 Leo Ungar
Leo Ungar David Sanders2
David Sanders2 Ailin Barseghian
Ailin Barseghian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 30 August 2018
Sec. Cardiovascular Epidemiology and Prevention
Volume 5 - 2018 | https://doi.org/10.3389/fcvm.2018.00116
Familial hypercholesterolemia (FH) is a common heritable condition in which mutations of genes governing cholesterol metabolism result in elevated LDL levels and accelerated atherosclerosis. The treatment of FH focuses on lipid lowering drugs to decrease patients' cholesterol levels and reduce their risk of cardiovascular events. Even with optimal medical therapy, some FH patients will develop coronary atherosclerosis, suffer myocardial infarction, and require revascularization. Yet, the revascularization of FH patients has not been widely studied. Here we review FH, identify unanswered questions in the interventional management of FH patients, and explore barriers and opportunities for answering these questions. Further research is needed in this neglected but important topic in interventional cardiology.
Familial hypercholesterolemia (FH) is among the most common heritable cardiovascular conditions, affecting up to 1/250 people worldwide and an estimated 1.3 million people in the United States (1). This genetic abnormality of lipid metabolism is estimated to have an incidence of 1 in 200 to 1 in 500, (2) and has a higher prevalence in certain ethnic groups, including Ashkenazy Jews, French Canadians, Lebanese, and Afrikaners, in which the incidence can be as high as 1 in 67 (3). FH is also burdensome, resulting in accelerated atherosclerosis and increased cardiovascular morbidity and mortality. Yet, it remains widely underdiagnosed, (1) often undertreated, (1) and, with regard to its revascularization management, extremely understudied. As a result, although many patients with FH undergo percutaneous interventions, the optimal approach to these patients is unknown. Here, we briefly review FH (section FH in Review), including its pathophysiology, clinical presentation, diagnostic criteria, and management; identify key unanswered questions related to revascularization in FH (section Interventional Cardiology of Fh: Unanswered Questions), related both to whether and how to intervene; and explore barriers to answering these questions (section Barriers to the Study of Intervention in FH), including a lack of focus on FH patients in existing studies in interventional cardiology, and a lack of focus on intervention in existing studies of FH; and call for further research in this neglected but important area of interventional cardiology.
Normally, 70% of LDL is cleared from plasma by LDL receptors on hepatocytes (Figure 1) (2). LDL binds to LDL receptors, which promote the cellular uptake of apolipoprotein B (ApoB) and ApoE containing lipoproteins (2). The LDL receptor, now bound to LDL, is endocytosed into the hepatocyte (4). The LDL receptor is then either recycled to the cell surface or directed by the PCSK9 peptide to degradation by lysosomes (5).
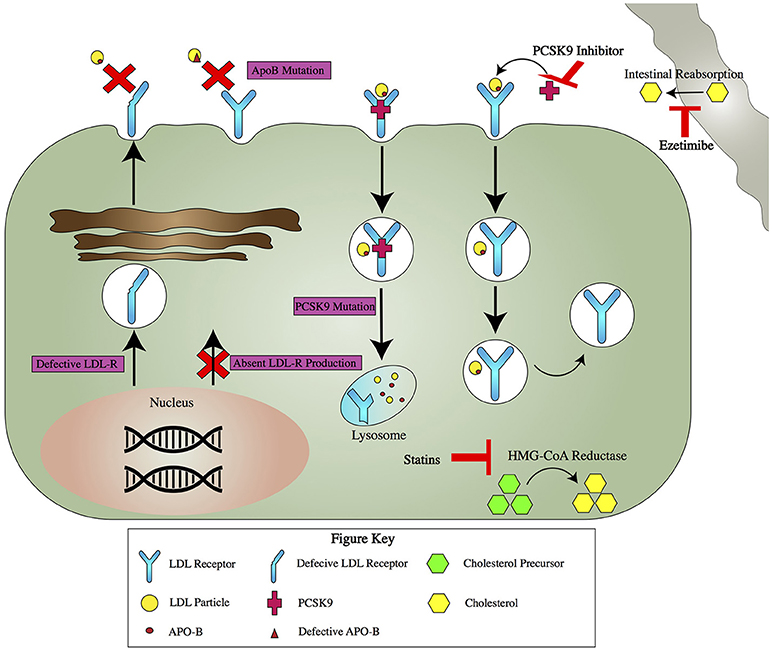
Figure 1. Pathogenetics of FH and Mechanism of Action for Key Drug Therapies for FH. Pathogenetics of FH: There are four major pathogenetic causes of FH (purple boxes). LDL-R gene mutations can result in production of a defective LDL-R that ineffectively binds LDL or a null allele that leads to no LDL-R production. Mutations in ApoB lead to impaired binding with the LDL-R. PCSK9 mutations lead to increased degradation of the LDL-R. Key Drug Therapies: Several drugs act via different mechanisms to lower serum LDL (red T's). Statins inhibit HMG-CoA reductase, the enzyme responsible for the rate limiting step in cholesterol production. Ezetimibe acts away from the hepatocyte, blocking cholesterol absorption in the gut. PCSK9 inhibitors bind PCSK9. This leads to increased recycling of LDL-R and ultimately more available receptors to take up LDL from the serum.
The pathophysiology of FH results from mutations in genes governing cholesterol processing (Figure 1). Three such genes are those coding for LDL receptors, ApoB, or PCSK9 (6). LDL gene mutations are the most common pathogenic mutations in FH, and greater than 1200 LDL gene mutations have been described (7). There are multiple ways to categorize these mutations, but the simplest divides them into two groups, one resulting in a defective LDL receptor, and a second resulting in a null allele which leads to no LDL receptor production. ApoB mutations also cause FH by leading to elevated LDL levels, but by contrast they do this by interfering with the binding of ApoB to the LDL receptor (8). PCSK9 mutations have different mechanisms but all result in increased degradation of the LDL receptor, which results in less LDL endocytosis and higher LDL levels (9). Less common pathogenic FH gene mutations have been described in genes coding for ApoE, LDL receptor adaptor protein, lysosomal acid lipase, and other proteins (10–12).
FH is just one type of heritable condition that results in disordered lipid metabolism. Each of the several disorders is characterized by its specific lipoprotein abnormality and the responsible genetic mutations. FH is the only one that is not associated with abnormal triglycerides. The most common of these heritable dyslipidemias is familial combined hyperlipidemia (FCH), which has an estimated prevalence of between 0.5 and 2% in the general population (13). FCH is a genetically complex disease that can result in significant elevation of both LDL and triglycerides (13). Similar lipid abnormalities, with elevations in LDL and triglycerides, are found in familial dysbetalipoproteinemia, a rare disorder that results from overabundance of ApoE2 protein (14). Both familial hyperchylomicronemia syndrome and primary simple hypertriglyceridemia result in much greater elevation in triglycerides than LDL (15). Familial hyperchylomicronemia, the result of lipoprotein lipase deficiency, is exceedingly rare but leads to severe elevations in triglycerides and pancreatitis (15, 16).
There are two forms of familial hypercholesterolemia: heterozygous familial hypercholesterolemia (HeFH), in which only one of two alleles in a key gene possesses a pathogenic mutation; and homozygous hypercholesterolemia (HoFH), the rarer genotype, in which both alleles at a locus are mutated.
The clinical consequences of both HeFH and HoFH result from the early and sustained elevation in LDL, leading to premature atherosclerosis and cardiovascular mortality. Children with HeFH are asymptomatic, but their LDL levels may range from 190 to 400 mg/dL (17). Atherosclerosis is accelerated, with subclinical disease detectable by adolescence and premature myocardial infarction possible as early as the third decade of life (18, 19). Without statin treatment, by age 60, the risk of clinically significant CAD may be greater than 50% for men and 30% for women (20). Additionally, patients may have characteristic non-cardiovascular manifestations including tendon xanthomas and corneal arcus (21, 22).
HoFH results in an even more severe phenotype. LDL levels may rise to greater than 500 mg/dL, (23) and atherosclerosis and cardiovascular events can begin as early as the first or second decades of life (24). Atheromatous deposits are extensive, and can involve the carotid arteries, cerebral vasculature, and the root of the aorta, leading to supravalvular aortic stenosis (25). Without treatment, patients with HoFH generally do not survive past 30 years (23).
There are three sets of diagnostic criteria for FH: the MED PED, Simon Broome, and Dutch Criteria (Table 1). MED PED is the simplest, using only a patient's age, LDL, and family history. The Simon Broome Criteria also takes into account physical findings and data from genetic testing. The Dutch Criteria includes the most variables and introduces a scoring system in which different features confer a certain number of points, and the likeliness of having FH is determined based on the sum of points. Recently, an app has been developed that in which patients' attributes can be plugged in to determine whether they can be diagnosed with FH by each of these three criteria (26).
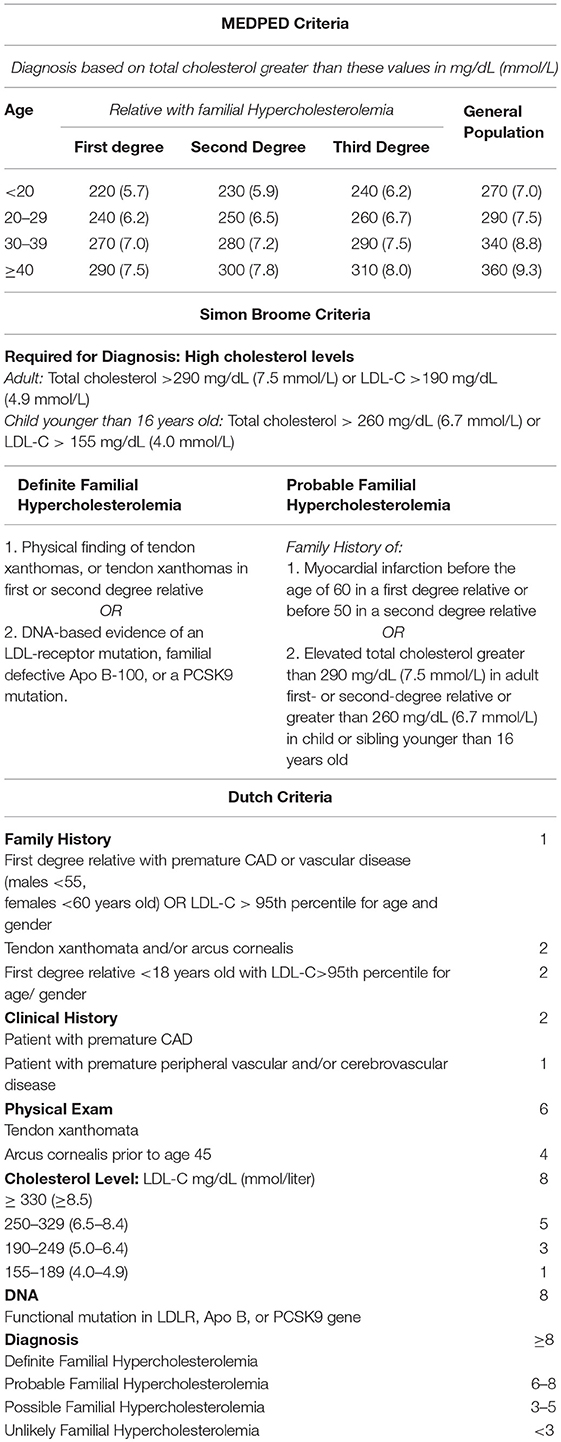
Table 1. Diagnostic Criteria for FH.
Currently, the management of FH focuses almost exclusively on the use of medications to lower the LDL (Figure 1). First line therapy is a high intensity statin, with a goal of reducing LDL by >50% (27). If this goal is not achieved with statin therapy alone, ezetimibe can be added (28, 29).
If a >50% reduction in LDL is achieved but LDL remains above 100 in patients with no history of clinically significant atherosclerosis, or above 70 in patients with a history of atherosclerosis, it is recommended to add ezetimibe if <25% additional LDL lowering is required, or a proprotein convertase subtilisin/kexin type 9 (PCSK9) monoclonal antibody if >25% LDL reduction is needed (30). PCSK9 normally binds to LDL receptors and causes their degradation. PCSK9 inhibitors, such as alirocumab and evolocumab, work by inhibiting PCSK9 and decreasing LDL receptor degradation, which results in an increased number of LDL receptors, which in turn results in increased resorption of LDL from the bloodstream and lower LDL levels (31, 32).
Non-medication therapies include lipophoresis, which is beneficial in HoFH, (33) and, as a last resort, liver transplant, which has also been used in HoFH including preemptively before vascular complications arise (34, 35).
Novel therapeutics are in development. Mipomersen works by the novel mechanism mRNA inhibition, (36) binding to the mRNA of Apo B-100, resulting in mRNA degradation by ribonuclease H1 and decreased Apo B-100 production. Apo B-100 is an integral part of the formation of VLDL, and decreasing Apo B-100's production results in a lowering of LDL (36). Another novel therapeutic agent, lomitapide, also works via inhibition of VLDL formation. Lomitapide prevents VLDL production by inhibiting microsomal triglyceride transfer protein, which is essential in the assembly of VLDL by the liver (37). Both mipomersen and lomitapide have been FDA approved for the treatment of HoFH and are currently under investigation for use in HeFH (38, 39) A third agent that is still under investigation is inclisiran, a small interfering RNA (siRNA) that inhibits PCSK9 production by selectively silencing the translation of the messenger RNAs for PCSK9 (40–42).
LDL lowering in FH reduces atherosclerosis and improves outcomes. In FH patients on lipid lowering therapy, LDL levels have been correlated with coronary artery disease severity by CT coronary angiography (43). Lipid lowering by medical therapy combined with lipophoresis has been associated with plaque regression as assessed by coronary angiography and coronary intravascular ultrasound (44). Lipid lowering also reduces cardiovascular adverse events, and if goal LDL levels are reached, FH patients can achieve a risk of MI similar to that of the general population (1, 45, 46).
The goal of medical therapy is primary or secondary prevention, to reduce the risk of a patient having either a first or second atherosclerotic event (47, 48). Even with optimal therapy, however, some patients will develop coronary artery disease, suffer myocardial infarction, and require revascularization. Yet, the interventional care of FH patients remains understudied. We identify key questions facing the interventional cardiologist caring for FH patients, discuss some of the challenges in answering these questions, and call for further research.
The initial question facing interventionalists, for patients with and without FH alike, is always whether and when to intervene (Table 2). In STEMI(49, 50) and NSTE-ACS(51) primary percutaneous intervention is not controversial, with multiple studies demonstrating the efficacy of primary PCI in those settings. Nor should it be controversial in STEMI and NSTE-ACS patients with FH. In other settings, however, the benefits of percutaneous intervention are less clear, rendering it ambiguous whether intervention should be pursued.

Table 2. Revascularization in FH: Unanswered Questions, Barriers to Study, and Future Directions.
There is a longstanding and ongoing debate about the benefits of revascularization in stable angina. The COURAGE Trial demonstrated that angiography-guided PCI in stable angina was not superior to optimal medical therapy (OMT) (52). Conversely, FAME-2 showed FFR-guided PCI in stable angina was superior to OMT with regard to a primary composite endpoint that included all-cause mortality, non-fatal MI, or unplanned hospitalization with urgent revascularization (53). However, the only one of the three individual components of this composite outcome that PCI significantly reduced was urgent revascularization, causing some to argue against even FFR-guided PCI for stable angina on the grounds that it is tantamount just to performing a PCI to prevent a PCI (54). PCI is also often thought to reduce symptoms in stable angina, but that was called into question more recently in the ORBITA trial, which randomized patients with stable angina to PCI vs. a sham procedure stimulating PCI, and which demonstrated no benefit of PCI relative to the sham procedure with regard to symptoms or function (55).
The issue here is whether these data are generalizable to FH. None of these studies were limited to a FH population, or resulted in secondary analyses examining an FH subpopulation. The generalizability of these data to FH patients is therefore unclear. It may be that FH patients have more (or less) benefit from revascularization in stable angina, and further studies of FH patients specifically would be required to clarify this issue.
A second consideration is angiographic and FFR- or iFR-guided PCI. The question here is how are outcomes impacted by not intervening on lesions that are angiographically intermediate but not physiological significant by FFR or iFR? In non-FH patients, the FAME trials demonstrated that FFR-guided PCI is at least on par with angiographic-guided PCI (non-inferior with regard to certain outcomes, superior with regard to others), (53, 56, 57) and more recently DEFINE-FLAIR (58) and iFR-SWEDEHEART (59) showed outcomes with iFR are equivalent to those with FFR, arguing that iFR guidance, too, is likely superior to angiography alone, even after five years of follow-up (60). These trials did not have subanalyses of an FH population, however, so their implications for FH patients are unclear. Theoretically, one might imagine that, given that FH patients often get clinically significant atherosclerosis younger than do non-FH patients,(18, 19) plaques could progress more quickly in FH patients than in non-FH patients, especially if FH patients are statin non-adherent and their LDL remains high. That hypothesis, if true, could provide reason to question whether hemodynamic biomarkers of plaque significance provide the same reassurance for long-term follow-up in FH, both for prognosis and for treatment effect (60).
The second question facing interventionists is, once it has been decided that revascularization should be performed, how should it performed? Two considerations we address here include PCI vs. CABG, and whether bioabsorbable stents may have a unique role to play in intervention in very young HoFH patients.
In non-FH patients, PCI vs. CABG has been studied extensively (61–65). Essentially, differences between outcomes for PCI and CABG have not been demonstrated, except in a few key situations in which CABG has been associated with improved outcomes as compared to PCI, including: patients with diabetes and 3-vessel disease, and patients with or without diabetes with 3-vessel disease and intermediate or high SYNTAX scores (66, 67). A theory is that in patients with more severe disease, the benefit of CABG results from bypassing not only the lesions that are culprit at the time of intervention, but other lesions that may develop in the future.
This logic could be applied to FH. FH patients, just like diabetics and patients with multivessel disease, may be more likely to have future lesions, and so CABG may have additional benefit in bypassing the lesions of both today and tomorrow. On the other hand, against CABG for FH patients may be that they are often younger at the time of their MI, and venous grafts are known to close off, at a rate of approximately 50% graft failure by 10–15 years, (68–74) potentially making them less viable for younger, otherwise healthy patients who may require their revascularization to be longer-lasting. These theoretical considerations should be viewed as hypothesis generating, and studies should be performed comparing PCI to CABG in FH patients.
The second consideration is, if PCI is performed, what sort of stent should be inserted, especially in HoFH patients who can have MIs even as young children? In general, drug-eluting stents (DES) have become the stent type of choice, as landmark trials demonstrated that they have decreased rates of in-stent thrombosis and restenosis as compared to bare metal stents (75, 76). These stents remain in the coronaries permanently, which is usually not an issue, but a theoretical concern, perhaps unique to HoFH, is that even young children can have myocardial infarctions and require revascularization, and stents that are appropriately sized for child-sized coronaries may not remain so after the child, and their coronaries, grow.
A new technology may help with this issue. Bioabsorbable stents are made of a special polymer which, over time, dissolves completely, leaving no residuum in the artery in which it was implanted (77). This technology initially generated excitement and investment, and gained FDA-approval in 2016 (78). After further studies demonstrated lower efficacy and higher thrombotic rates with bioabsorbables, (79) however, in 2017 they were pulled from the market by their manufacturer. One very specific area in which they may have special utility is in percutaneous intervention in children. Notably, a slew of case studies have been reported in which PCI using a bioabsorbable stent was performed on a child with FH, with the hope that its unique capacity to entirely dissolve would yield special benefit to the pediatric patient whose coronary arteries are still growing by circumventing an eventual size mismatch between stent and artery (80, 81). This creative idea thus far is the topic of case reports only, but more longitudinal study of these patients could reveal whether there is any actual evidence of this hypothetical, if nevertheless plausible, advantage.
As noted above, data on the interventional cardiology of FH are lacking. Here we propose two reasons why this might be as well as two possible opportunities going forward (Table 2).
First, most of the studies that have been performed previously do not have the granularity to identify FH patients. Trials generally assess their study drug's impact on particular patient populations by preidentifying specific patient populations in advance of the trial (such as diabetic patients), and then making sure to identify which patients in the study overall have the condition of interest. Regrettably, many clinical trials do not collect the data that would allow investigators to identify which study-patients have FH, specifically family history, key physical exam findings (tendon xanthoma and arcus cornealis), and genetic testing. We suggest that future trials include FH patients among their preselected subpopulations for further study, and collect the data necessary to identify them.
A possible barrier to this approach is that performing genetic analysis is costly, ranging from $620 to 1,485 according to one report, (82) and doing so for all trial participants would likely be cost-prohibitive. A possible workaround would be to collect all the relevant data besides the genetics (specifically family history and physical exam findings of tendon xanthoma and arcus cornealis) and proceed as best as possible with the available information, which could still yield diagnoses, by Dutch and/or Simon-Broome Criteria, of possible or probable FH in many cases (3). The optimal way to design trials to identify FH patients with high sensitivity and specificity while avoiding prohibitive cost increases merits further consideration.
Second, the studies that do focus specifically on the FH subpopulation have focused more on medical therapy and neglected to look at outcomes after percutaneous intervention. The PCSK9 trials, to their credit, examined outcomes in FH patients, but these obviously investigate a medical rather than an interventional therapy (83). The CASCADE FH Registry is a promising, longitudinal, observational study which as of 1/8/2017 had enrolled 3,960 patients across the US (84). Despite the CASCADE FH registry yielding multiple reports on diverse topics in FH, (85–87) to our knowledge, it has not yet reported on outcomes after intervention, and exactly what data it is collecting on interventions in FH patients is unclear. Neither have any other FH registries, to our knowledge, reported analyses of outcomes with interventional therapies, specifically. We suggest that these focused prospective studies of the FH patients, both randomized studies of particular therapies and observational registries, collect data on interventions and report outcomes after FH patients have undergone intervention.
FH is a genetic disease of lipid metabolism that is common and burdensome but underdiagnosed and undertreated (1). Current management is primarily medical, includes therapeutics that are both established (statins, ezetimibe) and novel (PCSK9 inhibitors, mipomersen, lomitapide), and focuses on lipid lowering for primary and secondary prevention of cardiovascular events. Revascularization in FH, however, is extremely understudied. As a result, although many patients with FH undergo percutaneous coronary interventions, the optimal interventional approach to these patients is unknown. Key unanswered questions relate to both whether (In stable angina?) and how (CABG vs. PCI? Bioabsorbable stents?) to intervene. Barriers to answering these questions include a lack of focus on FH patients in existing studies in interventional cardiology, and insufficient concentration on percutaneous intervention in existing studies of FH. Further research on coronary revascularization in FH is needed to guide the interventional management of FH patients.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. European atherosclerosis society consensus, familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. (2013) 34:3478–90a. doi: 10.1093/eurheartj/eht273
2. Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, et al. The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association. Circulation (2015) 132:2167–92. doi: 10.1161/CIR.0000000000000297
3. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. (2004) 160:407–20. doi: 10.1093/aje/kwh236
4. Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. (2009) 29:431–8. doi: 10.1161/ATVBAHA.108.179564
5. Kosenko T, Golder M, Leblond G, Weng W, Lagace TA. Low density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (PCSK9) in human plasma and inhibits PCSK9-mediated low density lipoprotein receptor degradation. J Biol Chem. (2013) 288:8279–88. doi: 10.1074/jbc.M112.421370
6. Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. (2003) 111:1795–803. doi: 10.1172/JCI200318925
7. Usifo E, Leigh SE, Whittall RA, Lench N, Taylor A, Yeats C, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet. (2012) 76:387–401. doi: 10.1111/j.1469-1809.2012.00724.x
8. Boren J, Ekstrom U, Agren B, Nilsson-Ehle P, Innerarity TL. The molecular mechanism for the genetic disorder familial defective apolipoprotein B100. J Biol Chem. (2001) 276:9214–8. doi: 10.1074/jbc.M008890200
9. Lambert G, Sjouke B, Choque B, Kastelein JJ, Hovingh GK. The PCSK9 decade. J Lipid Res. (2012) 53:2515–24. doi: 10.1194/jlr.R026658
10. Rashidi OM, Alama MN, Awan ZA. Interpreting the mechanism of APOE (p.Leu167del) Mutation in the incidence of familial hypercholesterolemia; an in-silico approach. Open Cardiovasc Med J. (2017) 11:84–93. doi: 10.2174/1874192401711010084
11. Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science (2001) 292:1394–8. doi: 10.1126/science.1060458
12. Chora JR, Alves AC, Medeiros AM, Mariano C, Lobarinhas G, Guerra A, et al. Lysosomal acid lipase deficiency: a hidden disease among cohorts of familial hypercholesterolemia? J Clin Lipidol. (2017) 11:477–484.e2. doi: 10.1016/j.jacl.2016.11.002
13. Gaddi A, Cicero AF, Odoo FO, Poli AA, Paoletti R. Practical recommendations for familial combined hyperlipidemia diagnosis and management: an update. Vascu Health Risk Manage. (2007) 4:877–86. doi: 10.2174/1567270010704030229
14. Kei A, Miltiadous G, Bairaktari E, Hadjivassiliou M, Cariolou M, Elisaf M. Dysbetalipoproteinemia: two cases report and a diagnostic algorithm. World J Clin Cases (2015) 3:371–6. doi: 10.12998/wjcc.v3.i4.371
16. Stroes E, Moulin P, Parhofer KG, Rebours V, Lohr JM, Averna M. Diagnostic algorithm for familial chylomicronemia syndrome. Atheroscler Suppl. (2017) 23:1–7. doi: 10.1016/j.atherosclerosissup.2016.10.002
17. Civeira FH. International panel on management of familial, guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis (2004) 173:55–68. doi: 10.1016/j.atherosclerosis.2003.11.010
18. Gidding SS, Bookstein LC, Chomka EV. Usefulness of electron beam tomography in adolescents and young adults with heterozygous familial hypercholesterolemia. Circulation (1998) 98:2580–3. doi: 10.1161/01.CIR.98.23.2580
19. Mabuchi H, Koizumi J, Shimizu M, Takeda R. Development of coronary heart disease in familial hypercholesterolemia. Circulation (1989) 79:225–32. doi: 10.1161/01.CIR.79.2.225
20. Stone NJ, Levy RI, Fredrickson DS, Verter J. Coronary artery disease in 116 kindred with familial type II hyperlipoproteinemia. Circulation (1974) 49:476–88. doi: 10.1161/01.CIR.49.3.476
21. Oosterveer DM, Versmissen J, Yazdanpanah M, Defesche JC, Kastelein JJ, Sijbrands EJ. The risk of tendon xanthomas in familial hypercholesterolaemia is influenced by variation in genes of the reverse cholesterol transport pathway and the low-density lipoprotein oxidation pathway. Eur Heart J. (2010) 31:1007–12. doi: 10.1093/eurheartj/ehp538
22. Winder AF, Jolleys JC, Day LB, Butowski PF. Corneal arcus, case finding and definition of individual clinical risk in heterozygous familial hypercholesterolaemia. Clin Genet. (1998) 54:497–502. doi: 10.1111/j.1399-0004.1998.tb03770.x
23. Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. a position paper from the consensus panel on familial hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. (2014) 35:2146–57. doi: 10.1093/eurheartj/ehu274
24. Al-Shaikh AM, Abdullah MH, Barclay A, Cullen-Dean G, McCrindle BW. Impact of the characteristics of patients and their clinical management on outcomes in children with homozygous familial hypercholesterolemia. Cardiol Young (2002) 12:105–12. doi: 10.1017/S1047951102000240
26. Thomas IG, Kiraly KJ, Thomas GS. iPhone apps for heterozygous familial hypercholesterolemia. J Clin Lipidol. (2013) 7:689. doi: 10.1016/j.jacl.2013.07.002
27. Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet (2001) 357:577–81. doi: 10.1016/S0140-6736(00)04053-8
28. Saltijeral A, L. Perez de Isla, Alonso R, Muniz O, Diaz-Diaz JL, Fuentes F, et al. Attainment of LDL cholesterol treatment goals in children and adolescents with familial hypercholesterolemia. The SAFEHEART follow-up registry. Rev Esp Cardiol. (2017) 70:444–50. doi: 10.1016/j.rec.2016.10.010
29. Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. (2008) 358:1431–43. doi: 10.1056/NEJMoa0800742
30. Lloyd-Jones DM, Morris PB, Ballantyne CM, Birtcher KK, Daly DD Jr, DePalma SM, et al. Focused Update of the 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the american college of cardiology task force on expert consensus decision pathways. J Am Coll Cardiol. (2017) 70:1785–822. doi: 10.1016/j.jacc.2017.07.745
31. Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. (2007) 282:18602–12. doi: 10.1074/jbc.M702027200
32. Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. (2015) 36:2996–3003. doi: 10.1093/eurheartj/ehv370
33. Wang A, Richhariya A, Gandra SR, Calimlim B, Kim L, Quek RG, et al. Systematic review of low-density lipoprotein cholesterol apheresis for the treatment of familial hypercholesterolemia. J Am Heart Assoc. (2016) 5:e003294. doi: 10.1161/JAHA.116.003294
34. Castilla Cabezas JA, Lopez-Cillero P, Jimenez J, Fraga E, Arizon JM, Briceno J, et al. Role of orthotopic liver transplant in the treatment of homozygous familial hypercholesterolemia. Rev Esp Enferm Dig. (2000) 92:601–8.
35. Maiorana A, Nobili V, Calandra S, Francalanci P, Bernabei S, El Hachem M, et al. Preemptive liver transplantation in a child with familial hypercholesterolemia. Pediatr Transpl. (2011) 15:E25–9. doi: 10.1111/j.1399-3046.2010.01383.x
36. Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet (2010) 375:998–1006. doi: 10.1016/S0140-6736(10)60284-X
37. Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. (2007) 356:148–56. doi: 10.1056/NEJMoa061189
38. Parizo J, Sarraju A, Knowles JW. Novel Therapies for Familial Hypercholesterolemia. Curr Treat Options Cardiovasc Med. (2016) 18:64. doi: 10.1007/s11936-016-0486-2
39. Stein EA, Dufour R, Gagne C, Gaudet D, East C, Donovan JM, et al. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation (2012) 126:2283–92. doi: 10.1161/CIRCULATIONAHA.112.104125
40. Nishikido T, Ray KK. Inclisiran for the treatment of dyslipidemia. Expe Opin Invest Drugs (2018) 27:287–94. doi: 10.1080/13543784.2018.1442435
41. Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med. (2017) 376:1430–40. doi: 10.1056/NEJMoa1615758
42. Ray KK, Stoekenbroek RM, Kallend D, Leiter LA, Landmesser U, Wright RS, et al. Effect of an siRNA Therapeutic Targeting PCSK9 on atherogenic lipoproteins: pre-specified secondary end points in ORION 1. Circulation (2018). doi: 10.1161/CIRCULATIONAHA.118.034710. [Epub ahead of print].
43. Neefjes LA, Ten Kate GJ, Alexia R, Nieman K, Galema-Boers AJ, Langendonk JG, et al. Accelerated subclinical coronary atherosclerosis in patients with familial hypercholesterolemia. Atherosclerosis (2011) 219:721–7. doi: 10.1016/j.atherosclerosis.2011.09.052
44. Matsuzaki M, Hiramori K, Imaizumi T, Kitabatake A, Hishida H, Nomura M, et al. Intravascular ultrasound evaluation of coronary plaque regression by low density lipoprotein-apheresis in familial hypercholesterolemia: the low density lipoprotein-apheresis coronary morphology and reserve trial (LACMART). J Am Coll Cardiol. (2002) 40:220–7. doi: 10.1016/S0735-1097(02)01955-1
45. Huijgen R, Hutten BA, Kindt I, Vissers MN, Kastelein JJ. Discriminative ability of LDL-cholesterol to identify patients with familial hypercholesterolemia: a cross-sectional study in 26,406 individuals tested for genetic FH. Circ Cardiovasc Genet. (2012) 5:354–9. doi: 10.1161/CIRCGENETICS.111.962456
46. Starr B, Hadfield SG, Hutten BA, Lansberg PJ, Leren TP, Damgaard D, et al. Development of sensitive and specific age- and gender-specific low-density lipoprotein cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clin Chem Lab Med. (2008) 46:791–803. doi: 10.1515/CCLM.2008.135
47. Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation (2011) 124:2202–7. doi: 10.1161/CIRCULATIONAHA.111.042523
48. Neil A, Cooper J, Betteridge J, Capps N, McDowell I, Durrington P, et al. Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study. Eur Heart J. (2008) 29:2625–33. doi: 10.1093/eurheartj/ehn422
49. O'Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos JA, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. (2013) 61:e78–140. doi: 10.1016/j.jacc.2012.11.019
50. Levine GN, Bates ER, Blankenship JC, Bailey SR, Bittl JA, Cercek B, et al. 2015 ACC/AHA/SCAI focused update on primary percutaneous coronary intervention for patients With ST-elevation myocardial infarction: an update of the 2011 ACCF/AHA/SCAI Guideline for percutaneous coronary intervention and the 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction. J Am Coll Cardiol. (2016) 67:1235–50. doi: 10.1016/j.jacc.2015.10.005
51. Amsterdam EA, Wenger NK, Brindis RG, Casey DE Jr, Ganiats TG, Holmes DR Jr, et al. 2014 AHA/ACC guideline for the management of patients with Non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. (2014) 64:e139–e228. doi: 10.1016/j.jacc.2014.09.017
52. Boden WE, O'Rourke RA, Teo KK, Hartigan PM, Maron DJ, Kostuk WJ, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med. (2007) 356:1503–16. doi: 10.1056/NEJMoa070829
53. De Bruyne B, Pijls NH, Kalesan B, Barbato E, Tonino PA, Piroth Z, et al. Fractional flow reserve-guided PCI versus medical therapy in stable coronary disease. N Engl J Med. (2012) 367:991–1001. doi: 10.1056/NEJMoa1205361
54. Boden WE. Which is more enduring–FAME or COURAGE? N Engl J Med. (2012) 367:1059–61. doi: 10.1056/NEJMe1208620
55. Al-Lamee R, Thompson D, Dehbi HM, Sen S, Tang K, Davies J, et al. Percutaneous coronary intervention in stable angina (ORBITA): a double-blind, randomised controlled trial. Lancet (2017) 391:31–40. doi: 10.1016/S0140-6736(17)32714-9
56. Tonino PA, De Bruyne B, Pijls NH, Siebert U, Ikeno F, van't Veer M, et al. Fractional flow reserve versus angiography for guiding percutaneous coronary intervention. N Engl J Med. (2009) 360:213–24. doi: 10.1056/NEJMoa0807611
57. Zimmermann FM, De Bruyne B, Pijls NH, Desai M, Oldroyd KG, Park SJ, et al. Rationale and design of the fractional flow reserve versus angiography for multivessel evaluation (FAME) 3 Trial: a comparison of fractional flow reserve-guided percutaneous coronary intervention and coronary artery bypass graft surgery in patients with multivessel coronary artery disease. Am Heart J. (2015) 170:619–26.e2. doi: 10.1016/j.ahj.2015.06.024
58. Davies JE, Sen S, Dehbi HM, Al-Lamee R, Petraco R, Nijjer SS, et al. Use of the Instantaneous Wave-free Ratio or Fractional Flow Reserve in PCI. N Engl J Med. (2017) 376:1824–34. doi: 10.1056/NEJMoa1700445
59. Gotberg M, Christiansen EH, Gudmundsdottir IJ, Sandhall L, Danielewicz M, Jakobsen L, et al. Instantaneous wave-free ratio versus fractional flow reserve to guide PCI. N Engl J Med. (2017) 376:1813–23. doi: 10.1056/NEJMoa1616540
60. van Nunen LX, Zimmermann FM, Tonino PA, Barbato E, Baumbach A, Engstrom T, et al. Fractional flow reserve versus angiography for guidance of PCI in patients with multivessel coronary artery disease (FAME): 5-year follow-up of a randomised controlled trial. Lancet (2015) 386:1853–60. doi: 10.1016/S0140-6736(15)00057-4
61. Park SJ, Ahn JM, Kim YH, Park DW, Yun SC, Lee JY, et al. Trial of everolimus-eluting stents or bypass surgery for coronary disease. N Engl J Med. (2015) 372:1204–12. doi: 10.1056/NEJMoa1415447
62. Stone GW, Sabik JF, Serruys PW, Simonton CA, Genereux P, Puskas J, et al. Everolimus-eluting stents or bypass surgery for left main coronary artery disease. N Engl J Med. (2016) 375:2223–35. doi: 10.1056/NEJMoa1610227
63. Farkouh ME, Domanski M, Sleeper LA, Siami FS, Dangas G, Mack M, et al. Strategies for multivessel revascularization in patients with diabetes. N Engl J Med. (2012) 367:2375–84. doi: 10.1056/NEJMoa1211585
64. Velazquez EJ, Lee KL, Deja MA, Jain A, Sopko G, Marchenko A, et al. Coronary-artery bypass surgery in patients with left ventricular dysfunction. N Engl J Med. (2011) 364:1607–16. doi: 10.1056/NEJMoa1100356
65. Serruys PW, Morice MC, Kappetein AP, Colombo A, Holmes DR, Mack MJ, et al. Percutaneous coronary intervention versus coronary-artery bypass grafting for severe coronary artery disease. N Engl J Med. (2009) 360:961–72. doi: 10.1056/NEJMoa0804626
66. Fihn SD, Blankenship JC, Alexander KP, Bittl JA, Byrne JG, Fletcher BJ, et al. 2014 ACC/AHA/AATS/PCNA/SCAI/STS focused update of the guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines, and the American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. (2014) 64:1929–49. doi: 10.1016/j.jacc.2014.07.017
67. m. Authors/Task Force, Windecker S, Kolh P, Alfonso F, Collet JP, Cremer J, et al. 2014 ESC/EACTS Guidelines on myocardial revascularization: The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS)Developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J. (2014) 35:2541–619. doi: 10.1093/eurheartj/ehu278
68. Parang P, Arora R. Coronary vein graft disease: pathogenesis and prevention. Can J Cardiol. (2009) 25:e57–62. doi: 10.1016/S0828-282X(09)70486-6
69. Loop FD, Golding LR, MacMillan JP, Cosgrove DM, Lytle BW, Sheldon WC. Coronary artery surgery in women compared with men: analyses of risks and long-term results. J Am Coll Cardiol. (1983) 1:383–90. doi: 10.1016/S0735-1097(83)80064-3
70. Grondin CM, Campeau L, Lesperance J, Enjalbert M, Bourassa MG. Comparison of late changes in internal mammary artery and saphenous vein grafts in two consecutive series of patients 10 years after operation. Circulation (1984) 70:I208–12.
71. FitzGibbon GM, Leach AJ, Keon WJ, Burton JR, Kafka HP. Coronary bypass graft fate. Angiographic study of 1,179 vein grafts early, one year, and five years after operation. J Thorac Cardiovasc Surg. (1986) 91:773–8.
72. Goldman S, Zadina K, Moritz T, Ovitt T, Sethi G, Copeland JG, et al. Long-term patency of saphenous vein and left internal mammary artery grafts after coronary artery bypass surgery: results from a department of veterans affairs cooperative study. J Am Coll Cardiol. (2004) 44:2149–56. doi: 10.1016/j.jacc.2004.08.064
73. Desai ND, Cohen EA, Naylor CD, Fremes SE. Radial artery patency study, a randomized comparison of radial-artery and saphenous-vein coronary bypass grafts. N Engl J Med. (2004) 351:2302–9. doi: 10.1056/NEJMoa040982
74. Cho KR, Kim JS, Choi JS, Kim KB. Serial angiographic follow-up of grafts one year and five years after coronary artery bypass surgery. Eur J Cardiothorac Surg. (2006) 29:511–6. doi: 10.1016/j.ejcts.2005.12.026
75. Moses JW, Leon MB, Popma JJ, Fitzgerald PJ, Holmes DR, O'Shaughnessy C, et al. Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N Engl J Med. (2003) 349:1315–23. doi: 10.1056/NEJMoa035071
76. Stone GW, Ellis SG, Cox DA, Hermiller J, O'Shaughnessy C, Mann JT, et al. One-year clinical results with the slow-release, polymer-based, paclitaxel-eluting TAXUS stent: the TAXUS-IV trial. Circulation (2004) 109:1942–7. doi: 10.1161/01.CIR.0000127110.49192.72
77. Ormiston JA, Serruys PW. Bioabsorbable coronary stents. Circ Cardiovasc Interv. (2009) 2:255–60. doi: 10.1161/CIRCINTERVENTIONS.109.859173
78. Rizik DG, Shah MG, Burke RF. First US experience following FDA approval of the ABBOTT vascular bioresorbable vascular scaffold for the treatment of coronary artery disease. Catheter Cardiovasc Interv. (2016) 88:899–901. doi: 10.1002/ccd.26708
79. Sorrentino S, Giustino G, Mehran R, Kini AS, Sharma SK, Faggioni M, et al. Everolimus-eluting bioresorbable scaffolds versus everolimus-eluting metallic stents. J Am Coll Cardiol. (2017) 69:3055–66. doi: 10.1016/j.jacc.2017.04.011
80. Ravat H, Garekar S, Changela V. Left Main coronary angioplasty of a 9-year-old child with bioresorbable vascular scaffold [corrected]. Catheter Cardiovasc Interv. (2017) 89:867–71. doi: 10.1002/ccd.26896
81. Nazif TM, Kalra S, Ali ZA, Karmpaliotis D, Turner ME, Starc TJ, et al. Percutaneous coronary intervention with bioresorbable scaffolds in a young child. JAMA Cardiol. (2017) 2:430–4. doi: 10.1001/jamacardio.2016.4954
82. Carlson B. Familial hypercholesterolemia captures gene test controversies. Biotechnol Healthcare (2010) 7:8–9.
83. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. (2017) 376:1713–22. doi: 10.1056/NEJMoa1615664
84. O'Brien EC, Roe MT, Fraulo ES, Peterson ED, Ballantyne CM, Genest J, et al. Rationale and design of the familial hypercholesterolemia foundation cascade screening for awareness and detection of familial hypercholesterolemia registry. Am Heart J. (2014)167:342–9.e17. doi: 10.1016/j.ahj.2013.12.008
85. Ahmad ZS, Andersen RL, Andersen LH, O'Brien EC, Kindt I, Shrader P, et al. US physician practices for diagnosing familial hypercholesterolemia: data from the CASCADE-FH registry. J Clin Lipidol. (2016) 10:1223–9. doi: 10.1016/j.jacl.2016.07.011
86. deGoma EM, Ahmad ZS, O'Brien EC, Kindt I, Shrader P, Newman CB, et al. Treatment gaps in adults with heterozygous familial hypercholesterolemia in the United States: data from the CASCADE-FH Registry. Circ Cardiovasc Genet. (2016) 9:240–9. doi: 10.1161/CIRCGENETICS.116.001381
Keywords: familial hypercholesterolemia (FH), percutaneous coronary intervention (PCI), statins, PCSK9 inhibitors, revascularization in familial hypercholesterolemia, familial hypercholesterolemia registry, LDL receptor, FH diagnostic criteria
Citation: Ungar L, Sanders D, Becerra B and Barseghian A (2018) Percutaneous Coronary Intervention in Familial Hypercholesterolemia Is Understudied. Front. Cardiovasc. Med. 5:116. doi: 10.3389/fcvm.2018.00116
Received: 26 March 2018; Accepted: 08 August 2018;
Published: 30 August 2018.
Edited by:
Michael D. Shapiro, Oregon Health & Science University, United StatesReviewed by:
Eugene Yang, University of Washington, United StatesCopyright © 2018 Ungar, Sanders, Becerra and Barseghian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ailin Barseghian, YmFyc2VnaGFAdWNpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.