng.2480
52. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. (2015) 47:1121–30. doi: 10.1038/ng.3396
53. Myocardial Infarction G, Investigators CAEC, Stitziel NO, Stirrups KE, Masca NG, Erdmann J, et al. Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med. (2016) 374:1134–44. doi: 10.1056/NEJMoa1507652
54. Nioi P, Sigurdsson A, Thorleifsson G, Helgason H, Agustsdottir AB, Norddahl GL, et al. Variant ASGR1 associated with a reduced risk of coronary artery disease. N Engl J Med. (2016) 374:2131–41. doi: 10.1056/NEJMoa1508419
55. Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NG, Jansen H, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. (2017) 69:823–36. doi: 10.1016/j.jacc.2016.11.056
56. Howson JMM, Zhao W, Barnes DR, Ho WK, Young R, Paul DS, et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat Genet. (2017) 49:1113–9. doi: 10.1038/ng.3874
57. Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. (2017) 49:1392–7. doi: 10.1038/ng.3914
58. van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. (2018) 122:433–43. doi: 10.1161/CIRCRESAHA.117.312086
59. Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE (2012) 7:e30733. doi: 10.1371/journal.pone.0030733
60. Barrett SP, Salzman J. Circular RNAs: analysis, expression and potential functions. Development (2016) 143:1838–47. doi: 10.1242/dev.128074
61. Holdt LM, Kohlmaier A, Teupser D. Molecular roles and function of circular RNAs in eukaryotic cells. Cell Mol Life Sci. (2018) 75:1071–98. doi: 10.1007/s00018-017-2688-5
62. Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature (2011) 470:264–8. doi: 10.1038/nature09753
63. Mehta NN. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Circ Cardiovasc Genet. (2011) 4:327–9. doi: 10.1161/CIRCGENETICS.111.960443
64. Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet. (2017) 49:1602–12. doi: 10.1038/ng.3963
65. Svensson PA, Wahlstrand B, Olsson M, Froguel P, Falchi M, Bergman RN, et al. CDKN2B expression and subcutaneous adipose tissue expandability: possible influence of the 9p21 atherosclerosis locus. Biochem Biophys Res Commun. (2014) 446:1126–31. doi: 10.1016/j.bbrc.2014.03.075
66. Li WQ, Pfeiffer RM, Hyland PL, Shi J, Gu F, Wang Z, et al. Genetic polymorphisms in the 9p21 region associated with risk of multiple cancers. Carcinogenesis (2014) 35:2698–705. doi: 10.1093/carcin/bgu203
67. Kong Y, Sharma RB, Ly S, Stamateris RE, Jesdale WM, Alonso LC. CDKN2A/B T2D genome-wide association study risk SNPs impact locus gene expression and proliferation in human islets. Diabetes (2018) 67:872–84. doi: 10.2337/db17-1055
68. Almontashiri NA, Antoine D, Zhou X, Vilmundarson RO, Zhang SX, Hao KN, et al. 9p21.3 coronary artery disease risk variants disrupt TEAD transcription factor-dependent transforming growth factor beta regulation of p16 expression in human aortic smooth muscle cells. Circulation (2015) 132:1969–78. doi: 10.1161/CIRCULATIONAHA.114.015023
69. Congrains A, Kamide K, Katsuya T, Yasuda O, Oguro R, Yamamoto K, et al. CVD-associated non-coding RNA, ANRIL, modulates expression of atherogenic pathways in VSMC. Biochem Biophys Res Commun. (2012) 419:612–6. doi: 10.1016/j.bbrc.2012.02.050
70. Zhou X, Han X, Wittfeldt A, Sun J, Liu C, Wang X, et al. Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-kappaB pathway. RNA Biol. (2016) 13:98–108. doi: 10.1080/15476286.2015.1122164
71. Bochenek G, Hasler R, El Mokhtari NE, Konig IR, Loos BG, Jepsen S, et al. The large non-coding RNA ANRIL, which is associated with atherosclerosis, periodontitis and several forms of cancer, regulates ADIPOR1, VAMP3 and C11ORF10. Hum Mol Genet. (2013) 22:4516–27. doi: 10.1093/hmg/ddt299
72. Bai Y, Nie S, Jiang G, Zhou Y, Zhou M, Zhao Y, et al. Regulation of CARD8 expression by ANRIL and association of CARD8 single nucleotide polymorphism rs2043211 (p.C10X) with ischemic stroke. Stroke (2014) 45:383–8. doi: 10.1161/STROKEAHA.113.003393
73. Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, et al. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature (2013) 494:497–501. doi: 10.1038/nature11884
74. Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell (2010) 38:662–74. doi: 10.1016/j.molcel.2010.03.021
75. Arslan S, Berkan O, Lalem T, Ozbilum N, Goksel S, Korkmaz O, et al. Long non-coding RNAs in the atherosclerotic plaque. Atherosclerosis (2017) 266:176–81. doi: 10.1016/j.atherosclerosis.2017.10.012
76. Kojima Y, Downing K, Kundu R, Miller C, Dewey F, Lancero H, et al. Cyclin-dependent kinase inhibitor 2B regulates efferocytosis and atherosclerosis. J Clin Invest. (2014) 124:1083–97. doi: 10.1172/JCI70391
77. Sato K, Nakagawa H, Tajima A, Yoshida K, Inoue I. ANRIL is implicated in the regulation of nucleus and potential transcriptional target of E2F1. Oncol Rep. (2010) 24:701–7. doi: 10.3892/or_00000910
78. Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature (2010) 464:409–12. doi: 10.1038/nature08801
Keywords: lncRNA (long non-coding RNA), circRNA, GWAS (genome-wide association study), eQTL analysis, transcription, splicing, tumor suppressor proteins, cardiovascular diseases
Citation: Holdt LM and Teupser D (2018) Long Noncoding RNA ANRIL: Lnc-ing Genetic Variation at the Chromosome 9p21 Locus to Molecular Mechanisms of Atherosclerosis. Front. Cardiovasc. Med. 5:145. doi: 10.3389/fcvm.2018.00145
Received: 12 May 2018; Accepted: 01 October 2018;
Published: 06 November 2018.
Edited by:
Jeanette Erdmann, Universität zu Lübeck, GermanyReviewed by:
Yuqi Zhao, University of California, Los Angeles, United StatesClint L. Miller, University of Virginia, United States
Copyright © 2018 Holdt and Teupser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Teupser, ZGFuaWVsLnRldXBzZXJAbWVkLnVuaS1tdWVuY2hlbi5kZQ==