 Stephen C. Kolwicz Jr.
Stephen C. Kolwicz Jr.- Heart and Muscle Metabolism Laboratory, Health and Exercise Physiology Department, Ursinus College, Collegeville, PA, United States
Research has demonstrated that the high capacity requirements of the heart are satisfied by a preference for oxidation of fatty acids. However, it is well known that a stressed heart, as in pathological hypertrophy, deviates from its inherent profile and relies heavily on glucose metabolism, primarily achieved by an acceleration in glycolysis. Moreover, it has been suggested that the chronically lipid overloaded heart augments fatty acid oxidation and triglyceride synthesis to an even greater degree and, thus, develops a lipotoxic phenotype. In comparison, classic studies in exercise physiology have provided a basis for the acute metabolic changes that occur during physical activity. During an acute bout of exercise, whole body glucose metabolism increases proportionately to intensity while fatty acid metabolism gradually increases throughout the duration of activity, particularly during moderate intensity. However, the studies in chronic exercise training are primarily limited to metabolic adaptations in skeletal muscle or to the mechanisms that govern physiological signaling pathways in the heart. Therefore, the purpose of this review is to discuss the precise changes that chronic exercise training elicits on cardiac metabolism, particularly on substrate utilization. Although conflicting data exists, a pattern of enhanced fatty oxidation and normalization of glycolysis emerges, which may be a therapeutic strategy to prevent or regress the metabolic phenotype of the hypertrophied heart. This review also expands on the metabolic adaptations that chronic exercise training elicits in amino acid and ketone body metabolism, which have become of increased interest recently. Lastly, challenges with exercise training studies, which could relate to several variables including model, training modality, and metabolic parameter assessed, are examined.
Introduction
The physiological benefits of exercise training have long been appreciated. Research has demonstrated enormous cardiovascular benefits including decreased blood pressure in hypertensive individuals (1), improved glycemic control in diabetics (2), improved blood lipid profiles (3), and improved quality of life in heart failure patients (4). Exercise has also been shown to have beneficial effects on the vasculature including improvements in endothelial function (5) and atherosclerosis and plaque stability (6, 7). Recent evidence has indicated that exercise may increase cardiac myocyte proliferation (8, 9), even after myocardial infarction (9). Therefore, exercise prescription remains an essential component of cardiac rehabilitation in patients after myocardial infarction, coronary artery by-pass grafting (CABG) surgery, and heart failure with reduced ejection fraction (HFrEF) (10–12). Surprisingly, although exercise intolerance is a primary manifestation of heart failure with preserved ejection fraction (HFpEF), a disproportional amount of research has been performed on this population (13). Despite the well-accepted benefits of exercise training in diseased population, the precise molecular adaptations that exercise elicits on the system are still not understood. Because of this, the National Institutes of Health (NIH) recently established a common fund aimed at identification of the molecular benefits that occur due to chronic exercise (14). Several excellent reviews on the cardiovascular adaptations that result from chronic exercise training have been published recently (15–17). However, these articles are limited in the discussion of the adaptations that occur in the cardio-metabolic pathways. Therefore, the purpose of this review is to summarize the existing literature that report adaptations in cardiac metabolism that result from chronic exercise training. For this review, the focus will be primarily limited to the adaptations that govern myocardial substrate utilization. Systemic adaptations, particularly contributing to oxygen delivery via enhanced coronary blood flow, are reviewed elsewhere (18, 19).
Metabolic Remodeling in the Pathological Heart
A myriad of studies elucidated the major substrates that supply the substantial energy requirements of the incessantly contracting heart in both health and disease. In the healthy myocardium, the literature demonstrates that fatty acids supply approximately 50–70% of the necessary substrates to fuel continual ATP resynthesis (20). Moreover, it is well accepted that during ischemic conditions as well as the development of pathological hypertrophy, the metabolic profile of the heart converts to a glucose-dependent phenotype, where glycolysis is markedly upregulated (21). The increased reliance on glucose as a fuel is matched by a decline in fatty acid oxidation present in both compensated and decompensated hypertrophy (22). Conversely, conditions of lipid overload such as diabetes and obesity, subjects the heart to a condition where the supply of fatty acids exceeds oxidation, leading to the development of cardio-lipotoxicity (23–25). In a diseased state, the chronic deviation from the inherent cardio-metabolic profile may result in the loss of metabolic flexibility that contributes to the development of cardiac dysfunction (26). Therefore, novel strategies that target metabolic therapies for the treatment of cardiac pathologies is a focus of several research initiatives.
Exercise Training and Cardiac Disease
Exercise training has long been known to elicit positive adaptations in both healthy and diseased populations. Up until the early 1950s, 4–6 weeks of complete bed rest was the traditional treatment for myocardial infarction (27). However, the controversial ideas of Herman Hellerstein, followed by seminal publications in the 1960s from Naughton (28) and Saltin (29) as well as Hellerstein (30), provided the foundation for the development of modern cardiac rehabilitation programs. Since then, numerous studies investigating the consequences of exercise-training, within the context of cardiac rehabilitation, on mortality, risk factors, and psychosocial factors have been conducted and are reviewed elsewhere (27).
The American Heart Association (AHA) declared physical activity as a major modifiable risk factor for cardiovascular disease (31). Moreover, low cardiorespiratory fitness levels are associated with an elevated mortality risk from cardiovascular disease (32). To this end, the AHA, the American College of Cardiology, and the American College of Sports Medicine put forth specific recommendations for the inclusion of cardiorespiratory exercise at a moderate-intensity for 30–40 min at 3–5 times per week (33, 34). Exercise training results in a condition of chronic volume overload, which induces myocardial remodeling and increased end-diastolic volume. In addition, myocardial contractility is also enhanced, reducing the end-systolic volume. As a result, the major physiological adaptations of exercise training is an increased stroke volume at rest (35). Because cardiac output is unchanged at rest, an additional side effect of chronic exercise training is a reduction in resting heart rate. Since heart failure is defined as an inability of the heart to maintain cardiac output to match systemic metabolic demands, exercise training, due to its ability to modify both stroke volume and heart rate, may be a promising therapeutic intervention.
Numerous studies tested the ability of chronic exercise training to elicit positive benefits in both animal models of heart failure as well as in patients with HFpEF or HFrEF. Additional efforts have been undertaken to determine the effectiveness of pre-operative exercise training for improving outcomes from cardiac surgery (36–38). In smaller studies of patients with dilated cardiomyopathy, 5–8 months of exercise training at a moderate intensity was sufficient to improve exercise performance and left ventricular function (39, 40). In addition, positive changes in metabolism were also noted with improved oxidative metabolism (40) or a tendency to augment myocardial phosphocreatine levels (39). Recently, a meta-analysis of 7 studies in patients with HFpEF determined that exercise capacity, diastolic function, and quality of life measures were all significantly increased with exercise training (41). In addition, the Exercise Training in Diastolic Heart Failure (Ex-DHF) reported improvements in exercise capacity and diastolic function (42). The elevation in exercise performance measures with exercise training are also echoed in studies of HFrEF patients (43–45). Despite positive changes in cardiac function in small population studies, larger studies including the Exercise Rehabilitation Trial (EXERT), the Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training (HF-ACTION), and the Exercise Training Meta-Analysis of Trials in Patients with Chronic Heart failure (ExTraMATCH) do not consistently find improvements in cardiac function or mortality (46–48). In accordance, inconsistent findings in changes in cardiac function are also observed in animal models as ejection fraction or diastolic function may increase (49–51), decrease (52), or remain unchanged (53). Several factors may contribute to the discrepant findings. In human studies, the presence of additional co-morbidities, varied disease etiologies, and unknown side effects of medications could hamper improvements in exercise performance. In animal studies, sex differences and training protocols clearly contribute to conflicting reports as discussed later in this review. This certainly highlights the need for additional studies that account for these numerous confounding factors.
The Exercise Physiologist's Take on Metabolism
The typical undergraduate exercise physiology textbook discusses major concepts regarding bioenergetics pathways during exercise (54). One primary focus is the time and intensity dependent contributions of the three major energy pathways. First, the phosphagen system, the PCr to ATP reaction regulated by creatine kinase, resynthesizes ATP during immediate, high intensity work. Second, the short-term lactic acid system, relies on glycogen-dependent glycolysis to fuel intermediate, moderate to high intensity activity (55). Third, the long-term aerobic system requires efficient oxidative metabolism to support moderately intense, long-duration exercise. In parallel to the energy systems, careful consideration of the oxygen demands and time course of oxygen uptake are also necessary. In the early course of exercise, an increase in the cellular energetic demand occurs, requiring increased oxygen uptake. However, despite constant intensity, there is a slight delay (up to several minutes) in oxygen uptake to match the steady-state metabolic demands. This phenomenon, deemed the “oxygen deficit,” represents the mismatch between total oxygen uptake and the steady-state oxygen requirement (56). During this time, ATP resynthesis is supported by both the immediate and short-term energy systems (i.e., PCr and glycolysis). In time, the oxygen uptake matches the oxygen demand and the steady-state metabolic needs are met primarily by long-term aerobic metabolism. Research has shown that exercise-trained individuals have a reduced “oxygen deficit” and reach steady-state, aerobic metabolism at a faster rate compared to sedentary counterparts (57). In other words, trained individuals have an increased capacity to utilize oxidative pathways to fuel exercise. The enhanced metabolic capacity of the system is likely furnished by a combination of augmented oxygen delivery and improved biochemical processes.
One long-standing dogma is that the failing heart is a metabolically comprised organ the contributes dysfunctional status, representing an “engine out of fuel” (58). Coinciding with this concept, a heart subjected to pressure-overload hypertrophy could be paralleled to the initial phase of intense exercise where a new steady state aerobic metabolism has not yet been achieved. In this case, the “oxygen deficit” is initially compensated by phosphocreatine and glycolysis. Indeed, accelerated glycolysis is a hallmark of the pathological myocardium (59) and alterations in the PCr/ATP ratio have been reported (60, 61). In this scenario, the hypertrophied heart would require a strategy to achieve steady state aerobic metabolism and return to its preferred fatty acid oxidation. Based on the ability to reduce the “oxygen deficit” and promote aerobic metabolism, perhaps exercise training could serve as a suitable intervention.
Cardiac Metabolism in Response to Exercise
When interpreting findings of exercise-based research, it is important to elucidate the acute vs. chronic responses of the physiological stress of exercise. Acute exercise, a single bout typically lasting from several minutes to hours, results in a host of cardiovascular and biochemical changes that return to baseline in a short time after cessation of the activity. Conversely, chronic exercise, or exercise training, refers to repeated bouts of acute exercise that occur over an extended period time (i.e., weeks, months, years) that result in distinct cardiovascular and biochemical adaptations that can be present for a prolonged duration. In some instances, the changes between acute and chronic exercise may be in opposition. For example, heart rate increases with acute exercise but tends to decrease with chronic exercise training. Therefore, it is critical to make these distinctions.
Past research has clearly delineated the changes that occur in the systemic usage of glucose and fatty acids in response to both acute and chronic exercise Likewise, research performed in the field of cardiac metabolism largely uncovered the relationship between the utilization of glucose and fatty acids, particularly during acute and chronic pathological stress. Although the exercise literature explored skeletal muscle metabolism of both amino acids and ketone bodies, these substrates are just starting to gain prominence in cardiac metabolism. In the ensuing section, an attempt to merge the two fields of exercise and cardiac metabolism is taken in order to understand adaptations that occur in metabolic pathways of the heart in response to chronic exercise training.
Changes in Glucose and Fatty Acid Metabolism
The systemic usage of glucose and fatty acids in the response to acute exercise has been well established by the scientific literature primarily by measuring the respiratory exchange ratio (RER) or respiratory quotient (RQ). RER or RQ is a ratio of the output of carbon dioxide divided by the intake of oxygen. RER values of 1.0 represent carbohydrate (i.e., glucose) while 0.7 represent fatty acids. It is suggested that the typical human has a resting RER of 0.85 representing a mixture of fuel usage (62, 63). During the early course of an exercise bout, the RER value rapidly approaches 1.0, proportionate to intensity, indicating a rapid utilization of glucose, presumably by the contracting skeletal muscle (64). This abrupt increase in glucose uptake and oxidation during exercise has been observed in the human heart as well in perfused hearts during acute workload (65, 66). Moreover, a significant portion of the myocardial glucose utilization is supplied by endogenous glycogen stores (67, 68), which is similar to observations made in skeletal muscle (69). If exercise intensity is moderate enough and continues for an extended duration, the RER value will return to values closer to 0.70, indicating a greater percentage of fatty acid usage (64). This coincides with elevated plasma fatty acid concentrations due to enhanced adipose tissue lipolysis (70). In summary, the relative usage of glucose vs. fatty acids during acute exercise is based on the intensity and duration of the activity.
Past research in exercise physiology has determined the systemic adaptations that result from engagement in long-term exercise training programs. These findings generally show that chronic endurance exercise training results in an increased capacity to oxidize fatty acids at rest and during sub-maximal exercise, partly due to skeletal muscle adaptations (64, 71, 72). However, the metabolic adaptations that occur in the heart in response to chronic exercise are still not completely elucidated. Despite numerous studies investigating various aspects of metabolic responses to exercise training, a disproportionate number of studies over the last 20 years directly assessed changes in myocardial substrate utilization. Due to the logistical and technological challenges with performing these analyses in humans, most of these studies were performed in rodent models and relied on data obtained from gene expression analysis or enzymatic activity assays. However, several of the studies did utilize more traditional methods of analyzing cardiac metabolism including, isotopic tracing techniques in isolated perfused hearts and positron emission tomography (PET). Table 1 summarizes the major findings of these chronic exercise-training studies in non-diseased mice.
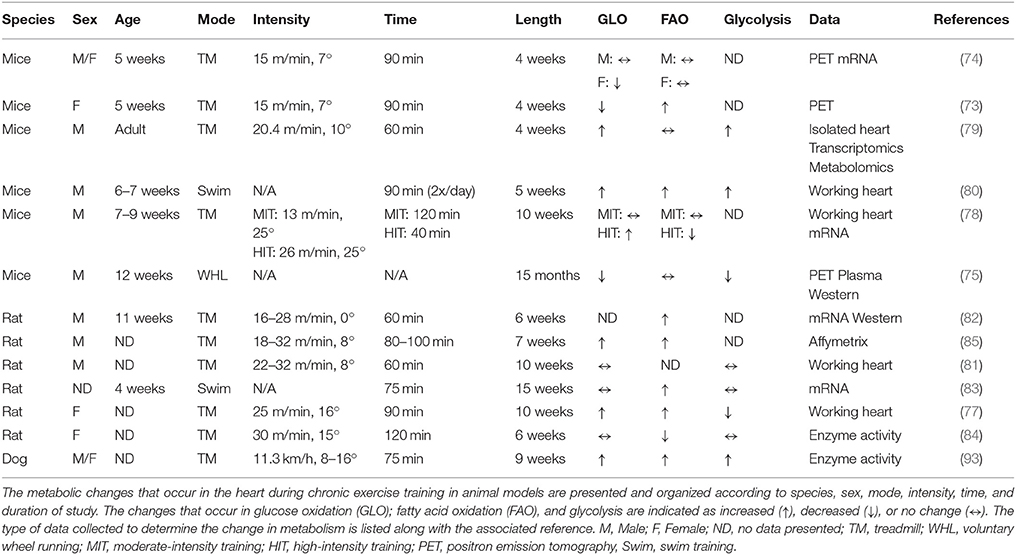
Table 1. Cardio-metabolic effects of chronic exercise training in healthy animals.
Based on the data presented in Table 1, it is difficult to determine the exact changes that occur in myocardial substrate utilization due to chronic exercise training. Using small animal PET scanning, glucose uptake was found to be decreased (73–75) or unchanged (74) while fatty acid uptake was likewise unaltered (74, 75) or increased (76). Using isolated rodent heart perfusions, exercise training resulted in an elevation (77–80) or no change (78, 81) in glucose oxidation while glycolysis was increased (79, 80), decreased (77) or unchanged (81). With likewise inconsistencies, fatty acid oxidation was found to be increased (77, 80), decreased (78), or unaffected (78, 79) by exercise training. The duration of training in the above studies largely ranged from 4 to 10 weeks using both mice and rodents. However, these differences do not appear to account for the lack of agreement in the data. Interestingly, 5 weeks of swim training resulted in significant increases in glucose oxidation, fatty acid oxidation, and glycolysis suggesting that this mode of exercise might be preferable for eliciting metabolic adaptations (80). However, 15 weeks of swim training in rats did not result in similar changes (83). Divergent results were also reported in females (77, 84). One notable finding is the overall decrease or no change in metabolic parameters in mice subjected to 15 months of wheel running (75), which may suggest a potential aging effect or a specific requirement to monitor the intensity of exercise.
Unfortunately, potential mechanistic targets for modulation of cardiac metabolism are also lacking from the current literature primarily due to limited exploration of the associated pathways. The findings are summarized in Figure 1. Detailed transcriptomic and metabolomics analyses of exercise-trained mouse hearts yielded minimal changes except for a significant upregulation of phosphofructokinase 2 (PFK2), accounting for glycolytic remodeling (79). Swim training in mice enhanced citrate synthase (CS) and hydroxyacyl-coenzyme A dehydrogenase (HADH) activity and led to increased expression of the carnitine palmitoyltransferase I (CPT1), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), and subunits of the electron transport chain (80). Treadmill training in mice also increased CPT1b expression as well as regulators of lipid metabolism, peroxisome proliferator-activated receptor (PPARα) and sterol regulatory element-binding protein 1c (SREBP1c) (82). In addition, gene expression of CD36 was shown to be upregulated (85). However, other studies found no change in PPARα (75), CD36 (83), or CPT1b (83). Overall, these studies might suggest the need to standardize training protocols including the mode, time and duration of training, and other requirements, such as intensity, in order for more solid conclusions to be drawn.

Figure 1. Changes in cardiometabolic pathways with chronic exercise trainingx. There are limited reports that detailed the changes that occur in the metabolic pathways associated with myocardial substrate utilization. Summarized are the reported changes in enzymes or genes that are involved in the processes: Fatty acid (FA) uptake, lipogenesis, FA oxidation, mitochondrial function, and glycolysis. CD36, cluster of differentiation 36; CS, citrate synthase; CPT1b, carnitine palmitoyltransferase 1b; DGAT1, diacylglycerol acyltransferase 1; ETC, electron transport chain; FABP, fatty acid binding protein; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HADH, hydroxyacyl-coenzyme A dehydrogenase; PPARα, peroxisome proliferator-activated receptor, alpha; PGC1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PFK2, phosphofructokinase 2; PK, pyruvate kinase; PFK; SCD1, stearoyl CoA desaturase 1; SREBP1c, sterol regulatory element-binding protein 1c.
High intensity interval training (HIIT) has traditionally been utilized to increase exercise performance in athletes but has gained mainstream and clinical attention recently. HIIT incorporates exercises that require near-maximal efforts for seconds to minutes interspersed with frequent longer duration rest periods (86). Although the physiological benefits of HIIT are apparent, there is debate whether HIIT is superior to the commonly recommended moderate-intensity continuous training (MICT) (76, 87, 88). Moreover, HIIT has been considered for patients with heart disease [for review see 89) and is the focus of a current randomized controlled study in the United Kingdom (90). However, a recent evaluation of 261 patients with heart failure found no benefit of HIIT over MICT on left ventricular dimensions or aerobic capacity (91). In regards to cardiac substrate utilization, HIIT training in mice led to a reduction in myocardial fatty acid oxidation and increased reliance of glucose oxidation, which was associated with a decrease in the expression of PPARα and increase in glycolytic genes (78), a metabolic profile more similar to the pathological heart. Furthermore, 4 weeks of HIIT was not as effective as MICT in reducing fibrosis or enhancing angiogenesis in hypertensive rats (92). Certainly, additional research in both animal and human models is needed before any conclusions can be reached.
Despite the overall variability in the reported metabolic adaptations, the most common reported change was elevated fatty acid oxidation by isolated perfused heart experiments (77, 80), gene expression (79, 82, 83, 85), or enzymatic activities (93). These potential findings are significant based on the known down-regulation of fatty acid oxidation that occurs in the pathologically hypertrophied heart (60, 94). Our recent work demonstrated that increasing myocardial fatty acid oxidation, via cardiac-specific deletion of acetyl CoA carboxylase 2 (ACC2), prevented the impairments in fatty acid oxidation that occurred during pressure-overload hypertrophy by transverse aortic constriction (TAC) or chronic angiotensin II treatment (60, 95). In addition to prevention of the metabolic remodeling process, systolic (60) or diastolic (95) function was maintained. Overall, these data suggested that targeting myocardial fatty acid oxidation was a promising therapeutic intervention. Since the above exercise training studies appear to indicate a potential to elicit positive adaptations in myocardial fatty acid oxidation, perhaps, exercise training either as a primary or secondary intervention might demonstrate likewise results as in the ACC2 mouse studies.
In addition to the decrements observed in oxidation of exogenous fatty acids, a decreased ability of hypertrophied hearts to oxidize endogenous fatty acids, from triacylglycerol (TAG) stores, also exists (96). Interestingly, recent work demonstrated that provision of unsaturated fatty acids improved endogenous fatty acid oxidation and cardiac function parameters in isolated perfused hypertrophied hearts (97). In conjunction, enhancing myocardial triacylglycerol turnover via diacylglycerol acyltransferase 1 (DGAT1) overexpression was sufficient to prevent impaired functional recovery from ischemia (98) and prevent cardiac dysfunction due to lipotoxicity (99, 100). Previous research showed that several genes in the TAG pathway are enhanced in exercise-trained skeletal muscle (101, 102) which also appears to hold true in trained cardiac muscle (82, 99). In this regard, exercise training might be beneficial in upregulating both exogenous and endogenous fatty acid metabolism and aid in the treatment of cardiac dysfunction, although additional research is certainly required in support of this hypothesis.
There are limited reports supporting the hypothesis that exercise training may prevent the appearance of the fetal metabolic profile in pathological cardiac hypertrophy. There is also a paucity of data investigating the effects of exercise training on the modulation of cardiac metabolism in the diabetic heart, which has been reviewed recently (103). Studies performed in rats revealed that treadmill running led to a distinct cardio-metabolic gene profile compared to aortic banding (82) or myocardial infarction (85). Specifically, genes involved in endogenous lipid metabolism (82) or beta-oxidation (85) were upregulated in hearts from trained rats. However, these studies did not test the interventional effects of training in the pathological models. However, endurance exercise training was effective in normalization of genes associated with glucose or fatty acid metabolism in spontaneously hypertensive rats (83). Likewise, exercise training was sufficient to partially normalize glycolytic, beta-oxidation, or mitochondrial enzymatic activities in volume overloaded rat hearts due to aortic regurgitation (104). Despite these findings, future research in this area is certainly warranted.
The Importance of Lactate Metabolism in Exercise
As discussed previously, there is a significant upregulation of glycolysis in skeletal muscle during the early course of an exercise bout, which is proportional to intensity. As a result, plasma lactate concentrations can increase 3- to 5-fold (105). Because of its omnivorous capacity, the heart can readily utilize the excess lactate to satisfy energetic demands. Previous studies demonstrated myocardial oxidation of lactate is significant and may be proportional to exogenous concentration within a physiological range (106, 107) or during elevated workloads (108). Interestingly, this elevated concentration of lactate can supplant fatty acid oxidation in the heart (107), providing a mechanism to preferentially oxidize the surplus lactate generated during intense activity.
Amino Acid Metabolism and Exercise
In the exercise literature, amino acids are generally considered a necessary nutrient for the post-exercise recovery period, providing necessary substrate for skeletal muscle repair. Despite numerous studies, the promotion of exercise capacity with amino acid supplementation, particularly with branched-chain amino acids (BCAAs), is still debated (109). Recent evidence has correlated cardiovascular disease with elevated plasma BCAAs levels (110). Moreover, disruption of BCAA catabolism via genetic deletion of the mitochondrial localized 2C-type serine-threonine protein phosphatase (PP2cm) has been linked to heart failure (111), cardiac dysfunction after myocardial infarction (112), and impaired functional recovery from ischemia (113). However, the contribution of amino acid to overall cardiac metabolism has generally been considered minimal, equating to less than 5% of the total, although studies directly testing this assertion are limited (114). Likewise, the metabolic adaptations in the cardiac amino acid pathway after exercise training require additional exploration.
Ketone Body Metabolism and Exercise
The contribution of ketone bodies to both cardiac and systemic metabolism has become of increased interest in the last several years. Recent work observed an increase in the enzyme, mitochondrial β-hydroxybutyrate dehydrogenase (BDH1), which coincided with elevated plasma levels of β-hydroxybutyrate (BHB) in both rodent and human models of heart failure (115, 116). In addition, increased measures of BHB oxidation in isolated perfused hearts was also noted (115). These studies suggested that an increased reliance on ketone body metabolism could be an additional hallmark of metabolic remodeling in the failing heart; however, whether this is an adaptive or maladaptive response remains to be seen (117). Of note, plasma ketone body concentrations and myocardial uptake are also increased in Type II diabetic patients without cardiac dysfunction (118), suggesting that the pathological consequence is due to increased availability. Indeed, it is known that ketone body uptake and oxidation in brain, heart, and skeletal muscle is proportional to the delivery (119).
In contrast to the pathological conditions of heart failure and diabetes, nutritional provision of ketone bodies in the form of ketone esters appears to improve exercise performance in both humans and rodents (120, 121), and is likely to gain increased scrutiny in the athletic performance field. In humans, ketone body ester supplementation decreased the reliance of skeletal muscle metabolism on glucose, evidenced by decreased glycolytic intermediates and blood lactate accumulation (120). The ketone body supplement also appeared to promote oxidation of intramuscular triacylglycerol during exercise (120). Interestingly, rodents fed a ketone body ester diet had improved cardiac energetics when exposed to acute isoproterenol stimulation (121). From these limited studies, the metabolic effect of ketone bodies has the potential to reduce reliance on glycolysis, promote endogenous lipid metabolism, and preserve energetics in actively working muscle. However, more research is needed to support these observations.
So, does exercise training result in any metabolic adaptations of the ketone body pathway in the heart? In essence, the answer remains unknown. There are limited reports of ketone body metabolism in exercise with one report demonstrating that 14-weeks of training in rats did not significantly change activities of enzymes associated with ketone body utilization (122). Interestingly, the cardiac activities of various ketone body enzymes, including BDH1, were 2- to 5-fold higher than that of slow-red oxidative (i.e., Type I) skeletal muscle (122), suggesting a relatively high robustness of myocardial ketone body metabolism. Overall, cardiac oxidation of ketone bodies has been suggested to be relatively minor (10–20%) in healthy hearts under physiological concentrations (114, 123). In skeletal muscle, activities of enzymes involved in ketone body hydrolysis have been reported to be up-regulated with exercise training which corresponds to both increased uptake and oxidation of ketone bodies in trained vs. untrained skeletal muscle, for review see Evans et al. (124). Whether exercise training also confers increased capacity of cardiac ketone body metabolism remains relatively unexplored.
Challenges With Exercise Training Research
Research statistics reveal that ~43% of adults in the United States (~31% worldwide) are physical inactive, defined as performing less than 30 min of moderately intense activity on 5 days per week or 20 min of highly intense activity on 3 days per week (125). Therefore, the potential population for exercise related studies might be biased toward active individuals. Because of this, most of the existing literature focused on populations that were easily recruited, i.e., athletes in various academic institutions. In addition, performing molecular based inquiries requires invasive data collection techniques, such as blood draws and muscle biopsies, which tend to make participation in the study less attractive.
Because of the above challenges, exercise-training studies in animals, particularly rodents, are preferable. Beyond the translational difficulties, numerous other factors need to be considered. Several different modalities of exercise are often employed: swim training, treadmill running, and voluntary wheel running. All of these have their advantages and disadvantages. For example, with swim training, appropriate temperature control of the “pool” is critical. In addition, constant monitoring to avoid mortality due to drowning is necessary. Rodents tend to have unpredictable behaviors during swimming, (i.e., “floating”) which can make monitoring intensity difficult (126). In treadmill running, many researchers employ an electric shock grid to “motivate” the animals. This presents potential ethical issues and may confound results particularly since the sedentary animals do not receive this same stimulus. However, less aversive motivational techniques exist, which can eliminate this concern (127). Voluntary wheel running eliminates the “forced” aspect of exercise but results in the inability to monitor intensity closely and requires single housing of the animals (128). However, voluntary wheel running may be preferred to treadmill particularly considering the reproducibility in evaluation of endurance exercise (127). Despite these issues, all of these modalities are frequently used in the exercise literature for training protocols and endurance capacity tests.
Regardless of the specific modality used, researchers must also consider general parameters of exercise prescription, namely intensity, frequency, and duration. Treadmill running presents an advantage by allowing the researcher to set a constant running speed that is equivalent to intensity. Although the animals need to be monitored closely to ensure adherence to the exercise period, some mouse strains have varied inherent running capabilities, termed critical running speed, which needs consideration (129). In general, the majority of exercise training studies employ a frequency of 5 days per week. Duration for treadmill running typically last for 60 min per session whereas swim training may encompass two 90-min sessions, for a total of 3 h per day (80). Furthermore, the length of the exercise training treatment period is traditionally from 4 weeks (130) to 12 weeks (131). Although frequency and duration are usually similar to humans, the determination of exercise intensity is difficult. Therefore, a biochemical marker documenting that a training effect has been achieved is necessary. Citrate synthase activity, a surrogate marker of mitochondrial density, in skeletal muscle is often used (80, 132). It should be noted that acute effects of exercise might persist for up to 24 h (64), so it is advisable to adjust the harvesting of animal tissues accordingly. A final challenge with conducting exercise-training research is critical in studies that use bioengineered mice. It is more frequently noted that the mouse strain can greatly influence the treatment response, including high fat feeding (133) and pressure-overload hypertrophy (134). This is also true for exercise as recent studies demonstrate a profound difference in exercise performance in a variety of mouse strains. Of note, the FVB/NJ, commonly used in transgenic colonies, significantly outperform the frequently used strain for knockout models, the C57BL/6J (130, 135, 136). Further complicating matters, there also appears to be a sexual dimorphism as female mice exhibit greater exercise performance and capacity (137–139) and more pronounced physiological hypertrophy (74, 137, 138). Therefore, careful planning of exercise training studies is definitely required.
Conclusions and Perspectives
One potential critique with exercise training research is the inability of dissecting a specific mechanism due to the intricate systemic interactions that are caused by the exercise treatment. However, any pathological model used in research ultimately affects the entire system, so focusing on the outcomes of any particular organ is viable in the research setting. Several studies discussed above reported various positive outcomes in response to exercise training. However, additional research is required to conclude whether exercise training prevents or reverses cardiac function in models of pathological hypertrophy. Furthermore, although the precise metabolic adaptations that occur in the heart from chronic exercise training are not definitive, some evidence suggests that fatty acid oxidation may be enhanced, although it is not clear whether this represents a change in substrate preference or an increase in the metabolic pathway. However, promoting myocardial fatty acid oxidation, particularly in diseased models, is still debated (140, 141). Therefore, more in-depth research focusing on the cardio-metabolic adaptations that result from exercise training may uncover a novel therapeutic intervention to combat the metabolic derangements that occur in the pathological heart.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Funding
This work was supported by a Scientist Development Grant from the American Heart Association (Grant number 14SDG18590020).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cornelissen VA, Smart NA. Exercise training for blood pressure: a systematic review and meta-analysis. J Am Heart Assoc. (2013) 2:e004473. doi: 10.1161/JAHA.112.004473
2. Marwick TH, Hordern MD, Miller T, Chyun DA, Bertoni AG, Blumenthal RS, et al. Exercise training for type 2 diabetes mellitus: impact on cardiovascular risk: a scientific statement from the American Heart Association. Circulation (2009) 119:3244–62. doi: 10.1161/CIRCULATIONAHA.109.192521
3. Lavie CJ, Arena R, Swift DL, Johannsen NM, Sui X, Lee DC, et al. Exercise and the cardiovascular system: clinical science and cardiovascular outcomes. Circ Res. (2015) 117:207–19. doi: 10.1161/CIRCRESAHA.117.305205
4. Ostman C, Jewiss D, Smart NA. The effect of exercise training intensity on quality of life in heart failure patients: a systematic review and meta-analysis. Cardiology (2017) 136:79–89. doi: 10.1159/000448088
5. Ashor AW, Lara J, Siervo M, Celis-Morales C, Oggioni C, Jakovljevic DG, et al. Exercise modalities and endothelial function: a systematic review and dose-response meta-analysis of randomized controlled trials. Sports Med. (2015) 45:279–96. doi: 10.1007/s40279-014-0272-9
6. Palmefors H, DuttaRoy S, Rundqvist B, Borjesson M. The effect of physical activity or exercise on key biomarkers in atherosclerosis–a systematic review. Atherosclerosis (2014) 235:150–61. doi: 10.1016/j.atherosclerosis.2014.04.026
7. Pellegrin M, Miguet-Alfonsi C, Bouzourene K, Aubert JF, Deckert V, Berthelot A, et al. Long-term exercise stabilizes atherosclerotic plaque in ApoE knockout mice. Med Sci Sports Exerc. (2009) 41:2128–35. doi: 10.1249/MSS.0b013e3181a8d530
8. Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D, et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell (2010) 143:1072–83. doi: 10.1016/j.cell.2010.11.036
9. Vujic A, Lerchenmuller C, Wu TD, Guillermier C, Rabolli CP, Gonzalez E, et al. Exercise induces new cardiomyocyte generation in the adult mammalian heart. Nat Commun. (2018) 9:1659. doi: 10.1038/s41467-018-04083-1
10. Menezes AR, Lavie CJ, Milani RV, Forman DE, King M, Williams MA. Cardiac rehabilitation in the United States. Prog Cardiovasc Dis. (2014) 56:522–9. doi: 10.1016/j.pcad.2013.09.018
11. Ambrosy AP, Cerbin LP, DeVore AD, Greene SJ, Kraus WE, O'Connor CM, et al. Aerobic exercise training and general health status in ambulatory heart failure patients with a reduced ejection fraction-findings from the heart failure and a controlled trial investigating outcomes of exercise training (HF-ACTION)trial. Am Heart J. (2017) 186:130–8. doi: 10.1016/j.ahj.2016.12.017
12. Balady GJ, Williams MA, Ades PA, Bittner V, Comoss P, Foody JM, et al. Core components of cardiac rehabilitation/secondary prevention programs: 2007 update: a scientific statement from the American Heart Association Exercise, Cardiac Rehabilitation, and Prevention Committee, the Council on Clinical Cardiology; the Councils on Cardiovascular Nursing, Epidemiology and Prevention, and Nutrition, Physical Activity, and Metabolism; and the American Association of Cardiovascular and Pulmonary Rehabilitation. Circulation (2007) 115:2675–82. doi: 10.1161/CIRCULATIONAHA.106.180945
13. Ades PA, Keteyian SJ, Balady GJ, Houston-Miller N, Kitzman DW, Mancini DM, et al. Cardiac rehabilitation exercise and self-care for chronic heart failure. JACC Heart Fail. (2013) 1:540–7. doi: 10.1016/j.jchf.2013.09.002
14. Neufer PD, Bamman MM, Muoio DM, Bouchard C, Cooper DM, Goodpaster BH, et al. Understanding the cellular and molecular mechanisms of physical activity-induced health benefits. Cell Metab. (2015) 22:4–11. doi: 10.1016/j.cmet.2015.05.011
15. Roh J, Rhee J, Chaudhari V, Rosenzweig A. The role of exercise in cardiac aging: from physiology to molecular mechanisms. Circ Res. (2016) 118:279–95. doi: 10.1161/CIRCRESAHA.115.305250
16. Vega RB, Konhilas JP, Kelly DP, Leinwand LA. Molecular mechanisms underlying cardiac adaptation to exercise. Cell Metab. (2017) 25:1012–26. doi: 10.1016/j.cmet.2017.04.025
17. Wei X, Liu X, Rosenzweig A. What do we know about the cardiac benefits of exercise? Trends Cardiovasc Med. (2015) 25:529–36. doi: 10.1016/j.tcm.2014.12.014
18. Bruning RS, Sturek M. Benefits of exercise training on coronary blood flow in coronary artery disease patients. Prog Cardiovasc Dis. (2015) 57:443–53. doi: 10.1016/j.pcad.2014.10.006
19. Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. (2008) 88:1009–86. doi: 10.1152/physrev.00045.2006
20. Kolwicz SCJr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. (2013) 113:603–16. doi: 10.1161/CIRCRESAHA.113.302095
21. Kolwicz SC Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. (2011) 90:194–201. doi: 10.1093/cvr/cvr071
22. Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res. (2011) 90:234–42. doi: 10.1093/cvr/cvr015
23. Bayeva M, Sawicki KT, Ardehali H. Taking diabetes to heart–deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J Am Heart Assoc. (2013) 2:e000433. doi: 10.1161/JAHA.113.000433
24. Lehrke M, Marx N. Diabetes mellitus and heart failure. Am J Med. 130:S40–50. doi: 10.1016/j.amjmed.2017.04.010
25. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
26. Ritterhoff J, Tian R. Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res. (2017) 113:411–21. doi: 10.1093/cvr/cvx017
27. Kachur S, Chongthammakun V, Lavie CJ, De Schutter A, Arena R, Milani RV, et al. Impact of cardiac rehabilitation and exercise training programs in coronary heart disease. Prog Cardiovasc Dis. (2017) 60:103–14. doi: 10.1016/j.pcad.2017.07.002
28. Naughton J, Lategola MT, Shanbour K. A physical rehabilitation program for cardiac patients: a progress report. Am J Med Sci. (1966) 252:545–53.
29. Saltin B, Blomqvist G, Mitchell JH, Johnson RL Jr, Wildenthal K, Chapman CB. Response to exercise after bed rest and after training. Circulation (1968) 38(5 Suppl.):1–78.
31. Artinian NT, Fletcher GF, Mozaffarian D, Kris-Etherton P, Van Horn L, Lichtenstein AH, et al. Interventions to promote physical activity and dietary lifestyle changes for cardiovascular risk factor reduction in adults: a scientific statement from the American Heart Association. Circulation (2010) 122:406–41. doi: 10.1161/CIR.0b013e3181e8edf1
32. Laukkanen JA, Zaccardi F, Khan H, Kurl S, Jae SY, Rauramaa R. Long-term change in cardiorespiratory fitness and all-cause mortality: a population-based follow-up study. Mayo Clin Proc. (2016) 91:1183–8. doi: 10.1016/j.mayocp.2016.05.014
33. Eckel RH, Jakicic JM, Ard JD, de Jesus JM, Houston Miller N, Hubbard VS, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. (2014) 63(25 Pt B):2960–84. doi: 10.1016/j.jacc.2013.11.003
34. Garber CE, Blissmer B, Deschenes MR, Franklin BA, Lamonte MJ, Lee IM, et al. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. (2011) 43:1334–59. doi: 10.1249/MSS.0b013e318213fefb
35. Rivera-Brown AM, Frontera WR. Principles of exercise physiology: responses to acute exercise and long-term adaptations to training. PMR (2012) 4:797–804. doi: 10.1016/j.pmrj.2012.10.007
36. Achttien RJ, Staal JB, van der Voort S, Kemps HM, Koers H, Jongert MW, et al. Exercise-based cardiac rehabilitation in patients with coronary heart disease: a practice guideline. Neth Heart J. (2013) 21:429–38. doi: 10.1007/s12471-013-0467-y
37. Richardson K, Levett DZH, Jack S, Grocott MPW. (2017). Fit for surgery? Perspectives on preoperative exercise testing and training. Br J Anaesth. 119(Suppl. 1):i34–43. doi: 10.1093/bja/aex393
38. Valkenet K, van de Port IG, Dronkers JJ, de Vries WR, Lindeman E, Backx FJ. The effects of preoperative exercise therapy on postoperative outcome: a systematic review. Clin Rehabil. (2011) 25:99–111. doi: 10.1177/0269215510380830
39. Beer M, Wagner D, Myers J, Sandstede J, Kostler H, Hahn D, et al. Effects of exercise training on myocardial energy metabolism and ventricular function assessed by quantitative phosphorus-31 magnetic resonance spectroscopy and magnetic resonance imaging in dilated cardiomyopathy. J Am Coll Cardiol. (2008) 51:1883–91. doi: 10.1016/j.jacc.2007.09.075
40. Stolen KQ, Kemppainen J, Ukkonen H, Kalliokoski KK, Luotolahti M, Lehikoinen P, et al. Exercise training improves biventricular oxidative metabolism and left ventricular efficiency in patients with dilated cardiomyopathy. J Am Coll Cardiol. (2003) 41:460–7. doi: 10.1016/S0735-1097(02)02772-9
41. Dieberg G, Ismail H, Giallauria F, Smart NA. Clinical outcomes and cardiovascular responses to exercise training in heart failure patients with preserved ejection fraction: a systematic review and meta-analysis. J Appl Physiol. (2015) 119:726–33. doi: 10.1152/japplphysiol.00904.2014
42. Edelmann F, Gelbrich G, Dungen HD, Frohling S, Wachter R, Stahrenberg R, et al. Exercise training improves exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction: results of the Ex-DHF (Exercise training in Diastolic Heart Failure) pilot study. J Am Coll Cardiol. (2011) 58:1780–91. doi: 10.1016/j.jacc.2011.06.054
43. Ismail H, McFarlane JR, Dieberg G, Smart NA. Exercise training program characteristics and magnitude of change in functional capacity of heart failure patients. Int J Cardiol. (2014) 171:62–5. doi: 10.1016/j.ijcard.2013.11.045
44. Ismail H, McFarlane JR, Nojoumian AH, Dieberg G, Smart NA. Clinical outcomes and cardiovascular responses to different exercise training intensities in patients with heart failure: a systematic review and meta-analysis. JACC Heart Fail. (2013) 1:514–22. doi: 10.1016/j.jchf.2013.08.006
45. Smart N, Haluska B, Jeffriess L, Case C, Marwick TH. Cardiac contributions to exercise training responses in patients with chronic heart failure: a strain imaging study. Echocardiography (2006) 23:376–82. doi: 10.1111/j.1540-8175.2006.00224.x
46. McKelvie RS, Teo KK, Roberts R, McCartney N, Humen D, Montague T, et al. Effects of exercise training in patients with heart failure: the Exercise Rehabilitation Trial (EXERT). Am Heart J. (2002) 144:23–30. doi: 10.1067/mhj.2002.123310
47. Piepoli MF, Davos C, Francis DP, Coats AJ, ExTra MC. Exercise training meta-analysis of trials in patients with chronic heart failure (ExTraMATCH). BMJ (2004) 328:189. doi: 10.1136/bmj.37938.645220.EE
48. van Tol BA, Huijsmans RJ, Kroon DW, Schothorst M, Kwakkel G. Effects of exercise training on cardiac performance, exercise capacity and quality of life in patients with heart failure: a meta-analysis. Eur J Heart Fail. (2006) 8:841–50. doi: 10.1016/j.ejheart.2006.02.013
49. de Waard MC, Duncker DJ. Prior exercise improves survival, infarct healing, and left ventricular function after myocardial infarction. J Appl Physiol. (2009) 107:928–36. doi: 10.1152/japplphysiol.91281.2008
50. Hiemstra JA, Veteto AB, Lambert MD, Olver TD, Ferguson BS, McDonald KS, et al. Chronic low-intensity exercise attenuates cardiomyocyte contractile dysfunction and impaired adrenergic responsiveness in aortic-banded mini-swine. J Appl Physiol. (2018) 124:1034–44. doi: 10.1152/japplphysiol.00840.2017
51. Lachance D, Plante E, Bouchard-Thomassin AA, Champetier S, Roussel E, Drolet MC, et al. Moderate exercise training improves survival and ventricular remodeling in an animal model of left ventricular volume overload. Circ Heart Fail. (2009) 2:437–45. doi: 10.1161/CIRCHEARTFAILURE.108.845487
52. Schultz RL, Swallow JG, Waters RP, Kuzman JA, Redetzke RA, Said S, et al. Effects of excessive long-term exercise on cardiac function and myocyte remodeling in hypertensive heart failure rats. Hypertension (2007) 50:410–6. doi: 10.1161/HYPERTENSIONAHA.106.086371
53. van Deel ED, de Boer M, Kuster DW, Boontje NM, Holemans P, Sipido KR, et al. Exercise training does not improve cardiac function in compensated or decompensated left ventricular hypertrophy induced by aortic stenosis. J Mol Cell Cardiol. (2011) 50:1017–25. doi: 10.1016/j.yjmcc.2011.01.016
54. McArdle WD, Katch FI, Katch VL. (2016). Essentials of Exercise Physiology. V ed. Philadelphia, PA: Wolters Kluwer.
55. Greenberg CC, Jurczak MJ, Danos AM, Brady MJ. Glycogen branches out: new perspectives on the role of glycogen metabolism in the integration of metabolic pathways. Am J Physiol Endocrinol Metab. (2006) 291:E1–8. doi: 10.1152/ajpendo.00652.2005
56. Stainsby WN, Barclay JK. Exercise metabolism: O 2 deficit, steady level O 2 uptake and O 2 uptake for recovery. Med Sci Sports (1970) 2:177–81.
57. Demarle AP, Slawinski JJ, Laffite LP, Bocquet VG, Koralsztein JP, Billat VL. Decrease of O(2) deficit is a potential factor in increased time to exhaustion after specific endurance training. J Appl Physiol. (2001) 90:947–53. doi: 10.1152/jappl.2001.90.3.947
58. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
59. Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, et al. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension (2004) 44:662–7. doi: 10.1161/01.HYP.0000144292.69599.0c
60. Kolwicz SCJr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. (2012) 111:728–38. doi: 10.1161/CIRCRESAHA.112.268128
61. Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation (1997) 96:2190–6.
62. Goedecke JH, St Clair Gibson A, Grobler L, Collins M, Noakes TD, Lambert EV. Determinants of the variability in respiratory exchange ratio at rest and during exercise in trained athletes. Am J Physiol Endocrinol Metab. (2000) 279:E1325–34. doi: 10.1152/ajpendo.2000.279.6.E1325
63. Hunschede S, El Khoury D, Antoine-Jonville S, Smith C, Thomas S, Anderson GH. Acute changes in substrate oxidation do not affect short-term food intake in healthy boys and men. Appl Physiol Nutr Metab. (2015) 40:168–77. doi: 10.1139/apnm-2014-0188
64. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. (2013) 17:162–84. doi: 10.1016/j.cmet.2012.12.012
65. Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J Clin Invest. (1988) 82:2017–25. doi: 10.1172/JCI113822
66. Goodwin GW, Taegtmeyer H. Improved energy homeostasis of the heart in the metabolic state of exercise. Am J Physiol Heart Circ Physiol. (2000) 279:H1490–501. doi: 10.1152/ajpheart.2000.279.4.H1490
67. Goodwin GW, Ahmad F, Doenst T, Taegtmeyer H. Energy provision from glycogen, glucose, and fatty acids on adrenergic stimulation of isolated working rat hearts. Am J Physiol. (1998) 274(4 Pt 2):H1239-47.
68. Goodwin GW, Taylor CS, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. (1998) 273:29530–9.
69. Jensen TE, Richter EA. Regulation of glucose and glycogen metabolism during and after exercise. J Physiol. (2012) 590:1069–76. doi: 10.1113/jphysiol.2011.224972
70. Horowitz JF, Klein S. Lipid metabolism during endurance exercise. Am J Clin Nutr. (2000) 72(2 Suppl.):558S−63S. doi: 10.1093/ajcn/72.2.558S
71. Coyle EF. Substrate utilization during exercise in active people. Am J Clin Nutr. (1995) 61(4 Suppl.):968S−79S.
72. Klein S, Coyle EF, Wolfe RR. Fat metabolism during low-intensity exercise in endurance-trained and untrained men. Am J Physiol. (1994) 267(6 Pt 1):E934–40. doi: 10.1152/ajpendo.1994.267.6.E934
73. Foryst-Ludwig A, Kreissl MC, Benz V, Brix S, Smeir E, Ban Z, et al. Adipose Tissue Lipolysis Promotes Exercise-induced Cardiac Hypertrophy Involving the Lipokine C16:1n7-Palmitoleate. J Biol Chem. (2015) 290:23603–15. doi: 10.1074/jbc.M115.645341
74. Foryst-Ludwig A, Kreissl MC, Sprang C, Thalke B, Bohm C, Benz V, et al. Sex differences in physiological cardiac hypertrophy are associated with exercise-mediated changes in energy substrate availability. Am J Physiol Heart Circ Physiol. (2011) 301:H115–22. doi: 10.1152/ajpheart.01222.2010
75. Monleon D, Garcia-Valles R, Morales JM, Brioche T, Olaso-Gonzalez G, Lopez-Grueso R, et al. Metabolomic analysis of long-term spontaneous exercise in mice suggests increased lipolysis and altered glucose metabolism when animals are at rest. J Appl Physiol. (2014) 117:1110–9. doi: 10.1152/japplphysiol.00585.2014
76. Foster C, Farland CV, Guidotti F, Harbin M, Roberts B, Schuette J, et al. The effects of high intensity interval training vs steady state training on aerobic and anaerobic capacity. J Sports Sci Med. (2015) 14:747–55.
77. Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, et al. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol. (2004) 287:H1055–63. doi: 10.1152/ajpheart.00925.2003
78. Hafstad AD, Boardman NT, Lund J, Hagve M, Khalid AM, Wisloff U, et al. High intensity interval training alters substrate utilization and reduces oxygen consumption in the heart. J Appl Physiol. (2011) 111:1235–41. doi: 10.1152/japplphysiol.00594.2011
79. Gibb AA, Epstein PN, Uchida S, Zheng Y, McNally LA, Obal D, et al. Exercise-induced changes in glucose metabolism promote physiological cardiac growth. Circulation (2017) 136:2144–57. doi: 10.1161/CIRCULATIONAHA.117.028274
80. Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, et al. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol Cell Biol. (2014) 34:3450–60. doi: 10.1128/MCB.00426-14
81. Broderick TL, Poirier P, Gillis M. Exercise training restores abnormal myocardial glucose utilization and cardiac function in diabetes. Diabetes Metab Res Rev. (2005) 21:44–50. doi: 10.1002/dmrr.479
82. Dobrzyn P, Pyrkowska A, Duda MK, Bednarski T, Maczewski M, Langfort J, et al. Expression of lipogenic genes is upregulated in the heart with exercise training-induced but not pressure overload-induced left ventricular hypertrophy. Am J Physiol Endocrinol Metab. (2013) 304:E1348–58. doi: 10.1152/ajpendo.00603.2012
83. Iemitsu M, Miyauchi T, Maeda S, Sakai S, Fujii N, Miyazaki H, et al. Cardiac hypertrophy by hypertension and exercise training exhibits different gene expression of enzymes in energy metabolism. Hypertens Res (2003) 26:829–37. doi: 10.1291/hypres.26.829
84. Terblanche SE, Gohil K, Packer L, Henderson S, Brooks GA. The effects of endurance training and exhaustive exercise on mitochondrial enzymes in tissues of the rat (Rattus norvegicus). Comp Biochem Physiol A Mol Integr Physiol. (2001) 128:889–96. doi: 10.1016/S1095-6433(00)00344-5
85. Strom CC, Aplin M, Ploug T, Christoffersen TE, Langfort J, Viese M, et al. Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. FEBS J. (2005) 272:2684–95. doi: 10.1111/j.1742-4658.2005.04684.x
86. Laursen PB, Jenkins DG. The scientific basis for high-intensity interval training: optimising training programmes and maximising performance in highly trained endurance athletes. Sports Med. (2002) 32:53–73. doi: 10.2165/00007256-200232010-00003
87. Gillen JB, Martin BJ, MacInnis MJ, Skelly LE, Tarnopolsky MA, Gibala MJ. Twelve weeks of sprint interval training improves indices of cardiometabolic health similar to traditional endurance training despite a five-fold lower exercise volume and time commitment. PLoS ONE (2016) 11:e0154075. doi: 10.1371/journal.pone.0154075
88. Whyte LJ, Ferguson C, Wilson J, Scott RA, Gill JM. Effects of single bout of very high-intensity exercise on metabolic health biomarkers in overweight/obese sedentary men. Metabolism (2013) 62:212–9. doi: 10.1016/j.metabol.2012.07.019
89. Ribeiro PA, Boidin M, Juneau M, Nigam A, Gayda M. High-intensity interval training in patients with coronary heart disease: prescription models and perspectives. Ann Phys Rehabil Med. (2017) 60:50–7. doi: 10.1016/j.rehab.2016.04.004
90. McGregor G, Nichols S, Hamborg T, Bryning L, Tudor-Edwards R, Markland D, et al. High-intensity interval training versus moderate-intensity steady-state training in UK cardiac rehabilitation programmes (HIIT or MISS UK): study protocol for a multicentre randomised controlled trial and economic evaluation. BMJ Open (2016) 6:e012843. doi: 10.1136/bmjopen-2016-012843
91. Ellingsen O, Halle M, Conraads V, Stoylen A, Dalen H, Delagardelle C, et al. High-intensity interval training in patients with heart failure with reduced ejection fraction. Circulation (2017) 135:839–49. doi: 10.1161/CIRCULATIONAHA.116.022924
92. Holloway TM, Bloemberg D, da Silva ML, Simpson JA, Quadrilatero J, Spriet LL. High intensity interval and endurance training have opposing effects on markers of heart failure and cardiac remodeling in hypertensive rats. PLoS ONE (2015) 10:e0121138. doi: 10.1371/journal.pone.0121138
93. Stuewe SR, Gwirtz PA, Agarwal N, Mallet RT. Exercise training enhances glycolytic and oxidative enzymes in canine ventricular myocardium. J Mol Cell Cardiol. (2000) 32:903–13. doi: 10.1006/jmcc.2000.1131
94. Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest. (2000) 105:1723–30. doi: 10.1172/JCI9056
95. Choi YS, de Mattos AB, Shao D, Li T, Nabben M, Kim M, et al. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J Mol Cell Cardiol. (2016) 100:64–71. doi: 10.1016/j.yjmcc.2016.09.001
96. O'Donnell JM, Fields AD, Sorokina N, Lewandowski ED. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J Mol Cell Cardiol. (2008) 44:315–22. doi: 10.1016/j.yjmcc.2007.11.006
97. Lahey R, Wang X, Carley AN, Lewandowski ED. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation (2014) 130:1790–9. doi: 10.1161/CIRCULATIONAHA.114.011687
98. Kolwicz SC Jr, Liu L, Goldberg IJ, Tian R. Enhancing cardiac triacylglycerol metabolism improves recovery from ischemic stress. Diabetes (2015) 64:2817–27. doi: 10.2337/db14-1943
99. Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, et al. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. (2009) 284:36312–23. doi: 10.1074/jbc.M109.049817
100. Liu L, Yu S, Khan RS, Homma S, Schulze PC, Blaner WS, et al. Diacylglycerol acyl transferase 1 overexpression detoxifies cardiac lipids in PPARgamma transgenic mice. J Lipid Res. (2012) 53:1482–92. doi: 10.1194/jlr.M024208
101. Alsted TJ, Nybo L, Schweiger M, Fledelius C, Jacobsen P, Zimmermann R, et al. Adipose triglyceride lipase in human skeletal muscle is upregulated by exercise training. Am J Physiol Endocrinol Metab. (2009) 296:E445–53. doi: 10.1152/ajpendo.90912.2008
102. Ikeda S, Miyazaki H, Nakatani T, Kai Y, Kamei Y, Miura S, et al. Up-regulation of SREBP-1c and lipogenic genes in skeletal muscles after exercise training. Biochem Biophys Res Commun. (2002) 296:395–400. doi: 10.1016/S0006-291X(02)00883-5
103. Hafstad AD, Boardman N, Aasum E. How exercise may amend metabolic disturbances in diabetic cardiomyopathy. Antioxid Redox Signal. (2015) 22:1587–605. doi: 10.1089/ars.2015.6304
104. Lachance D, Dhahri W, Drolet MC, Roussel E, Gascon S, Sarrhini O, et al. Endurance training or beta-blockade can partially block the energy metabolism remodeling taking place in experimental chronic left ventricle volume overload. BMC Cardiovasc Disord. (2014) 14:190. doi: 10.1186/1471-2261-14-190
105. Kemppainen J, Fujimoto T, Kalliokoski KK, Viljanen T, Nuutila P, Knuuti J. Myocardial and skeletal muscle glucose uptake during exercise in humans. J Physiol. (2002) 542(Pt 2):403–12. doi: 10.1113/jphysiol.2002.018135
106. Chatham JC, Gao ZP, Forder JR. Impact of 1 wk of diabetes on the regulation of myocardial carbohydrate and fatty acid oxidation. Am J Physiol. (1999). 277(2 Pt 1): E342–51.
107. Schonekess BO. Competition between lactate and fatty acids as sources of ATP in the isolated working rat heart. J Mol Cell Cardiol. (1997) 29:2725–33. doi: 10.1006/jmcc.1997.0504
108. Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. (1994) 267(2 Pt 2): H742–50. doi: 10.1152/ajpheart.1994.267.2.H742
109. Campbell B, Kreider RB, Ziegenfuss T, La Bounty P, Roberts M, Burke D, et al. (2007). International society of sports nutrition position stand: protein and exercise. J Int Soc Sports Nutr. 4:8. doi: 10.1186/1550-2783-4-8
110. Shah SH, Bain JR, Muehlbauer MJ, Stevens RD, Crosslin DR, Haynes C, et al. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ Cardiovasc Genet. (2010) 3:207–14. doi: 10.1161/CIRCGENETICS.109.852814
111. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation (2016) 133:2038–49. doi: 10.1161/CIRCULATIONAHA.115.020226
112. Wang W, Zhang F, Xia Y, Zhao S, Yan W, Wang H, et al. Defective branched chain amino acid catabolism contributes to cardiac dysfunction and remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol. (2016) 311:H1160–9. doi: 10.1152/ajpheart.00114.2016
113. Li T, Zhang Z, Kolwicz SC Jr, Abell L, Roe ND, Kim M, et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to Ischemia-reperfusion injury. Cell Metab. (2017) 25:374–85. doi: 10.1016/j.cmet.2016.11.005
114. Lopaschuk GD, Ussher JR. Evolving concepts of myocardial energy metabolism: more than just fats and carbohydrates. Circ Res. (2016) 119:1173–6. doi: 10.1161/CIRCRESAHA.116.310078
115. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The failing heart relies on ketone bodies as a fuel. Circulation (2016) 133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
116. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation (2016) 133:706–16. doi: 10.1161/CIRCULATIONAHA.115.017545
117. Kolwicz SC Jr, Airhart S, Tian R. Ketones step to the plate: a game changer for metabolic remodeling in heart failure? Circulation (2016) 133:689–91. doi: 10.1161/CIRCULATIONAHA.116.021230
118. Mizuno Y, Harada E, Nakagawa H, Morikawa Y, Shono M, Kugimiya F, et al. The diabetic heart utilizes ketone bodies as an energy source. Metabolism (2017) 77:65–72. doi: 10.1016/j.metabol.2017.08.005
119. Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. (2013) 304:H1060–76. doi: 10.1152/ajpheart.00646.2012
120. Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. (2016) 24:256–68. doi: 10.1016/j.cmet.2016.07.010
121. Murray AJ, Knight NS, Cole MA, Cochlin LE, Carter E, Tchabanenko K, et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. (2016) 30:4021–32. doi: 10.1096/fj.201600773R
122. Winder WW, Baldwin KM, Holloszy JO. Enzymes involved in ketone utilization in different types of muscle: adaptation to exercise. Eur J Biochem. (1974) 47:461–7
123. Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation (2009) 119:2818–28. doi: 10.1161/CIRCULATIONAHA.108.832915
124. Evans M, Cogan KE, Egan B. Metabolism of ketone bodies during exercise and training: physiological basis for exogenous supplementation. J Physiol. (2017) 595:2857–71. doi: 10.1113/JP273185
125. Hallal PC, Andersen LB, Bull FC, Guthold R, Haskell W, Ekelund U, et al. Global physical activity levels: surveillance progress, pitfalls, and prospects. Lancet (2012) 380:247–57. doi: 10.1016/S0140-6736(12)60646-1
126. Kregel KC, Allen DL, Booth FW, Fleshner MR, Henrikson EJ, Musch TI, et al. Exercise Protocols Using Rats and Mice. Resource Book for the Design of Animal Exercise Protocols. Bethesda, MD: American Physiological Society (2006).
127. Knab AM, Bowen RS, Moore-Harrison T, Hamilton AT, Turner MJ, Lightfoot JT. Repeatability of exercise behaviors in mice. Physiol Behav. (2009) 98:433–40. doi: 10.1016/j.physbeh.2009.07.006
128. Goh J, Ladiges W. Voluntary wheel running in mice. Curr Protoc Mouse Biol. (2015) 5:283–90. doi: 10.1002/9780470942390.mo140295
129. Billat VL, Mouisel E, Roblot N, Melki J. Inter- and intrastrain variation in mouse critical running speed. J Appl Physiol. (2005)98:1258–63. doi: 10.1152/japplphysiol.00991.2004
130. Gibb AA, McNally LA, Riggs DW, Conklin DJ, Bhatnagar A, Hill BG. FVB/NJ mice are a useful model for examining cardiac adaptations to treadmill exercise. Front Physiol. (2016) 7:636. doi: 10.3389/fphys.2016.00636
131. Kolwicz SC, MacDonnell SM, Renna BF, Reger PO, Seqqat R, Rafiq K, et al. Left ventricular remodeling with exercise in hypertension. Am J Physiol Heart Circ Physiol. (2009) 297:H1361–8. doi: 10.1152/ajpheart.01253.2008
132. Vigelso A, Andersen NB, Dela F. The relationship between skeletal muscle mitochondrial citrate synthase activity and whole body oxygen uptake adaptations in response to exercise training. Int J Physiol Pathophysiol Pharmacol. (2014) 6:84–101.
133. Nguyen S, Shao D, Tomasi LC, Braun A, de Mattos ABM, Choi YS, et al. The effects of fatty acid composition on cardiac hypertrophy and function in mouse models of diet-induced obesity. J Nutr Biochem. (2017) 46:137–42. doi: 10.1016/j.jnutbio.2017.05.009
134. Garcia-Menendez L, Karamanlidis G, Kolwicz S, Tian R. Substrain specific response to cardiac pressure overload in C57BL/6 mice. Am J Physiol Heart Circ Physiol. (2013) 305:H397–402. doi: 10.1152/ajpheart.00088.2013
135. Avila JJ, Kim SK, Massett MP. Differences in Exercise Capacity and Responses to Training in 24 Inbred Mouse Strains. Front Physiol. (2017) 8:974. doi: 10.3389/fphys.2017.00974
136. Massett MP, Berk BC. Strain-dependent differences in responses to exercise training in inbred and hybrid mice. Am J Physiol Regul Integr Comp Physiol. (2005) 288:R1006–13. doi: 10.1152/ajpregu.00476.2004
137. Dworatzek E, Mahmoodzadeh S, Schubert C, Westphal C, Leber J, Kusch A, et al. Sex differences in exercise-induced physiological myocardial hypertrophy are modulated by oestrogen receptor beta. Cardiovasc Res. (2014) 102:418–28. doi: 10.1093/cvr/cvu065
138. Konhilas JP, Maass AH, Luckey SW, Stauffer BL, Olson EN, Leinwand LA. Sex modifies exercise and cardiac adaptation in mice. Am J Physiol Heart Circ Physiol. (2004) 287:H2768–76. doi: 10.1152/ajpheart.00292.2004
139. McMullan RC, Kelly SA, Hua K, Buckley BK, Faber JE, Pardo-Manuel de Villena F, et al. Long-term exercise in mice has sex-dependent benefits on body composition and metabolism during aging. Physiol Rep. (2016) 4:e13011. doi: 10.14814/phy2.13011
140. Abdurrachim D, Luiken JJ, Nicolay K, Glatz JF, Prompers JJ, Nabben M. Good and bad consequences of altered fatty acid metabolism in heart failure: evidence from mouse models. Cardiovasc Res. (2015) 106:194–205. doi: 10.1093/cvr/cvv105
Keywords: exercise training, lipid metabolism, metabolic remodeling, fatty acid oxidation, exercise adaptation, heart failure, cardiac hypertrophy
Citation: Kolwicz SC Jr (2018) An “Exercise” in Cardiac Metabolism. Front. Cardiovasc. Med. 5:66. doi: 10.3389/fcvm.2018.00066
Received: 16 March 2018; Accepted: 17 May 2018;
Published: 07 June 2018.
Edited by:
Thomas Pulinilkunnil, Dalhousie University, CanadaReviewed by:
Martin Sénéchal, University of New Brunswick Fredericton, CanadaSuowen Xu, University of Rochester, United States
Jan F. C. Glatz, Maastricht University, Netherlands
Copyright © 2018 Kolwicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen C. Kolwicz Jr, c2tvbHdpY3pAdXJzaW51cy5lZHU=